
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reverse ADOR: reconstruction of UTL zeolite from layered IPC-1P†
Ondřej
Veselý
a,
Pavla
Eliášová
a,
Russell E.
Morris
 *b and
Jiří
Čejka
*a
*b and
Jiří
Čejka
*a
aDepartment of Physical and Macromolecular Chemistry & Charles University Center of Advanced Materials, Faculty of Science, Charles University, Hlavova 8, 12843 Prague, Czech Republic. E-mail: jiri.cejka@natur.cuni.cz
bSchool of Chemistry, EaStChem, University of St. Andrews, North Haugh, St. Andrews, Fife KY16 9ST, UK
First published on 2nd April 2021
Abstract
The assembly–disassembly–organisation–reassembly (ADOR) process has led to the discovery of numerous zeolite structures, albeit limited to materials with decreased pore size in relation to the parent germanosilicate zeolite. This limitation stems from the rapid decrease in d-spacing upon hydrolysis (disassembly). Nevertheless, we have artificially increased the d-spacing of layered IPC-1P by intercalating organic species. Furthermore, we have reconstructed double four rings (D4R) between layers, thus transforming IPC-1P back into the parent UTL zeolite. This reconstruction has provided not only germanosilicate but also a new, high-silica UTL zeolite (Si/Ge = 481). Therefore, our “reverse ADOR” opens up new synthetic routes towards promising extra-large-pore zeolite-based materials with new chemical compositions.
1. Introduction
Zeolites are crystalline silicate-based microporous materials. The micropore size of common zeolites is similar to the kinetic diameters of small organic molecules.1 For this reason, zeolites are extensively used in separation and shape-selective catalysis processes. For example, they are commonly applied as heterogeneous catalysts in petrochemistry.2–4 Moreover, zeolites have also been modified to catalyse biomass conversion5–7 and fine chemical synthesis,8,9 highlighting the wide range of industrial uses of these materials.Zeolites are commonly prepared by solvothermal (mostly hydrothermal) synthesis10 in the presence of structure directing agents (SDAs) and mineralising agents (OH− or F−).11 While the hydrothermal method is versatile and easy to perform, its mechanism remains difficult to generalise. As a result, new zeolites are often discovered by trial and error. In contrast, the assembly–disassembly–organisation–reassembly (ADOR) method has been developed to prepare new zeolites and to predict their structure based on theoretical calculations and experimental conditions.12,13 The ADOR process exploits labile Ge-rich double-four ring (D4R) units in germanosilicates, such as UTL or *CTH.14,15 The structure of these zeolites consists of Si-rich layers connected by Ge-rich D4Rs. Upon selective hydrolysis of D4Rs, layered materials are formed, thus preserving their original structure. Subsequently, these layers undergo topotactic condensation to a new 3D structure and hence a new zeolite. Accordingly, the ADOR is a tool for the rational design of new zeolites by controlled 3D-2D-3D transformation.14
The ADOR process was first studied on the UTL germanosilicate, which hydrolyses to layered IPC-1P.16 Further manipulations of IPC-1P layers have resulted in a whole new family of materials (PCR, OKO, *PCS, IPC-7, IPC-9 and IPC-10; Fig. 1).17–20 The IPC materials contain the same Si-rich layers as the parent UTL but differ in the connections between layers and consequently in pore size. The average pore size of IPC materials, 12-ring and 10-ring, is usually smaller than that of the parent structure (14-ring). However, no UTL or other structure with 14-ring pores has been formed after full 3D-2D-3D transformation until now.
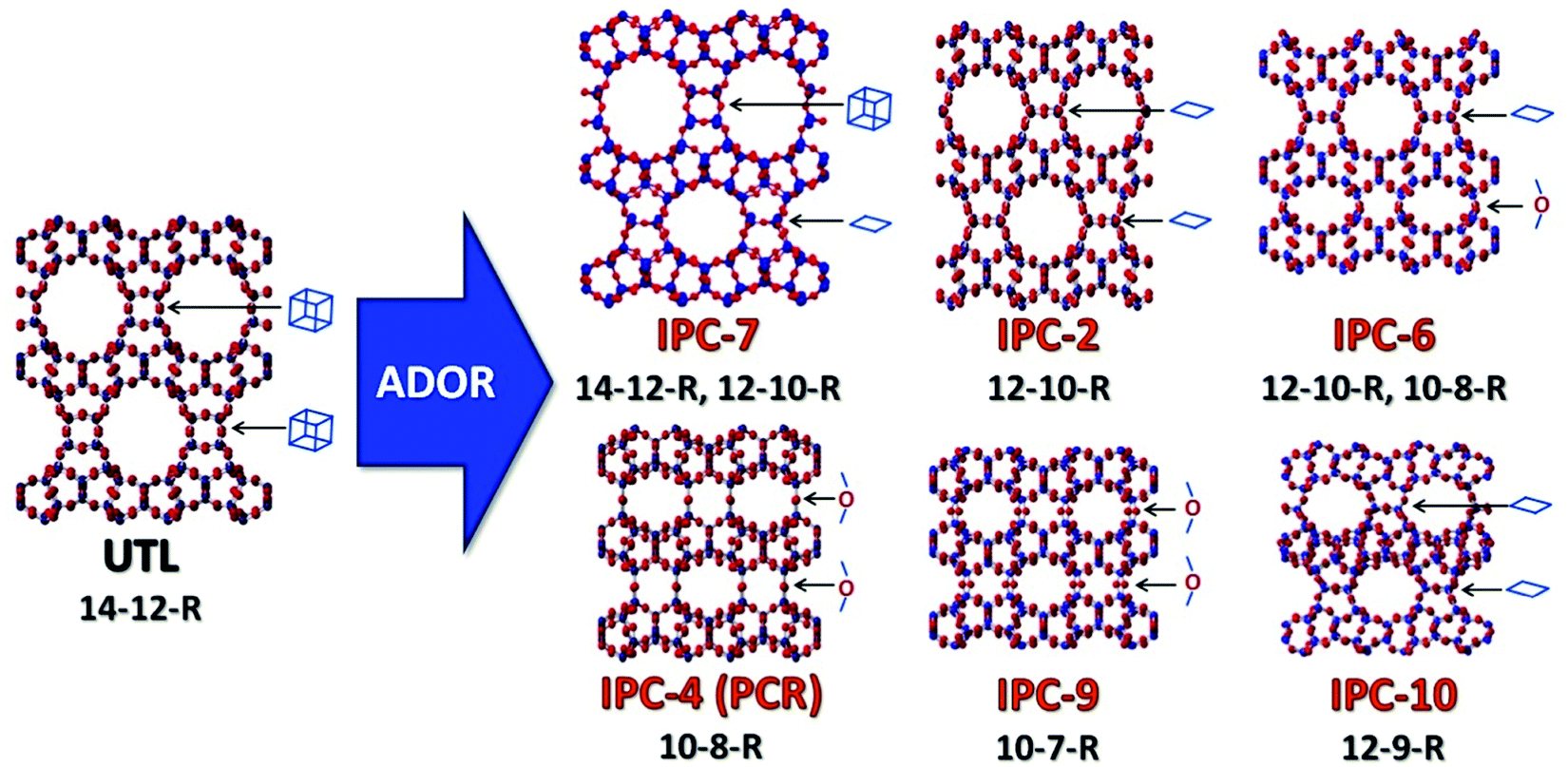 | ||
| Fig. 1 Zeolite UTL and its daughter structures prepared using the ADOR method, highlighting the connecting units between the layers in the different materials. | ||
The kinetics of UTL hydrolysis has been described in detail by Henkelis et al.21UTL rapidly disassembles into IPC-1P, thereby decreasing the interlayer distance (as measured by the d200-spacing in XRD) from 1.45 nm to 1.05 nm. As the IPC-1P layers slowly rearrange, an IPC-2P with 1.18 nm d-spacing is formed, but this IPC-2P is never fully reconverted into UTL under such conditions.22 Conversely, Xu et al. succeeded in doing so when using another Ge-rich UTL, which was transformed back to UTL through isomorphous substitution of Ge by Si. This Ge-rich UTL, however, still contained the SDA. The SDA molecules were necessary to preserve the structure throughout the process, otherwise UTL would quickly disassemble into layered IPC-1P. Once the layered precursor was formed, the UTL structure could not be restored,23 so UTL restoration from layered IPC-1P remains a challenge. Wu et al. have also reported a 2D–3D transformation of the layered material MCM-22P.24,25 After expanding the interlayer distance by intercalating organic agents, they connected the expanded layers through silanes, thus forming MCM-IEZ. These authors used a similar approach to transform the layered HUS-2 to ECNU-19. However, in both MCM-IEZ and ECNU-19, the layers were not connected by D4R units, which are present in UTL, but instead by individual Si linkages. Therefore, by definition, these materials are not zeolites.26
Considering the above, in this study we have developed a method for reversing the ADOR process and fully restoring the UTL zeolite from layered IPC-1P for the first time. In this approach, UTL reconstruction relies on the formation of D4R units between layers. D4Rs form either in a favourable ratio of Si and Ge or in the presence of additional agents, such as fluorides. The process results in a UTL zeolite with Si/Ge ratios ranging from 8 to 400, depending on synthesis design.
2. Experimental section
2.1 Synthesis of the structure-directing agent (SDA) for UTL
(6R,10S)-6,10-Dimethyl-5-anizosporo[4.5]decane hydroxide was used as the SDA to synthetize the UTL germanosilicate.27 In total, 60 mL of 1,4-dibromobutane, 82.9 g of K2CO3 and 500 mL of acetonitrile were mixed in a round-bottom flask. Subsequently, 67 mL of 2,6-dimethylpiperidine was added dropwise, and the mixture was heated to 85 °C and kept refluxing overnight. The acetonitrile was evaporated, and the solid product was dissolved in ethanol. Insoluble compounds were filtered off. Ethanol was evaporated to create an almost saturated solution. Then, the product was precipitated by adding diethyl ether, filtered off, washed with ether, and dried under vacuum overnight. The identity of the SDA structure was confirmed by 1H NMR spectroscopy. The product was ion exchanged to the hydroxide form using the Ambersep 900(OH) ion exchange resin.2.2 UTL synthesis
UTL was prepared using the hydrothermal method.28 Germanium dioxide was dissolved in SDA solution in water. Then, silica (Cab–O–Sil M5) was added to the solution, and the mixture was stirred at room temperature until completely dissolved. The resulting mixture, with a molar composition of 0.67SiO2:0.33GeO2:0.4SDA:33.3H2O, was charged into a 1000 mL Parr autoclave and heated to 175 °C for 6 days with agitation (200 rpm). The solid product was recovered by filtration, washed out with a copious amount of distilled water and dried in the oven at 60 °C. The SDA was removed by calcination in air at 550 °C for 6 h, with a temperature ramp of 1 °C min−1.2.3 IPC-1P synthesis
Calcined UTL zeolite was hydrolysed in 1 M CH3COOH (250 mL per g of sample) at 85 °C for 16 h. The product was isolated by filtration, washed with water and dried at 60 °C.2.4 Intercalation
IPC-1P was treated with a 20% solution of tetrabutylammonium hydroxide (TBAOH) for 6 hours at room temperature.27 The product was recovered by centrifugation and washed with distilled water to neutral pH. The intercalated precursor (IPC-1TBA) was dried at 60 °C overnight.2.5 D4R restoration
In total, 0.1 g of IPC-1TBA was added to a 25 mL Teflon-lined steel autoclave with 5 mL of 1.25 M HCl in ethanol and the respective sources of silicon and germanium (see Table 1). The autoclave was heated to 170 °C for 20 hours. The product was filtered off, washed with ethanol and dried at 60 °C. The TBA was removed by calcination in air at 550 °C for 6 h, with a temperature ramp of 1 °C min−1.| Sample | (EtO)2Me2Si | (EtO)2Me4Si2O | Me8Si4O4 | POSS | (MeO)4Ge |
|---|---|---|---|---|---|
| (EtO)2Me2Si = Diethoxydimethylsilane. (EtO)2Me4Si2O = 1,3-diethoxy-1,1,3,3-tetramethyl-disiloxane. Me8Si4O4 = 2,4,6,8-tetramethylcyclotetrasiloxane. POSS = octamethylsilsesquioxane. (MeO)4Ge = germanium methoxide. | |||||
| rec Si1 | 65.8 mg | — | — | — | — |
| rec Si2 | — | 49.4 mg | — | — | — |
| rec Si4 | — | — | 26.7 mg | — | — |
| rec Si8 | — | — | — | 29.8 mg | — |
rec Si–Ge (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
49.4 mg | — | — | — | 21.9 mg |
| rec Si–Ge (1:1) |
32.9 mg | — | — | — | 43.7 mg |
| rec Si–Ge (1:3) |
16.5 mg | — | — | — | 65.6 mg |
| rec Ge | — | — | — | — | 87.3 mg |
2.6 Characterisation
The crystalline structure of the samples was determined by X-ray powder diffraction (XRD) on a Bruker AXS D8 Advance diffractometer with a Vantec-1 detector in the Bragg–Brentano geometry using Cu Kα radiation (1.54056 Å).High-resolution transmission electron microscopy (HRTEM) images and energy dispersive X-ray (EDX) spectra were acquired under a JEOL NEOARM 200 F microscope with a Schottky-type field emission gun at an accelerating voltage of 200 kV. The samples were dispersed onto the carbon-coated copper grids before the measurements.
Ar adsorption/desorption isotherms were collected at −186 °C on a 3Flex (Micromeritics) static volumetric apparatus. All samples were degassed on a SmartVac Prep (Micromeretics) at 300 °C under vacuum for 8 h before the sorption measurements. The surface area was calculated using the BET method and adsorption data on a relative pressure range of p/p°0 = 0.05–0.25. The t-plot method was applied to determine the micropore volume (Vmic). The adsorbed amount at a relative pressure of p/p°0 = 0.975 reflects the total micropore volume and interparticle adsorption (Vtot). The pore size distributions were calculated using the Horwath–Kawazoe method.
3. Results and discussion
3.1 Intercalation
The d-spacing between the respective layers in the UTL structure is 1.44 nm (which corresponds to reflection at 6.14° in X-ray diffraction using Cu Kα radiation). Hydrolysis of the D4R units in UTL results in the layered material IPC-1P. IPC-1P consists of silica-rich layers with a d-spacing of 1.05 nm (corresponding to reflection at 8.41°). The first challenge in reconverting IPC-1P into UTL was to increase the spacing between IPC-1P layers to 1.44 nm; however, the distance between IPC-1P layers cannot spontaneously increase to the original value (ref. 21; Fig. S1, ESI†). Hence, we adjusted this distance by intercalation. Intercalation, or swelling, of layered silicates and zeolites commonly involves surfactants, such as cetyltrimethylammonium hydroxide.29–31 However, their long hydrocarbon chains are flexible, and their structure between the layers is sensitive to the pH of the environment. Because this may produce disorder and irregularities in the reconstructed material,32 we decided to intercalate the layers with more rigid species, such as the tetrabutylammonium cation (TBA+).27 TBA+ intercalation into IPC-1P shifted the interlayer 200 reflection from XRD from 8.4° to 6.37°, which is relatively close to 6.14° (Fig. 2) – the position of the 200 reflection of the UTL material.33 Accordingly, TBA+ is suitable for increasing the d-spacing of IPC-1P to a distance close to that of the UTL zeolite.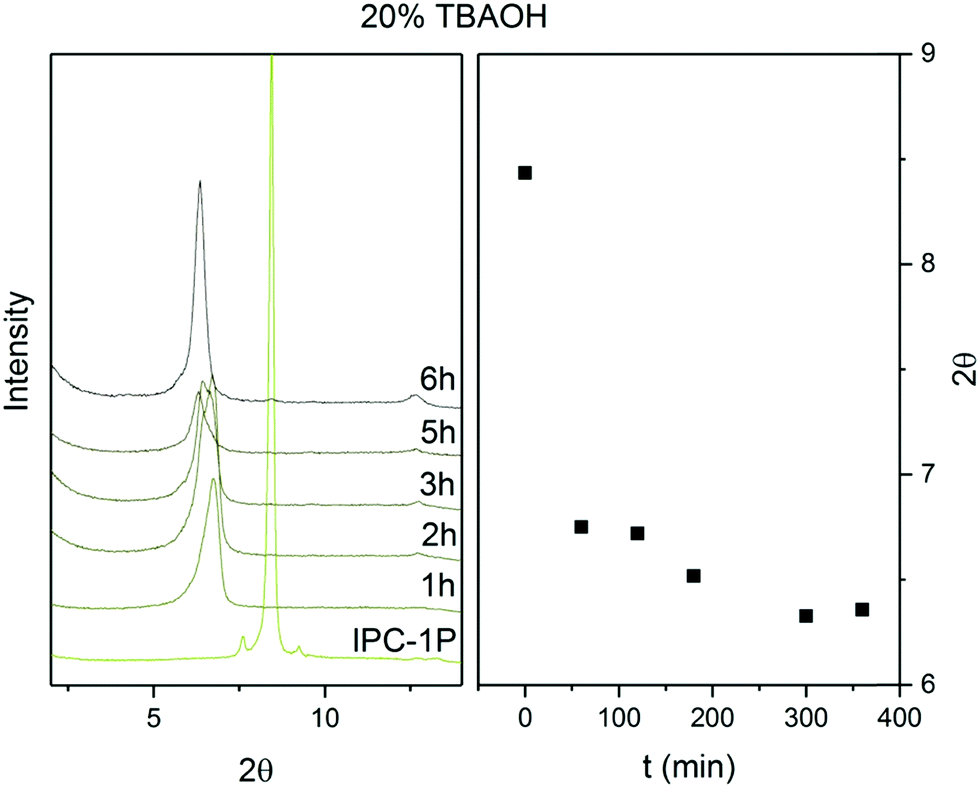 | ||
| Fig. 2 Powder XRD patterns and positions of 200 diffraction lines of IPC-1P intercalated with TBA+ over time. | ||
To optimise the intercalation time, we performed a kinetic experiment with a 20% TBAOH solution. This solution has a basic pH, which is necessary to deprotonate the IPC-1P layers, to form silanolates, and to break the H-bonds between the layers.34 However, high pH also causes desilication,35–38 creating defects and mesopores in the material, which loses layer crystallinity. To preserve the crystallinity of the IPC-1P layers, we shortened the intercalation time. Fig. 2 shows the stabilisation of the 200 reflection at 6.37° for 6 hours. After 6 hours, the 200 peak position remains constant under the conditions of the treatment, thus indicating that the intercalation has ended.
3.2 Reconstruction
The aim, and the main challenge, of this study was to restore the UTL structure by reconstructing D4R units between IPC-1TBA layers. Numerous D4R-containing zeolites, including UTL, crystallise mainly as germanosilicates; therefore, we reconstructed UTL using silicon and germanium alkoxides and their combination for their good reactivity and solubility (see Section 3.3). After silicon and germanium incorporation, the 200 diffraction shifted from 6.37° to 6.16° (Fig. 3). New diffraction lines also appeared at 6.99, 7.35, 8.27, 9.55, 16.7 and 17.7°. These diffraction lines match the XRD pattern of the UTL structure. Moreover, these reflections remained unchanged after calcining the sample to remove the TBA+. These findings suggest that the reconstructed UTL is stable without the support of organic agents.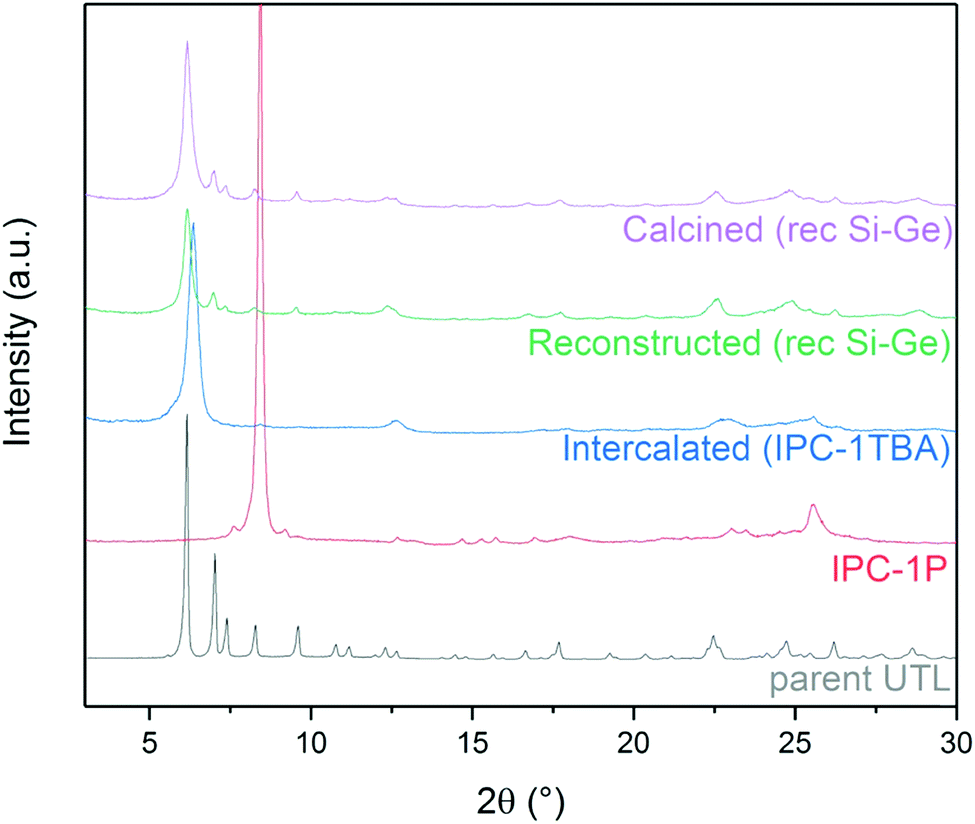 | ||
| Fig. 3 Powder XRD patterns of individual stages in the IPC-1P-to-UTL reconstruction, using the rec Si–Ge (1:1) sample as an example. | ||
The STEM image (Fig. 4) confirmed that the distance between the layers is 1.41 nm and that the layers are visibly connected as a three-dimensional framework. However, STEM also revealed that the treatments caused some etching of the crystal, thereby forming mesopores in the zeolite. The formation of these mesopores is further supported by the changes in textural properties outlined in Table 2.
 | ||
| Fig. 4 STEM image of the reconstructed UTL sample. | ||
| BET (m2 g−1) | S ext (m2 g−1) | V tot (cm3 g−1) | V mic (cm3 g−1) | Si/Ge | |
|---|---|---|---|---|---|
| Parent UTL | 620 | 52 | 0.27 | 0.25 | 4.47 |
| Reconstructed UTL | 808 | 188 | 0.48 | 0.15 | 7.50 |
3.3 Ge content effect
Germanium can stabilise D4R units in zeolites.39,40 The results shown above illustrate the structure directing effect of germanium on UTL reconstruction. We investigated how the germanium content affects the result in a series of similar reconstruction experiments with varying Si:Ge molar ratios (we use the “Si:Ge” notation to express the ratio of Si and Ge sources that we added to the reconstruction mixture to differentiate this ratio from the overall Si/Ge ratio of bulk samples).
XRD showed a broad diffraction peak at 7° (Fig. 5) in the sample reconstructed without germanium. The broad peak is caused by a non-uniform interlayer distance, thus implying that the layers lost their ideal ordering upon calcination. The loss of ordering likely stems from unsuccessful or incomplete reconstruction of the D4R units that connect individual layers. The samples reconstructed with both Si and Ge resulted in UTL structures. The peaks in XRD are less pronounced in the samples with 1:3 and 3:1 Si:Ge, mainly in the region from 5 to 10 °C. These patterns suggest that the interlayer ordering contains some defects. The sample with 1:1 Si:Ge produced the powder XRD pattern closest to that of the parent UTL zeolite. When we used only germanium for the reconstruction, the structure also collapsed upon calcination. Moreover, the diffraction pattern also contained new peaks at 25.7, 35.7, 37.7 and 39.2° belonging to germanium oxide. The germanium oxide species also appeared in the STEM image of the sample (Fig. S2, ESI†), which had a very low micropore volume (0.07 cm3 g−1; Table 3). The formation of germanium oxide may result from the high reactivity of germanium methoxide, which forms the oxide before it can be incorporated into the framework. Alternatively, the purely Ge-based D4Rs may be unstable under the current conditions, but this assumption requires further investigation, which is beyond the scope of this article.
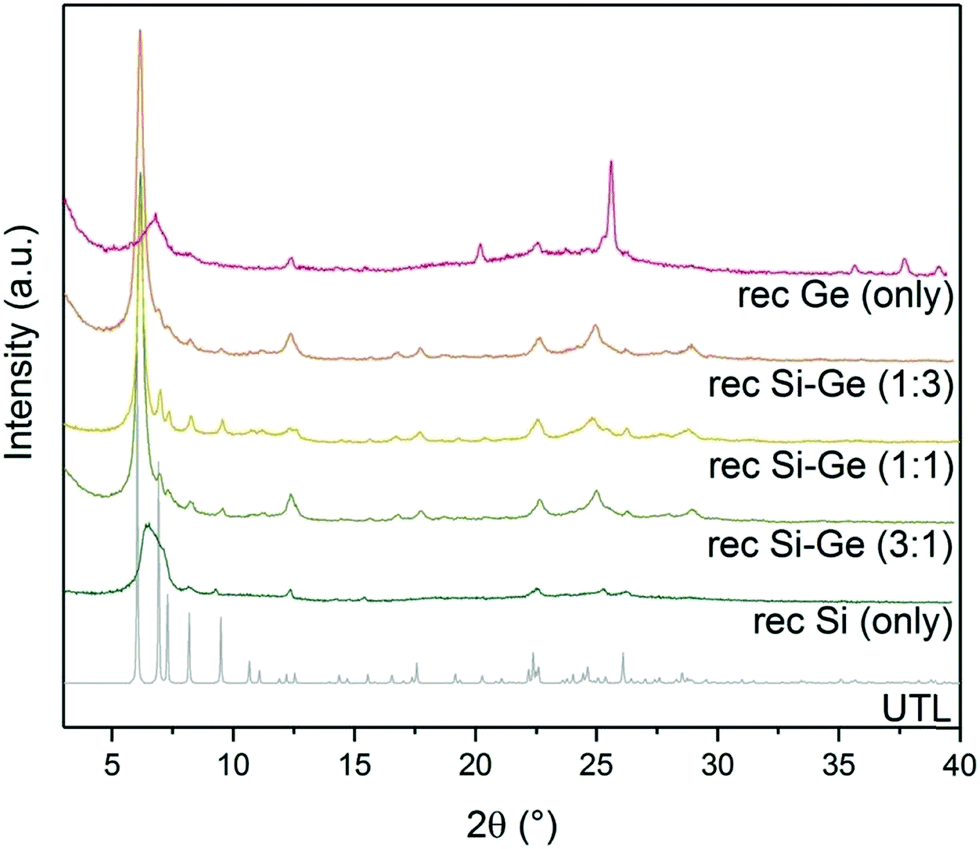 | ||
| Fig. 5 Powder XRD patterns of calcined samples after reconstruction with different Si:Ge compositions. | ||
| BET (m2 g−1) | S ext (m2 g−1) | V tot (cm3 g−1) | V mic (cm3 g−1) | |
|---|---|---|---|---|
| UTL | 620 | 52 | 0.27 | 0.25 |
| rec Si (only) | 645 | 334 | 0.48 | 0.12 |
| rec Si–Ge (3:1) |
550 | 156 | 0.35 | 0.12 |
| rec Si–Ge (1:1) |
808 | 188 | 0.48 | 0.15 |
| rec Si–Ge (1:3) |
555 | 165 | 0.36 | 0.11 |
| rec Ge (only) | 344 | 139 | 0.34 | 0.07 |
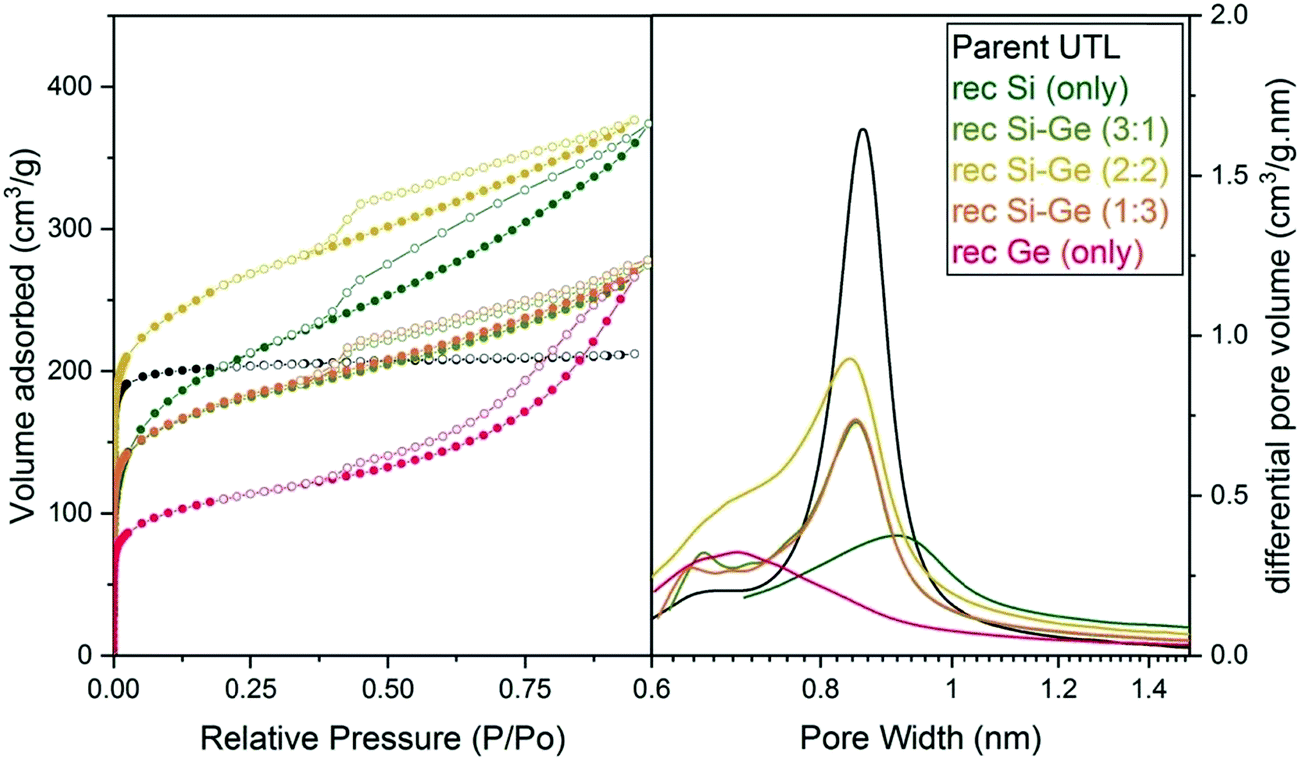 | ||
| Fig. 6 Argon adsorption–desorption isotherms (left) and H–K micropore size distributions (right) of samples after reconstruction with different Si:Ge compositions. | ||
The micropore volume of the reconstructed UTL samples ranges from 0.11 to 0.15 cm3 g−1 (Fig. 6) and is thus smaller than the micropore volume of the parent UTL (0.25 cm3 g−1). These poor textural properties may be caused by incomplete D4R reconstruction because intercalated TBA+ occupy some D4R positions. Consequently, D4R reconstruction does not proceed until the intercalant is removed. To test this hypothesis, we performed D4R reconstruction with 1:1 Si:Ge, as described above, calcined the sample and repeated the reconstruction under the same conditions. However, we observed only negligible differences in powder XRD patterns (Fig. S3, ESI†) and adsorption behaviour (Fig. S4, ESI†). Nevertheless, the pore volume of the samples decreased after the second reconstruction cycle (Table S1, ESI†) due to further damage under such harsh conditions. This experiment disproved that TBA+ significantly hinders D4R reconstruction.
| BET (m2 g) | S ext (m2 g−1) | V tot (cm3 g−1) | V mic (cm3 g−1) | |
|---|---|---|---|---|
| UTL | 620 | 52 | 0.27 | 0.25 |
| Pure Si | 691 | 341 | 0.56 | 0.05 |
| Si:Ge 1:1 |
555 | 281 | 0.46 | 0.06 |
| Pure Ge | 280 | 101 | 0.26 | 0.07 |
Considering the presence of germanium in its D4R, we theorised that the reconstructed UTL may be able to undergo hydrolysis to IPC-1P. To test this hypothesis, we performed a hydrolysis of the reconstructed sample in water and in solution of acetic acid at 85 °C. In both cases the 200 reflection on the XRD pattern shifted to higher angles (Fig. S5, ESI†). The shift suggests that the material transformed during the hydrolysis. However, neither of the experiments produced the IPC-1P, probably due to low germanium content or uneven germanium distribution which prevented complete disassembly into layers.
3.4 Stabilisation by fluorides
Fluoride anions can also stabilise D4R units in zeolites. In fact, numerous D4R-containing extra-large-pore zeolites have been prepared in fluoride medium.39,41,42 Based on the above, we assessed the effect of fluorides on D4R reconstruction. We performed another set of experiments with varying Si:Ge contents, with and without ammonium fluoride.The powder XRD patterns (Fig. 7) of samples reconstructed with Si or Ge alone changed significantly after adding ammonium fluoride. The samples that were synthesized with fluoride showed 200 diffraction at 6.16°, similar to UTL. This diffraction remained unchanged even after calcination. Other reflections, characteristic of the UTL, also appeared at 6.99, 7.35, 8.27 and 9.55° in the sample reconstructed solely with Ge in fluoride-containing medium. However, the powder diffraction pattern of this sample also contained the peaks of germanium oxide. The sample reconstructed with Si alone showed a similar change after adding fluorides, but the other diffraction lines were significantly less intense. The XRD pattern of the sample prepared with 1:1 Si:Ge shows no significant difference after adding ammonium fluoride.
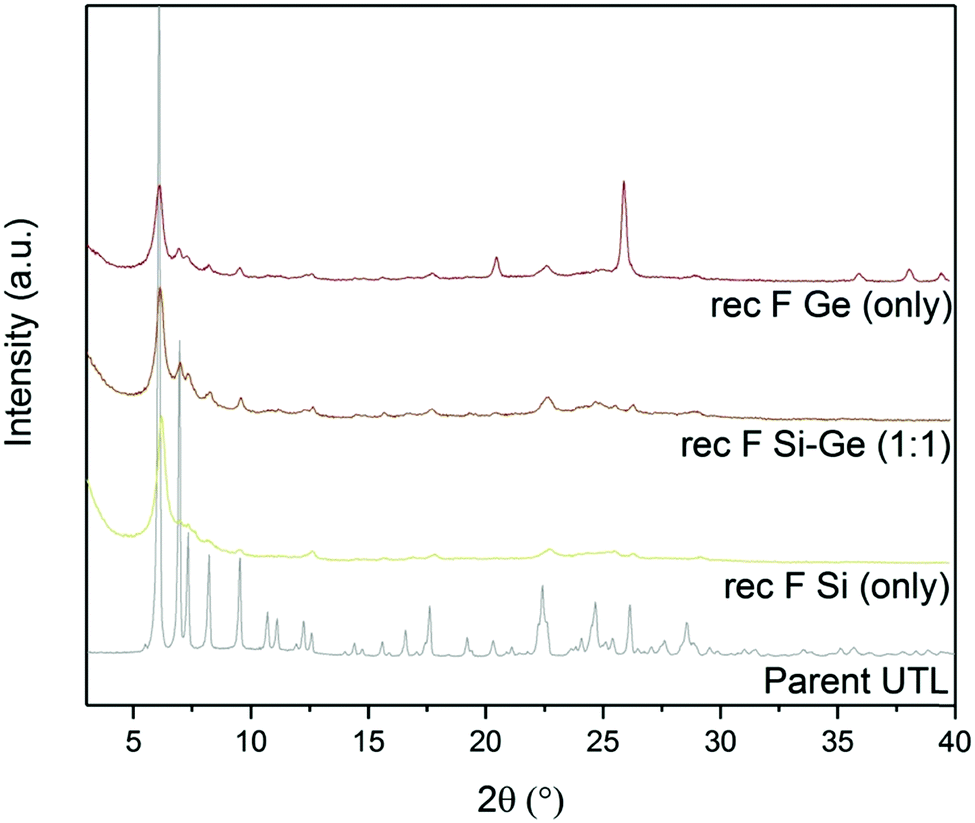 | ||
| Fig. 7 Powder XRD patterns of calcined samples after D4R reconstruction with different Si:Ge compositions, with and without NH4F. | ||
Structural changes caused by fluoride addition to the mixture are also identified in the pore-size distribution. The shape of the distribution curves (Fig. 8) of Ge- and Si-only samples narrowed down. Simultaneously, the maxima of the distribution shifted to 0.84 nm, near the 0.85 nm of the parent UTL. However, all samples prepared in fluoride media had micropore volumes smaller than 0.07 cm3 g−1 (Table 4) far lower than that of the parent UTL (0.25 cm3 g−1; Table 2). This decrease may originate from framework etching by fluoride anions in solution, leading to partial amorphisation of the material (Fig. S7, ESI†) and/or subsequent pore blockage by amorphous framework debris.
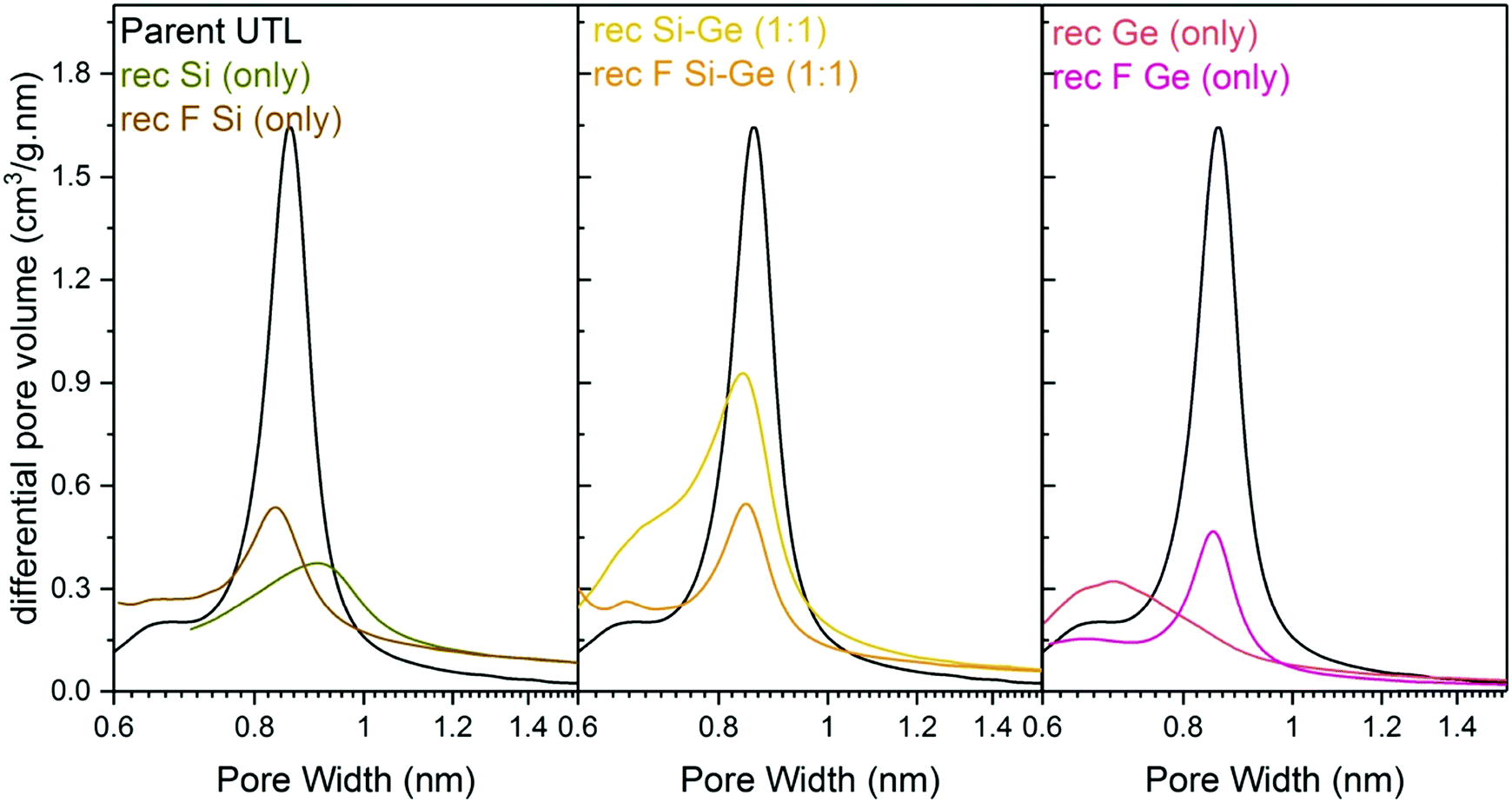 | ||
| Fig. 8 H–K micropore size distributions of samples after reconstruction with different Si:Ge compositions, with and without NH4F. | ||
In summary, while fluoride anions stabilise D4R units, they simultaneously damage the material, which accounts for the poor micropore volumes of the samples.
3.5 Structure of the silicon source
D4R formation solely from silane molecules is unfavourable without fluoride anions to stabilise these units. For this reason, we investigated whether other Si sources would form D4R units, even without using germanium or fluorides, e.g., diethoxydimethylsilane (Si1), 1,3-diethoxy-1,1,3,3-tetramethyl-disiloxane (Si2), 2,4,6,8-tetramethylcyclotetrasiloxane (Si4) and octamethylsilsesquioxane (Si8) as Si sources. The broad diffraction peak at 7° in both Si1 and Si2 samples (Fig. 9) implies that their relative layer arrangements lack order and that their structure collapsed after calcination. In contrast, samples Si4 and Si8 retained a very narrow 200 diffraction peak at 6.16°, even after the calcination. The other diffractions at 6.99, 7.35, 8.27 and 9.55° remained unchanged but were less intense than in the germanium-containing sample, as shown in Fig. 3. This lower intensity may arise from the higher silicon content of the samples Si4 and Si8.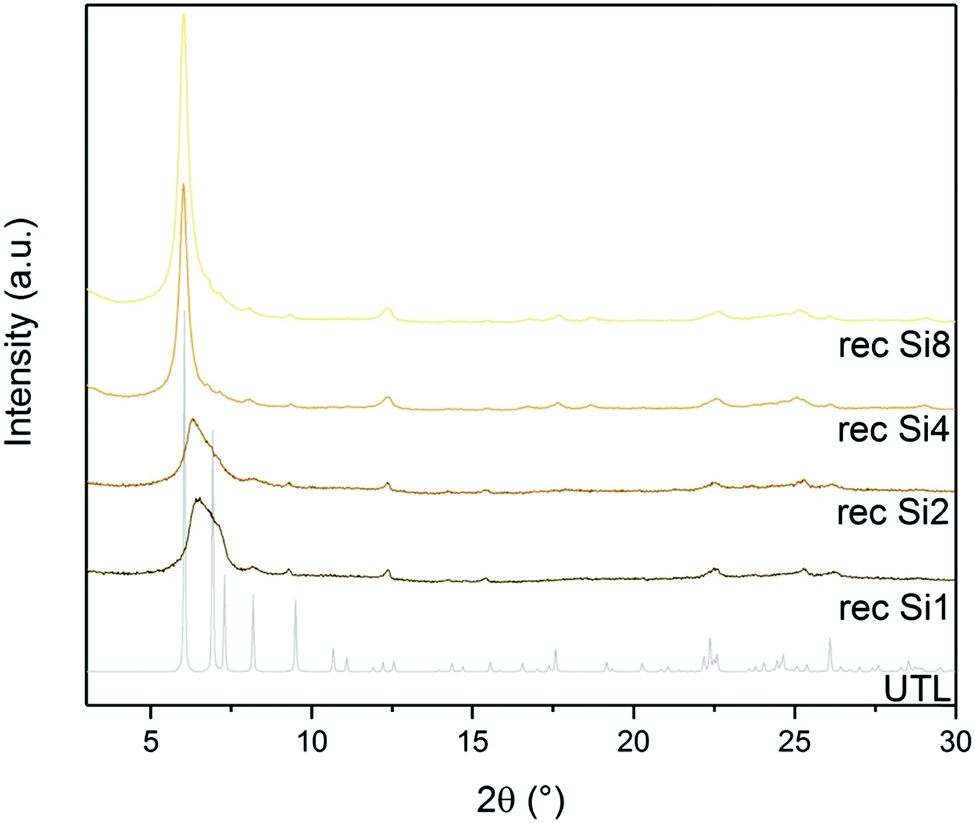 | ||
| Fig. 9 Powder XRD patterns of calcined samples after D4R reconstruction with different Si sources. | ||
STEM measurements (Fig. 10) further confirmed the structure and interlayer spacing, showing 1.41 nm distances between individual layers. This distance is in line with the UTL structure. We analysed the composition of the sample Si4 by EDX. The sample has a Si/Ge ratio of 481. This is an interesting result because D4R-containing zeolites or any extra-large pore zeolite with such a high Si content are seldom prepared without any hetero-element (such as Ge) or fluorides.43
 | ||
| Fig. 10 STEM image of the reconstructed UTL sample Si4. | ||
3.6 Tuning the textural properties
Regardless of their elemental composition, all the UTL samples that were reconstructed in this study had lower micropore volumes, ranging from 0.11 to 0.15 cm3 g−1, than their parent UTL sample (0.25 cm3 g−1). Their lower pore volumes stem from their decreased crystallinity inflicted during the intercalation of the samples with TBA+. The IPC-1TBA crystal shown in Fig. 11 is severely damaged after intercalation because the treatment resulted in formation of defects and mesopores in the crystal. However, this is not necessarily a disadvantage of the method as the formation of defects may be beneficial because mesopores can enhance diffusion in catalytic applications.44 | ||
| Fig. 11 TEM image of the damaged IPC-1TBA sample crystal. | ||
The high pH of the treatment solution causes etching but is needed for TBA+ intercalation. Under optimal conditions, intercalation would proceed at the lowest pH possible to minimise the damage to the crystals, albeit high enough to support TBA+ intercalation. To find the optimal conditions, we intercalated IPC-1P with 40, 20, 10, 5 and 2% TBAOH solutions and monitored the position of the 200 diffraction (Fig. 12). The position of 200 peak in the samples treated with 40, 20 and 10% TBAOH was identified at 6.37°. The powder XRD pattern of the sample treated with 5% TBAOH also contained a peak at 6.37° but another, minor diffraction appeared at 7.67°. Treatment with 2% TBAOH resulted in three diffractions at 6.37, 7.67 and 8.4°, indicating incomplete intercalation. In summary, 5% is the minimum TBAOH concentration required for successful TBA+ intercalation into IPC-1P.
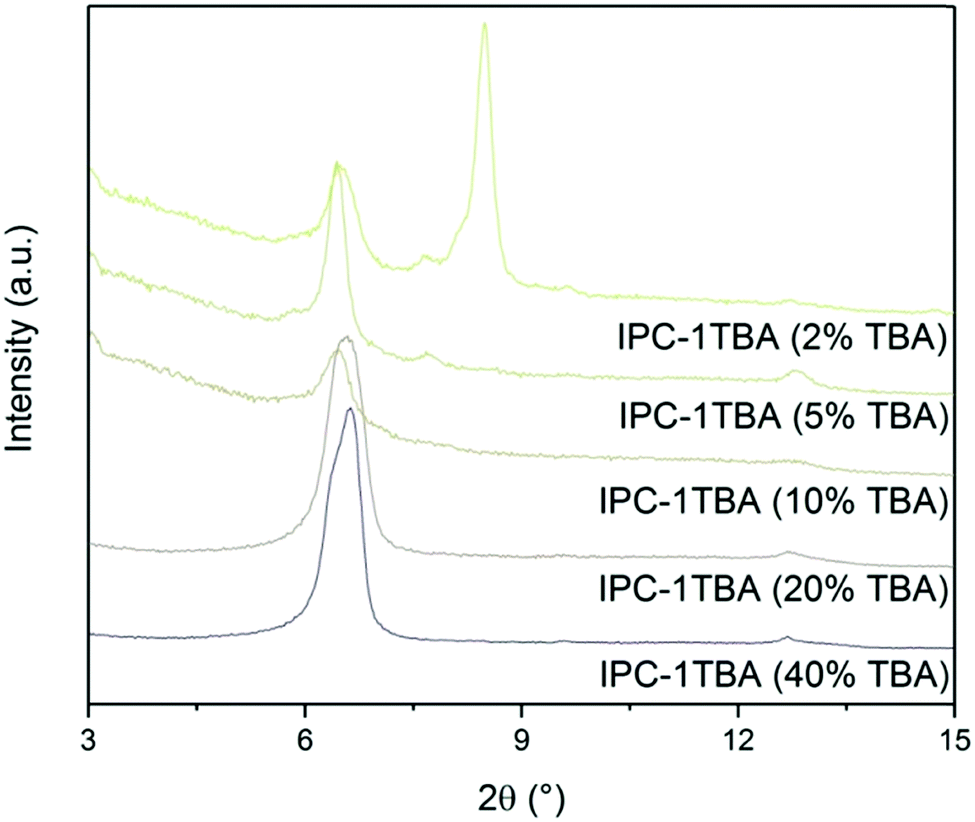 | ||
| Fig. 12 Powder XRD patterns of IPC-1P samples intercalated with TBAOH of varying concentrations. | ||
We reconstructed all samples with 1:1 Si:Ge, including the sample treated with a 2% TBAOH, and subsequently characterised them by adsorption. All isotherms showed large adsorbed amounts at a relative pressure bellow 0.1 (Fig. 13) – filling of micropores. At higher pressures, the intake decreased in all samples; however, the flat plateau observed in the parent UTL sample was not found, suggesting that the samples contain not only micropore but also some mesopores. Nevertheless, the slope of the isotherm above the relative pressure p/p0 of 0.1 decreases with the decrease in the concentration of the TBAOH solution. The lower hydroxide concentration mitigates the damage and produces samples with fewer defects.
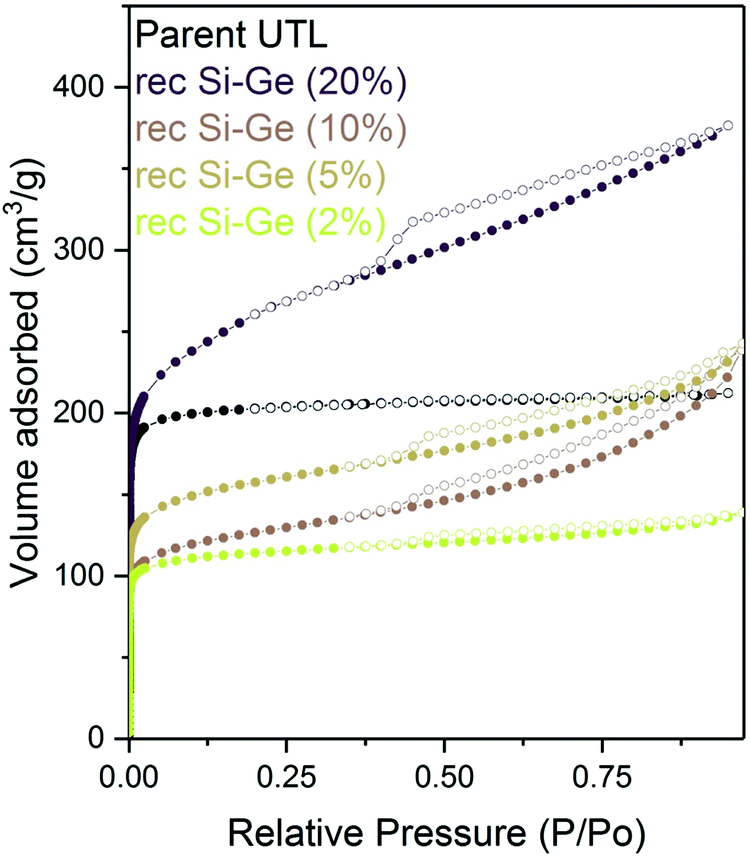 | ||
| Fig. 13 Argon adsorption–desorption isotherms of reconstructed samples previously intercalated with TBAOH of varying concentrations. | ||
4. Conclusions
Until now, the ADOR method was limited to decreasing the interlayer distance and, consequently, the pore size of the parent zeolite during the process. In this study, we overcome this limitation by intercalating organic species between IPC-1P layers formed by UTL hydrolysis. TBAOH intercalation increases the d-spacing to 1.41 nm, the distance of the original UTL structure. The UTL structure is then restored using varying Si and Ge ratios to rebuild the D4R units between the intercalated IPC-1P layers, and the formation of D4Rs is promoted in the presence of an optimal Ge content or fluoride ions. Moreover, the UTL zeolite can also be restored using structurally more complex compounds, such as cyclotetrasiloxanes or polyhedral silsesquioxanes. In summary, the “Reverse ADOR” produces zeolites with a wide range of various compositions, ranging from the common UTL with a 7.5 Si/Ge to the new high-silica UTL with 481 Si/Ge.While the intercalation treatment inherently causes the formation of mesopores in the zeolite crystals, we can mitigate its impact by tuning the treatment conditions. Furthermore, these mesopores may, in turn, be advantageous in future catalytic applications. Therefore, the “Reverse ADOR” method opens up opportunities for incorporating other elements towards producing novel zeolite-based catalysts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the support of the Czech Science Foundation to the project EXPRO (19-27551X) and OP VVV “Excellent Research Teams”, project no. CZ.02.1.01/0.0/0.0/15_003/0000417 – CUCAM. REM thanks the European Research Council for the provision of an Advanced Grant (ADOR – 787073). The authors acknowledge Dr Michal Mazur (Charles University) for the STEM imaging, Dr Martin Kubů (Charles University) for the Ar adsorption measurements, and Dr Carlos V. Melo for editing the manuscript.References
- J. Čejka, H. van Bekkum, A. Corma and F. Schueth, Introduction to Zeolite Molecular Sieves, Elsevier, Amsterdam, 2007 Search PubMed.
- J. Čejka, E. R. Morris and P. Nachtigall, Zeolites in Catalysis: Properties and Applications, The Royal Society of Chemistry, 2017 Search PubMed.
- W. Vermeiren and J. P. Gilson, Impact of zeolites on the petroleum and petrochemical industry, Top. Catal., 2009, 52, 1131–1161 CrossRef CAS.
- J. Prech, P. Pizarro, D. P. Serrano and J. Cejka, From 3D to 2D zeolite catalytic materials, Chem. Soc. Rev., 2018, 47, 8263–8306 RSC.
- T. Ennaert, J. Van Aelst, J. Dijkmans, R. De Clercq, W. Schutyser, M. Dusselier, D. Verboekend and B. F. Sels, Potential and challenges of zeolite chemistry in the catalytic conversion of biomass, Chem. Soc. Rev., 2016, 45, 584–611 RSC.
- P. Sudarsanam, E. Peeters, E. V. Makshina, V. I. Parvulescu and B. F. Sels, Advances in porous and nanoscale catalysts for viable biomass conversion, Chem. Soc. Rev., 2019, 48, 2366–2421 RSC.
- E. Taarning, C. M. Osmundsen, X. Yang, B. Voss, S. I. Andersen and C. H. Christensen, Zeolite-catalyzed biomass conversion to fuels and chemicals, Energy Environ. Sci., 2011, 4, 793–804 RSC.
- M. Shamzhy, M. Opanasenko, P. Concepción and A. Martínez, New trends in tailoring active sites in zeolite-based catalysts, Chem. Soc. Rev., 2019, 48, 1095–1149 RSC.
- L.-H. Chen, M.-H. Sun, Z. Wang, W. Yang, Z. Xie and B.-L. Su, Hierarchically Structured Zeolites: From Design to Application, Chem. Rev., 2020, 120, 11194–11294 CrossRef CAS PubMed.
- C. S. Cundy and P. A. Cox, The Hydrothermal Synthesis of Zeolites: History and Development from the Earliest Days to the Present Time, Chem. Rev., 2003, 103, 663–702 CrossRef CAS PubMed.
- J. Yu, Synthesis of Zeolites, in Studies in Surface Science and Catalysis, ed. J. Čejka, H. van Bekkum, A. Corma and F. Schüth, Elsevier, 2007, pp. 39–103 Search PubMed.
- P. Eliášová, M. Opanasenko, P. S. Wheatley, M. Shamzhy, M. Mazur, P. Nachtigall, W. J. Roth, R. E. Morris and J. Čejka, The ADOR mechanism for the synthesis of new zeolites, Chem. Soc. Rev., 2015, 44, 7177–7206 RSC.
- D. S. Firth, S. A. Morris, P. S. Wheatley, S. E. Russell, A. M. Z. Slawin, D. M. Dawson, A. Mayoral, M. Opanasenko, M. Položij, J. Čejka, P. Nachtigall and R. E. Morris, Assembly–Disassembly–Organization–Reassembly Synthesis of Zeolites Based on cfi-Type Layers, Chem. Mater., 2017, 29, 5605–5611 CrossRef CAS.
- M. Opanasenko, M. Shamzhy, Y. Wang, W. Yan, P. Nachtigall and J. Čejka, Synthesis and Post-Synthesis Transformation of Germanosilicate Zeolites, Angew. Chem., Int. Ed., 2020, 59, 19380–19389 CrossRef CAS PubMed.
- C. J. Heard, L. Grajciar, F. Uhlík, M. Shamzhy, M. Opanasenko, J. Čejka and P. Nachtigall, Zeolite (In)Stability under Aqueous or Steaming Conditions, Adv. Mater., 2020, 32, 2003264 CrossRef CAS PubMed.
- W. J. Roth, O. V. Shvets, M. Shamzhy, P. Chlubna, M. Kubu, P. Nachtigall and J. Čejka, Postsynthesis Transformation of Three-Dimensional Framework into a Lamellar Zeolite with Modifiable Architecture, J. Am. Chem. Soc., 2011, 133, 6130–6133 CrossRef CAS PubMed.
- W. J. Roth, P. Nachtigall, R. E. Morris, P. S. Wheatley, V. R. Seymour, S. E. Ashbrook, P. Chlubna, L. Grajciar, M. Polozij, A. Zukal, O. Shvets and J. Čejka, A family of zeolites with controlled pore size prepared using a top-down method, Nat. Chem., 2013, 5, 628–633 CrossRef CAS PubMed.
- P. S. Wheatley, P. Chlubná-Eliášová, H. Greer, W. Zhou, V. R. Seymour, D. M. Dawson, S. E. Ashbrook, A. B. Pinar, L. B. McCusker, M. Opanasenko, J. Čejka and R. E. Morris, Zeolites with Continuously Tuneable Porosity, Angew. Chem., 2014, 126, 13426–13430 CrossRef.
- S. A. Morris, G. P. M. Bignami, Y. Tian, M. Navarro, D. S. Firth, J. Čejka, P. S. Wheatley, D. M. Dawson, W. A. Slawinski, D. S. Wragg, R. E. Morris and S. E. Ashbrook, In situ solid-state NMR and XRD studies of the ADOR process and the unusual structure of zeolite IPC-6, Nat. Chem., 2017, 9, 1012–1018 CrossRef CAS PubMed.
- M. Mazur, P. S. Wheatley, M. Navarro, W. J. Roth, M. Položij, A. Mayoral, P. Eliášová, P. Nachtigall, J. Čejka and R. E. Morris, Synthesis of ‘unfeasible’ zeolites, Nat. Chem., 2016, 8, 58–62 CrossRef CAS PubMed.
- S. E. Henkelis, M. Mazur, C. M. Rice, P. S. Wheatley, S. E. Ashbrook and R. E. Morris, Kinetics and Mechanism of the Hydrolysis and Rearrangement Processes within the Assembly–Disassembly–Organization–Reassembly Synthesis of Zeolites, J. Am. Chem. Soc., 2019, 141, 4453–4459 CrossRef CAS PubMed.
- S. E. Henkelis, M. Mazur, C. M. Rice, G. P. M. Bignami, P. S. Wheatley, S. E. Ashbrook, J. Čejka and R. E. Morris, A procedure for identifying possible products in the assembly–disassembly–organization–reassembly (ADOR) synthesis of zeolites, Nat. Protoc., 2019, 14, 781–794 CrossRef CAS PubMed.
- H. Xu, J.-G. Jiang, B. Yang, L. Zhang, M. He and P. Wu, Post-Synthesis Treatment gives Highly Stable Siliceous Zeolites through the Isomorphous Substitution of Silicon for Germanium in Germanosilicates, Angew. Chem., Int. Ed., 2014, 53, 1355–1359 CrossRef CAS PubMed.
- W. J. Roth, W. Makowski, B. Marszalek, P. Michorczyk, W. Skuza and B. Gil, Activity enhancement of zeolite MCM-22 by interlayer expansion enabling higher Ce loading and room temperature CO oxidation, J. Mater. Chem. A, 2014, 2, 15722–15725 RSC.
- P. Wu, J. Ruan, L. Wang, L. Wu, Y. Wang, Y. Liu, W. Fan, M. He, O. Terasaki and T. Tatsumi, Methodology for Synthesizing Crystalline Metallosilicates with Expanded Pore Windows Through Molecular Alkoxysilylation of Zeolitic Lamellar Precursors, J. Am. Chem. Soc., 2008, 130, 8178–8187 CrossRef CAS PubMed.
- B. Yang, J.-G. Jiang, H. Xu, H. Wu and P. Wu, Synthesis of Large-Pore ECNU-19 Material (12 × 8-R) via Interlayer-Expansion of HUS-2 Lamellar Silicate, Chin. J. Chem., 2018, 36, 227–232 CrossRef CAS.
- M. Mazur, P. Chlubná-Eliášová, W. J. Roth and J. Čejka, Intercalation chemistry of layered zeolite precursor IPC-1P, Catal. Today, 2014, 227, 37–44 CrossRef CAS.
- O. V. Shvets, A. Zukal, N. Kasian, N. Žilková and J. Čejka, The Role of Crystallization Parameters for the Synthesis of Germanosilicate with UTL Topology, Chem. – Eur. J., 2008, 14, 10134–10140 CrossRef CAS PubMed.
- W. J. Roth, Cation size effects in swelling of the layered zeolite precursor MCM-22-P, Pol. J. Chem., 2006, 80, 703–708 CAS.
- P. Chlubna, W. J. Roth, A. Zukal, M. Kubu and J. Pavlatova, Pillared MWW zeolites MCM-36 prepared by swelling MCM-22P in concentrated surfactant solutions, Catal. Today, 2012, 179, 35–42 CrossRef CAS.
- M. Shamzhy, M. Mazur, M. Opanasenko, W. J. Roth and J. Cejka, Swelling and pillaring of the layered precursor IPC-1P: tiny details determine everything, Dalton Trans., 2014, 43, 10548–10557 RSC.
- M. Opanasenko, W. O. N. Parker, M. Shamzhy, E. Montanari, M. Bellettato, M. Mazur, R. Millini and J. Čejka, Hierarchical Hybrid Organic–Inorganic Materials with Tunable Textural Properties Obtained Using Zeolitic-Layered Precursor, J. Am. Chem. Soc., 2014, 136, 2511–2519 CrossRef CAS PubMed.
- C. Baerlocher and L. B. McCusker, in, Database of Zeolite Structures, http://www.iza-structure.org/databases/.
- W. J. Roth and J. C. Vartuli, Preparation of exfoliated zeolites from layered precursors: The role of pH and nature of intercalating media, in Nanoporous Materials Iii, ed. A. Sayari and M. Jaroniec, 2002, pp. 273–279 Search PubMed.
- B. Gil, Ł. Mokrzycki, B. Sulikowski, Z. Olejniczak and S. Walas, Desilication of ZSM-5 and ZSM-12 zeolites: Impact on textural, acidic and catalytic properties, Catal. Today, 2010, 152, 24–32 CrossRef CAS.
- D. Verboekend and J. Perez-Ramirez, Desilication Mechanism Revisited: Highly Mesoporous All-Silica Zeolites Enabled Through Pore-Directing Agents, Chem. – Eur. J., 2011, 17, 1137–1147 CrossRef CAS PubMed.
- M. Kubů, M. Opanasenko and M. Shamzy, Modification of textural and acidic properties of SVR zeolite by desilication, Catal. Today, 2014, 227, 26–32 CrossRef.
- M. Kubů, M. Opanasenko and D. Vitvarová, Desilication of SSZ-33 zeolite – Post-synthesis modification of textural and acidic properties, Catal. Today, 2015, 243, 46–52 CrossRef.
- T. Blasco, A. Corma, M. J. Díaz-Cabañas, F. Rey, J. A. Vidal-Moya and C. M. Zicovich-Wilson, Preferential Location of Ge in the Double Four-Membered Ring Units of ITQ-7 Zeolite, J. Phys. Chem. B, 2002, 106, 2634–2642 CrossRef CAS.
- P. Kamakoti and T. A. Barckholtz, Role of Germanium in the Formation of Double Four Rings in Zeolites, J. Phys. Chem. C, 2007, 111, 3575–3583 CrossRef CAS.
- J. Jiang, J. L. Jorda, M. J. Diaz-Cabanas, J. Yu and A. Corma, The Synthesis of an Extra-Large-Pore Zeolite with Double Three-Ring Building Units and a Low Framework Density, Angew. Chem., Int. Ed., 2010, 49, 4986–4988 CrossRef CAS PubMed.
- A. Corma, M. J. Díaz-Cabañas, J. Jiang, M. Afeworki, D. L. Dorset, S. L. Soled and K. G. Strohmaier, Extra-large pore zeolite (ITQ-40) with the lowest framework density containing double four- and double three-rings, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13997–14002 CrossRef CAS PubMed.
- M. Fischer and L. Freymann, Local Distortions in a Prototypical Zeolite Framework Containing Double Four-Ring Cages: The Role of Framework Composition and Organic Guests**, ChemPhysChem, n/a.
- D. Schneider, D. Mehlhorn, P. Zeigermann, J. Kärger and R. Valiullin, Transport properties of hierarchical micro–mesoporous materials, Chem. Soc. Rev., 2016, 45, 3439–3467 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00212k |
| This journal is © The Royal Society of Chemistry 2021 |