DOI:
10.1039/D1MA00254F
(Paper)
Mater. Adv., 2021,
2, 4685-4693
The fluorescence turn-off mechanism of a norbornene-derived homopolymer – an Al3+ colorimetric and fluorescent chemosensor†
Received
24th March 2021
, Accepted 14th May 2021
First published on 14th May 2021
Abstract
A deep understanding of fluorescence turn-off or turn-on mechanisms is fundamental in designing highly effective sensor molecules. However, these mechanisms are subtle as multiple factors may affect the fluorescence signals. Herein, for the first time, we systematically investigated the turn-off fluorescence mechanism upon adding Al3+ cation to a norbornene-derived homopolymer (PNDMIP-DASA). The corresponding monomer (NDMIP-DASA) showed fluorescence enhancement upon the addition of Al3+ ions to THF/DIW solution, while fluorescence turn-off is observed for PNDMIP-DASA. Both NDMIP-DASA and PNDMIP-DASA showed Al3+ detection ability with a low detection limit of 27.60 nM and 2.94 nM, respectively, even in the presence of metal ions. Time-dependent density functional theory (TD-DFT) calculations showed that the PNDMIP-DASA sensor exhibits a large displacement of the potential energy curve between the ground state and the excited state, resulting in an emission shift from the UV-vis to NIR regions. In addition, the oscillator strength of the S1 to S0 transition is close to zero. These results explained the fluorescence sensing mechanism of the turn-off phenomenon. The excellent agreement between calculations and experimental observations also suggests that computational chemistry is a powerful tool to aid the molecular design and engineering of fluorescent compounds.
1. Introduction
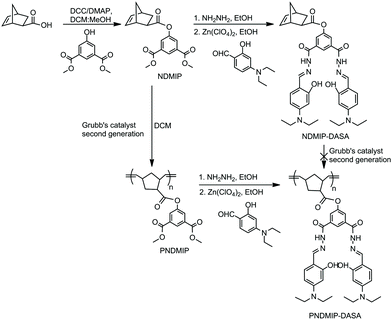
Aluminum, the third most abundant element and the most abundant metallic element in the earth's crust, has been exploited in various fields, such as food additives, beverage packaging, cooking utensils, paper industry, textile industry, and medicines. It is also present in ambient, occupational, airborne particulates and drinking water (due to its use in municipal water treatment). The aluminum-free metal cation Al3+ is highly reactive and the excess accumulation of Al3+ damages the central nervous system in humans.1 Aluminum toxicity can cause Alzheimer's disease, Parkinson's disease, breast cancer, bone softening, impaired lung function, fibrosis, chronic renal failure, etc.1–5 Therefore, the development of chemosensors for Al3+ is very important in controlling its concentration both in environmental and in biological systems. The detection of Al3+ is quite difficult due to its poor coordination and strong hydration compared to other transition metals (Cu2+, Pb2+, Hg2+, Zn2+, etc.).6,7 The fluorescence method is the most attractive and highly sensitive method for detecting low concentrations of ions and neutral analysts.8–10 In recent years, significant progress has been made in detecting Al3+ using a fluorescent chemosensor.11–16 Because of the individual photophysical properties and the strong binding abilities to Al3+ ions, Schiff base derivatives have been widely investigated as fluorescent chemosensors for Al3+ detection.17–25 Most of the Schiff base-derived fluorescent chemosensors are small molecules. However, compared to the “turn-on” type, the “turn-off” type fluorescent sensors for Al3+ detection were rarely reported in the literature, which usually have a lower detection limit. Recently, polymer-based fluorescent chemosensors have attracted a wide range of interest due to their simplicity, signal amplification, a combination of different outputs, and ease of fabrication into devices.10,26–31 Therefore, the combination of Schiff base derivatives and polymers can be an efficient pathway to develop fluorescent chemosensors for the selective detection of Al3+. To prepare polymer-based sensors for Al3+, a norbornene-derived homopolymer was synthesized from a norbornene monomer (NDMIP-DASA) (Scheme 1). NDMIP-DASA was designed with two main moieties, norbornene and Schiff base moieties, which provide an ideal framework for a specific coordinate to Al3+ due to its ability to contribute a coordinate environment of −CONH, −C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, and −OH groups.6 High selectivity for Al3+ over other metal ions was observed for both the NDMIP-DASA monomer and the PNDMIP-DASA homopolymer. Interestingly, we had found that the monomeric sensor showed turn-on fluorescence after adding an Al3+ cation and its homopolymer responded with turn-off fluorescence to the presence of Al3+ in the microenvironment. The limit of detection achieved for PNDMIP-DASA is 2.94 nM which is ten times lower than that of monomer NDMIP-DASA (27.60 nM). A deep understanding of the fluorescence turn-off or turn-on mechanism is very important to understand the difference between monomers and polymers. This issue is of fundamental significance for the design of optimized metal ion detection sensors. Herein, we systematically investigated the fluorescence turn-off mechanism after adding an Al3+ cation into the polymer sensor. To the best of our knowledge, the fluorescence turn-off for the Al3+ sensor was explored a little. For a detailed understanding of the sensing mechanism, NMR titration and time-dependent density functional theory (TD-DFT) calculations were carried out which revealed the fluorescence turn-off or fluorescence enhancement mechanism of the sensors in the presence of Al3+.
N, and −OH groups.6 High selectivity for Al3+ over other metal ions was observed for both the NDMIP-DASA monomer and the PNDMIP-DASA homopolymer. Interestingly, we had found that the monomeric sensor showed turn-on fluorescence after adding an Al3+ cation and its homopolymer responded with turn-off fluorescence to the presence of Al3+ in the microenvironment. The limit of detection achieved for PNDMIP-DASA is 2.94 nM which is ten times lower than that of monomer NDMIP-DASA (27.60 nM). A deep understanding of the fluorescence turn-off or turn-on mechanism is very important to understand the difference between monomers and polymers. This issue is of fundamental significance for the design of optimized metal ion detection sensors. Herein, we systematically investigated the fluorescence turn-off mechanism after adding an Al3+ cation into the polymer sensor. To the best of our knowledge, the fluorescence turn-off for the Al3+ sensor was explored a little. For a detailed understanding of the sensing mechanism, NMR titration and time-dependent density functional theory (TD-DFT) calculations were carried out which revealed the fluorescence turn-off or fluorescence enhancement mechanism of the sensors in the presence of Al3+.
 |
| | Scheme 1 Synthetic routes of NDMIP-DASA and PNDMIP-DASA. | |
2. Material and methods
2.1 Materials
Exo-5-norbornenecarboxylic acid (Nor-COOH) (97%), N,N′-dicyclohexylcarbodiimide (DCC), 4-(dimethylamino)pyridine (DMAP), zinc perchlorate hexahydrate (Zn(ClO4)2·6H2O), hydrazine monohydrate (NH2NH2·H2O, 64%), aluminum nitrate nonahydrate (Al(NO3)3·9H2O), DMSO-d6, and CDCl3 were purchased from Sigma-Aldrich. Ethyl vinyl ether, tetrahydrofuran (THF), dichloromethane (DCM), diethyl ether, and methanol were purchased from Acros Organic. Dimethyl 5-hydroxyisophthalate (DMIP) and 4-(diethylamino)salicylaldehyde (DASA) were purchased from Fluorochem. Grubbs catalyst, 2nd Generation (98%), was purchased from AK Scientific, Inc. All other chemicals were of analytical grade and used without further purification.
2.2 Synthesis of NDMIP
Exo-5-norbornenecarboxylic acid (142 mg, 1.0 mmol), dimethyl 5-hydroxyisophthalate (231 mg, 1.1 mmol), DCC (227 mg, 1.1 mmol) and DMAP (12 mg, 0.1 mmol) were dissolved in 10 mL of DCM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH (9:1). After stirring for 24 h at room temperature under nitrogen, the white precipitate was filtered out. The reaction mixture was extracted with 0.1 N HCl. The combined organic layers were washed with brine solution and dried over anhydrous Na2SO4. The solvent was removed to yield NDMIP (yield 92%) as a white solid. 1H NMR (600 MHz, CDCl3) (Fig. S1, ESI,†δ): 8.25 (d, 1H), 7.75 (d, 2H), 6.13 (m, 2H), 3.94 (s, 3H), 3.12 (s, 1H), 2.95 (s, 1H), 2.27 (m, 1H), 1.95 (m, 1H), 1.12 (t, 3H) 0.84 (m, 1H). FT-IR (KBr, cm−1) (Fig. S2, ESI†): 3361, 3012, 2964, 2929, 2850, 1793, 1727, 1705, 1620, 1601, 1431, 1359, 1301, 1267, 1009, 756, 445.
:MeOH (9:1). After stirring for 24 h at room temperature under nitrogen, the white precipitate was filtered out. The reaction mixture was extracted with 0.1 N HCl. The combined organic layers were washed with brine solution and dried over anhydrous Na2SO4. The solvent was removed to yield NDMIP (yield 92%) as a white solid. 1H NMR (600 MHz, CDCl3) (Fig. S1, ESI,†δ): 8.25 (d, 1H), 7.75 (d, 2H), 6.13 (m, 2H), 3.94 (s, 3H), 3.12 (s, 1H), 2.95 (s, 1H), 2.27 (m, 1H), 1.95 (m, 1H), 1.12 (t, 3H) 0.84 (m, 1H). FT-IR (KBr, cm−1) (Fig. S2, ESI†): 3361, 3012, 2964, 2929, 2850, 1793, 1727, 1705, 1620, 1601, 1431, 1359, 1301, 1267, 1009, 756, 445.
2.3 Synthesis of PNDMIP
NDMIP (100 mg, 0.26 mmol) and second-generation Grubbs catalyst (2.1–4.3 mg) were dissolved in dry DCM (5 mL) under nitrogen. The reaction mixture was stirred for 24 h and stopped by the addition of 10 mL ethyl vinyl ether. The resulting polymer was isolated by precipitating in cold ether and dried under vacuum. 1H NMR (600 MHz, DMSO-d6) (Fig. S3, ESI,†δ): 8.25 (d, 2H), 7.75 (d, 2H), 6.62 (d, 1H), 5.75 (s, 1H), 2.96 (s, 1H), 2.34 (m, 1H), 1.97 (m, 1H), 0.85 (m, 1H). 1.24 (m, 1H), 1.12 (t, 3H) 0.84 (m, 1H). FT-IR (KBr, cm−1) (Fig. S4, ESI†): 3361, 3012, 2964, 2929, 2850, 1793, 1727, 1704, 1620, 1600, 1500, 1431, 1359, 1301, 1267, 1184, 1008, 756, 445.
2.4 Synthesis of NDMIP-DASA and PNDMIP-DASA
NDMIP or PNDMIP (0.5 mmol) was dissolved in 10 mL of ethanol. Hydrazine (NH2NH2, 64%, 3 mmol) was added dropwise into the mixture and refluxed for 24 h. 213 mg of 4-(diethylamino)salicylaldehyde (DASA, 1.1 mmol) in 10 mL ethanol along with 2–3 mg of zinc perchlorate were added to the reaction mixture. The color of the solution changed immediately to yellow. The precipitates separated out within 20 min and were filtered, washed with methanol and dried under vacuum to obtain yellow solid with 85% yield. The molecular weight of the homopolymer PNDMIP-DASA was measured by gel permeation chromatography (GPC) using PMMA standards. NDMIP-DASA: 1H NMR (600 MHz, CDCl3) (Fig. S5, ESI,†δ): 11.80 (s, 1H), 8.45 (d, 2H), 8.09 (d, 1H), 7.10 (d, 2H), 6.26 (m, 2H), 1.55 (m, 1H), 1.20 (t, 3H), 0.84 (m, 1H); FT-IR (KBr, cm−1) (Fig. S2, ESI†): 2972, 2929, 2873, 2362, 1631, 1594, 1558, 1515, 1454, 1411, 1375, 1351, 1230, 1191, 1132, 1080, 966, 823, 788, 709, 650. PNDMIP-DASA: 1H NMR (600 MHz, DMSO-d6) (Fig. 1a, δ): 11.48 (s, 1H), 11.24 (s, 1H), 8.60 (d, 1H), 7.43 (d, 1H), 6.36 (d, 1H), 6.12 (d, 1H), 6.04 (d, 1H), 5.75 (s, 1H), 2.62 (m, 1H), 1.10 (t, 3H), 0.84 (m, 1H); FT-IR (KBr, cm−1) (Fig. S6, ESI†): 2972, 2929, 2897, 2362, 1631, 1594, 1558, 1514, 1483, 1454, 1411, 1375, 1352, 1299, 1284, 1247, 1230, 1191, 1132, 1095, 1014, 966, 833, 789, 761 709, 650, 621.
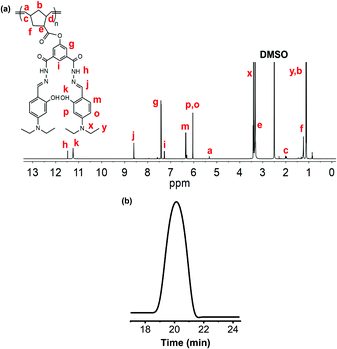 |
| | Fig. 1
1H NMR analysis of PNDMIP-DASA (a); GPC chromatogram of PNDMIP-DASA, Mn = 56 kDa; PDI = 1.13 (b). | |
2.5 Complexation of PNDMIP-DASA–Al3+
10 mL solution of Al(NO3)3·9H2O (7.5 mg, 0.04 mmol) in distilled water was added dropwise into a stirring 5 mL suspension of PNDMIP-DASA (50 mg, 0.02 mmol) over 30 min. The mixture was then stirred for another half an hour. The reaction mixture was concentrated and precipitated out in an ice bath. Dark brown precipitates were collected and dried under vacuum. 1H NMR (600 MHz, DMSO-d6, δ): 8.60 (d, 1H), 7.43 (d, 1H), 6.36 (d, 1H), 6.12 (d, 1H), 6.04 (d, 1H), 5.75 (s, 1H), 2.62 (m, 1H), 1.10 (t, 3H), 0.84 (m, 1H); FT-IR (KBr, cm−1): 2972, 2929, 2897, 2362, 1631, 1594, 1558, 1514, 1483, 1454, 1411, 1375, 1352, 1312, 1299, 1284, 1247, 1230, 1191, 1132, 1095, 1014, 966, 823, 779, 761 709, 650.
2.6 Characterization
Nuclear magnetic resonance (NMR).
1H NMR spectroscopy was carried out on a Bruker 600 MHz spectrometer using CDCl3 and DMSO-d6 as solvents. The 1H NMR spectra of the solutions were calibrated to tetramethylsilane as an internal standard (δH 0.00).
Gel permeation chromatography (GPC).
Molecular weights and PDIs were measured by Waters gel permeation chromatography in THF relative to polystyrene standards on systems equipped with a Waters Model 1515 HPLC pump and Waters Model 2414 refractive index detector at 40 °C with a flow rate of 1 mL min−1.
Fourier transform infrared (FT-IR).
FT-IR spectra were obtained using an FT-IR Shimadzu Tracer-100 spectrometer at a nominal resolution of 4 cm−1.
Ultraviolet (UV) spectroscopy.
UV-vis absorption measurements were carried out on a Jasco V-730 UV-vis spectrometer, with a scan rate of 100 nm min−1.
Fluorescence spectroscopy.
Fluorescence emission spectra were recorded on a fluorescence spectrometer (Jasco spectrofluorometer FP-8350).
2.7 Computational details
All the calculations in this study were carried out using the Gaussian 09 package.32 The geometries of sensor M, sensor D, and their complexes were optimized using the density functional B3LYP in all calculations.33 The polarized split-valence triple-ζ 6-31G(d) basis set was employed for all the atoms.34 For all optimized structures, frequency calculations were carried out to ensure the optimized geometries represent the local minima in the absence of imaginary frequencies. To choose a more reliable DFT functional for calculating the theoretical UV-vis spectra, we performed benchmark calculations for the monomer NDMIP-DASA sensor, which considered different DFT functionals such as B3LYP, CAM-B3LYP, M062X, ωB97XD, and PBE0 in the THF solvent.33,35–38 The conductor-like polarizable continuum model (CPCM) was used to calculate the solvent effects.39 The calculated adsorption energies along with the experimental valuate are given in Table S1 (ESI†). The benchmark results show that the B3LYP method provides better performance in predicting the adsorption maximum than the other functionals. Hence, we have used the TD-DFT calculations by the B3LYP functional to simulate the excitations of the corresponding sensors and emission spectra according to the optimized structures in the S1 excited state.
3. Results and discussion
3.1 Synthesis and characterization
To obtain our desired probe with fluorescence activity, NDMIP was first prepared by the coupling reaction between exo-5-norbornenecarboxylic acid and dimethyl 5-hydroxyisophthalate (DMIP) using N,N′-dicyclohexylcarbodiimide (DCC) and 4-(dimethylamino)pyridine (DMAP) (Scheme 1). The formation of NDMIP was confirmed by 1H NMR and FTIR spectroscopy. From the 1H NMR, the signals at δ 6.13 ppm (m, 2H) corresponded to the norbornene olefinic protons, while the signals at δ 3.94 ppm and δ 7.75–8.25 ppm were due to the methyl group and the aromatic ring of DMIP, respectively (Fig. S1, ESI†). The FTIR spectra of NDMIP (Fig. S2, ESI†) showed a very strong absorption band at 1728, 1705 cm−1 which corresponded to the stretching vibration of CO, 1620 cm−1 (CC), and 1601 cm−1 (C–C of the aromatic ring); this indicated the formation of NDMIP. The NDMIP-DASA monomer was synthesized by a Schiff base condensation reaction (Scheme 1) and characterized by 1H NMR and FTIR spectroscopic techniques (Fig. S2 and S5, ESI†). The homopolymerization of NDMIP-DASA was carried out using second-generation Grubbs catalyst via ring-opening metathesis polymerization (ROMP).
In the course of our study on the ROMP of the NDMIP-DASA monomer, we found that with non-protected amino groups, the homopolymer was unsuccessful with the second-generation Grubbs catalyst due to the incompatibility of amines with ruthenium.40 Therefore, the post-polymerization of NDMIP by second-generation Grubbs catalyst (with different ratios) was performed to obtain PNDMIP at room temperature in the DCM solvent. The new signals were observed at δ 5.29–5.35 ppm (CH protons of the polymer peak) and norbornene olefinic protons disappeared at δ 6.13 ppm, indicating the formation of the product (Fig. S3, ESI†).
The PNDMIP-DASA homopolymer was synthesized by a Schiff base condensation reaction with PNDMIP hydrazine and DASA in the presence of Zn(ClO4)2 to obtain our final macromolecular probe. The fruitful post-polymerization reaction of PNDMIP-DASA was confirmed by the 1H NMR analysis, where characteristic peaks at δ 11.48 ppm (NH), 11.24 ppm (OH), 6.04–8.60 ppm (CH protons of the aromatic ring), and 5.29–5.35 ppm (CH protons of the polymer peak) were generated (Fig. 1a), which indicated the formation of PNDMIP-DASA. The molecular weight of the homopolymer PNDMIP-DASA was measured by gel permeation chromatography (GPC) in THF relative to polystyrene standards. The PNDMIP-DASA homopolymers with different molecular weights were synthesized for the comparison of sensing towards Al3+. The observed molecular weights (Mn) and polydispersity index (PDI) are summarized in Table S2 (ESI†) with Mn ∼ 24000, 39000, and 56000, and a very narrow PDI. The molecular weight distribution is monomodal and narrow (Mw/Mn = 1.13), indicating that the polymerization was well controlled (Fig. 1b).
3.2 Colorimetric and fluorogenic spectral responses
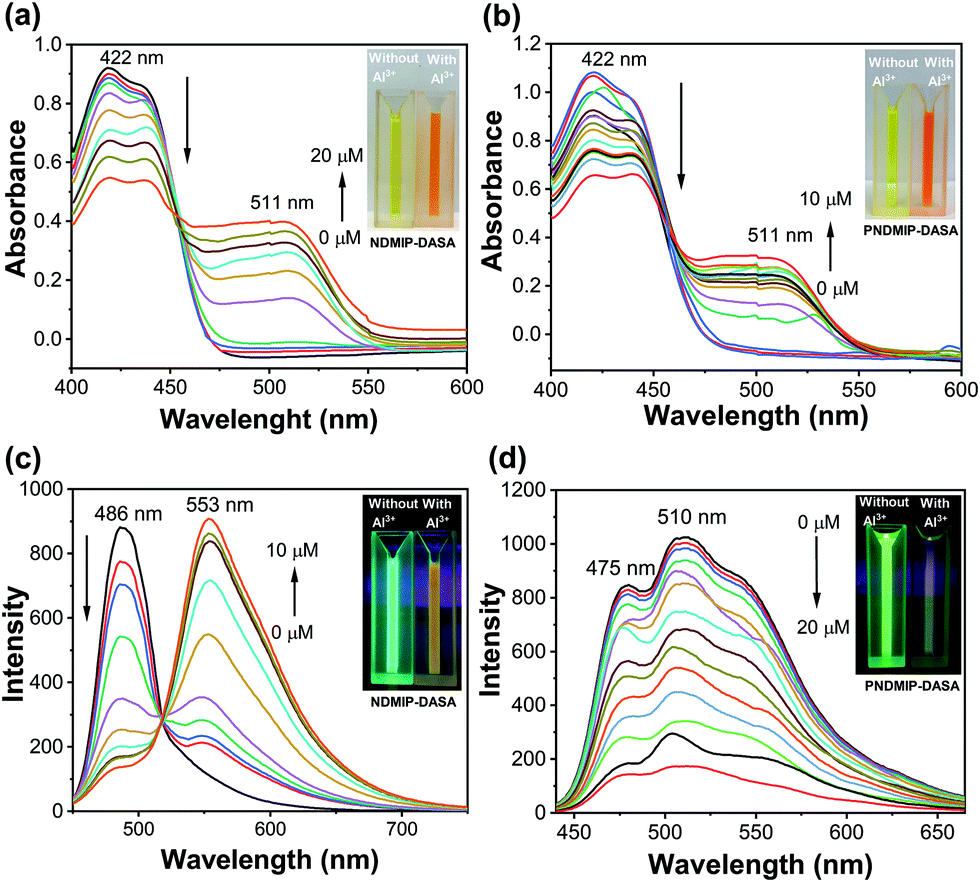
Next, to understand the photophysical properties of NDMIP-DASA and PNDMIP-DASA in THF/DIW 2:8 solutions, UV-vis absorption and fluorescence spectroscopies were performed. The UV-vis absorption spectrum of the NDMIP-DASA monomer exhibited an absorption band centered at 422 nm in THF/DIW solution, as shown in Fig. 2a. Upon the addition of Al3+, the formation of a new band was observed at λmax of 511 nm and increased gradually. In addition, the absorbance at 422 nm decreased concomitantly with the appearance of isosbestic points at 453 nm. A color change was observed from bright yellow to orange (inset, Fig. 2a and b) which corroborated the shifts of these absorption bands. Similar behavior was observed for the PNDMIP-DASA homopolymers (Fig. 2b). The NDMIP-DASA and PNDMIP-DASA were further investigated for their fluorogenic properties in THF/DIW solution with the excitation wavelength at 422 nm (Fig. 2c and d). In the case of NDMIP-DASA, a strong fluorescence at λem of 486 nm was observed. The NDMIP-DASA exhibited fluorescence enhancement upon the addition of Al3+ centered at 553 nm. Upon the addition of Al3+, the intensity at 486 nm decreased gradually with the appearance of a new peak at λmax 553 nm (Fig. 2c). On the other hand, the PNDMIP-DASA exhibited a strong fluorescence at λem 475 and 510 nm (Fig. 2d). More interestingly, upon the addition of Al3+, the fluorescence intensity was decreased upon the gradual addition of Al3+, and there is no new emission at λmax 553 nm. The PNDMIP-DASA with different molecular weights exhibited a similar turn-off fluorescence trend upon the addition of Al3+ ions (Fig. S7, ESI†).
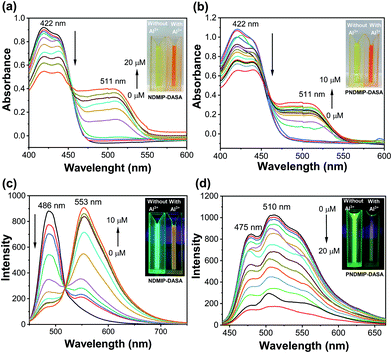 |
| | Fig. 2 Changes of UV-vis (a and b) and fluorescence spectra (c and d) of NDMIP-DASA and PNDMIP-DASA (6.5 ppm, THF/DIW 2:8 solution) upon graduation of Al3+, respectively. | |
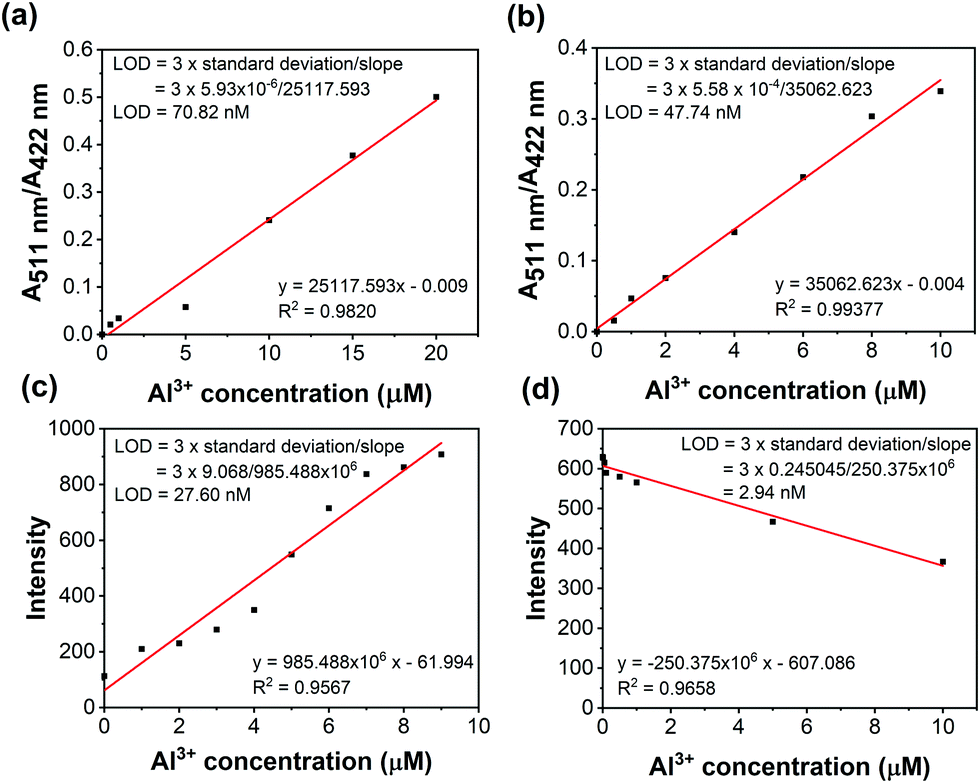
The absorption intensity ratio (A511nm/A422nm) and the fluorescence intensity plot are shown in Fig. 3. The linear relationships indicated the detection of Al3+ quantitatively by ratiometric absorption and fluorescence methods. Furthermore, the limit of detection (LOD) was calculated from the ratiometric absorption and fluorescence methods as shown in Fig. 3 and Table S3 (ESI†).
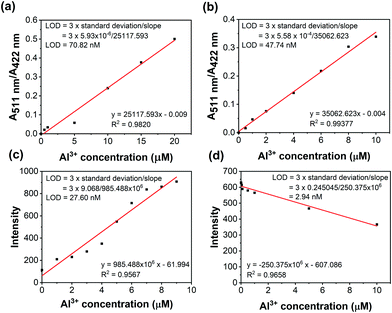 |
| | Fig. 3 Colorimetric titration (a and b) and fluorescence titration (c and d) of NDMIP-DASA and PNDMIP-DASA (6.5 ppm, THF/DIW 2:8 solution) against Al3+, respectively. | |
3.3 Computational studies
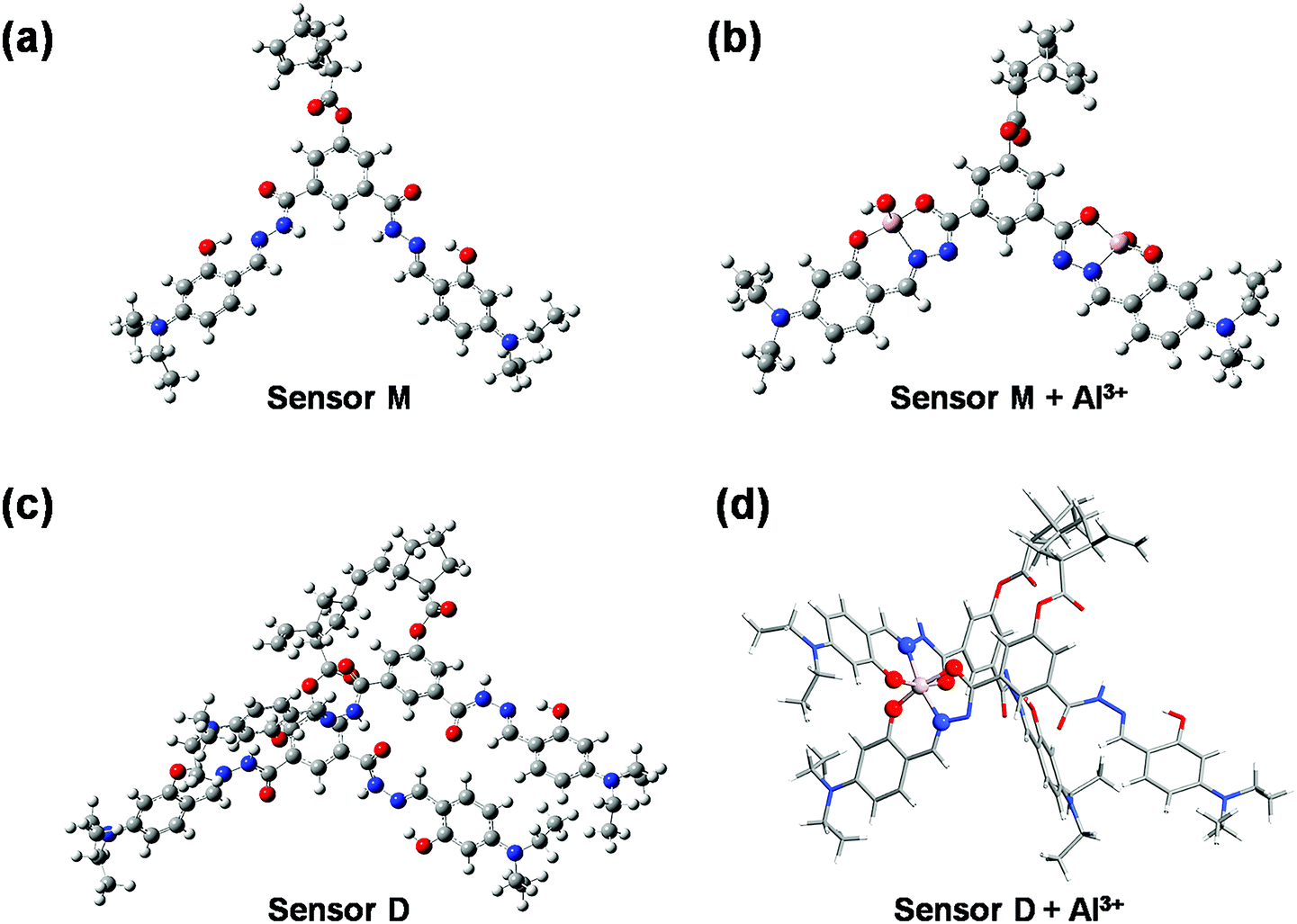
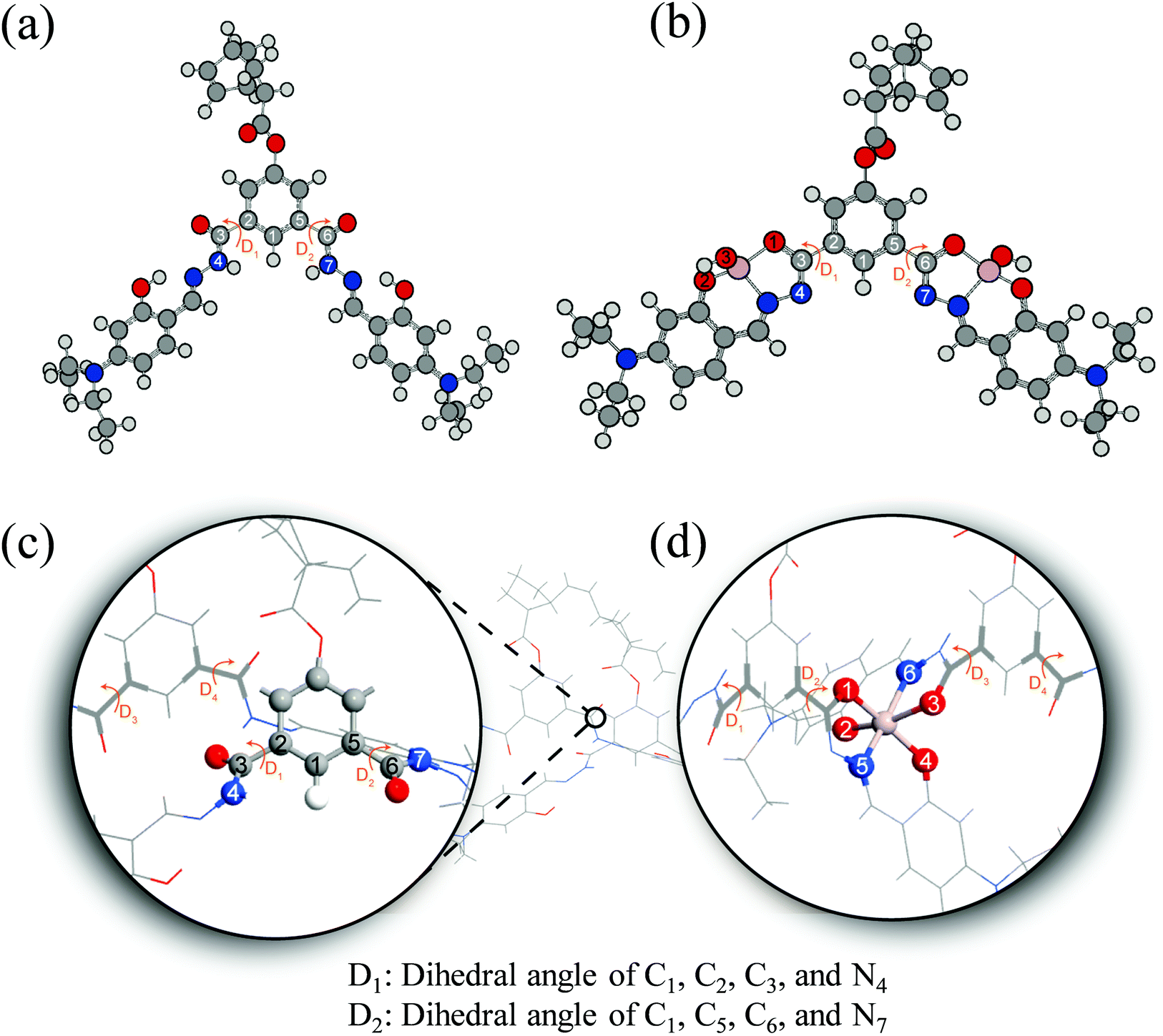
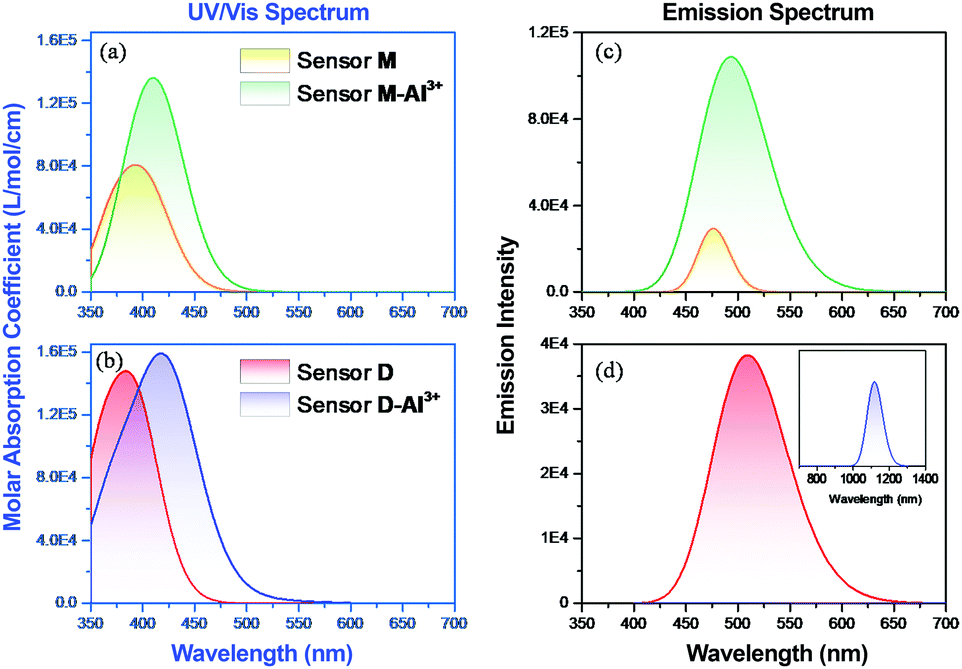
To understand the fluorescence sensing mechanism of the monomer NDMIP-DASA and polymer PNDMIP-DASA sensors toward Al3+ ions, we first considered the monomer NDMIP-DASA (M) structure with the corresponding aluminum-coordinated complex M–Al3+, as shown in Fig. 4, which illustrated the complex formed by two coordinated bonds between NDMIP-DASA and Al3+ ions. The selected geometrical parameters of the optimized sensors and the stable complexes are summarized in Tables S4 and S5 of the ESI.† We used the dihedral angles of D1–D4, as shown in Fig. 5, to illustrate the conjugation between the central benzene with side trains. The dihedral angle value is close to 0° or 180°, indicating better conjugation. M's optimized structure showed a distorted configuration with a nonplanar dihedral angle, D1, of C1–C2–C3–N4 (Fig. 4a). After the complexation with Al3+ cation, sensor M coordinated with Al3+ through the nitrogen and oxygen atoms of the Schiff base moiety and the oxygen atom of the OH-group from the solvent (Fig. 4b).
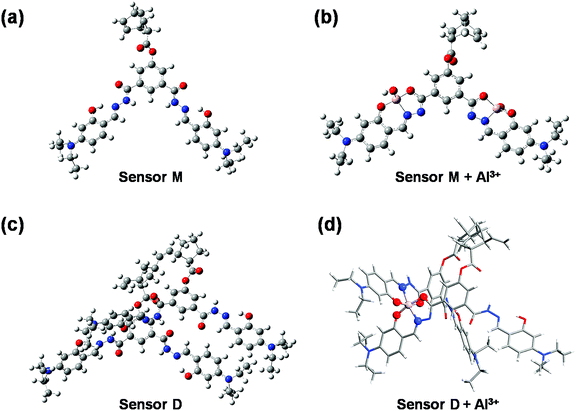 |
| | Fig. 4 The optimized structures of NDMIP-DASA (M) and PNDMIP-DASA (D) with and without Al3+ cation. | |
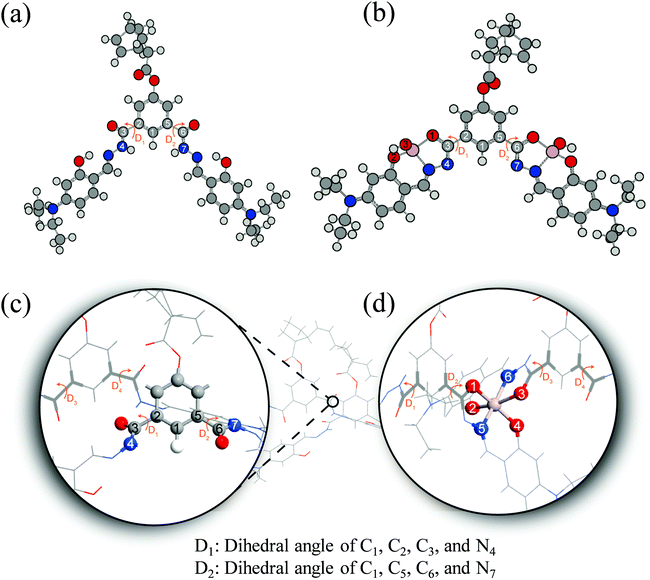 |
| | Fig. 5 Illustration of monomer M (a), M–Al3+ (b), dimer D (c), and D–Al3+ (d). | |
Furthermore, we investigated the polymer PNDMIP-DASA sensor and its interaction with Al3+ cation. Then, we considered two units of the polymer PNDMIP-DASA as a dimer sensor model, denoted as D. As compared to the structure of sensor M, sensor D shows a larger distorted configuration with a dihedral angle of 46.84° (Table S5, ESI†). After complexation, the optimized sensor D showed six coordination numbers of four oxygen and two nitrogen atoms with Al3+ cation. The complex D–Al3+ formed better conjugation with a dihedral angle, D1, of 6.99°. We used the time-dependent density functional theory (TD-DFT) calculations to investigate the fluorescence turn-off mechanism by adding Al3+ cation to the polymer sensor. We recorded the UV-vis absorption spectra and emission spectra for both sensor M and sensor D in the absence and presence of the interaction with Al3+ cation, as shown in Fig. 6. The corresponding excitation energies and molecular orbital (MO) compositions of vertical transitions for these complexes are summarized in Table 1. As shown in Fig. 6a and b, without the interaction with Al3+ cation, the simulated absorption spectra corresponding to sensors M and D showed a strong absorption band in the visible region. The maximum absorption wavelength (λmax) of sensors M and D were 408 and 396 nm, respectively. After complexation, the λmax values of sensors M and D became 420 and 434 nm, red-shifted by 12 and 38 nm, respectively. Furthermore, Fig. 6c and d show the simulated emission spectra of sensors M and D with and without the interaction with Al3+ cation. For the case of sensor M, as shown in Fig. 6c, sensor M and its complex M-Al3+ possessed emission at 476 and 481 nm, respectively. The emission wavelengths of sensor M and M–Al3+ were shorter than their absorption wavelengths, indicating that the potential energy curve of the excited state was shifted compared with the ground state. Fig. 6d shows that the emission band of sensor D appears at 509 nm, while the emission band of composite D–Al3+ was at 1118 nm. The calculation results explain that the experiment did not observe the fluorescence of composite D–Al3+ because its emission band was not in the visible region but the near-infrared (NIR) region. To further study the sensing mechanism of sensors M and D for Al3+ cation, the TD-DFT calculations were used to compare the geometric differences between the ground state (S0) and the excited state (S1) of sensors M and D, and the frontier molecular orbital (FMO) contribution of sensors M and D. All the main structural parameters are listed in Tables S4 and S5 (ESI†). We can see from Table S4 (ESI†) that the dihedral angle of D1 for sensor M is 29.21°. Photoirradiation changed the non-planar structure in the S0 state to a nearly planar structure (11.71°) in the S1 state. In the case of complex M–Al3+, the change of the dihedral angle is similar to that of sensor M. In addition, Table S4 (ESI†) demonstrates that the Al–O and Al–N bond distances of complex M–Al3+ were similar in the S0 and S1 states.
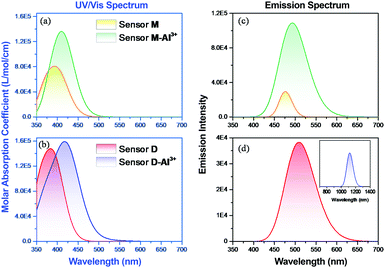 |
| | Fig. 6 DFT-computed (a and b) UV-vis and (c and d) emission spectra of sensors M and D with and without the complexation of Al3+ cation. | |
Table 1 Calculated absorption energy (λ in nm), oscillator strength (f in a.u.), and the corresponding MO transition of sensor M, sensor D and the complexes M–Al3+ and D–Al3+
|
|
Transition |
λ (nm) |
f
|
Composition |
CI (%) |
|
M
|
S0 → S1 |
408 |
0.545 |
HOMO → LUMO |
73.2 |
| HOMO−1 → LUMO |
24.0 |
|
M–Al3+* |
S0 → S1 |
420 |
1.384 |
HOMO → LUMO |
91.8 |
| HOMO−1 → LUMO+1 |
5.5 |
|
D
|
S0 → S3 |
396 |
1.285 |
HOMO−1 → LUMO+1 |
65.5 |
| HOMO−2 → LUMO |
14.6 |
| HOMO → LUMO+1 |
6.1 |
|
D–Al3+* |
S0 → S5 |
434 |
0.715 |
HOMO−3 → LUMO |
83.8 |
| HOMO → LUMO+1 |
11.1 |
In addition, we calculated the frontier molecular orbital (FMO) contribution of sensor M and M–Al3+ in the excited state (S1) to understand the red-shift phenomenon in the emission spectra (Fig. 6c) upon the complexation of Al3+ cation. The frontier molecular orbitals corresponding to the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of sensors M and D as well as their complexes with Al3+ cation are illustrated in Fig. 7. In the S1 state of sensor M, the LUMO → HOMO transition mainly dominates the S1 → S0 transition at the 476 nm emission, which was contributed by charge transfer from the central phenyl group to the Schiff base moiety as shown in Fig. 7. The energy difference between the HOMO and the LUMO of sensor M was 2.60 eV, as shown in Table 2. In the case of complex M–Al3+, the HOMO and LUMO energy gap slightly decreased to 2.57 eV due to the complex formation with a charge transfer from the phenyl group toward the aforementioned metal-binding site and the imine bond. As compared to the obtained UV-vis spectra, it can be seen that both sensor M and M–Al3+ showed a small shift to a longer wavelength between the absorption and the fluorescence spectra. The result indicated that a small Stokes shift occurred because of the geometry distortion from the non-planar structure in the S0 state to a nearly planar structure in the S1 state (Table S4, ESI†). Meanwhile, we did a similar calculation of the case of sensor D in the S1 state. We can see from Table S5 that the structural transformation was small, and sensor D maintained a non-planar structure in the S1 state. However, the structural transformation of complex D–Al3+ in the S1 state was significant. The structural transformation of complex D–Al3+ was attributed to the changes of the dihedral angles and the Al–O bond distances between the S0 and S1 states, as shown in Table S5 (ESI†). The large structural transformation would lead to a large displacement of the potential energy curve in the excited state with respect to the ground state, resulting in a large Stokes shift between the absorption and the emission spectra. Fig. 7 shows that the LUMO → HOMO transition of sensor D was similar to that of sensor M due to the charge transfer from the central phenyl group to the Schiff base moiety with an emission of 509 nm. However, after the complexation with Al3+ cation, the contributions of the LUMO and HOMO were from different atoms, causing the emission of S1 → S0 to have a tiny oscillator strength of 0.0004. In addition, the energy gap between the HOMO and the LUMO significantly decreased after complex formation, making the emission shift to the NIR region at 1118 nm. The NIR emission and the small oscillator strength influenced the fluorescence behavior of complex D–Al3+, which made it difficult to be observed.
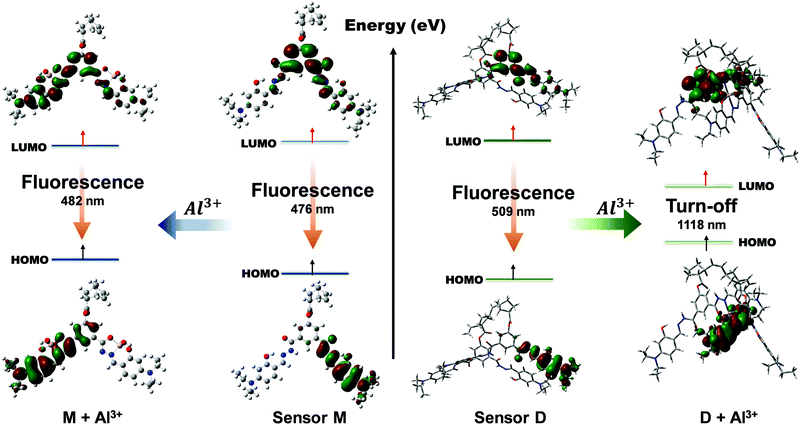 |
| | Fig. 7 Frontier molecular orbitals (HOMO and LUMO) of sensor M and its complex M–Al3+ (left) as well as sensor D and its complex D–Al3+ (right). | |
Table 2 Calculated emission energy (λ in nm), oscillator strength (f in a.u.), and the corresponding MO transition of sensor M, sensor D and the complexes M–Al3+ and D–Al3+
|
|
Transition |
E (eV) |
λ (nm) |
f
|
Composition |
CI (%) |
|
M
|
S1 → S0 |
2.60 |
476 |
0.217 |
LUMO to HOMO |
98.5 |
|
M–Al3+*
|
S1 → S0 |
2.57 |
481 |
1.580 |
LUMO to HOMO |
92.9 |
|
D
|
S1 → S0 |
2.43 |
509 |
0.567 |
LUMO to HOMO |
97.3 |
|
D–Al3+*
|
S1 → S0 |
1.10 |
1118 |
0.004 |
LUMO to HOMO |
99.5 |
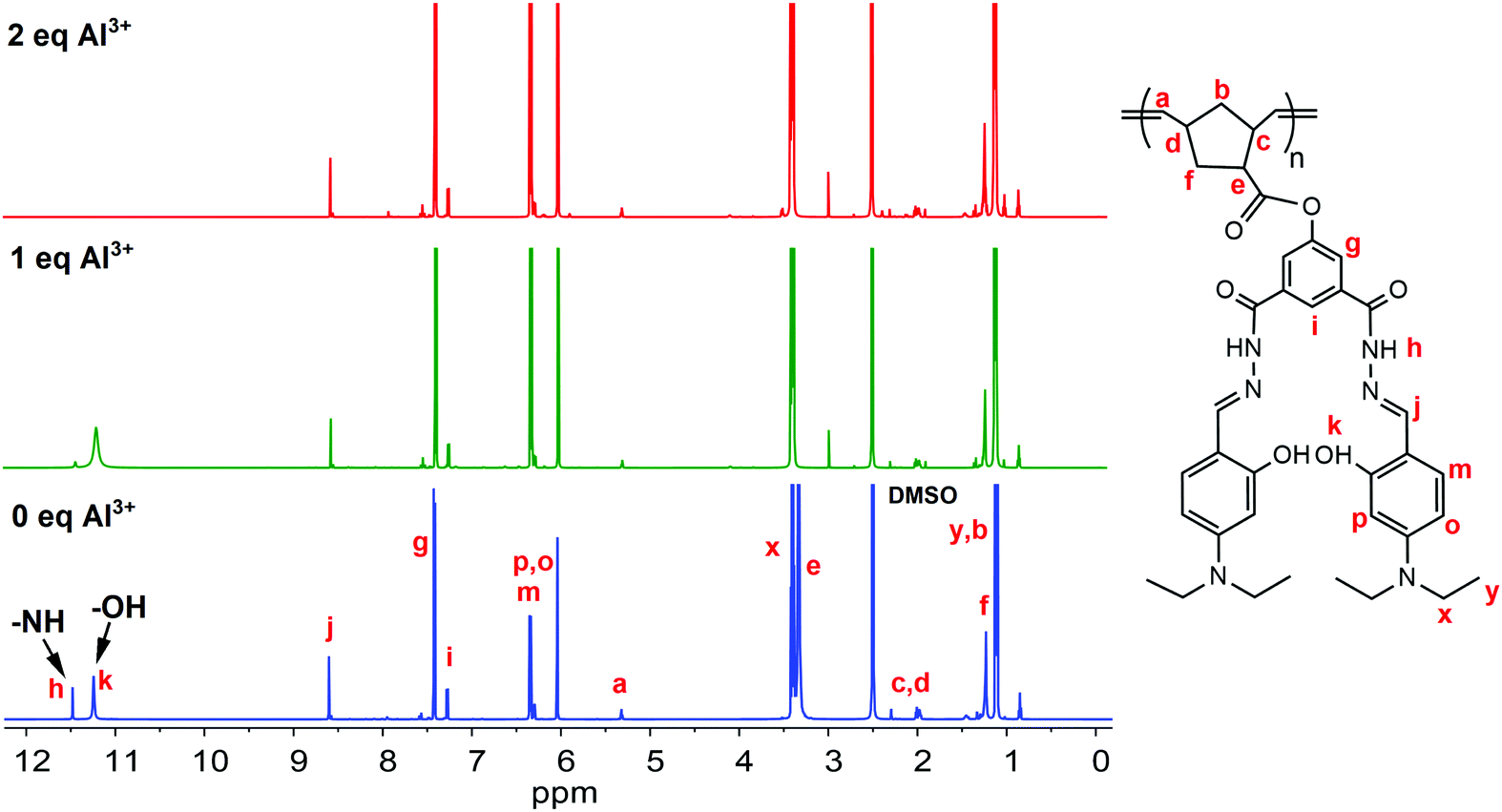
A significant benchmark that an outstanding sensor should reach is high selectivity with excellent sensitivity. However, in this report, we mainly focused on understanding the turn-off fluorescence mechanism towards Al3+ ions. The reason behind the fluorescence sensing selectivity of PNDMIP-DASA toward Al3+ in the presence of other metal ions would be our future report. In the next stage, a Job's plot experiment was conducted to derive the binding stoichiometry of NDMIP-DASA and PNDMIP-DASA with Al3+ cation. As displayed in Fig. S8 (ESI†), the NDMIP-DASA–Al3+ complex exhibited the 1:1 binding mode, whereas the PNDMIP-DASA exhibited the 1:2 binding mode with Al3+ ions. 1H NMR titrations were performed in DMSO-d6 to investigate the binding motif between Al3+ and the PNDMIP-DASA polymer. As shown in Fig. 8, by the addition of Al3+, the peak of –NH at δ 11.48 ppm and –OH at δ 11.24 ppm completely disappeared at 2 eq. of Al3+. The signal of proton –CHN and the aromatic proton in the vicinity of the –OH group showed a slight downward shift, whereas the other positions remain unchanged. These results further supported the proposed binding phenomenon, as discussed by using computational data. The pH effect on the absorption of NDMIP-DASA and PNDMIP-DASA in the presence of Al3+ was also investigated and shown in Fig. S9 (ESI†).
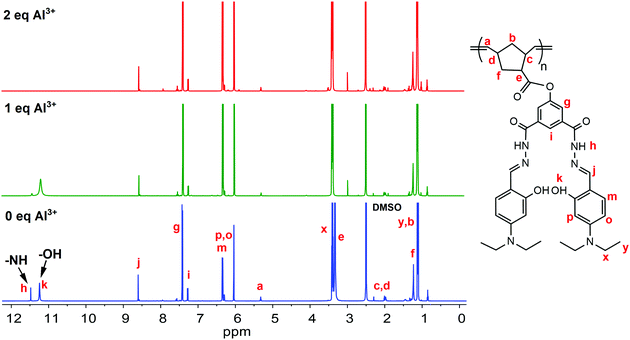 |
| | Fig. 8 Changes in 1H NMR of PNDMIP-DASA (6.5 ppm) upon the addition of Al3+. | |
4. Conclusions
In conclusion, we have reported a norbornene-derived hydrazone-based NDMIP-DASA monomer, and its homopolymer PNDMIP-DASA which consists of –CONH, –CN, and –OH groups to understand the turn-off fluorescence mechanism towards Al3+ ions. NDMIP-DASA and PNDMIP-DASA exhibited excellent sensitivity towards Al3+. The limit of detection of NDMIP-DASA and PNDMIP-DASA towards Al3+ ions was 27.60 nM and 2.94 nM, respectively. Sensor M, sensor D, and complex M–Al3+ showed a small structural transformation after photoirradiation, causing the small displacement of the potential energy curve between the ground state and the excited state. The small displacement of the potential energy curve resulted in a small Stokes shift between the absorption and emission spectra in the UV-vis region. However, the complex D–Al3+ structure in the S1 state showed a large distortion compared to the ground state. The large displacement of the potential energy curve in the S1 state decreased the energy difference between the S0 and S1 states, resulting in an emission shift from the UV-vis to NIR regions. In addition, the oscillator strength of the S1 to S0 transition was close to zero. These results made it difficult to find the emission of complex D–Al3+ in the UV-vis region. This computational study provided new insights into the fluorescence sensing of the turn-off mechanism.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Trong-Nghia Le thanks NTUST for research fellowships. N. Vijayakameswara Rao thanks NTUST for start-up funding. The authors thank the National Center of High-Performance Computing (NCHC) for donating computer facilities.
Notes and references
- K. Klotz, W. Weistenhöfer, F. Neff, A. Hartwig, C. van Thriel and H. Drexler, Dtsch. Ärztebl. Int., 2017, 114, 653 Search PubMed.
- B. Wang, W. Xing, Y. Zhao and X. Deng, Environ. Toxicol. Pharmacol., 2010, 29, 308–313 Search PubMed.
- C. C. Willhite, N. A. Karyakina, R. A. Yokel, N. Yenugadhati, T. M. Wisniewski, I. M. Arnold, F. Momoli and D. Krewski, Crit. Rev. Toxicol., 2014, 44, 1–80 Search PubMed.
- Y. K. Jang, U. C. Nam, H. L. Kwon, I. H. Hwang and C. Kim, Dyes Pigm., 2013, 99, 6–13 Search PubMed.
- Y. W. Choi, G. J. Park, Y. J. Na, H. Y. Jo, S. A. Lee, G. R. You and C. Kim, Sens. Actuators, B, 2014, 194, 343–352 Search PubMed.
- S. Sharma, G. Dubey, B. S. Sran, P. V. Bharatam and G. Hundal, ACS Omega, 2019, 4, 18520–18529 Search PubMed.
- L. Wang, J. Yang, H. Wang, C. Ran, Y. Su and L. Zhao, Sensors, 2019, 19, 2423 Search PubMed.
- C.-T. Chen and W.-P. Huang, J. Am. Chem. Soc., 2002, 124, 6246–6247 Search PubMed.
- K. P. Carter, A. M. Young and A. E. Palmer, Chem. Rev., 2014, 114, 4564–4601 Search PubMed.
- H. N. Kim, Z. Guo, W. Zhu, J. Yoon and H. Tian, Chem. Soc. Rev., 2011, 40, 79–93 Search PubMed.
- Y. Wang, L. J. Hou, Y.-B. Wu, L. L. Shi, Z. Shang and W. J. Jin, J. Photochem. Photobiol., A, 2014, 281, 40–46 Search PubMed.
- D. Kara, A. Fisher and S. J. Hill, J. Environ. Monit., 2007, 9, 994–1000 Search PubMed.
- H. M. Park, B. N. Oh, J. H. Kim, W. Qiong, I. H. Hwang, K.-D. Jung, C. Kim and J. Kim, Tetrahedron Lett., 2011, 52, 5581–5584 Search PubMed.
- M. Ahmed, M. Faisal, A. Ihsan and M. M. Naseer, Analyst, 2019, 144, 2480–2497 Search PubMed.
- T.-J. Jia, W. Cao, X.-J. Zheng and L.-P. Jin, Tetrahedron Lett., 2013, 54, 3471–3474 Search PubMed.
- H. Xiao, K. Chen, N. Jiang, D. Cui, G. Yin, J. Wang and R. Wang, Analyst, 2014, 139, 1980–1986 Search PubMed.
- S. Kim, J. Y. Noh, K. Y. Kim, J. H. Kim, H. K. Kang, S.-W. Nam, S. H. Kim, S. Park, C. Kim and J. Kim, Inorg. Chem., 2012, 51, 3597–3602 Search PubMed.
- R. Purkait, C. Patra, A. D. Mahapatra, D. Chattopadhyay and C. Sinha, Sens. Actuators, B, 2018, 257, 545–552 Search PubMed.
- J. W. Jeong, B. A. Rao and Y.-A. Son, Sens. Actuators, B, 2015, 208, 75–84 Search PubMed.
- G. Balamurugan, S. Velmathi, N. Thirumalaivasan and S. P. Wu, Analyst, 2017, 142, 4721–4726 Search PubMed.
- D. Sarkar, P. Ghosh, S. Gharami, T. K. Mondal and N. Murmu, Sens. Actuators, B, 2017, 242, 338–346 Search PubMed.
- G.-Q. Wang, J.-C. Qin, C.-R. Li and Z.-Y. Yang, Spectrochim. Acta, Part A, 2015, 150, 21–25 Search PubMed.
- Y. Li, Q. Niu, T. Wei and T. Li, Anal. Chim. Acta, 2019, 1049, 196–212 Search PubMed.
- S. K. Shoora, A. K. Jain and V. K. Gupta, Sens. Actuators, B, 2015, 216, 86–104 Search PubMed.
- W.-H. Ding, W. Cao, X.-J. Zheng, D.-C. Fang, W.-T. Wong and L.-P. Jin, Inorg. Chem., 2013, 52, 7320–7322 Search PubMed.
- D. T. McQuade, A. E. Pullen and T. M. Swager, Chem. Rev., 2000, 100, 2537–2574 Search PubMed.
- S. W. Thomas, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339–1386 Search PubMed.
- T. M. Swager, Acc. Chem. Res., 1998, 31, 201–207 Search PubMed.
- B. Wang and M. R. Wasielewski, J. Am. Chem. Soc., 1997, 119, 12–21 Search PubMed.
- S. Sarkar and R. Shunmugam, ACS Appl. Mater. Interfaces, 2013, 5, 7379–7383 Search PubMed.
- S. Bhattacharya, V. N. Rao, S. Sarkar and R. Shunmugam, Nanoscale, 2012, 4, 6962–6966 Search PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 Search PubMed.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 Search PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 Search PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 Search PubMed.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 Search PubMed.
- S. Sutthasupa, M. Shiotsuki, H. Matsuoka, T. Masuda and F. Sanda, Macromolecules, 2010, 43, 1815–1822 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data and supplementary figures including 1H NMR, FT-IR spectra, and computational details. See DOI: 10.1039/d1ma00254f |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Kuan-Yu
Lin‡
,
Kuan-Yu
Lin‡