
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceV-shaped pyranylidene/triphenylamine-based chromophores with enhanced photophysical, electrochemical and nonlinear optical properties†
Sergio
Gámez-Valenzuela
a,
David
Neusser
b,
Carlos
Benitez-Martin
 cd,
Francisco
Najera
cd,
Juan A.
Guadix
de,
Carlos
Moreno-Yruela
fg,
Belén
Villacampa
h,
Rocío
Ponce Ortiz
a,
Sabine
Ludwigs
b,
Raquel
Andreu
*f and
M. Carmen
Ruiz Delgado
*a
cd,
Francisco
Najera
cd,
Juan A.
Guadix
de,
Carlos
Moreno-Yruela
fg,
Belén
Villacampa
h,
Rocío
Ponce Ortiz
a,
Sabine
Ludwigs
b,
Raquel
Andreu
*f and
M. Carmen
Ruiz Delgado
*a
aDepartment of Physical Chemistry, University of Malaga, Campus de Teatinos s/n, 29071, Malaga, Spain. E-mail: carmenrd@uma.es
bIPOC – Functional Polymers, Institute of Polymer Chemistry, University of Stuttgart, Pfaffenwaldring 55, D-70569 Stuttgart, Germany
cDepartamento de Química Orgánica, Universidad de Málaga-IBIMA, Campus de Teatinos s/n, Málaga 29071, Spain
dCentro Andaluz de Nanomedicina y Biotecnología-BIONAND, Parque Tecnológico de Andalucía, c/Severo Ochoa, 35, 29590 Campanillas, Málaga 29071, Spain
eDepartamento de Biología Animal, Facultad de Ciencias, Universidad de Málaga-IBIMA, Campus de Teatinos s/n, Málaga 29071, Spain
fInstituto de Nanociencia y Materiales de Aragón (INMA)-Departamento de Química Orgánica, CSIC-Universidad de Zaragoza, Zaragoza 50009, Spain. E-mail: randreu@unizar.es
gCenter for Biopharmaceuticals & Department of Drug Design and Pharmacology, Faculty of Health and Medical Sciences, University of Copenhagen, Universitetsparken 2, DK-2100 Copenhagen, Denmark
hInstituto de Nanociencia y Materiales de Aragón (INMA)-Departamento de Física de la Materia Condensada, CSIC-Universidad de Zaragoza, Zaragoza 50009, Spain
First published on 25th May 2021
Abstract
We report the synthesis and comprehensive study of two chromophores based on 4H-pyranylidene moiety as a part of the π-conjugated spacer. Triphenylamine (TPA) acts as donor and tricarbonitrile-based electron-accepting groups complete these V-shaped D–A–D architectures (A, acceptor; D, donor). Their electrochemical, photophysical and nonlinear optical properties are analyzed in detail by using a joint experimental and theoretical approach. The two chromophores exhibit near-infrared fluorescence, large Stokes shift, enhanced emission in tetrahydrofuran/water mixtures and good photostability. Additionally, the dimerization of triphenylamine groups to tetraphenylbenzidine (TPB) takes place upon electrochemical and chemical oxidation showing their peculiar electrochemical behavior and film formation capabilities. Interestingly, high molecular first hyperpolarizabilities and two-photon absorption cross-sections were found, highlighting their potential applications in electro-optical devices. Overall, our work demonstrates that these near-infrared (NIR) fluorescent chromophores are versatile materials with a myriad of applications ranging from optoelectronics to biological applications.
Introduction
Organic push–pull molecules represent a subclass of π-conjugated systems that are end-capped with electron donor(s) and electron acceptor(s). The D–π–A arrangement allows the direct interaction of the donor (D) and the acceptor (A) through the π-system, so-called intramolecular charge transfer (ICT). Their versatility and well-defined and tunable structures, which give rise to predictable properties, represent a main advantage keeping them into focus for different applications: nonlinear optics (NLO),1 dye-sensitized solar cells (DSSCs),2 bulk-heterojunction solar cells (BHJSCs),3 organic light-emitting diodes (OLEDs),4 two-photon absorbers,5 or near-infrared absorbing dyes (NIR).6In addition to the ordinary linear D–π–A systems, push–pull chromophores may also adopt quadrupolar (D–π–A–π–D or A–π–D–π–A), tripodal (D–(π–A)3 or A–(π–D)3) or even more extraordinary arrangements, which may be pictured as uppercase letters (molecules with shape similar to H, V, X or Y).7 As far as the field of NLO is concerned, non-centrosymmetry is an imperative requirement for applications based on second order processes.8 On the other hand, for two-photon absorption (2PA) (a third order process) a high degree of π conjugation is desirable. Following the arrangements above mentioned, branching donors and acceptors to a π-conjugated backbone contributes in a great extent to enhance the 2PA cross-sections. Thus, D–π–D, A–π–A, D–A–D or A–D–A architectures are pointed as ideal platforms for developing two-photon absorption active compounds.5
Triphenylamine (TPA) and 4H-pyranylidene are building blocks often used in the design of D–π–A systems in different research areas. Thus, the use of TPA is based on its strong electron donor character and excellent transporting capabilities, together with its special propeller-like structure.9 Its nonplanar conformation is beneficial for restraining intermolecular aggregation as well as for obtaining amorphous materials. Its electrochemical behavior has been extensively studied due to the dimerization ability as a result of oxidative coupling,10–13 which is also observed in polymer systems.14 TPA derivatives have been investigated and applied in a variety of areas, such as organic photovoltaics,11,15 DSSC and perovskite solar cells,15 2PA16–18 materials or OLEDs.19
On the other hand, 4H-pyranylidene can act as a proaromatic donor unit,20 but also as a part of the π-bridge of a push–pull system, with one C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of the spacer in the ring, and opening the possibility of introducing strong acceptor groups on its 4 position. With this architecture, materials with interesting properties, such as NLO activity21 or good performance as dyes in DSSCs have been developed.22
C bond of the spacer in the ring, and opening the possibility of introducing strong acceptor groups on its 4 position. With this architecture, materials with interesting properties, such as NLO activity21 or good performance as dyes in DSSCs have been developed.22
Furthermore, both entities (TPA and 4H-pyranylidene) have demonstrated to lead to derivatives with large fluorescence yields.19,23 This fact, together with the possibility of becoming highly emissive upon aggregation in poor solvents24 and the achievement of large two photon-absorption cross sections17,25–27 make these type of compounds good candidates for the development of highly efficient probes for biological imaging.25,28,29
Within this context, we present a comprenhensive study of two V-shaped D–A–D systems, which have been designed according to this approach: (i) the propeller-like TPA is the donor moiety; (ii) 4H-pyranylidene acts as a part of the π-conjugated bridge; (iii) strong polycyano acceptors like 1,1,3-tricyano-2-phenylpropene30 and 2-dicyanomethylene-3-cyano-4,5,5-trimethyl-2,5-dihydrofuran (TCF)31 are attached to the pyran unit (4 position) giving rise to chromophores 1 and 2, respectively (Fig. 1). The electronic and steric effect of cyano groups is also important in the development of systems with great color tunability and enhanced emission upon aggregation.24
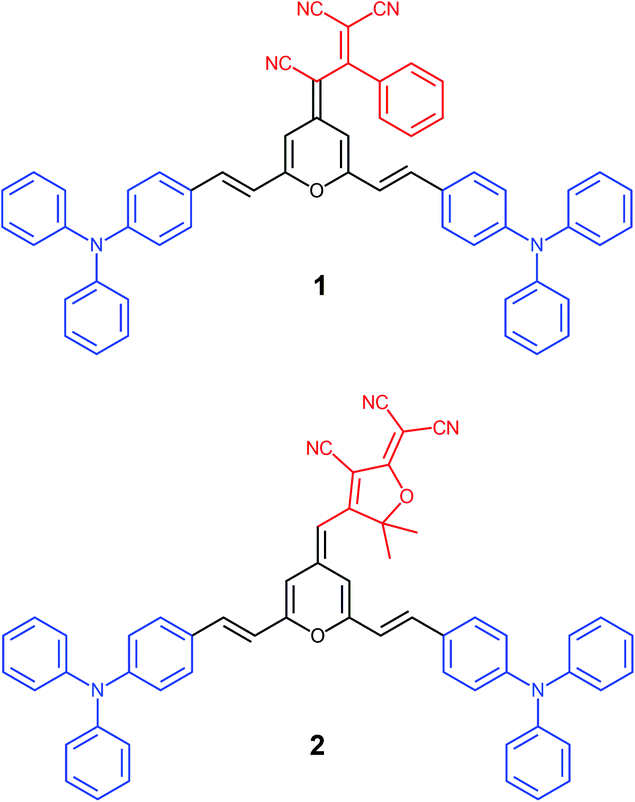 | ||
| Fig. 1 Chemical structures of the two chromophores under study. | ||
The excellent potential of these two conjugated systems in different fields will be studied by means of a complementary experimental and theoretical approach. On the one hand, the prominent ICT from the external branched units to the central acceptor group are expected to provide both second-order NLO and suitable two-photon absorption (2PA) properties; on the other hand, due to the presence of two fluorescent moieties, they will be studied as luminescent materials, paying particular attention to the effect of biological-compatible aqueous solutions on luminescence. Their photostability together with the 2PA fluorescence cell imaging have been investigated. Eventually, and having in mind the electrochemical behavior of TPA derivatives, an extensive study of this aspect has been carried out.
Results and discussion
1. Synthesis
Target conjugated systems 1 and 2 were prepared by Knœvenagel-type condensation between the commercially available p-diphenylaminobenzaldehyde (3) (2 equiv.) and the 2,6-dimethyl-4H-pyran-4-ylidene derivatives 432 and 532 respectively (Scheme 1). Compound 1 was prepared slightly modifying the reaction conditions and purification previously described.29 In addition to the described characterization, mp, IR and elemental analysis have been also included (see ESI†). The attainment of small amounts of the corresponding mono-substituted derivatives and subsequent repeated chromatographic purification on silica gel (for compound 2) contribute to explain the fact that chromophores were obtained in low-moderate yields. The analysis of the 3JHH coupling constants allows us to infer that the formed CHCH bond has an E configuration. It is noteworthy to remark that signals corresponding to aromatic protons are overlapped for both compounds.
 | ||
| Scheme 1 Synthesis of chromophores 1 and 2. | ||
2. Structural features
Fig. 2 displays the top and lateral views of the DFT-optimized ground-state structures for 1 and 2. As we observe, the outermost phenyl rings of the TPA units are largely distorted by ∼45° from the rest of the π-conjugated backbone which shows an almost coplanar arrangement; note that very small dihedral angles are predicted between the vinyl-pyran unit and the adjacent phenyl ring, with θ3, θ4 and θ3′, θ4′ values of ∼0°–5°. However, when we focus on the distinctive chemical electron-acceptor units, notable geometrical differences are found: (i) in compound 1, the phenyl and external dicyanovinyl group of the acceptor moiety is appreciably distorted from planarity (with θ5 and θ6 values of 45° and 32°, respectively), in good agreement with X-ray structures of other push–pull systems bearing the same acceptor,29,32 and (ii) the TCF acceptor unit of 2 shows an almost planar disposition with respect to the pyran unit.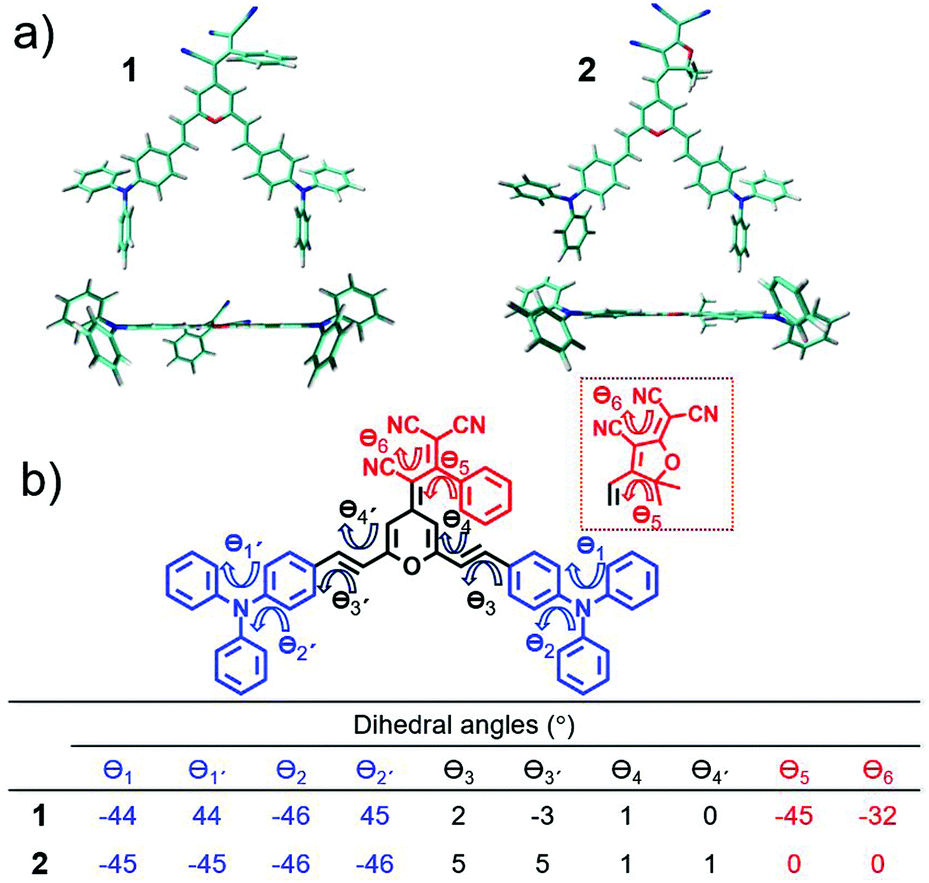 | ||
| Fig. 2 (a) Top and lateral views of the DFT-optimized structures for 1 and 2 systems at the PCM-M06-2X/6-31G** level using CH2Cl2 as solvent. (b) DFT-calculated intering dihedral angles (θ) along the conjugated backbones of 1 and 2 using the same level of theory. | ||
We now make use of the Mülliken atomic charges and bond length alternation (BLA) values that allow for a deeper understanding of the ground-state polarization of the chromophores. Note that the BLA values are related to the degree of bond length alternation, δr, which equals 0 for benzene while values ∼0.10 are found in fully quinoid rings.33 As seen in Fig. 3, the two chromophores exhibit a decreased degree of bond length alternation for pyran (δr = 0.065 for 1 and δr = 0.069 for 2) and a lengthening of the pyran exocyclic C4–Cexo bond length (1.413 Å for 1 and 1.403 Å for 2) when compared to that reported for a fully quinoidal 2,2,6,6-tetraphenylbipyranylidene (with δr = 0.11 and C4–Cexo bond length of 1.385 Å)34 together with less alternated internal phenyl rings when compared to the external ones. These structural parameters reflect a degree of mixing between the neutral form and the charge-separated zwitterionic one with a more (less) aromatized pyran spacer (internal phenyl rings). As seen in Fig. 3, Mülliken population analysis confirms these trends indicating a negative charge over the acceptor group (−0.569 e in 1 and −0.534 e in 2) which is around 2 times higher than the sum of the positive charge over the two TPA donor groups (+0.260 e in 1 and +0.258 e in 2), while the π-spacer is largely polarized bearing around 70% (+0.310 e in 1 and +0.276 e in 2) of the total positive charge. Moreover, it is worth highlighting that the positive charge located in the pyran ring (together with vinyl unit) is of the same order35 or even larger36,37 to those encountered for other D–π–A derivatives with the 4H-pyranylidene as main donor. Thus, these data account for a significant polarization of the π-conjugated structure in these V-shaped chromophores.
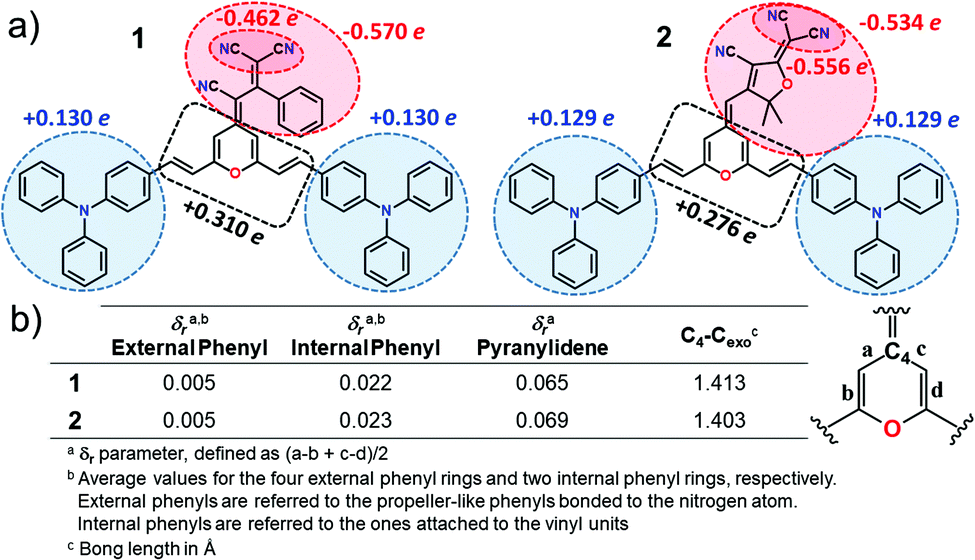 | ||
| Fig. 3 (a) Mülliken atomic charges on various molecular domains and (b) structural parameters for chromophores 1 and 2 calculated at the PCM-M06-2X/6-31G** level using CH2Cl2 as solvent. | ||
IR and Raman spectra can provide useful information about the degree of charge polarization in the ground state.38–40 In this sense, the frequency of the ν(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) is very sensitive to the total charge borne by the nitrile groups, downshifting upon ground-state polarization. Indeed, taken the nonconjugated malononitrile (CH2(CN)2) as reference, for which the ν(CN) band is recorded at 2270 cm−1, the low frequency values for the corresponding IR absorption in our chromophores (at 2213 and 2188 cm−1 for 1 and 2217 and 2206 cm−1 for 2, see Fig. 4a) reveal an intramolecular charge-transfer (ICT) from the TPA groups towards the central electron-withdrawing moiety.41–43 On the other hand, it is interesting to note that the Raman bands in the 1640–1590 cm−1 region, which corresponds to ν(CC/C–C) stretching of the π-conjugated spacer (vinyl and pyran) and the TPA donor groups, slightly changes on going from 1 to 2 (Fig. 4b and Fig. S5, S6, ESI†). The downshift of the ν(CC) mode in the pyran moiety when going from 2 to 1 (from 1639 to 1632 cm−1) can be ascribed to the different electronic nature of the group directly linked to the pyran exocyclic bond (a CH group in 2 and a C(CN) group in 1) which results in a more positive charge localization over the pyran unit in 1 than in 2 as seen in Fig. 3a. However, a shift towards lower frequencies of the ν(CC) stretching on the vinyl groups (from 1622 in 1 to 1619 cm−1 in 2) and a larger negative charge on the peripheral C(CN)2 groups (−0.462 e in 1 to −0.556 e in 2) is found when comparing 1 with 2. Theoretical IR and Raman spectra are in good agreement with the experimental results (Fig. S3–S6, ESI†) and the vibrational eigenvectors nicely support the assignments. Overall, these results reveal similar ground-state polarization in both systems with a slightly larger contribution of the charge-separated zwitterionic form in 2 due to the insertion of the TCF acceptor.
N) is very sensitive to the total charge borne by the nitrile groups, downshifting upon ground-state polarization. Indeed, taken the nonconjugated malononitrile (CH2(CN)2) as reference, for which the ν(CN) band is recorded at 2270 cm−1, the low frequency values for the corresponding IR absorption in our chromophores (at 2213 and 2188 cm−1 for 1 and 2217 and 2206 cm−1 for 2, see Fig. 4a) reveal an intramolecular charge-transfer (ICT) from the TPA groups towards the central electron-withdrawing moiety.41–43 On the other hand, it is interesting to note that the Raman bands in the 1640–1590 cm−1 region, which corresponds to ν(CC/C–C) stretching of the π-conjugated spacer (vinyl and pyran) and the TPA donor groups, slightly changes on going from 1 to 2 (Fig. 4b and Fig. S5, S6, ESI†). The downshift of the ν(CC) mode in the pyran moiety when going from 2 to 1 (from 1639 to 1632 cm−1) can be ascribed to the different electronic nature of the group directly linked to the pyran exocyclic bond (a CH group in 2 and a C(CN) group in 1) which results in a more positive charge localization over the pyran unit in 1 than in 2 as seen in Fig. 3a. However, a shift towards lower frequencies of the ν(CC) stretching on the vinyl groups (from 1622 in 1 to 1619 cm−1 in 2) and a larger negative charge on the peripheral C(CN)2 groups (−0.462 e in 1 to −0.556 e in 2) is found when comparing 1 with 2. Theoretical IR and Raman spectra are in good agreement with the experimental results (Fig. S3–S6, ESI†) and the vibrational eigenvectors nicely support the assignments. Overall, these results reveal similar ground-state polarization in both systems with a slightly larger contribution of the charge-separated zwitterionic form in 2 due to the insertion of the TCF acceptor.
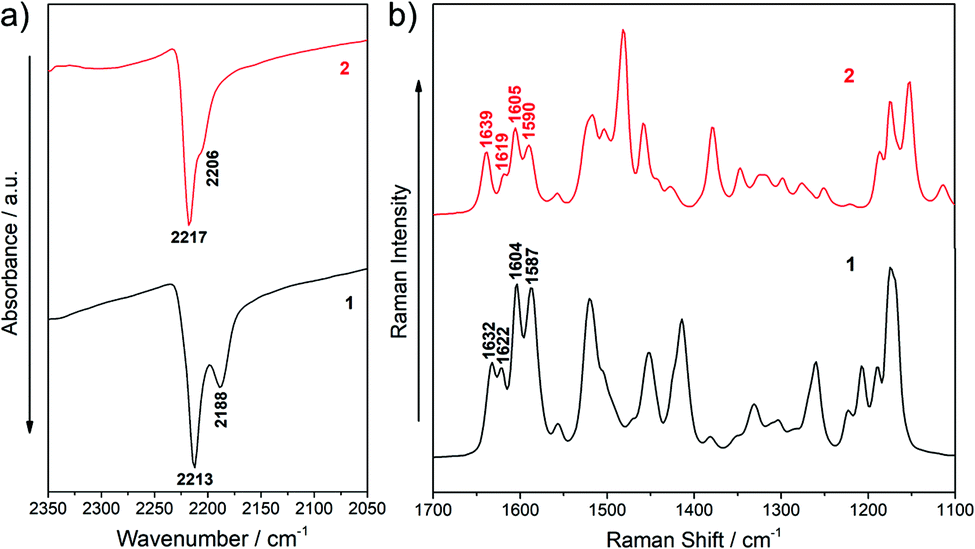 | ||
| Fig. 4 (a) IR and (b) Raman spectra of chromophores 1 and 2. | ||
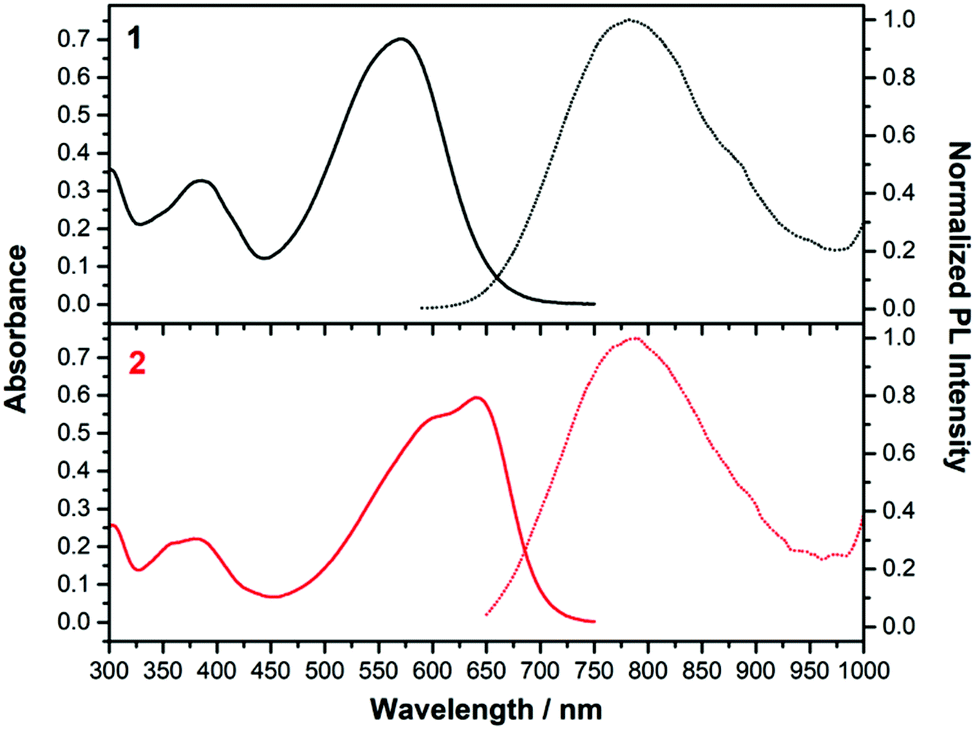 | ||
| Fig. 5 Absorption (left, solid line) and emission (right, dashed line) spectra of 1 (black) and 2 (red) in CH2Cl2. | ||
3. Photophysical properties
Fig. 5 displays the absorption and emission spectra of 1 and 2 in CH2Cl2, and Table 1 collects the main photophysical parameters. The absorption spectra show a broad and strong band in the Vis-NIR spectral region which is red-shifted when going from 1 to 2 (570 nm for 1 and 641 nm for 2). As well, emission bands are centered ca. 800 nm for both compounds in the considered solvent.According to TD-DFT calculations, the lowest-energy absorption band results from the overlap of the S0 → S1 and S0 → S2 electronic transitions, which are assigned to different combinations of HOMO → LUMO and HOMO−1 → LUMO one-electron excitations (see Fig. 6); this is in accordance with the structured two-peak profile of the absorption band in 2. Due to the nearly symmetric structures of these D–A–D systems, HOMO and HOMO−1 energy levels are very close in energy, being delocalized over the branched units (i.e., TPA and vinyl units) while LUMO is delocalized over the π-spacer (i.e., pyran and vinyl units) and the acceptor group. Therefore, the S0 → S1 and S0 → S2 electronic transitions imply an electron density redistribution from the peripheral TPA donor units towards the central acceptor group, thus displaying ICT character. Interestingly, the insertion of a stronger acceptor group in 2 causes a lower energy LUMO while the HOMO is slightly affected, thus decreasing the HOMO–LUMO gap, in agreement with the trends shown by the optical gaps (Eg) in Table 1, and with electrochemical data (see Section 4).
 | ||
| Fig. 6 DFT-calculated frontier molecular orbital energies and topologies for 1 and 2, at the PCM-M06-2X/6-31G** level using CH2Cl2 as solvent. The S0 → S1 and S0 → S2 electronic transitions are also shown as solid and dashed black arrows, respectively. | ||
| Compound | Solvent | λ abs (nm) | λ em (nm) | E g,op (eV) | λ em − λabs (nm) | Stokes shiftd (cm−1) | Φ |
|---|---|---|---|---|---|---|---|
| a Lowest-energy electronic absorption maxima. b Emission fluorescence maxima. c Optical band gap estimated from the tangent to the low energy edge of the absorption band. d Stokes shift as the difference between the wavelength of the emission and absorption maxima. e Fluorescence emission quantum yield, 15% error. | |||||||
| 1 | CH2Cl2 | 570 | 782 | 1.90 | 212 | 4756 | 0.035 |
| Toluene | 545 | 665 | 1.94 | 120 | 3311 | 0.009 | |
| 2 | CH2Cl2 | 641 | 784 | 1.77 | 143 | 2846 | 0.008 |
| Toluene | 594 | 735 | 1.76 | 141 | 3230 | 0.002 | |
Significant Stokes shifts were observed for both chromophores. The differences between the emission fluorescence maxima (λem) and the lowest-energy electronic absorption maxima (λmax) are 4756 and 2486 cm−1 in CH2Cl2 for 1 and 2 respectively. As predicted by TD-DFT (Fig. S8, ESI†), this can be associated to the significant geometrical changes found upon S1 → S0 transition due to the twisted TPA units. This effect is even more pronounced in 1 due to the distorted acceptor group, and a larger Stokes shift is found accordingly.
Furthermore, the absorption and fluorescence emission spectra were studied in different solvents (Fig. S9, ESI†). Both absorption and emission spectra display a marked redshift in parallel to the increase of the solvent polarity, with a more pronounced effect in emission. It is worth noting that large Stokes shifts were appreciated for both compounds regardless of the considered solvent. Treatment of the data according to the Lippert–Mataga equation44–46 yielded a dipole moment change (Δμ) between the ground and the excited state of 19.6 and 15.2 D for 1 and 2 respectively. This is in accordance with the higher dipolar moment calculated in the excited state when compared to the ground state (Table S1, ESI†) and with the stronger positive solvatochromism observed in emission when compared to absorption. This solvent-dependent behaviour denotes the presence of an ICT in these chromophores, and is more outstanding in 2 in line with its more polarized structure; this is in very good agreement with the Raman data and DFT-calculated structural features.
Remarkable photostability in CH2Cl2 aerated solutions under the irradiation of UV light was determined for 1 and 2 (Fig. S10, ESI†).47 In addition, the emission spectra of these chromophores were also examined in different tetrahydrofuran/water mixtures (Fig. S11, ESI†). As the water fraction is higher, a notable increment in the fluorescence intensity was observed for both dyes. This fluorescence enhancement could be ascribed to a combination of intra- and intermolecular effects, since the twisted TPA moieties might restrict the face-to-face intermolecular interactions in the aggregate state favoring the radiative deactivation pathway. These joint features (being dispersable in aqueous media and remaining fluorescent, large Stokes shifts and suitable photostability) suggest potential application of these derivatives in biological fields. Indeed, the utility of compound 1 for confocal fluorescence bioimaging (1P microscopy) in a highly proliferative cell line has been previously reported.29
4. Electrochemical studies and spectroelectrochemical Properties
In order to experimentally determine the HOMO and LUMO levels of the chromophores, cyclic voltammetry measurements in CH2Cl2 were performed. Similar oxidation potentials with slightly lower reduction potentials are found in 2 with respect to 1, resulting in similar HOMO levels (−5.64 eV for 1 and −5.65 eV for 2) and slightly different LUMO levels (−3.88 eV for 1 and −3.93 eV for 2). This trend is in very good accordance with the DFT-calculated HOMO and LUMO energy levels (see Fig. 6). The unique V-shape structure of the chromophores 1 and 2 containing a central acceptor unit surrounded by two TPA donor units also gives rise to interesting properties regarding intermolecular oxidative coupling reactions. The bridging between TPA units is explored now via either electrochemical or chemical doping approaches. We focus here on the first approach, for the second one see Supporting Information for more details. Both chromophores were electrochemically deposited onto indium tin oxide (ITO) substrates from solutions containing the monomers in CH2Cl2. The corresponding cyclic voltammograms recorded during electrodeposition are shown in Fig. 7a and b. The obtained CV data in combination with an observed deposition of material on the working electrode suggests an electrochemical polymerization process between the TPA units of the chromophores.11,13,14 The increasing peak currents for each consecutive cycle can be explained by the increasing amount of redox active material on the electrode. | ||
| Fig. 7 (a) and (b) Electrochemical oxidative coupling and deposition of 1 and 2via multiple cycling on ITO substrates (0.1 M TBAPF6 in CH2Cl2, 20 mV s−1, C(monomer) = 0.5 × 10−4 mol L−1) to generate cl-1 and cl-2. (c) to (f) In situ spectroelectrochemistry of electrochemically polymerized films on ITO substrates in monomer free solution (0.1 M TBAPF6 in MeCN, 20 mV s−1) of cl-1 (c) and cl-2 (d). UV-Vis-NIR absorption spectra during charging cl-1 (e) and cl-2 (f), while in (c) and (d) the corresponding evolutions of characteristic absorption bands are given as function of potential. Measurements are performed in a three-electrode setup using Pt as counter electrode and Ag/AgCl as reference electrode. | ||
Fig. 7c–f present the results of spectroelectrochemical measurements of electrochemically polymerized films of 1 and 2, abbreviated as cl-1 and cl-2 in the following in monomer free electrolyte. Both polymer films show fully reversible and stable CVs (Fig. 7c and d). cl-2 has a higher onset of oxidation when compared to cl-1, which can be explained by the stronger electron acceptor group in molecule 2. While the electrochemical polymerization was performed in CH2Cl2 to ensure good solubility of the pure chromophore in the electrolyte, the spectroelectrochemical characterization in monomer free solution displayed in Fig. 7c–f was done in MeCN; thus, the reason for additional features in the CV of cl-2 in MeCN when compared to that recorded in CH2Cl2 might be ascribed to the different polarity of the used electrolytes which can lead to peak splitting in CV.48,49 Moreover, the individual peaks could also be interpreted in the context of partially interacting redox sites as a consequence to the unique shape of the chromophores having two identical redox groups on each side of the molecule.50,51 The interaction can cause slight shifts in the oxidation potentials of the individual redox sites, which might interact over the conjugated bridging acceptor unit in the center of the molecular structure.
Fig. 7c and d further display the evolution of absorption intensity of three distinct wavelengths sampled in the spectroelectrochemical experiment. According to literature11,13,14 the blue band at around 350 nm can be assigned to a neutral tetraphenylbenzidine (TPB) dimer and the red band at wavelengths above 700 nm to the dication of the TPB dimer. The exact identification of the intermediate radical cation species is difficult since an overlap of absorption signals with the dication complicates a clear assignment for these species. Still, the band at 456 nm (cl-1) and 479 nm (cl-2) most likely belong to the intermediate radical cation species and are also included in green in the peak evolution graphs. Note that this is also in good agreement with the evolution of the TD-DFT calculated absorption spectra for TPB dimers upon oxidation (Fig. S13, ESI†). All characteristic absorption bands are summarized in Table 2.
| λ neutral (nm) | λ radical cation (nm) | λ dication (nm) | |
|---|---|---|---|
| cl-1 | 346 | 325, 456 | 322, 450, 704 |
| cl-2 | 352 | 343, 479 | 325, 718 |
At the starting point of the CVs, the neutral band is at maximum absorption confirming the polymer films are in their neutral state (Fig. 7e and f). With increasing electrochemical potential, the neutral band decreases in favor of the green and at higher potentials in favor of the red (dication) band. Following this observation, the films are being transferred from neutral state into the dication passing through an intermediate state herein referred to as radical cation. This process is fully reversed during the reverse scan of the cycle. Still, remaining absorption intensity in the neutral state at wavelengths of 576 nm (cl-1) and 663 nm (cl-2) at the position of each of the chromophores charge transfer bands indicates that there might be still TPA monomers present in the films (Fig. S13, ESI†). Nevertheless, the spectroelectrochemical measurements prove a successful electrochemical polymerization of both chromophores from solution and help to identify the charged species of the produced TPB dimer units inside the films.
5. Nonlinear optical properties
We now investigate both experimentally and theoretically the molecular second-order NLO response (μβ) and the two-photon absorption (2PA) properties of chromophores 1 and 2 in order to establish structure-NLO properties relationships. Finally, we demonstrate the application of these V-shaped chromophores in 2P-excited fluorescence bioimaging (i.e., under two-photon excitation conditions).5.1 Molecular hyperpolarizabilty
Table 3 collects the second-order NLO properties (i.e., μβ values) of chromophores 1 and 2 measured by electric field-induced second harmonic generation (EFISHG) at 1907 nm in CH2Cl2 solution, together with the DFT-calculated data. Zero frequency μβ0 values have been extrapolated by means of the two level model,52 considering the lowest-energy electronic absorption maxima in CH2Cl2 solution (see Table 1). Different electronic transitions contribute to the observed absorption bands, therefore μβ0 values thus estimated should be treated with caution. For the sake of comparison, Disperse Red 1 (DR1), a common benchmark for organic NLO chromophores has been measured under the same experimental conditions as for 1 and 2. The obtained μβ0 value of ca. 510 × 10−48 esu is quite close to the value reported for DR1 by Dirk et al.53| Compound | Experimental | Theoretical | |
|---|---|---|---|
| μβ (10−48 esu) | μβ 0 (10−48 esu) | μβ 0 (10−48 esu) | |
| a Measured by EFISH in CH2Cl2 at 1.9 μm (experimental uncertainty less than ±15%). b Experimental μβ0 values calculated using the two level model. c Calculated at the M06-2X/6-31G** level in CH2Cl2. | |||
| 1 | 2550 | 1490 | 4871 |
| 2 | 3500 | 1710 | 5628 |
The fact that our chromophores exhibit positive μβ0 values reflects that the contribution of the charge-separated zwitterionic form is higher in the excited state than in the ground state, thus resulting in an increased dipole moment change upon excitation as predicted by TD-DFT calculations (Table S1, ESI†); this is in accordance with the positive solvatochromic behaviour for both chromophores discussed above. Interestingly, compound 2 exhibits a higher μβ0 value than 1 in consonance with the more polarized structure and more efficient ICT character upon the insertion of the stronger electron acceptor TCF group. While theoretical results overestimate the experimental values, they reproduce the observed trends. Finally, it could also be instructive to compare the NLO properties of the compounds herein reported to those of related derivatives, although they are somewhat restricted by the different experimental setups used in the measurements. With these strong polycyano acceptors, only diethylaminophenyl analogues32 are described, and interestingly, the measured μβ0 value of 2 is larger than that reported for the equivalent.32 Concerning the influence of the cyano-acceptor on position 4 of the 4H-pyranylidene, compounds 1 and 2 present a higher NLO activity that the analogue with a dicyanovinylene end,54 due to the more efficient electron-withdrawing character for the acceptors used here. On the other hand, chromophores featuring quite different acceptor/donor moieties have been reported with lower21 or higher μβ0 values.55
5.2 Two photon-absorption properties
The 2PA cross-sections of 1 and 2 were investigated between 700 and 1000 nm by the two-photon-excited fluorescence method,56,57 and are represented in Fig. 8. Accounting the photophysical properties and the prior TD-DFT calculations (Fig. S9 and S17, ESI†), in this spectral region the transitions to the first two excited states can not be observed. Instead, higher excited states are reached upon the biphotonic excitation (see Fig. S19, ESI† for comparison between 1PA and 2PA spectra). Compound 2, having a more prominent ICT character, shows significant 2PA cross section values of up to 326 and 203 GM at 790 and 890 nm, respectively. Furthermore, 2PA properties are even interesting in wavelengths longer than 900 nm, where the cross section is in general above 100 GM. On the other hand, compound 1 displays somewhat reduced 2PA properties when comparing with 2. However, adequate and remarkable 2PA cross-section values were determined at 720 and 830 nm for this dye (103 and 53 GM respectively). For comparison of our derivatives with other V-shaped compounds with 2PA properties, see Table S3 (ESI†). | ||
| Fig. 8 Two-Photon cross sections (σ2PA) of compounds 1 and 2 (black and red lines correspondingly, 10 μM solutions in toluene). The inset shows the dependence of the two-photon excited fluorescence intensity on the laser power with λexc,2P = 890 nm for 1 and λexc,2P = 790 nm for 2. The slopes (1.9 (r2 = 0.9994) and 2.0 (r2 = 0.9965) for compounds 1 and 2 respectively) are in accordance with the occurrence of two-photon absorption. | ||
The inherent advantages of the 2PA process, derived from the square dependence of the process on the excitation light intensity, can be also exploited in a wide range of solid-related applications, e.g., optical limiting, 3D data storage, microfabrication and so on.5,58,59 Bearing in mind, we decided to examine the 2PA properties in the cl-1 and cl-2 electrochemically polymerized films obtained from the two chromophores under study (Fig. S20 and S21, ESI†). It is worth noting that despite the push–pull systems of the dyes being altered by the polymerization process, the 2P-excited emission is conserved and the biphotonic ocurrence was confirmed in both cases. Interestingly, fluorescence of cl-2 is blue shifted when compared with that of cl-1. Changes in the excitation spectra are also noted, where the 2PA regime is bounded to the region above 800 nm. Thus, these good results seem to indicate that these derivatives, in the polymerized form, are potential candidates to some of the applications mentioned previously.
5.3 Two photon-absorption fluorescence cell imaging
Currently, there is a strong need for organic compounds whose activation takes place in the NIR region of the spectrum with optimal tissue penetration and minimizing background interference and phototoxicity.5,60–62 The high 2PA cross-sections, large Stokes shift and NIR emission wavelengths of 1 and 2 are clear advantages for their use in biomedical applications. In fact, chromophore 1 was successfully applied for labeling proliferative cells.29 Since the two-photon properties of 2 are enhanced with respect to 1, even more interesting results are expected.For this approach, we selected a healthy cell line consisting of mouse embryonic fibroblast (MEF) cells. The fluorescence properties of chromophores 1 and 2 within living MEFs were analyzed by 2P-microscopy. After incubating MEFs cells with these compounds, these were visualized upon 2P-excitation at 720 and 740 nm respectively, and fluorescence was detected in both cases between 450 nm and 700 nm using a HyD non-descanned detector. As seen in Fig. 9, live treated MEFs with chromophores 1 and 2 exhibited fluorescence at localized cytoplasmic regions. Thus, our results demonstrate that chromophores 1 and 2 are successfully applied as 2P fluorescent probes for bioimaging.
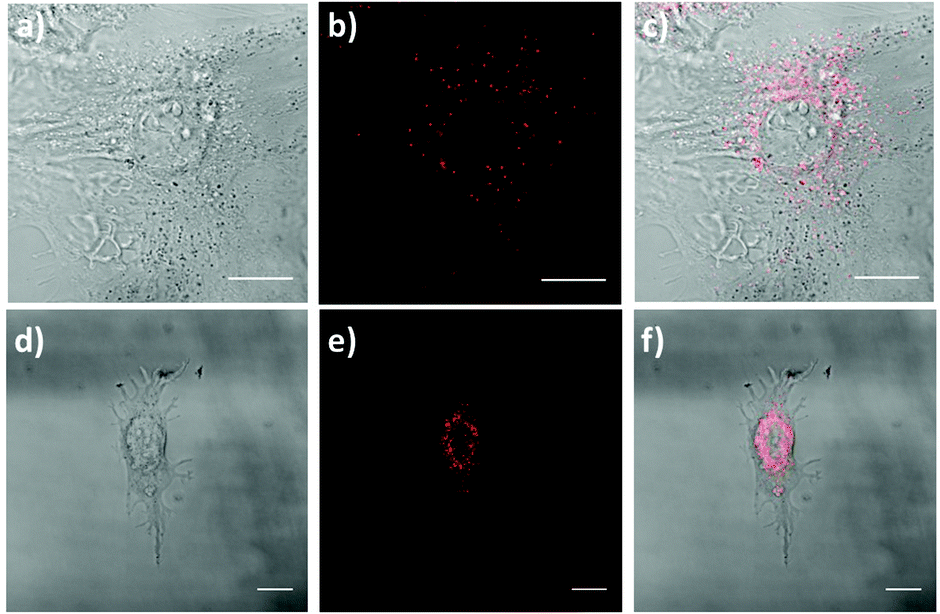 | ||
| Fig. 9 Fluorescence images of subcellular localization of 1 (a–c, λexc,2P = 720 nm, λem = 450–700 nm) and 2 (d–f, λexc,2P = 740 nm, λem = 450–700 nm) in MEF cells. (a and d) Brightfield in MEF cells. Fluorescence images of compound 1 (b) and compound 2 (e). Brightfield and fluorescence merged images of compound 1 (c) and compound 2 (f). Scale bar: 15 μm. | ||
Conclusions
Two V-shaped D–A–D systems with 4H-pyranylidene as a part of the π-conjugated bridge, propeller-like TPA donor units, and tricarbonitrile-based acceptor groups have been successfully developed. The two chromophores exhibit interesting photophysical properties (i.e., NIR emission, large Stokes shift, enhanced emission in tetrahydrofuran/water mixtures and good photostability). A slightly larger polarization of the π-conjugated structure is found in chromophore 2 due to the insertion of the TCF acceptor group, as proved by Raman spectroscopy and DFT calculations. In line with this, even more remarkable 2PA cross-section values and molecular first hyperpolarizabilities were found for 2 suggesting their potential for electrooptical and biological applications. In fact, the utility of the two chromophores for two-photon fluorescence bioimaging has been successfully demonstrated. On the other hand, the peculiar electrochemical behavior of these V-shaped chromophores results in the dimerization of the triphenylamino groups to tetraphenylbenzidine upon electrochemical and chemical oxidation. Importantly, the two-photon excited emission properties are preserved in the polymer films, which highlights their potential for other solid-state applications such as optical limiting, or 3D data storage.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work at the University of Málaga was funded by the MICINN (PID2019-110305GB-I00, PID2019-104293GB-I00, RTI2018-095410-B-I00, EuroNanoMed 2019 PCI2019-111825-2), the Institute of Health Carlos III (ISCIII) RETIC ARADYAL (RD16/0006/0012) and by the Junta de Andalucıa (P09-FQM-4708, UMA18-FEDERJA-080, UMA18-FEDERJA-007, PIER-0084-2019). The work at the University of Zaragoza was funded by the MICINN (PID2019-104307GB-I00/AEI/10.13039/501100011033) and Gobierno de Aragón (E47_20R). The work at the University of Stuttgart was funded by the German Science Foundation (DFG) through the project “LU 1445/7-1, project number 416982273” on electrooptical hybrid modulators, C. Malacrida is acknowledged for discussions. S. G.-V. thanks the MINECO for a FPU predoctoral fellowship (FPU17/04908) and CB-M for FPU fellowship (FPU16/02516). Computer resources, technical expertise and assistance provided by the SCBI (Supercomputing and Bioinformatics) centre of the University of Málaga are gratefully acknowledged. We thank the Vibrational spectroscopy lab (EVI) of the Research Central Services (SCAI) of the University of Málaga and John Pearson (BIONAND) for help with laser confocal microscopy analysis. We gratefully acknowledge the ICTS “NANBIOSIS” facilities, more specifically the U28 Unit of the Andalusian Centre for Nanomedicine & Biotechnology (BIONAND), for their help with the 2PA characterization and the microscopy studies.References
- L. R. Dalton, P. A. Sullivan and D. H. Bale, Chem. Rev., 2010, 110, 25–55 Search PubMed.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 Search PubMed.
- K. A. Mazzio and C. K. Luscombe, Chem. Soc. Rev., 2015, 44, 78–90 Search PubMed.
- Y. Ohmori, Laser Photonics Rev., 2010, 4, 300–310 Search PubMed.
- M. Pawlicki, H. A. Collins, R. G. Denning and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 3244–3266 Search PubMed.
- G. Qian and Z. Y. Wang, Chem. – Asian J., 2010, 5, 1006–1029 Search PubMed.
- M. Klikar, P. Solanke, J. Tydlitát and F. Bureš, Chem. Rec., 2016, 16, 1886–1905 Search PubMed.
- L. R. Dalton, P. Günter, M. Jazbinsek, O. P. Kwon and P. A. Sullivan, Organic Electro-Optics and Photonics: Molecules, Polymers, and Crystals, Cambridge University Press, Cambridge, 2015 Search PubMed.
- Z. Ning and H. Tian, Chem. Commun., 2009, 5483–5495 Search PubMed.
- O. Yurchenko, D. Freytag, L. zur Borg, R. Zentel, J. Heinze and S. Ludwigs, J. Phys. Chem. B, 2012, 116, 30–39 Search PubMed.
- P. Blanchard, C. Malacrida, C. Cabanetos, J. Roncali and S. Ludwigs, Polym. Int., 2019, 68, 589–606 Search PubMed.
- P. Reinold, K. Bruchlos and S. Ludwigs, Polym. Chem., 2017, 8, 7351–7359 Search PubMed.
- C. Malacrida, Y. Lu, K. Dirnberger, S. Gámez-Valenzuela, M. C. Ruiz Delgado and S. Ludwigs, J. Mater. Chem. C, 2020, 8, 15393–15405 Search PubMed.
- O. Yurchenko, J. Heinze and S. Ludwigs, ChemPhysChem, 2010, 11, 1637–1640 Search PubMed.
- J. Wang, K. Liu, L. Ma and X. Zhan, Chem. Rev., 2016, 116, 14675–14725 Search PubMed.
- C. Allain, F. Schmidt, R. Lartia, G. Bordeau, C. Fiorini-Debuisschert, F. Charra, P. Tauc and M.-P. Teulade-Fichou, ChemBioChem, 2007, 8, 424–433 Search PubMed.
- Y. Liu, M. Kong, Q. Zhang, Z. Zhang, H. Zhou, S. Zhang, S. Li, J. Wu and Y. Tian, J. Mater. Chem. B, 2014, 2, 5430–5440 Search PubMed.
- R. Whitby, A. Kay and M. C. Simpson, in Three-Dimensional Microfabrication Using Two-Photon Polymerization, ed. T. Baldacchini, William Andrew Publishing, 2nd edn, 2020, pp. 101–141 Search PubMed.
- W. Z. Yuan, Y. Gong, S. Chen, X. Y. Shen, J. W. Y. Lam, P. Lu, Y. Lu, Z. Wang, R. Hu, N. Xie, H. S. Kwok, Y. Zhang, J. Z. Sun and B. Z. Tang, Chem. Mater., 2012, 24, 1518–1528 Search PubMed.
- R. Andreu, L. Carrasquer, S. Franco, J. Garín, J. Orduna, N. Martínez de Baroja, R. Alicante, B. Villacampa and M. Allain, J. Org. Chem., 2009, 74, 6647–6657 Search PubMed.
- R. Andreu, E. Galán, J. Garín, V. Herrero, E. Lacarra, J. Orduna, R. Alicante and B. Villacampa, J. Org. Chem., 2010, 75, 1684–1692 Search PubMed.
- S. G. Awuah, J. Polreis, J. Prakash, Q. Qiao and Y. You, J. Photochem. Photobiol., A, 2011, 224, 116–122 Search PubMed.
- Z. Wang, M. Wang, J. Peng, Y. Xie, M. Liu, W. Gao, Y. Zhou, X. Huang and H. Wu, J. Phys. Chem. C, 2019, 123, 27742–27751 Search PubMed.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 Search PubMed.
- R. Chennoufi, H. Bougherara, N. Gagey-Eilstein, B. Dumat, E. Henry, F. Subra, S. Bury-Moné, F. Mahuteau-Betzer, P. Tauc, M.-P. Teulade-Fichou and E. Deprez, Sci. Rep., 2016, 6, 21458 Search PubMed.
- M.-X. Liu, L.-L. Ma, X.-Y. Liu, J.-Y. Liu, Z.-L. Lu, R. Liu and L. He, ACS Appl. Mater. Interfaces, 2019, 11, 42975–42987 Search PubMed.
- Y. M. Poronik, V. Hugues, M. Blanchard-Desce and D. T. Gryko, Chem. – Eur. J., 2012, 18, 9258–9266 Search PubMed.
- M. Jiang, X. Gu, J. W. Y. Lam, Y. Zhang, R. T. K. Kwok, K. S. Wong and B. Z. Tang, Chem. Sci., 2017, 8, 5440–5446 Search PubMed.
- X. Wang, Z. Guo, S. Zhu, Y. Liu, P. Shi, H. Tian and W.-H. Zhu, J. Mater. Chem. B, 2016, 4, 4683–4689 Search PubMed.
- S. Alías, R. Andreu, M. J. Blesa, S. Franco, J. Garín, A. Gragera, J. Orduna, P. Romero, B. Villacampa and M. Allain, J. Org. Chem., 2007, 72, 6440–6446 Search PubMed.
- G. Melikian, F. P. Rouessac and C. Alexandre, Synth. Commun., 1995, 25, 3045–3051 Search PubMed.
- R. Andreu, L. Carrasquer, J. Garín, M. J. Modrego, J. Orduna, R. Alicante, B. Villacampa and M. Allain, Tetrahedron Lett., 2009, 50, 2920–2924 Search PubMed.
- C. Dehu, F. Meyers and J. L. Brédas, J. Am. Chem. Soc., 1993, 115, 6198–6206 Search PubMed.
- D. Chasseau, J. Gaultier, C. Hauw, R. Fugnitto, V. Gianis and H. Strzelecka, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1982, 38, 1629–1631 Search PubMed.
- N. Martínez de Baroja, J. Garín, J. Orduna, R. Andreu, M. J. Blesa, B. Villacampa, R. Alicante and S. Franco, J. Org. Chem., 2012, 77, 4634–4644 Search PubMed.
- E. Galán, R. Andreu, J. Garín, L. Mosteo, J. Orduna, B. Villacampa and B. E. Diosdado, Tetrahedron, 2012, 68, 6427–6437 Search PubMed.
- A. B. Marco, N. Martínez de Baroja, S. Franco, J. Garín, J. Orduna, B. Villacampa, A. Revuelto and R. Andreu, Chem. – Asian J., 2015, 10, 188–197 Search PubMed.
- M. C. Ruiz Delgado, J. Casado, V. Hernández, J. T. López Navarrete, J. Orduna, B. Villacampa, R. Alicante, J.-M. Raimundo, P. Blanchard and J. Roncali, J. Phys. Chem. C, 2008, 112, 3109–3120 Search PubMed.
- J. Casado, M. C. Ruiz Delgado, M. C. Rey Merchán, V. Hernández, J. T. López Navarrete, T. M. Pappenfus, N. Williams, W. J. Stegner, J. C. Johnson, B. A. Edlund, D. E. Janzen, K. R. Mann, J. Orduna and B. Villacampa, Chem. – Eur. J., 2006, 12, 5458–5470 Search PubMed.
- C. C. Ferrón, M. C. Ruiz Delgado, O. Gidron, S. Sharma, D. Sheberla, Y. Sheynin, M. Bendikov, J. T. López Navarrete and V. Hernández, Chem. Commun., 2012, 48, 6732–6734 Search PubMed.
- T. Takenaka, Spectrochim. Acta, Part A, 1971, 27, 1735–1752 Search PubMed.
- A. Girlando and C. Pecile, Spectrochim. Acta, Part A, 1973, 29, 1859–1878 Search PubMed.
- M. S. Khatkale and J. P. Devlin, J. Chem. Phys., 1979, 70, 1851–1859 Search PubMed.
- E. Lippert, Z. Naturforsch., A: Phys. Sci., 1955, 10, 541–545 Search PubMed.
- N. Mataga, Y. Kaifu and M. Koizumi, Bull. Chem. Soc. Jpn., 1955, 28, 690–691 Search PubMed.
- N. Mataga, Y. Kaifu and M. Koizumi, Bull. Chem. Soc. Jpn., 1956, 29, 465–470 Search PubMed.
- Note that compound 1 was previously demonstrated to display better photoestability to that recorded to the commercially NIR dye ICG. See ref. 29.
- S. Arnaboldi, T. Benincori, A. Penoni, L. Vaghi, R. Cirilli, S. Abbate, G. Longhi, G. Mazzeo, S. Grecchi, M. Panigati and P. R. Mussini, Chem. Sci., 2019, 10, 2708–2717 Search PubMed.
- C. Malacrida, A. H. Habibi, S. Gámez-Valenzuela, I. Lenko, P. Simón Marqués, A. Labrunie, J. Grolleau, J. T. López Navarrete, M. C. Ruiz Delgado, C. Cabanetos, P. Blanchard and S. Ludwigs, ChemElectroChem, 2019, 6, 4215–4228 Search PubMed.
- E. Quartapelle Procopio, T. Benincori, G. Appoloni, P. R. Mussini, S. Arnaboldi, C. Carbonera, R. Cirilli, A. Cominetti, L. Longo, R. Martinazzo, M. Panigati and R. Pò, New J. Chem., 2017, 41, 10009–10019 Search PubMed.
- M. Hasegawa, K. Kobayakawa, H. Matsuzawa, T. Nishinaga, T. Hirose, K. Sako and Y. Mazaki, Chem. – Eur. J., 2017, 23, 3267–3271 Search PubMed.
- D. R. Kanis, M. A. Ratner and T. J. Marks, Chem. Rev., 1994, 94, 195–242 Search PubMed.
- C. W. Dirk, H. E. Katz, M. L. Schilling and L. A. King, Chem. Mater., 1990, 2, 700–705 Search PubMed.
- C. R. Moylan, S. Ermer, S. M. Lovejoy, I. H. McComb, D. S. Leung, R. Wortmann, P. Krdmer and R. J. Twieg, J. Am. Chem. Soc., 1996, 118, 12950–12955 Search PubMed.
- S.-S. P. Chou and C.-Y. Yu, Synth. Met., 2004, 142, 259–262 Search PubMed.
- F. Terenziani, C. Katan, E. Badaeva, S. Tretiak and M. Blanchard-Desce, Adv. Mater., 2008, 20, 4641–4678 Search PubMed.
- M. Rumi and J. W. Perry, Adv. Opt. Photon., 2010, 2, 451–518 Search PubMed.
- S. R. M. A. K.-S. Lee, Photoresponsive Polymers I, Springer, Berlin, Heidelberg, 1st edn, 2008 Search PubMed.
- G. S. He, L.-S. Tan, Q. Zheng and P. N. Prasad, Chem. Rev., 2008, 108, 1245–1330 Search PubMed.
- C. Benitez-Martin, J. A. Guadix, J. R. Pearson, F. Najera, J. M. Perez-Pomares and E. Perez-Inestrosa, Sens. Actuators, B, 2019, 284, 744–750 Search PubMed.
- C. Benitez-Martin, J. A. Guadix, J. R. Pearson, F. Najera, J. M. Perez-Pomares and E. Perez-Inestrosa, ACS Sens., 2020, 5, 1068–1074 Search PubMed.
- C. S. Lim and B. R. Cho, Tetrahedron, 2015, 71, 8219–8249 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and theoretical details, synthesis, DFT-calculations, photostability, spectroelectrochemistry and chemical doping and 2P-fluorescence spectra/imaging. See DOI: 10.1039/d1ma00415h |
| This journal is © The Royal Society of Chemistry 2021 |