
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEvaluating the effect of synthesis, isolation, and characterisation variables on reported particle size and dispersity of drug loaded PLGA nanoparticles†
Bruna C.
Garms‡
 ab,
Hamish
Poli‡
a,
Darcy
Baggley
a,
Felicity Y.
Han
cd,
Andrew K.
Whittaker
bd,
Anitha
A
*a and
Lisbeth
Grøndahl
*ad
ab,
Hamish
Poli‡
a,
Darcy
Baggley
a,
Felicity Y.
Han
cd,
Andrew K.
Whittaker
bd,
Anitha
A
*a and
Lisbeth
Grøndahl
*ad
aSchool of Chemistry and Molecular Biosciences, University of Queensland, Brisbane, QLD 4072, Australia. E-mail: l.grondahl@uq.edu.au; a.sudheeshkumar@uq.edu.au
bARC Centre of Excellence in Convergent Bio-Nano Science and Technology, University of Queensland, Brisbane, QLD 4072, Australia
cSchool of Biomedical Sciences, University of Queensland, Brisbane, QLD 4072, Australia
dAustralian Institute for Bioengineering and Nanotechnology, University of Queensland, Brisbane, QLD 4072, Australia
First published on 5th August 2021
Abstract
Poly(lactic-co-glycolic acid) (PLGA) nanoparticles have been extensively studied as carriers of a wide variety of drugs. However, the variability in protocols and reporting in nanoscience represents a barrier to advances in new therapies. Recent literature has highlighted the challenge of reproducibility in biomedical research. In this study involving three researchers performing the experiments we therefore verified the repeatability and replicability of PLGA nanoparticles fabricated by the emulsion solvent evaporation method. Furthermore, we investigated how variables during synthesis, isolation, and characterisation of PLGA particles affect the reported particle size and drug encapsulation. Synthesis parameters including emulsification technique, type of polymer, and type of surfactant were investigated. In the isolation process, we tested the effect of washing and filtration. Dynamic light scattering (DLS) was used to determine the number weighted mean, z-average, and polydispersity index (PDI). Direct visualization of the particles was achieved by scanning and transmission electron microscopies. Changes in the source of energy input to the system using a sonic probe and homogeniser showed the most pronounced effect on particle size and PDI. This work has demonstrated that particle properties are affected by multiple variables involved in the synthesis and isolation processes. We provided advice for PLGA-based nanoparticle fabrication and drug encapsulation quantification as well as the minimum required information to be reported allowing reproducibility.
Introduction
Poly(D,L-lactic-co-glycolic acid) (PLGA) is a class of synthetic polymers that has been extensively studied for drug delivery.1 These statistical co-polymers have tuneable properties that allow the rate of drug release to be tailored to a required application. This is achieved by e.g. varying polymer properties such as molecular weight, nature of the end group, and monomer ratio (L![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :G).1,2 The polymer properties and material dimensions determine the degradation behaviour.3 The degradation products are metabolised via the Krebs cycle and cleared by the body4 and several studies have reported their biocompatibility in many biological tissues.1,4–8 There are many examples in the literature using PLGA-based materials for delivery of therapeutics to promote either fast or prolonged drug release with applications in cancer treatment, pain management, immunotherapy, and more recently, in the treatment of pulmonary tuberculosis, urinary tract infections, migraine and glaucoma.5,6,9–15
:G).1,2 The polymer properties and material dimensions determine the degradation behaviour.3 The degradation products are metabolised via the Krebs cycle and cleared by the body4 and several studies have reported their biocompatibility in many biological tissues.1,4–8 There are many examples in the literature using PLGA-based materials for delivery of therapeutics to promote either fast or prolonged drug release with applications in cancer treatment, pain management, immunotherapy, and more recently, in the treatment of pulmonary tuberculosis, urinary tract infections, migraine and glaucoma.5,6,9–15
There are several techniques that are used for the preparation of PLGA particles including single and double emulsion solvent evaporation (ESE), nanoprecipitation, spray-drying, microfluidics, and hydrogel templating.2,16–19 These techniques enable the synthesis of a wide range of PLGA nano- and sub-micron sized particles. The ideal size and polydispersity of particles vary according to the application. In brain cancer therapy, for example, monodispersed particles below 100 nm in diameter can facilitate tissue permeation when locally administered and are ideal to reach infiltrative cancer cells.20 For intravenous administration in cancer therapy, PLGA particles of size in the range of 120–150 nm showed improved tumour accumulation and enhanced tumour inhibition rate in mouse breast tumour models (4T1-bearing nude mice).21 In contrast, submicron particles (500 nm) were demonstrated to be optimal to promote a sustained release of ketamine and hydromorphone for over a month, and are suitable for pain management.22
The choice of the PLGA nanoparticles (NP) fabrication technique is driven by the application, physico–chemical characteristics of the therapeutic to be encapsulated, and the required size and drug release rate. Among the available techniques, the encapsulation of small hydrophobic molecules into NPs is commonly achieved using the single ESE technique (Fig. 1).4,16,23,24 The advantage of this technique is the ability to control particle size and polydispersity as well as tuning the release kinetics of the encapsulated compound.16,25,26 An oil in water (O/W) emulsion is prepared from the polymer and the drug dissolved in a volatile organic solvent and a surfactant in water. To form nano-sized droplets from the O/W emulsion, high energy input is required, and this is typically facilitated using a homogeniser or sonic probe. The suspension is left under stirring to allow evaporation of the organic solvent and the NPs are collected by ultracentrifugation. This technique can be easily scaled up and allows control of particle size.
 | ||
| Fig. 1 Key variables involved in PLGA NP fabrication by the single emulsion solvent evaporation (ESE) technique. | ||
Several processing parameters in the fabrication and isolation of particles by the single ESE technique may affect the size and yield of the NPs as well as the drug encapsulation efficiency. Some variables that have previously been investigated with regards to drug release rate, particle size, and degradation include the L:G ratio of PLGA and concentrations of polymer, drug, and surfactant.26–28 For example, polymers with high lactic acid content produce NPs with increased crystallinity, resulting in a slower rate of degradation of the NPs and therefore a slower drug release.29 Regarding drug encapsulation, it was found that at a lower surfactant concentration, NPs can form agglomerates and have reduced encapsulation efficiency.28 In a review, Jenjob et al. compared single ESE to other emulsion techniques available for NP fabrication and showed that the choice of the technique can affect the morphology and the size distribution of the particles.30 In another study, the use of a co-surfactant was shown to produce particles with reduced size compared to the use of a single surfactant.26 Hernandez-Giottonini et al. investigated variables in the isolation process and found that an increase in centrifugation speed allows the collection of smaller NPs with narrower PDI and that the presence of cryoprotectant is critical for maintaining size after lyophilisation.27 However, there are few reports evaluating other variables involved in the synthesis and isolation of PLGA NPs.17,27,28
Although there is extensive literature published on PLGA NPs for biomedical application, only 13 medicinal products have translated from research to clinical use.31–33 This limited success has been associated with the lack of standard protocols during pre-clinical evaluation which is fundamental to determine the safety and quality of the product.31,34–36 The variability in protocols and reporting in nanoscience represents a major barrier to advances in new therapies.37 Recent literature has highlighted the challenge of reproducibility in biomedical research. While not a new concept, the “3R”s comprising repeatability, replicability, and reproducibility, were recently adapted and reinforced in biointerface science.38 Within the context of our work, repeatability refers to results obtained from an experiment performed by one researcher with the same experimental setup. To be replicable, the same results have to be obtained by a different researcher when using the same experimental setup. Reproducibility refers to an experiment performed by two or more researchers using different measuring systems that will produce the same results.37,39
With the aim of improving reproducibility overall in the field, this study provides advice for PLGA-based NP fabrication and drug encapsulation quantification. The repeatability and replicability of PLGA NP formulation were verified by three researchers using the same experimental set-up. To investigate the effect of variables in synthesis, isolation, and characterisation on the reported particle size and drug encapsulation, we specifically targeted variables not previously investigated in detail. The outcome of the synthesis was evaluated using three different PLGA polymers (L:G of 1:1) with different end groups and glass transition temperature (Tg), two different surfactants, variable energy input, and two energy sources. The isolation parameters evaluated complements previous work and we investigated the effect of washing and filtration on both particle size and surfactant content. From the evaluation of particle size and particle size distribution using a series of techniques, we highlight the minimum information that should be reported. Overall, this study complements previous work17,27,28 and highlights how particle properties are affected by multiple variables involved in the synthesis and isolation processes.
Materials and methods
Materials
Three different types of poly(D,L-lactide-co-glycolide) (PLGA) (L:G = 1:1) were used. PLGA-E1 (Resomer® RG 502, ester terminated, MW = 7–17 kg mol−1, Tg = 42–46 °C) and PLGA-A1 (Resomer® RG 502 H, acid terminated, MW = 7–17 kg mol−1, Tg = 42–46 °C) were obtained from Sigma Aldrich while PLGA-E2 (50:50, ester terminated, inherent viscosity 0.15–0.25 dL g−1, Tg = 28 °C) was obtained from DURECT Corporation, Birmingham, AL, USA. Bovine serum albumin (BSA, ≥96%, fatty acid-free) was obtained from Sigma Aldrich and poly(vinyl alcohol) (PVA, MW = 25 kg mol−1, 88% hydrolysed) was obtained from Polysciences Inc. Curcumin was obtained from Merck Millipore and Rhodamine B (RhB, ≥95%, HPLC) from Sigma Aldrich. Ultrapure (UP) water (18.2 Ω cm) was obtained from a VWR International PureLab Flex ELGA purification system, absolute ethanol was obtained from Thermo Fisher Scientific, Inc., dichloromethane (DCM, anhydrous, ≥98%), tetrahydrofuran (THF, anhydrous, ≥99.9%), and dimethyl sulfoxide (DMSO, anhydrous, ≥99.9%) were obtained from Merck. Deuterated DMSO (≥99.9 atom % D) was obtained from Sigma Aldrich.
PLGA particle fabrication
The organic phase was prepared by dissolving PLGA (20 mg mL−1) in DCM (0.5 mL) and this was added dropwise to a surfactant solution containing PVA (10 mg mL−1) in UP water (2 mL) whilst stirring. The emulsion was sonicated in an ice bath using a Branson Digital Sonifier 450 with a 1/8′′ tapered microtip operated at 20% amplitude (179 μm) for 2 min. The resulting particle suspension was added dropwise into 40 mL of UP water and magnetically stirred for 3 h in a fume hood to allow evaporation of the organic solvent. The suspension was centrifuged at the relative centrifugal force (rcf) = 76500 × g (25000 rpm) for 1 h maintained at 17 °C on a Beckman Coulter Avanti HP-20 with a JA25.50 fixed angle rotor. The pellet was washed twice by resuspending in UP water and centrifuged as described above. The washed particle pellet was either resuspended in 1 mL of UP water and stored at 4 °C or freeze-dried for 48 hours on a Thermo Savant Micro Modulyo −115 Freeze Dryer and collected for further analysis.
Variables
A series of parameters were varied during particle synthesis and isolation. The surfactant type was changed to a surfactant solution containing BSA (30 mg mL−1) in UP water (2 mL). The sonication amplitude and time were varied as follows: the amplitude was varied from 10% (116 μm) to 20% (179 μm) at a constant time of 2 min. In addition, the time was varied from 20 s to 2 min at a constant amplitude of 20%. The emulsification type was investigated using a homogeniser (T25 ULTRA-TURRAX® homogenizer; IKA®-WERKE, Staufen, Germany) operated at 20000 rpm for 5 min. The effect of washing the particle pellet was assessed by comparing particles prepared with 2 wash cycles and particles prepared without washing. The effect of filtration of the particles after centrifugation was evaluated using a 0.45 μm polypropylene syringe filter.
When a particular parameter was tested, samples are named according to the polymer (e.g. PLGA-A1), surfactant (BSA or PVA), energy source (sonic probe (S), or homogeniser (H)), and drug encapsulated. For example, particles prepared from PLGA-A1 emulsified by homogeniser with BSA and loaded with curcumin are labelled as PLGA-A1-BSA-H-cur. All other variables are described in the text and figure captions.
Drug encapsulation
Curcumin or RhB was dissolved in DCM and added to the polymer solution where the drug/polymer mass ratio was 1:10 (curcumin and RhB), 2:10 and 5:10 (RhB). The organic phase was added dropwise into the aqueous phase containing PVA where the polymer/surfactant mass ratio was 1:20.
Direct and indirect methods were used to determine curcumin encapsulation. In the indirect method, the supernatant and washing liquid were collected during isolation and washing, combined, and diluted to 100 mL in a volumetric flask. The washing liquid was kept at 4 °C if it needed to be stored for longer than 1 hour. The concentration of the drug in this solution was evaluated by ultraviolet-visible (UV-Vis) spectroscopy at 424 nm for curcumin or 555 nm for RhB (Aligent Technologies Cary 60 UV-visible spectrophotometer using a 1 cm quartz cuvette) against a calibration curve prepared by adding 200 μL of curcumin solution dissolved in absolute ethanol to a supernatant mock solution of PVA in UP water (0.2 mg mL−1). The calibration curve (Fig. 7A) was linear over the concentration range of 0–20 μg mL−1 for curcumin. For quantification of RhB, only the indirect method was used and the calibration curve was linear over a range of 0–5 μg mL−1.
In the direct method, accurately weighed freeze-dried samples (analytical balance, XS205 DualRange, Mettler Toledo Ltd) were dissolved in a THF/DMSO solution (2:1) and centrifuged at 3000g for 5 min to remove the polymer that had precipitated. The supernatant was transferred quantitatively to a volumetric flask and filled to the measure. The curcumin concentration was quantified immediately after extraction by spectroscopy at 424 nm against a calibration curve prepared from curcumin dissolved in THF/DMSO (2:1). This linear calibration curve is displayed in Fig. 7B). Nuclear magnetic resonance (NMR) was done on a Bruker Avance III 500 MHz spectrometer. 1H NMR of dissolved lyophilised particles in DMSO-d6. Data were processed with either Bruker Topspin software or MestReNova software.
The drug encapsulation efficiency (EE%), loading capacity (LC%), and particle yield (Yield%) were calculated using the equations shown in Fig. 2. The mean values and the standard deviation are reported (n = 3 for curcumin, n = 2 for RhB).
 | ||
| Fig. 2 Indirect and direct methods for drug quantification and equations used for the determination of drug encapsulation efficiency (EE%), loading capacity (LC%), and particle yield (Yield%) using both direct and indirect methods. Symbols used: mdrug = mass of drug initially used, mpolymer = mass of polymer initially used, mdrug(e) = mass of drug (encapsulated), mdrug(f) = mass of drug (free) and mNP = mass of drug loaded NPs. | ||
Characterisation
Nanoparticle tracking analysis
Nanoparticle tracking analysis (NTA) was performed using a Malvern Nanosight LM 10 cell (λLaser = 405 nm) and sCMOS camera using Nanosight Software (v3.2). NP suspensions were diluted to 0.00001 vol% in UP water and injected with a syringe (1 mL) controlled by a syringe pump. Measurements were performed in triplicate. Camera settings such as camera level, shutter, and gain were optimised before capture recording. Constant capture settings included frames per second = 25, duration = 60 s, temperature = 25 °C, viscosity = 0.9 cP (water), syringe pump speed 100 μL min−1. Particles were then analysed at an optimal detection threshold. Size distribution curves were developed as a concentration (particles per mL) distribution across bin widths (5 nm).Electron microscopy
The morphology of the NPs was studied using a field emission microscope (JEOL JSM-7100F), a scanning electron microscope (SEM, Hitachi SU3500, Hitachi High Technologies America, Inc.), and a high-resolution transmission electron microscope (TEM, Hitachi HT7700 120 kV). Prior to SEM analysis, a droplet of the particle suspension (approximately 0.25 mg mL−1) was placed on a piranha treated silica wafer and air-dried before mounting onto SEM stubs and platinum-coated (15 nm thickness) in a vacuum using a sputter coater (Quorum Q150TS platinum coater, Quorum Technologies Inc.). Prior to TEM analysis, a droplet of the particle suspension (approximately 2.5 mg mL−1) was placed on a carbon film-covered copper grid (200 mesh) and then negatively stained with 2 wt% phosphotungstic acid solution. A manual approach to NPs size determination was used for measurement of the images captured at magnifications of 10000× for SEM and 30000× or 80000× for TEM. A circle drawn using ImageJ software was placed on each NP which displayed well-defined edges to determine the area which allowed the calculation of the diameter of the NP. The scale bar of the images was used to calibrate the measured diameter to nanometres. Agglomerates and NPs found at the edge of the image were not included in the measurements. All NPs of the image were counted, and in cases where <100 particles were found, more than one image was used for particle size determination.
X-Ray photoelectron spectroscopy
X-Ray photoelectron spectroscopy (XPS) was used to evaluate the surface composition, using a Kratos Axis ULTRA X-Ray Photoelectron Spectrometer incorporating a 165 mm hemispherical electron energy analyzer. The incident radiation was Monochromatic Al Kα X-rays (1.486.6 eV) at 150 W (15 kV, 10 mA). Survey scans were taken at an analyser pass energy of 160 eV and narrow scans at 20 eV. Survey scans were carried out over 1200–0 eV binding energy range with 1.0 eV steps and a dwell time of 100 ms. Narrow scans were run with 0.05 eV steps and 250 ms dwell time. The base pressure in the analysis chamber was 10−9 torr and during sample analysis 10−8 torr. Atomic concentrations and peak fitting of the narrow scans were calculated using the CasaXPS version 2.3.14 software and a Shirley baseline with Kratos library Relative Sensitivity Factors. All narrow scans were charge corrected to the C*–C peak at 285.0 eV.Data analysis
The data are presented as mean (±standard deviation) unless otherwise specified. Statistical differences between groups were evaluated for various data sets (GraphPad Prism™, v8.0.1; GraphPad Software Inc., La Jolla, CA). A multiple two-tailed unpaired t-test was used to perform an analysis between two samples and curves were treated with a D’Agostino & Pearson normality test. To compare three or more groups, statistical differences were evaluated with a 1-way or 2-way analysis of variance (ANOVA) followed by the multiple comparisons test. For comparing the means of each studied variable the statistical significance criterion was p ≤ 0.05.Results and discussion
Emulsion-based fabrication techniques are favoured for formulation of PLGA NPs for drug delivery applications because of the ease of processing and optimising. It is well established that a 50:50 lactic acid to glycolic acid ratio (L:G = 1:1) achieves the fastest rate of degradation and that the rate can be reduced by increasing the proportion of either lactic or glycolic acid.42 This work used 1:1 L:G ratio to capture the most common type of PLGA polymer used in NP fabrication.43–47 The choice of surfactant is important for controlling the stability and the size of the NPs. This work used PVA as the main surfactant as it represents a commonly used non-ionic surfactant, part of the family of surfactants that has been used for PLGA NP stabilisation also including Tween, Pluronic, and PEG.48 Additionally, protein based NP stabilisation represents a novel method to improve the biocompatibility of drug delivery systems and as such, BSA has established precedence49 and was therefore selected for this study as a comparison to PVA.
Repeatability and replicability of PLGA formulation were investigated in this study (data provided in ESI,† Fig. S1). The first “R”, repeatability, was evaluated by one researcher. Among eight replications of PLGA-E1-PVA-S NPs, the number weighted mean and z-average of the particles were 138 and 180 nm, respectively, and the standard deviation was less than 5 (ESI,† Fig. S1A). This result demonstrated consistency in the fabrication process of NPs and a low batch-to-batch variation. In addition, the PDI of the NPs remained below 0.2, confirming their narrow dispersity in size. To ensure replicability of the study, three researchers in the same laboratory followed the same experimental set-up to fabricate PLGA-A1-PVA-S NPs and no significant difference was observed between the resulting NPs (see ESI,† Fig. S1B). Based on this established repeatability and replicability of PLGA NP fabrication in the current study, evaluation of the effect of various parameters (e.g. within synthesis, isolation, and characterisation) on NP size and size distribution could be pursued.
Common techniques for characterisation of nanoparticles
Recent literature has reinforced the importance of standardization of NP characterisation and the minimum information reporting to improve reproducibility in nano-science research.31,37,39 A range of techniques has been used to report the size and morphology of NPs, such as electron microscopy, light scattering, and particle tracking analysis.50,51 The most commonly used technique is DLS, as it allows the determination of the particle size distribution (PSD) in a rapid manner and with minimum sample preparation. However, DLS is considered a low-resolution technique, and further comparative high-resolution techniques are recommended.31,36 In this study, DLS was used for determining standard PSD of all samples, and electron microscope (data described below) and NTA (data included in Fig. 4) were used for comparison and to provide a better understanding of the NP properties.The characterisation by TEM, SEM, and DLS of two NP types, differing with regards to the PLGA end group and surfactant (BSA or PVA) are shown in Fig. 3. The characterisation by all three techniques was in both cases done on the same sample to minimize the effect of any minor batch-to-batch variation. The electron microscope images of PLGA-E1-PVA-S and PLGA-A1-BSA-S illustrate that the NPs presented spherical morphology and smooth surfaces. In Fig. 3A and D, TEM histograms showed that the PLGA-E1-PVA-S NPs displayed particles in a size range of 30–140 nm and a mean particle size of 64 ± 22 nm. The use of BSA as a surfactant, resulted in smaller particle size, with a size range of 30–100 nm (except for four large particles >150 nm) and a mean particle size of 54 ± 35 nm. While a more narrow size range was also observed for PLGA-A1-BSA-S compared to PLGA-E1-PVA-S in the histogram generated by SEM (Fig. 3B and E), the mean diameter was not significantly different and yielded a mean particle size of approximately 115 nm for both samples. The number mean size distribution obtained by DLS measurements (Fig. 3C and F) showed a broader distribution of NP size compared to the results obtained from electron microscopy. The number weighted mean particle size of PLGA-E1-PVA-S and PLGA-A1-BSA-S were 144 ± 8 nm and 77 ± 10 nm, respectively (z-average values are reported in Table S1, ESI†).
 | ||
| Fig. 3 Characterisation of PLGA-E1-PVA-S (left column) and PLGA-A1-BSA-S NPs (right column). (A) and (D) TEM micrographs at 30000 and 80000 × magnification were used for generation of the histograms where n = 155 for (A) and n = 119 for (D). Inserts show TEM image at 150 000 × magnification. (B) and (E) SEM micrographs at 20,000 × magnification were used for generation of the histograms where n = 125 for (B) and n = 131 for (E). Insets show SEM images at 20000 × magnification. (C) and (F) are histograms of NPs number percent determined by DLS. The error is the standard deviation of the distribution (n given above for each sample) and the size refers to the diameter of particles. | ||
It is apparent when comparing the values obtained from the different techniques, that analysis of SEM images generates a larger average particle size compared to TEM. DLS measurements for the PLGA-E1-PVA-S NPs correlate better with SEM than TEM, however, for PLGA-A1-BSA-S, there is a better correlation with TEM than SEM. Contributing to these differences is the inherent different information derived from the different techniques. DLS measures the hydrodynamic radius of a particle50 while the electron microscope techniques image the particles in the dry state and under vacuum. Furthermore, obtaining high-resolution SEM images of the PLGA NPs was challenging due to the polymer being degraded under the electron beam. Furthermore, NP aggregation was observed to be more pronounced in SEM than TEM when comparing samples prepared at a fixed concentration. This resulted in a lack of particles less than 50 nm in size being identified in the SEM images while in the TEM images, particles of 30 nm could be observed. This presents as an inherently biased measurement of the SEM technique. Other studies comparing particle sizing methods have likewise used TEM as a ‘reference’ technique.52 Furthermore, the broader PSD observed in DLS compared to TEM has previously been documented for monodisperse samples and is inherent in the DLS measurement.52–54 Therefore, TEM is considered as a preferable high-resolution option for evaluation of NP morphology and size due to the high magnification achievable and the simpler sample preparation including no requirement for sample coating.
As mentioned above, due to its simplicity, particle sizing is often performed using light scattering techniques such as DLS and to a lesser extent NTA. Whilst DLS characterises size from the decay of light scattering as a result of Brownian motion, NTA records the light scattering of NPs to allow tagging and tracking of their movement due to Brownian motion. From the movement of NPs in both methods, the software can then determine a diffusion coefficient and calculate a hydrodynamic radius of the particle from the Stokes–Einstein equation. Size measurements from DLS can be inaccurate if the NP sample has a high PDI because the intensity of scattered light is proportional to the sixth power of the particle size. Larger particles will have a greater intensity and be incorrectly interpreted as being an accurate representation of the particle size within the bulk.52 Alternatively, NTA uses light scattering to identify the NPs, it does not use the scattered light intensity to determine the particle size which means it is more robust against the presence of large aggregates. A comparison of data obtained from DLS and NTA is included in Fig. 4A. The raw data produced by DLS is known as the correlogram which is a representation of the decaying intensity of scattered light as particles move due to Brownian motion. The size can then be determined and reported in two ways; (1) the z-average as a singular value derived from a single exponential analysis of the decaying correlogram curve and (2) a multi-exponential analysis of the decaying correlogram curve. The latter provides a population distribution which can be represented as an intensity, number or volume weighted PSD.
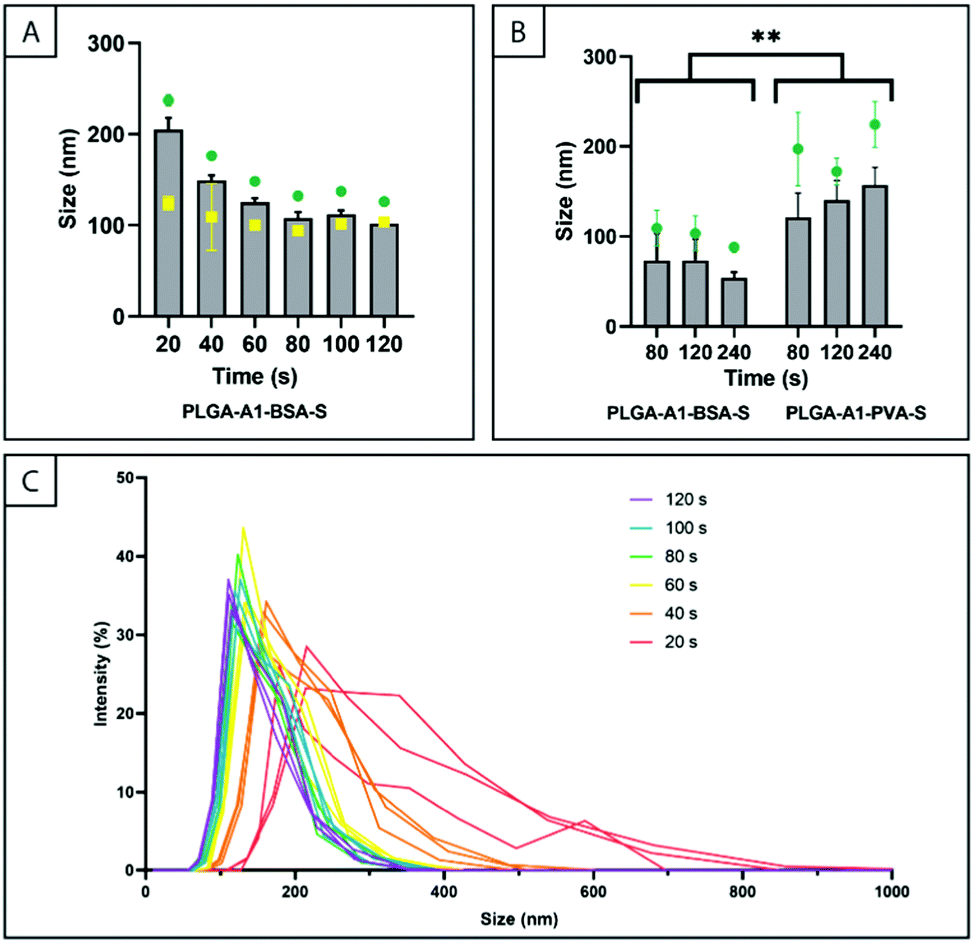 | ||
Fig. 4 The effect of surfactant and sonication time on the size of PLGA particles. The number weighted mean is represented by columns ( ), z-average is represented by ), z-average is represented by  , as measured by DLS, and the mean particle size determined by NTA is represented by , as measured by DLS, and the mean particle size determined by NTA is represented by  . (A) Effect of sonication time (20–120 s) on the particle size (diameter) for PLGA-A1-BSA-S NPs (sonication amplitude of 20%); (B) effect of sonication time (80–240 s) on the particle size (diameter) for PLGA-A1-BSA-S and PLGA-A1-PVA-S NPs (sonication amplitude of 20%); (C) intensity plots from DLS data corresponding to the data in (A). The error bars represent the standard deviation of replicate samples (n = 3). ** represents statistical significance of p < 0.01 as calculated from two-way ANOVA. It should be noted that the data in figures (A) and (B) were measured on different instruments. . (A) Effect of sonication time (20–120 s) on the particle size (diameter) for PLGA-A1-BSA-S NPs (sonication amplitude of 20%); (B) effect of sonication time (80–240 s) on the particle size (diameter) for PLGA-A1-BSA-S and PLGA-A1-PVA-S NPs (sonication amplitude of 20%); (C) intensity plots from DLS data corresponding to the data in (A). The error bars represent the standard deviation of replicate samples (n = 3). ** represents statistical significance of p < 0.01 as calculated from two-way ANOVA. It should be noted that the data in figures (A) and (B) were measured on different instruments. | ||
Reporting only the mean particle size from a PSD can be prone to misrepresentation when the distribution diverges from Gaussian form. Obtaining an accurate number weighted PSD from DLS data requires input of refractive indices but is the more robust presentation and should therefore be used when possible for polydisperse (PDI > 0.1) samples to calculate a number weighted mean particle size rather than reporting the z-average. Faria et al., in a guideline for minimal information reporting in nanoscience research, likewise states that reporting only the z-average size is insufficient to characterize NPs for biological application.37 Therefore, when reporting DLS measurements, accompanying PSD curves (intensity, number, or volume) should be included. To establish precedence, in this work, all PSD curves not included within the main manuscript can be found in ESI† (Fig. S2 for DLS data and Fig. S3 for NTA data). This includes individual curves for the triplicate samples evaluated. Furthermore, in order to demonstrate the difference between the z-average and number weighted mean particle size as determined by DLS, both have been reported in the current study in most figures. Finally, included in the ESI,† is a table (Table S1) of values obtained from DLS measurements for the majority of the samples of this study.
Effect of the variables in the fabrication process
As outlined in Fig. 1, the NP fabrication method ESE has a number of variables which all potentially can affect the particle size. Using a sonic probe to provide the energy input and a single polymer type (PLGA-A1), the effect of surfactant type, sonication time, and sonication amplitude was evaluated. Considering the high energy input from the sonic probe, this process is done in an ice bath. It was confirmed in the current study that the temperature of the reaction mixture did not exceed 9 °C (details provided in ESI,† Table S2) which is well below the Tg of the polymers used. Assessment of the sonication amplitude was evaluated by comparing NPs produced with a sonication emulsification amplitude of 10, 15, and 20% (constant time of 120 seconds and BSA as the surfactant). Using DLS, these settings were found to produce NPs with z-average particle sizes of 160, 140, and 120 nm, respectively. This inverse relationship between sonication amplitude and particle size has been explained by Taurozzi et al. and is related to the acoustic power as described by eqn (1).55 | (1) |
The effect of sonication time was analysed by DLS (Fig. 4A and B) and NTA (Fig. 4A). The effect of sonication time over the range of 20–120 seconds on particle size was investigated using both DLS and NTA and the data are shown in Fig. 4A. The DLS data illustrated a decrease in the number weighted mean particle size for particles prepared using a sonication time of 20 s (205 ± 13 nm) compared to 120 s (102 ± 1 nm) and the z-average values follow the same trend. The data obtained by NTA showed a similar reduction in the mean particle size over time, however, the initial mean particle size after 20 seconds was found to be 124 ± 7 nm which was over 80 nm different from the number weighted mean particle size measured by DLS. However, after 120 seconds the mean particle size measured by NTA had converged to 103 ± 2 nm which was similar to the number weighted mean particle size measured by DLS. Considering the different bias of the two methods,52 these values would indicate that the PSD for particles prepared with a sonication time of 20 seconds is very broad and not mono-modal. This is supported by the change in the PDI which is reduced from 0.288 for a sonication time of 20 s to 0.131 for a sonication time of 120 s and it is also clearly illustrated in the intensity plots generated from the DLS data presented in Fig. 4C. This work highlights that when low polydispersity is required for NPs produced using ultrasonication induced emulsification, the duration of ultrasonic exposure can be increased to afford narrow PSDs and the specific details should be established for individual laboratories.
The effect of sonication time from 80 to 240 seconds on the particle size was evaluated separately (using a different zeta sizer) for particles prepared with either PVA or BSA as the surfactant (Fig. 4B). The size of BSA stabilized particles did not change significantly with sonication time (p > 0.05) and the number weighted mean particle size was measured to be 73 ± 31 nm after sonication for 80 s and 54 ± 6 nm after 240 s. A sonication time of 80 s resulted in a number weighted mean particle size of PVA stabilized NPs of 120 ± 30 nm. The particle size was not affected by sonication time (p > 0.05) where 120 s and 240 s yielded particles of 140 ± 22 nm and 157 ± 19 nm, respectively. Thus, for both BSA and PVA stabilised particles, there was no significant effect of sonication time within the range of 80–240 seconds. The apparent difference in number weighted mean particle size between Fig. 4A and B, for similarly fabricated particles is likely due to the difference in DLS resolution aforementioned in the methods section. The data in Fig. 4B indicate a significant difference in the number weighted mean particle size for particles that were fabricated with different surfactant type (PVA and BSA) and different surfactant concentration, but otherwise identical procedure (p < 0.01). It is difficult to isolate the key variable causing this difference since the surfactants differ both in molecular size and in composition. Shkodra-Pila et al. and Menon et al. demonstrated that PLGA particle size does not increase significantly when increasing the MW of the surfactant PVA. However, there has been significant research illustrating the reduction of PLGA particle size when increasing the surfactant concentration (PVA, 0.5–5% w/v).56 Therefore it is possible that the higher concentration of BSA (30 mg mL−1) compared to PVA (10 mg mL−1) contributed to the smaller particle size of PLGA particles produced with the former, although the different surfactant type may also have contributed.
Another aspect of the NP fabrication process which may affect the PSD is the choice of energy source (Fig. 1). In the current study, this was investigated by the use of a homogeniser and a sonic probe for particles produced using PVA as surfactant and two different polymers (PLGA-E1 and PLGA-E2) as well as with and without drug encapsulation. The evaluation is based on data obtained by DLS (z-average and number weighted mean). Comparing the particle size of empty NPs prepared using ultrasonication and using the two different polymers revealed a significant difference in particle size (z-average and number weighted mean p ≤ 0.05) with number weighted mean of 169 ± 7 nm and 139 ± 1 nm for PLGA-E2-PVA-S and PLGA-E1-PVA-S, respectively. However, a small increase of only 30 nm was observed. When using the homogeniser the number weighted mean were 149 ± 3 nm and 177 ± 19 nm for PLGA-E2-PVA-H and PLGA-E1-PVA-H, respectively, which were not significantly different (p ≥ 0.05). This demonstrates that the Tg of the polymer has only a minor effect on the particle size of empty NPs and only when using sonication to provide the energy input.
For NPs produced using the polymer PLGA-E1, the particle size of empty NPs was significantly different when using the sonic probe compared to the homogeniser to provide the energy input (p ≤ 0.0001 and p ≤ 0.05 for z-average and number weighted mean particle size). Although the use of the homogeniser in fabrication of empty NPs led to a size increase of less than 100 nm compared to the use of ultrasonication, the population of the NPs was more heterogeneous (PDI = 0.2 for PLGA-E1-PVA-H compared to PDI < 0.1 for PLGA-E1-PVA-S) (Fig. 5A).
 | ||
| Fig. 5 Size distribution (diameter) of nano and sub-micron particles. (A) Size by intensity (%) of PLGA-E1-PVA-H NPs and (B) PLGA-E1-PVA-H-cur particles by DLS; (C) FE-SEM image of PLGA-E1-PVA-H-cur at an accelerating voltage of 1 kV, a working distance of 5.9 mm and 10000× magnification. For (A) and (B) each size distribution curve represent a different sample. | ||
In regards to the effect of drug loading, in the case of NPs prepared using the sonic probe, an increase in z-average by 39 and 32 nm (p ≥ 0.05) for PLGA-E1-PVA-S-cur and PLGA-E2-PVA-S-cur was observed after encapsulation but no increase in number weighted mean. The result agrees with a recent study in which the polymer mass was kept constant while varying the initial drug amount where it was found that the effect on particle size was observed at a high drug:polymer ratios of 1:2 or greater.25 For the PLGA-E1-PVA-H-cur particles, a large heterogeneity in particle population was observed compared to the PLGA-E1-PVA-S-cur particles. Furthermore, particles prepared by use of the homogenizer displayed high variability between runs when characterized by DLS as illustrated in ESI,† Fig. S4. This indicates that the composition of the sample changes faster than the time of the DLS run and that sedimentation was possibly occurring. This renders DLS an unsuitable technique for particle size determination for this system.54,57 In this case, the use of high-resolution electron microscopy techniques is recommended, as showed in Fig. 5C. Parot et al. have likewise reported a lack in the accuracy of the DLS technique for evaluating the particle size of NPs larger than 250 nm and with a high PDI.57 The data demonstrate that the particle polydispersity can be manipulated by the choice of energy input where the use of a homogenizer affords the production of mixtures of micron and submicron particles. Previously, the use of such polydisperse particles have been reported to result in prolonged drug release (>21 days).58 It is thus recommended when prolonged treatment is required.25 In contrast, for the synthesis of small and monodisperse NPs, aiming to achieve fast release of the drug, it is recommended to use ultrasonication rather than homogenisation.
Effect of the variables in the isolation process
There are many research papers in the literature that evaluate the influence of parameters of particle synthesis on the PSD,17,26–30,59 and within such studies where the particle isolation process can be considered constant, these parameters can be evaluated. However, without a full description of the isolation process, it is not meaningful to compare data from different research papers. One recent study on PLGA NP synthesis has included evaluation of centrifugation speed and cryo-protectants.27 This section will therefore focus on the effects of the NP isolation processes of washing of the NP pellet and filtration of the NP suspensions while a general description is provided for the effect of centrifugation.The way the centrifugation is carried out and the type of centrifuge used greatly influence the fraction of particles isolated in accordance with Stoke's law, which states that the velocity at which a droplet moves is proportional to the square of its radius. The equation in Fig. 6A can be used to calculate the centrifugation time required to isolate particles of radius, r, at an angular velocity of the centrifuge, ω.60,61 The equation requires the consideration of the distance between centrifugal axis and the sample that experiences the centrifugal force which varies between different centrifuges. Most experimental procedures for isolating NPs reported in the literature specify the revolutions per minute (rpm), however, the centrifugal force experienced by the particles is different between different machines and rotors. It has been previously demonstrated that the size of NPs collected is a function of the rcf, as by increasing the force applied from 9391 × g to 37565 × g (10000 and 20000 rpm) the overall size of NPs collected after washing decreased.27 Consequently, in order to improve accuracy and reproducibility (3R), it is recommended that authors report the rcf as a minimum standard when describing the centrifugation protocol.
 | ||
Fig. 6 (A) Equation used to calculate the centrifuge time (tc) required to pelletize a NP suspension of defined radius (r). Where η = viscosity of liquid, ρ = density of particle, ρ0 = density of supernatant, ω = angular velocity of centrifuge, r1 = distance from surface of the liquid to centrifuge axis and r2 = distance from bottom of centrifuge tube to centrifuge axis. The z-average ( ) and number weighted mean particle size ( ) and number weighted mean particle size ( ) of particles that were (B) unwashed and washed (PLGA-A1-PVA-S), and (C) unfiltered and filtered with a 0.45 μm polypropylene syringe filter (PLGA-A1-PVA-S). The error bars represent the standard deviation of replicate samples (n = 3) and size refers to particle diameter. Accompanying intensity weighted PSD curves for (B) and (C) are shown in (D) and (E), respectively. ) of particles that were (B) unwashed and washed (PLGA-A1-PVA-S), and (C) unfiltered and filtered with a 0.45 μm polypropylene syringe filter (PLGA-A1-PVA-S). The error bars represent the standard deviation of replicate samples (n = 3) and size refers to particle diameter. Accompanying intensity weighted PSD curves for (B) and (C) are shown in (D) and (E), respectively. | ||
An important parameter in particle isolation is the number of centrifugation cycles also referred to as washes. After the first cycle, particles are commonly resuspended in water in order to wash the particles. This is typically done two or three times and serves to remove excess surfactant to avoid potential cytotoxicity.62Fig. 6B shows the particle size of PLGA-E1-PVA-S after being washed twice compared to the absence of a washing process and Fig. 6D shows the PSD in corresponding intensity plots. Unwashed NPs measured by DLS were found to have a number weighted mean particle size of 136 ± 18 nm while NPs subjected to washing had a number weighted particle size of 124 ± 15 nm. No significant difference in the particle size was observed between washed and unwashed samples (p ≥ 0.05). These results agree with a similar study applying a centrifugation protocol of 37565 × g (20000 rpm) where the overall size of NP remained consistent after three washing cycles with a 16 nm increase in the size of PLGA NPs as a result of washing.27 They also observed that by increasing the centrifugation speed this variation in size between washing cycles was minimised which agrees with the results in our study where the centrifugation cycles were done at 76500 × g (25000 rpm).27 Therefore, we recommend the washing process of NPs for biomedical application and highlight that the process does not affect size under the studied conditions.
Considering that the washing procedure for NPs to some extent is done in order to remove excess surfactant, we evaluated changes in the relative amounts of the PLGA and PVA in PLGA-A1-PVA-S NPs before and after one and two washing cycles. This was done using X-ray photoelectron spectroscopy (XPS) as a surface sensitive technique of a lyophilised NP pellet as well as 1H NMR as a bulk technique of a dissolved NP pellet. By evaluating the C 1s and O 1s narrow scans of the pure polymers (PLGA-A1 and PVA) as well as NP pellets, the presence of PVA in the NPs was evident from the [C–O]/[C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O] atomic ratio where the value for the NPs fell within that of the pure polymers (Table 1 and Fig. S5A, ESI†). It was also evident from this data, that there was no detectable change in the relative amounts of PLGA and PVA at the surface when comparing data for a NP pellet before washing with that of a NP pellet after two washing cycles.
O] atomic ratio where the value for the NPs fell within that of the pure polymers (Table 1 and Fig. S5A, ESI†). It was also evident from this data, that there was no detectable change in the relative amounts of PLGA and PVA at the surface when comparing data for a NP pellet before washing with that of a NP pellet after two washing cycles.
| ([C–O])/([CO]) from C 1s |
([C–O])/([CO]) from O 1s |
|
|---|---|---|
| PLGA | 1.0 ± 0.2 | 1.0 ± 0.2 |
| PVA | 6.2 ± 0.8 | 4.6 ± 0.7 |
| PLGA-A1-PVA-S | 1.8 ± 0.3 | 1.6 ± 0.2 |
| PLGA-A1-PVA-S-W | 2.1 ± 0.3 | 1.5 ± 0.2 |
Spek et al. previously studied quantification of PLGA and PVA at the surface and in the bulk of NPs prepared using different PLGA–PEG copolymers.63 That study found ratios of PLGA to PVA at the NP surface close to 10% PVA, in agreement with the results in the current study, which indicate a composition of 10–15% PVA for the washed and unwashed NP samples. 1H NMR was utilized to quantify the relative amount of PLGA and PVA in the bulk of unwashed and washed NP samples based on integrals of peaks from each of the polymers as shown in the ESI† (Fig. S5B). Using this method of quantification, the PVA in the bulk appeared to remain constant after one wash and to decrease after two washes by 6% which is considered non-significant and within the error of the measurement. This data thus indicate that the washing step has only minor or no effect on the PVA content. It should be noted, that washing of the NPs is of course an important step to include when working with drug-loaded NPs in order to remove any drug that is adsorbed to the particle surface rather than encapsulated.
Fig. 6C presents the particle size of PLGA-A1-PVA-S before and after syringe filtration (using a 0.45 μm polypropylene syringe filter), which was done after the washing steps described above. Fig. 6E shows the PSD in corresponding intensity plots. Unfiltered and filtered NPs were both found to have a z-average of 170 ± 10 nm, as well as similar values for the number weighted mean particle size (140 ± 10 nm and 140 ± 20 nm for unfiltered and filtered NPs). No effect on the PDI was observed between unfiltered (PDI = 0.06) and filtered (PDI = 0.07) samples. Microfiltration has been suggested as an effective method for removal of large aggregates and submicron sized particles thereby reducing the polydispersity and increasing the reliability of DLS measurements.64 In accordance with this, in our work, the filtration of NPs samples produced no significant effect on the particle size which can be related to the NPs displaying a monodisperse PSD already after the washing steps. Despite this, we concur with Hackley and Clogston on their stance on particle purification and suggest that the effect of filtration during the isolation of NPs be evaluated.
Drug encapsulation
Curcumin has previously been studied in single drug encapsulation and co-encapsulation with other drugs using PLGA NPs.43–46,65 Although there is extensive literature on the use of curcumin either as a therapeutic compound or a drug model, there are several limitations regarding its encapsulation and release studies.65–71 Considering the challenges of chemical instability and poor solubility in water, we have chosen curcumin as a challenging drug model to investigate the variables involved in the fabrication of PLGA NPs that might affect drug encapsulation. Specifically, we have investigated the effect of the polymer type and energy input on curcumin encapsulation as well as different methods for determination of the drug encapsulation. Due to the high binding affinity of curcumin with BSA (K = 3.3 (±0.8) × 104 M−1)72 we restricted our drug encapsulation study to the use of PVA as the surfactant. Caution must be applied when using proteins for stabilization of drug loaded NP formulations to avoid binding of drug molecules to the surfactant, both in solution and at the surface of the NPs as this can reduce encapsulation efficiency and loading capacity.Determination of EE% and LC% using spectrophotometric methods is known to be dependent on the solvent system, additives and their respective concentrations. Standard curves therefore must be prepared in the solvent system corresponding to that of the unknown sample. This is illustrated with the standard curves used for the indirect method (Fig. 7A) and the direct method (Fig. 7B) in the current study. Furthermore, Fig. 7C illustrates that despite the fixed curcumin concentration there is a change in absorbance of curcumin (0.8 mg/100 mL, λmax = 408–415 nm) caused by an increasing PVA concentration (5–30 mg/100 mL) where the absorbance value increases from 0.674 to 0.825. Considering the large excess of PVA used in the particle synthesis we, therefore, recommend the addition of PVA at the initial concentration (20 mg/100 mL) when producing in the standard curve used for the indirect method, which was also done in the current study. This highlights one of the difficulties of the use of indirect determination of EE% and LC% using spectrophotometric methods. The increase in curcumin absorbance with respect to PVA concentration was also shown by Shah et al., where the molar extinction coefficient of PVA-bound-curcumin was found to increase by 50% compared to free curcumin.73 Using the relationship of Beer Lambert's law, the higher molar extinction coefficient accounts for the increased absorbance of PVA-bound-curcumin.
 | ||
| Fig. 7 (A) a standard curve of curcumin in water and PVA, where [PVA] = 20 mg/100 mL, and a line of best fit of A = (0.0465 ± 0.0007) × ccur (n = 1); (B) a standard curve of curcumin in 2:1 THF/DMSO where the line of best fit was A = (0.0514 ± 0.0005) × ccur and the error bars represent the standard deviation of replicate samples (n = 3); (C) the UV-visible absorbance spectrum of curcumin (0.8 mg/100 mL) and variable concentrations of PVA as indicated (5–30 mg/100 mL); (D) 1H NMR spectrum (500 MHz) of PLGA-A1-PVA-S-cur pellet in DMSO-d6 (n = 1). The peak/s shown in orange and green belong to curcumin and PLGA, respectively. The integral ratio between a series of curcumin and PLGA peaks can then be used to calculate the encapsulation efficiency (equation and sample calculation is provided in ESI†). | ||
Loading capacity and encapsulation efficiency were evaluated using both direct and indirect methods and the resulting data are reported in Table 2. Using the direct method of determining the drug content in lyophilised NPs, the EE% and LC% of NPs fabricated with the sonic probe as an energy source are in a range of 44–58% and 6–9%, respectively. Furthermore, no significant difference in curcumin encapsulation was observed between the three polymers (differing with regards to Tg and end-group) (p ≥ 0.05). This level of curcumin encapsulation mimics that of a recently reported study using similar fabrication conditions, where a prolonged sonication time of 10 min at 90% amplitude was applied.45 Specifically, we obtained an EE% that was 10% higher than the reported in the recent study and the LC% was the same in the two studies.45 This high curcumin encapsulation efficiency has previously been attributed to intermolecular interactions between the drug and the polymer.43 Similar values for LC% of PLGA-E1 and PLGA-E2 were obtained by the direct method when the homogeniser was used as the source of energy (Table 2). However, it was found that the curcumin EE% was 10–20% lower than obtained when the sonic probe was used.
:10a
| Polymer | Direct method | Indirect method | |||
|---|---|---|---|---|---|
| EE (%) | LC (%) | EE (%) | LC (%) | Yield (%) | |
|
a (n = 3 for curcumin, n = 2 for RhB).
b Polymer/drug ratio 2:10.
|
|||||
| PLGA-E1-PVA-S-cur | 58 ± 9 | 6 ± 2 | 36 ± 11 | 4 ± 1 | 88 ± 11 |
| PLGA-E2-PVA-S-cur | 46 ± 9 | 7 ± 2 | 38 ± 8 | 6 ± 1 | 58 ± 4 |
| PLGA-A1-PVA-S-cur | 44 ± 1 | 9 ± 1 | 53 ± 1 | 11 ± 1 | 95 ± 7 |
| PLGA-E1-PVA-S-RhB | — | — | 7 ± 1 | 1 ± 1 | 74 ± 5 |
| PLGA-E1-PVA-S-RhB2b | — | — | 38 ± 4 | 12 ± 4 | 52 ± 11 |
| PLGA-E1-PVA-H-cur | 32 ± 7 | 6 ± 2 | 42 ± 6 | 8 ± 2 | 49 ± 14 |
| PLGA-E2-PVA-H-cur | 36 ± 5 | 5 ± 1 | 57 ± 7 | 7 ± 1 | 70 ± 5 |
| PLGA-E1-PVA-H-RhB | — | — | 10 ± 4 | 3 ± 1 | 35 ± 5 |
| PLGA-E1-PVA-H-RhB2b | — | — | 7 ± 7 | 3 ± 3 | 43 ± 1 |
In the indirect approach, the concentration of the free drug is measured and the difference from the initial drug mass is used to estimate drug encapsulation. This method is often used as it does not require the destruction of the NPs. The EE% and LC% obtained by this method were significantly different from the values obtained by the direct method for PLGA-E2-PVA-H-cur (p ≤ 0.05) and PLGA-A1-PVA-S-cur (p ≤ 0.001). In both cases, the indirect method estimated a higher EE% compared to the direct method. This result can be attributed to curcumin loss by degradation during the fabrication process in agreement with previously reported 80% curcumin degradation within 4 hours at 24 °C.71 In general, when a direct method cannot be used, it is recommended that measurements of the supernatant are taken immediately after fabrication to avoid overestimation of EE% and LC% and that the time gap between NPs washing (or curcumin extraction) and absorbance measurement should be reported to account for the curcumin degradation during these steps.
The indirect method was used to quantify the encapsulation of an alternative drug model, Rhodamine B (RhB) at polymer/drug ratios of 1:10, 2:10, and 5:10. The overall yield of PLGA-RhB NPs fabricated using ultrasonication was higher than when using homogeniser. It was observed that increasing the drug/polymer ratio of NPs prepared using ultrasonication to 2:10, promotes an improvement in EE% and LC%. However, no further improvement was observed when increasing the drug/polymer ratio to 5:10. Differently, in particles prepared using homogeniser, the variation in polymer/drug ratio did not show an effect in EE% and LC%, and were approximately 7% and 3%, respectively. Regardless of the energy source applied, curcumin presented a higher encapsulation efficiency than RhB. Considering that the single ESE technique is designed for the encapsulation of hydrophobic compounds, this difference and be attributed to the difference in the water solubility of the drugs.4 The solubility of RhB is more than 10-fold higher than curcumin, therefore, during the evaporation process the excess of RhB can interact with the water phase leading to the decrease of drug entrapment by the NPs.69
As an alternative to the use of spectrophotometric methods (including e.g. HPLC) for evaluating the EE% as well as the amount of drug loaded into the NP, we explored the use of 1H NMR of a lyophilised PLGA-A1-PVA-S-cur pellet dissolved in DMSO-d6. This method is similar in principle to the direct method outlined in Fig. 2 as the amount of drug relative to polymer within the NP can be determined from the 1H NMR spectrum. Specifically, the EE% can be determined by comparing the mass ratio of drug to polymer as determined from the 1H NMR data, with the mass ratio used in the NP synthesis. Fig. 7D shows the resulting 1H NMR spectrum where the curcumin peaks in the aromatic region are compared to the LA (–CH) peak at δ 5.2 ppm using the equations and integration shown in ESI† Fig. S6. Based on this, a mean EE% of 73 ± 10% was determined. Furthermore, the mass ratio of drug to polymer (mdrug(e)/mpolymer) expressed as a percentage was found to be 7 ± 1%. This offers a different method for evaluating the amount of drug loaded into the NP. The EE% obtained by 1H NMR is significantly larger than that determined by UV-Vis based quantification for the PLGA-A1-PVA-S-cur NPs presented in Table 2. An important consideration for the use of this method is the diminishing accuracy of EE% when the yield (%) is reduced. However, it is in general considered more accurate as curcumin degradation will affect the spectrophotometric method to a much larger extent. Determination of EE% using NMR is advised for drug loaded NPs where the direct method is feasible and the drug cannot be quantified either as a result of degradation or lacking a chromophore.
Conclusions
This work has presented a systematic study demonstrating the repeatability and replicability of PLGA NP synthesis using the ESE technique as well as the influence of various controlled fabrication variables during the synthesis and isolation processes. To determine size, DLS (z-average, number mean, PDI), NTA, and electron microscopy (PSD) were used. Among the methods, TEM presented as a preferred option for determining size and morphology of particles and it is therefore recommended that this be used in combination with the common technique DLS. When reporting DLS measurements, we support previous literature recommending providing PSD curves (intensity, number, or volume) in order to improve reproducibility.We investigated the effect of polymer type, surfactant type, energy source, the magnitude of energy input, filtration, washing, and drug encapsulation on the size of NPs. The source of energy and magnitude of the energy input as well as the choice of surfactant showed to have the most significant influence on NP size. In general, it is recommended that ultrasonication is used when NPs of low polydispersity and a mean diameter below 200 nm are required and that the sonication time and amplitude are optimized in individual laboratories. If on the other hand, NPs of high polydispersity are required to obtain a desired drug release profile, the use of a homogenizer to provide the energy input is recommended. In regards to the choice to the choice of surfactant a smaller particle size was obtained in the current study using BSA, however, we caution its use due to high affinity to hydrophobic drug molecules. We, therefore, recommend the use of PVA which is commonly used in the literature.
In our work, filtration and washing have shown no influence on the size of NPs. However, it is important to note that the conditions of washing and whether filtration was required should be reported in order to allow direct comparison to the literature. Previous studies have demonstrated the effect of centrifugation and we likewise recommend reporting the rcf as a minimum standard when describing the centrifugation protocol in order to improve reproducibility.
The use of spectrophotometric methods for drug quantification by either direct or indirect methods requires the standard curve prepared using the same solvent system and with the same additives. Thus, for the presented protocol, PVA must be added to the solution when using the indirect method. In an alternative approach to spectrophotometric methods we evaluated the use of 1H NMR for determining the amount of drug encapsulated and the EE%. The use of this method is advised for drug loaded NPs where the direct method is feasible and the drug cannot be quantified either as a result of degradation or if it is lacking a chromophore.
In summary, ESE presented as a robust method for PLGA NP fabrication. In this study, we demonstrated that properties can potentially be adjusted to the required application by modification of multiple variables involved in the synthesis and guideline for PLGA-based NP fabrication and drug encapsulation quantification was provided.
Author contributions
Conceptualization, L. G. and A. A.; methodology, B. C. G., H. P., A. A. and L. G.; validation, L. G., A. W., F. H. and A. A.; formal analysis, B. C. G., H. P., D. B.; resources, L. G., A. W. and A. A.; data curation, B. C. G., H. P., D. B.; writing—original draft preparation, B. C. G., H. P., L. G., D. B.; writing—review and editing, B. C. G., H. P., F. H., A. W., A. A., L. G.; supervision, L. G. and A. A.; project administration, L. G. All authors have read and agreed to the published version of the manuscript. B. C. G. and H. P. equally contributed to this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was received from the National Health and Medical Research Council (NHMRC), Australia, for the Early Career Research Fellowship; APP1123340, 2017–2021 (A. A.), The University of Queensland for UQ Research Training scholarship (B. C. G.) and the Australian Government RTP scholarship (H. P.). The authors acknowledge the Australian Research Council (CE140100036) and the National Health and Medical Research Council (APP1107723) for support of this research. The authors thank Asim Mushtaq for technical support collecting TEM images and Alexandra Mutch for technical support for collecting the XPS data. The authors acknowledge the facilities and the scientific and technical assistance of the Australian Microscopy & Microanalysis Research Facility at the Centre for Microscopy and Microanalysis, The University of Queensland, Australia.References
- D. Ding and Q. Zhu, Recent advances of PLGA micro/nanoparticles for the delivery of biomacromolecular therapeutics, Mater. Sci. Eng., C, 2018, 92, 1041–1060 CrossRef CAS PubMed.
- F. Y. Han, K. J. Thurecht, A. K. Whittaker and M. T. Smith, Bioerodable PLGA-Based Microparticles for Producing Sustained-Release Drug Formulations and Strategies for Improving Drug Loading, Front. Pharmacol., 2016, 7, 185 Search PubMed.
- B. Laycock, M. Nikolić, J. M. Colwell, E. Gauthier, P. Halley, S. Bottle and G. George, Lifetime prediction of biodegradable polymers, Prog. Polym. Sci., 2017, 71, 144–189 CrossRef CAS.
- Y. Dang and J. Guan, Nanoparticle-based drug delivery systems for cancer therapy, Smart Mater. Med., 2020, 1, 10–19 CrossRef.
- S. Rezvantalab, N. I. Drude, M. K. Moraveji, N. Güvener, E. K. Koons, Y. Shi, T. Lammers and F. Kiessling, PLGA-Based Nanoparticles in Cancer Treatment, Front. Pharmacol., 2018, 9, 1260 CrossRef CAS PubMed.
- F. Qi, J. Wu, H. Li and G. Ma, Recent research and development of PLGA/PLA microspheres/nanoparticles: A review in scientific and industrial aspects, Front. Chem. Sci. Eng., 2018, 13(1), 1–14 Search PubMed.
- S. Haque, C. W. Pouton, M. P. McIntosh, D. B. Ascher, D. W. Keizer, M. R. Whittaker and L. M. Kaminskas, The impact of size and charge on the pulmonary pharmacokinetics and immunological response of the lungs to PLGA nanoparticles after intratracheal administration to rats, Nanomedicine, 2020, 30, 102291 CrossRef CAS.
- Y. Luo, L. Yang, P. Feng, H. Qiu, X. Wu, S. Lu, M. Zhou, L. Xu and Y. Zhu, Pranoprofen Nanoparticles With Poly(L-Lactide)-b-Poly(Ethylene Glycol)-b-Poly(L-Lactide) as the Matrix Toward Improving Ocular Anti-inflammation, Front. Bioeng. Biotechnol., 2020, 8, 581621 CrossRef PubMed.
- X. Pan, X. Liu, X. Zhuang, Y. Liu and S. Li, Co-delivery of dexamethasone and melatonin by drugs laden PLGA nanoparticles for the treatment of glaucoma, J. Drug Delivery Sci. Technol., 2020, 60, 102086 CrossRef CAS.
- T. L. Phaam and D. W. Kim, Poly(lactic-co-glycolic acid) nanomaterial-based treatment options for pain management: a review, Nanomedicine, 2020, 15(19), 1897–1913 CrossRef PubMed.
- S. Shah, D. Cristopher, S. Sharma, M. Soniwala and J. Chavda, Inhalable linezolid loaded PLGA nanoparticles for treatment of tuberculosis: Design, development and in vitro evaluation, J. Drug Delivery Sci. Technol., 2020, 60, 102013 CrossRef CAS.
- Z. Zhang, D. Li, X. Ma, X. Li, Z. Guo, Y. Liu and S. Zheng, Carboxylated nanodiamond-mediated NH2-PLGA nanoparticle-encapsulated fig polysaccharides for strongly enhanced immune responses in vitro and in vivo, Int. J. Biol. Macromol., 2020, 165, 1331–1345 CrossRef CAS PubMed.
- J. Yang, L. Luo, Y. Oh, T. Meng, G. Chai, S. Xia, D. Emmert, B. Wang, C. G. Eberhart, S. Lee, W. J. Stark, L. M. Ensign, J. Hanes and Q. Xu, Sunitinib malate-loaded biodegradable microspheres for the prevention of corneal neovascularization in rats, J. Controlled Release, 2020, 327, 456–466 CrossRef CAS PubMed.
- W. K. Lau, D. Dharmasena, H. Horsley, N. V. Jafari, J. Malone-Lee, E. Stride, M. Edirisinghe and J. L. Rohn, Novel antibiotic-loaded particles conferring eradication of deep tissue bacterial reservoirs for the treatment of chronic urinary tract infection, J. Controlled Release, 2020, 328, 490–502 CrossRef CAS PubMed.
- E. Barcia, V. Sandoval, A. Fernandez-Carballido and S. Negro, Flunarizine-loaded microparticles for the prophylaxis of migraine, J. Drug Delivery Sci. Technol., 2020, 60, 102012 CrossRef CAS.
- M. K. Anwer, M. Mohammad, E. Ezzeldin, F. Fatima, A. Alalaiwe and M. Iqbal, Preparation of sustained release apremilast-loaded PLGA nanoparticles: in vitro characterization and in vivo pharmacokinetic study in rats, Int. J. Nanomed., 2019, 14, 1587–1595 CrossRef CAS PubMed.
- C. Busatto, J. Pesoa, I. Helbling, J. Luna and D. Estenoz, Effect of particle size, polydispersity and polymer degradation on progesterone release from PLGA microparticles: Experimental and mathematical modeling, Int. J. Pharm., 2018, 536(1), 360–369 CrossRef CAS PubMed.
- S. A. Damiati, D. Rossi, H. N. Joensson and S. Damiati, Artificial intelligence application for rapid fabrication of size-tunable PLGA microparticles in microfluidics, Sci. Rep., 2020, 10(1), 19517 CrossRef CAS PubMed.
- N. Lababidi, C. V. Montefusco-Pereira, C. de Souza Carvalho-Wodarz, C.-M. Lehr and M. Schneider, Spray-dried multidrug particles for pulmonary co-delivery of antibiotics with N-acetylcysteine and curcumin-loaded PLGA-nanoparticles, Eur. J. Pharm. Biopharm., 2020, 157, 200–210 CrossRef CAS PubMed.
- E. Nance, C. Zhang, T.-Y. Shih, Q. Xu, B. S. Schuster and J. Hanes, Brain-Penetrating Nanoparticles Improve Paclitaxel Efficacy in Malignant Glioma Following Local Administration, ACS Nano, 2014, 8(10), 10655–10664 CrossRef CAS PubMed.
- F. Hu, R. Zhang, W. Guo, T. Yan, X. He, F. Hu, F. Ren, X. Ma, J. Lei and W. Zheng, PEGylated-PLGA Nanoparticles Coated with pH-Responsive Tannic Acid–Fe(III) Complexes for Reduced Premature Doxorubicin Release and Enhanced Targeting in Breast Cancer, Mol. Pharmaceutics, 2021, 18(6), 2161–2173 CrossRef CAS.
- M. Zhu, A. K. Whittaker, X. Jiang, R. Tang, X. Li, W. Xu, C. Fu, M. T. Smith and F. Y. Han, Use of Microfluidics to Fabricate Bioerodable Lipid Hybrid Nanoparticles Containing Hydromorphone or Ketamine for the Relief of Intractable Pain, Pharm. Res., 2020, 37(10), 211 CrossRef CAS.
- G. Baidya, R. Tiwary, M. Mudassir, N. Singh, S. Saha, K. Chosdol, S. Sinha and P. Chattopadhyay, Passive internalization and active extrusion determines PLGA-nanoparticle concentration in cancer cell lines, Nanomedicine, 2020, 15(23), 2229–2239 CrossRef CAS PubMed.
- Y. Malinovskaya, P. Melnikov, V. Baklaushev, A. Gabashvili, N. Osipova, S. Mantrov, Y. Ermolenko, O. Maksimenko, M. Gorshkova, V. Balabanyan, J. Kreuter and S. Gelperina, Delivery of doxorubicin-loaded PLGA nanoparticles into U87 human glioblastoma cells, Int. J. Pharm., 2017, 524(1–2), 77–90 CrossRef CAS PubMed.
- C. Isely, M. A. Hendley, K. P. Murphy, S. Kader, P. Annamalai, E. Jabbari and R. M. Gower, Development of microparticles for controlled release of resveratrol to adipose tissue and the impact of drug loading on particle morphology and drug release, Int. J. Pharm., 2019, 568, 118469 CrossRef CAS.
- N. Sharma, P. Madan and S. Lin, Effect of process and formulation variables on the preparation of parenteral paclitaxel-loaded biodegradable polymeric nanoparticles: A co-surfactant study, Asian J. Pharm. Sci., 2016, 11(3), 404–416 CrossRef.
- K. Y. Hernández-Giottonini, R. J. Rodríguez-Córdova, C. A. Gutiérrez-Valenzuela, O. Peñuñuri-Miranda, P. Zavala-Rivera, P. Guerrero-Germán and A. Lucero-Acuña, PLGA nanoparticle preparations by emulsification and nanoprecipitation techniques: effects of formulation parameters, RSC Adv., 2020, 10(8), 4218–4231 RSC.
- K. Kızılbey, Optimization of Rutin-Loaded PLGA Nanoparticles Synthesized by Single-Emulsion Solvent Evaporation Method, ACS Omega, 2019, 4(1), 555–562 CrossRef.
- H. K. Makadia and S. J. Siegel, Poly Lactic-co-Glycolic Acid (PLGA) as biodegradable controlled drug delivery carrier, Polymers, 2011, 3(3), 1377–1397 CrossRef CAS.
- R. Jenjob, T. Phakkeeree, F. Seidi, M. Theerasilp and D. Crespy, Emulsion Techniques for the Production of Pharmacological Nanoparticles, Macromol. Biosci., 2019, 19(6), e1900063 CrossRef.
- F. Caputo, J. Clogston, L. Calzolai, M. Rösslein and A. Prina-Mello, Measuring particle size distribution of nanoparticle enabled medicinal products, the joint view of EUNCL and NCI-NCL. A step by step approach combining orthogonal measurements with increasing complexity, J. Controlled Release, 2019, 299, 31–43 CrossRef CAS.
- J. Ghitman, E. I. Biru, R. Stan and H. Iovu, Review of hybrid PLGA nanoparticles: Future of smart drug delivery and theranostics medicine, Mater. Des., 2020, 193, 108805 CrossRef CAS.
- H. Zhong, G. Chan, Y. Hu, H. Hu and D. Ouyang, A comprehensive map of FDA-approved pharmaceutical products, Pharmaceutics, 2018, 10(4), 263 CrossRef CAS PubMed.
- S. Gioria, F. Caputo, P. Urbán, C. M. Maguire, S. Bremer-Hoffmann, A. Prina-Mello, L. Calzolai and D. Mehn, Are existing standard methods suitable for the evaluation of nanomedicines: some case studies, Nanomedicine, 2018, 13(5), 539–554 CrossRef CAS PubMed.
- J.-B. Coty and C. Vauthier, Characterization of nanomedicines: A reflection on a field under construction needed for clinical translation success, J. Controlled Release, 2018, 275, 254–268 CrossRef CAS PubMed.
- S. R. D'Mello, C. N. Cruz, M.-L. Chen, M. Kapoor, S. L. Lee and K. M. Tyner, The evolving landscape of drug products containing nanomaterials in the United States, Nat. Nanotechnol., 2017, 12(6), 523–529 CrossRef.
- M. Faria, M. Björnmalm, K. J. Thurecht, S. J. Kent, R. G. Parton, M. Kavallaris, A. P. R. Johnston, J. J. Gooding, S. R. Corrie, B. J. Boyd, P. Thordarson, A. K. Whittaker, M. M. Stevens, C. A. Prestidge, C. J. H. Porter, W. J. Parak, T. P. Davis, E. J. Crampin and F. Caruso, Minimum information reporting in bio–nano experimental literature, Nat. Nanotechnol., 2018, 13(9), 777–785 CrossRef CAS PubMed.
- H. E. Plesser, Reproducibility vs. Replicability: A brief history of a confused terminology, Front. Neuroinform., 2018, 11, 76 CrossRef PubMed.
- S. L. McArthur, Repeatability, Reproducibility, and Replicability: Tackling the 3R challenge in biointerface science and engineering, Biointerphases, 2019, 14(2), 020201 CrossRef PubMed.
- A. Borah, S. C. Pillai, A. K. Rochani, V. Palaninathan, Y. Nakajima, T. Maekawa and D. S. Kumar, GANT61 and curcumin-loaded PLGA nanoparticles for GLI1 and PI3K/Akt-mediated inhibition in breast adenocarcinoma, Nanotechnology, 2020, 31(18), 185102 CrossRef CAS PubMed.
- A. Lucero-Acuña, J. J. Jeffery, E. R. Abril, R. B. Nagle, R. Guzman, M. D. Pagel and E. J. Meuillet, Nanoparticle delivery of an akt/pdk1 inhibitor improves the therapeutic effect in pancreatic cancer, Int. J. Nanomed., 2014, 9(1), 5653–5665 Search PubMed.
- R. A. Miller, J. M. Brady and D. E. Cutright, Degradation rates of oral resorbable implants (polylactates and polyglycolates): Rate modification with changes in PLA/PGA copolymer ratios, J. Biomed. Mater. Res., 1977, 11(5), 711–719 CrossRef CAS PubMed.
- M. A. Akl, A. Kartal-Hodzic, T. Oksanen, H. R. Ismael, M. M. Afouna, M. Yliperttula, A. M. Samy and T. Viitala, Factorial design formulation optimization and in vitro characterization of curcumin-loaded PLGA nanoparticles for colon delivery, J. Drug Delivery Sci. Technol., 2016, 32, 10–20 CrossRef CAS.
- R. Mukhopadhyay, R. Sen, B. Paul, J. Kazi, S. Ganguly and M. C. Debnath, Gemcitabine Co-Encapsulated with Curcumin in Folate Decorated PLGA Nanoparticles a Novel Approach to Treat Breast Adenocarcinoma, Pharm. Res., 2020, 37(3), 56 CrossRef CAS.
- S. Shahgordi, M. Sankian, Y. Yazdani, K. Mashayekhi, S. Hasan Ayati, M. Sadeghi, M. Saeidi and M. Hashemi, Immune responses modulation by curcumin and allergen encapsulated into PLGA nanoparticles in mice model of rhinitis allergic through sublingual immunotherapy, Int. Immunopharmacol., 2020, 84, 106525 CrossRef CAS PubMed.
- M. A. Vakilinezhad, A. Amini, T. Dara and S. Alipour, Methotrexate and Curcumin co-encapsulated PLGA nanoparticles as a potential breast cancer therapeutic system: In vitro and in vivo evaluation, Colloids Surf., B, 2019, 184, 110515 CrossRef CAS PubMed.
- M. M. Yallapu, B. K. Gupta, M. Jaggi and S. C. Chauhan, Fabrication of curcumin encapsulated PLGA nanoparticles for improved therapeutic effects in metastatic cancer cells, J. Colloid Interface Sci., 2010, 351(1), 19–29 CrossRef CAS PubMed.
- B. Shkodra-Pula, C. Grune, A. Traeger, A. Vollrath, S. Schubert, D. Fischer and U. S. Schubert, Effect of surfactant on the size and stability of PLGA nanoparticles encapsulating a protein kinase C inhibitor, Int. J. Pharm., 2019, 566, 756–764 CrossRef CAS PubMed.
- P. Jiang, D. Yu, W. Zhang, Z. Mao and C. Gao, Influence of bovine serum albumin coated poly(lactic-co-glycolic acid) particles on differentiation of mesenchymal stem cells, RSC Adv., 2015, 5(51), 40924–40931 RSC.
- A. Schavkan, C. Gollwitzer, R. Garcia-Diez, M. Krumrey, C. Minelli, D. Bartczak, S. Cuello-Nuñez, H. Goenaga-Infante, J. Rissler, E. Sjöström, G. B. Baur, K. Vasilatou, A. G. Shard, Correction and A. Schavkan, et al., Number Concentration of Gold Nanoparticles in Suspension: SAXS and spICPMS as Traceable Methods Compared to Laboratory Methods, Nanomaterials, 2019, 9, 502 CrossRef CAS PubMed , Nanomaterials 2019, 9(8), 1060.
- A. George, P. A. Shah and P. S. Shrivastav, Natural biodegradable polymers based nano-formulations for drug delivery: a review, Int. J. Pharm., 2019, 561, 244–264 CrossRef CAS PubMed.
- W. Anderson, D. Kozak, V. A. Coleman, Å. K. Jämting and M. Trau, A comparative study of submicron particle sizing platforms: accuracy, precision and resolution analysis of polydisperse particle size distributions, J. Colloid Interface Sci., 2013, 405, 322–330 CrossRef CAS.
- F. Giorgi, J. M. Curran, D. Gilliland, R. La Spina, M. Whelan and E. A. Patterson, Limitations of Nanoparticles Size Characterization by Asymmetric Flow Field-Fractionation Coupled with Online Dynamic Light Scattering, Chromatographia, 2021, 84(2), 199–206 CrossRef CAS.
- F. Caputo, D. Mehn, J. D. Clogston, M. Rösslein, A. Prina-Mello, S. E. Borgos, S. Gioria and L. Calzolai, Asymmetric-flow field-flow fractionation for measuring particle size, drug loading and (in)stability of nanopharmaceuticals. The joint view of European Union Nanomedicine Characterization Laboratory and National Cancer Institute – Nanotechnology Characterization Laboratory, J. Chromatogr. A, 2021, 1635 Search PubMed.
- J. S. Taurozzi, V. A. Hackley and M. R. Wiesner, Ultrasonic dispersion of nanoparticles for environmental, health and safety assessment – issues and recommendations, Nanotoxicology, 2011, 5(4), 711–729 CrossRef CAS PubMed.
- S. K. Sahoo, J. Panyam, S. Prabha and V. Labhasetwar, Residual polyvinyl alcohol associated with poly (D,L-lactide-co-glycolide) nanoparticles affects their physical properties and cellular uptake, J. Controlled Release, 2002, 82(1), 105–114 CrossRef CAS.
- J. Parot, F. Caputo, D. Mehn, V. A. Hackley and L. Calzolai, Physical characterization of liposomal drug formulations using multi-detector asymmetrical-flow field flow fractionation, J. Controlled Release, 2020, 320, 495–510 CrossRef CAS PubMed.
- F. Y. Han, K. J. Thurecht, A.-L. Lam, A. K. Whittaker and M. T. Smith, Novel Polymeric Bioerodable Microparticles for Prolonged-Release Intrathecal Delivery of Analgesic Agents for Relief of Intractable Cancer-Related Pain, J. Pharm. Sci., 2015, 104(7), 2334 CrossRef CAS PubMed.
- M. Halayqa and U. Domańska, PLGA biodegradable nanoparticles containing perphenazine or chlorpromazine hydrochloride: effect of formulation and release, Int. J. Mol. Sci., 2014, 15(12), 23909–23923 CrossRef PubMed.
- K. T. Whitby, A Rapid General Purpose Centrifuge Sedimentation Method for Measurement of Size Distribution of Small Particles—Apparatus and Method, J. Air Pollut. Control Assoc., 1955, 5(2), 120–132 CAS.
- R. Jenjob, T. Phakkeeree, F. Seidi, M. Theerasilp and D. Crespy, Emulsion Techniques for the Production of Pharmacological Nanoparticles, Macromol. Biosci., 2019, 19(6), 1900063 CrossRef PubMed.
- N. Sharma, P. Madan and S. Lin, Effect of process and formulation variables on the preparation of parenteral paclitaxel-loaded biodegradable polymeric nanoparticles: a co-surfactant study, Asian J. Pharm. Sci., 2016, 11(3), 404–416 CrossRef.
- S. Spek, M. Haeuser, M. M. Schaefer and K. Langer, Characterisation of PEGylated PLGA nanoparticles comparing the nanoparticle bulk to the particle surface using UV/vis spectroscopy, SEC, 1H NMR spectroscopy, and X-ray photoelectron spectroscopy, Appl. Surf. Sci., 2015, 347, 378–385 CrossRef CAS.
- V. A. Hackley and J. D. Clogston, Measuring the Hydrodynamic Size of Nanoparticles in Aqueous Media Using Batch-Mode Dynamic Light Scattering, Characterization of Nanoparticles Intended for Drug Delivery, 2010, pp. 35–52 Search PubMed.
- M. A. Akl, A. Kartal-Hodzic, T. Suutari, T. Oksanen, I. M. Montagner, A. Rosato, H. R. Ismael, M. I. Afouna, P. Caliceti, M. Yliperttula, A. M. Samy, F. Mastrotto, S. Salmaso and T. Viitala, Real-Time Label-Free Targeting Assessment and in Vitro Characterization of Curcumin-Loaded Poly-lactic-co-glycolic Acid Nanoparticles for Oral Colon Targeting, ACS Omega, 2019, 4(16), 16878–16890 CrossRef CAS PubMed.
- K. M. Nelson, J. L. Dahlin, J. Bisson, J. Graham, G. F. Pauli and M. A. Walters, The Essential Medicinal Chemistry of Curcumin, J. Med. Chem., 2017, 60(5), 1620–1637 CrossRef CAS PubMed.
- O. Naksuriya, M. J. van Steenbergen, J. Sastre Torano, S. Okonogi and W. E. Hennink, A Kinetic Degradation Study of Curcumin in Its Free Form and Loaded in Polymeric Micelles, AAPS J., 2016, 18(3), 777–787 CrossRef CAS.
- O. D. Okagu, O. Verma, D. J. McClements and C. C. Udenigwe, Utilization of insect proteins to formulate nutraceutical delivery systems: Encapsulation and release of curcumin using mealworm protein-chitosan nano-complexes, Int. J. Biol. Macromol., 2020, 151, 333–343 CrossRef CAS PubMed.
- K. Suresh and A. Nangia, Curcumin: Pharmaceutical solids as a platform to improve solubility and bioavailability, CrystEngComm, 2018, 20(24), 3277–3296 RSC.
- Q. Guo, J. Su, X. Shu, F. Yuan, L. Mao, J. Liu and Y. Gao, Production and characterization of pea protein isolate-pectin complexes for delivery of curcumin: Effect of esterified degree of pectin, Food Hydrocolloids, 2020, 105, 105777 CrossRef CAS.
- Z. Liu, Q. Wu, J. He, F. Vriesekoop and H. Liang, Crystal-Seeded Growth of pH-Responsive Metal–Organic Frameworks for Enhancing Encapsulation, Stability, and Bioactivity of Hydrophobicity Compounds, ACS Biomater. Sci. Eng., 2019, 5(12), 6581–6589 CrossRef CAS PubMed.
- P. Bourassa, C. D. Kanakis, P. Tarantilis, M. G. Pollissiou and H. A. Tajmir-Riahi, Resveratrol, Genistein, and Curcumin Bind Bovine Serum Albumin, J. Phys. Chem. B, 2010, 114(9), 3348–3354 CrossRef CAS PubMed.
- C. P. Shah, B. Mishra, M. Kumar, K. I. Priyadarsini and P. N. Bajaj, Binding studies of curcumin to polyvinyl alcohol/polyvinyl alcohol hydrogel and its delivery to liposomes, Curr. Sci., 2008, 95(10), 1426–1432 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00410g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |