
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCation disorder and octahedral distortion control of internal electric field, band bending and carrier lifetime in Aurivillius perovskite solid solutions for enhanced photocatalytic activity†
Jaideep
Malik
a,
Shubham
Kumar
a,
Priya
Srivastava
 b,
Monojit
Bag
bc and
Tapas Kumar
Mandal
*ac
b,
Monojit
Bag
bc and
Tapas Kumar
Mandal
*ac
aDepartment of Chemistry, Indian Institute of Technology Roorkee, Roorkee 247 667, India. E-mail: tapas.mandal@cy.iitr.ac.in
bDepartment of Physics, Indian Institute of Technology Roorkee, Roorkee 247 667, India
cCentre of Nanotechnology, Indian Institute of Technology Roorkee, Roorkee 247 667, India
First published on 7th June 2021
Abstract
The overall photocatalytic activity shown by a semiconductor photocatalyst stems from a complex interplay of several critical factors that includes light absorption, carrier recombination dynamics, charge transfer resistance, lifetime of photogenerated charge carriers and surface adsorption. While most of the factors are intrinsic to a semiconductor controlled by its structure and composition, the overall activity is determined by the dominance of a single or multiple factors. The present work examines the competing roles of these factors in the photocatalytic activity of a closely related series of Aurivillius perovskite solid solutions, Bi6−xSrxTi3+xFe2−xO18 (x = 0.75, 0.5, and 0.25). The cation disorder in the intergrowth structure and octahedral distortion that control the internal electric fields in these compounds alter multiple factors responsible for enhanced photocatalytic activity. The five-layer Aurivillius perovskites forming in the non-centrosymmetric F2mm space group with highly distorted octahedra at the terminal position and least distorted octahedra at the central perovskite layer are controlled by a second-order Jahn–Teller effect of Ti4+. While a greater extent of dye adsorption leads to faster photocatalytic degradation in Bi5SrTi4FeO18 and other Aurivillius perovskites, Bi5.5Sr0.5Ti3.5Fe1.5O18 (x = 0.5) shows inferior photocatalytic activity as compared to Bi5.75Sr0.25Ti3.25Fe1.75O18 (x = 0.25) and Bi5.25Sr0.75Ti3.75Fe1.25O18 (x = 0.75) despite having the highest dye adsorption in the series. The highest rate of photocatalytic RhB degradation in the Fe1.75 compound is due to its longest average carrier lifetime in the series, sluggish e−–h+ recombination and lower charge transfer resistance. Furthermore, the enhanced activity observed under external bias is understood to have originated from a greater upward band bending effect, which acts as thermal barrier for e−–h+ recombination and facilitates effective charge separation and transfer. The work elucidates the delicate interplay of the competing factors affecting the photocatalytic activity and establishes the origin of non-uniform and enhanced photocatalytic activity across the homologous solid solution series of layered perovskites. The protocol will help breaking compositional limits in uncovering new solid-solution compounds with enhanced activity by appropriate substitutions and further enhancement under external bias for faster removal and degradation of pollutants.
Introduction
Increasing energy demands, growing population and depleting fossil fuel reserves compel us to look for renewable and clean resources of energy. With abundant sunlight reaching Earth's surface throughout the year, harvesting solar energy, a clean and renewable resource, can be the ultimate solution.1 One requires efficient devices to harness the abundant solar energy. Semiconductor photocatalysis have shown immense potential in energy generation and environmental remediation using solar energy.2,3 Photocatalysts with extended visible-light absorption can harvest maximum sunlight and may have wide ranging applications in the area of hydrogen production by water splitting,4 degradation or complete mineralization of organic pollutants in wastewater5,6 and hydrogen peroxide generation.7,8Since the electrochemical decomposition of water at the TiO2 electrode by Fujishima and Honda in 1972,9 research based on semiconductor photocatalysis for the development of new photocatalysts has surged. For the last several decades, metal–oxide based semiconductor materials have been widely used for photocatalytic degradation.10,11 Most of them are TiO2 based semiconductors due to their availability, stability and non-hazardous nature.12,13 However, they cannot be used under visible-light due to their large band gaps falling mainly in the UV region making them “inefficient” as the UV region only accounts for 3–5% of the incoming solar energy. Thus, novel visible-light-driven photocatalysts absorbing in the visible region with a wider absorption range to utilize maximum amounts of sunlight is indispensable. Many visible-light-driven photocatalysts were studied in recent years with layered compounds getting huge attraction.5,14–27 Recently, photocatalyst designs with self-healing internal electric fields have been demonstrated exhibiting accelerated photocatalysis.28 Moreover, a gradient doping of P in CdS was realized to create a built-in internal electric field to facilitate charge carrier extraction and enhance H2 evolution.29 Similarly, a heterojunction design with a graded solid–solution interface in MnxCd1−xS facilitated efficient separation and transfer of charge carriers due to a gradient built-in electric field resulting excellent photocatalytic hydrogen generation.30
Compounds with the perovskite structure, especially the layered variants have shown tremendous research thrust in visible-light-driven photocatalysis.5,19,31,32 The layered compounds, largely Aurivillius phases, attract a lot of attention for their improved separation of charge carriers due to the presence of internal electric fields33 resulting in enhanced photocatalytic activity. The Aurivillius phases are a part of a large family of layered perovskites with regular fluorite-like (Bi2O2)2+ layers sandwiched between the perovskite (An−1BnO3n+1)2− blocks, where n represents the number of corner connected octahedral layers.34,35 There are many reports of Aurivillius phases related to the study of their ferroelectric and dielectric properties36–40 but only some of them were studied for their photocatalytic properties.20,32,41–44 Out of these, a very limited number of semiconductor photocatalysts are active in the visible region of the solar spectrum. The layered compounds are modified by substitution at the A-site as well as the B-sites of the perovskite block to extend their absorption to the visible region. The compositional substitution in layered Aurivillius phases were studied most intensely for n = 1 members of the family, namely, Bi2WO6 and Bi2MoO6.45–51 On the contrary, higher order members (with n = 2, 4 and 5) were limited and few were studied for their photocatalytic activities.5,19,31,52–54 Aurivillius phases, which always contain Bi, are noteworthy because the presence of Bi pushes the top of the valence band upward and often shifts the absorption edge in the higher wavelength or visible region when compared with their non-bismuth counterparts, thus enhancing the catalytic activities of the compounds under visible-light irradiation. Recently, five-layer Bi6Ti3Fe2O18 intrigued researchers towards its optical and ferroelectric properties, and the rare earth (La, Sm, Gd, and Dy) substituted Bi6Ti3Fe2O18 for their magnetic and magnetoelectric properties.55,56
Considering that the five-layer Bi6Ti3Fe2O18 crystallizes in a non-centrosymmetric space group and substitution at the A-site as well as the B-sites of the perovskite would alter the strength of internal electric fields, we envisaged various compositions Bi6−xSrxTi3+xFe2−xO18 (x = 0.75, 0.5, and 0.25) that fall between the two end members Bi6Ti3Fe2O18 and Bi5SrTi4FeO18. The compositions are attained by altering the amount of Fe with respect to Ti and counterbalancing the charge by replacing Bi with Sr. It is believed that the compositional manipulation would change the extent of octahedral distortion, the strength of internal electric field and charge carrier lifetime in addition to subtle changes in the band structure of the compounds, and thus help understanding the role of various factors affecting photocatalytic activity in the closely related solid-solution series. The interplay of various factors responsible for photocatalytic activity and their relative dominance or suppression in affecting the photocatalytic activity of Bi6−xSrxTi3+xFe2−xO18 (x = 0.75, 0.5, and 0.25) are reported. The study highlights the effect of photocatalytic activity in the presence of bias voltage and its consequences under simulated solar radiation.
Experimental
Materials and synthesis
For the synthesis, Bi2O3 (≥98%), SrCO3 (≥99.9%), TiO2 (99.8%) and FeC2O4·2H2O (99%) were obtained from Sigma-Aldrich and used directly as supplied. All analytical grade reagents were used where required without any further purification in all our experiments and Millipore (Bedford, MA, USA) water used for preparing all the dye solutions.The five-layered Aurivillius phases, Bi6−xSrxTi3+xFe2−xO18 (x = 1.0, 0.75, 0.5, 0.25, and 0.0), were prepared by conventional solid-state reactions. For this, stoichiometric amounts of Bi2O3, SrCO3, TiO2 and FeC2O4·2H2O were ground in an agate mortar for 1 h and the powder mixture was fired at 780 °C for 2 h. Then, the mixture was ground again and pressed into pellets of 12 mm diameter. These pellets were reheated at 1000 °C for different times with intermediate grinding as given in Table S1 (ESI†).
Characterization
In the earlier stages of structure refinement, scale factor and cell parameters were refined followed by background coefficients along with zero shift. In the successive runs shape and other profile parameters were refined systematically to avert any divergence. Positional parameters were refined just before the thermal parameters in the later stages of the refinement based on the atomic weights of elements, i.e., by choosing the heavier one first while positional parameters of oxygen were refined last as it is the lightest atom, although its thermal parameters were left unchanged to avoid unrecoverable divergence. Finally, refinement by varying the occupancy of atoms was performed to examine cation disorder by fixing refined positions and thermal parameters of the atoms.
Photocatalytic activity study
| Adsorption (%) = (1 − C/C0) × 100 | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm to separate suspended catalyst and recording the absorbance of the centrifugate on a Shimadzu 2450 UV-visible spectrophotometer. A blank experiment was also performed under similar conditions to account for any correction due to self-degradation of the dye.
000 rpm to separate suspended catalyst and recording the absorbance of the centrifugate on a Shimadzu 2450 UV-visible spectrophotometer. A blank experiment was also performed under similar conditions to account for any correction due to self-degradation of the dye.
Electrochemical measurements
Electrochemical impedance spectroscopy (EIS) studies were carried out on a Multi Autolab/M204 electrochemical workstation (Metrohm Autolab B.V., Netherlands) with a three-electrode assembly. For this, Pt wire was used as the counter electrode, Ag/AgCl was used as the reference electrode and the photocatalyst material coated on ITO was used as the working electrode in a 10 mM K3[Fe(CN)6] solution containing 0.1 M KCl as the electrolyte while the EIS study under illumination at biased voltage was carried out on a Zahner Electrochemical Workstation in the frequency range of 100 mHz to 1 MHz under the light intensity of 400 W m−2 at 0 and 1 V in a two-electrode system. For this, Pt wire was used as the counter electrode, the photocatalyst coated ITO was used as the working electrode and tetrabutylammonium hexafluorophosphate (TBAPF6) in dichloromethane (DCM) was used as the liquid electrolyte. Working electrodes were prepared by mixing compounds with 2% of PVDF (polyvinylidene difluoride) in NMP (N-methyl-2-pyrrolidone) to make a slurry, which was then coated on ITO and dried overnight at 60 °C. Experimental data were fitted with equivalent electrical circuits using ZView® software from Scribner Associates to estimate various parameters.Results and discussion
PXRD analysis
The observed PXRD patterns (Fig. 1) of Bi6−xSrxTi3+xFe2−xO18 (x = 1.0, 0.75, 0.5, 0.25, and 0.0) indicate the formation of a five-layer Aurivillius phase which is in agreement with the JCPDS PDF #21-0101 and earlier data reported in the literature for five-layer phases.19,59 All the reflections present in a pattern are indexable in the F2mm (no. 42) space group. The least squares refined unit cell parameters (Table S2 in ESI†) show an increase in the c-parameter for the compounds in the series with increasing Fe from 1.25 to 1.75.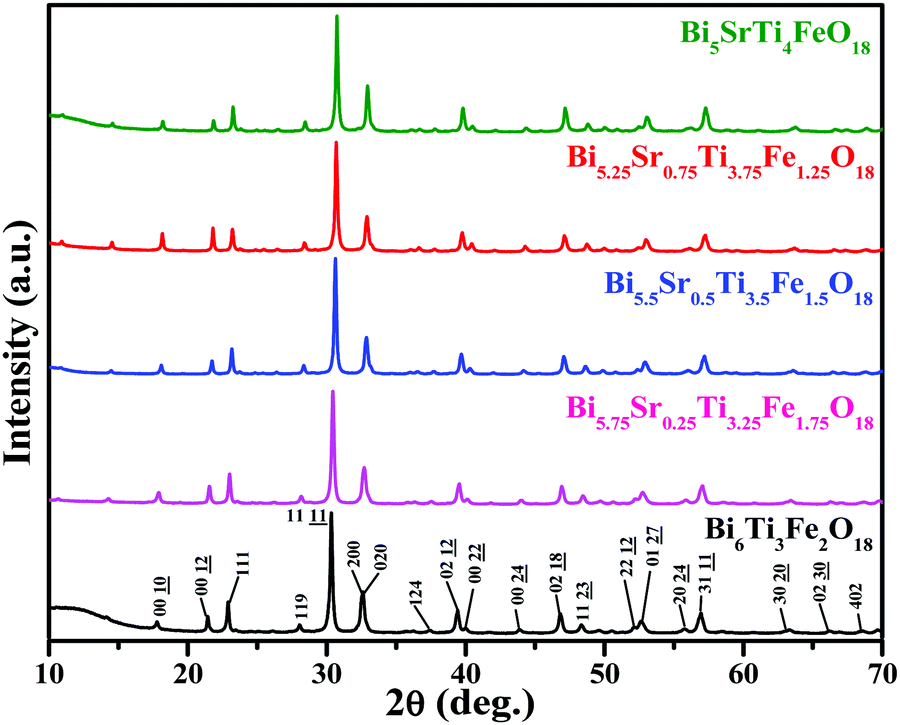 | ||
| Fig. 1 PXRD patterns of Bi6−xSrxTi3+xFe2−xO18 (x = 1.0, 0.75, 0.5, 0.25, 0.0). | ||
Rietveld structure refinements of the compounds are carried out using the FullProf program suite using the model structure of Bi6Ti3Fe2O18, where one complete layer of Fe is confined to the central perovskite layer and the remaining Fe (0.25, 0.5 and 0.75) are statistically distributed over the terminal Ti(1) and pre-terminal Ti(2)-sites. During the trial runs with all Fe distributed over all the octahedral sites and Sr over the A-sites of perovskite as well as the [Bi2O2]2+ layers, the refinement resulted in higher values of the reliability factors. The observed, calculated and difference profiles with the lowest χ2 value are shown in Fig. 2. The refined atomic positions, occupancies and thermal parameters are given in the Tables S3–S5 (ESI†) and the crystal structure drawn from the refined parameters for a representative member, Bi5.75Sr0.25Ti3.25Fe1.75O18, is shown in Fig. 2. The Aurivillius perovskites form in the non-centrosymmetric F2mm space group with the occupation of one Fe in the central octahedral layer while the remaining Fe's are statistically distributed over the pre-terminal and terminal octahedral layers (ignored in the structural presentation). The distortions of octahedra are visible for regular FeO6 octahedra at the central layer and distorted Ti/FeO6 octahedra at the pre-terminal and terminal positions with out-of-centre displacement of the Ti/Fe.
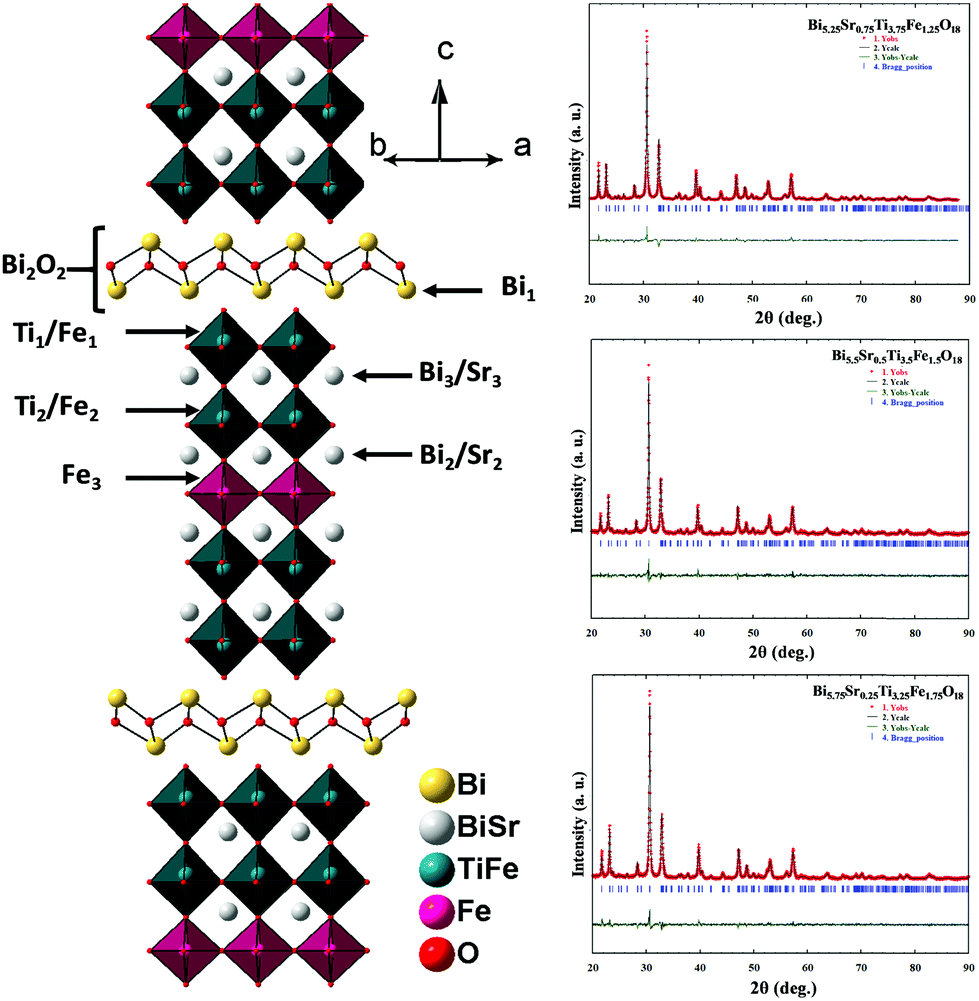 | ||
| Fig. 2 Rietveld refinement profiles of Bi5.25Sr0.75Ti3.75Fe1.25O18, Bi5.5Sr0.5Ti3.5Fe1.5O18 and Bi5.75Sr0.25Ti3.25Fe1.75O18 (right panel). Observed (+), calculated (−) and difference (bottom) profiles are shown, Vertical bars represent Bragg positions. The crystal structure of Bi5.75Sr0.25Ti3.25Fe1.75O18 constructed from the refined parameters is shown in the left panel. | ||
The B-cation displacement in the pre-terminal and terminal octahedra give rise to the short and long bond sequence along the c-axis direction (see later for detail). This also leads to the buckling of in-plane bonds at the pre-terminal and terminal octahedra. Because of this, the centre of symmetry of BO6 octahedra is lost, leading to the development of local non-zero polarization at the octahedra while in a centrosymmetric structure, the dipoles cancel each other resulting in the loss of overall polarization. In compounds with non-centrosymmetric space groups the resultant dipoles persist causing permanent polarization in the compounds.
FE-SEM, EDS and elemental mapping analysis
The FE-SEM images of Bi6−xSrxTi3+xFe2−xO18 (x = 0.75, 0.5, 0.25) (Fig. S2 in the ESI†) show aggregated particles dominated by plate-like crystallites for all the compounds with the particle size ranging from hundreds of nanometers to few micrometers. The features are same throughout as seen in the images taken at several regions of the sample. The average calculated elemental ratios from EDS are in good agreement with the nominal compositions within admissible error limits. Elemental mapping is performed to look at the elemental distribution in Bi6−xSrxTi3+xFe2−xO18 (x = 0.75, 0.50, 0.25) in different areas of the powder samples. The EDS elemental maps indicate a homogeneous distribution of the constituent elements in all the cases as shown in Fig. 3. An increase in the quantity of Bi and Fe and decrease in the quantity of Sr and Ti as x varies from 0.75 to 0.25 can be clearly seen in the elemental maps. | ||
| Fig. 3 SEM images and corresponding EDS elemental maps for Bi5.25Sr0.75Ti3.75Fe1.25O18 (a–e), Bi5.5Sr0.5Ti3.5Fe1.5O18 (f–j) and Bi5.75Sr0.25Ti3.25Fe1.75O18 (k–o). | ||
HR-TEM and EDS analysis
The lattice periodicity and crystalline structure is further analysed with TEM. The cross-sectional HR-TEM image (Fig. 4a) reveals the intergrowth of Aurivillius structure with [Bi2O2]2+ units interleaving between the five-layer perovskite slabs as seen clearly by the contrast of lattice fringes. A lattice distance of 4.91 nm between the marked fringes corresponds to the c-parameter periodicity of Bi5.75Sr0.25Ti3.25Fe1.75O18 and is in agreement with the calculated distance obtained from the PXRD. Moreover, the SAED pattern (Fig. 4b) shows single crystalline diffraction patterns and some of the bright spots are indexable corresponding to specific hkl planes allowed by the F2mm space group and in agreement with the PXRD data. | ||
| Fig. 4 (a) HR-TEM image showing periodic lattice fringes in the c-direction (the inset shows lattice periodicity in the ab-plane) and (b) SAED pattern of Bi5.75Sr0.25Ti3.25Fe1.75O18. | ||
UV-vis DRS analysis
Fig. S3 (ESI†) shows the UV-vis DRS in absorption term and the corresponding Tauc plots for the compounds. The compounds show two-step absorption feature with stronger and sharper absorption at the lower wavelength side, while the higher wavelength edge is more stretched. This is attributed to the electronic band structure of the compounds. It is presumed that the valence band (VB) states of these Aurivillius phases are made up of Fe (3d), Bi (6s) and O (2p) hybridized states, while the conduction band (CB) states are mostly made up of Ti (3d) and Bi (6p) states.60 The extension of absorption edges in the visible region is clear with increasing amount of Fe in the compounds and this corresponds to the build-up of Fe (3d) states on the top of valence band. Subsequently, the higher energy edge is also red-shifted consistent with the decreasing trend of band gaps in the compounds. Band gap energies of these compounds are determined by the Tauc plots (see Table S2 and Fig. S3 in the ESI†). The plots suggested an indirect band gap transition as (αhν)1/nvs. hν was better fitted for n = 2. It can be claimed that the UV-vis absorption features do not change significantly in the series Fe1.25 to Fe1.75, although more Fe (3d) states build up with increased density of states (DOS) contribution near the top of the valence band.PL and FLS analysis
The PL spectra of the compounds (Fig. 5) recorded at an excitation wavelength of 355 nm give a broad emission (425–510 nm) that peaks around 470 nm. Intensity of PL spectra gives indirect information about charge carrier separation in Bi6−xSrxTi3+xFe2−xO18 (x = 1–0). A higher PL intensity suggests greater radiative e−–h+ recombination leaving fewer charge separated carriers61 that can take part in the photocatalytic activity. Thus, one can expect photocatalytic activity in the order, Bi5.75Sr0.25Ti3.25Fe1.75O18 > Bi6Ti3Fe2O18 > Bi5.5Sr0.5Ti3.5Fe1.5O18 > Bi5.25Sr0.75Ti3.75Fe1.25O18 > Bi5SrTi4FeO18, by comparing the PL intensities (Fig. 5a) although the final activity order is different depending on the dominance of other competing factors.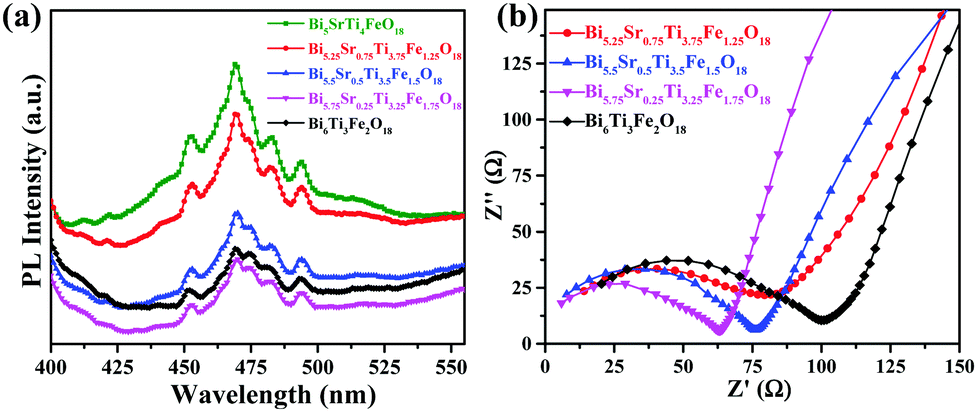 | ||
| Fig. 5 (a) PL spectra (λex = 355 nm) of Bi6−xSrxTi3+xFe2−xO18 (x = 1, 0.75, 0.5, 0.25 and 0.0). (b) EIS Nyquist plots of Bi6−xSrxTi3+xFe2−xO18 (x = 0.75, 0.5, 0.25 and 0.0). | ||
FLS analysis tells us about the lifetime of photogenerated charge species. The radiative lifetimes and their percentage contributions are obtained by fitting the FLS decay spectra (Fig. 6). An enhancement in decay lifetime is observed for Bi5.75Sr0.25Ti3.25Fe1.75O18 with a relatively stretched exponential decay of fluorescence intensity. The average lifetime (τavg) is calculated using a three exponential function using the following formula:62
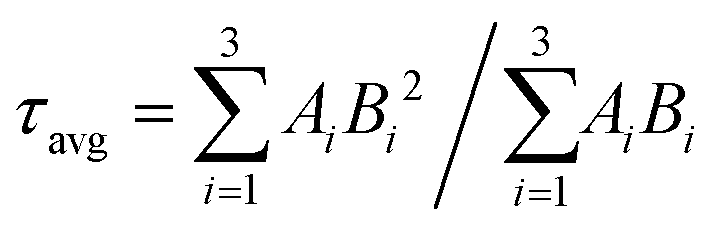 | (2) |
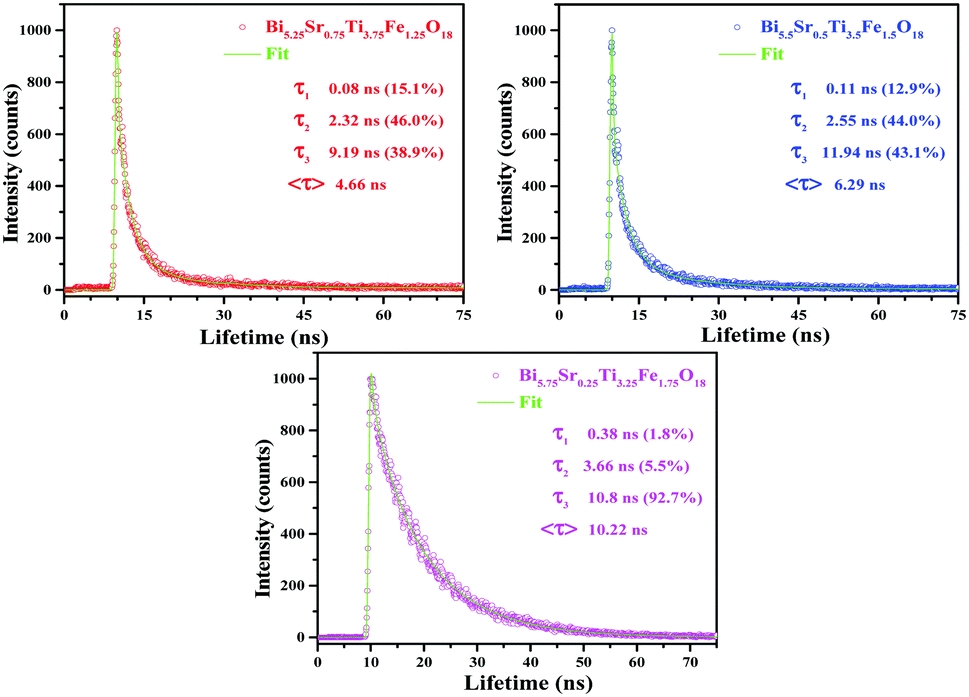 | ||
| Fig. 6 FLS spectra of Bi6−xSrxTi3+xFe2−xO18 (x = 0.75, 0.50 and 0.25). | ||
EIS analysis
EIS measurements are carried out to study the electrical properties at the interface such as charge transfer resistance (RCT) to the flow of photogenerated charge carriers. The corresponding EIS Nyquist plots (Fig. 5b) of the compounds give a semicircle in the higher frequency side signifying the RCT and a tail at lower frequency range implies the Warburg impedance contribution. Bi5.75Sr0.25Ti3.25Fe1.75O18 indicate a smaller semicircle as compared to others implying a smaller RCT. The estimated RCT values for Bi5.75Sr0.25Ti3.25Fe1.75O18, Bi5.5Sr0.5Ti3.5Fe1.5O18, Bi5.25Sr0.75Ti3.75Fe1.25O18 and Bi6Ti3Fe2O18 are 58.7, 68.3, 79.9 and 83.6 Ω, respectively. This would help the easy transfer of electron and holes at Bi5.75Sr0.25Ti3.25Fe1.75O18 semiconductor surfaces, implying improved activity compared to other compounds with higher RCT.The EIS analysis of Bi5.75Sr0.25Ti3.25Fe1.75O18 under light illumination and an external bias is carried out to understand the actual resistance to charge-transfer and e−–h+ pair recombination dynamics during photocatalysis. We observed a lower RCT at 1 V applied bias compared to that at 0 V under simulated solar illumination (see Fig. S4a in the ESI†). This suggests that the compound would show higher photocatalytic activity under the applied bias. The effect of applied voltage on the photocatalytic activity is studied and discussed in the next section. The charge transfer resistance (RCT), double layer capacitance (Cdl) and Warburg parameters of all the samples under external bias have been estimated for comparison by fitting the impedance data using a conventional R–C circuit and Warburg impedance (Fig. S5–S7 and Table S6 in the ESI†).
Photocatalytic activity
Photocatalytic activities of the semiconductor photocatalysts are estimated by way of dye degradation. For this, we have selected aqueous solutions of RhB dye as a model system for photocatalysis and the quantity of catalyst used was determined by dosage optimization results (Fig. S1, ESI†). The compounds, Bi6−xSrxTi3+xFe2−xO18 (x = 0.75, 0.5, 0.25), showed very efficient photocatalytic activity toward complete degradation of RhB at pH 2 within 20 to 25 minutes of sunlight irradiation (Fig. 7a). The UV-vis absorption data (Fig. S8, ESI†) also show clear-cut fast kinetics for Bi5.75Sr0.25Ti3.25Fe1.75O18, whereas the compounds with Fe1.25 and Fe1.5 show similar kinetics. Moreover, there is negligible or no degradation of RhB at pH 7 and 11 (Fig. S9, ESI†) and no self-degradation or photolysis of the dye in the sunlight at any of the pH investigated (Fig. 7 and Fig. S9, ESI†).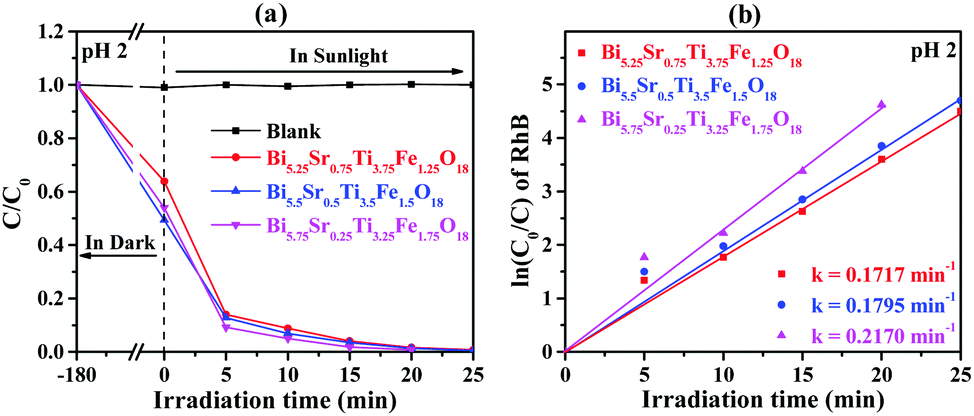 | ||
| Fig. 7 (a) C0/C vs. irradiation time for RhB at pH 2 and (b) corresponding ln(C0/C) as a function of irradiation time over Bi6−xSrxTi3+xFe2−xO18 (x = 0.75, 0.5, and 0.25). | ||
The kinetics of the photocatalytic RhB degradation over Bi6−xSrxTi3+xFe2−xO18 (x = 0.75, 0.50, 0.25) was fitted with a Langmuir–Hinshelwood rate expression.42
| ln(C0/C) = kt | (3) |
The effect of applied voltage on the photocatalytic activity is studied by the degradation of RhB at 0 and 1 V applied bias for five minutes under simulated solar light. At 1 V bias, Bi5.75Sr0.25Ti3.25Fe1.75O18 degrades ∼64% of dye while only ∼44% dye is degraded at 0 V after five minutes of irradiation (see Fig. S4b in the ESI†). The enhanced activity is correlated with the additional band bending effects (see later section) in the presence of an external electric field, in addition to the already existing internal electric fields in the compounds. It is believed that under the external electric field the ferroelectric domains and local surface dipoles in powder samples preferentially orient in the direction of the field improving the carrier separation efficiency. Moreover, the enhanced thermal barrier due to an upward band bending also reduces the chances of e−–h+ recombination.
Detection of reactive species
A series of scavenger tests were performed in the presence of ammonium oxalate (AO, h+ scavenger), benzoquinone (BQ, O2˙− scavenger) and tertiary butyl alcohol (t-BuOH, ˙OH scavenger) to ascertain the reactive species responsible for the photocatalytic RhB degradation over Bi5.75Sr0.25Ti3.25Fe1.75O18. The effect of these scavengers on the RhB degradation efficiency of Bi5.75Sr0.25Ti3.25Fe1.75O18 at pH 2 is shown in Fig. 8. Addition of AO to the dye–photocatalyst suspension greatly reduces the photodegradation of RhB as compared to that in the absence of any scavenger. Moreover, addition of BQ to the dye-photocatalyst suspension causes a significant reduction in RhB degradation, while the addition of t-BuOH to the dye-photocatalyst suspension caused reduction in photodegradation only to a small extent at this pH. Thus, the above results indicate that h+ and O2˙− are key active species involved in the photocatalytic degradation of RhB over Bi5.75Sr0.25Ti3.25Fe1.75O18 in the acidic medium and ˙OH plays a negligible role in the degradation.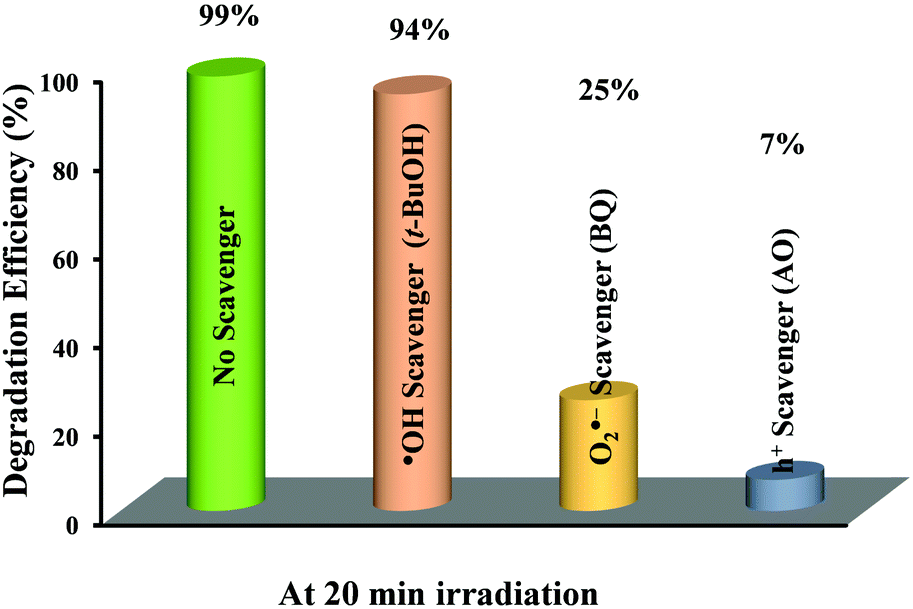 | ||
| Fig. 8 Effect of different scavengers on the degradation efficiency of RhB over Bi5.75Sr0.25Ti3.25Fe1.75O18 under sunlight irradiation. | ||
Stability and reusability studies
Whether the Aurivillius photocatalysts are reusable and stable at ambient conditions or not is important for their repeated use. The reusability study of Bi5.75Sr0.25Ti3.25Fe1.75O18 for five successive cycles (Fig. S10a, ESI†) shows ∼98% of dye degradation even in the 5th cycle almost without any change in its ability. The slight decrease (∼2%) in the activity is due to the inevitable loss of catalysts during the experiment. The stability of Bi5.75Sr0.25Ti3.25Fe1.75O18 is also assessed by PXRD, HR-TEM and XPS analysis of the compound recovered after the 5th cycle. PXRD patterns (Fig. S10b, ESI†) show retention of phase purity with sharp peaks affirming the unchanged crystallinity of the compound. The HR-TEM analysis (Fig. S10c, ESI†) of post-catalytic samples also reaffirmed preservation of good crystallinity without any expansion or contraction in lattice parameters, as the distances between fringes remained intact before and after the photocatalysis. Similarly, post-catalytic PXRD of the Fe1.25 and Fe1.5 analogues also demonstrated good catalyst stability under the reaction conditions (Fig. S11, ESI†).Fig. 9 represents the XPS data of Bi5.75Sr0.25Ti3.25Fe1.75O18 recorded before and after the photocatalytic studies. While the survey scan shows surface presence of all the elements, the high-resolution elemental scan shows that all the elements in Bi5.75Sr0.25Ti3.25Fe1.75O18 are chemically stable. High-resolution XPS of Bi 4f (Fig. 9b) show spin–orbit coupled Bi 4f7/2 and Bi 4f5/2 states at the binding energy (B.E.) of 158.8 and 164.1 eV, respectively, confirming the presence of Bi in +3 oxidation state. Similarly, Ti 2p3/2 and Ti 2p1/2 states with B.E. of 457.7 and 462.8 eV (Fig. 9d) confirm the presence of Ti4+. In the same region, an additional peak of Bi 4d3/2 at 465.6 eV is also seen consistent with the Bi3+ state. In the Fe 2p spectra (Fig. 9e), the spin–orbit coupled states, 2p3/2 and 2p1/2, appear at the B.E. of 710.2 and 723.7 eV, respectively, which corroborates well with the presence of Fe3+ in the compound before and after photocatalysis. Likewise, Sr 3d XPS (Fig. 9c) also show spin–orbit coupled doublet of 3d5/2 and 3d3/2 states corresponding to the presence of Sr2+.
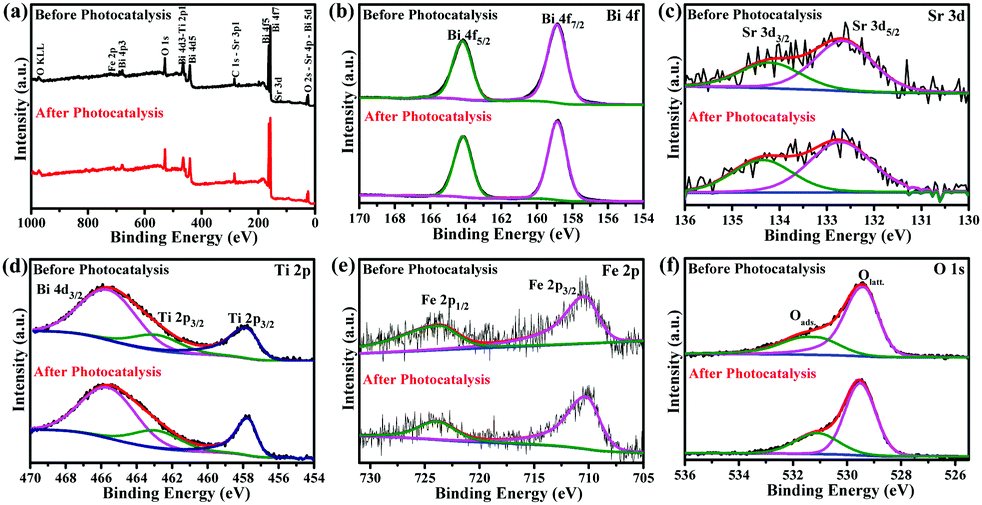 | ||
| Fig. 9 (a) Survey and (b–f) high-resolution XPS of Bi 4f (b), Sr 3d (c), Ti 2p (d), Fe 2p (e) and O 1s (f) of Bi5.75Sr0.25Ti3.25Fe1.75O18, before and after five cycles of photocatalytic RhB degradation at pH 2. | ||
The O 1s XPS peaks at 529.3–529.5 and 531.1 eV (Fig. 9f) normally correspond to the lattice and adsorbed or hydroxylated oxygens, respectively. While the intensity of lattice oxygen peaks appeared to remain same before and after the photocatalysis, the peak intensity at 531.1 eV appeared to have increased slightly. This generally corresponds to an increase in adsorbed oxygen or hydroxylated species and it is natural to have this increase due to the continuous exposure of the compound to the acidic aqueous atmosphere in several catalytic cycles. Considering the harsh environment at pH 2, post-catalytic PXRD, HR-TEM and XPS studies indicate that the Aurivillius compounds are robust under the photocatalytic reaction condition and are stable to photoleaching or photocorrosion and phase decomposition. Moreover, the intact chemical state of the transition element (Fe in this case) indicates the pure photocatalytic nature of the dye degradation process and the absence of any redox catalytic processes during degradation.
Adsorption
It is well established and proved from earlier findings that adsorption of dye at the catalyst surface plays a vital role in photocatalytic degradation. Hence to determine the effect of adsorption on the photocatalytic activity, RhB adsorption tests are performed over Bi6−xSrxTi3+xFe2−xO18 (x = 0.75, 0.5, and 0.25) under acidic, neutral and alkaline pH. The adsorption results show the highest adsorption at acidic pH (Fig. 10a) and order varies as Bi5.5Sr0.5Ti3.5Fe1.5O18 > Bi5.75Sr0.25Ti3.25Fe1.75O18 > Bi5.25Sr0.75Ti3.75Fe1.25O18. The photocatalytic activity order of the compounds, however, did not follow the adsorption order, in contrast to earlier reports. This brings out the important role of other competing factors in determining the overall activity of these closely related Aurivillius solid-solution phases.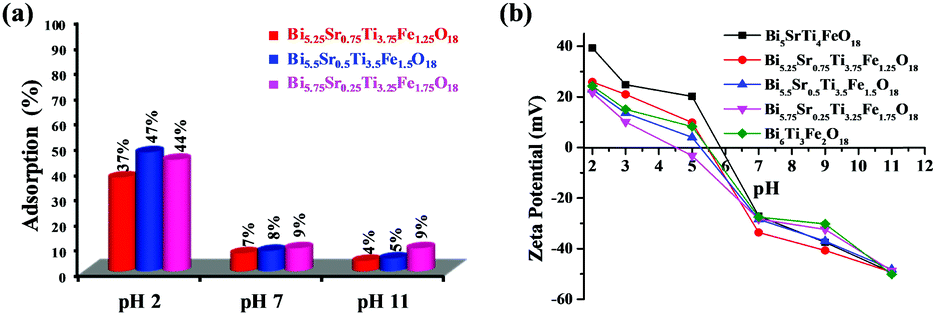 | ||
| Fig. 10 (a) Adsorption percentage of RhB dye over of Bi6−xSrxTi3+xFe2−xO18 (x = 0.75, 0.50 & 0.25) at different pH values. (b) ζ-Potential of Bi6−xSrxTi3+xFe2−xO18 (x = 1–0) catalysts at different pH. | ||
ζ-Potential study
As the surface charge of catalysts is an important factor for dye adsorption on the catalyst, the ζ-potential is measured at different pH for the compounds (Fig. 10b). The highest negative ζ-potential for the compounds is observed at alkaline pH and it gradually decreases from more negative to less negative and finally reaches positive values in the acidic medium (pH 2). The following trend is due to the action of H+ and OH− ions on the surface oxygen or hydroxyl groups attached to the surface of the oxide catalysts.5 The potential of zero charge (pzc) arises at a lower pH for Fe1.75 as compared to those of others. This indicates a larger intrinsic negative surface charge.Mechanism of photocatalytic activity
The mechanism of photocatalytic degradation can be interpreted from the energy level diagram (ELD) presented in Fig. 11. All the potentials shown in Fig. 11 are with reference to normal hydrogen electrode (NHE) potentials. The flat-band potential (Efb) of −0.511 V vs. SCE calculated for Bi5.75Sr0.25Ti3.25Fe1.75O18 with help of Mott–Schottky plots (see Fig. S12 in ESI†) corresponds to −0.269 V w.r.t. NHE. As Efb is very close to the bottom of ECB(1) in n-type semiconductors, Efb is considered as ECB(1) (−0.269 V) and EVB (1.811 V) is calculated using the band gap (2.08 eV) from the expression given below.| EVB = ECB + Eg | (4) |
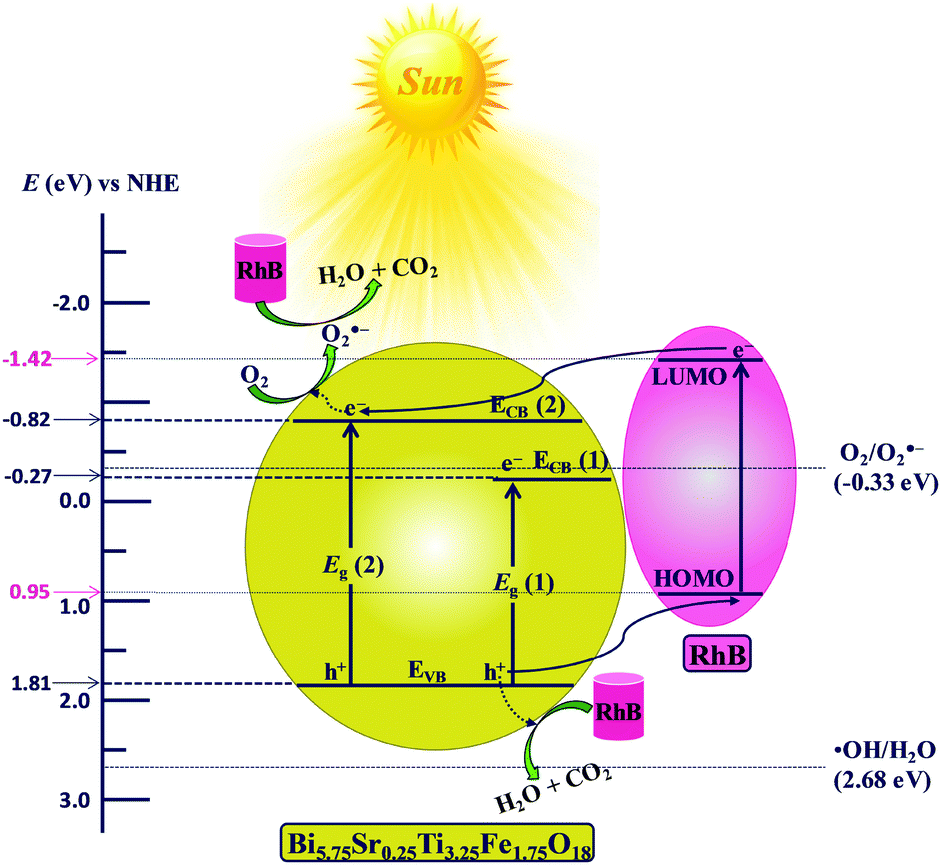 | ||
| Fig. 11 Schematic energy level diagram of Bi5.75Sr0.25Ti3.25Fe1.75O18 with respect to the potential of ˙OH/H2O (E˙OH/H2O), O2/O2˙− (EO2/O2˙−) and the HOMO–LUMO levels of RhB (NHE scale). | ||
Similarly, the position of the higher energy edge, ECB(2) (−0.819 V) is calculated using the energy of higher band edge (2.62 eV). The positions for CB and VB edges for Bi6−xSrxTi3+xFe2−xO18 (x = 0.0, 0.75, 0.5 and 0.25) calculated using the flat band potentials (see Table S7 in ESI†) are summarized in Table 1 and are shown in Fig. S13 of the ESI.† It is evident from the ELD (Fig. 11) that ECB(2) of Bi5.75Sr0.25Ti3.25Fe1.75O18 (−0.82 V vs. NHE) is suitable for generation of the O2˙− (EO2/O2˙− = −0.33 V vs. NHE),63 therefore, the photoelectrons from ECB(2) could be transferred to adsorbed oxygens to produce O2˙− radicals. In addition, the holes generated in VB can also be transported directly to HOMO of adsorbed RhB molecules and thereby degrade it. However, the VB potential (+1.81 V vs. NHE) is not suitable for generation of •OH radicals (E˙OH/H2O = +2.68 V vs. NHE)61 by oxidizing H2O molecules. The above findings are consistent with the scavenger studies showing O2˙− and h+ as the major reactive species responsible for the RhB degradation. The overall mechanism of O2˙− and h+ mediated RhB degradation do not vary across the series of compounds given the close and unaltered positioning of their VB and CB edges.
| Compound | Valence band, EVB (V) | Conduction band | |
|---|---|---|---|
| E CB(1) (V) | E CB(2) (V) | ||
| Bi5.25Sr0.75Ti3.75Fe1.25O18 | 1.797 | −0.333 | −0.863 |
| Bi5.5Sr0.5Ti3.5Fe1.5O18 | 1.954 | −0.156 | −0.686 |
| Bi5.75Sr0.25Ti3.25Fe1.75O18 | 1.811 | −0.269 | −0.819 |
| Bi6Ti3Fe2O18 | 1.850 | −0.230 | −0.809 |
The role of the crystal structure in the photocatalytic activity of the Aurivillius phase perovskites is evaluated. The catalyst surface charge can be influenced by an unsymmetrical bond polarization due to its non-centrosymmetric crystal structure (space group, F2mm). The octahedral distortion parameter64,65 (Δo) of the compounds are calculated for all the Fe/TiO6 octahedra to analyse the role of structural distortion on the RhB adsorption on the catalyst surface. The octahedral distortion parameter is defined as, 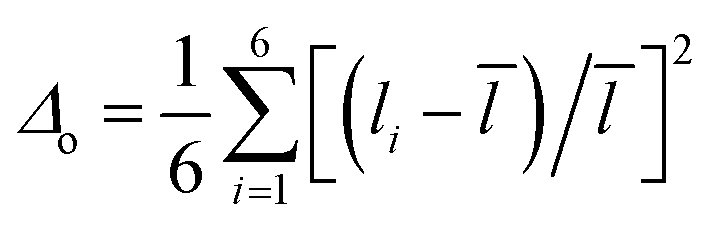 where, the sum is over six bonds of the octahedra,
where, the sum is over six bonds of the octahedra, ![[l with combining macron]](https://www.rsc.org/images/entities/i_char_006c_0304.gif) is the average bond distance and li are individual bond distances. The structure with central, pre-terminal and terminal octahedra along with their bond distances for Bi6−xSrxTi3+xFe2−xO18 (x = 0.75, 0.5, and 0.25) are shown in Fig. S14 (ESI†) and the calculated Δo values are given in Table S8 of the ESI.†
is the average bond distance and li are individual bond distances. The structure with central, pre-terminal and terminal octahedra along with their bond distances for Bi6−xSrxTi3+xFe2−xO18 (x = 0.75, 0.5, and 0.25) are shown in Fig. S14 (ESI†) and the calculated Δo values are given in Table S8 of the ESI.†
The highest Δo value of 0.0115 is obtained for the terminal octahedra of Bi5.5Sr0.5Ti3.5Fe1.5O18, while relatively smaller Δo values of 0.0106 and 0.0076 are found for Bi5.75Sr0.25Ti3.25Fe1.75O18 and Bi5.25Sr0.75Ti3.75Fe1.25O18, respectively. As reported in previous studies that the Δo values gradually decreases in the order, terminal > pre-terminal > central octahedral layer for the five-layer Aurivillius phase perovskites in a given series of compounds, is consistent with the second-order Jahn–Teller (SOJT) distortion of d0 cations in these compounds.66–68 The increase in local octahedral distortion enhances the extent of adsorption of dye molecules on the catalyst surface. Furthermore, the axial distortion parameter, defined as,  , where the li are the two axial bond lengths in an octahedra parallel to the crystallographic c-axis, is also calculated (Table S8, ESI†). Interestingly, the variation of Δax across the octahedral layers for any given compound also follows the order of distortion similar to Δo, being highest for the terminal and lowest in the central octahedral layer. It is shown that the crystal chemical correlation can be exploited in the catalytic activity enhancement by designing oxides with increased axial octahedral distortion in a homologous series, wherein the frontier band structure and photoluminescence properties largely remain unaffected due to compositional modifications at the A-sites of the perovskites largely by alkali and alkaline earth metals. However, the subsequent B-cation substitution, i.e., Ti by Fe in the present series also modifies the charge carrier concentration, charge transfer resistance and carrier lifetime that alters the expected photocatalytic activity solely based on dye adsorption.
, where the li are the two axial bond lengths in an octahedra parallel to the crystallographic c-axis, is also calculated (Table S8, ESI†). Interestingly, the variation of Δax across the octahedral layers for any given compound also follows the order of distortion similar to Δo, being highest for the terminal and lowest in the central octahedral layer. It is shown that the crystal chemical correlation can be exploited in the catalytic activity enhancement by designing oxides with increased axial octahedral distortion in a homologous series, wherein the frontier band structure and photoluminescence properties largely remain unaffected due to compositional modifications at the A-sites of the perovskites largely by alkali and alkaline earth metals. However, the subsequent B-cation substitution, i.e., Ti by Fe in the present series also modifies the charge carrier concentration, charge transfer resistance and carrier lifetime that alters the expected photocatalytic activity solely based on dye adsorption.
It is clear from previous reports that Bi5SrTi4FeO18 exhibits higher activity compared to that of Bi6Ti3Fe2O18 and the extent of dye adsorption is a crucial factor for the enhanced photocatalysis. The introduction of one Sr in place of lone pair active Bi in the five-layer perovskite modifies the overall octahedral distortion of the perovskite blocks and enhances the electric polarization at the terminal octahedra ensuing enhanced surface charge and dye adsorption. While an increase in the photocatalytic activity is observed on increasing amount of Fe in Bi5SrTi4FeO18 to Fe1.75, it decreases sharply in Bi6Ti3Fe2O18 (compound without Sr). Interestingly, the compound with Fe1.5 shows the highest extent of dye adsorption among all the compounds in the series but does not show the highest activity. Therefore, it brings out the role of other competing factors into play when it comes to the overall rate of photocatalytic dye degradation. The compound with Fe1.25 shows the slowest degradation activity due to its relatively high rate of carrier recombination and shorter carrier lifetime and high RCT together with smaller extent of dye adsorption. In the case of the Fe1.5 compound, the highest extent of dye adsorption is observed but it shows moderate activity due to its higher rate of carrier recombination (increased PL intensity), high RCT and lower lifetime of charge carriers as compared to the Fe1.75 compound. Despite showing an intermediate dye adsorption, the Fe1.75 compound shows the highest activity due to the combined effect of reduced carrier recombination, higher carrier lifetime and enhanced charge transport (smallest RCT). It is presumed that the dominance of the above factors has originated from the internal electric fields. In addition to ferroelectric domain polarization, the polar surface terminations due to intergrowth structure of two oppositely charged motifs (fluorite like layers and perovskite blocks) would give rise to an in-built electric field in the compounds. Moreover, the local octahedral distortions of terminal octahedra at surfaces and surface adsorption would modify the fields. The built-in electric field is believed to be altered by the compositional disorder of Sr at the [Bi2O2]2+ layer. Structure refinements enabling Sr disorder at the [Bi2O2]2+ layer indicate the presence of a higher extent of Sr disorder with increasing Sr content in the compounds, i.e., Fe1.75 < Fe1.5 < Fe1.25 (Table S9, ESI†). An increase in Sr disorder would decrease the charges on the structural motifs, thereby reducing the internal field. The Sr disorder in the [Bi2O2]2+ layer would also cause an expansion of the in-plane lattice due to larger size of Sr. This is evident in the case of Bi5.25Sr0.75Ti3.75Fe1.25O18, the sample with highest Sr content in the series.
Furthermore, enhanced photocatalytic activity of Fe1.75 is observed under a biased voltage. The EIS with visible-light illumination and photocatalytic activity of the compound under the external bias can be correlated with an upward band bending effect (Fig. 12). The initial upward band bending effect is due to the internal electric field present in the non-centrosymmetric Aurivillius perovskite phase, while further band bending results with the application of an external electric field. The external field (1 V) results in partial reversal of ferroelectric domain polarization in the powder samples. The enhanced internal field thus helps in better charge-carrier separation and reduced recombination as the thermal barrier to e−–h+ recombination increases with the increased band bending. It is observed that the impedance is enhanced on the application of a negative bias (Fig. S15, ESI†) and the positive bias condition favours the charge separation and transfer. Despite the samples being powder, the suggested polarization under positive bias is attributed to arise from a preferred orientation of the crystallites in the electrode due to plate-like crystal habits of the layered Aurivillius compounds.
 | ||
| Fig. 12 Schematic diagram showing band bending effect of n-type Bi5.75Sr0.25Ti3.25Fe1.75O18 due to the internal electric field (left) and with the externally applied electric field (right). | ||
Moreover, the additional band bending (Fig. 12, right) can also enable the participation of the ECB(1) level for the generation of O2˙−, which is forbidden in the absence of an external field. In addition, the bias voltage reduces the probability of trapping and recombination in the semiconductor bulk and increases the hole mobility substantially than that in the absence of an external field. The above factors enhance the photocatalytic activity of Fe1.75 compound substantially under the external bias. The idea can be applied appropriately for engineering the right kind of band bending and enhancement of photocatalytic activity of a semiconductor catalyst with an external electric field by compositional control of cation disorder and octahedral distortion.
Conclusions
Five-layer Aurivillius perovskites, Bi6−xSrxTi3+xFe2−xO18 (x = 0.75, 0.5, 0.25 and 0.0), with different Ti/Fe and adjusted Bi/Sr ratios were synthesized to study the competing effects of various parameters affecting photocatalytic activity. The compounds crystallized with orthorhombic structure in the non-centrosymmetric F2mm space group. The Fe1.75 compound with smaller Sr and larger Fe content in the series have exhibited the highest rate of RhB degradation despite having relatively weak dye adsorption. The octahedral distortion of the SOJT type is found to increase the adsorption capacity of the compounds, being highest in the Fe1.5 compound. Scavenger results and energy level positioning supported h+ and O2˙− mediated RhB degradation. A lower charge-transfer resistance (RCT) and enhanced carrier lifetime in Fe1.75 led to an enhanced photocatalytic activity. An external bias of 1 V led to a further decrease in RCT, and enhanced band bending effect in the compound reduced the thermal barrier to e−–h+ recombination and enhanced the charge-separation efficiency. The enhanced charge-separation was presumed to originate from partial ferroelectric polarization reversal effects of the powder samples that augment the built-in electric fields. A lower cation disorder at the [Bi2O2] layer of the intergrowth structure in Bi5.75Sr0.25Ti3.25Fe1.75O18 is believed to have resulted from a higher internal electric field and played an important role in enhancing its activity. The work demonstrates that the cation disorder and octahedral distortion can be controlled via compositional manipulations to achieve higher activities by manipulating internal electric fields. Moreover, the barrier to charge transfer may also be overcome by application of external bias to enhance the photocatalytic activity. Further work involving measurements of polarization and carrier density on single crystals of the compounds would provide quantitative details of internal fields and carrier dynamics for new design strategies of highly efficient photocatalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SERB, Government of India, through grant number EMR/2016/003029, financially supported this work. J. M. thanks the University Grants Commission (UGC), S. K. thanks SERB and the Ministry of Human Resources and Development (MHRD) and P. S. thanks MHRD, Government of India, for providing research fellowships. The authors are thankful to the Department of Metallurgical and Materials Engineering and the Institute Instrumentation Centre (IIC), IIT Roorkee, for providing the facilities.Notes and references
- J. Goldemberg and T. B. Johansson, World energy assessment: overview 2004 update, 2004 Search PubMed.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- H. Fujito, H. Kunioku, D. Kato, H. Suzuki, M. Higashi, H. Kageyama and R. Abe, J. Am. Chem. Soc., 2016, 138, 2082–2085 CrossRef CAS PubMed.
- G. Naresh and T. K. Mandal, ACS Appl. Mater. Interfaces, 2014, 6, 21000–21010 CrossRef CAS.
- C. Wang, X. Zhang, X. Song, W. Wang and H. Yu, ACS Appl. Mater. Interfaces, 2016, 8, 5320–5326 CrossRef CAS PubMed.
- Y. Shiraishi, S. Kanazawa, Y. Kofuji, H. Sakamoto, S. Ichikawa, S. Tanaka and T. Hirai, Angew. Chem., Int. Ed., 2014, 53, 13454–13459 CrossRef CAS PubMed.
- M. Teranishi, R. Hoshino, S. Naya and H. Tada, Angew. Chem., Int. Ed., 2016, 2, 2–7 Search PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- M. A. Fox and M. T. Dulay, Chem. Rev., 1993, 93, 341–357 CrossRef CAS.
- O. Legrini, E. Oliveros and A. M. Braun, Chem. Rev., 1993, 93, 671–698 CrossRef CAS.
- B. Li, C. Lai, G. Zeng, L. Qin, H. Yi, D. Huang, C. Zhou, X. Liu, M. Cheng, P. Xu, C. Zhang, F. Huang and S. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 18824–18836 CrossRef CAS PubMed.
- S. Dong, J. Feng, M. Fan, Y. Pi, L. Hu, X. Han, M. Liu, J. Sun and J. Sun, RSC Adv., 2015, 5, 14610–14630 RSC.
- J. Yoshimura, Y. Ebina, J. Kondo, K. Domen and A. Tanaka, J. Phys. Chem., 1993, 97, 1970–1973 CrossRef CAS.
- H. G. Kim, P. H. Borse, J. S. Jang, C. W. Ahn, E. D. Jeong and J. S. Lee, Adv. Mater., 2011, 23, 2088–2092 CrossRef CAS PubMed.
- W. S. Choi, M. F. Chisholm, D. J. Singh, T. Choi, G. E. Jellison and H. N. Lee, Nat. Commun., 2012, 3, 686–689 CrossRef PubMed.
- J. Boltersdorf and P. A. Maggard, ACS Catal., 2013, 3, 2547–2555 CrossRef CAS.
- W. Bai, C. Chen, J. Yang, Y. Zhang, R. Qi, R. Huang, X. Tang, C.-G. Duan and J. Chu, Sci. Rep., 2015, 5, 17846 CrossRef PubMed.
- G. Naresh and T. K. Mandal, ACS Sustainable Chem. Eng., 2015, 3, 2900–2908 CrossRef CAS.
- J. W. Tang, Z. G. Zou and J. H. Ye, Catal. Lett., 2004, 92, 53–56 CrossRef CAS.
- H. G. Kim, D. W. Hwang and J. S. Lee, J. Am. Chem. Soc., 2004, 126, 8912–8913 CrossRef CAS.
- T. K. Mandal, T. Sivakumar, S. Augustine and J. Gopalakrishnan, Mater. Sci. Eng., B, 2005, 121, 112–119 CrossRef.
- F. E. Osterloh, Chem. Mater., 2008, 20, 35–54 CrossRef CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- H. Zhang, G. Chen and D. W. Bahnemann, J. Mater. Chem., 2009, 19, 5089–5121 RSC.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- L. Zhang, Y. Man and Y. Zhu, ACS Catal., 2011, 1, 841–848 CrossRef CAS.
- B. Dai, Y. Yu, Y. Chen, H. Huang, C. Lu, J. Kou, Y. Zhao and Z. Xu, Adv. Funct. Mater., 2019, 29, 1807934 CrossRef.
- H. Huang, B. Dai, W. Wang, C. Lu, J. Kou, Y. Ni, L. Wang and Z. Xu, Nano Lett., 2017, 17, 3803–3808 CrossRef CAS PubMed.
- H. Huang, Z. Wang, B. Luo, P. Chen, T. Lin, M. Xiao, S. Wang, B. Dai, W. Wang, J. Kou, C. Lu, Z. Xu and L. Wang, Nano Energy, 2020, 69, 104410 CrossRef CAS.
- B. Muktha, M. H. Priya, G. Madras and T. N. G. Row, J. Phys. Chem. B, 2005, 109, 11442–11449 CrossRef CAS PubMed.
- Y. Shimodaira, H. Kato, H. Kobayashi and A. Kudo, J. Phys. Chem. B, 2006, 110, 17790–17797 CrossRef CAS PubMed.
- L. Li, P. a Salvador and G. S. Rohrer, Nanoscale, 2014, 6, 24–42 RSC.
- E. C. Subbarao, J. Am. Ceram. Soc., 1962, 45, 166–169 CrossRef CAS.
- M. S. Haluska and S. T. Misture, J. Solid State Chem., 2004, 177, 1965–1975 CrossRef CAS.
- B. Frit and J. P. Mercurio, J. Alloys Compd., 1992, 188, 27–35 CrossRef CAS.
- M. A. Rodriguez, T. J. Boyle, C. D. Buchheit, R. G. Tissot, C. A. Drewien, B. A. Hernandez and M. O. Eatough, Integr. Ferroelectr., 1997, 14, 201–210 CrossRef CAS.
- S. Sun, Y. Huang, G. Wang, J. Wang, Z. Fu, R. Peng, R. J. Knize and Y. Lu, Nanoscale, 2014, 6, 13494–13500 RSC.
- M. Reddyprakash, S. K. Rout, A. Satapathy, T. P. Sinha and S. M. Sariful, Ceram. Int., 2016, 42, 8798–8803 CrossRef CAS.
- T. W. Surta, A. Manjón-Sanz, E. K. Qian, R. H. Mansergh, T. T. Tran, L. B. Fullmer and M. R. Dolgos, Chem. Mater., 2017, 29, 7774–7784 CrossRef CAS.
- H. Kato, M. Hori, R. Konta, Y. Shimodaira and A. Kudo, Chem. Lett., 2004, 33, 1348–1349 CrossRef CAS.
- H. Fu, C. Pan, W. Yao and Y. Zhu, J. Phys. Chem. B, 2005, 109, 22432–22439 CrossRef CAS PubMed.
- X. Zhao, T. Xu, W. Yao, C. Zhang and Y. Zhu, Appl. Catal., B, 2007, 72, 92–97 CrossRef CAS.
- Y. Sasaki, H. Nemoto, K. Saito and A. Kudo, J. Phys. Chem. C, 2009, 113, 17536–17542 CrossRef CAS.
- N. Tian, Y. Zhang, H. Huang, Y. He and Y. Guo, J. Phys. Chem. C, 2014, 118, 15640–15648 CrossRef CAS.
- B. V. Kumar, M. D. Prasad and M. Vithal, Mater. Lett., 2015, 152, 200–202 CrossRef CAS.
- A. Etogo, R. Liu, J. Ren, L. Qi, C. Zheng, J. Ning, Y. Zhong and Y. Hu, J. Mater. Chem. A, 2016, 4, 13242–13250 RSC.
- Z. Dai, F. Qin, H. Zhao, J. Ding, Y. Liu and R. Chen, ACS Catal., 2016, 6, 3180–3192 CrossRef CAS.
- P. Dumrongrojthanath, A. Phuruangrat, S. Thongtem and T. Thongtem, Mater. Lett., 2017, 209, 501–504 CrossRef CAS.
- C. Yu, Z. Wu, R. Liu, D. D. Dionysiou, K. Yang, C. Wang and H. Liu, Appl. Catal., B, 2017, 209, 1–11 CrossRef CAS.
- L. Wang, Z. Wang, L. Zhang and C. Hu, Chem. Eng. J., 2018, 352, 664–672 CrossRef CAS.
- X. Li, Z. Ju, F. Li, Y. Huang, Y. Xie, Z. Fu, R. J. Knize and Y. Lu, J. Mater. Chem. A, 2014, 2, 13366 RSC.
- R. Hailili, Z. Wang, M. Xu, Y. Wang, X. Gong, T. Xu and C. Wang, J. Mater. Chem. A, 2017, 5, 21275–21290 RSC.
- L. Mi, Y. Feng, L. Cao, M. Xue, C. Qin, Y. Huang, L. Qin and H. J. Seo, J. Nanopart. Res., 2018, 20, 2 CrossRef.
- N. V. Prasad and G. S. Kumar, Mater. Sci. Eng., B, 2004, 108, 194–199 CrossRef.
- N. V. Prasad and G. S. Kumar, J. Magn. Magn. Mater., 2000, 213, 349–356 CrossRef CAS.
- W. Łasocha and K. Lewinski, J. Appl. Crystallogr., 1994, 27, 437–438 CrossRef.
- J. Rodríguez-Carvajal, Phys. B, 1993, 192, 55–69 CrossRef.
- J.-B. Li, Y. P. Huang, G. H. Rao, G. Y. Liu, J. Luo, J. R. Chen and J. K. Liang, Appl. Phys. Lett., 2010, 96, 222903 CrossRef.
- S. Sun, W. Wang, H. Xu, L. Zhou, M. Shang and L. Zhang, J. Phys. Chem. C, 2008, 112, 17835–17843 CrossRef CAS.
- K. Fujihara, S. Izumi, T. Ohno and M. Matsumura, J. Photochem. Photobiol., A, 2000, 132, 99–104 CrossRef CAS.
- L. Jiang, X. Yuan, G. Zeng, J. Liang, Z. Wu, H. Yu, D. Mo, H. Wang, Z. Xiao and C. Zhou, J. Colloid Interface Sci., 2019, 536, 17–29 CrossRef CAS PubMed.
- P. M. Wood, FEBS Lett., 1974, 44, 22–24 CrossRef CAS.
- S. Rani, G. Naresh and T. K. Mandal, Dalton Trans., 2020, 49, 1433–1445 RSC.
- M. E. Fleet, Mineral. Mag., 1976, 40, 531–533 CrossRef.
- M. Kunz and I. David Brown, J. Solid State Chem., 1995, 115, 395–406 CrossRef CAS.
- Ismunandar, T. Kamiyama, A. Hoshikawa, Q. Zhou, B. J. Kennedy, Y. Kubota and K. Kato, J. Solid State Chem., 2004, 177, 4188–4196 CrossRef CAS.
- G. Naresh, J. Malik, V. Meena and T. K. Mandal, ACS Omega, 2018, 3, 11104–11116 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00304f |
| This journal is © The Royal Society of Chemistry 2021 |