
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cobalt nanoparticle catalysed graphitization and the effect of metal precursor decomposition temperature†
Stuart J.
Goldie
 ,
Shan
Jiang
and
Karl S.
Coleman
*
,
Shan
Jiang
and
Karl S.
Coleman
*
Department of Chemistry, Durham University, South Road, Durham, DH1 3LE, UK. E-mail: k.s.coleman@durham.ac.uk
First published on 13th April 2021
Abstract
Porous carbon foams hold great promise for supercapacitors and next generation energy storage materials but greater control over the formation of the pore structure would aid the development of these materials. High temperature graphitization has been investigated as a sustainable method of producing these technologically important materials and the addition of transition metals is known to promote the formation of graphitic carbon and potentially control the pore structure, however the effect of different metal precursors has rarely been examined. Using different cobalt salts, specifically Co(OAc)2, CoCl2 and Co(NO3)2, in a dextran/TritonX-45 aerogel graphitization, the foams produced were analyzed using Raman spectroscopy, XRD, thermal analysis, gas sorption and various electron microscopy techniques. These revealed that when using salts with low thermal stability such as Co(NO3)2 and Co(OAc)2 the metal nanoparticles are formed rapidly and become trapped in the carbon matrix causing uniform graphitization. In contrast, when stable salts are used such as CoCl2 the carbon decomposes before metal reduction and large metal crystals, microns in size, are formed. This control of nanoparticle size through understanding the thermal stability of metal precursors should be general to other widely used reagents to inform the rational design and production of future functional materials.
Introduction
Porous carbon foams are attracting interest for important energy storage applications in battery and supercapacitor devices, as well as potential photo and electro-catalyst supports thanks to the low density, high surface area and desirable conductivities possible,1–5 and because graphitic and amorphous carbon foams can be produced from sustainable biomass.6 In contrast, self-assembly from graphene oxide dispersions readily produces high surface area structures but the oxidised graphene used to form the dispersion contains defects in the sp2 carbon network reducing many of the most useful properties.7–10 Alternatively, chemical vapour deposition onto porous templates has produced high quality few-layer graphene foams, however this requires an expensive synthesis and produces low volumes of material.11–14Graphitization, the high temperature conversion of carbon materials into graphite, has been applied to porous and templated carbon materials for decades and represents a facile method of converting biomass or polymer structures into porous graphitic carbon.15–17 Such methods have been applied to form hard carbon anodes for sodium and lithium ion batteries and supercapacitor electrodes.18–20 The transformation of carbon into graphite is known to occur spontaneously, but only at very high temperatures often exceeding 2000 °C.17 The presence of transition metals reduces the temperature at which this process happens thanks to dissolution/precipitation by which the carbon forms a high temperature super saturated solution in the metal (or in some cases stable metal carbide) that precipitates forming graphite crystals.21,22 Recently there have been many attempts to produce porous graphitic materials from polymers or biomass, mixed either with metal particles or metal salts that form metals in situ.17,23–27 In a study of this process Sevilla et al. proposed the conversion proceeded by the decomposition of the metal precursor and carbon material to produce metal, or metal oxide, nanoparticles embedded in an amorphous carbon structure; these nanoparticles then convert the surrounding carbon into graphitic crystallites.25,28
Many carbon based materials can in principle be graphitized but the most common approaches use: polymer xerogels from emulsion polymerization to form a polymer foam with transition metal salts dissolved in the aqueous phase;23,25,29–32 insoluble carbon feedstocks, often biomass, into which metal salt solutions are soaked;19,28,33–35 and solutions of both metal salts and carbon feedstock that form well mixed materials when dried.22,26,36,37 The most common catalyst precursors are transition metal salts, often of Fe, Ni or Co, that decompose at elevated temperatures to form the catalytic metal.38,39 The mixed foam is then heated under an inert atmosphere to high temperatures, usually exceeding 800 °C, causing decomposition into elemental carbon. An example of a single solution prepared from soluble sugars and cobalt salts, followed by drying and heating is shown in Scheme 1. During graphitization carbon will undergo rearrangement to adopt the hexagonal layers of graphene and graphite whilst functional groups are lost as gases, commonly CO2, CO, H2 and light hydrocarbons; causing the structure to shrink and pores to form.22 The resulting graphitized foam can be washed to remove metal particles and structural templates.
 | ||
| Scheme 1 Preparation of dextran and TritonX-45 gel containing Co(II) salts, photograph of dried aerogel in a recrystallised alumina crucible and a photograph of the carbonized foam after heat treatment. | ||
Structural templates or other mechanisms for controlling the porous structure are required because energy storage devices like supercapacitors require hierarchical porous structures with both micropores (smaller than 2 nm) to provide high surface areas for charge storage, and meso (>2 nm, <50 nm) and macro pores (>50 nm) to allow electrolyte infiltration and diffusion.40 Hard structural templates like zeolites and silica nanoparticles are effective at controlling the resulting porosity, although, harsh washing is generally required to remove such templates;31,41–44 alternatively soft template polymers and biomaterials have been used that require little to no washing, however, the relationship between initial gel structure and final porous carbon is still uncertain. Recently studies using gelatin with metal salts have found the interaction between the metals and the biopolymer in the initial foaming stage can have an impact on the macro porosity and structure of the final foam.45 The metals exhibit a change in coordination environment when mixed with gelatin complicated by the pH and temperature of gel formation. The method of foam production is also reported to be important, slow heat drying produces collapsed pores whereas freeze drying the foams rapidly, produces high surface area materials.46
Comparisons of different transition metals as catalysts have also been undertaken, generally Cu is found to perform poorly due to its low carbon solubility, whereas Fe, Ni and Co have comparable performance producing carbon materials with different aromatic carbon content and pore structures depending on production method.22,23,47 To investigate the graphitization process we utilize a dextran sugar based gel previously shown to be an effective template for hard metal foams used for CVD growth with cobalt salts.48 In contrast to other investigations we vary the cobalt salts used rather than the identity of the metal and probe the relationship between the reactive salts used and the carbon foam produced. Whilst many transition metals are effective at catalyzing graphene growth in this way, cobalt nanoparticles are interesting for other applications including water splitting electrocatalysts.49
Experimental
Materials
Cobalt(II) nitrate hexahydrate ACS reagent ≥ 98%, cobalt(II) acetate tetrahydrate ACS reagent ≥ 98.0%, dextran from Leuconostoc mesenteroides (average mol wt 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500000–2800000) and TritonTM X-45 were all used as purchased from Sigma Aldrich. Cobalt(II) chloride hexahydrate was used as purchased from Alfa Aesar. Pureshield argon (99.998%) and hydrogen (99.995%) were used as provided by BOC. 6 M hydrochloric acid was prepared from hydrochloric acid S.G 1.18 purchased from Fisher Scientific. Furnace tube washing and waste oil analysis was done with acetone AR (99.5%) from Fisher Scientific.
500000–2800000) and TritonTM X-45 were all used as purchased from Sigma Aldrich. Cobalt(II) chloride hexahydrate was used as purchased from Alfa Aesar. Pureshield argon (99.998%) and hydrogen (99.995%) were used as provided by BOC. 6 M hydrochloric acid was prepared from hydrochloric acid S.G 1.18 purchased from Fisher Scientific. Furnace tube washing and waste oil analysis was done with acetone AR (99.5%) from Fisher Scientific.
Synthesis of gel
Gels were prepared in similar fashion to Khan and Mann;50 dextran was gently stirred into water with the salt to form a viscous liquid to which TritonTM X-45 was added and the mixture then stirred with a mechanical stirrer at 600 rpm to form a homogeneous mixture. In every case 4 g of salt was used and the other reagents were added to maintain a consistent mass ratio (1:2:2) between the cobalt metal, dextran and TritonTM respectively; water was added to keep a constant ratio (1:2.5) between sugar and water respectively. The wet gel was placed into an alumina boat and freeze dried (SP Scientific BenchTop Pro) for at least 24 hs at ∼4 Pa to produce a hard foam.
Synthesis of graphene foam
An alumina boat containing the foam was placed inside a quartz worktube (I.D 29 mm) inside a Carbolite tube furnace (MTF 12/38/400). The system was purged with argon (800 mL min−1) for 30 min then hydrogen (8.72 mL min−1) was added to the flow all at atmospheric pressure; argon gas flow measured by volumetric flow meter whilst hydrogen controlled by a Brooks 5850 TR Series mass controller in totalizer mode. The furnace was heated to 1000 °C at 10 °C min−1 and held at this temperature for one hour before it was allowed to cool.Extraction and washing
Both the quartz worktube and resulting carbonized foam were washed with acetone (∼200 mL) to remove oily residues then vacuum filtered and air dried. The oily by-product was isolated by removal of solvent in vacuo and then re-dissolved in acetone (10 μL mL−1) for GC–MS analysis. The carbonized foam was washed with 6 M hydrochloric acid for 42 h before being filtered through a 0.02 μm membrane and washed with copious high purity water until washings were neutral; these were diluted into one litre of high purity water using a volumetric flask for ICP elemental analysis. Before any analysis of solid material all samples were dried in a vacuum oven for at least 24 hs (60 °C, <1 mbar). A note on terminology, when a distinction must be made ‘carbonized’ refers to carbon/metal foams before acid washing whilst ‘washed’ refers to carbon post acid wash.Characterization
Results and discussion
Herein we discuss the role of different cobalt salts on the morphology and graphitization of carbon foams. Firstly, a wet dextran/TritonXTM-45/cobalt gel was prepared and freeze dried to form a solid material as illustrated in Scheme 1. This was heated in a mildly reducing atmosphere to generate a carbon and metal foam and then washed with HCl to isolate the final graphene-based foam. A dextran based gel was chosen as a starting point because it is simple to produce and the high oxygen content has been linked to an even distribution of cations throughout the structure.37,50 Acetate, chloride and nitrate cobalt salts were chosen for their availability and high water solubility.Probing the carbon produced
The large area Raman microanalysis and powder XRD of the carbonized materials clearly show the significant effect the cobalt has; the metal free control is exclusively amorphous carbon as shown by the lack of a 2D Raman peak in Fig. 1, and a very broad XRD peak centred around 10.5°, Fig. 2.55 In contrast the cobalt containing materials show signs of graphitic domains within the structure giving rise to the strong 002 in-plane reflection in the XRD pattern and a 2D Raman signal, attributed to the resonant scattering from the sp2 network in graphene sheets.56 Despite this, the carbon materials produced using different cobalt salts as catalyst precursors have different degrees of graphitization. The XRD patterns (Fig. 2) show a wide 002 reflection at 2θ = 12.2° and in the case of CoCl2 a sharper reflection at a greater 2θ value, 2θ = 12.4°. Using the graphitization models summarized by Ōya and Marsh the CoCl2 derived material can be described as G-effect graphitization, in which regions of highly crystalline graphite form within an amorphous carbon structure.57 It's also supported by the Raman map data (Fig. 1b); the amorphous carbon with a very low I2D/IG ratio but ID/IG ≈ 1.0 dominates much of the map however there are points with Raman spectra more consistent with graphite, exhibiting low ID/IG ratios and I2D/IG ≈ 0.4.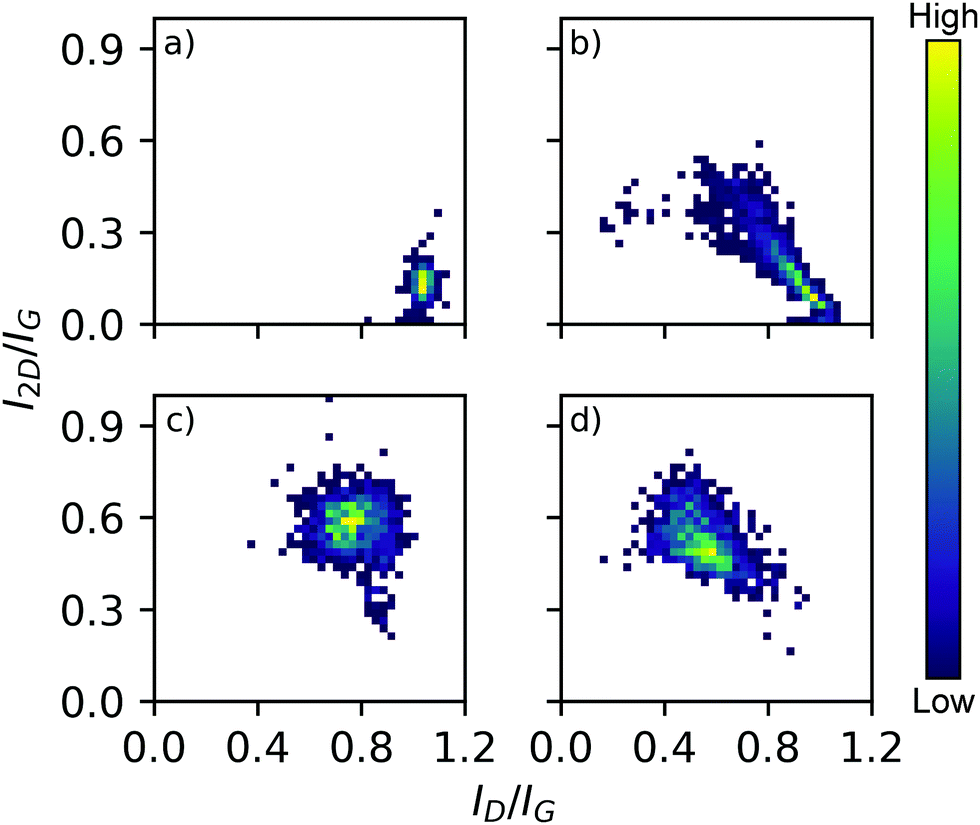 | ||
| Fig. 1 3D bivariate histograms displaying Raman map data from carbonized samples derived from salts (a) metal free control, (b) CoCl2, (c) Co(OAc)2, (d) Co(NO3)2. Graphitic ID/IG and I2D/IG peak intensity ratios are on each axis and histogram frequency denoted with the heat scale shown. Representative spectra in ESI,† Fig. S12. | ||
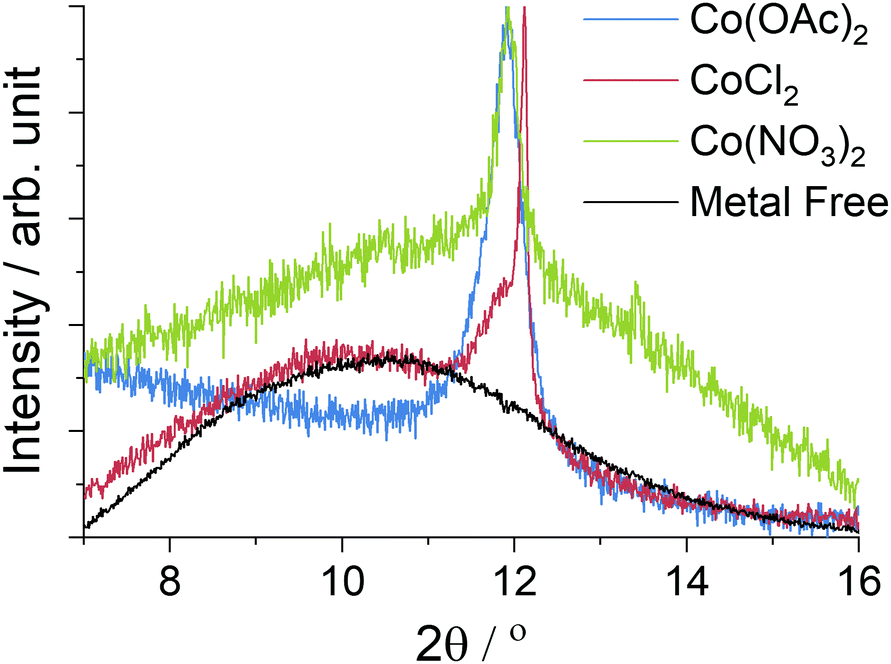 | ||
| Fig. 2 Powder XRD data from the carbonized materials recorded with a Mo source (λ = 0.7093 Å), the data is the same as Fig. 4 but focused on the graphite 002 peak which appears weak due to the high metal content. Peak intensities have been normalized, whilst the metal free control is shown with reduced intensity for clarity. | ||
In contrast, the Co(OAc)2 and Co(NO3)2 catalyst precursors produce materials better described by Ts-effect graphitization caused by finely divided catalyst particles that produce many turbostratic graphitic domains lacking long range 3D stacking. The XRD patterns of such materials generally have a wider 002 reflection with a 2θ value lower than the G-effect peak which is consistent with the observed patterns and peak position at 2θ = 12.2°. This shift of 0.2° as measured with a Mo X-ray source, while small, agrees with the reported shift of 0.5° when measured with Cu radiation (1.5406 Å).57 A distribution of small graphitic domains is also consistent with the Raman map data (Fig. 1c and d); both Co(OAc)2 and Co(NO3)2 derived foams show a I2D/IG ratio higher than expected from pristine graphite (Fig. S13, ESI†), 0.6 and 0.45 respectively but an increased ID/IG of 0.75 and 0.55 consistent with smaller graphite flakes.
Thermal analysis in air (Fig. S1, ESI†) reveals the CoCl2 derived foam to contain two domains, one very similar to the amorphous carbon control with an onset at 580 °C and a small phase of graphitic carbon accounting for 9% of the mass that burnt at a higher temperature, onset at 650 °C. The Co(OAc)2 in contrast shows only a single mass loss with an onset of 625 °C consistent with the graphitic carbon present; the Co(NO3)2 derived foam burns at a lower temperature starting at 350 °C, however this is the result of residual metal nanoparticles catalysing the combustion of the carbon surrounding them.31
Viewing the metal particles
Electron micrographs of the carbonized materials show very clearly the difference in metal particles produced by in situ reduction when using different cobalt salts. When CoCl2 is used as a precursor the metal is found in large, highly crystalline particles microns in size decorated over the surface from which graphite layers can grow following the dissolution–precipitation mechanism.21 EDX mapping (Fig. 3b) highlights the metal content concentrated exclusively into the crystals whose sharp edges and regular shapes indicate high crystallinity, a feature supported by the clear XRD pattern (Fig. 4) obtained from the carbonized material before acid washing. Further imaging after acid washing (Fig. 3c) reveals the carbon shells grown on the metal surface can actually maintain their structure even after the metal is removed from inside. Whilst it might be supposed that a complete carbon coating of the metal would protect it from reacting with the acid, the absence of any XRD diffraction peaks (Fig. S11, ESI†) or mass remaining after thermal analysis (Fig. S1, ESI†) shows that HCl etching is effective at removing the metal. The cracks and gaps observed by SEM in the carbon shell support this incomplete carbon growth from the metal that allows some acid penetration. | ||
| Fig. 3 Scanning electron micrographs of CoCl2 derived foam after carbonization. (a) Carbonized foam before acid washing showing metal particles; (b) composite SE image and EDX map showing cobalt (blue) and carbon (red) distribution; (c) after acid washing showing carbon shells (imaged at 4 kV), a wider angle image is available in ESI;† (d) after acid washing imaged at 10 keV causing the slight transparency of the thin carbon shells. | ||
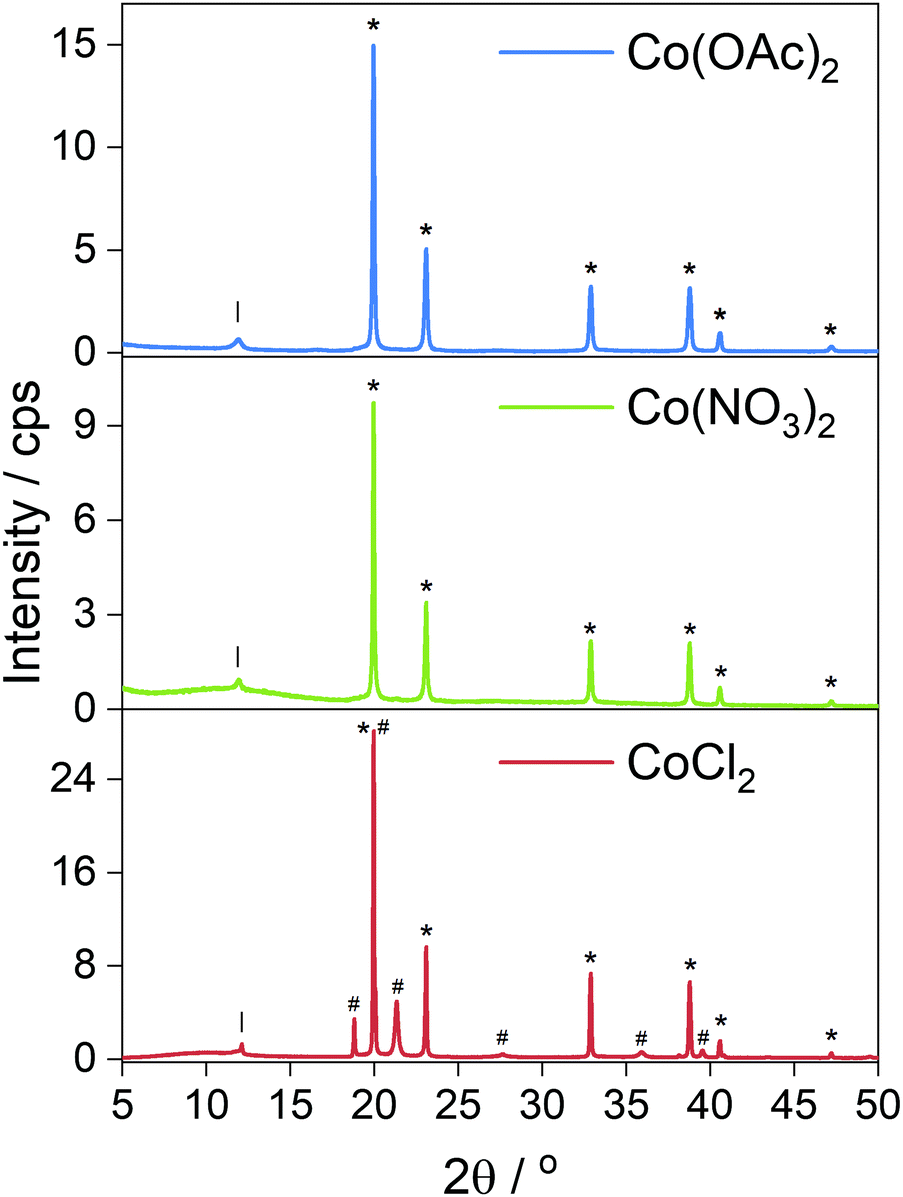 | ||
| Fig. 4 Powder XRD patterns of carbonized foams containing Co metal produced in situ; Co(OAc)2 and Co(NO3)2 salts produced metal with exclusively ccp reflections (*). The CoCl2 salt produced both hcp (#) and ccp (*) phases. Full index available in ESI.† | ||
SEM struggles to resolve the nanoparticles present in the Co(OAc)2 and Co(NO3)2 derived samples whilst the TEM images clearly show large numbers of spherical particles scattered throughout the carbon matrix (Fig. 5). These particle size distributions are shown in Fig. S17 and S18 (ESI†). Neither follow a normal distribution, instead containing a tail extending to larger particles sizes, but the Co(NO3)2 derived foam produced nanoparticles with a mean diameter of 10 ± 3 nm whilst the Co(OAc)2 sample produced smaller nanoparticles 2.2 ± 0.6 nm in diameter. Larger metal particles greater than 100 nm in diameter were also observed in the Co(NO3)2 sample although they were substantially less common than the nanoparticles. These particles are observed in SEM images and EDX elemental mapping (Fig. 5b); although they are poorly resolved because the nanoparticles are smaller than the electron interaction volume at electron energy levels sufficient to excite the cobalt.
 | ||
| Fig. 5 Electron micrographs of the carbonized foams produced from the Co(NO3)2 (a–d) and Co(OAc)2 (e and f) salts. (a) SEM with metal present, (b) composite secondary electron image and EDX map showing cobalt (blue) and carbon (red) distribution, (c) HRTEM image of a single metal particle with carbon growth visible around the edge, (d) HRTEM image after washing showing nanoparticles and the vacant sites left by other particles highlighted with arrows, (e) SEM image of carbon surface with metal present, (f) TEM of carbon sheets with nanoparticle observed on the surface. | ||
The impact of these nanoparticles on carbon graphitization has been studied and generally the same dissolution/precipitation mechanism is thought to be responsible for carbon growth, however, the significant curvature of the metal nanoparticles can induce strain in the graphite sheets or cause incomplete coverage.21,25 The HRTEM images of the carbon around these particles and left behind after removal support these conclusions (Fig. 5d); lines are observed in circles around the particle sites indicating graphite layers grown outwards before forming a larger matrix of graphitic carbon domains crosslinked together. A selected area electron diffraction (SAED) pattern (Fig. S25, ESI†) collected from one of these regions shows clear rings with a very faint hexagonal pattern of spots visible; the spots indicate that crystalline graphite layers are present, however, there is a significant proportion of randomly oriented sheets of carbon.
Analysis of surface area
It was expected that such coverage of the metals by inert carbon would impede their removal by mineral acids. However, the foam produced from the reduction of CoCl2 was not impacted due to cracks and gaps within the carbon; in contrast the Co(NO3)2 precursor formed smaller nanoparticles, some of which were completely covered by a thin layer of carbon growth. This protected some of the nanoparticles from the acid wash; there was an 8 wt% difference between the expected cobalt content and actual mass reduction following HCl washing. In addition even after acid washing metal particles were observed in the TEM images (Fig. 5d) and the thermal analysis shows a 3.8% residue at 1000 °C (Fig. S1, ESI†) and catalytic combustion at lower temperatures.31,58 In contrast, the Co(OAc)2 reduction produced very small nanoparticles that could freely migrate out of the structure; gentle dispersion in ethanol was sufficient to remove these and TEM images show the otherwise clean carbon grid decorated with metal nanoparticles.Gas sorption analysis was performed on these materials as shown in Fig. S29 (ESI†). The small nanoparticles and porous structure of the Co(OAc)2 have the highest BET surface area as measured by N2 adsorption (Table 1), although the anomalously high reading before any washing was probably caused by removal of the nanoparticles during the sample preparation. In stark contrast the Co(NO3)2 sample had a BET surface area of 39 m2 g−1 before washing but increased substantially to 103 m2 g−1 despite the wash step being insufficient to removal all the nanoparticles. The smallest change upon removal of the metal was the CoCl2 sample, probably due to the large particles covered in smooth graphite crystallites that will contribute less to the surface area than the amorphous regions. The N2 isotherm shows a very significant hysteresis consistent with liquid nitrogen trapped in narrow channels by capillary forces (Fig. S29, ESI†). SEM images of the Co(NO3)2 also show regions of a more fibrous network with metal particles trapped in the carbon (Fig. S16, ESI†).
| Cobalt precursor | Surface area/m2 g−1 | |
|---|---|---|
| Carbon & metal | Acid washed | |
| Co(OAc)2 | 96 | 144 |
| CoCl2 | 52 | 96 |
| Co(NO3)2 | 39 | 103 |
The role of different salts on the initial foaming process has been investigated and was reported to affect the macro structure of the material.45 This effect was observed here as the Co(NO3)2 containing gel expanded to a greater volume during freeze drying than the other cobalt salt containing gels. This does not appear to have any influence on the micro-structure since the surface area for the Co(NO3)2 based foam is smaller than the other samples although the low density fibrous material seen in SEM images may be influenced by the interaction between the salt and dextran chains.
Role of metal reduction
The reduction of the cobalt salts in a hydrogen atmosphere was investigated with thermal analysis (Fig. 6) coupled to mass spectrometry; see ESI† for mass spectrometry data. CoCl2·2H2O is a very stable salt undergoing no significant reaction until 500 °C when it rapidly decomposes into the metal, evolving HCl as the chlorine is lost.59 The Co(OAc)2 undergoes a two-step reduction beginning at 250 °C, evolving CO2 as the temperature increases before only the metal remains. Co(NO3)2 is the least stable salt first melting at 55 °C and losing the water of hydration; at 165 °C the nitrogen begins to decompose via NOx species to leave the metal mostly reduced at 400 °C.60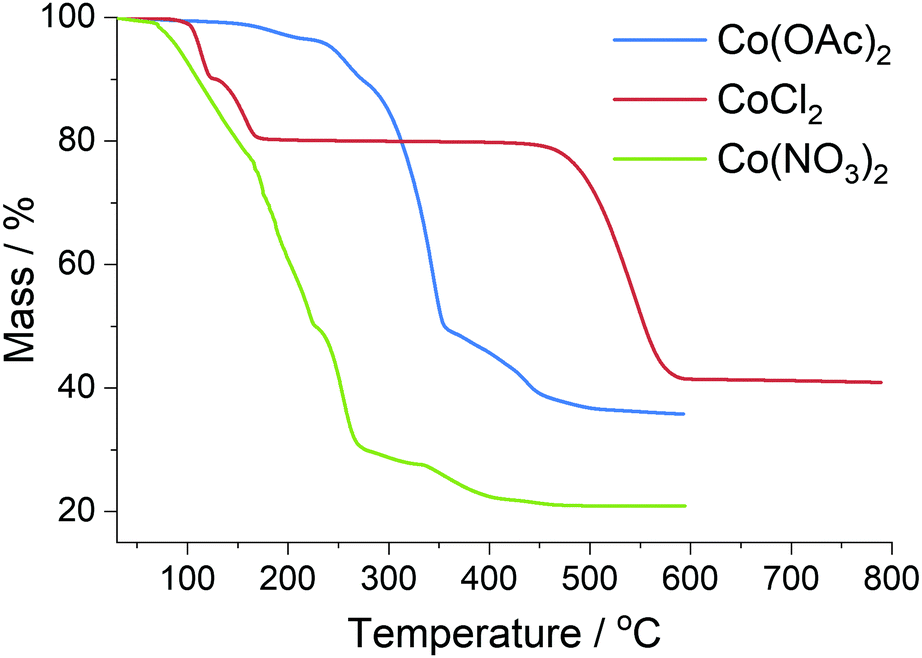 | ||
| Fig. 6 Thermal analysis of metal salts used in preparation of carbon foams, completed in a reducing atmosphere. | ||
The presence of reactive gases generated inside the foam material may explain some of the morphology observed. Significant concentrations of oxidizing NOx gases from the Co(NO3)2 may effectively etch some of the carbon leaving a more open structure as observed with SEM and explaining the much smaller increase in percentage carbon content (Table 2). This release of gases would be expected before graphitization could occur, producing the open macroporous structure; but with individual nanoparticles later trapped by graphitization. In contrast, the CO2 and related by-products from the Co(OAc)2 do not cause additional mass loss when compared with the salt free control and the SEM shows a more dense, solid structure at low magnification levels (Fig. S14, ESI†).
| Sample | Solid residue wt% | Carbon wt% before heat | Carbon wt% after heat |
|---|---|---|---|
| *Carbon content of OAc ligand not included. | |||
| Control | 10.865(4) | ||
| Co(OAc)2* | 24.610(2) | 32.7(4) | 40.61(4) |
| CoCl2 | 28.934(3) | 35.4(3) | 44.69(9) |
| Co(NO3)2 | 17.799(3) | 30.9(4) | 32.09(9) |
The carbon produced from the CoCl2 precursor is the heaviest of those measured, however, neither thermal analysis (Fig. S2, ESI†) nor EDX spectra (Fig. S28, ESI†) reveal any chlorine remaining in the carbon foam to account for this greater mass. GC–MS analysis of the oily by-products revealed significant quantities of TritonXTM fragments including many that had undergone nucleophilic substitution with the reactive HCl present (see ESI† information). However, it is worth noting that even tightly bound into clay structures, TritonXTM is known to completely desorb and vaporize by 500 °C.61 This suggests the TritonXTM vapours condensed on the cold quartz tube outside the furnace hot zone and then HCl gas flowing from the reduction of the CoCl2 reacted with the alcohol groups to produce the compounds observed, and the different masses remaining do indeed reflect the different quantities of carbon remaining after graphitization.
Given the limited evidence for the direct reaction of gases produced from the decomposition of the cobalt salts with the carbon foam, we suggest the temperature of reduction and carbon environment influences the size and distribution of the metal nanoparticles produced; and thus these nanoparticles are the key to determining the graphitization process that follows at higher temperatures.
Considering the carbon foam produced with CoCl2 as a catalyst precursor, previous studies suggest that sugar based carbon undergoes pyrolysis into amorphous carbon between 300–600 °C which would be before any of the salt underwent reduction.36,37 It follows that this would produce an open pore structure of amorphous carbon that allows significant migration of the cobalt at high temperature to anneal into micron sized crystalline particles. These large particles then graphitize the carbon around them in a highly ordered manner leaving more disordered carbon in the rest of the structure, consistent with the G model of catalytic graphitization and the characterization completed in this study.
The Co(OAc)2 and Co(NO3)2 salts both decompose at lower temperatures when the carbon foam will still contain many functional groups and retain much of its original structure. The large number of oxygen functional groups may aid in evenly dispersing the cobalt salt which, combined with the regular structure, results in localized nanoparticles that are trapped inside the carbon matrix and mostly unable to anneal into larger particles. This hypothesis is fully consistent with the microscopy and XRD data collected and would cause the Ts model graphitization observed from these samples. In a previous study with polymer aerogels Fu et al. found that cobalt nanoparticles began forming from Co(NO3)2 ion exchanged into a charged polymer matrix at 450 °C but carbon layers were not observed until 600 °C.25 Whilst the carbon structure's interactions with the charged cobalt cations are very different between our materials, the finding of different temperatures influencing nanoparticle formation is clearly significant.
The difference between the Co(NO3)2 and Co(OAc)2 salts can be explained by a mixture of temperature and reactive gas formation: the lower temperature melt and decomposition of the Co(NO3)2 followed by etching of the carbon matrix by NOx allows the cobalt to coalesce to a greater extent forming larger nanoparticles within the structure. The Co(OAc)2 in contrast does not have this space and the metal becomes trapped in very small nanoparticles distributed throughout the structure, close enough together that channels form between them after graphitization of the surrounding carbon. The larger particles from Co(NO3)2 appear to be large enough for more complete local graphitization completely enveloping the nanoparticles, protecting them from solvent and acid extraction. In both cases however, graphene formation is relatively uniform throughout the macrostructure on account of the regular presence of cobalt nanoparticles, although the highly curved surface of the nanoparticle produced strained graphene sheets with the significant ID/IG ratio observed.56
Conclusions
Mesoporous graphene foams have been produced in a single step reduction of dextran, TritonX-45TM and three common cobalt salts. The cobalt salts are shown to reduce in situ to form catalytic cobalt nanoparticles, and the temperature of this reduction could be used to control the graphitization and morphology of the resulting foam. This could be applicable to the production of graphitic foams for use in energy storage devices, where the compromise between high surface area carbon and more ordered graphitic regions is key. Salts that undergo reduction at low temperatures like Co(OAc)2 and Co(NO3)2 are shown to form crystalline nanoparticles, trapped within the carbon structure. These cause uniform graphene growth throughout the material and template porous voids around themselves that can be accessed by removing the metal. More thermally stable salts like CoCl2 only form the catalytic metal particles after the carbon support has decomposed around them, leaving large void spaces that allow for larger crystalline particles. These are very effective at graphite growth in close proximity but leave amorphous carbon in much of the structure.Abbreviations
BET, Brunauer, Emmett, and Teller adsorption model; ccp, cubic close packed; EDX, energy dispersive X-ray spectroscopy; GC–MS, gas chromatography–mass spectrometry; hcp, hexagonal close packed; HRTEM, high resolution transmission electron microscopy; SAED, specific area electron diffraction; SEM, scanning electron microscopy; TEM, transmission electron microscopy; TGA, thermal gravimetric analysis; XRD, X-ray diffraction.Throughout ‘foam’ is used to refer to a porous 3D structure of solid material separated by open void spaces irrespective of production method.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a grant from the Engineering and Physical Sciences Research Council (EPSRC), UK (Ref number EP/N509462/1). This research used the GJ Russell Microscopy Facility at Durham University and the authors wish to thank Mr L. Bowen and Dr B. Mendis for their assistance with electron microscopy.Notes and references
- D. A. C. Brownson and C. E. Banks, Phys. Chem. Chem. Phys., 2012, 14, 8264–8281 RSC.
- C. Xu, B. Xu, Y. Gu, Z. Xiong, J. Sun and X. S. Zhao, Energy Environ. Sci., 2013, 6, 1388 RSC.
- N. Zhang, M.-Q. Yang, S. Liu, Y. Sun and Y.-J. Xu, Chem. Rev., 2015, 115, 10307–10377 CrossRef CAS PubMed.
- V. Kumar, K.-H. Kim, J.-W. Park, J. Hong and S. Kumar, Chem. Eng. J., 2017, 315, 210–232 CrossRef CAS.
- L. Jiang and Z. Fan, Nanoscale, 2014, 6, 1922–1945 RSC.
- Y. Li, Y. Lu, Q. Meng, A. C. S. Jensen, Q. Zhang, Q. Zhang, Y. Tong, Y. Qi, L. Gu, M.-M. Titirici and Y.-S. Hu, Adv. Energy Mater., 2019, 9, 1902852 CrossRef CAS.
- M. T. H. Aunkor, I. M. Mahbubul, R. Saidur and H. S. C. Metselaar, RSC Adv., 2016, 6, 27807–27828 RSC.
- K. Hu, X. Xie, T. Szkopek and M. Cerruti, Chem. Mater., 2016, 28, 1756–1768 CrossRef CAS.
- L. Stobinski, B. Lesiak, A. Malolepszy, M. Mazurkiewicz, B. Mierzwa, J. Zemek, P. Jiricek and I. Bieloshapka, J. Electron Spectrosc. Relat. Phenom., 2014, 195, 145–154 CrossRef CAS.
- S. Pei and H.-M. Cheng, Carbon N. Y., 2012, 50, 3210–3228 CrossRef CAS.
- Z. Chen, W. Ren, L. Gao, B. Liu, S. Pei and H.-M. Cheng, Nat. Mater., 2011, 10, 424–428 CrossRef CAS PubMed.
- K. Chen, L. Shi, Y. Zhang and Z. Liu, Chem. Soc. Rev., 2018, 47, 3018–3036 RSC.
- S. Drieschner, M. Weber, J. Wohlketzetter, J. Vieten, E. Makrygiannis, B. M. Blaschke, V. Morandi, L. Colombo, F. Bonaccorso and J. A. Garrido, 2D Mater., 2016, 3, 45013 CrossRef.
- L. Shi, K. Chen, R. Du, A. Bachmatiuk, M. H. Rümmeli, K. Xie, Y. Huang, Y. Zhang and Z. Liu, J. Am. Chem. Soc., 2016, 138, 6360–6363 CrossRef CAS PubMed.
- R. W. Pekala and C. T. Alviso, MRS Online Proc. Libr., 1992, 270, 3 CrossRef CAS.
- S. Jun, S. H. Joo, R. Ryoo, M. Kruk, M. Jaroniec, Z. Liu, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 2000, 122, 10712–10713 CrossRef CAS.
- F. J. Maldonado-Hódar, C. Moreno-Castilla, J. Rivera-Utrilla, Y. Hanzawa and Y. Yamada, Langmuir, 2000, 16, 4367–4373 CrossRef.
- M. Zhong, J. Yan, H. Wu, W. Shen, J. Zhang, C. Yu, L. Li, Q. Hao, F. Gao, Y. Tian, Y. Huang and S. Guo, Fuel Process. Technol., 2020, 198, 106241 CrossRef CAS.
- D. Sun, X. Yu, X. Ji, Z. Sun and D. Sun, J. Alloys Compd., 2019, 805, 327–337 CrossRef CAS.
- K. Wang, Y. Cao, X. Wang, P. R. Kharel, W. Gibbons, B. Luo, Z. Gu, Q. Fan and L. Metzger, Energy, 2016, 101, 9–15 CrossRef CAS.
- F. J. Derbyshire, A. E. B. Presland and D. L. Trimm, Carbon N. Y., 1975, 13, 111–113 CrossRef CAS.
- Q. Yan, J. Li, X. Zhang, E. B. Hassan, C. Wang, J. Zhang and Z. Cai, J. Nanopart. Res., 2018, 20, 223 CrossRef.
- W. Kiciński, M. Bystrzejewski, M. H. Rümmeli and T. Gemming, Bull. Mater. Sci., 2014, 37, 141–150 CrossRef.
- F. Su, J. Zeng, X. Bao, Y. Yu, J. Y. Lee and X. S. Zhao, Chem. Mater., 2005, 17, 3960–3967 CrossRef CAS.
- R. Fu, T. F. Baumann, S. Cronin, G. Dresselhaus, M. S. Dresselhaus and J. H. Satcher, Langmuir, 2005, 21, 2647–2651 CrossRef CAS PubMed.
- Q. Yan, J. Li, X. Zhang, J. Zhang and Z. Cai, Nanomater. Nanotechnol., 2018, 8, 184798041881895 CrossRef.
- C. J. Thambiliyagodage, S. Ulrich, P. T. Araujo and M. G. Bakker, Carbon N. Y., 2018, 134, 452–463 CrossRef CAS.
- M. Sevilla, C. Sanchís, T. Valdés-Solís, E. Morallón and A. B. Fuertes, J. Phys. Chem. C, 2007, 111, 9749–9756 CrossRef CAS.
- T. Hyeon, S. Han, Y.-E. Sung, K.-W. Park and Y.-W. Kim, Angew. Chem., Int. Ed., 2003, 42, 4352–4356 CrossRef CAS PubMed.
- S. Han, Y. Yun, K.-W. Park, Y.-E. Sung and T. Hyeon, Adv. Mater., 2003, 15, 1922–1925 CrossRef CAS.
- A.-H. Lu, W.-C. Li, E.-L. Salabas, B. Spliethoff and F. Schüth, Chem. Mater., 2006, 18, 2086–2094 CrossRef CAS.
- J. Yuan, C. Giordano and M. Antonietti, Chem. Mater., 2010, 22, 5003–5012 CrossRef CAS.
- Y. Liu, Y. Guan and K. Zhang, J. Anal. Appl. Pyrolysis, 2019, 138, 196–202 CrossRef CAS.
- S. Glatzel, Z. Schnepp and C. Giordano, Angew. Chem., Int. Ed., 2013, 52, 2355–2358 CrossRef CAS PubMed.
- G. Zhong, L. Ma, C. Yan, P. Zhuang and X. Ma, Chem. Eng. J., 2020, 398, 125545 CrossRef CAS.
- M. Sevilla, C. Sanchís, T. Valdés-Solís, E. Morallón and A. B. Fuertes, Carbon N. Y., 2008, 46, 931–939 CrossRef CAS.
- M. Sevilla and A. B. Fuertes, Mater. Chem. Phys., 2009, 113, 208–214 CrossRef CAS.
- A. Ōya and S. Ōtani, Carbon N. Y., 1979, 17, 131–137 CrossRef.
- W. Weisweiler, N. Subramanian and B. Terwiesch, Carbon N. Y., 1971, 9, 755–761 CrossRef CAS.
- T. Liu, F. Zhang, Y. Song and Y. Li, J. Mater. Chem. A, 2017, 5, 17705–17733 RSC.
- W. Chen, S. Li, C. Chen and L. Yan, Adv. Mater., 2011, 23, 5679–5683 CrossRef CAS PubMed.
- Y. Li, K. Yan, H.-W. Lee, Z. Lu, N. Liu and Y. Cui, Nat. Energy, 2016, 1, 15029 CrossRef CAS.
- PCT Int. Appl., The Regents of the University of California, USA, 2017, p. 20 Search PubMed.
- Z. Ma, T. Kyotani and A. Tomita, Chem. Commun., 2000, 2365–2366 RSC.
- A. E. Danks, M. J. Hollamby, B. Hammouda, D. C. Fletcher, F. Johnston-Banks, S. E. Rogers and Z. Schnepp, J. Mater. Chem. A, 2017, 5, 11644–11651 RSC.
- Z. Schnepp, M. Thomas, S. Glatzel, K. Schlichte, R. Palkovits and C. Giordano, J. Mater. Chem., 2011, 21, 17760–17764 RSC.
- J. Hoekstra, A. M. Beale, F. Soulimani, M. Versluijs-Helder, J. W. Geus and L. W. Jenneskens, J. Phys. Chem. C, 2015, 119, 10653–10661 CrossRef CAS.
- M. K. Tynan, D. W. Johnson, B. P. Dobson and K. S. Coleman, Nanoscale, 2016, 8, 13303–13310 RSC.
- S.-X. Guo, Y. Liu, A. M. Bond, J. Zhang, P. Esakki Karthik, I. Maheshwaran, S. Senthil Kumar and K. L. N. Phani, Phys. Chem. Chem. Phys., 2014, 16, 19035–19045 RSC.
- F. Khan and S. Mann, J. Phys. Chem. C, 2009, 113, 19871–19874 CrossRef CAS.
- S. J. Goldie, S. Bush, J. A. Cumming and K. S. Coleman, ACS Appl. Nano Mater., 2020, 3, 11229–11239 CrossRef CAS.
- B. H. Toby and R. B. Von Dreele, J. Appl. Crystallogr., 2013, 46, 544–549 CrossRef CAS.
- E. A. Owen and D. M. Jones, Proc. Phys. Soc., London, Sect. B, 1954, 67, 456 CrossRef.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671 CrossRef CAS PubMed.
- Z. Q. Li, C. J. Lu, Z. P. Xia, Y. Zhou and Z. Luo, Carbon N. Y., 2007, 45, 1686–1695 CrossRef CAS.
- A. C. Ferrari and D. M. Basko, Nat. Nanotechnol., 2013, 8, 235–246 CrossRef CAS PubMed.
- A. Ōya and H. Marsh, J. Mater. Sci., 1982, 17, 309–322 CrossRef.
- J. W. Long, M. Laskoski, T. M. Keller, K. A. Pettigrew, T. N. Zimmerman, S. B. Qadri and G. W. Peterson, Carbon, 2010, 48, 501–508 CrossRef CAS.
- J. R. Partington and R. P. Towndrow, Trans. Faraday Soc., 1939, 35, 553–559 RSC.
- T. Cseri, S. Békássy, G. Kenessey, G. Liptay and F. Figueras, Thermochim. Acta, 1996, 288, 137–154 CrossRef CAS.
- C. Breen, G. Thompson and M. Webb, J. Mater. Chem., 1999, 9, 3159–3165 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Thermal analysis, XRD indexing, Raman spectra, EM images and particles sizing, gas sorption and GC–MS. See DOI: 10.1039/d1ma00125f |
| This journal is © The Royal Society of Chemistry 2021 |