
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanostructural analysis of distinct nucleation sites in pathological mineralization
Emilio Satoshi
Hara
 *a,
Susumu
Oozawa
b,
Noriyuki
Nagaoka
c,
Masahiro
Okada
a,
Shingo
Kasahara
b and
Takuya
Matsumoto
a
*a,
Susumu
Oozawa
b,
Noriyuki
Nagaoka
c,
Masahiro
Okada
a,
Shingo
Kasahara
b and
Takuya
Matsumoto
a
aDepartment of Biomaterials, Okayama University Graduate School of Medicine, Dentistry and Pharmaceutical Sciences, 2-5-1 Shikata-cho, Kita-ku, Okayama-shi, Okayama-ken, 700-8558, Japan. E-mail: haraemilio@okayama-u.ac.jp; Fax: +81 86 235 6669; Tel: +81 86 235 6667
bDepartment of Cardiovascular Surgery, Okayama University Graduate School of Medicine, Dentistry and Pharmaceutical Sciences, Okayama, Japan
cOkayama University, Dental School, Advanced Research Center for Oral and Craniofacial Sciences, Japan
First published on 3rd June 2021
Abstract
Pathological mineralization, such as that in atherosclerosis or following transplantation of biomaterials, is associated with millions of deaths worldwide. A deeper understanding of the mechanisms underlying mineral nucleation in pathological mineralization will facilitate the development of novel bioinspired materials/techniques as therapeutics for either disease prevention or bone tissue reconstruction. The objective of this study was to perform a systematic and ultrastructural analysis of the nucleation sites of atherosclerotic mineralization from the viewpoint of material science. Histological staining, ultrastructural analysis, and selected area elemental mapping of human atherosclerotic tissues revealed two different nucleation sites in a collagenous and a lipidic area, respectively, comprising phospholipidic large vesicles (average 827 ± 470 nm) and non-vesicular phospholipids. In both sites, the initial minerals were identified to be amorphous calcium phosphate (ACP), which then transformed into hydroxyapatite (HAp). Mineralization assays in vitro was performed by incubating non-vesicular low- (LDL) and high- (HDL) density lipoprotein and cholesterol in 2 mM CaCl2 for 2 days, without additional phosphate. Calcium ions in the formed minerals, measured using atomic absorption (AA) spectroscopy, showed that only LDL was mineralized. We then evaluated the mineralization of the major phospholipids, phosphatidylserine (PS) and phosphatidylcholine (PC) under the same conditions. The results of AA and mineral dry weight showed that PS was more abundantly mineralized than PC, most likely due to its higher hydrolysis rate. Similar to the in vivo findings, PS mineralization initially formed ACP, which then crystallized into HAp. Taken together, these results indicated that atherosclerosis involves two distinct nucleation sites and that non-vesicular phospholipids were one of the major nucleation sites for atherosclerotic mineral formation.
1. Introduction
Biomineralization is the process through which living organisms form mineralized materials such as bones and teeth with extraordinary mechanical properties. In physiological mineralization of bones and teeth, the nucleation sites, the formation of initial amorphous calcium phosphate (ACP) and its transformation into apatite have been largely studied.1,2 However, there are only few studies evaluating the initial mineralization process of pathological mineralization from a material science perspective.Pathological mineralization can occur spontaneously (e.g., atherosclerosis) or be induced by transplantation of biomaterials.3 In particular, atherosclerosis is a pathological mineralization in the coronary arteries involved in several diseases including stroke, which causes millions of deaths worldwide annually.4–6 Numerous studies have associated atherosclerotic mineralization with genetic factors, cell activities (e.g., differentiation, apoptosis) and cell-released factors, microenvironmental factors (e.g., extracellular matrix composition, pH), and systemic signals.4,7–12 Previous studies have suggested that the process of atherosclerotic mineralization would be similar to that of bone formation, where cardiovascular cells (e.g., vascular smooth muscle cells and valve interstitial cells) differentiate into osteoblast-like cells, and subsequently secrete extracellular vesicles (also known as matrix vesicles (MVs) of <200 nm) that act as the nucleation site for mineral deposition in the vascular tissues.4,11,13,14 Macrophages (also referred to as foam cells when they contain high levels of low-density lipids and cholesterol), and macrophage-derived MVs have also been associated with atherosclerotic mineralization.15,16
In addition to the cellular and molecular analyses of the mechanisms underlying atherosclerosis development, analysis of the chemical and molecular mechanisms of mineral nucleation sites and crystal growth from the perspective of material science is crucial for a more integrative understanding of the mineralization process in atherosclerotic pathologies.9 For instance, findings of high-resolution microcomputed tomography have shown that the size of atherosclerotic microcalcifications is usually ∼15 μm.17 Nanostructure studies of atherosclerotic tissues have revealed that crystalline hydroxyapatite spheres, associated with atherosclerosis, differ structurally from bone apatite.9 However, the structure of nucleation sites during atherosclerotic tissue mineralization and mineral maturation has remained obscure.
Previously, we have performed a systematic analysis of the very initial mineral formation site in physiological mineralization during intramembranous and endochondral ossification.2,18 The results revealed the distinct nucleation sites in these processes. Intramembranous ossification starts inside MVs secreted by osteoblasts, whereas endochondral ossification starts from chondrocyte membrane nanofragments as nucleation sites.
In this study, the same systematic analysis of the nucleation sites in human atherosclerotic specimens was performed with scanning electron microscopy with energy-dispersive X-ray spectroscopy (SEM-EDS) and scanning-transmission electron microscopy (STEM) with EDS. We identified mineralization in collagenous and lipidic areas, with micrometer-sized phospholipidic large vesicles and non-vesicular phospholipids, respectively, as distinct nucleation sites for mineral formation. Moreover, atomic absorption (AA) spectroscopy revealed that low- (LDH) and high-density lipoproteins (HDL), cholesterol, phosphatidylserine (PS), and phosphatidylcholine (PC) had different mineralization abilities in vitro.
2. Results
2.1. Histomorphological analysis of the mineralized atherosclerotic tissue
Cross-sections of fresh human atherosclerotic tissues embedded in resin were histologically stained with hematoxylin and eosin (HE) and Villanueva–Goldner (VG) stain to detect calcified tissues. Fig. 1A and B show extensive mineralized areas throughout the outer collagenous layer of smooth muscle cells and another mineralized area in adipose tissue below the collagenous layer visualized as intense green VG staining.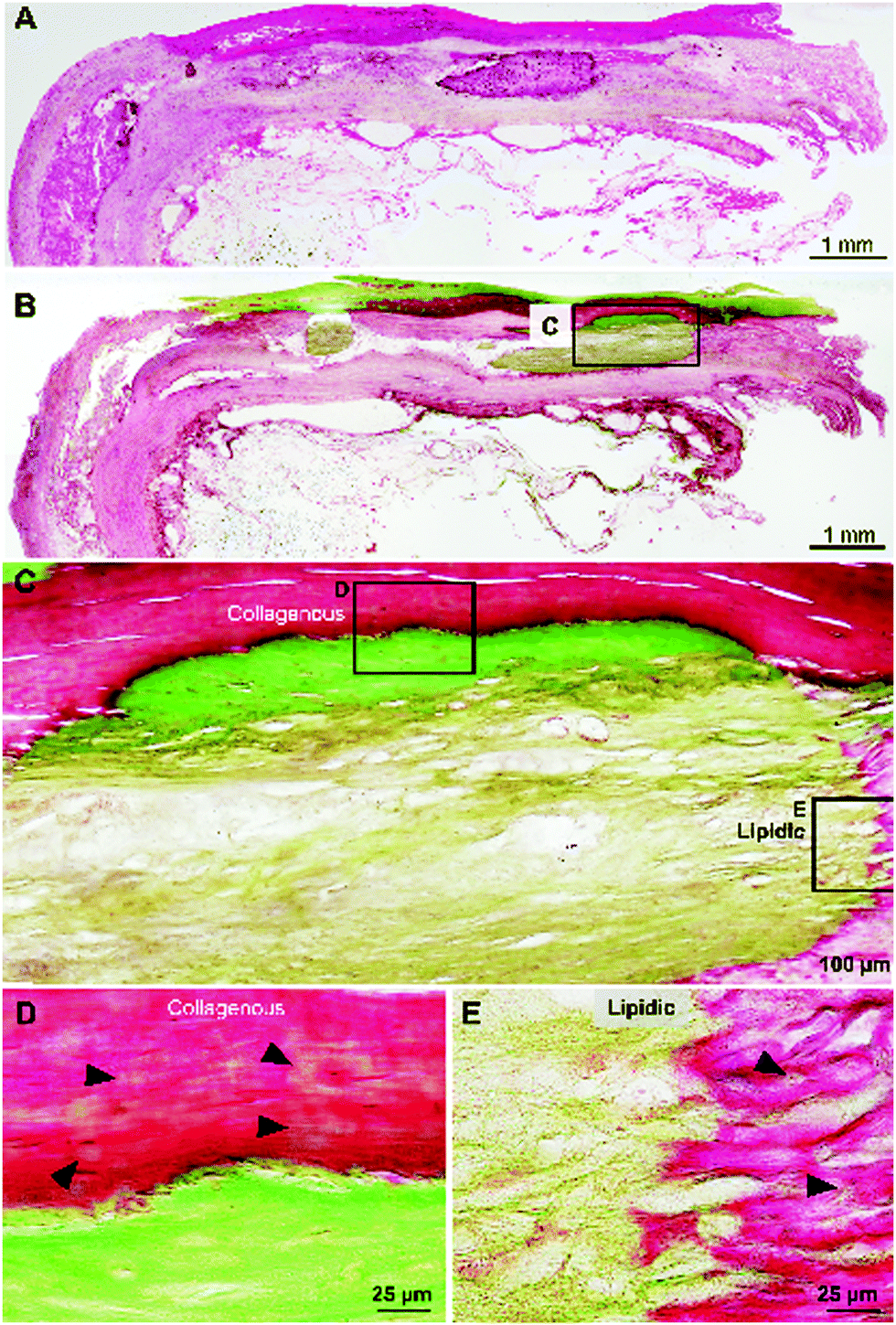 | ||
| Fig. 1 Histological analysis of atherosclerotic calcification. (A and B) Low magnification of HE- and VG-stained calcification site. (C) Higher magnification of box in (B). Calcification sites differ between collagenous and lipidic areas. Higher magnification of calcified collagenous (D) and lipidic (E) areas. Arrowheads in (D and E) indicate initial mineral formation around calcified areas stained green with VG. HE, hematoxylin and eosin; VG, Villanueva–Goldner. | ||
At a higher magnification (Fig. 1C–E), the differences in the mineralized collagenous (Fig. 1D) and adipose (Fig. 1E) tissues are evident, suggesting two different types of mineralization processes in atherosclerosis. Importantly, note the initial mineralization sites (Fig. 1D and E arrowheads) in the surroundings of the densely calcified area.
2.2. Structural analysis of the mineralized atherosclerotic tissue
Minerals were ultrastructurally analyzed using SEM-EDS and STEM-EDS. Fig. 2A–C shows microcalcifications in the dense collagenous area as a stratified region corresponding to the area shown in Fig. 1D. The average diameter of the calcified spheres was 870 ± 470 nm (range, 500–2200 nm, Fig. 2G), which is considerably larger than the osteoblast-secreted MVs (<200 nm). The STEM findings revealed spheres of minerals with clear surrounding boundaries that initially formed in the collagen area (Fig. 3–5). | ||
| Fig. 2 Field emission (FE)-SEM images of the initially formed minerals in atherosclerotic calcification. (A–F) FE-SEM images taken by backscattered electron imaging (BEI) mode of the (A–C) collagenous and (D–F) lipidic calcification sites. (A and D) Low magnification images. White areas in the top-left indicate the mature mineral blocks. (B and E) Higher magnification images of the red squares in (A and B), respectively. (C and F) SEM images taken by secondary electron imaging (SEI) mode of the minerals after NaClO treatment. Minerals show high sphericity with a smooth surface and irregular shapes and surfaces in the collagenous and lipidic areas, respectively. (G) Diameter of minerals formed in collagenous area inside vesicles and in lipidic environment. High standard deviation of minerals in lipidic environment indicates mineral heterogeneity. (H) Frequency of minerals according to size. High frequency of minerals at 200–500 and ≥2300 nm were responsible for large standard deviation of mineral size in lipidic area (G). | ||
 | ||
| Fig. 3 STEM HAADF images of atherosclerotic calcification and elemental mapping. Different nucleation sites in (A–D) collagenous and (E–H) lipidic calcification sites. Arrowheads (A) indicate collagen fibers. Red dotted lines show the selected area for elemental (Ca and P) analysis. Elemental map shows levels of (B and F) calcium, (E and F) phosphorus, and (D and H) osmium. Osmium binds to carbon double bonds in lipids and thus indirectly indicate lipid levels. Small high-contrast areas in lipidic regions with abundant phosphorus and osmium (phospholipids) are not yet mineralized. | ||
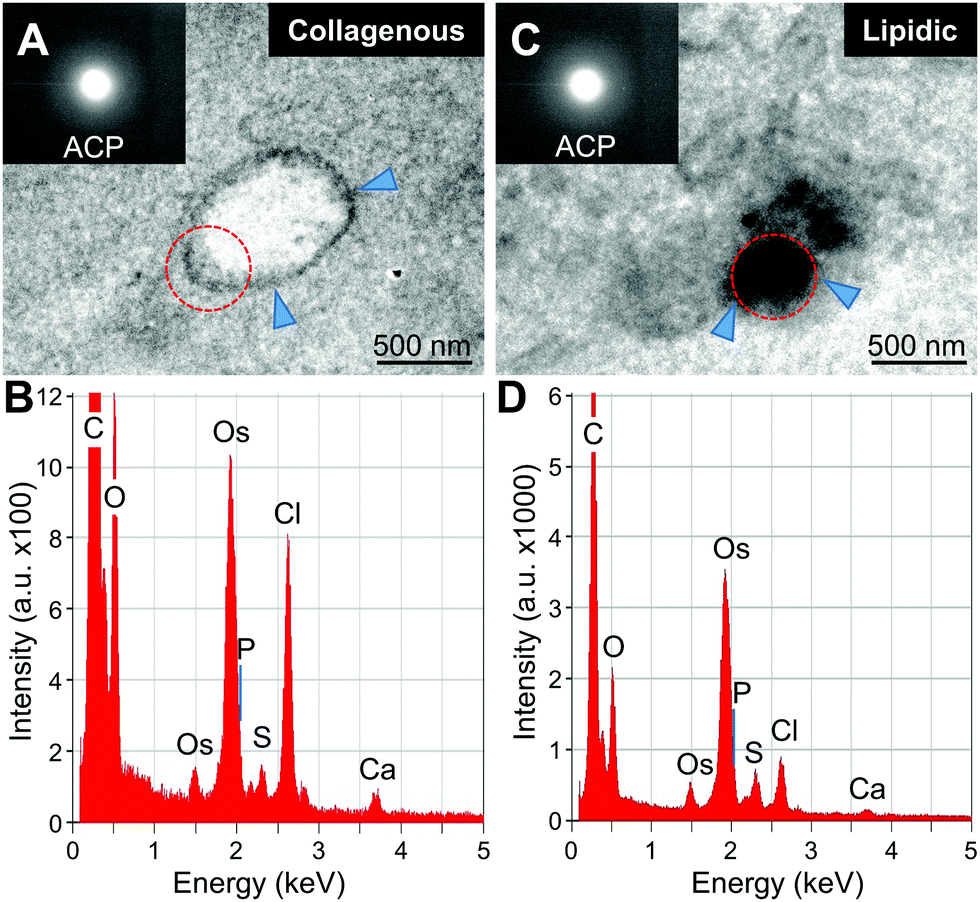 | ||
| Fig. 4 STEM-EDS and TEM/electron diffraction analyses of the initial minerals. (A and C) STEM images of the initial minerals inside large vesicles and in the lipidic matrix. The minerals apparently start to form inside the vesicles in the collagenous area (A), but randomly in the lipidic area (C). Insets show the diffuse electron diffraction pattern of the minerals indicating the presence of amorphous calcium phosphate (ACP). (B and D) EDS analysis confirmed the presence of calcium (Ca) and phosphorus (P) in the initially mineralized area (arrowheads). Red dotted lines show the selected area for EDS analysis. | ||
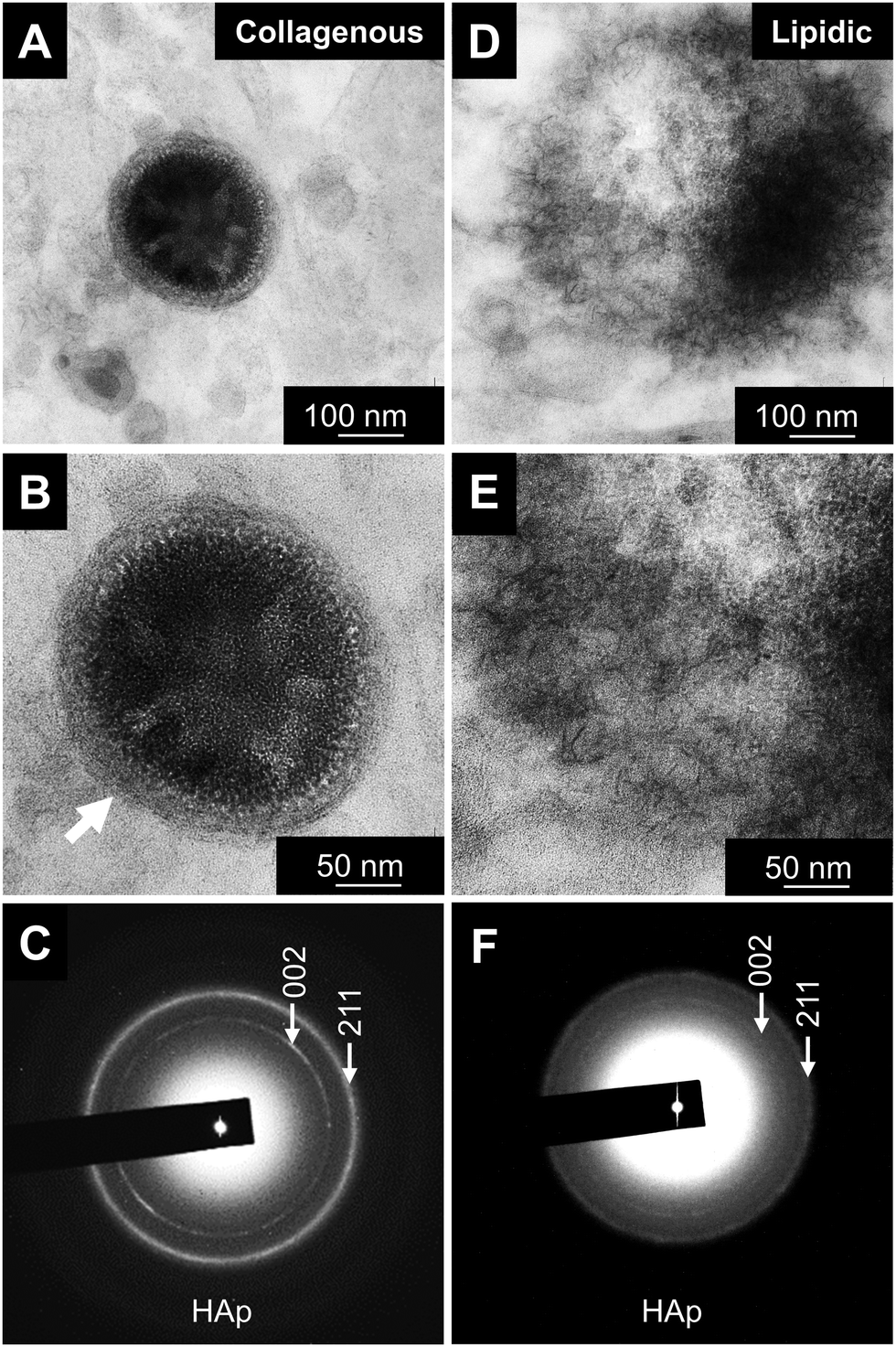 | ||
| Fig. 5 Electron diffraction analysis of atherosclerotic minerals. Nucleation sites in (A and B) collagenous and (D and E) lipidic calcification sites visualized by TEM. (B) and (E) are higher magnification of (A) and (D), respectively. Arrows, lipidic layer of large vesicles in area of collagenous calcification. (C and F) Electron diffraction of areas shown in (B) and (E), respectively. Vesicle boundaries are absent in lipidic calcification sites, where minerals might freely grow. Selected area elemental mapping identified the minerals as hydroxyapatite (HAp). | ||
In contrast, minerals that formed in adipose tissues had irregular surfaces and were surrounded by sparse adipose tissue (Fig. 2D and E), like the histological findings (Fig. 1E). The average diameter of the calcified spheres was 1215 ± 980 nm (range, 250 to 3500 nm, Fig. 2G). This suggested rapid, random mineral formation and unrestrained growth occurs in the lipidic environment compared with that inside vesicles.
2.3. Elemental analysis of nucleation sites
Minerals were analyzed in more detail using STEM-EDS (Fig. 3). In the collagenous area, the minerals begin to form inside the large vesicles, which are surrounded by large amounts of collagen fibers. The high intensity of phosphorus and osmium, which binds to carbon double-bounds, would represent the phospholipid composition of the vesicles (Fig. 3C and D). That is, the vesicles were nucleation sites for mineral formation in the collagen-rich area.In contrast, adipose tissues did not contain vesicles (Fig. 3E–H). Instead, the minerals appeared to have randomly formed in the lipid-sparse tissue. Small areas of high contrast in high-angle annular dark-field (HAADF) images represent initial mineralization sites where levels of calcium were still low or undetectable. Their presence suggest that they are likely to continue to mineralize and grow during subsequent stages (Fig. 3F). Abundant phosphorus and osmium suggested that the mineralizing matrix is rich in phospholipids meaning that non-vesicular phospholipids are the nucleation sites for mineral formation in the lipidic area.
Qualitative analysis of minerals was then performed with STEM-EDS and transmission electron microscopy (TEM) and electron diffraction (Fig. 4 and 5). The initial minerals grown both inside the vesicles and in the lipidic matrix produced diffuse rings in the diffraction pattern, indicating the presence of amorphous calcium phosphate (ACP, Fig. 4A and C, insets). Apparently, the minerals start to form in the inner part of the large vesicles. On the other hand, the minerals in the lipidic area nucleate randomly. EDS-based elemental analysis confirmed the presence of calcium and phosphorus (Fig. 4B–D). Further analysis of minerals at a more mature stage indicated the presence of crystalline hydroxyapatite (HAp, Fig. 5). The results were consistent with the semi-quantitative analysis of Ca/P molar ratio, which were 1.5 and 1.35 for the initial HAp found in collagenous and lipidic areas, respectively. These results also suggest that ACP could be the precursor for HAp formation in atherosclerotic mineralization. Of note, the minerals inside the vesicles in the collagenous area presented a denser and more compact structure compared to the counterparts in the lipidic area (Fig. 5A and D).
2.4. Analysis of phospholipid mineralization in vitro
Since in vitro reproduction of mineralization in large vesicles containing lipoprotein components (e.g., apolipoproteins, triglycerides) is challenging, this study aimed to clarify the mineralization of non-vesicular phospholipids. We firstly assayed mineralization in vitro using LDL, HDL, and cholesterol by incubating the molecules in 2 mM CaCl2 for 2 days, without additional phosphates. LDL and HDL is known to contain high amounts of phospholipids, while cholesterol itself has no phosphate in its structure. Consistently, Fig. 6A and B shows that LDL mineralized considerably more than HDL, while cholesterol did not.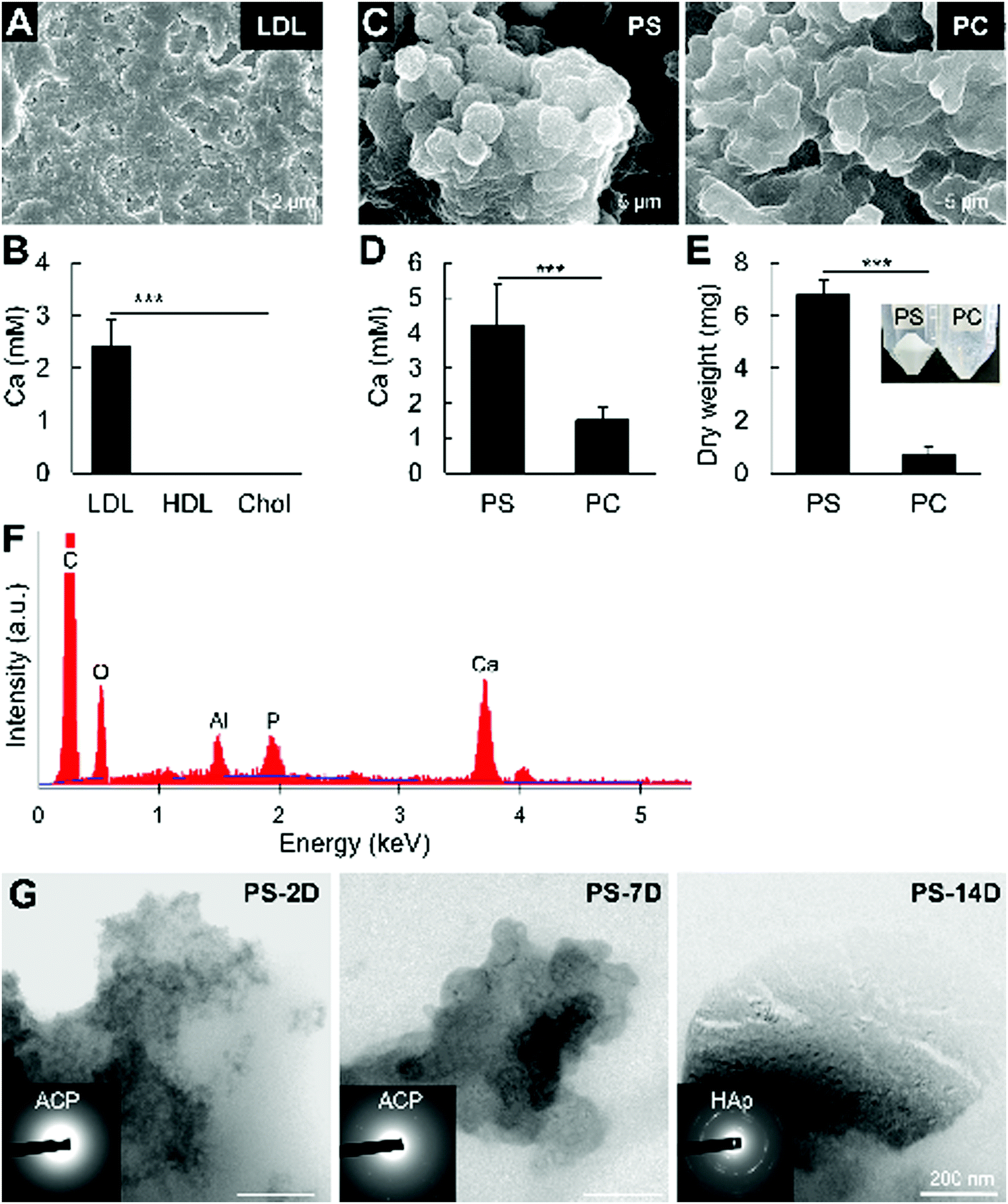 | ||
| Fig. 6 Mineralization of LDL, HDL, cholesterol, and phospholipids in vitro. (A and C) Scanning electron micrographs of minerals formed by calcification of LDL, PS and PC. (B and D) Quantified calcium in the minerals formed from (B) LDL, HDL, and cholesterol (Chol), (D) PS and PC after incubation with 2 mM CaCl2 for 2 days. Mineralization of LDL is obvious, whereas HDL and cholesterol did not mineralize. PS is more abundantly mineralized than PC. (E) Dry weight of the minerals formed by PS and PC calcification. Inset shows the formed minerals after PS and PC calcification. (F) Representative EDS analysis of the minerals formed from PS and PC calcification, after mineral treatment with NaClO. (G) TEM photographs and electron diffraction analysis (insets) of the minerals formed by PS calcification after 2 (2D), 7 (7D) and 14 days (14D) of incubation in 2 mM CaCl2. For B, ***p < 0.001, one-way ANOVA, Tukey test. For (D and E), ***p < 0.001, Students’ t-test. | ||
Low-density lipoproteins mainly comprise cholesterol, phospholipids and lipoproteins. Therefore, we analyzed the mineralization of PS and PC, which have been identified in LDL samples19 and are the major phospholipid components of the cellular membrane. Although both phospholipids mineralized, PS showed a more prevalent mineralization compared to PC, as measured by AA and dry weight (Fig. 6C–E). The presence of calcium and phosphate was confirmed by EDS analysis of the minerals after NaClO treatment (Fig. 6F). Similar to the in vivo findings, electron diffraction analysis indicated that the minerals formed from PS calcification were initially ACP, which then crystallized into HAp after 14 days of incubation in 2 mM CaCl2(Fig. 6G). These results indicate that non-vesicular phospholipids mineralize rapidly, and strongly suggest their involvement in atherosclerotic mineralization.
3. Discussion
Atherosclerotic mineralization involves a complex sequence of events that leads to the irreversible damage and deterioration of the arteries and cardiac valves. A deeper understanding of the mechanisms underlying mineral formation in atherosclerotic tissues from a material science perspective is therefore critically important to determine the nucleation sites as well as the processes of mineral growth and maturation, and therefore, to accelerate the development of novel methods and techniques to prevent and treat atherosclerosis. By understanding the process of pathological mineralization, new insights can also be obtained for development of bioinspired materials for bone tissue reconstruction.We previously analyzed the initial stages of mineral formation during intramembranous and endochondral ossification from biological and material science perspectives.2,18 The results of the systematic and multidisciplinary approaches revealed different nucleation sites for bone formation. Initial mineralization in mouse calvaria starts on embryonic day 14 with MVs secreted from cells as nucleation sites for intramembranous ossification.18 MVs are <200 nm in diameter, rich in mineralizing factors (e.g., annexin V) and are secreted by osteoblasts via exocytosis.20 Initial mineralization in the mouse femur epiphysis was shown to occur at postnatal day 6. In this process, instead of matrix vesicles, we found that chondrocyte burst-derived cell membrane nanofragments21 were the nucleation site for mineral formation. In fact, cell membrane nanofragments isolated from cultured cells mineralized within 1 day, whereas live cells required 14 days to mineralize.22 These findings differed from the notion that minerals precipitate and grow in gaps between collagen fibrils. By using such a multidisciplinary approach, a deeper understanding of the mechanisms of bone formation could be achieved.
Here, we clarified distinct nucleation sites of mineral formation in a collagenous and lipidic microenvironment of atherosclerotic human tissues. Vesicles found in the collagenous environment were remarkably larger than the MVs secreted by osteoblasts (870 nm vs. <200 nm, respectively), but within the size range of chylomicrons (>800 nm)23 and apoptotic bodies (≈1000 nm).24 A previous study also found that circulating plasma vesicles isolated from stroke patients were approximately 1105 ± 240 nm.25 Of note, no cells were located around the large vesicles in the collagenous area indicating that the origins differed between these vesicles and the small MVs secreted by osteoblasts during bone formation.20
Lipids are known to play critical roles as nucleation sites for mineral formation in atherosclerosis. Others have suggested that circulating oxidized lipids carry mineralizing factors which would then facilitate mineralization in atherosclerotic regions.26,27 However, the exact process of oxidized lipid or lipoprotein mineralization in vitro has not been confirmed. In addition to circulating oxidized lipids, free DNA in mineralization sites remaining after cell apoptosis and lysis can precipitate calcium phosphate crystals in arteries.28 In addition to DNA, cell fragments could be sites of mineral formation in vitro.29 Cell fragments obtained by different methods were shown to mineralize within 2 days.29 The activity of alkaline phosphatase, which is known to be a major phosphatase associated with bone formation, immediately increased after cell lysis, indicating that it could be also the main enzyme involved in the mineralization of cell fragments.29 Plasma membrane fragments were shown to be particularly important sites for mineral nucleation during initial bone formation.2,22
Qualitative analysis of the minerals identified the initial formation of ACP, which then transformed into HAp inside the vesicles and in the lipidic matrix. These findings are consistent with previous report detecting the presence of ACP in bones2 and atherosclerotic minerals.18,30 The minerals were larger in the lipidic than in the collagenous area, possibly because phospholipid mineralization does not involve vesicles, which could restrict mineral growth. A microenvironment that is rich in dense collagen fibers could be another factor limiting mineral growth.
Furthermore, environmental pH critically affects the fibrillogenesis of collagen as well as mineral formation and growth. Collagen molecules form fibrils with diameters of 85 and ∼200 nm at pH 6.6 and 6.9–8.0, respectively.31 The rate of fibrillogenesis also increases with increasing pH within this range. Environmental pH also affects ion dissolution and thus calcium phosphate nucleation. Long minerals should form at acidic pH whereas spherical minerals are more prominent at alkaline pH due to the high precipitability of Ca and P ions and isotropic or weak-anisotropic crystal growth.22 Although in this study, the microenvironmental conditions could not be analyzed in detail, further systematic evaluation of these factors is crucial to reveal the mechanisms of atherosclerosis development.
In this study, we compared the in vitro mineralization of major lipidic materials associated with atherosclerosis development, and found that only LDL has elements that mineralize in vitro within 2 days in a solution without phosphate ions. We compared the mineralization of the LDL lipidic components, i.e., cholesterol, and the phospholipids, PS and PC. Cholesterol should not mineralize due to the absence of a phosphate group, whereas phospholipid hydrolysis should facilitate calcium phosphate reactions and the formation of calcified material.32 In fact, mineralization assays clarified, for the first time, the higher mineralization ability of PS compared to PC. Previous studies have also demonstrated the mineralization of PS in vesicles or micelles.33,34 However, in these studies, PS was incubated in solutions rich in calcium and phosphate ions, which only indirectly indicates the role of PS as a nucleation site but does not show its direct role as a mineralizing agent. Moreover, these studies failed to characterize the minerals formed from PS. Herein, we demonstrated that the phosphate group of PS directly participate in the formation of ACP, which then transformed into HAp after 14 days of incubation. The likely faster hydrolysis rate of PS could be the major factor associated with this finding. Meanwhile, the higher percentage of PC in the phospholipid fraction of HDL19,35 could be one possible reasoning associated with its lower mineralization compared to LDL.
In conclusion, this study showed that large vesicles and phospholipids are distinct nucleation sites in collagenous and lipidic environments. Phospholipids are important factors in pathological mineralization. PS mineralized more abundantly than PC.
4. Materials and methods
4.1. Sample preparation
Human samples were obtained from patients undergoing surgical aortic valve replacement at Okayama University Hospital, under the approval of the Ethics Committee of Okayama University Hospital (RIN 1909-015). Samples were immediately fixed in 10% formalin for histological and ultrastructural analyses.Formalin-fixed samples were first scanned using micro-computed tomography (micro-CT, SkyScan 1174, SkyScan, Aartselaar, Belgium) at a resolution of 6.5 μm to detect mineralized areas. Mineralized tissues were cut into 2 mm2 slices, dehydrated with a graded series of ethanol, acetone, and ethylene oxide. The tissues were then embedded in a methyl methacrylate resin. Specimens were then sliced into ultrathin sections (∼200 μm) and polished to a thickness of 30–40 μm using a micro-grinder before staining with HE and VG to detect calcified tissue. Resin embedding and staining procedures were processed by KAC Co., Ltd (Kyoto, Japan).
Alternatively, the mineralized tissues were embedded in EPON 812 resin, as described.2,22 Briefly, specimens were washed with phosphate-buffered saline and fixed with 3% potassium ferrocyanide and 2% aqueous osmium tetroxide for 1 h on ice. The specimens were then washed thoroughly for 15 min and incubated with fresh 1% thiocarbohydrazide for 20 min at room temperature. The specimens were washed thoroughly for 15 min, incubated with 1% osmium for 30 min at room temperature, then washed again for 15 min. The specimens were dehydrated using a sequence of ethanol and acetone, then embedded in EPON 812 resin. The specimens were cross-sectioned and polished by argon ion etching before ultrastructural analysis using SEM, TEM, and EDS.
For analysis of the crystal ultrastructure, the formalin-fixed samples were submitted to NaClO treatment for elimination of organic components for at least 12 h, at room temperature. The remaining inorganic crystals were then washed thoroughly with MilliQ pure water, dehydrated with ethanol and dried at room temperature, coated with osmium (Neoc-Pro, Meiwafosis Co. Ltd, Tokyo, Japan), before observation by an SEM.
4.2. Electron microscopy, EDS interfacial analysis and electron diffraction
Resin-embedded specimens were polished by an argon ion etching (SM-090101 Cross Section Polisher; JEOL, Tokyo, Japan) and observed by a field emission-SEM (FE-SEM; JSM-6701F, JEOL) operated at 5 kV using an annular semiconductor detector, as reported.2,36 Images were taken by backscattered electron imaging (BEI) mode.After FE-SEM observation, the resin-embedded specimens were sectioned in slices of 80 nm to 100 nm in thickness with a diamond knife, and coated with carbon before observation in a scanning transmission electron microscope (STEM; JEM-2100F, JEOL) equipped with a probe Cs-corrector (CEOS, Heidelberg, Germany) and an EDS (JEM-2300T, JEOL), as described previously.36
STEM-EDS analysis was performed using a STEM operated at 200 kV using a current density of 40 pA cm−2. The probe forming Cs corrector enabled sub-Å resolution STEM imaging (minimum probe size: 0.09 nm). For EDS, an electron spot with a 0.3 nm diameter was applied. A drift correction was performed each minute to avoid any possible drifts that might have occurred at the nano-scale during the acquisition of STEM-EDS multi-elemental mapping. High-angle annular dark-field (HAADF) images were taken with a 167–228 mrad detector. The same microscope was also operated without the scanning mode.
The standardless quantification of Ca/P molar ratio was performed after corrections for atomic number effects (Z), absorption (A) and fluorescence (F).
4.3. Mineralization assays in vitro
Human plasma-derived LDL (molecular weight: 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 750000; total cholesterol ≥4000 mg dL−1) and HDL (molecular weight: 175000–360000; total cholesterol ≥1500 mg dL−1) were purchased from Lee BioSolutions (Maryland Heights, MO, USA). Cholesterol and phosphatidylcholine (PC) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Phosphatidylserine (PS) was purchased from Carbosynth Ltd (Berkshire, United Kingdom). Cholesterol was dissolved in chloroform to a final concentration of 30 μM. PS and PC were dissolved in chloroform to a final concentration of 100 mM.
750000; total cholesterol ≥4000 mg dL−1) and HDL (molecular weight: 175000–360000; total cholesterol ≥1500 mg dL−1) were purchased from Lee BioSolutions (Maryland Heights, MO, USA). Cholesterol and phosphatidylcholine (PC) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Phosphatidylserine (PS) was purchased from Carbosynth Ltd (Berkshire, United Kingdom). Cholesterol was dissolved in chloroform to a final concentration of 30 μM. PS and PC were dissolved in chloroform to a final concentration of 100 mM.
Cholesterol, LDL, and HDL (∼30 μM) were spread on glass-bottomed dishes, dried, then incubated in 2 mM CaCl2 for 2 days at 37 °C under a 5% CO2, humidified atmosphere. Samples were collected in 1.5 mL tubes and washed using a benchtop centrifuge at 12000 rpm for 5 min. Precipitated minerals were suspended in 0.1 M HNO3; calcium ions were then quantified using a simultaneous multi-element analytical AA spectrophotometer (AA, Hitachi Z-9000, Tokyo, Japan). The mineralization of PS and PC (100 mM each) was likewise analyzed.
The mineralization of PS and PC was also assessed by direct measurement of the dry weight of the formed minerals. The two phospholipids (PS and PC, 100 mM, 200 μL) were spread in 10 cm glass dishes, left to dry, and incubated in 2 mM CaCl2 (20 mL) for 2 days. The mineralized PS and PC were then collected in a centrifuge tube and treated with NaClO for at least 4 h for lipid degradation. The dispersion was then centrifuged at 10000 × g for 10 min for isolation of the minerals. The minerals were centrifugally-washed (10000 × g) twice with water and 100% ethanol, and dried in vacuum. The dry weight of the minerals was measured using a benchtop scale.
4.4. Image analysis
Analysis of mineral sizes was performed with ImageJ (NIH, Bethesda, MD, USA) based on at least 65 images taken by SEM and STEM. The raw data were used to draw graphs with the mean ± standard deviation and frequency distribution. Trend lines in the frequency distribution graphs were defined by a polynomial of degree 4.4.5. Statistical analysis
Analysis of the differences between groups was performed with unpaired Students’ t-test or one-way analysis of variance (ANOVA) followed by a Tukey post hoc correction test, when appropriate. Prism 5 software (GraphPad Software, La Jolla, CA, USA) was used for the analyses. The level of significance was set as: *p < 0.05, **p < 0.01, ***p < 0.001.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research was supported by JSPS Grants-in-Aid for Scientific Research Grant Number JP20H04534.References
- R. E. Wuthier and G. F. Lipscomb, Front. Biosci., 2011, 16, 2812–2902 Search PubMed.
- E. S. Hara, M. Okada, N. Nagaoka, T. Hattori, T. Kuboki, T. Nakano and T. Matsumoto, ACS Biomater. Sci. Eng., 2018, 4, 617–625 Search PubMed.
- F. J. Schoen, H. Harasaki, K. M. Kim, H. C. Anderson and R. J. Levy, J. Biomed. Mater. Res., 1988, 22, 11–36 Search PubMed.
- A. L. Durham, M. Y. Speer, M. Scatena, C. M. Giachelli and C. M. Shanahan, Cardiovasc. Res., 2018, 114, 590–600 Search PubMed.
- P. Libby, J. E. Buring, L. Badimon, G. K. Hansson, J. Deanfield, M. S. Bittencourt, L. Tokgozoglu and E. F. Lewis, Nat. Rev. Dis. Primers, 2019, 5, 56 Search PubMed.
- C. O. Johnson, M. Nguyen, G. A. Roth, E. Nichols, T. Alam, D. Abate, F. Abd-Allah, A. Abdelalim, H. N. Abraha, N. M. Abu-Rmeileh, O. M. Adebayo, A. M. Adeoye, G. Agarwal, S. Agrawal, A. N. Aichour, I. Aichour, M. T. E. Aichour, F. Alahdab, R. Ali, N. Alvis-Guzman, N. H. Anber, M. Anjomshoa, J. Arabloo, A. Arauz, J. Arnlov, A. Arora, A. Awasthi, M. Banach, M. A. Barboza, S. L. Barker-Collo, T. W. Barnighausen, S. Basu, A. B. Belachew, Y. M. Belayneh, D. A. Bennett, I. M. Bensenor, K. Bhattacharyya, B. Biadgo, A. Bijani, B. Bikbov, M. S. Bin Sayeed, Z. A. Butt, L. Cahuana-Hurtado, J. J. Carrero, F. Carvalho, C. A. Castaneda-Orjuela, F. Castro, F. Catala-Lopez, Y. Chaiah, P. P. C. Chiang, J. Y. J. Choi, H. Christensen, D. T. Chu, M. Cortinovis, A. A. M. Damasceno, L. Dandona, R. Dandona, A. Daryani, K. Davletov, B. De Courten, V. De la Cruz-Gongora, M. G. Degefa, S. D. Dharmaratne, D. Diaz, M. Dubey, E. E. Duken, D. Edessa, M. Endres, E. J. A. Faraon, F. Farzadfar, E. Fernandes, F. Fischer, L. S. Flor, M. Ganji, A. K. Gebre, T. G. Gebremichael, B. Geta, K. E. Gezae, P. S. Gill, E. V. Gnedovskaya, H. Gomez-Dantes, A. C. Goulart, G. Grosso, Y. M. Guo, R. Gupta, A. Haj-Mirzaian, A. Haj-Mirzaian, S. Hamidi, G. J. Hankey, H. Y. Hassen, S. I. Hay, M. I. Hegazy, B. Heidari, N. A. Herial, M. A. Hosseini, S. Hostiuc, S. S. N. Irvani, S. M. S. Islam, N. Jahanmehr, M. Javanbakht, R. P. Jha, J. B. Jonas, J. J. Jozwiak, M. Jurisson, A. Kahsay, R. Kalani, Y. Kalkonde, T. A. Kamil, T. Kanchan, A. Karch, N. Karimi, H. Karimi-Sari, A. Kasaeian, T. D. Kassa, H. Kazemeini, A. T. Kefale, Y. S. Khader, I. A. Khalil, E. A. Khan, Y. H. Khang, J. Khubchandani, D. Kim, Y. J. Kim, A. Kisa, M. Kivimaki, A. Koyanagi, R. K. Krishnamurthi, G. A. Kumar, A. Lafranconi, S. Lewington, S. S. Li, W. D. Lo, A. D. Lopez, S. Lorkowski, P. A. Lotufo, M. T. Mackay, M. Majdan, R. Majdzadeh, A. Majeed, R. Malekzadeh, N. Manafi, M. A. Mansournia, M. M. Mehndiratta, V. Mehta, G. Mengistu, A. Meretoja, T. J. Meretoja, B. Miazgowski, T. Miazgowski, T. R. Miller, E. M. Mirrakhimov, B. Mohajer, Y. Mohammad, M. Mohammadoo-Khorasani, S. Mohammed, F. Mohebi, A. H. Mokdad, Y. Mokhayeri, G. Moradi, L. Morawska, I. M. Velasquez, S. M. Mousavi, O. S. S. Muhammed, W. Muruet, M. Naderi, M. Naghavi, G. Naik, B. R. Nascimento, R. I. Negoi, C. T. Nguyen, L. H. Nguyen, Y. L. Nirayo, B. Norrving, J. J. Noubiap, R. Ofori-Asenso, F. A. Ogbo, A. T. Olagunju, T. O. Olagunju, M. O. Owolabi, J. D. Pandian, S. Patel, N. Perico, M. A. Piradov, S. Polinder, M. J. Postma, H. Poustchi, V. Prakash, M. Qorbani, A. Rafiei, F. Rahim, K. Rahimi, V. Rahimi-Movaghar, M. Rahman, M. A. Rahman, C. Reis, G. Remuzzi, A. M. N. Renzaho, S. Ricci, N. L. S. Roberts, S. R. Robinson, L. Roever, G. Roshandel, P. Sabbagh, H. Safari, S. Safari, S. Safiri, A. Sahebkar, S. S. Zahabi, A. M. Samy, P. Santalucia, I. S. Santos, J. V. Santos, M. M. S. Milicevic, B. Sartorius, A. R. Sawant, A. E. Schutte, S. G. Sepanlou, A. Shafieesabet, M. A. Shaikh, M. Shams-Beyranvand, A. Sheikh, K. N. Sheth, K. Shibuya, M. Shigematsu, M. J. Shin, I. Shiue, S. Siabani, B. H. Sobaih, L. A. Sposato, I. Sutradhar, P. A. Sylaja, C. E. I. Szoeke, B. J. T. Ao, M. H. Temsah, O. Temsah, A. G. Thrift, M. Tonelli, R. Topor-Madry, B. X. Tran, K. B. Tran, T. C. Truelsen, A. G. Tsadik, I. Ullah, O. A. Uthman, M. Vaduganathan, P. R. Valdez, T. J. Vasankari, R. Vasanthan, N. Venketasubramanian, K. Vosoughi, G. T. Vu, Y. Waheed, E. Weiderpass, K. G. Weldegwergs, R. Westerman, C. D. A. Wolfe, D. Z. Wondafrash, G. L. Xu, A. Yadollahpour, T. Yamada, H. Yatsuya, E. M. Yimer, N. Yonemoto, M. Yousefifard, C. H. Yu, Z. Zaidi, M. Zamani, A. Zarghi, Y. Q. Zhang, S. Zodpey, V. L. Feigin, T. Vos, C. J. L. Murray and G. S. Collaborators, Lancet Neurol., 2019, 18, 439–458 Search PubMed.
- S. Pechlivanis, A. A. Mahabadi, P. Hoffmann, M. M. Nothen, M. Broecker-Preuss, R. Erbel, S. Moebus, A. Stang and K. H. Jockel, BMC Med. Genet., 2020, 21, 62 Search PubMed.
- H. Pan, C. Xue, B. J. Auerbach, J. Fan, A. C. Bashore, J. Cui, D. Y. Yang, S. B. Trignano, W. Liu, J. Shi, C. O. Ihuegbu, E. C. Bush, J. Worley, L. Vlahos, P. Laise, R. A. Solomon, E. S. Connolly, A. Califano, P. A. Sims, H. Zhang, M. Li and M. P. Reilly, Circulation, 2020, 142(21), 2060–2075 Search PubMed.
- S. Bertazzo, E. Gentleman, K. L. Cloyd, A. H. Chester, M. H. Yacoub and M. M. Stevens, Nat. Mater., 2013, 12, 576–583 Search PubMed.
- S. E. New, C. Goettsch, M. Aikawa, J. F. Marchini, M. Shibasaki, K. Yabusaki, P. Libby, C. M. Shanahan, K. Croce and E. Aikawa, Circ. Res., 2013, 113, 72–77 Search PubMed.
- A. Leszczynska, A. O'Doherty, E. Farrell, J. Pindjakova, F. J. O'Brien, T. O'Brien, F. Barry and M. Murphy, Stem Cells, 2016, 34, 913–923 Search PubMed.
- M. R. Hosen, P. R. Goody, A. Zietzer, G. Nickenig and F. Jansen, Antioxid. Redox Signaling, 2020, 33, 621–644 Search PubMed.
- T. M. Doherty, K. Asotra, L. A. Fitzpatrick, J. H. Qiao, D. J. Wilkin, R. C. Detrano, C. R. Dunstan, P. K. Shah and T. B. Rajavashisth, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 11201–11206 Search PubMed.
- D. A. Chistiakov, V. A. Myasoedova, A. A. Melnichenko, A. V. Grechko and A. N. Orekhov, BioMed Res. Int., 2017, 2017, 7463590 Search PubMed.
- F. Wang, Z. Zhang, A. Fang, Q. Jin, D. Fang, Y. Liu, J. Wu, X. Tan, Y. Wei, C. Jiang and X. Song, Front. Immunol., 2018, 9, 3127 Search PubMed.
- M. Hamada, M. Nakamura, M. T. Tran, T. Moriguchi, C. Hong, T. Ohsumi, T. T. Dinh, M. Kusakabe, M. Hattori, T. Katsumata, S. Arai, K. Nakashima, T. Kudo, E. Kuroda, C. H. Wu, P. H. Kao, M. Sakai, H. Shimano, T. Miyazaki, P. Tontonoz and S. Takahashi, Nat. Commun., 2014, 5, 3147 Search PubMed.
- A. Kelly-Arnold, N. Maldonado, D. Laudier, E. Aikawa, L. Cardoso and S. Weinbaum, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 10741–10746 Search PubMed.
- Y. Kunitomi, E. S. Hara, M. Okada, N. Nagaoka, T. Kuboki, T. Nakano, H. Kamioka and T. Matsumoto, J. Biomed. Mater. Res., Part A, 2019, 107, 1021–1030 Search PubMed.
- J. E. Vance and D. E. Vance, J. Biol. Chem., 1986, 261, 4486–4491 Search PubMed.
- T. Iwayama, T. Okada, T. Ueda, K. Tomita, S. Matsumoto, M. Takedachi, S. Wakisaka, T. Noda, T. Ogura, T. Okano, P. Fratzl, T. Ogura and S. Murakami, Sci. Adv., 2019, 5, eaax0672 Search PubMed.
- E. S. Hara, M. Okada, N. Nagaoka, T. Hattori, L. M. Iida, T. Kuboki, T. Nakano and T. Matsumoto, Integr. Biol., 2018, 10, 57–66 Search PubMed.
- E. S. Hara, M. Okada, T. Kuboki, T. Nakano and T. Matsumoto, J. Mater. Chem. B, 2018, 6, 6153–6161 Search PubMed.
- A. Demichelis, C. Divieto, L. Mortati and M. Sassi, IEEE International Symposium on Medical Measurements and Applications (MeMeA) Proceedings, 2015, pp. 622–626.
- B. W. Sodar, A. Kittel, K. Paloczi, K. V. Vukman, X. Osteikoetxea, K. Szabo-Taylor, A. Nemeth, B. Sperlagh, T. Baranyai, Z. Giricz, Z. Wiener, L. Turiak, L. Drahos, E. Pallinger, K. Vekey, P. Ferdinandy, A. Falus and E. I. Buzas, Sci. Rep., 2016, 6, 24316 Search PubMed.
- G. Serrano-Heras, I. Díaz-Maroto, B. Castro-Robles, B. Carrión, A. B. Perona-Moratalla, J. Gracia, S. Arteaga, F. Hernández-Fernández, J. García-García, O. Ayo-Martín and T. Segura, Biol. Proced. Online, 2020, 22, 17 Search PubMed.
- M. Ahotupa, Free Radical Res., 2017, 51, 439–447 Search PubMed.
- K. Uchida, Free Radical Biol. Med., 2004, 37, S5 Search PubMed.
- R. Coscas, M. Bensussan, M. P. Jacob, L. Louedec, Z. Massy, J. Sadoine, M. Daudon, C. Chaussain, D. Bazin and J. B. Michel, Atherosclerosis, 2017, 259, 60–67 Search PubMed.
- M. N. Akhter, E. S. Hara, K. Kadoya, M. Okada and T. Matsumoto, Int. J. Mol. Sci., 2020, 21(15), 5327 Search PubMed.
- I. Perrotta and E. Perri, Microsc. Microanal., 2017, 23, 1030–1039 Search PubMed.
- Y. P. Li, A. Asadi, M. R. Monroe and E. P. Douglas, Mater. Sci. Eng., C, 2009, 29, 1643–1649 Search PubMed.
- U. Ravnskov, M. de Lorgeril, D. M. Diamond, R. Hama, T. Hamazaki, B. Hammarskjold, N. Hynes, M. Kendrick, P. H. Langsjoen, L. Mascitelli, K. S. McCully, H. Okuyama, P. J. Rosch, T. Schersten, S. Sultan and R. Sundberg, Expert Rev. Clin. Pharmacol., 2018, 11, 959–970 Search PubMed.
- M. A. E. Cruz, C. R. Ferreira, C. B. Tovani, F. A. de Oliveira, M. Bolean, L. Caseli, S. Mebarek, J. L. Millan, R. Buchet, M. Bottini, P. Ciancaglini and A. P. Ramos, J. Struct. Biol., 2020, 212(2), 107607 Search PubMed.
- A. L. Boskey, W. Ullrich, L. Spevak and H. Gilder, Calcif. Tissue Int., 1996, 58, 45–51 Search PubMed.
- S. Rauschert, A. Gázquez, O. Uhl, F. F. Kirchberg, H. Demmelmair, M. Ruíz-Palacios, M. T. Prieto-Sánchez, J. E. Blanco-Carnero, A. Nieto, E. Larqué and B. Koletzko, Lipids Health Dis., 2019, 18, 20 Search PubMed.
- M. Inokoshi, K. Yoshihara, N. Nagaoka, M. Nakanishi, J. De Munck, S. Minakuchi, K. Vanmeensel, F. Zhang, Y. Yoshida, J. Vleugels, I. Naert and B. Van Meerbeek, J. Dent. Res., 2016, 95, 102–109 Search PubMed.
| This journal is © The Royal Society of Chemistry 2021 |