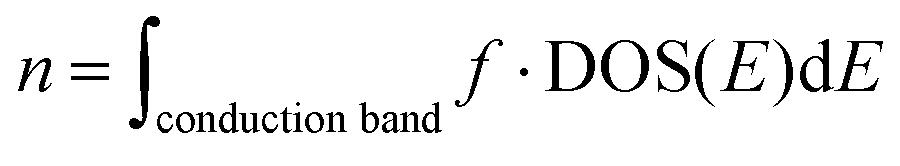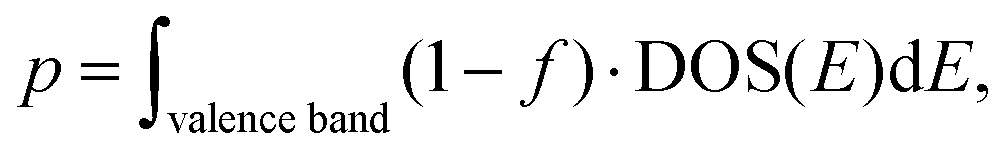DOI:
10.1039/D1MA00328C
(Paper)
Mater. Adv., 2021,
2, 4694-4701
Can T-carbon serve as a Li storage material and a Li battery anode?†‡
Received
10th April 2021
, Accepted 28th May 2021
First published on 28th May 2021
Abstract
Carbon can form a wide variety of bulk structures. The possibility of carbon-based materials to be utilized in future energy applications has attracted extensive attention. T-carbon is a newly discovered diamond-like phase with an acetylene-bond supported hollow structure. This feature inspires us to consider its possibility of serving as a high-capacity Li storage material and a Li battery anode. Density functional theory calculations are performed to investigate the structure evolution, energy spectrum, stability, and electrode potential of Li-loaded T-carbon. The adsorption of T-carbon to Li atoms originates from the valence electrons of Li filling into the antibonding π* orbitals of acetylene bonds. The maximum theoretical Li capacity is three times that of graphite (1116 mA h g−1). At low Li density, too strong adsorption is disadvantageous to the release of Li and the discharge of the anode. At high Li density, Li atoms push against each other and fight against the adsorption of T-carbon. Such an advantageous issue leads to a decrease in anode potential. The significant difference of Li adsorption between low and high density makes a “dead zone” in which only half of the Li capacity could be useful for the discharge of the anode.
I. Introduction
As a fundamental element on the earth, carbon possesses the unique ability to form a variety of complex structures. With sp3, sp2, and sp hybridization, carbon-based structures exhibit chemical and biological diversity. Besides graphite and diamond in nature, people have synthesized many new carbon allotropes including fullerenes,1 carbon nanotubes,2 and graphene.3 With the development of synthesis technology, a large number of carbon allotropes with sp bonds or sp–sp2 and sp–sp3 combinations have been obtained.4–8 Beyond that, theoretical predictions on new carbon crystalline phases, e.g. M-carbon,9 bct C4 carbon,10 penta-graphene,11 BC14 penta-diamond,12 BCO-C16,13 and the graphyne family14 have been proposed. For a long time, carbon materials have been widely utilized in energy technology. Carbon matrices (amorphous carbon and many other forms) and carbides15,16 are always used as catalyst supports. The utilization of newly discovered carbon allotropes (e.g. graphdiyne) and carbides15,17 in emerging energy applications such as catalysis, Li-ion batteries, solar cells and hydrogen storage has been paid much attention.18–24
In 2011, T-carbon, a new diamond-like carbon allotrope (space group Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m), was theoretically proposed.25 The structure of T-carbon is constituted by substituting each atom in diamond by a carbon tetrahedron which connects with another tetrahedron by an acetylene bond. The calculated results show that T-carbon possesses a much lower density of 1.50 g cm−3 and a Vickers hardness of 61.1 GPa (smaller than that of diamond (93.7 GPa) but comparable with that of cubic boron nitride). Recently, T-carbon has been successfully prepared by picosecond laser irradiation on carbon nanotubes26 and plasma-enhanced chemical vapor deposition.27 The potential applications of T-carbon in photocatalysis, solar cells, adsorption, energy storage, supercapacitors, aerospace materials, electronic devices are widely concerned.28,29 With a hollow structure, T-carbon is expected to be a promising material for Li storage. Its excellent carrier mobility29 and mechanical ductility30 ensure favorable properties for use as the electrode in Li-ion batteries.
m), was theoretically proposed.25 The structure of T-carbon is constituted by substituting each atom in diamond by a carbon tetrahedron which connects with another tetrahedron by an acetylene bond. The calculated results show that T-carbon possesses a much lower density of 1.50 g cm−3 and a Vickers hardness of 61.1 GPa (smaller than that of diamond (93.7 GPa) but comparable with that of cubic boron nitride). Recently, T-carbon has been successfully prepared by picosecond laser irradiation on carbon nanotubes26 and plasma-enhanced chemical vapor deposition.27 The potential applications of T-carbon in photocatalysis, solar cells, adsorption, energy storage, supercapacitors, aerospace materials, electronic devices are widely concerned.28,29 With a hollow structure, T-carbon is expected to be a promising material for Li storage. Its excellent carrier mobility29 and mechanical ductility30 ensure favorable properties for use as the electrode in Li-ion batteries.
In this paper, density functional theory (DFT) calculations are employed to investigate the possibility of T-carbon serving as the material for Li storage and a Li battery anode. We carry out DFT-based Monte Carlo simulations to build the structure and energy spectrum of Li-loaded T-carbon. Molecular dynamics (MD) simulations are performed to confirm the structural stability. The convex hull of the energy spectrum is built to predict the structure evolution in the Li loading process. C–Li bonding and the mechanism of the change of Li adsorption strength in the loading process are analyzed. The anode potential exhibits a significant difference between the case of low and high Li density, which makes a “dead zone” in which only half of the Li capacity could be useful for the discharge of the anode. This work provides basic guidance for future studies on the possibility of utilizing other carbon allotropes in emerging energy applications.
II. Computational methods
DFT calculations are performed with the projector augmented wave (PAW) method31,32 as implemented in the Vienna ab initio simulation package (VASP).33–36 The electron exchange and correlation are described at the level of Perdew–Burke–Ernzerhof (PBE) functional.37 The correction of van der Waals interactions is treated by the DFT-D3 method with Becke–Jonson damping.38,39 A plane-wave basis set is used with a kinetic energy cutoff of 500 eV. The Brillouin-zone integration for the 1 × 1 × 1 (2 × 2 × 2) T-carbon supercell is performed with an 8 × 8 × 8 (4 × 4 × 4) Γ-centered Monkhorst–Pack grid. For electronic structure calculations, a 25 × 25 × 25 Γ-centered Monkhorst–Pack grid is used. The convergence of total energy is considered to be achieved until the total energy difference of two iterated steps is less than 10−5 eV. Geometry relaxations are performed until all the atomic forces are below 0.001 eV Å−1. The climbing image nudged elastic band method40–42 was used to find the minimum energy paths and barriers of Li migrations in T-carbon.
To verify the DFT parameters, we perform computations on graphite as the anode of Li battery. The main phases of Li-loaded graphite are C12Li1 (ESI,‡ Fig. S1(a), called as stage II in ref. 43) and C6Li1 (Fig. S1(b), ESI,‡ called stage I in ref. 43). For the two phases, the calculated Li adsorption energy (eqn (1) and Fig. S1(c), ESI‡) and the anode potential of graphite (see Section III.3. and Fig. S1(d), ESI‡) are close to the results in ref. 43.
For a certain number of Li atoms loading on the T-carbon supercell, the structure evolution is carried out using the Metropolis Monte Carlo method for the sampling of the energy spectrum. In every random move, each Li atom is moved with a displacement less than 1.5 Å, and then the structure is fully relaxed. Every relaxed configuration is accepted according to a Gibbs distribution on the DFT total energy within an effective temperature of 4000 K. The structural stability is then verified by molecular dynamics (MD) simulations using the Nose–Hoover thermostat.
III. Results and discussion
III.1. Preliminary understanding
The structure of T-carbon is shown in the ESI,‡ Fig. S2(a) (8 C atoms in one unit cell). T-carbon possesses a diamond-like architecture with acetylene bonds connecting neighboring C tetrahedrons. The energy bands and density of state (DOS) are shown in the ESI,‡ Fig. S2(b) and (c). It is known that the PBE functional always underestimates the bandgap. Thus, we further calculate electronic structures using the hybrid Heyd–Scuseria–Ernzerhof (HSE06) functional.44,45 T-carbon presents a semiconducting feature with the calculated band gap of T-carbon being 2.23 (3.18) eV at the level of PBE (HSE06). Its hollow cage structure allows Li atoms to be stored in it.
To obtain a simple view of basic information, we first investigate Li adsorption in a 1 × 1 × 1 T-carbon cell. To measure the strength of Li adsorption in T-carbon, we employ the Li adsorption energy
| | | Ead = [E(T-carbon–Lin) − E(T-carbon) − nE(Li)]/n | (1) |
relative to the Li bulk. Here,
E(T-carbon) is the total energy of the T-carbon cell,
E(Li) is the energy per Li atom in the bulk bcc phase, and
E(T-carbon–Li
n) is the total energy of the T-carbon cell with
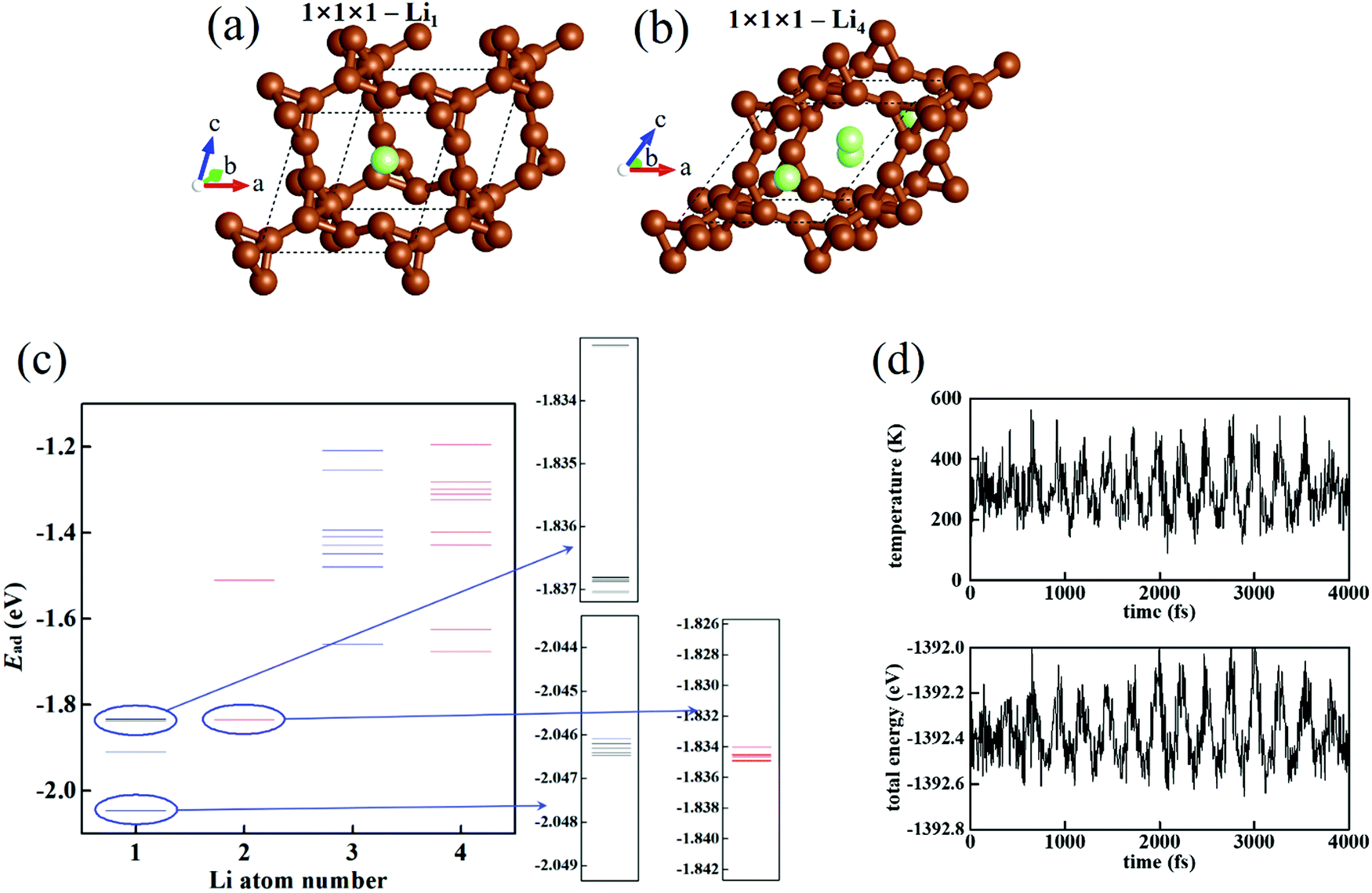
n Li atoms. For a single Li atom, the preferred position of Li is located in the C-cage (
Fig. 1(a)). The structure of T-carbon–Li
4 (C
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
:
Li = 2
:
1) is shown in
Fig. 1(b), in which the Li–Li distance (2.4–3.3 Å) is shorter or closer than that of the Li bulk (3.0 Å). More Li atoms inserted into T-carbon may enlarge and destroy the C skeleton. So, this is considered as the state of maximum Li density. If T-carbon is used as the Li-ion battery anode (with the discharge process Li → Li
+ + e), the maximum Li capacity is three times that of graphite (1116 mA h g
−1). The structural stability of 1 × 1 × 1-T-carbon–Li
4 is confirmed by MD simulations at 300 K (ESI,
‡ Movie S1). The evolution of temperature and total energy (
Fig. 1(d)) fluctuating near the average values exhibits the stability of the system. We utilize Monte Carlo simulations to find the structure and energy spectrum of Li
n with
n = 1–4 (
Fig. 1(c), where each line denotes the Li adsorption energy
Ead of an individual structure). It can be seen that the
Ead of the most stable structure gradually increases with the Li number
n, showing a gradually saturated carrying of T-carbon to Li atoms. In 1 × 1 × 1-T-carbon–Li
4, C–Li binding causes structure distortion making the lattice become a low-symmetry system and leading to a relaxation of the structure with extra energy decrease. Therefore, the most stable structure of Li
4 possesses a lower energy than the most stable structure of Li
3. The Li-rich T-carbon is then stabilized by the distortion effect.
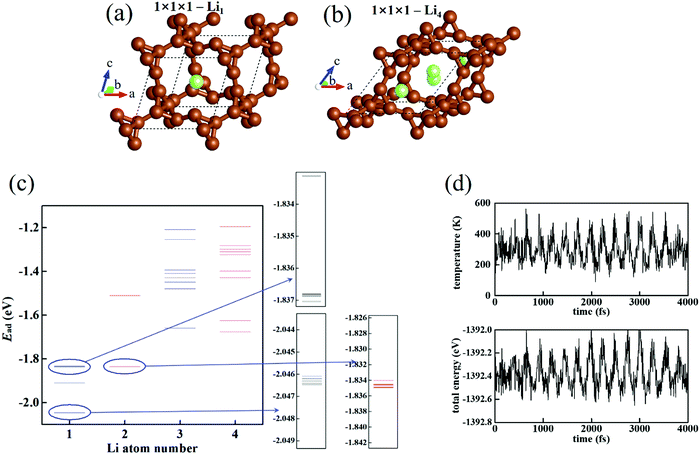 |
| | Fig. 1 (a) The most stable structure of 1 × 1 × 1 T-carbon cell with Li1. (b) The most stable structure of 1 × 1 × 1 T-carbon cell with Li4. (c) The energy spectrum of 1 × 1 × 1 T-carbon cell with Lin. Some parts of Li1 and Li2 are enlarged. (d) The evolution of temperature and total energy in the MD simulation of 1 × 1 × 1 T-carbon cell with Li4 at 300 K. | |
III.2. Li distribution and the convex hull of the energy spectrum
To realize a more realistic spectrum of Li distribution in T-carbon, we turn to the structure search in a larger 2 × 2 × 2 T-carbon cell. The most stable structure of Li4 in 2 × 2 × 2 T-carbon (Fig. 2(a)) exhibits partial segregation with more Li atoms aggregating together. The aggregated Li atoms may obtain lower energy. But for more Li atoms (Fig. 2(b) for Li8 and Fig. 2(c) for Li16), it is closer to a good distribution because the energy would be high if too many Li atoms aggregate in a small corner. The lattice of 2 × 2 × 2-T-carbon–Li16 shows structure distortion with a tilted unit cell. The extra structure relaxation leads to the lowest energy and the T-carbon–Li system is then stabilized. This distortion can be known as the structural phase transition. For 2 × 2 × 2-T-carbon–Lin with n ≥ 16, Li atoms are filled into this new phase. But the repulsion of denser Li causes an increase in the Li adsorption energy. As we inferred before, 1 × 1 × 1-Li4 is the state of maximum Li storage, and for a 2 × 2 × 2 T-carbon cell the maximum is Li32 (Fig. 2(d)). MD simulations confirm the structural stability of 2 × 2 × 2-Li32 (ESI,‡ Movie S2), with the evolution of temperature and total energy always fluctuating near the average values (Fig. 2(e)). A full energy spectrum for Lin with n = 1–32 is obtained by Monte Carlo simulations (Fig. 2(f)). It can be seen that the most stable structure of Li16 has the lowest energy, while the most stable energies of both sides Li1 and Li32 are high. In a complex energy spectrum, we are mainly concerned with the lowest energy structure of every Lin. The phases of Lin1 (with an energy E(n1)) and Lin2 (with an energy E(n2)) may mix with ratios f and 1 − f, and the average energy per formula is| | | Ē = fE(n1) + (1 − f)E(n2). | (2) |
In comparison with the structure of the Li number n = fn1 + (1 − f)n2, if E(n) is lower than Ē, the structure of Lin would be the actually formed phase in the evolution of the T-carbon anode. However, in Fig. 2(f) the structures between Li1 and Li16 all have higher energy than Ē (see the red line). So, we infer that in the realistic charging process, the anode would be composed of a mixture of Li1 and Li16. Similarly, the structures between Li16 and Li32 all have higher energy than the average energy of Li16 and Li32 (f: 1 − f). So in the subsequent charging process, the anode would be composed of a mixture of Li16 and Li32.
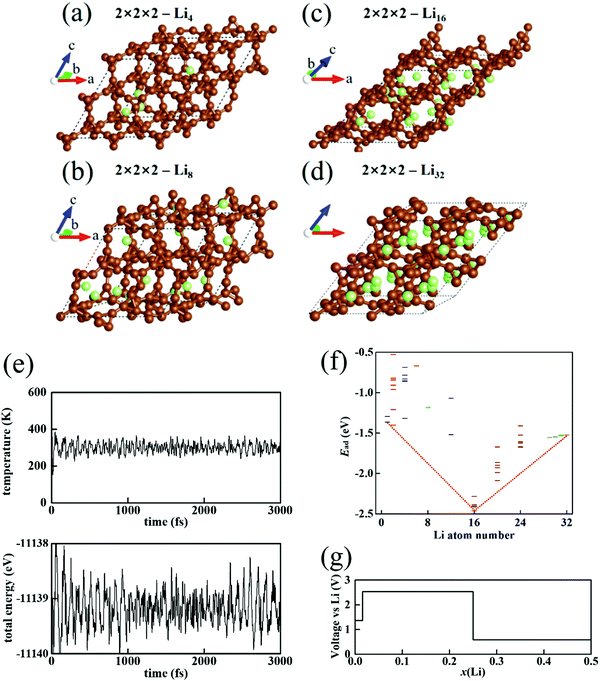 |
| | Fig. 2 The most stable structure of a 2 × 2 × 2 T-carbon cell with (a) Li1, (b) Li8, (c) Li16, and (d) Li32. (e) The evolution of temperature and total energy in the MD simulations of a 2 × 2 × 2 T-carbon cell with Li32 at 300 K. (f) The energy spectrum and convex hull of a 2 × 2 × 2 T-carbon cell with Lin. (g) The anode voltage of T-carbon as the anode of Li battery. | |
III.3. Anode potential of Li-loaded T-carbon
In this section, we will estimate the electrode potential of T-carbon serving as the anode of a Li battery. Generally, the ideal maximum electrode potential can be estimated by the change of Gibbs free energy in the process. In the last section, we inferred that in the charge or discharge process, the phases 2 × 2 × 2-Li1 and 2 × 2 × 2-Li16 (or 2 × 2 × 2-Li16 and 2 × 2 × 2-Li32) could coexist. For a phase 2 × 2 × 2-Lin2 changing into 2 × 2 × 2-Lin1 during discharge, the reaction on the side of the T-carbon anode is| | | 2 × 2 × 2-Lin2 → 2 × 2 × 2-Lin1 + (n2 − n1)Li+ + (n2 − n1)e. | (3) |
Generally, the electrode potential is measured relative to the Li bulk. If the Li bulk is taken as the cathode, the reaction isThe change of Gibbs free energy in the whole process 2 × 2 × 2-Lin2 → 2 × 2 × 2-Lin1 + (n2 − n1) Li(bulk) is then| | | ΔG = G(2 × 2 × 2-Lin1) + (n2 − n1)G(Li bulk) − G(2 × 2 × 2-Lin2). | (5) |
Here, we approximately take free energy G as the DFT total energy, and the anode potential then reads V = ΔG/e (e is the elementary charge).
The discharge of the T-carbon anode can be generally divided into three stages,
| | | 2 × 2 × 2-Li32 → 2 × 2 × 2-Li16 + 16Li+ + 16e, | (6) |
| | | 2 × 2 × 2-Li16 → 2 × 2 × 2-Li1 + 15Li+ + 15e, | (7) |
and
| | | 2 × 2 × 2-Li1 → T-carbon + Li+ + e. | (8) |
In the calculation results (
Fig. 2(g),
x(Li) = 0–0.5 is the ratio of Li
:
C), we can see that the anode voltages of these three stages are very different. Since the Li adsorption energy
Ead in 2 × 2 × 2-Li
16 is much lower than the
Ead in 2 × 2 × 2-Li
32, the process (6) is easy to proceed with because the energy goes down. This leads to a lower anode voltage. When the discharge reaches a certain degree, process (7) occurs thereupon. Since the
Ead in 2 × 2 × 2-Li
1 is much higher than the
Ead in 2 × 2 × 2-Li
16, the process (7) proceeds with difficulty and then the anode voltage increases. The anode voltage of the first stage (6) is close to the anode voltage of graphite anode (about 0 V). But the voltage of the second stage (7) is too high. Therefore, we infer that (7) is disadvantageous to the Li battery. The exploitable part is (6), in which the Li load is
x(Li) = 0.25–0.50. Only a half of Li storage in T-carbon could be usable for battery discharge. Regarding 2 × 2 × 2-Li
16 as the substrate, the maximum load (16 more Li within) corresponds to a Li capacity of 487 mA h g
−1, which is about 1.3 times the capacity of graphite.
III.4. Electronic structure and the conductivity
To use T-carbon as the anode of a Li battery, the electronic conductivity should be paid attention to. The widely used graphite is conductive, but pure T-carbon is a semiconductor with weaker conductivity. Li loading may enhance the conductivity of T-carbon. To investigate the effect of Li, we calculate the projected density of states (PDOS). In 2 × 2 × 2-Li1, Li bonds with C atoms and a part of Li electrons fill into C 2p orbitals (Fig. 3(a)). This leads to an energy level broadening near the Fermi level, and make some contribution to conductivity. A doping level locates at about 1 eV above the Fermi level. With more Li atoms filling into T-carbon, more electrons of Li fill into C 2p orbitals. At the maximum Li loading, i.e. 2 × 2 × 2-Li32, the extra electrons just fill a whole band, and a new small bandgap appears at the Fermi level (Fig. 3(b)). There should gradually exhibit a change between the case of Li1 and Li32, which is discussed in the following text.
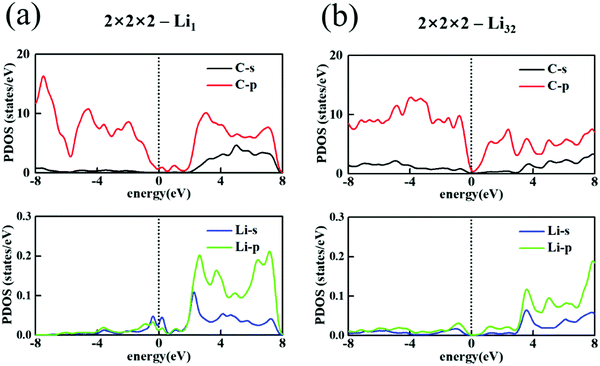 |
| | Fig. 3 PDOS of 2 × 2 × 2 T-carbon cell with (a) Li1 and (b) Li32. | |
The electronic conductivity of bulk materials depends on electron–phonon relaxation and the electronic distribution in the bands. The conductivity tensor reads
| | 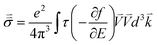 | (9) |
where
E is the electron energy,
τ is the electron–phonon relaxation time,
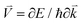
is the electron velocity, and
f = 1/(1 + exp[(
E −
EF)/
kBT]) is the Fermi–Dirac distribution. We know that ∂
f/∂
E is a delta-like function that is sharp near the Fermi energy
EF. In
Fig. 4, we plot the density of states (DOS) in 2 × 2 × 2-Li
n along with the outline of function ∂
f/∂
E at
T = 300 K. For 2 × 2 × 2-Li
1 and 2 × 2 × 2-Li
8 (
Fig. 4(a) and (b)), the contribution of DOS in the peak of ∂
f/∂
E is small, and the conductivity would be low. For 2 × 2 × 2-Li
16 and 2 × 2 × 2-Li
24 (
Fig. 4(c) and (d)), DOS in the peak of ∂
f/∂
E is much larger and the conductivity would be higher. For 2 × 2 × 2-Li
32, since the electrons of Li fill a whole hand, a gap forms near the Fermi energy and the electronic structure is semiconducting (a bandgap of 0.30 eV). The electron and hole density in the intrinsic semiconductor are
| | 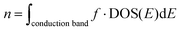 | (10) |
and
| | 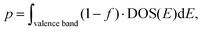 | (11) |
respectively. The calculated full carrier density
n +
p in 2 × 2 × 2-Li
32 is 2.2 × 10
17 cm
−3. Such a high carrier density ensures 2 × 2 × 2-Li
32 to be conductive. Overall, we infer that Li-rich T-carbon has enough carriers for the anode of the Li battery to be conductive.
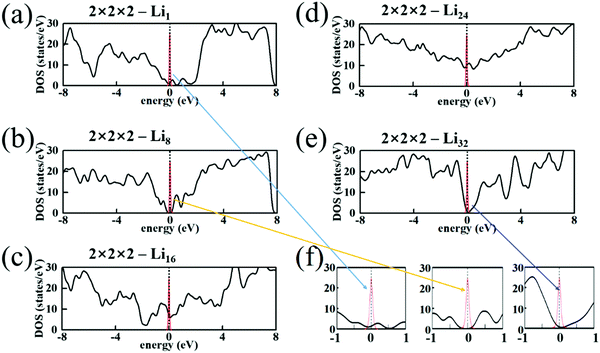 |
| | Fig. 4 DOS of 2 × 2 × 2 T-carbon cell with (a) Li1, (b) Li8, (c) Li16, (d) Li24, (e) Li32. Some parts are enlarged in (f). The red curves outline the function −∂f/∂E (f is the Fermi–Dirac distribution function at 300 K). | |
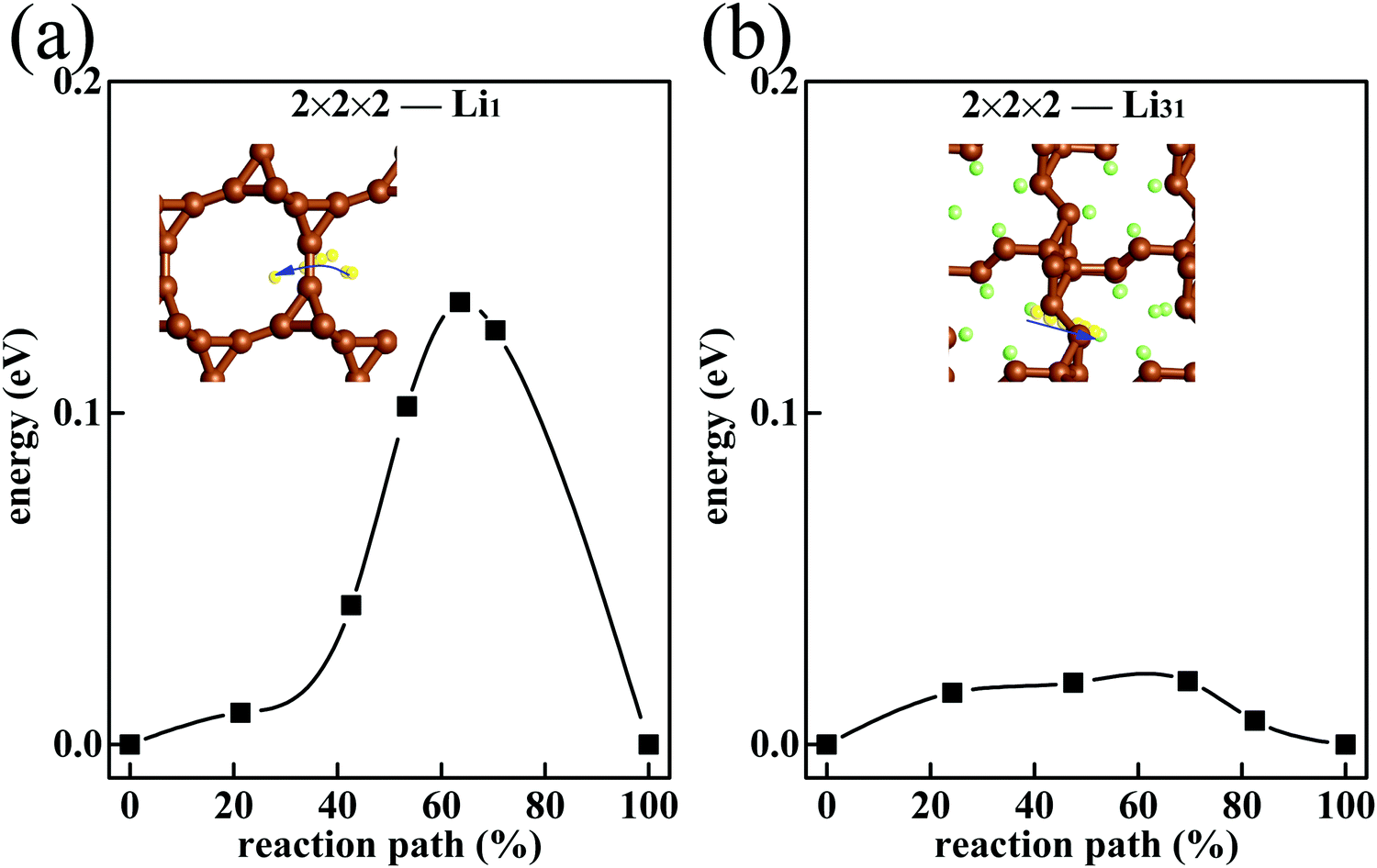
To investigate the ionic conductivity, the migration barriers of Li in T-carbon are investigated. In the initial charging process, low-concentration Li atoms form the 2 × 2 × 2-Li1 phase, in which Li atoms can wander in the free space of T-carbon. Fig. 5(a) exhibits the migration process of the Li atom from one adsorption site to the neighboring site. The migration barrier Eb(Li1) = 0.13 eV is smaller than the Li diffusion barrier on pristine graphene (about 0.3 eV),46,47 graphene with a point defect (about 0.4–0.5 eV)46 and graphite (about 0.5 eV).48 This promises a faster Li diffusion in T-carbon.
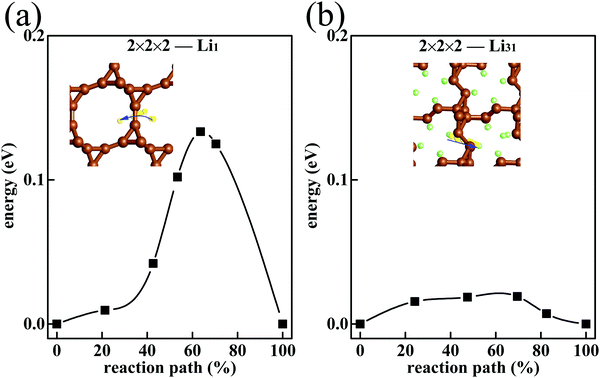 |
| | Fig. 5 The migration barrier of Li atom in 2 × 2 × 2 T-carbon cell with (a) Li1 and (b) Li31. The migratory Li atom is shown in yellow. | |
In the final charging process, the system is saturated with Li (the 2 × 2 × 2-Li32 phase). Thus, we consider the Li migration in the state of 2 × 2 × 2-Li31 which is close to the saturation. Since 2 × 2 × 2-Li31 is less filled than 2 × 2 × 2-Li32, in 2 × 2 × 2-Li31 a Li atom can migrate to a nearing vacancy. Such processes proceed successively, and then the Li atoms migrate. In 2 × 2 × 2-Li31, a Li atom is affected by other Li in all directions, and the chemical environment is more isotropic. So, the Li migration barrier in 2 × 2 × 2-Li31Eb(Li31) = 0.02 eV (Fig. 5(b)) is much lower than in T-carbon with low-concentration Li. In Li-filled T-carbon, the easier migration is beneficial to charging and discharging at high Li density. As the anode of the Li ion battery, T-carbon is better than graphite with faster Li ion migration.
IV. Conclusions
The combination of T-carbon and Li is systematically studied by DFT calculations and Monte Carlo simulations. T-carbon could serve as a Li storage and Li ion battery anode. Possible energy-favorable phases are C64Li, C64Li16 and C64Li32. The maximum Li capacity is 1116 mA h g−1. The T-carbon–Li system prefers to form the mixture of these stable phases rather than other unstable phases. In the discharge process of the T-carbon anode, the transition C64Li32 → C64Li16 → C64Li causes a significant change in anode potential due to the difference of Li adsorption energy in these phases. The anode potential of the first stage C64Li32 → C64Li16 + 16Li+ + 16e is close to the anode potential of the graphite anode, while the anode potential of the second stage C64Li16 → C64Li + 15Li+ + 15e is high and disadvantageous to the Li battery. This leads to a “dead zone” in which only half of the Li capacity could be usable (with an effective capacity of 487 mA h g−1). Li storage in T-carbon contributes extra electrons into C π2p orbitals and ensures its conductivity serving as an anode. Our work may provide guidance to the future study on the electrodes of Li, Na or K ion batteries in emerging energy applications.
Data availability
The raw/processed data required to reproduce these findings cannot be shared at this time as the data also form part of an ongoing study.
Conflicts of interest
The authors declare that they have no conflict of interest.
Acknowledgements
This work is supported by the Natural Science Basic Research Program of Shaanxi (No. 2021JM-117 & 2021JQ-185), the Fundamental Research Funds for the Central Universities (No. XJS200503) and the Postdoctoral Research Project of Shaanxi Province (No. 2018BSHEDZZ68).
References
- H. W. Kroto, J. R. Heath, S. C. O’Brien, R. F. Curl and R. E. Smalley, C60: buckminsterfullerene, Nature, 1985, 318, 162 Search PubMed.
- S. Iijima, Helical microtubules of graphitic carbon, Nature, 1991, 354, 56 Search PubMed.
- K. Novoselov, A. Geim, S. V. Morozov, D. Jiang, Y. Zhang and S. V. Dubonos,
et al., Electric Field Effect in Atomically Thin Carbon Films, Science, 2004, 306, 666 Search PubMed.
- F. Diederich and M. Kivala, All-Carbon Scaffolds by Rational Design, Adv. Mater., 2010, 22, 803 Search PubMed.
- A. Hirsch, The era of carbon allotropes, Nat. Mater., 2010, 9, 868 Search PubMed.
- G. Li, Y. Li, H. Liu, Y. Guo, Y. Lia and D. Zhua, Architecture of graphdiyne nanoscale films, Chem. Commun., 2010, 46, 3256 Search PubMed.
- Y. Li, L. Xu, H. Liu and Y. Li, Graphdiyne and Graphyne: from Theoretical Predictions to Practical Construction, Chem. Soc. Rev., 2014, 43, 2572 Search PubMed.
- H. Yan, P. Yu, G. Han, Q. Zhang, L. Gu and Y. Yi,
et al., High-Yield and Damage-free Exfoliation of Layered Graphdiyne in Aqueous Phase, Angew. Chem., Int. Ed., 2019, 58, 746 Search PubMed.
- Q. Li, Y. Ma, A. R. Oganov, H. Wang, H. Wang and Y. Xu,
et al., Superhard Monoclinic Polymorph of Carbon, Phys. Rev. Lett., 2009, 102, 175506 Search PubMed.
- K. Umemoto, R. M. Wentzcovitch, S. Saito and T. Miyake, Body-Centered Tetragonal C4: a Viable sp3 Carbon Allotrope, Phys. Rev. Lett., 2010, 104, 125504 Search PubMed.
- S. Zhang, J. Zhou, Q. Wang, X. Chen, Y. Kawazoe and P. Jena, Penta-graphene: a new carbon allotrope, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 2372 Search PubMed.
- J.-T. Wang, C. Chen and H. Mizuseki, Body centered cubic carbon BC14: an all-sp3 bonded full-fledged pentadiamond, Phys. Rev. B, 2020, 102, 184106 Search PubMed.
- J.-T. Wang, H. Weng, S. Nie, Z. Fang, Y. Kawazoe and C. Chen, Body-Centered Orthorhombic C16: a Novel Topological Node-Line Semimetal, Phys. Rev. Lett., 2016, 116, 195501 Search PubMed.
- D. Malko, C. Neiss, F. Viñes and A. Görling, Competition for Graphene: graphynes with Direction-Dependent Dirac Cones, Phys. Rev. Lett., 2012, 108, 086804 Search PubMed.
- H. Chen, Y. Hua, N. Luo, X. He, Y. Li and Y. Zhang,
et al., Lithiation Abilities of SiC Bulks and Surfaces: a First-Principles Study, J. Phys. Chem. C, 2020, 124, 7031 Search PubMed.
- Y. Shao, J. Liu, Y. Wang and Y. Lin, Novel catalyst support materials for PEM fuel cells: current status and future prospects, J. Mater. Chem., 2009, 19, 46 Search PubMed.
- H. Vrubel and X. Hu, Molybdenum Boride and Carbide Catalyze Hydrogen Evolution in both Acidic and Basic Solutions, Angew. Chem., Int. Ed., 2012, 51, 12703 Search PubMed.
- Z. Zuo and Y. Li, Emerging Electrochemical Energy Applications of Graphdiyne, Joule, 2019, 3, 899 Search PubMed.
- Y. Fang, Y. Xue, L. Hui, H. Yu, Y. Liu and C. Xing,
et al., In situ growth of graphdiyne based heterostructure: toward efficient overall water splitting, Nano Energy, 2019, 59, 591 Search PubMed.
- K. Srinivasu and S. K. Ghosh, Graphyne and Graphdiyne: promising Materials for Nanoelectronics and Energy Storage Applications, J. Phys. Chem. C, 2012, 116, 5951 Search PubMed.
- C. Li, J. Li, F. Wu, S.-S. Li, J.-B. Xia and L.-W. Wang, High Capacity Hydrogen Storage in Ca Decorated Graphyne: a First-Principles Study, J. Phys. Chem. C, 2011, 115, 23221 Search PubMed.
- J. He, N. Wang, Z. Cui, H. Du, L. Fu and C. Huang,
et al., Hydrogen substituted graphdiyne as carbon-rich flexible electrode for lithium and sodium ion batteries, Nat. Commun., 2017, 8, 1172 Search PubMed.
- X. Ren, X. D. Li, Z. Yang, X. Wang, J. J. He and K. Wang,
et al., Tailoring Acetylenic Bonds in Graphdiyne for Advanced Lithium Storage, ACS Sustainable Chem. Eng., 2020, 8, 2614 Search PubMed.
- X. D. Li, N. Wang, J. J. He, Z. Yang, Z. Y. Tu and F. H. Zhao,
et al., Designing the efficient lithium diffusion and storage channels based on graphdiyne, Carbon, 2020, 162, 579 Search PubMed.
- X.-L. Sheng, Q.-B. Yan, F. Ye, Q.-R. Zheng and G. Su, T-Carbon: a Novel Carbon Allotrope, Phys. Rev. Lett., 2011, 106, 155703 Search PubMed.
- J. Zhang, R. Wang, X. Zhu, A. Pan, C. Han and X. Li,
et al., Pseudo-topotactic conversion of carbon nanotubes to T-carbon nanowires under picosecond laser irradiation in methanol, Nat. Commun., 2017, 8, 683 Search PubMed.
- K. Xu, H. Liu, Y.-C. Shi, J.-Y. You, X.-Y. Ma and H.-J. Cui,
et al., Preparation of T-carbon by plasma enhanced chemical vapor deposition, Carbon, 2020, 157, 270 Search PubMed.
- G. Qin, K.-R. Hao, Q.-B. Yan, M. Hu and G. Su, Exploring T-carbon for energy applications, Nanoscale, 2019, 11, 5978 Search PubMed.
- P.-P. Sun, L. Bai, D. R. Kripalani and K. Zhou, A new carbon phase with direct bandgap and high carrier mobility as electron transport material for perovskite solar cells, npj Comput. Mater., 2019, 5, 9 Search PubMed.
- L. Bai, P.-P. Sun, B. Liu, Z. Liu and K. Zhou, Mechanical behaviors of T-carbon: a molecular dynamics study, Carbon, 2018, 138, 357 Search PubMed.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 Search PubMed.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 Search PubMed.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 Search PubMed.
- G. Kresse and J. Furthmüller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a planewave basis set, Comput. Mater. Sci., 1996, 6, 15 Search PubMed.
- G. Kresse and J. Hafner,
Ab initio molecular dynamics for liquid metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 Search PubMed.
- G. Kresse and J. Hafner,
Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H–Pu, J. Chem. Phys., 2010, 132, 154104 Search PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456 Search PubMed.
- G. Mills and H. Jónsson, Quantum and thermal effects in H2 dissociative adsorption: evaluation of free energy barriers in multidimensional quantum systems, Phys. Rev. Lett., 1994, 72, 1124 Search PubMed.
- G. Mills, H. Jónsson and G. K. Schenter, Reversible work transition state theory: application to dissociative adsorption of hydrogen, Surf. Sci., 1995, 324, 305 Search PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, A climbing image nudged elastic band method for finding saddle points and minimum energy paths, J. Chem. Phys., 2000, 113, 9901 Search PubMed.
- K. Persson, Y. Hinuma, Y. S. Meng, A. van der Ven and G. Ceder, Thermodynamic and kinetic properties of the Li-graphite system from first-principles calculations, Phys. Rev. B, 2010, 82, 125416 Search PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, Hybrid functionals based on a screened Coulomb potential, J. Chem. Phys., 2003, 118, 8207–8215 Search PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, Erratum: hybrid functionals based on a screened Coulomb potential, J. Chem. Phys., 2006, 124, 219906 Search PubMed.
- L.-J. Zhou, Z. F. Hou and L.-M. Wu, First-Principles Study of Lithium Adsorption and Diffusion on Graphene with Point Defects, J. Phys. Chem. C, 2012, 116, 21780 Search PubMed.
- X. F. Fan, W. T. Zheng and J. L. Kuo, Adsorption and diffusion of Li on pristine and defective graphene, ACS Appl. Mater. Interfaces, 2012, 4, 2432 Search PubMed.
- K. Toyoura, Y. Koyama, A. Kuwabara, F. Oba and I. Tanaka, First-principles approach to chemical diffusion of lithium atoms in a graphite intercalation compound, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 214303 Search PubMed.
Footnotes |
| † PACS: 61.50.-f; 65.40.gk. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00328c |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Xi-Mei
Li
and
Xi
Chen
*,
Xi-Mei
Li
and
Xi
Chen


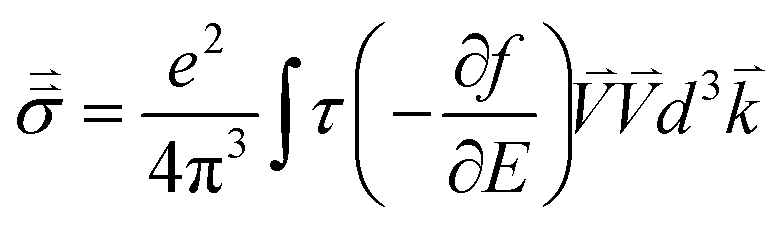
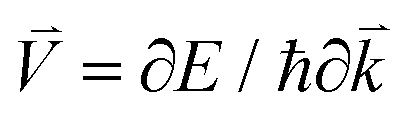 is the electron velocity, and f = 1/(1 + exp[(E − EF)/kBT]) is the Fermi–Dirac distribution. We know that ∂f/∂E is a delta-like function that is sharp near the Fermi energy EF. In Fig. 4, we plot the density of states (DOS) in 2 × 2 × 2-Lin along with the outline of function ∂f/∂E at T = 300 K. For 2 × 2 × 2-Li1 and 2 × 2 × 2-Li8 (Fig. 4(a) and (b)), the contribution of DOS in the peak of ∂f/∂E is small, and the conductivity would be low. For 2 × 2 × 2-Li16 and 2 × 2 × 2-Li24 (Fig. 4(c) and (d)), DOS in the peak of ∂f/∂E is much larger and the conductivity would be higher. For 2 × 2 × 2-Li32, since the electrons of Li fill a whole hand, a gap forms near the Fermi energy and the electronic structure is semiconducting (a bandgap of 0.30 eV). The electron and hole density in the intrinsic semiconductor are
is the electron velocity, and f = 1/(1 + exp[(E − EF)/kBT]) is the Fermi–Dirac distribution. We know that ∂f/∂E is a delta-like function that is sharp near the Fermi energy EF. In Fig. 4, we plot the density of states (DOS) in 2 × 2 × 2-Lin along with the outline of function ∂f/∂E at T = 300 K. For 2 × 2 × 2-Li1 and 2 × 2 × 2-Li8 (Fig. 4(a) and (b)), the contribution of DOS in the peak of ∂f/∂E is small, and the conductivity would be low. For 2 × 2 × 2-Li16 and 2 × 2 × 2-Li24 (Fig. 4(c) and (d)), DOS in the peak of ∂f/∂E is much larger and the conductivity would be higher. For 2 × 2 × 2-Li32, since the electrons of Li fill a whole hand, a gap forms near the Fermi energy and the electronic structure is semiconducting (a bandgap of 0.30 eV). The electron and hole density in the intrinsic semiconductor are