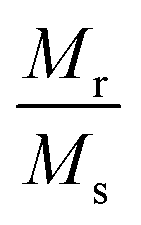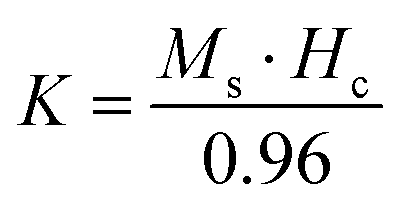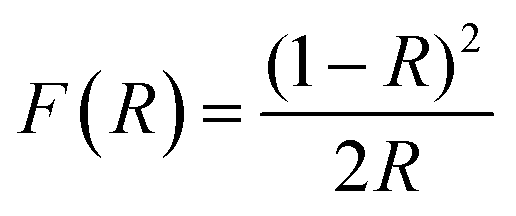DOI:
10.1039/D1MA00228G
(Paper)
Mater. Adv., 2021,
2, 4843-4858
Bandgap tuning of photo Fenton-like Fe3O4/C catalyst through oxygen vacancies for advanced visible light photocatalysis†
Received
15th March 2021
, Accepted 8th June 2021
First published on 9th June 2021
Abstract
Herein, low-cost Fe3O4/C nanocomposites have been prepared by a simplistic single-pot D-glucose-mediated hydrothermal reduction technique using a single iron precursor (FeCl3·6H2O) acquired from accumulated iron ore tailings. Two noticeably different samples having different crystallite sizes were fabricated by tuning the hydrothermal reduction method. To tune the hydrothermal reaction condition, two specifically different temperatures and reaction times were selected and a suitable justification has been provided for this selection. A qualitative rationalization for bandgap dependence on the crystallite size has been offered with the help of UV-visible differential reflectance spectroscopy (UV-DRS) and photoluminescence (PL) spectroscopy, which point to quantum confinement effects. Thermogravimetric analysis (TGA) data evidently quantify the total amount of carbon in the sample. X-ray photoelectron spectroscopy (XPS) and Rietveld analysis elucidate the dependence of crystallite size with the concentration of oxygen vacancy. The sample having adequate oxygen vacancies and favorable bandgap was established to be more efficient for the photodecomposition of Rhodamine B under visible light irradiation, which also outperformed standard TiO2 (Degussa P-25) with tremendous recyclability and structural stability. Furthermore, the magnetization measurements illustrate the superparamagnetic behavior of the sample, which is also found to be dependent on the crystallite size.
1. Introduction
World Health Organization (WHO) ascertained that about 1/4th of the global population are exposed to health and environmental problems associated to water pollution.1,2 In recent years, rapid industrialization and urbanization have resulted in elevated discharge of untreated toxic effluents in water resources and the successive occurrence of pollution of the water bodies at an exceptional rate.3 Therefore, it is essential to treat industrial waste water before its discharge into water resources through an efficient and environmentally benign approach. Advanced oxidation processes (AOPs) are considered as most efficient and environmentally benign approaches for the degradation of wastewater containing non-biodegradable organic contaminants.4,5 AOPs generate hydroxyl radicals (˙OH), an extremely strong oxidizing species with a high oxidation potential [E0 (˙OH/H2O) = +2.8VNHE], which are capable of oxidizing most recalcitrant pollutants non-selectively and rapidly.6,7 Among the existing AOPs, photo-assisted Fenton oxidation using inexpensive and benign semiconductor nanomaterials has become one of the most accepted approach because of its simplicity of technology and efficient performance.5,8 In a typical photo-Fenton degradation process, ferrous ions (FeII) present in the iron-based catalyst are oxidized to ferric ions (FeIII) and more hydroxyl radicals (˙OH) is produced with the help of hydrogen peroxide (H2O2), followed by the photochemical regeneration of ferrous ions (FeII) by the photo-reduction of ferric ions (FeIII) under ultraviolet-A or visible solar light.5,8
Efforts have been made to develop efficient iron-containing heterogeneous catalyst for organic pollutant degradation, such as iron oxides,9,10 Fe-clays,7,11 and iron-immobilized materials.12,13 Among these, iron oxides are low-cost, non-toxic, and environment-friendly compounds, which are abundantly available in nature and can be easily synthesized in laboratories.14 In addition, most of them reveal semiconductor properties and are more suitable for photocatalytic applications.14,15 Recently, magnetite (Fe3O4) was used to treat numerous organic pollutants by Fenton and photo-Fenton process due to its excellent chemical and magnetic properties.16,17 Separation of catalyst from the reaction medium is still an open issue, whereas magnetite undergoes easy magnetic separation from the reaction medium eliminating the generation of undesirable iron sludge at the end of the process.18,19
Magnetite (Fe3O4) has become one of the most accepted and extensively studied semiconductors, it displays both n- and p-type semiconductor behavior with a narrow bandgap (0.1 eV).20–22 Narrow bandgap energy allows the rapid recombination of the photo-generated electrons and holes, which significantly limits the production of reactive oxygen species (ROS).23 To date, the strategies developed to modify the electronic and optical properties of the Fe3O4 nanocrystal to lower the recombination rate through doping23–25 or developing composite heterojunctions26–28 or self-doping/defect engineering.16,29,30 Among them, the formation of oxygen vacancies on the particle surface as a self-doping/defect engineering technique has attained increasing attention in the photocatalysis field.16,31,32
However, previous work has established that the modification of the Fe3O4 nanocrystal size and morphology are expected to influence the optical and electronic properties because of the formation of surface defects and oxygen vacancies at the particle surface.33 It is evident that the generation of surface defects and oxygen vacancies varies with size and morphology of the nanocrystals, which could influence the photocatalytic properties. In general, the size, shape, crystal morphology, and the degree of crystallinity of the magnetite nanoparticles can be controlled by the synthetic method. Various synthetic methods have been reported in the literature to produce magnetite nanoparticles with desired physical and chemical properties, such as (i) co-precipitation of aqueous ferrous (Fe2+) and ferric (Fe3+) salt solution in an alkaline medium,34 (ii) sono-chemical method,35 (iii) hydrothermal synthesis,36 (iv) and thermal decomposition of an organic iron precursor at high temperature.37 However, all the above synthetic routes for preparing size- and shape-controlled magnetite nanoparticles require a complicated and expensive equipment setup for industrial-scale production. In order to overcome the drawbacks, hydrothermal method assisted by organic templates has become a promising method to prepare size- and shape-controlled magnetite nanoparticles.38–41 Organic templates, such as glucose (i.e., reducing sugars) has been used as a reductive agent that partially reduces the iron precursor from Fe3+ ions to Fe2+ ions to produce hydrophilic magnetic nanoparticles through the in situ approach.38,40 Further, glucose here acts as an bifunctional agent, i.e., both as a reducing agent and as a source of carbon for the formation of the carbonaceous shell over the Fe3O4 nanoparticles.42,43 In addition, carbon derived from the hydrothermal carbonization of glucose serves as a support to solidify the carboxylate groups, which plays a major role in obtaining solid ferricarboxylate complexes that show higher photocatalytic activity under visible light.44–46 Previous works have established that the carbon coating efficiency can be modified by varying the reaction parameters such as the glucose concentration and the hydrothermal reaction temperature and time.38,43,45 It has been established that varying the carbon coating efficiency will have a dissimilar effect on the nucleation and growth of the Fe3O4 nanoparticles and that the size and morphology of the Fe3O4 nanoparticles can be significantly controlled.38,43 The crystallite size and coating efficiency significantly influence the photocatalytic activity of Fe3O4/C nanocomposites due to the formation of surface defects and oxygen vacancies.29
Herein, we investigate why and how the size and coating efficiency of Fe3O4/C nanocomposites control their photocatalytic properties. Different sized Fe3O4/C nanocomposites have been synthesized using Fe3+ as the sole metal precursor obtained from iron ore tailings (IOTs) using low temperature hydrothermal method along with an organic template. The crystallite size of the Fe3O4 nanoparticles is controlled in a straightforward manner by tailoring the reaction time and temperature. The structure, morphology, optical, and magnetic properties of the synthesized Fe3O4/C nanocomposites (M1 and M2) were investigated and the surface defects and oxygen vacancies associated with each sample were elucidated by X-ray photoelectron spectroscopy (XPS) and Rietveld analysis. The photocatalytic activity of the Fe3O4/C nanocomposites has been measured in in terms of Rhodamine B dye degradation and catalyst recovery, and recyclability tests have been performed to establish the photocatalyst's stability. Finally, a feasible photocatalytic mechanism has been proposed that seeks to relate the extent of oxygen vacancies and crystallite size with the photocatalytic properties.
2. Experimental
2.1. Materials and preparation
2.1.1. General synthetic and analytical details.
All the chemicals are purchased from Merck India (Reagent: analytical grade, Solvent: HPLC water (Millipore)) and used without further purification. The iron ore tailings (IOT) were obtained from tailing dams of the Donimalai iron ore mines (Bellary-Hospet sector, Karnataka, India). Table 1 and Fig. S1 (ESI†) show the chemical composition and the mineral phases of the iron ore tailings performed using micro X-ray fluorescence spectroscopy analysis (μ-XRF) and X-ray diffraction (XRD) analysis.
Table 1 Chemical composition of iron ore tailings (wt%) from Donimalai iron ore mines (Bellary-Hospet sector, Karnataka, India) measured by μ-XRF
| Element |
Fe |
Al |
Si |
Mg |
Ca |
Mn |
Ti |
Cr |
V |
K |
P |
S |
| wt% |
70.92 |
10.67 |
17.15 |
0.32 |
0.05 |
0.09 |
0.34 |
0.02 |
0.02 |
0.17 |
0.22 |
0.02 |
2.1.2. Preparation of the Fe3O4/C nanocomposites.
The synthesis of Fe3O4/C nanocomposites from iron ore tailings by hydrothermal reduction process has been previously reported by our group.47 In a typical procedure, properly weighed IOTs are subjected to acid digestion with 250 mL of conc. hydrochloric acid at 110 °C for 6 h. After leaching, insoluble residues (SiO2 and Al2O3) were separated from the hydrolysate by filtration and the obtained acid soluble hydrolysate (FeCl3·6H2O) was stored properly in reagent bottles before use. Subsequently, 1.80 g anhydrous dextrose (10 mmol) was added to 10 mL of 5 mM FeCl3.6H2O hydrolysate under vigorous stirring, and the reaction mixture is maintained at pH 10 by the addition of 25% NH4OH solution. After vigorously stirring for 30 min, the reaction mixture was transferred to a 150 mL Teflon-lined stainless steel autoclave chamber and sealed. The autoclave was then heated in an oven under two distinctly different reaction conditions: (i) 96 h and 160 °C (sample M1) and (ii) 24 h and 200 °C (sample M2). After the hydrothermal reaction, the autoclave was naturally cooled to room temperature. The product was obtained by centrifugation (REMI-24), washed several times using distilled water (4 × 30 mL), and absolute ethanol (4 × 30 mL) to remove any further impurities. The obtained product was ultimately dried in a vacuum oven at 60 °C for 5 h to obtain the final black-brown solid.
2.2. Characterization
2.2.1. X-ray diffraction (XRD).
The XRD profiles were recorded using Ni-filtered CuKα radiation from a highly stabilized and automated PANalytical X-ray generator operated at 40 kV and 40 mA. The X-ray generator was coupled with a PW3071/60 bracket goniometer for sample mounting. Step scan data (step size 0.02° 2θ, counting time 2 s/step, depending on the peak intensity) were recorded for the entire angular range from 20° to 80° 2θ.
2.2.2. Rietveld analysis.
In the present work, microstructural characterization was performed by the whole profile fitting program, MAUD48 (Version 2.84) based on the Rietveld method.49,50 This method primarily deals with the modelling of a large portion (in most cases 20°–80° 2θ) of the experimental powder X-ray diffraction (PXRD) pattern by an analytical function to describe the profile whose intensity is directly related to each studied phase. The profile function is usually a pseudo-Voigt (pV) as it takes care of the effect of peak broadening caused by finite crystallite size and strain.51,52 True line broadening function and the symmetric part of the instrumental broadening may be expressed by a pV function as follows (eqn (1)).53,54| | 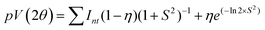 | (1) |
where S = (2θ − 2θ0)/HWHM, HWHM is the half width at half maxima of the X-ray peaks, η is the Gaussianity of the peak, θ0 is the Bragg angle, and Int is the scale parameter of the pV function. The Goodness of Fit (GoF) is determined through the weighted residual error (Rwp) and the expected error (Rexp) as follows (eqn (2)).| | 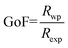 | (2) |
Refinement was carried out till convergence was reached and the value of GoF approached unity.
2.2.3. Transmission electron microscopy (TEM).
The synthesized samples were screened for their size and morphology using an ultra-high resolution field emission gun transmission electron microscope (UHR-FEG TEM, JEM-2100F, Jeol, Japan) operating at 200 kV. Data were processed using Gatan Microscopy Suite. Sample preparation for TEM characterization was executed by the droplet coating of ethanolic suspensions on carbon-coated Cu grids (Agar Scientific, 300 mesh).
2.2.4. X-ray photoelectron spectroscopy (XPS).
XPS measurements were carried out using a ThermoScientific K-Alpha system. The spectra were recorded using a focused monochromatized Mg Kα X-ray source (1253.6 eV) and Al Kα X-ray source (1486.8 eV). The binding energy was calibrated internally based on the C 1s line position.
2.2.5. Fourier-transform infrared spectroscopy (FTIR).
The Fourier transform infrared (FT-IR) spectra are recorded at the room temperature using JASCO FTIR-4000 model. FT-IR measurements of the samples are conducted in the transmission mode in the mid infrared range of 4000–400 cm−1.
2.2.6. Thermogravimetric analysis (TGA).
Thermogravimetric analysis (TGA) was performed on a TA instruments TGA 500, ranging from room temperature to 750 °C in air atmosphere using a ramp rate of 10 °C min−1.
2.2.7. Brunauer–Emmett–Teller (BET) surface area analysis.
The Brunaur–Emmett–Teller (BET) surface area of the samples was analyzed using nitrogen adsorption in a Quantachrome Autosorb-1 nitrogen-adsorption apparatus. The samples were degassed at 77 K before nitrogen adsorption measurements were commenced.
2.2.8. UV-Diffuse reflectance spectroscopy (UV-DRS).
The diffuse reflectance spectra were measured using a Varian Cary-50 UV-Vis spectrophotometer with a Harrick Video-Barrelino diffuse reflectance probe.
2.2.9. Photoluminescence spectroscopy.
The photoluminescence spectra were measured in an ethanolic solution (i.e., ethanolic solution obtained by sample dispersal in ethanol to form 1.0 × 10−5 M suspensions) on a PerkinElmer LS 55 fluorescence spectrometer.
2.2.10. Vibrating sample magnetometer.
To study the magnetic behavior of all the samples, a vibrating sample magnetometer (VSM) (Lakeshore-7144) was used. The magnetization values of the samples were recorded up to 1.5 Tesla of external magnetic fields at 300 K.
2.2.11. Photocatalytic experiments.
Photo-Fenton catalytic activities of the presented Fe3O4/C nanocomposites (M1 and M2) were investigated by the degradation of aqueous RhB solution with the addition of H2O2 under visible light obtained via a 100 W Xenon lamp fitted with an air mass 1.5 global (AM 1.5G) filter (Low-cost Solar Simulator-Royal Enterprise, 1 Sun illumination,100 mW cm−2). Typically, 5 × 10−1 g L−1 of the catalyst was added to 50 mL of 1 × 10−5 M aqueous RhB solution (pH 7) with continuous magnetic stirring. Before illumination, the suspension was stirred in dark for 45 min to ascertain the complete dispersion of the catalyst and to allow the adsorption/desorption equilibrium between the dye solution and the catalyst surface.55 After reaching the adsorption equilibrium, a 3.0 mL aliquot of the suspension was taken out and centrifuged. The λmax = 555 nm absorption of the RhB dye solution therein was determined to obtain the dye concentration before photocatalysis (C0). Afterward, H2O2 (0.15 mL) was added to the suspension and instantaneously irradiated under visible light irradiation. The RhB degradation was monitored in the experiments with respect to time (t) by UV-vis spectroscopy (λmax = 555 nm) to obtain the concentration (C) of the RhB dye as a function of time in subsequent aliquots. The reaction mixture was kept in an ice bath in order to avoid evaporation during photocatalysis and to avoid degradation caused by heating. Reference experiments, i.e., (i) without light irradiation in the presence of catalyst except with H2O2 (dark Fenton) and without H2O2 (adsorption study) and (ii) with light irradiation in the absence of catalyst but the presence of H2O2 were executed and the degradation efficiency was calculated to be consistent with the following eqn (3).| | | Degradation (%) = (1 − C/C0) × 100 | (3) |
Hydroxyl radicals (˙OH) generated throughout the photo-Fenton process were estimated by the fluorescence method using terephthalic acid (TA) as the probe molecule.56,57 Properly weighed 25.0 mg portion of the catalyst was dispersed in an aqueous solution of 30 mL TA (5 × 10−4 M), NaOH (2 × 10−3 M), and 0.15 mL H2O2. The resulting suspension was irradiated and at regular time intervals, 3.0 mL aliquots of the mixed suspension were collected and centrifuged (8000 rpm for 5 min). The maximum fluorescence emission intensity of the supernatant was measured with an excitation wavelength of 315 nm. This method relies on the fluorescence signal at 425 nm of 2-hydroxyterephthalic acid (TAOH) and was recorded using a PerkinElmer LS 55 fluorescence spectrometer.
3. Results and discussion
3.1. XRD pattern analysis by Rietveld method
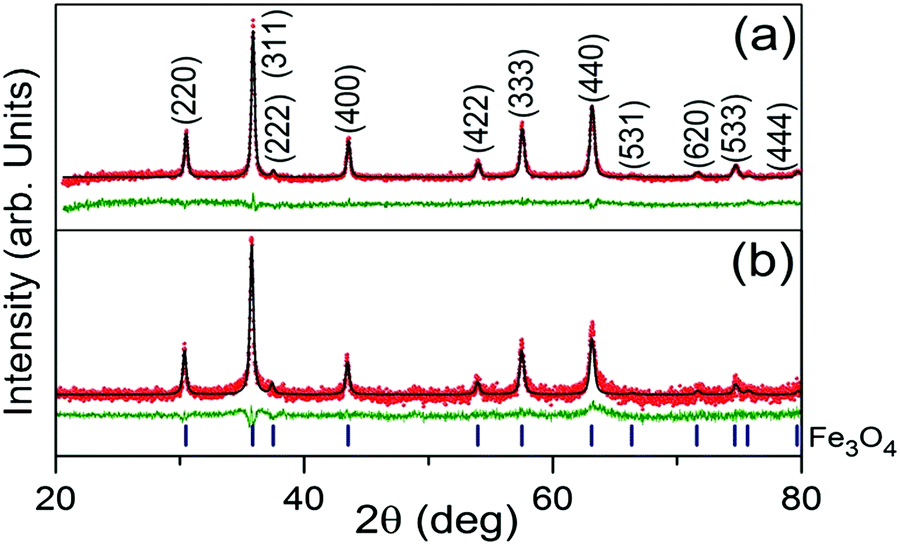
Rietveld refinement output of the XRD patterns of the Fe3O4/C nanocomposites (M1 and M2) are shown in Fig. 1a and b. From the analysis, it is observed that both the patterns consist of reflections only from crystalline cubic Fe3O4 (space group (no. 227) Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) and they are indexed accordingly. The absence of diffraction peaks of crystalline carbon implies that the carbonaceous coating over Fe3O4 is amorphous.46,58 Both the patterns (Fig. 1a and b) look almost similar, having almost similar peak positions and peak intensities. However, critical analysis by the Rietveld method reveals a slight modification in the coherently diffracting domain (crystallite) size and r.m.s. lattice strain (see Table 2). From Table 2, it is inferred that the larger crystallite size of M1 is a due to it being exposed to a temperature for a longer reaction time. Relatively, the lattice constant of M1 was found to be larger than that for M2; this increase in the lattice constant is apparently due to the reduction in the surface tension of the nanocrystallites with an increase in the crystallite size.59 Surficial oxygen vacancy plays an important role in determining the catalytic property of metal oxides. The calculated values of the oxygen vacancy show excellent agreement with the XPS observations; about ∼5.4% oxygen vacancies were established in the lattice of M1, whereas no oxygen vacancies were found in the M2 crystal lattice. However, the r.m.s lattice strain developed inside the crystal lattice of M1 and M2 was found to be 5.6223 × 10−4 and 3.8580 × 10−6, respectively. Comparatively, the higher value of r.m.s lattice strain in M1 may be attributed to the presence of a larger amount of point defects in the form of oxygen vacancies.60
m) and they are indexed accordingly. The absence of diffraction peaks of crystalline carbon implies that the carbonaceous coating over Fe3O4 is amorphous.46,58 Both the patterns (Fig. 1a and b) look almost similar, having almost similar peak positions and peak intensities. However, critical analysis by the Rietveld method reveals a slight modification in the coherently diffracting domain (crystallite) size and r.m.s. lattice strain (see Table 2). From Table 2, it is inferred that the larger crystallite size of M1 is a due to it being exposed to a temperature for a longer reaction time. Relatively, the lattice constant of M1 was found to be larger than that for M2; this increase in the lattice constant is apparently due to the reduction in the surface tension of the nanocrystallites with an increase in the crystallite size.59 Surficial oxygen vacancy plays an important role in determining the catalytic property of metal oxides. The calculated values of the oxygen vacancy show excellent agreement with the XPS observations; about ∼5.4% oxygen vacancies were established in the lattice of M1, whereas no oxygen vacancies were found in the M2 crystal lattice. However, the r.m.s lattice strain developed inside the crystal lattice of M1 and M2 was found to be 5.6223 × 10−4 and 3.8580 × 10−6, respectively. Comparatively, the higher value of r.m.s lattice strain in M1 may be attributed to the presence of a larger amount of point defects in the form of oxygen vacancies.60
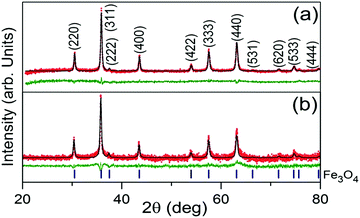 |
| | Fig. 1 Rietveld refinement output XRD patterns of Fe3O4/C nanocomposites synthesized at various reaction parameters; M1 (a) and M2 (b). The red scattered and black continuous lines represent the observed (IO) and simulated (IC) intensities, respectively. The green line plotted below each fitted pattern is the difference between the observed and calculated intensities (IO − IC). The blue markers below the XRD patterns indicate the 2θ positions of Fe3O4 for the entire 2θ range of 20°–80°. | |
Table 2 (Nano) structural parameters of the Fe3O4/C nanocomposites (M1 and M2) synthesized at various reaction conditions
| Sample |
Space group |
Lattice parameter (a), (Å) |
Particle size (nm) |
Oxygen vacancy (%) |
RMS lattice strain |
|
M
1
|
Fdm (Sp. Gr. No. 227) |
8.3493 |
44.5 |
5.4 |
5.6223 × 10−4 |
|
M
2
|
Fdm |
8.3329 |
38.2 |
0 |
3.8580 × 10−6 |
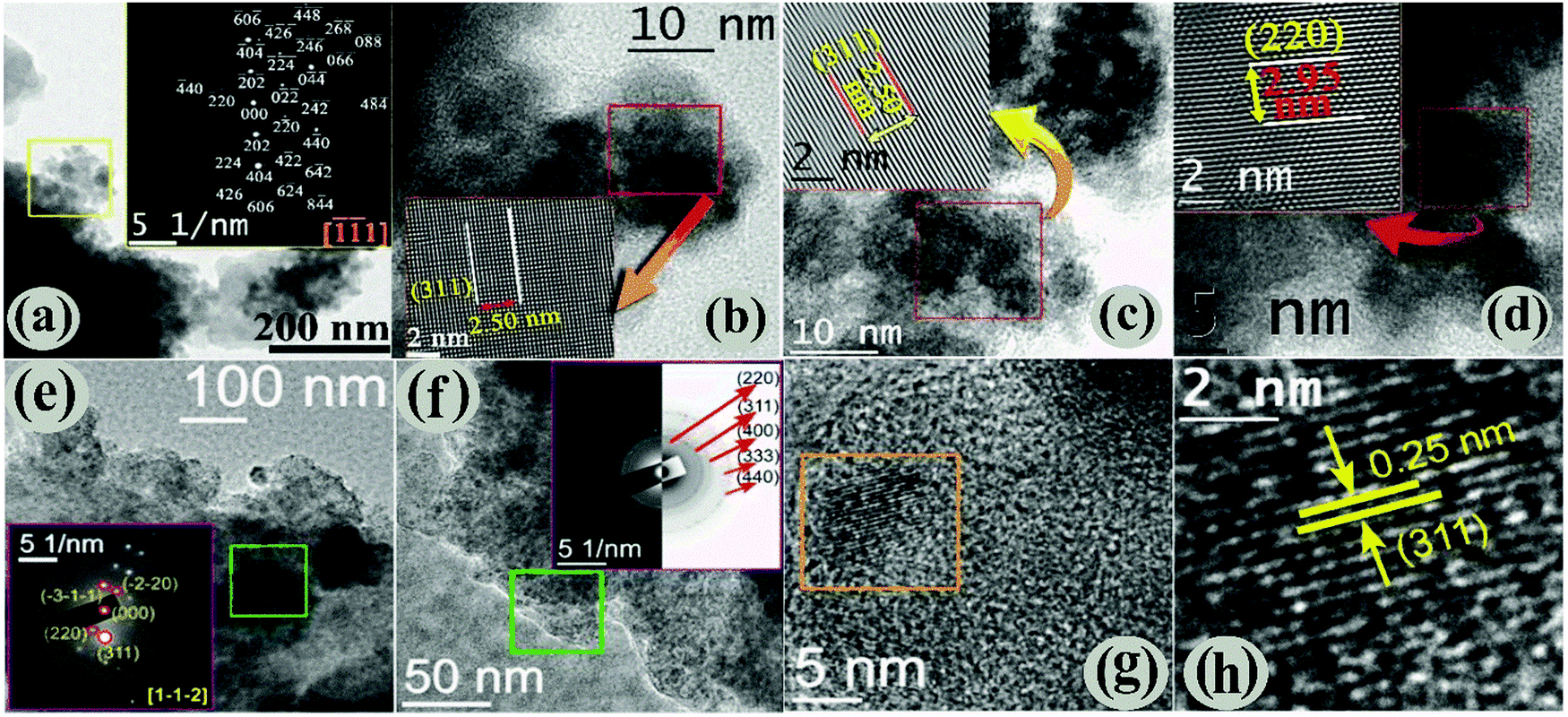
3.2. Morphological studies by TEM analysis
The morphology and microstructure of the Fe3O4/C nanocomposites (M1 and M2) were characterized by analyzing the HRTEM data. Preliminary HRTEM images (Fig. S3a and b, ESI†) suggest the formation of a core–shell-like structure for both the samples and further examination reveals the presence of an amorphous carbon shell (light grey) over Fe3O4 nanoparticles (dark grey); the existence of the amorphous carbon shell perhaps originates from carboxylate rich carbon.43,45 Both the samples show two distinctly different morphologies: platelets (ca. 0.5–1 μm diameter) capped by agglomerates of smaller particles, and the low-magnification HRTEM images show that the size of the smaller particles of M1 and M2 is about 10 nm (Fig. 2a and b) and 5 nm (Fig. 2e and f), respectively. However, the particles are not uniform in shape and size (polydisperse) and are extremely agglomerated in nature. The bright spots in the SAED pattern of M1 are unambiguously indexed (inset of Fig. 2a) using the characteristic interplanar distances of M1, considering the zone axis along [![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 1]. Fig. 2b–d shows high-magnification HRTEM images of M1, where the lattice planes are clearly visible (shown in the insets). The interplanar distance (measurement is taken for 10 consecutive lattice fringes) of the lattices was found to be ∼2.50 Å, which is equal to the value of the most intense d311 of the Fe3O4 phase. Similarly, the presence of the d220 cubic Fe3O4 phase is evidenced in Fig. 2d. The linearity of the lattice planes signifies a lack of any planar defects and twin faults. The low magnification HRTEM images of M2 are displayed in Fig. 2e and f. The SAED patterns are taken from the marked regions of Fig. 2e and f. The spot SAED pattern in Fig. 2e implies the presence of larger particles in the marked region. All the major diffraction spots are indexed to appear from the cubic Fe3O4 along the [1,,
1]. Fig. 2b–d shows high-magnification HRTEM images of M1, where the lattice planes are clearly visible (shown in the insets). The interplanar distance (measurement is taken for 10 consecutive lattice fringes) of the lattices was found to be ∼2.50 Å, which is equal to the value of the most intense d311 of the Fe3O4 phase. Similarly, the presence of the d220 cubic Fe3O4 phase is evidenced in Fig. 2d. The linearity of the lattice planes signifies a lack of any planar defects and twin faults. The low magnification HRTEM images of M2 are displayed in Fig. 2e and f. The SAED patterns are taken from the marked regions of Fig. 2e and f. The spot SAED pattern in Fig. 2e implies the presence of larger particles in the marked region. All the major diffraction spots are indexed to appear from the cubic Fe3O4 along the [1,,![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) ] zone axis. Nevertheless, the ring SAED pattern in Fig. 2f advocates the presence of smaller sized particles in the marked region. The presence of both types of SAED patterns evidently reflects size-inhomogeneity of the particles. The HRTEM image in Fig. 2g shows the presence of parallel lattice planes in the marked region. The interplanar distance (Fig. 2h) was found to be 0.25 nm, which matches to that of d311 of cubic Fe3O4. Overall, our data reveal that Fe3O4 nanoparticles are finely coated by carbon and the particle size of the samples (M1 and M2) are well connected with the hydrothermal carbonization (HTC) processing temperature and the time of D-glucose/dextrose. It is obvious that the carbon coating plays a prominent role in the structure of Fe3O4 nanoparticles. Therefore, the changes in the amount of carbon coating with the hydrothermal carbonization (HTC) processing temperature and time were investigated using thermogravimetric analysis (TGA) (see below).
] zone axis. Nevertheless, the ring SAED pattern in Fig. 2f advocates the presence of smaller sized particles in the marked region. The presence of both types of SAED patterns evidently reflects size-inhomogeneity of the particles. The HRTEM image in Fig. 2g shows the presence of parallel lattice planes in the marked region. The interplanar distance (Fig. 2h) was found to be 0.25 nm, which matches to that of d311 of cubic Fe3O4. Overall, our data reveal that Fe3O4 nanoparticles are finely coated by carbon and the particle size of the samples (M1 and M2) are well connected with the hydrothermal carbonization (HTC) processing temperature and the time of D-glucose/dextrose. It is obvious that the carbon coating plays a prominent role in the structure of Fe3O4 nanoparticles. Therefore, the changes in the amount of carbon coating with the hydrothermal carbonization (HTC) processing temperature and time were investigated using thermogravimetric analysis (TGA) (see below).
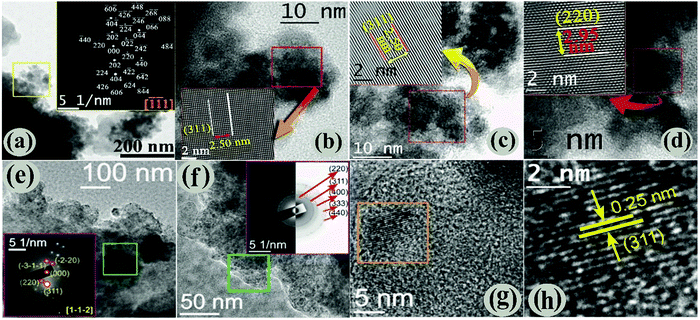 |
| | Fig. 2 (a) Low magnified TEM images with SAED pattern (insets), (b)–(d) high-magnified HRTEM images showing the characteristic interplanar spacing of M1. Low (e and f) and high (g and h) magnified HRTEM images of M2. The indexed SAED images taken from the marked areas are shown inset of the respective figures. | |
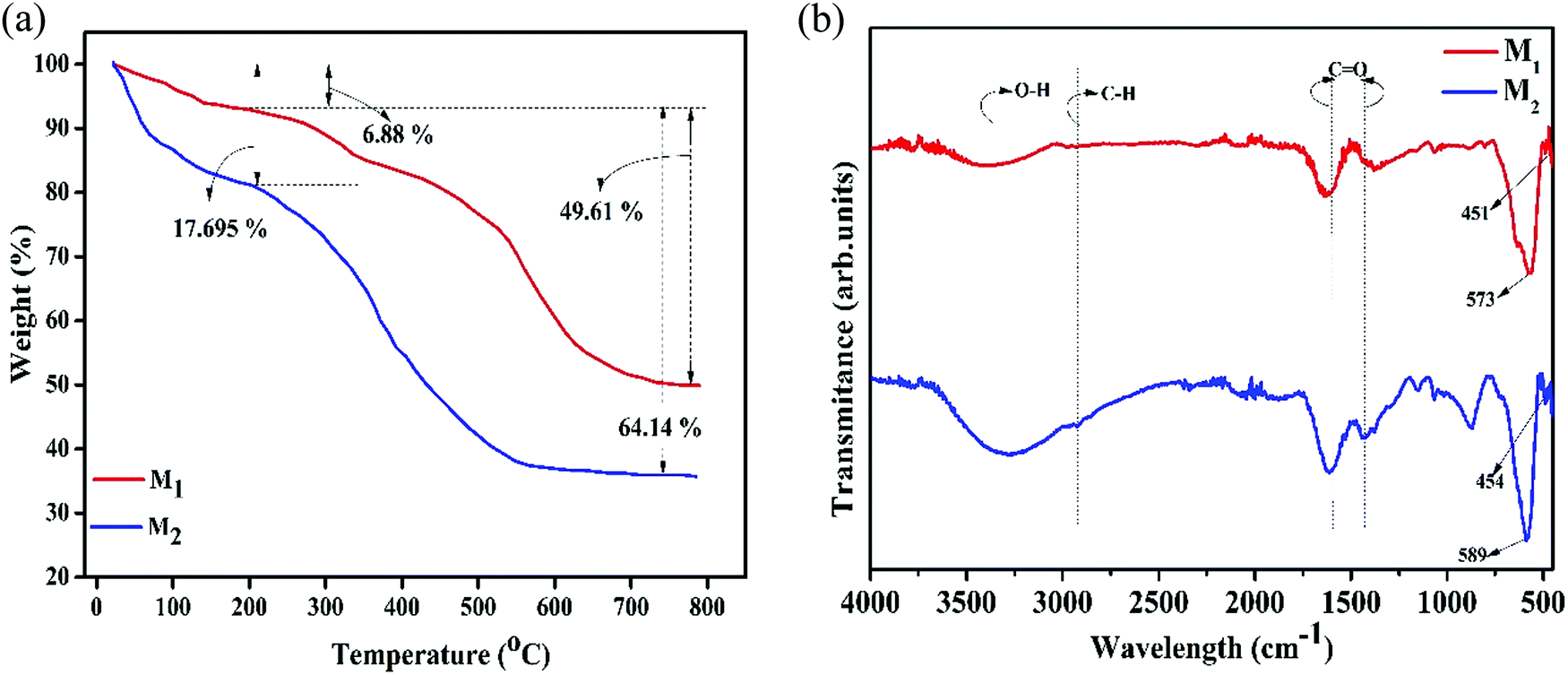
3.3. Thermogravimetric analysis (TGA)
TGA measurements under air atmosphere substantiate the presence of carbon coating on the surface of magnetite nanoparticles. Fig. 3a exhibits the TGA curves of the Fe3O4/C nanocomposites (M1 and M2), which exhibit two-step weight loss in the temperature range of 25–800 °C. The initial weight loss below 250 °C is probably due to the removal of moisture present in the samples whilst the maximum weight loss occurs in the range of 250–800 °C, which corresponds to evaporation and the subsequent decomposition of functional groups and amorphous carbon coating.40,45 The TGA results suggest that the carbon contents were 49.61 and 64.14% for M1 and M2, respectively, indicating the existence of dissimilar amounts of the coating agent on the surface of the particles.
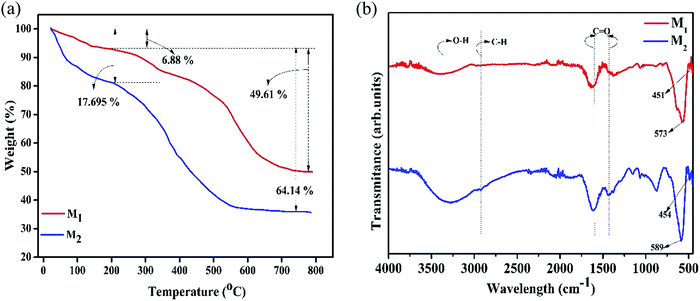 |
| | Fig. 3 (a) Thermogravimetric spectra (under air atmosphere with a scanning rate of 10 °C min−1 up to 800 °C) and (b) FTIR spectra (in the wide range of 400–4000 cm−1) of the synthesized Fe3O4/C nanocomposites (M1 and M2), respectively. | |
3.4. FTIR analysis
The presence of the coating agent on the surface of the Fe3O4 nanoparticles has been further supported by FTIR analysis (Fig. 3b). The bands at ∼400–650 cm−1 are the characteristic bands for the metal-oxygen (Fe–O) bond in Fe3O4; magnetite has a cubic inverse spinel structure and shows two typical absorption bands at 600 and 450 cm−1, which are attributed to the intrinsic stretching vibrations of the metal-oxygen (Fe–O) bond at the tetrahedral (Fetetra ↔ O, ν1) and octahedral site (Feocta ↔ O, ν2).61–63 However, it is evident that ν1 and ν2 shift to high wavenumbers in M2 compared to that in M1, which is associated with the size of the particles (Table 3).62 Further, absorption bands situated at ∼3600–3100 and ∼2925 cm−1 can be indexed to O–H and C–H bonds, respectively, which were recognized to be the hydroxyl stretching/bending vibrations of the surface hydroxyl groups and water and/or the retention of dextrose and/or its oxidation products (gluconic acid), respectively.38,39 The two noticeable peaks at 1441 and 1557 cm−1 agree well with the asymmetric and symmetric stretching vibrations of COO−, respectively, further substantiating a strong coordination of the carboxylate group with the Fe3+/Fe2+ cations on the Fe3O4 surface through an oxygen atom to form the ferric/ferrous carboxylate complexes, FeII(R-COO)2−nn and FeIII(R-COO)3−nn.38,46,64,65
Table 3 The value of vibrations of the Fe-O bond in the Fe3O4/C nanocomposites (M1 and M2) synthesized at various reaction parameters
| Sample |
ν
1 (cm−1) |
ν
2 (cm−1) |
Average crystallite size (nm), XRD |
|
M
1
|
451 |
573 |
44.5 |
|
M
2
|
454 |
589 |
38.2 |
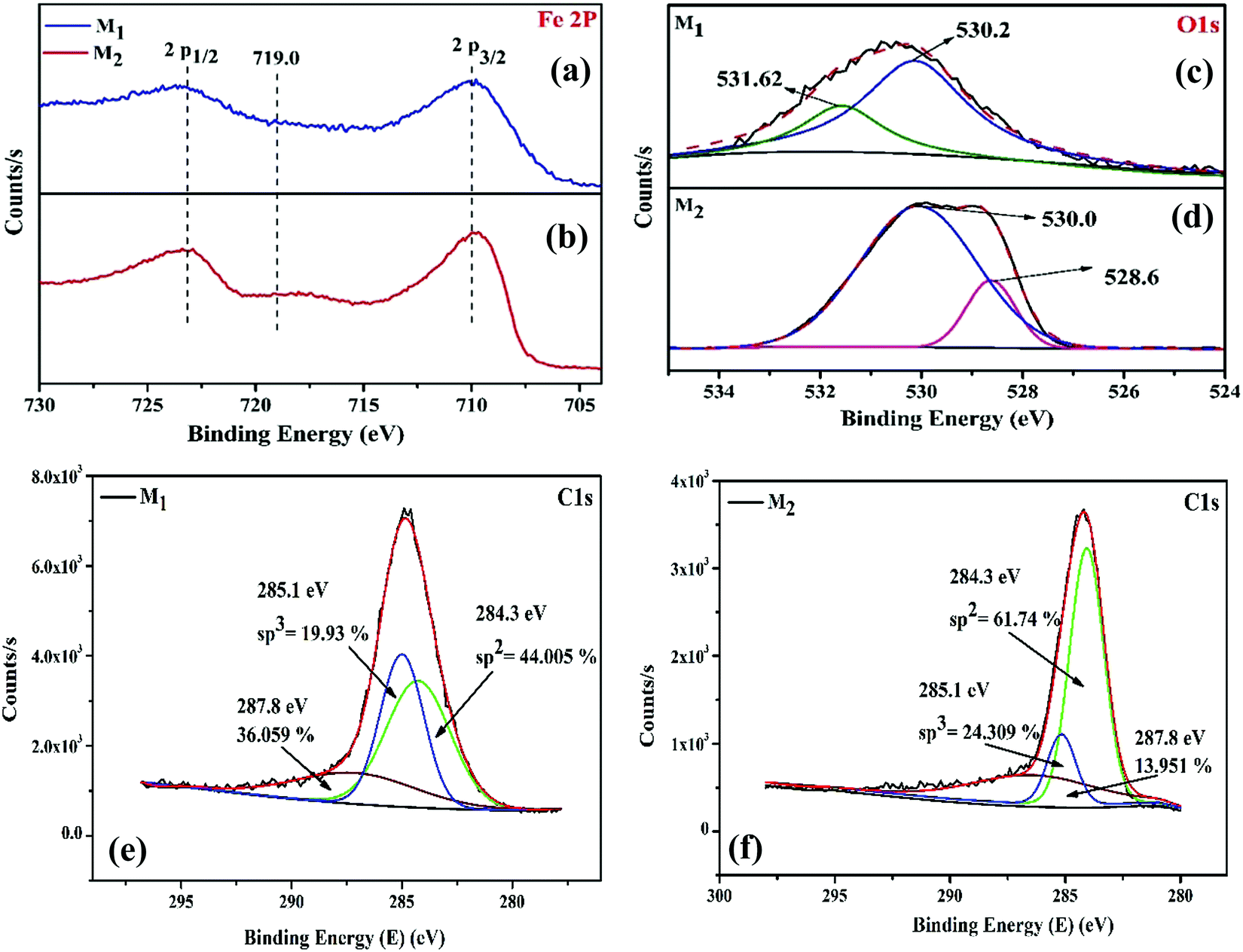
3.5. XPS analysis
Along with the structural and morphological changes, the reaction temperature and time significantly influence the surface composition and chemical oxidation states of surface and near-surface species. Therefore, to obtain a clear perspective into the nature of the interaction between the surface iron atoms and the carbon shell, X-ray photoelectron spectroscopy (XPS) was performed for the magnetite (M1 and M2) samples; the binding energies of the XPS peaks were corrected with carbon as a reference at 284.6 eV. Fig. 4a–f shows the XPS peaks characteristic of Fe 2p, O 1s, and C 1s levels of the Fe3O4/C nanocomposite (M1 and M2) samples. The Fe 2p core level (Fig. 4a and b) encompasses the doublet signifying Fe 2p1/2 and Fe 2p3/2, which is assigned to the characteristic binding energies of magnetite (respective Fe3+ and Fe2+ ions) and the existence of the broad Fe 2p peaks for both the samples (M1 and M2) establishes the presence of mixed iron oxide states (Fe2+ (2p3/2) and Fe2+ (2p1/2)).29 In addition, there is an existence of a minor but obvious peak at about 718.2 eV for M2, which is perhaps the satellite peak of Fe2+ and Fe3+ ions.66 The characteristic peak at about 719.0 eV for Fe3+ in the γ-Fe2O3 phase was not observed, authenticating the formation of pure Fe3O4.67 The deconvolution of O 1s XPS curves in Fig. 4c reveals that the O 1s peak is split into two peaks for M1; the most intense peak at 530.2 eV results from the lattice oxygen in Fe3O4,68 while the other peak at 531.6 eV having a shoulder results from the saturated surface oxygen vacancies on atmospheric exposure.29,30,69 Conversely, for M2, the O 1s spectrum (Fig. 4d) was deconvoluted into two peaks; a negative binding shift of 1.6 eV was observed and the corresponding binding energies for Fe–O and O–H were 528.6 and 530.0 eV, respectively.30
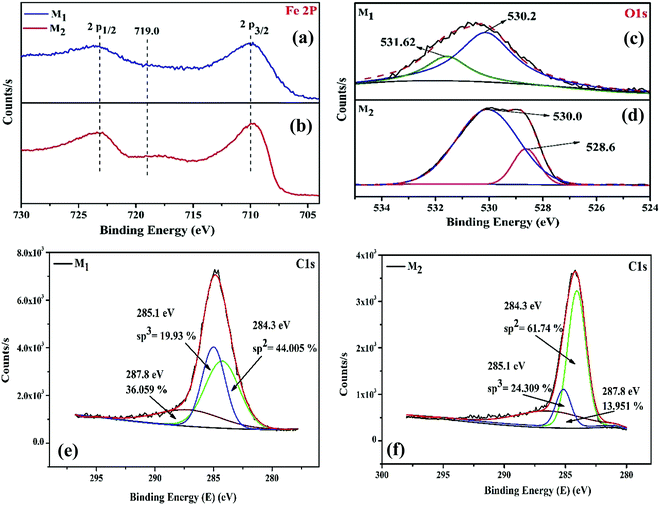 |
| | Fig. 4 (a and b) Fe 2p, (c and d) O 1s, and (e and f) C 1s core level XPS spectra of the as-synthesized Fe3O4/C nanocomposites (M1 and M2), respectively. | |
In order to investigate the carbon shell over the Fe3O4 nanoparticles, the C 1s XPS spectra (Fig. 4e and f) of M1 and M2 were studied. From the deconvoluted C 1s XPS spectra, it was revealed that the C 1s peak splits into three peaks for both the samples. In the two samples studied, the three peaks at 284.3, 285.1, and 287.8 eV correspond to graphitic carbon (sp2 bonded carbon), carbonyl groups (sp3 bonded carbon), and carboxyl/ester groups, respectively.58,70 The relative content of graphitic carbon, carbonyl, and carboxyl/ester groups in the carbon shells can be calculated based on the peak area and the obtained values are indexed in Fig. 4e and f. The graphitic carbon (sp2 bonded carbon) content gradually increased from 44.05% (M1) to 61.74% (M2). The sp3 bonded carbon content also steadily increased from 19.93% (M1) to 24.30% (M2). It is noticeable that by controlling the reaction temperature and time, the amount of sp2 and sp3 bonded carbon can be straightforwardly tuned. Furthermore, the amount of oxygen boned species is relatively high in M1 (36.05%) compared to that in M2 (13.951%), implying that M1 possesses abundant carboxylate groups compared to M2, which authenticates the presence of more ferric/ferrous carboxylate complexes, FeII(R-COO)2−nn and FeIII(R-COO)3−nn in M1 compared to that in M2.44,58
Based on the experimental findings and literature, it is evident that D-glucose functions as a bifunctional agent as follows: (i) under hydrothermal treatment in the presence of an enormous amount of reductive hydroxyl groups of D-glucose, Fe3+ ions are partly reduced to Fe2+ to generate Fe3O4 instead of α-Fe2O3.38,40 (ii) D-glucose also works as a coating agent and slows down the aggregation process, leading to the nucleation of much smaller-sized Fe3O4 nanoparticles.38,45 In detail, a large number of nucleated small magnetite crystallites with high surface energy tend to aggregate rapidly, which can be prevented by an in situ carbon coating around the Fe3O4 crystallites; the carbon coating was obtained from the hydrothermal carbonization of D-glucose. The above experimental findings establish that the carbon coating has a greater impact on obtaining magnetite nanoparticles. Control experiments were carried out by varying the reaction time and temperature while keeping the glucose concentration constant. These experiments prove that the reaction time and temperature play a vital role in obtaining final magnetite products. Fig. S2 (ESI†) exhibits the indexed XRD pattern of the as-synthesized Fe3O4/C nanocomposites at different reaction times (24, 48, 72, and 96 h) and the reaction temperature is 160 °C, whereas Fig. 1b exhibits the Fe3O4/C nanocomposites synthesized at 200 °C for 24 h. Previous scientific literature has proved that below 160 °C, D-glucose cannot be carbonized to carbon; even at 160 °C, the hydrothermal carbonization rate is very slow.71 From the XRD results (Fig. S2, ESI†), it is obvious that at low reaction time (24 h, 160 °C), no diffraction peak was observed; the reason might the very low concentration of carbon such that the nucleation rate of the iron species remains slow. On the other hand, when the reaction time is increased to 48 h (Fig. S2, ESI†), a small but considerable peak of magnetite is observed. Further increasing the reaction time to 72 and 96 h leads to the successful formation of magnetite nanoparticles; comparatively, the crystallinity of the magnetite increases with increasing reaction temperature. On the other hand, D-glucose at 200 °C undergoes rapid hydrothermal carbonization and yields a large amount of solid residue (i.e., hydrothermal carbon) at a reaction time of 24 h (see TGA); as a result, the nucleation rate of Fe3O4 nanocrystals is rapidly increased, which proves that carbon acts as a nucleating agent. According to the above experimental results, it is clear that the variation in the reaction time (24–96 h) and temperature (160–200 °C) has an obvious effect on magnetite crystallization and its particle size.
3.6. Static magnetic properties
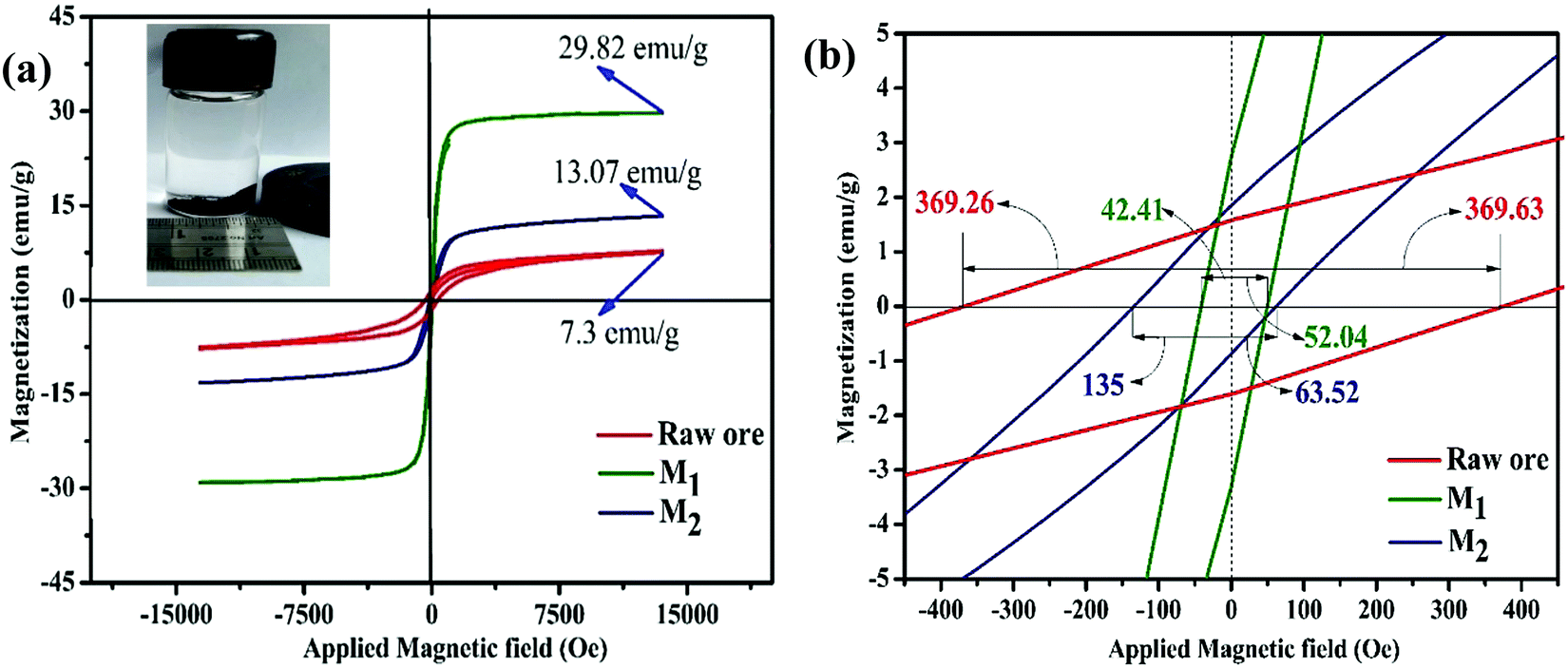
Room temperature magnetization (M) vs. the field strength (H) curve for iron ore tailings (IOTs) and Fe3O4/C nanocomposites (M1 and M2) are shown in Fig. 5a and b. The M–H curve of the IOTs evidently shows anti-ferromagnetic behavior.72 Both M1 and M2 show typical ferromagnetic behavior with high saturation magnetization and low coercivity. The saturation (Ms) magnetizations, remnant (Mr) magnetizations, and coercivity (Hc) values of the Fe3O4/C nanocomposites were calculated from the M–H curves and the results are summarized in Table 4. The calculated Ms values are 29.82 and 13.07 emu g−1 for M1 and M2, respectively, which indicates the large particle-sized M1 (Davg = 44.5 nm, XRD) has stronger saturation magnetization (Ms). The decrease in the Ms value for both M1 and M2 compared to that of bulk magnetite (92 emu g−1)73,74 can be attributed to either the finite size effect or the existence of organic coating agent (please see TGA above).38,75,76 The obtained low squareness ratio (Mr/Ms) and coercivity (Hc) values characterize the superparamagnetic nature of both M1 and M2. Moreover, in non-interacting superparamagnetic particles, the expected squareness ratio (Mr/Ms) value is 0.5, whereas in the case of interacting superparamagnetic particles, it is about 0.10, i.e., the dipole interaction reduces the squareness ratio (Mr/Ms) value due to a demagnetizing effect.77 The squareness ratio (Mr/Ms) values of M1 and M2 (Table 4) elucidate that M1 demonstrates the characteristic feature of interacting with superparamagnetics, whereas M2 shows characteristics of non-interacting superparamagnetics. The results confirms that M1 reacts rapidly to with increased the external magnetic field and allows easy magnetization and demagnetization with almost zero hysteresis loss; this magnetic property affirms sample M1 as a suitable material for nanoelectronic and spintronic applications.77 The anisotropy constant (K) values were computed using the relationship 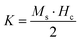 and the obtained values of the anisotropy constant are listed in Table 4. It is obvious that the values of K decrease with decreasing particle size, signifying that the presence of uniaxial anisotropy can be further ascribed to the surface effects and microstructural defects.18,40 Previous reports have established that the Ms Value of 7–22 emu g−1 is suitable for biomedical applications.78–80 Furthermore, due to its high magnetic response sensitivity, the as-synthesized Fe3O4/C nanocomposites may be used as magnetically separable catalyst for treating colored dyes and other organic pollutants in wastewater.
and the obtained values of the anisotropy constant are listed in Table 4. It is obvious that the values of K decrease with decreasing particle size, signifying that the presence of uniaxial anisotropy can be further ascribed to the surface effects and microstructural defects.18,40 Previous reports have established that the Ms Value of 7–22 emu g−1 is suitable for biomedical applications.78–80 Furthermore, due to its high magnetic response sensitivity, the as-synthesized Fe3O4/C nanocomposites may be used as magnetically separable catalyst for treating colored dyes and other organic pollutants in wastewater.
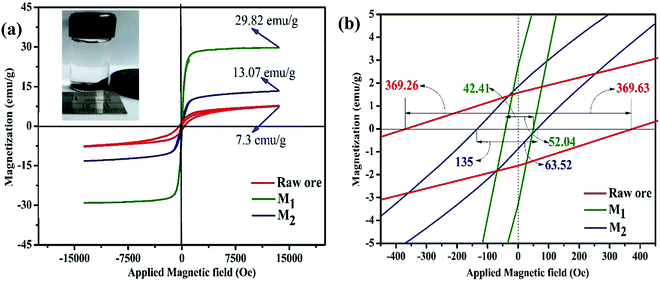 |
| | Fig. 5 (a) Room temperature M–H hysteresis loops of IOTs and synthesized Fe3O4/C nanocomposites (M1 and M2) (inset: separation of particles dispersed in hexane by a magnet); (b) respective magnified views at the origin of the M–H loops. | |
Table 4 Magnetic parameters of the as-synthesized Fe3O4/C nanocomposites (M1 and M2) and IOTs
| Sample |
Average crystallite size (D) (nm) XRD |
M
s (emu g−1) |
M
r (emu g−1) |
Squareness ratio 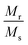
|
H
c (Oe) |
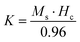
|
| IOTs |
276.5 |
7.3 |
1.569 |
0.215 |
369.63 |
2810.72 |
|
M
1
|
44.5 |
29.82 |
1.847 |
0.0619 |
52.04 |
1616.49 |
|
M
2
|
38.2 |
13.07 |
2.75 |
0.21040 |
63.52 |
864.79 |
3.7. Optical properties
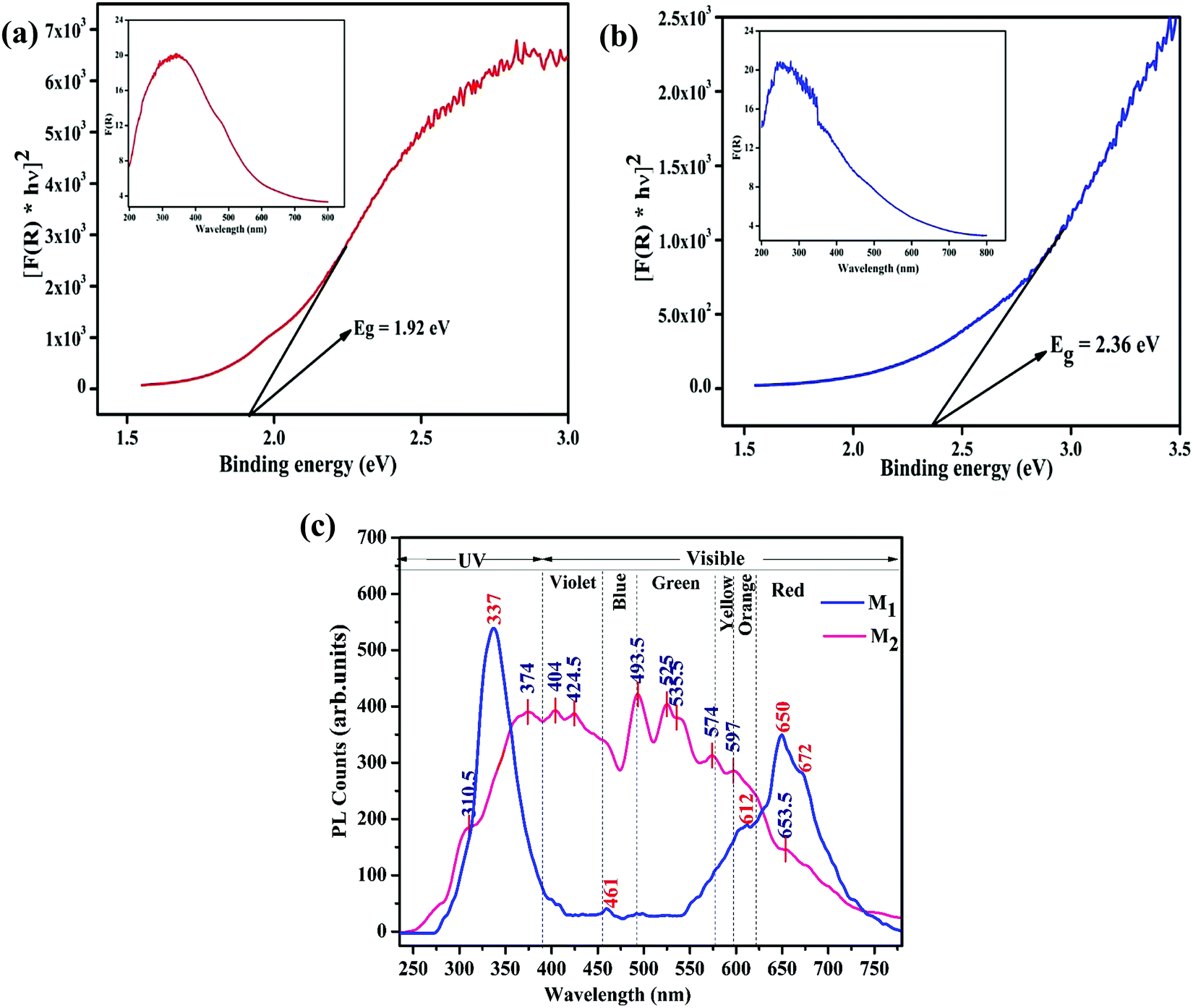
UV-vis diffuse reflectance spectroscopy (DRS) and photoluminescence (PL) spectroscopy was carried out to differentiate the optical properties of the Fe3O4/C nanocomposites (M1 and M2). The absorption spectra and bandgap energy (Eg) of the samples were calculated using the Kubelka–Munk (K–M) model.81 The following relation expresses the K–M equation at any wavelength.| | 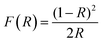 | (4) |
where F(R) is the K–M function and R is the reflectance. The inset of Fig. 6a and b shows the absorption spectra of M1 and M2 obtained from the diffuse reflectance spectra based on the Kubelka–Munk (K–M) theory. Comparatively, the absorption cut-off wavelength of M1 is higher than that for M2, suggesting that M1 can absorb visible light completely; consequently it might be a favorable photocatalytic material. The energy bandgap (Eg) of the Fe3O4/C nanocomposites (M1 and M2) can be estimated from the plot of [F(R) × hν]2vs. photon energy; the bandgap energies were found to be 1.92 and 2.36 eV for M1 and M2, respectively (Fig. 6a and b). It is interesting to note that the bandgap decreases from 2.36 to 1.92 eV for M1, suggesting that the bandgap has a prominent relationship with the particle size, which eventually indicates the appearance of quantum size effects for Fe3O4 nanocrystals and the smaller bandgap of M1 point out the prospects of using more visible light during photocatalysis.62,82Fig. 6c shows the photoluminescence (PL) spectra of M1 and M2 under excitation at λex = 250 nm. The PL spectra of M1 displays a strong excitonic emission band centered at 337 nm in the UV region, which is due to the recombination of free excitons,82 and the emission in the visible region (in the range of 550–700 nm) discloses its excitation peaks. The emission at the lower energy shoulder (ca. 672 nm, 1.79 eV) is attributable to the recombination of trapped electrons from the octahedral site to O (2p), or t2 → O (2p) on the tetrahedral sites.83 The PL peaks at 612 and 650 nm are attributed to interstitial oxygen vacancies.82,83 These data are consistent with the XPS analysis (see above), which points to oxygen being deficient in M1. Conversely, the PL spectra of M2 are found to be broad and asymmetric; the deconvolution of this broad peak exposes multiple emission bands. The PL peak at about 374 nm is attributed to the near band-edge emission of Fe3O4, which is due to the recombination of free excitons.82 The emission band at about 493 nm may be ascribed to surface defects and the emission band at about 565 nm is attributable to the radiative recombination of mobile electrons from t2g → eg (2.2 eV) on the octahedral sites in Fe3O4.82–84 It is interesting to note that M1 exhibit a stronger near-band-edge emission due to its large size and crystalline nature, while M2 show a stronger defect state emission due to its small size and high surface area, which evidently explains the quantum confinement effect in the reported Fe3O4/C nanocomposites.82 This shows that the optical properties of the Fe3O4/C nanocomposites change with respect to the particle size and finally, the energy of visible PL emission also corresponds well with the results obtained from the Tauc plot.
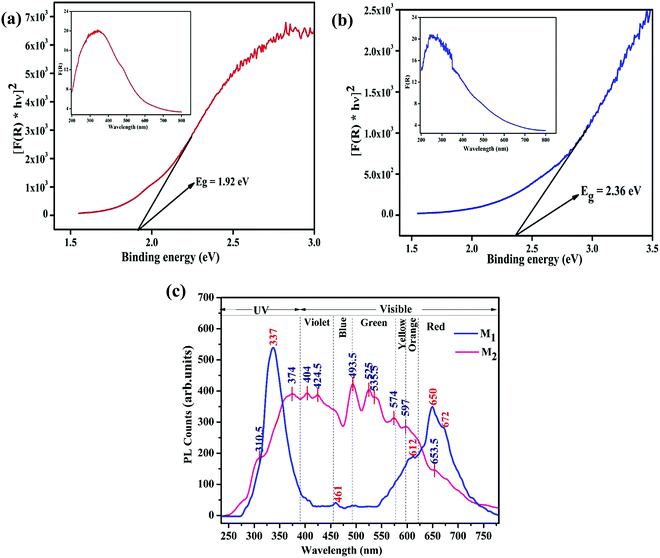 |
| | Fig. 6 (a) The Plot of [F(R) × hν]2 against hν showing the optical Eg estimated using the Kubelka–Munk function of the Fe3O4/C nanocomposites (a) M1 and (b) M2, respectively. The plots of F(R) vs. the photo energy for the estimation of the optical absorption edge energy of the products are shown inset of the respective figures. (c) Photoluminescence emission spectrum (excited at 250 nm) of the synthesized Fe3O4/C nanocomposites (M1 and M2), respectively. | |
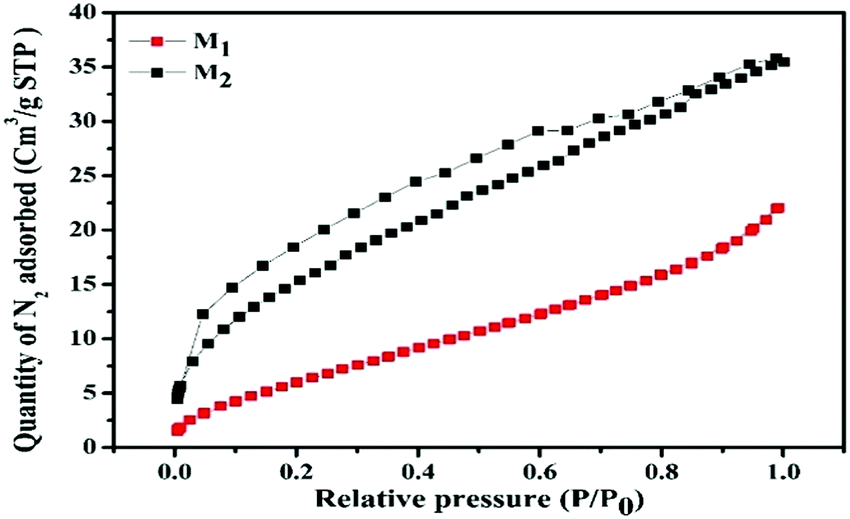
3.8. BET surface area analysis
On account of heterogeneous photocatalysis, the surface area plays a key role as a large surface area provides more surface active sites for the adsorption of reactant molecules, which improves the photocatalytic efficiency.85 The BET surface areas and pore size distributions of M1 and M2 were determined by measuring the nitrogen adsorption–desorption isotherms. The specific BET surface areas, pore volumes, and pore sizes of M1 and M2 are listed in Table 5. Fig. 7 illustrates that both the samples exhibit a type-IV isotherm with an H3-type hysteresis loop, representing the presence of an interconnected network of mesopores.86 Specific BET surface areas were found to be 25.980 and 59.639 m2 g−1 for M1 and M2, respectively. The findings were further recognized as per as the IUPAC classification by well-developed mesopores with mean pore diameters of 26.2 and 18.4 nm for M1 and M2, respectively.87 The superior surface area recorded for M2 may favor dye adsorption compared to M1 (please see the photocatalysis section below for details).
Table 5 Nitrogen sorption porosimetry studiesa of Fe3O4/C nanocomposites (M1 and M2)
| Photocatalyst |
S
BET (m2 g−1) |
Pore volume (cm3 g−1) |
Mean pore size (nm) |
|
Surface areas determined by the BET technique. Pore volume and mean pore diameters were obtained by applying the Barrett–Joyner–Halenda theory to the adsorption branch.
|
|
M
1
|
25.980 |
3.407 × 10−2 |
2.62251 × 10−10 |
|
M
2
|
59.639 |
5.487 × 10−2 |
1.84023 × 10−10 |
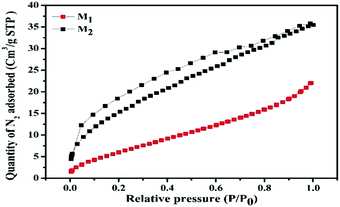 |
| | Fig. 7 Nitrogen adsorption–desorption isotherms (P and P0 are the equilibrium and the saturation pressures of N2 at the temperature of adsorption, respectively) of the synthesized Fe3O4/C nanocomposites (M1 and M2). | |
3.9. Visible light photocatalytic activity
In order to investigate the catalytic activity (both dark-Fenton and photo-Fenton) of the presented Fe3O4/C nanocomposites (M1 and M2), several experiments were conducted under different reaction conditions. For each experiment, 25 mg of each catalyst was used along with 50 mL of 1.0 × 10−5 M aqueous solution of RhB (pH 7) and the absorption band of RhB was witnessed at about 555 nm (λmax). Various blank experiments were also conducted by removing one or more components (H2O2, catalyst, and irradiation) in the photocatalytic experiments to prove their significant role toward RhB degradation. Based on the reference experiments, the following observations were recorded: (i) RhB does not experience any substantial degradation upon treatment with H2O2 under visible light irradiation for 180 min (Fig. S4a, ESI†); (ii) both the catalysts do not induce any noteworthy RhB degradation without H2O2 when irradiated under dark conditions (Fig. S4b and c, ESI†). Remarkably, under dark (adsorption) conditions, the discoloration rate of RhB reached 11.36 (M1) and 26.22 (M2)%. Earlier studies have established that the adsorption of dye molecules on the surface of the catalyst may improve the photodegradation efficacy.88 However, the adsorption process does not completely degrade the organic pollutant as it merely transforms the primary pollutants to secondary ones instead of degrading them.89 The study on RhB adsorption by Fe3O4/C nanocomposites (M1 and M2) shows that M2 (SBET = 59.639 m2 g−1) exhibited higher discoloration rate compared to M1 (SBET = 25.980 m2 g−1), which corroborates well with the fact that higher surface area favors higher RhB adsorption.
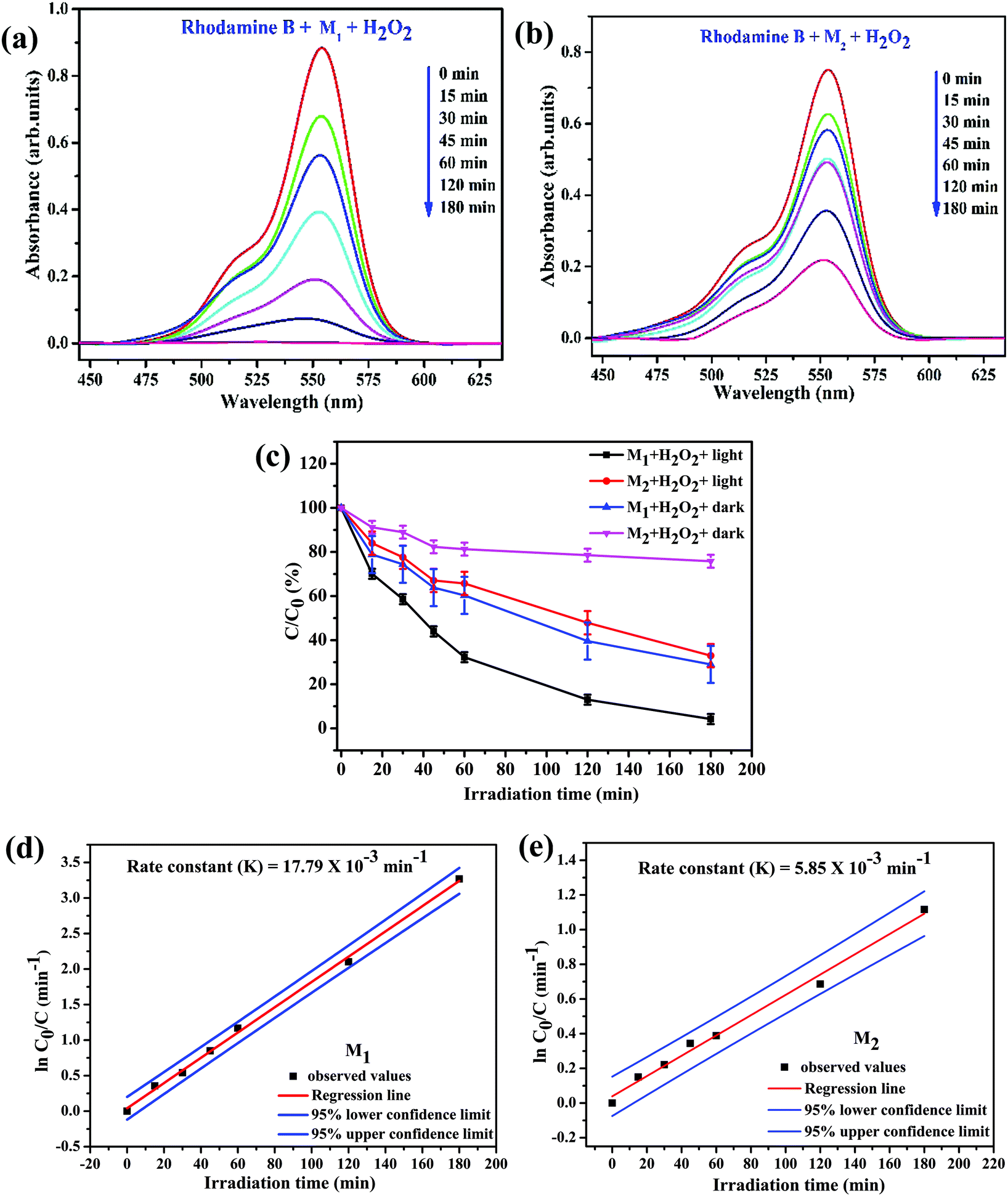
Fig. 8a and b illustrate the relationship between light absorbance and simulated solar irradiation time (t) for M1 and M2 with H2O2. It is witnessed that in the both cases, the absorption band gradually decreases with increasing irradiation time (t), though the rate of decrease is slightly greater in case of M1 when compared to M2 (please see Table 6 for detailed calculations). Reference experiments such as dark Fenton, without light irradiation in the presence of Fe3O4/C nanocomposite samples and H2O2 (Fig. S4d and e, ESI†) were also performed; the experiments exhibited a noteworthy discoloration of the dye. It was observed that in both dark-Fenton and photo-Fenton experiments that the discoloration of RhB in the presence of M1 is considerably higher than that in the presence of M2. Fig. 8c illustrates the decomposition rate for RhB λmax for M1 and M2 in the presence of H2O2 as a function of with and without simulated solar irradiation under identical conditions, respectively. Notably, the discoloration rate of RhB reached 70.99 (±8.4)% in the case of M1 and 24.20 (±2.914)% in the case of M2 under dark conditions in the presence of H2O2. The outcomes evidently ascertained that M1 demonstrates higher discoloration efficiency compared to M2, which is perhaps due to the fact that the Fe active sites may be covered by more carbon in M2 (please see TGA analysis above). This reveals that the carbon coating plays a crucial role in the dark-Fenton activity. From the XPS investigation, the amount of oxygen-bonded species was found to be 36.05 and 13.95% for M1 and M2, respectively. It is ascertained that M1 is rich in ferric/ferrous carboxylate complexes, FeII(R-COO)2−nn and FeIII(R-COO)3−nn compared to M2 since this complexation process is one of the reasons that M1 possesses higher discoloration efficiency under dark-Fenton conditions.45 The outcomes evidently ascertained the controlled generation of hydroxyl radicals (˙OH) by the catalytic dark-Fenton reactions. Remarkably, the discoloration rate of RhB reached 95.779 (±4.221) for M1 and 67.019 (±5.29)% for M2 in the case of photo-Fenton experiments.
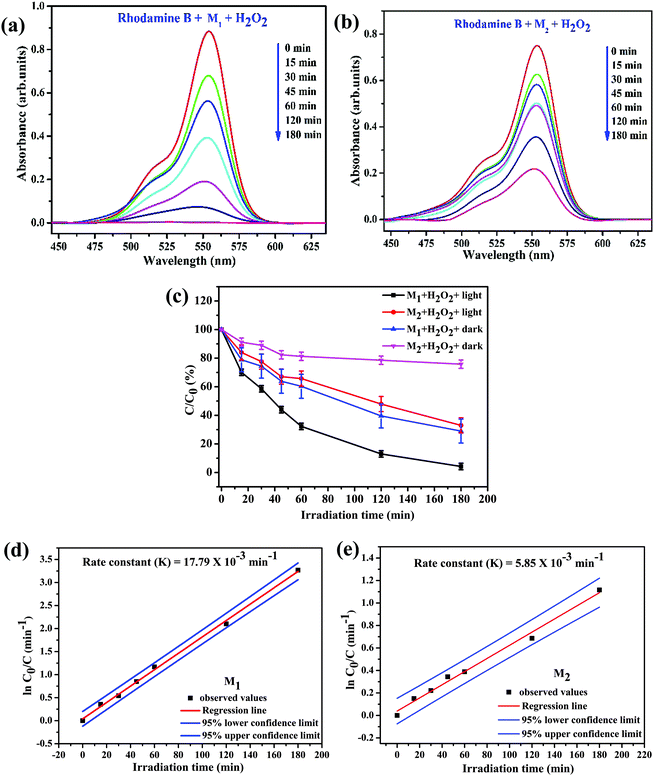 |
| | Fig. 8 Photolytically-induced changes to λmax (555 nm) for RhB (50 mL of 1.0 × 10−5 M aqueous solution) on treatment (a) with M1 (25.0 mg) and H2O2 (100 μL); (b) with M2 (25.0 mg) and H2O2 (100 μL); (c) plot of C/C0 (%) (where C0 and C are the concentrations of the dye before and after irradiation, respectively) for RhB as a function of the irradiation time in the presence of M1 and M2 under different conditions and plots of ln(C0/C) as a function of irradiation time in the presence of (d) M1 and (e) M2 under visible light irradiation and with H2O2. | |
Table 6 Degradation of RhB by Fe3O4/C nanocomposites (M1 and M2) synthesized at various reaction parameters
|
|
Samples |
Degradation efficiency (%) |
|
M
1
|
M
2
|
| Reaction condition |
Dark-Fenton |
70.99 ± 8.4 |
24.20 ± 2.9 |
| Photo-Fenton |
95.78 ± 4.2 |
67.02 ± 5.3 |
Our experimental findings showed that M1 (Davg (XRD) = 44.5 nm and SBET = 25.980 m2 g−1) reveals higher photocatalytic activity compared to M2 (Davg (XRD) = 38.2 nm and SBET = 59.639 m2 g−1). These findings disclose that the photocatalytic activity of the catalyst does not totally depend on the surface area of the catalyst, whereas the presence of surface defects and oxygen vacancies in the catalyst play a crucial role in the photocatalytic process. Based on the Rietveld analysis data, the amount of oxygen vacancy in the Fe3O4/C nanocomposite samples was found to be 5.40% for M1 and was almost nil for M2 (see Table 2). X-ray photoelectron spectroscopy (XPS) and photoluminescence (PL) spectroscopy show that the magnitude of oxygen vacancies is reasonably higher in M1 in comparison with that in M2 (please see the XPS and PL section above). It was ascertained that the presence of surface defects and oxygen vacancy in the catalyst facilitates the narrowing of the bandgap energy, which allows to restrain the recombination of the charge carriers and utilize visible light more effectively for photocatalysis, which are the core reasons behind the improved photo-Fenton activity of M1. All these observations evidently authenticate the synergetic effect of the catalyst, H2O2, and visible light for the degradation of RhB. It has been scrutinized that the degradation efficiency of RhB follows the order: M1/H2O2/light > M1/H2O2/dark > M2/H2O2/light > M2/H2O2/dark > H2O2/light.
To quantitatively evaluate the photocatalytic activities of these samples, the reaction rate constants (k) were calculated by adopting the pseudo-first-order kinetics model (eqn (5)), assuming low initial pollutant concentration.90,91
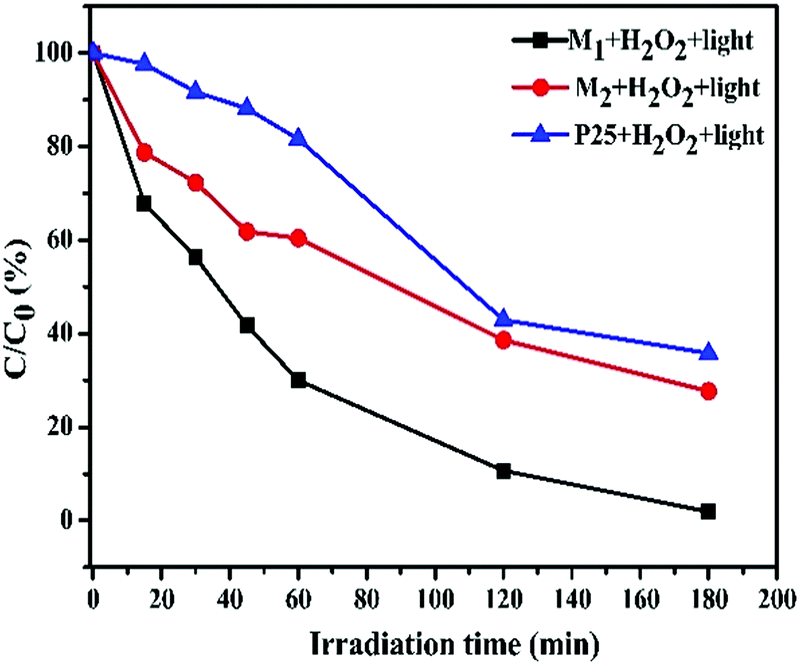
Plots of ln(C0/C) vs. irradiation time (t) are provided in Fig. 8d and e; the linear relationships pointing to each photodegradation follow first order kinetics. The apparent rate constants were estimated to be 17.79 × 10−3 and 5.85 × 10−3 min−1 for M1 and M2, respectively, which signifies that the photocatalytic activity of M1 is about 3 times higher than that of M2. The photocatalytic activities of M1 and M2 were compared with that of the standard TiO2 Degussa P25 powder. The results shown in Fig. S4f (ESI†) ascertain that P25 (25.0 mg) induces a steady decrease in the 555 nm absorption band of RhB with an efficiency of 64.21%, which is low compared to that of both M1 (98.07%) and M2 (72.31%) (Fig. 9).
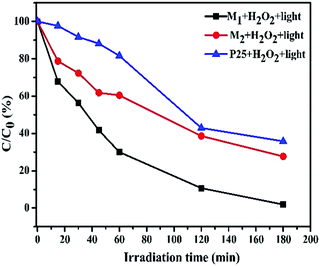 |
| | Fig. 9 A comparison of C/C0 (%) as a function of the irradiation time for RhB solutions treated with the synthesized Fe3O4/C nanocomposites (M1 and M2) and P25 (25.0 mg each) under the same conditions. | |
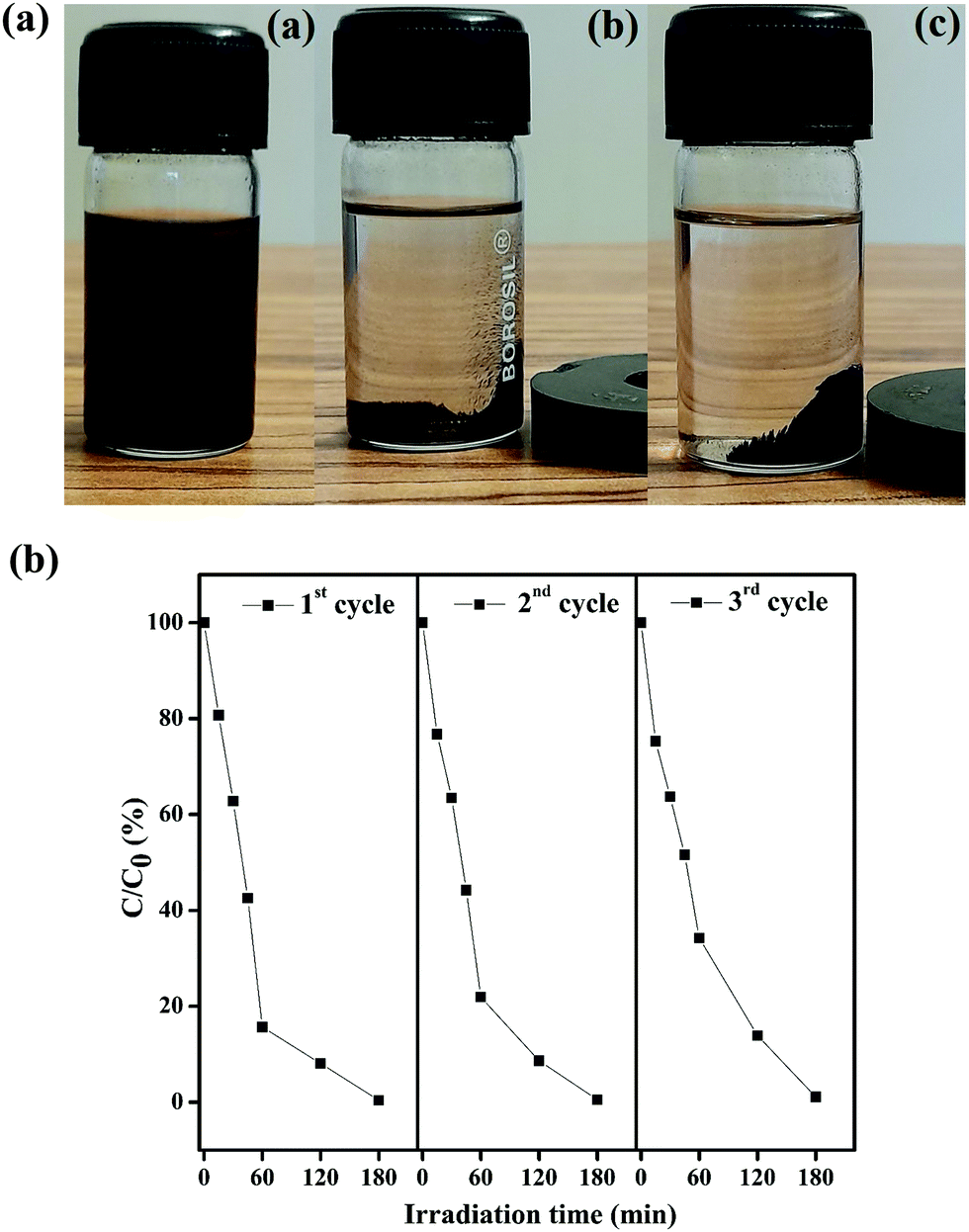
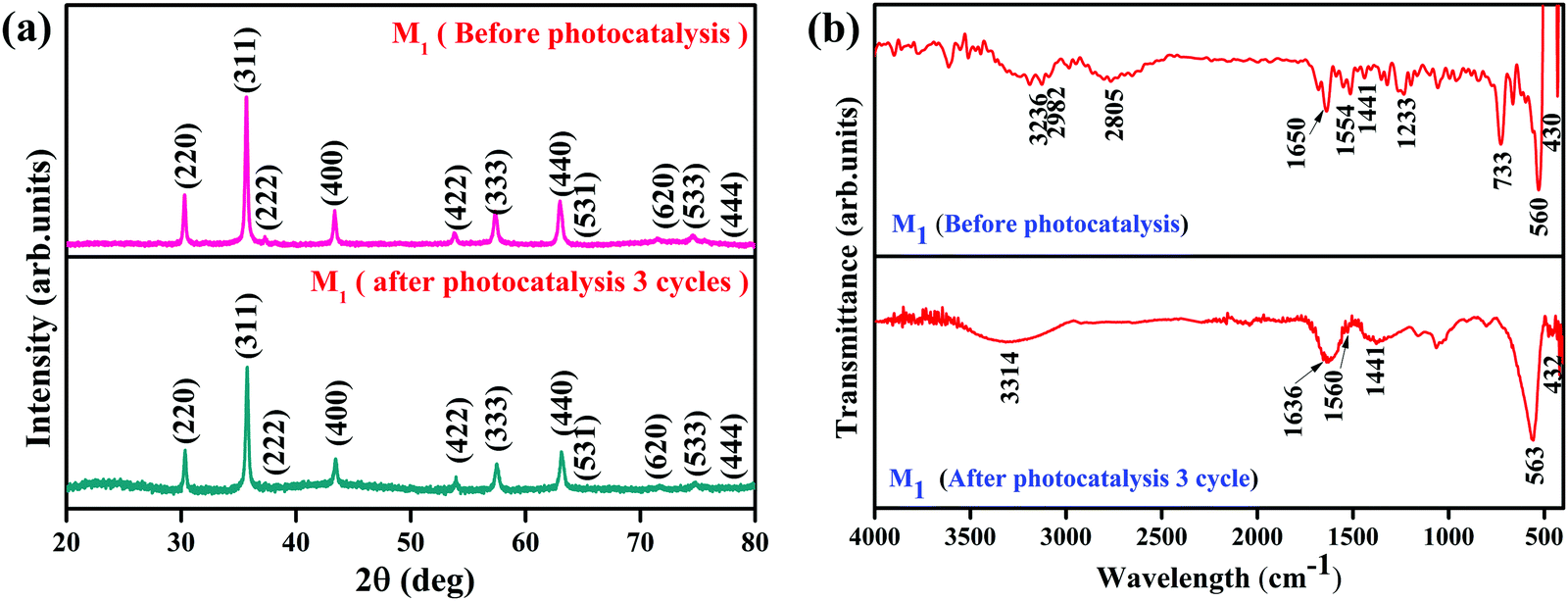
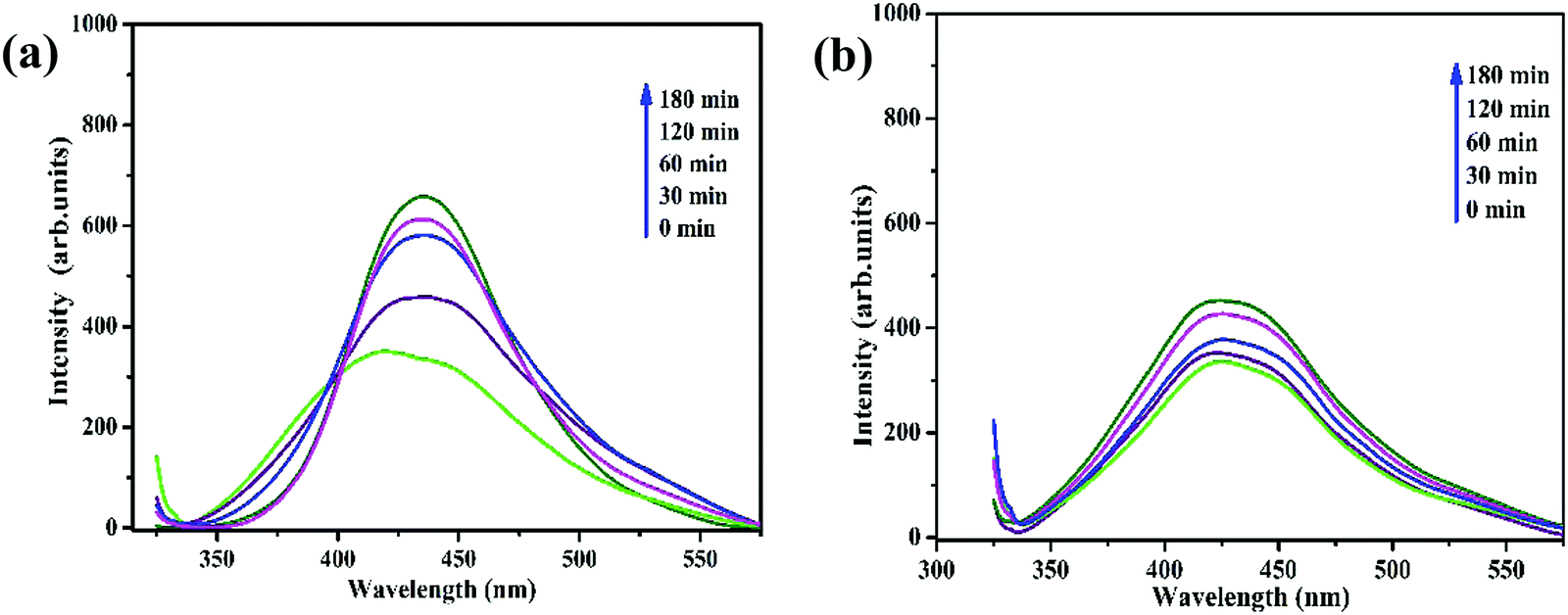
It is essential to investigate the stability of the photocatalytic performance and the reusability of the Fe3O4/C nanocomposite since this is an important factor from economic and environmental perspectives. To investigate the stability and reusability of our best reported photocatalyst (M1), cycling experiments for the photodegradation of RhB were conducted with the catalyst. After each recyclability test, the catalyst was recovered from the solution using an external magnetic field (Fig. 10a), washed with water and absolute ethanol, dried at 80 °C for 2–3 h, and then used for the following cycles. As shown in Fig. 10b, the degradation efficiency of M1 (Fe3O4/C nanocomposite) over RhB after the first, second, and third cycles was found to be 99.60, 99.48, and 98.904%, respectively. In addition, it was noted that about 2.7, 1.09, and 0.487% of the initial quantity of the catalyst was lost in each cycle. Overall, these data suggest that a slight reduction in the photocatalytic efficiency could be assigned to the inevitable loss of the catalyst during the recovery steps. We also consider that the reduced effectiveness of the catalyst after recycling is contributed to by the photo-bleaching of the catalyst surface.91,92 Further, Fig. 11a and b display the XRD patterns and FT-IR spectra of M1 (Fe3O4/C nanocomposite) before and after three RhB decomposition cycles, with the lack of noticeable changes in signifying that both the crystalline phase and structure remain intact.
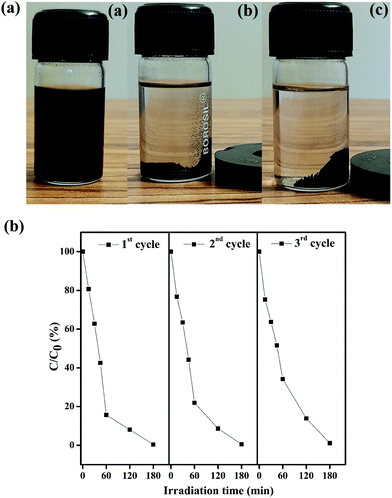 |
| | Fig. 10 (a) Suspension of Fe3O4/C nanoparticles before (a) and after 15 s (b) and 30 s (c) of separation by the use of an external magnetic bar. The latter clearly illustrates the facile recovery of the catalyst by an external magnetic field. (b) Reproducibility of the photocatalytic decomposition of RhB using fresh (cycle 1) and recycled (cycles 2 and 3) Fe3O4/C (M1) nanocomposites. | |
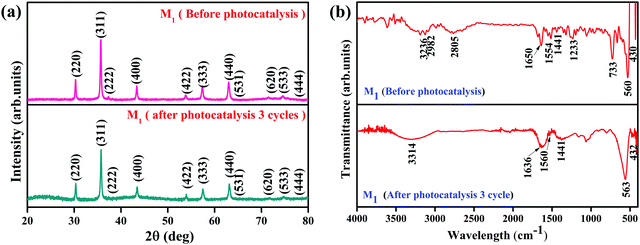 |
| | Fig. 11 (a) Indexed XRD pattern and (b) FTIR spectra of the Fe3O4/C (M1) nanocomposites before and after photocatalysis. | |
From the above experimental observations and literature, a probable photocatalytic mechanism for RhB degradation by the presented Fe3O4/C photocatalyst under visible light irradiation was proposed.44,93 The possible contributions to the exceptional photocatalytic performance of the Fe3O4/C semiconductor nanocomposites may originate from the synergistic effects between ferric/ferrous carboxylate complexes (FeII(R-COO)2−nn,FeIII(R-COO)3−nn) and Fe3O4 semiconductor nanoparticles, which is summarized in Scheme S1 (ESI†). Initially, the FeII(R-COO)2−nn complex on the surface of the Fe3O4/C nanocomposite catalyzes H2O2 to produce hydroxyl radicals (˙OH) via a typical Fenton reaction, as shown in eqn (6).5,44,94 The transformation of ferric to ferrous carboxylate complexes occurs chiefly without light in the presence of H2O2via dark Fenton reaction, as shown in eqn (7).94,95
| | | (FeII(R-COO)2−nn) + H2O2 → FeIII(R-COO)3−nn + OH− + ˙OH | (6) |
| | | FeIII(R-COO)3−nn + H2O2 → (FeII(R-COO)2−nn) + HO2˙ + H+ | (7) |
In the dark Fenton reaction, (FeIII(R-COO)3−nn) is reduced to (FeII(R-COO)2−nn) by consuming H2O2 and generating perhydroxyl radical (HO2˙), whose oxidation potential is moderately weak compare to that of the hydroxyl radical.5 Under simulated solar irradiation, FeIII(R-COO)3−nn is photo-reduced to (FeII(R-COO)2−nn), accompanied by the generation of ˙OH via the photo-Fenton reaction, as shown in eqn (8).44,57,96
| | | FeIII(R-COO)3−nn + H2O + hν (λ > 420 nm) → (FeII(R-COO)2−nn) + H+ + ˙OH | (8) |
Comparatively, the dark Fenton experiment (eqn (7)) consumes more H2O2 for the transformation of FeIII(R-COO)3−nn to (FeII(R-COO)2−nn), whereas in the photo-Fenton process, the transformation takes place principally without H2O2 consumption (eqn (8)).
Several studies have validated that the introduction of semiconductor materials into the photo-Fenton system can increase the degradation efficiency of the organic pollutants.57 Semiconductors such as TiO2,6,97 ZnO,98,99 and SnO285,100 have received huge attention due to their applications in the photocatalytic degradation of organic pollutants. Numerous studies have ascertained that Fe3O4 as a semiconductor (both n- and p-type) photocatalyst could efficiently degrade numerous organic pollutants.20,101
The photocatalytic properties of the semiconductors depends on the ability to generate electron–hole pairs and the formation of free radicals for secondary reactions. During photoexcitation, an electron in the valence band (VB) of the semiconductor is excited to the conduction band (CB) with the simultaneous generation of a hole in the VB, as shown in eqn (9).
| | | Fe3O4 + hν → Fe3O4 (eCB− + hVB+) | (9) |
Based on the literature, (FeIII(R-COO)3−nn) can act as an acceptor of photogenerated electrons from a semiconductor during the photocatalytic process (eqn (10)), which eventually slows down the electron–hole recombination process.57,102
| | | (FeIII(R-COO)3−nn) + Fe3O4 (eCB−) → (FeII(R-COO)2−nn) | (10) |
The regenerated (FeII(R-COO)2−nn) made the FeII/FeIII redox cycle logical, which confirms the interaction between the Fe3O4 core and the ferrous/ferric carboxylate complex shell (Scheme S1, ESI†). Simultaneously, the photoexcited electrons are trapped by H2O2 for co-generating ˙OH and OH−, and the holes left in the valence band react to form hydroxyl radicals (˙OH), which subsequently reacts with the adsorbed pollutant molecules (such as RhB) to produce oxidized species and/or decomposed products according to the following equations.
| | | Fe3O4 (eCB−) + H2O2 → ˙OH + OH− + Fe3O4 | (11) |
| | | Fe3O4 (hVB+) + OH− → Fe3O4 + ˙OH | (12) |
| | | Fe3O4 (eCB−) + O2 → Fe3O4 + ˙O2− | (13) |
| | | ˙O2− + H2O2 → ˙OH+ OH− + O2 | (14) |
| | | ˙O2− + H2O → HO2˙+ OH− | (15) |
| | | ˙OH/˙O2− + RhB → CO2 + H2O | (16) |
Xu et al.57 established that the hydroxyl radicals (˙OH) generated during photocatalysis act as the key active species in the degradation of organic contaminants. A terephthalic acid (TA) probe was employed to validate that the photocatalytic degradation of RhB by the current Fe3O4/C nanocomposite system by means of photoinduced ˙OH radical formation. Terephthalic acid, as a ˙OH radical scavenger, can react immediately with the ˙OH radical to produce 2-hydroxyterephthalic acid (2HTA), a highly fluorescent compound.56 The concentration of the photoinduced ˙OH radical can be measured by determining the concentration of 2HTA. Upon excitation at 315 nm, the highest intensity at 425 nm gradually increased with the irradiation time (Fig. 12a and b), verifying the photogeneration of ˙OH. From to Fig. 12a and b, it is apparent that the concentration of the generated ˙OH radicals increases evidently with M1 possibly due to the narrow (smaller) energy bandgap (band energy) (1.92 eV) compared with the bandgap of M1 (2.36 eV). The smaller bandgap of M1 indicates the possibility of utilizing more visible light for photocatalysis, which clearly establishes that the photocatalytic process proceeds more rapidly for M1 compared to M2. It was proposed that the size effect of the presented Fe3O4/C nanocomposites might be responsible for the photocatalytic property by comparing the optical properties and oxygen vacancies of the as-prepared samples.
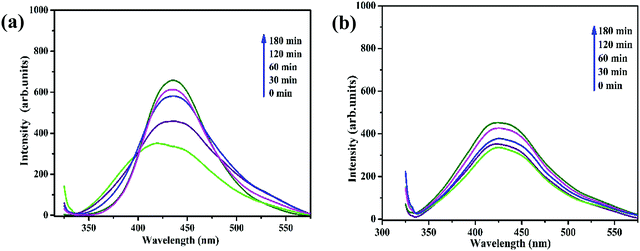 |
| | Fig. 12 Fluorescence spectral changes measured during illumination of Fe3O4/C nanocomposites, (a) M1 and (b) M2 with 150 μL of H2O2 in a basic solution of TA (excitation at 315 nm). | |
Literature has already established that O2− ions can escape from the host Fe3O4 lattice, leading to the generation of oxygen vacancies or defects (Vo).32,84 These oxygen vacancies in the Fe3O4 nanocrystals could serve as the active centers to capture the photo-induced electrons and efficiently slow down the recombination of photo-induced electrons and holes, thus significantly improving the photocatalytic activity.82,101 In the present study, the Fe3O4/C nanocomposite with larger crystallite size (M1) has more surface defects, whereas the Fe3O4/C nanocomposite with a comparatively smaller crystallite size (M2) does not display any surface defects (validated by XRD Rietveld analysis, X-ray photoelectron spectroscopy, and photoluminescence spectroscopy). In addition, the bandgap energy of the as-prepared Fe3O4/C nanocomposite decreases with increasing crystallite size (confirmed by UV-DRS analysis), which leads to the possibility of utilizing more visible light during photocatalysis. This shows that the reaction temperature and time have a significant effect on the structural, optical, and magnetic properties. Consequently, surface defects/oxygen vacancies are found to play an important role in the photocatalytic property, which may be probably initiated from the finite size effect of Fe3O4 and the interaction between the Fe3O4 nanoparticles and the carbon support.29,82 Thus, we conclude that magnetically recoverable Fe3O4/C nanocomposites with tunable size and high photocatalytic potential can used for wide environmental applications.
Conclusion
In this contribution, we report a straightforward, low-cost, and environmentally benign hydrothermal reduction route to synthesize glucose and gluconic acid-coated magnetite nanoparticles from an inexpensive iron precursor acquired from accumulated iron ore tailings. Two noticeably different samples having different crystallite sizes were produced by tailoring the reaction parameters (temperature and reaction time). Size-dependent surface defects and oxygen vacancy concentration of the as-prepared samples were examined by means of XPS and XRD/Rietveld analysis, respectively. UV-visible differential reflectance spectroscopy (UV-DRS) and photoluminescence (PL) analysis clearly established the relation of crystallite size and oxygen vacancy concentration with the bandgap energy. The results clearly confirmed that the crystallite size and oxygen vacancy concentration of the as-prepared samples have a considerable effect on the photodecomposition of Rhodamine B under visible light irradiation. The as-reported crystallite size and oxygen vacancy concentration-dependent photocatalyst could be serve as a helpful reference for developing environment-friendly photocatalysts from industrial waste materials.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
A. K. acknowledges support from the Department of Science and Technology (DST), Government of India through its INSPIRE Faculty Award Programme. A. K. also acknowledges support from the Royal Society's Newton International Fellowship Follow-on Funding scheme. S. S. acknowledges an RA-II Fellowships Grant from IACS, Kolkata. The Ministry of Human Resource Development (MHRD), India, financially supported the PhD of M. P. We gratefully acknowledge NMDC Ltd (Donimalai iron ore mines) for their consent to work inside their permit territory and for logistical help during the field trip. We equally express thankfulness toward Dr M. Sudarshan, Dr Souvik Chatterjee, and Dr Pallippuram Venkitaraman Rajesh (UGC-DAE Consortium for Scientific Research, Kolkata Centre) for their help with powder XRD analysis.
References
- M. Pera-Titus, V. García-Molina, M. A. Baños, J. Giménez and S. Esplugas, Appl. Catal., B, 2004, 47, 219–256 CrossRef CAS.
- A. N. Soon and B. Hameed, Desalination, 2011, 269, 1–16 CrossRef CAS.
- H. Bai, Z. Liu and D. D. Sun, Appl. Catal., B, 2012, 111, 571–577 CrossRef.
- C. R. Holkar, A. J. Jadhav, D. V. Pinjari, N. M. Mahamuni and A. B. Pandit, J. Environ. Manage., 2016, 182, 351–366 CrossRef CAS PubMed.
-
S. C. Ameta and R. Ameta, Advanced oxidation processes for wastewater treatment: emerging green chemical technology, Academic Press, 2018 Search PubMed.
- D. Du, W. Shi, L. Wang and J. Zhang, Appl. Catal., B, 2017, 200, 484–492 CrossRef CAS.
- F. F. Dias, A. A. Oliveira, A. P. Arcanjo, F. C. Moura and J. G. Pacheco, Appl. Catal., B, 2016, 186, 136–142 CrossRef CAS.
- P. K. Boruah, B. Sharma, I. Karbhal, M. V. Shelke and M. R. Das, J. Hazard. Mater., 2017, 325, 90–100 CrossRef CAS PubMed.
- J. He, X. Tao, W. Ma and J. Zhao, Chem. Lett., 2002, 86–87 CrossRef CAS.
- R. Matta, K. Hanna and S. Chiron, Sci. Total Environ., 2007, 385, 242–251 CrossRef CAS PubMed.
- D. Tabet, M. Saidi, M. Houari, P. Pichat and H. Khalaf, J. Environ. Manage., 2006, 80, 342–346 CrossRef CAS PubMed.
- S. Shin, H. Yoon and J. Jang, Catal. Commun., 2008, 10, 178–182 CrossRef CAS.
- X. Lv, Y. Xu, K. Lv and G. Zhang, J. Photochem. Photobiol., A, 2005, 173, 121–127 CrossRef CAS.
- C. Ruales-Lonfat, J. Barona, A. Sienkiewicz, M. Bensimon, J. Vélez-Colmenares, N. Benítez and C. Pulgarín, Appl. Catal., B, 2015, 166, 497–508 CrossRef.
- P. Xu, G. M. Zeng, D. L. Huang, C. L. Feng, S. Hu, M. H. Zhao, C. Lai, Z. Wei, C. Huang and G. X. Xie, Sci. Total Environ., 2012, 424, 1–10 CrossRef CAS PubMed.
- S. Rahim Pouran, A. A. Abdul Raman and W. M. A. Wan Daud, J. Cleaner Prod., 2014, 64, 24–35 CrossRef CAS.
- M. Minella, G. Marchetti, E. De Laurentiis, M. Malandrino, V. Maurino, C. Minero, D. Vione and K. Hanna, Appl. Catal., B, 2014, 154-155, 102–109 CrossRef CAS.
- P. Hu, T. Chang, W.-J. Chen, J. Deng, S.-L. Li, Y.-G. Zuo, L. Kang, F. Yang, M. Hostetter and A. A. Volinsky, J. Alloys Compd., 2019, 773, 605–611 CrossRef CAS.
- M. Munoz, Z. M. De Pedro, J. A. Casas and J. J. Rodriguez, Appl. Catal., B, 2015, 176, 249–265 CrossRef.
- C. Boxall, G. Kelsall and Z. Zhang, J. Chem. Soc., Faraday Transactions, 1996, 92, 791–802 RSC.
- L. Tan and T. Allen, Corros. Sci., 2009, 51, 2503–2507 CrossRef CAS.
-
E. C. Kroll, 1996.
- P. Mishra, S. Patnaik and K. Parida, Catal. Sci. Technol., 2019, 9, 916–941 RSC.
- Z. Zhu, Z. Lu, D. Wang, X. Tang, Y. Yan, W. Shi, Y. Wang, N. Gao, X. Yao and H. Dong, Appl. Catal., B, 2016, 182, 115–122 CrossRef CAS.
- R. Hassandoost, S. R. Pouran, A. Khataee, Y. Orooji and S. W. Joo, J. Hazard. Mater., 2019, 376, 200–211 CrossRef CAS PubMed.
- S.-a. He, W. Li, X. Wang, Q. Ma, M. Li, W. Xu, X.-c. Wang and C.-h. Zhao, Appl. Surf. Sci., 2020, 506, 144948 CrossRef CAS.
- S. Gao, C. Guo, J. Lv, Q. Wang, Y. Zhang, S. Hou, J. Gao and J. Xu, Chem. Eng. J., 2017, 307, 1055–1065 CrossRef CAS.
- S. Chidambaram, B. Pari, N. Kasi and S. Muthusamy, J. Alloys Compd., 2016, 665, 404–410 CrossRef CAS.
- L. Geng, B. Zheng, X. Wang, W. Zhang, S. Wu, M. Jia, W. Yan and G. Liu, ChemCatChem, 2016, 8, 805–811 CrossRef CAS.
- H. Jin, X. Tian, Y. Nie, Z. Zhou, C. Yang, Y. Li and L. Lu, Environ. Sci. Technol., 2017, 51, 12699–12706 CrossRef CAS PubMed.
- S. R. Pouran, A. Bayrami, A. A. Aziz, W. M. A. W. Daud and M. S. Shafeeyan, J. Mol. Liq., 2016, 222, 1076–1084 CrossRef.
- G. S. Parkinson, Z. K. Novotny, P. Jacobson, M. Schmid and U. Diebold, J. Am. Chem. Soc., 2011, 133, 12650–12655 CrossRef CAS PubMed.
- M. Rioult, D. Stanescu, E. Fonda, A. Barbier and H. L. N. Magnan, J. Phys. Chem. C, 2016, 120, 7482–7490 CrossRef CAS.
- K. Petcharoen and A. Sirivat, Mater. Sci. Eng., B, 2012, 177, 421–427 CrossRef CAS.
- R. Vijayakumar, Y. Koltypin, I. Felner and A. Gedanken, Mater. Sci. Eng., A, 2000, 286, 101–105 CrossRef.
- X. Wu, J. Tang, Y. Zhang and H. Wang, Mater. Sci. Eng., B, 2009, 157, 81–86 CrossRef CAS.
- H. Yao and Y. Ishikawa, J. Phys. Chem. C, 2015, 119, 13224–13230 CrossRef CAS.
- X. Sun, C. Zheng, F. Zhang, Y. Yang, G. Wu, A. Yu and N. Guan, J. Phys. Chem. C, 2009, 113, 16002–16008 CrossRef CAS.
- X. Sun, C. Zheng, F. Zhang, L. Li, Y. Yang, G. Wu and N. Guan, J. Phys. Chem. C, 2008, 112, 17148–17155 CrossRef CAS.
- A. Demir, R. Topkaya and A. Baykal, Polyhedron, 2013, 65, 282–287 CrossRef CAS.
- L. Hadian-Dehkordi and H. Hosseini-Monfared, Green Chem., 2016, 18, 497–507 RSC.
- S. Xuan, L. Hao, W. Jiang, X. Gong, Y. Hu and Z. Chen, Nanotechnology, 2007, 18, 035602 CrossRef PubMed.
- J. Zheng, Z. Liu, X. Zhao, M. Liu, X. Liu and W. Chu, Nanotechnology, 2012, 23, 165601 CrossRef CAS.
- T. Han, L. Qu, Z. Luo, X. Wu and D. Zhang, New J. Chem., 2014, 38, 942–948 RSC.
- Z. Luo, J. Wang, Y. Song, X. Zheng, L. Qu, Z. Wu and X. Wu, ACS Sustainable Chem. Eng., 2018, 6, 13262–13275 CrossRef CAS.
- Z. Luo, H. Tang, L. Qu, T. Han and X. Wu, CrystEngComm, 2012, 14, 5710–5713 RSC.
-
P. Muthaimanoj, S. Sain, E. Ghosh, K. J. Jenkinson, A. E. Wheatley, S. Mukhopadhyay and A. Kar, Submitted to journal.
-
L. Lutterotti, Copyright (c), 1997, 2008.
- H. Rietveld, Acta Crystallogr., 1967, 22, 151–152 CrossRef CAS.
- H. Rietveld, J. Appl. Crystallogr., 1969, 2, 65–71 CrossRef CAS.
- S. Sain, S. Patra and S. Pradhan, J. Phys. D: Appl. Phys., 2011, 44, 075101 CrossRef.
- S. Sain, A. Kar, A. Patra and S. K. Pradhan, CrystEngComm, 2014, 16, 1079–1090 RSC.
-
R. A. Young, The Rietveld method, International union of crystallography, 1993 Search PubMed.
- A. Chanda and M. De, J. Alloys Compd., 2000, 313, 104–114 CrossRef CAS.
- Y. Xu and C. H. Langford, Langmuir, 2001, 17, 897–902 CrossRef CAS.
- T. Tong, H. Zhang, J. Chen, D. Jin and J. Cheng, Catal. Commun., 2016, 87, 23–26 CrossRef CAS.
- T. Xu, R. Zhu, G. Zhu, J. Zhu, X. Liang, Y. Zhu and H. He, Appl. Catal., B, 2017, 212, 50–58 CrossRef CAS.
- T. Wu, Y. Liu, X. Zeng, T. Cui, Y. Zhao, Y. Li and G. Tong, ACS Appl. Mater. Interfaces, 2016, 8, 7370–7380 CrossRef CAS PubMed.
- Y. Qu, H. Yang, N. Yang, Y. Fan, H. Zhu and G. Zou, Mater. Lett., 2006, 60, 3548–3552 CrossRef CAS.
- B. Choudhury, P. Chetri and A. Choudhury, J. Exp. Nanosci., 2015, 10, 103–114 CrossRef CAS.
- W. Lei, Y. Liu, X. Si, J. Xu, W. Du, J. Yang, T. Zhou and J. Lin, Phys. Lett. A, 2017, 381, 314–318 CrossRef CAS.
- A. Radoń, A. Drygała, Ł. Hawełek and D. Łukowiec, Mater. Charact., 2017, 131, 148–156 CrossRef.
- M. Abboud, S. Youssef, J. Podlecki, R. Habchi, G. Germanos and A. Foucaran, Mater. Sci. Semicond. Process., 2015, 39, 641–648 CrossRef CAS.
- K. V. Shafi, A. Ulman, X. Yan, N.-L. Yang, C. Estournès, H. White and M. Rafailovich, Langmuir, 2001, 17, 5093–5097 CrossRef CAS.
- Z. Luo, J. Wang, L. Qu, J. Jia, S. Jiang, X. Zhou, X. Wu and Z. Wu, New J. Chem., 2017, 41, 12596–12603 RSC.
- S. Sathish and S. Balakumar, Mater. Chem. Phys., 2016, 173, 364–371 CrossRef CAS.
- J. A. Cuenca, K. Bugler, S. Taylor, D. Morgan, P. Williams, J. Bauer and A. Porch, J. Phys.: Condens. Matter, 2016, 28, 106002 CrossRef PubMed.
- D. Wilson and M. Langell, Appl. Surf. Sci., 2014, 303, 6–13 CrossRef CAS.
- S. Shaw, S. Alla, S. Meena, R. Mandal and N. Prasad, J. Magn. Magn. Mater., 2017, 434, 181–186 CrossRef CAS.
- Y. Liu, Y. Li, K. Jiang, G. Tong, T. Lv and W. Wu, J. Mater. Chem. C, 2016, 4, 7316–7323 RSC.
- C. Falco, N. Baccile and M.-M. Titirici, Green Chem., 2011, 13, 3273–3281 RSC.
-
F. Stacey, The physical principles of rock magnetism, Elsevier, 2012 Search PubMed.
- P. Guardia, B. Batlle-Brugal, A. Roca, O. Iglesias, M. D. P. Morales, C. Serna, A. Labarta and X. Batlle, J. Magn. Magn. Mater., 2007, 316, e756–e759 CrossRef CAS.
-
B. Cullity, Journal, 1972.
- A. Demortiere, P. Panissod, B. Pichon, G. Pourroy, D. Guillon, B. Donnio and S. Begin-Colin, Nanoscale, 2011, 3, 225–232 RSC.
- S. Xuan, Y.-X. J. Wang, J. C. Yu and K. Cham-Fai Leung, Chem. Mater., 2009, 21, 5079–5087 CrossRef CAS.
- C. Choodamani, G. Nagabhushana, B. Rudraswamy and G. Chandrappa, Mater. Lett., 2014, 116, 227–230 CrossRef CAS.
- M. Anbarasu, M. Anandan, E. Chinnasamy, V. Gopinath and K. Balamurugan, Spectrochim. Acta, Part A, 2015, 135, 536–539 CrossRef CAS PubMed.
- M. Srivastava, J. Singh, M. Yashpal, D. K. Gupta, R. Mishra, S. Tripathi and A. K. Ojha, Carbohydr. Polym., 2012, 89, 821–829 CrossRef CAS PubMed.
- G. Wang, Y. Chang, L. Wang, Z. Wei, J. Kang, L. Sang, X. Dong, G. Chen, H. Wang and M. Qi, J. Magn. Magn. Mater., 2013, 340, 57–60 CrossRef CAS.
- P. Kubelka and F. Munk, Z. Tech. Phys., 1931, 12, 259–274 Search PubMed.
- A. Manikandan, J. J. Vijaya, J. A. Mary, L. J. Kennedy and A. Dinesh, J. Ind. Eng. Chem., 2014, 20, 2077–2085 CrossRef CAS.
- M. Sadat, M. Kaveh Baghbador, A. W. Dunn, H. Wagner, R. C. Ewing, J. Zhang, H. Xu, G. M. Pauletti, D. B. Mast and D. Shi, Appl. Phys. Lett., 2014, 105, 091903 CrossRef.
- D. Shi, M. Sadat, A. W. Dunn and D. B. Mast, Nanoscale, 2015, 7, 8209–8232 RSC.
- A. Kar, J. Olszówka, S. Sain, S.-R. I. Sloman, O. Montes, A. Fernández, S. K. Pradhan and A. E. Wheatley, J. Alloys Compd., 2019, 810, 151718 CrossRef CAS.
- Z. ALOthman, Materials, 2012, 5, 2874–2902 CrossRef CAS.
- B. Kakavandi, A. Takdastan, N. Jaafarzadeh, M. Azizi, A. Mirzaei and A. Azari, J. Photochem. Photobiol., A, 2016, 314, 178–188 CrossRef CAS.
- M. Periyasamy, A. Saha, S. Sain, M. Mandal, U. Sengupta and A. Kar, J. Environ. Chem. Eng., 2020, 104604 CrossRef CAS.
- S. Guo, G. Zhang and J. Wang, J. Colloid Interface Sci., 2014, 433, 1–8 CrossRef CAS PubMed.
- Y. Feng, L. Li, M. Ge, C. Guo, J. Wang and L. Liu, ACS Appl. Mater. Interfaces, 2010, 2, 3134–3140 CrossRef CAS PubMed.
- A. Kar, S. Sain, D. Rossouw, B. R. Knappett, S. K. Pradhan and A. E. Wheatley, Nanoscale, 2016, 8, 2727–2739 RSC.
- Y. Yang, D. S. Kim, Y. Qin, A. Berger, R. Scholz, H. Kim, M. Knez and U. Gösele, J. Am. Chem. Soc., 2009, 131, 13920–13921 CrossRef CAS PubMed.
- X. S. Nguyen, G. Zhang and X. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 8900–8909 CrossRef CAS PubMed.
- X. Xue, K. Hanna, C. Despas, F. Wu and N. Deng, J. Mol. Catal., A, 2009, 311, 29–35 CrossRef CAS.
- Y. Baba, T. Yatagai, T. Harada and Y. Kawase, Chem. Eng. J., 2015, 277, 229–241 CrossRef CAS.
- Z. Luo, L. Qu, T. Han, Z. Zhang, X. Shao, X. Wu and Z. L. Chen, Eur. J. Inorg. Chem., 2014, 994–1000 CrossRef CAS.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed.
- C. B. Ong, L. Y. Ng and A. W. Mohammad, Renewable Sustainable Energy Rev., 2018, 81, 536–551 CrossRef CAS.
- Q. Liu, L. Zhou, L. Liu, J. Li, S. Wang, H. Znad and S. Liu, Composites, Part B, 2020, 200, 108345 CrossRef.
- M. Periyasamy and A. Kar, J. Mater. Chem. C, 2020, 8, 4604–4635 RSC.
- P. Mishra, S. Patnaik and K. Parida, Catal. Sci. Technol., 2019, 9, 916–941 RSC.
- T. Xu, R. Zhu, J. Zhu, X. Liang, Y. Liu, Y. Xu and H. He, Catal. Sci. Technol., 2016, 6, 4116–4123 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00228g |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *c
*c
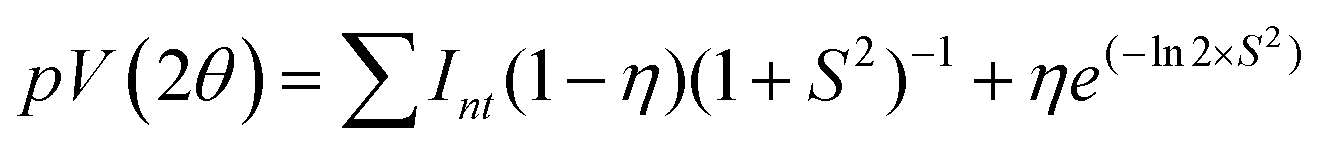
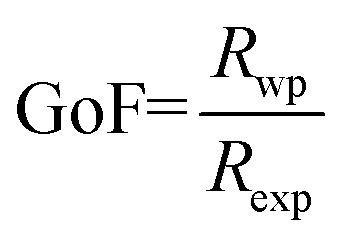
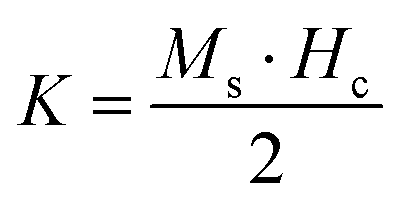 and the obtained values of the anisotropy constant are listed in Table 4. It is obvious that the values of K decrease with decreasing particle size, signifying that the presence of uniaxial anisotropy can be further ascribed to the surface effects and microstructural defects.18,40 Previous reports have established that the Ms Value of 7–22 emu g−1 is suitable for biomedical applications.78–80 Furthermore, due to its high magnetic response sensitivity, the as-synthesized Fe3O4/C nanocomposites may be used as magnetically separable catalyst for treating colored dyes and other organic pollutants in wastewater.
and the obtained values of the anisotropy constant are listed in Table 4. It is obvious that the values of K decrease with decreasing particle size, signifying that the presence of uniaxial anisotropy can be further ascribed to the surface effects and microstructural defects.18,40 Previous reports have established that the Ms Value of 7–22 emu g−1 is suitable for biomedical applications.78–80 Furthermore, due to its high magnetic response sensitivity, the as-synthesized Fe3O4/C nanocomposites may be used as magnetically separable catalyst for treating colored dyes and other organic pollutants in wastewater.