
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent progress in materials development for CO2 conversion: issues and challenges
Sourav
Ghosh
 a,
Arindam
Modak
a,
Arnab
Samanta
b,
Kanika
Kole
a and
Subhra
Jana
*ab
a,
Arindam
Modak
a,
Arnab
Samanta
b,
Kanika
Kole
a and
Subhra
Jana
*ab
aTechnical Research Centre, S. N. Bose National Centre for Basic Sciences, Block-JD, Sector-III, Salt Lake, Kolkata 700 106, India. E-mail: subhra.jana@bose.res.in; Tel: +91 33 2335 5706
bDepartment of Chemical, Biological & Macro-Molecular Sciences, S. N. Bose National Centre for Basic Sciences, Block - JD, Sector-III, Salt Lake, Kolkata 700 106, India
First published on 19th March 2021
Abstract
Increasing levels of CO2 emissions from our lifestyles impart a detrimental effect on human lives. To diminish the adverse effect of excessive CO2 in the atmosphere, porous architectures with inimitable microstructural features have been developed for the purposes of capture and storage. However, a critical element for ecological balance is reducing waste. This has further encouraged the research community to utilise captured CO2, since it is a cheap industrial waste. In this review, we have illustrated the development of strategies for CO2 utilization with an emphasis on CO2 capture, particularly highlighting the production of valuable chemicals. Numerous approaches have been elicited for the conversion of CO2 into value-added chemicals, such as cyclic carbonates, alcohols, dimethyl ether, olefins, aromatics and many more. For such aforementioned conversions, research interest has covered metal oxides (silica, zeolite, alumina and transition metal oxide), porous polymers, metal organic frameworks (MOFs), zeolitic imidazolate frameworks (ZIFs) and carbonaceous materials together with several metal NPs (NP). The most conventional state-of-the art strategy is immobilizing metallic or metal oxide NPs over porous networks, employing it as a catalyst for the production of agrochemicals, pharmaceuticals and fuels. The performance of the catalysts can be precisely tuned by morphological selectivity, surface area, thermal stability, elemental composition, mechanical inertness, ultra-fine porosity, etc. The efficiency of the catalyst has been further improved on the basis of yield of product, selectivity and reusability. This review demonstrates the latest tactics to fabricate different types of catalyst for CO2 fixation in order to achieve valuable chemicals with efficacy reaching towards commercial demand. In addition, future considerations to fabricate heterogeneous catalysts with microstructural diversity comprising potential active sites, are discussed, keeping an eye on the probable challenges in the context of transformation of CO2 to the value-added products.
 Sourav Ghosh | Dr Sourav Ghosh earned his BSc degree in chemistry from Calcutta University, and MSc degree in applied chemistry from Bengal Engineering and Science University, Shibpur. He received his PhD in 2016 from the CSIR-Central Glass and Ceramic Research Institute. He was awarded a SERB-National Postdoctoral Fellowship from the Department of Science & Technology, India, and started his research career as a postdoctoral fellow at the Indian Institute of Science Education and Research, Kolkata. He now works as a project scientist-C at the Technical Research Centre at S. N. Bose National Centre for Basic Sciences, India. His research efforts primarily focus on the design and utilization of porous materials for catalytic applications. |
 Arindam Modak | Dr Arindam Modak obtained his PhD degree from the Indian Association for the Cultivation of Science and subsequently completed his post-doctoral research in Japan and Israel for a couple of years. He worked as a project scientist-C at the S. N. Bose National Centre for Basic Sciences, Kolkata, and was also a recipient of the Chinese Academy of Science, CAS President's International Fellowship Initiative award in 2017 from China. He is interested in the rational design of novel porous materials for applications in catalysis and gas adsorption. |
 Arnab Samanta | Arnab Samanta completed his MSc Degree in chemistry at Jadavpur University, India. He is currently working as a senior research fellow at S. N. Bose National Centre for Basic Sciences, India. His present research area mainly focuses on the development of different transition metal-based nanomaterials and their applications in the field of catalysis. |
 Kanika Kole | Kanika Kole is currently working as a PhD Scholar at the S. N. Bose National Centre for Basic Sciences, India. She received her MSc degree in chemistry from Vidyasagar University, India, and BSc in chemistry from Burdwan University, India. Her research focuses on the development of inorganic–organic hybrid porous nanomaterials for CO2 adsorption and conversion. |
 Subhra Jana | Dr Subhra Jana obtained her PhD in chemistry from the Indian Institute of Technology, Kharagpur. She moved to Pennsylvania State University, USA, for her postdoc, then worked under the DST INSPIRE Faculty program at S. N. Bose National Centre for Basic Sciences, India. She remains here, working as a scientist-D at the Technical Research Centre. She was awarded the Young Associate Award from the Indian Academy of Sciences, Bangalore, and the SERB Women Excellence Award from the Department of Science & Technology, India. Her multi-disciplinary research focuses on the design and synthesis of alloys, intermetallics and hybrid nanocomposites for environmental remediation and heterogeneous catalysis. |
1. Introduction
The prompt rise of anthropogenic CO2 emissions has emerged as a main reason for climate change and has subsequently been instigated in global warming. Serious attention is needed to address the outcome of deforestation, fossil fuel consumption and industrialisation.1–4 To mitigate the severe impact of greenhouse gas emissions, the carbon capture strategy has turned out to be a key concern for storage and utilization purposes.5–7 In this context, the preparation of capture materials to be used as adsorbents, namely porous organic polymers (POPs), metal organic frameworks (MOFs), zeolite and zeolitic imidazolate frameworks (ZIFs), carbonaceous materials, and graphene based composites, has received remarkable attention in recent times. Furthermore, CO2 is known as the most economically abundant source of C1 feed stocks.8–10 Nowadays, the chemical conversion of CO2 to value-added chemicals has drawn incredible focus as we strive to develop a clean energy system. However, the thermodynamic stability of CO2 restricts its bulk scale implementation in the chemical industry.11 Notably, selective capture as well as sequestration of CO2 is less tricky than CO2 transformation, due to its non-polarity, weak acidity and kinetic inertness.12,13 As a consequence, high pressure and temperature are usually required to activate CO2 for chemical transformation. To eliminate the need for such drastic conditions, the fabrication of catalysts is absolutely necessary for industrial scalable production of value-added chemicals. Thus, a strategy for both CO2 adsorption and utilization is crucial to improve the conversion of CO2 into value-added products.Nowadays, CO2 capture and storage (CCS) technology has been treated as a key global target by the scientific community. Such a strategy can achieve the cost-effective production of a CO2 stream at high pressure, available for storage.14 Conversion of CO2 to industrially valuable products would be an alternative approach for CO2 capture and utilization (CCU) technology, unlocking several barriers to low-carbon emission energy production.15–18 Execution of CCU methods primarily includes CO2 diffusion and adsorption on the catalytic surfaces, followed by transformation into products and then desorption from active sites.19,20 The CO2 capture material could be adsorbent and/or catalytic. Thus, suitable adsorbents for CO2 capture are required for the implementation of both CCS and CCU technologies. The adsorption of CO2 usually takes place through either chemisorption via chemical interaction, or physisorption, involving weak van der Waals forces, electrostatic interactions, dipole–dipole forces, hydrophobic associations etc.21 The bond energy for chemisorbed species is about 60–418 kcal mol−1, thereby indicating permanent adsorption of toxic gas by a solid support. However, 8–41 kcal mol−1 is encountered for physisorption that elucidates an easy regeneration process for the solid adsorbent. The efficiency of adsorbents could be governed by several factors, such as adsorption capacity, sorption kinetics, heat of adsorption, selectivity, stability, etc. For gas adsorption technology, selectivity is an important parameter which depends onto the physicochemical features of gases, like quadruple moment, kinetic diameter, dipole moment and polarizability.
An advance has been achieved for the production of alternate fuels from CO2 hydrogenation. Among the C1 products, the direct synthetic route has gained immense interest, particularly for the preparation of methane (CH4), carbon monoxide (CO), methanol (CH3OH) and formic acid (HCOOH).22,23 Catalytic conversion of CO2 into CH4 can be tuned with gas hourly space velocity (GHSV) at atmospheric pressure. A high yield with surprising selectivity can be correlated with the theoretical equilibrium model and has been reported as an expedient method for CH4 production.24 Again, a reverse water gas shift (RWGS) reaction is regarded as the most suitable way to produce CO.25 The economic interest of methanol as an alternate fuel can never be suppressed in the commercial market. For the hydrogenation-steam reforming cycle, methanol is regarded as a scalable form of hydrogen under ambient conditions. The problem associated with storage, transport and distribution of hydrogen can be easily avoided by considering methanol as a liquid hydrogen carrier.26 Likewise, enormous effort has been observed for the fabrication of a catalytic framework consisting of ZrO2 with copper, for the production of methanol at atmospheric pressure.27 CO2 fixation can be further extended for the synthesis of formic acid by the catalytic reduction method.28 Again, the requirement of a catalyst is extremely essential for the supply of valuable chemical products containing more than one carbon (C2+). In this context, methanol is also well-established as an efficient intermediate for the preparation of dimethylether (DME), which is a valuable fuel for diesel engines.29 Not only DME, but several other chemicals such as higher alcohols, olefins, liquid fuels, etc. can be achieved by catalytic conversion of CO2. The intricacy regarding the formation of C2+ chemicals from CO2 is well-known due to the extremely large C–C coupling energy barrier.30 However, the high energy density and economic value of C2+ products compared to C1 chemicals have encouraged attention from industry. On other side, intensive enthusiasm has been observed for the conversion of epoxide into cyclic carbonate using CO2 at ambient conditions. The utility of CO2 as a carbonyl source is extremely useful from both the environmental and waste-material management point of view. Further, cyclic carbonate can be employed as a polar aprotic solvent in organic synthesis, a medium for lithium-ion batteries (LIB), and a precursor for polycarbonate.31 Moreover, the importance of cyclic carbonate as a core of organic drug molecules is well-known for pharmaceutical companies. The industrial synthesis of cyclic carbonate is carried out using phosgene of C1 origin, and 1,2-diol. However, the renowned toxicity of phosgene limits its use for the bulk scale production of cyclic carbonate in today's scenario.32 Other value-added products, hydroxy carbamates, have multiple applications in coating, substrate design, organic synthesis and in medicine.33–35 The commercial preparation of hydroxy carbamates involves a chemical reaction between cyclic carbonate and organic amine in the presence of an efficient catalyst.36 However, the proper design of a catalytic network is necessary for the production of such valuable chemicals. Still today, bulk scale synthesis of urea is one of the convincing chemical routes for CO2 fixation.37,38 In this comprehensive review, we have highlighted the latest developments and strategy, mostly in CO2 utilization with a brief emphasis on CO2 capture, particularly highlighting the production of valuable chemicals.
2. Carbon capture
CCS technology has turned out to be a cutting-edge research area in recent times. To reduce CO2 emissions from power plants, primarily three types of separation technique have been proposed: the pre-combustion method, post-combustion technique and oxyfuel combustion approach.39 For concentrated CO2 sources, conventional cryodistillation and sorption techniques are usually employed. Nevertheless, physical adsorption technology has been regarded as the most convenient at dilute levels, such as biogas, natural gas, syngas, etc.40 In addition, natural gas purification has tremendous economic importance in the commercial business market. Carbon dioxide capture from air under ambient conditions is quite tricky and noteworthy. Nowadays, direct air capture (DAC) is a well-known strategy, since it often deals with CO2 at ultra-dilute conditions.41,42 Several materials have shown potential for DAC purposes,43–49 such as carbonaceous adsorbents, zeolitic materials, MOFs, alkali metal carbonate, amine loaded solid adsorbents, etc. Several potential adsorbents for direct CO2 capture are presented in Fig. 1. DAC methods not only regulate the CO2 level in the atmosphere but also offer leakage insurance from CCS sites or geologic resource units. DAC methods can be operated anywhere and remove the costly mechanical set-up required for CO2 transport. A CCS approach could assist in reducing excessive CO2 emissions and reform the ecological milieu comprising soil, oceans and air in a sustainable way. | ||
| Fig. 1 Crystal structures of different (A) PC-POP,50 (B) MOF MUF-17,51 (C) usf-ZMOF,52 (D) UTSA-49,53 (E) COF-554 and (F) poly(benzimidazobenzophenanthroline) or BBL polymer.55 All the figures are reproduced with permission from the corresponding references. | ||
Traditionally, liquid amine-based technology is regarded as the most convenient method to capture CO2, which is a highly quadrupole gas.56 The excellent performance of this technology can be attributed to the high capacity, rapid kinetics, moderate cyclic loading and fast mass transfer rate. Among liquid amines, monoethanolamine (MEA) is the most widely employed for CO2 capture because it can achieve 90% adsorption capacity at atmospheric pressure.57–59 Apart from MEA, several other amines, e.g., diethanolamine (DEA), methyldiethanolamine (MDEA), diethylenetriamine (DETA), triethylenetetramine (TETA), aminoethylethanolamine (AEEA), have been employed for the same issue.60–63 In addition, polyamine is usually used to enhance the adsorption capacity via interaction with CO2 through multiple amino groups.64 The reaction trajectory can be proposed in two step modes, where liquid amine (R-NH2) interacts with CO2 to form zwitterion (R-NH2+-CO2−) intermediate species followed by conversion into carbamate (R-NH-CO2−) via instantaneous neutralisation by liquid amine or hydroxide ions or water molecules.65–67 The carbamate associated mechanistic portfolio was further proved via free energy calculations and kinetics studies by Xie et al.68 However, the probability of CO2 removal from the intermediate could not be omitted under the experimental scenario.69 Thus, control over the transformation route in the reaction path can be considered as an important parameter for adequate yield of product. However, the reaction footprint for CO2 fixation utilizing MEA was also reported by the formation of carbamic acid in an intermediate step that was subsequently turned into carbamate via an MEA assisted catalytic reaction.70 Although, numerous research has aimed to elucidate the actual mechanism, reaction steps with the distinct formation of transient intermediate species are still controversial from a chemical absorption and weak force interaction point of view. Even after wide interest, the high volatility and corrosive nature of liquid amine is considered a major bottleneck for this technology. Furthermore, the tedious regeneration process restricts the bulk scale execution of liquid amine-based adsorption technology in reality.71
On the contrary, low cost and sustainability have inspired the implementation of adsorption-based CO2 capture technology to overcome the practical problem. Amine grafting or amine impregnation over a solid support has been demonstrated as a preparative strategy to enhance the CO2 capture performance. Increasing the alkalinity of the solid support has been regarded as the main aim of such strategies due to the inherent acidic nature of CO2. Usually, an amine grafting approach renders a faster adsorption rate and long cyclic stability in comparison to an amine impregnation technique. In addition, the amount of amine loading is usually high for the impregnation process, thus inducing high diffusion resistance during the adsorption process.72 Song and co-workers designed a polyethylenimine (PEI) impregnated carbonaceous and silica-based mesoporous network, and demonstrated the influence of moisture, temperature and PEI loading towards CO2 adsorption capacity.73–76 Despite the decrease of surface area and porosity, CO2 adsorption performance was significantly enhanced with increasing PEI loading. Even after achieving high CO2 adsorption capacity, poor thermal stability limits the orthodox use of this strategy on a regular basis.77 Notably, the amine grafting strategy through silylation method has motivated researchers to overcome the limitation of the amine impregnation technique. In 1995, Leal et al. proposed (3-aminopropyl)triethoxysilane (APS) modified silica gel for the same issue and described the CO2 adsorption trajectory through the formation of ammonium bicarbonate in the presence of moisture and ammonium carbamate under anhydrous conditions.78 A tremendous amount of work was carried out by Jones and his group and they demonstrated several amine containing oxide supports as excellent adsorbents for CO2 capture under ambient air conditions.45,79 The amine grafting approach with (3-aminopropyl)triethoxysilane (APS), N-[(3-trimethoxysilyl)propyl]ethylenediamine (2N-APS) and N-[(3-trimethoxysilyl)propyl]diethylenetriamine (3N-APS), was further investigated on pore-swelled SBA-15, and the capacity compared with conventional SBA-15 and MCM-41.80 This study showed that a larger amount of amine can accommodate within the pores of SBA-15 without any blocking during the adsorption process. Again, aminosilane grafting was more facile in the larger pore size, and the highest CO2 adsorption performance was attained for 3N-APS. Jana and co-workers designed uniform dandelion-like mesoporous silica nanoflowers by a light driven synthetic approach with controllable surface area and porosity. By loading different amines on the surface of mesoporous silica nanoflowers, they designed several solid adsorbents which showed excellent performance towards CO2 capture for long term usage (Fig. 2).81 They also reported clay nanotubes based on several solid adsorbents loaded with different aminosilanes, for atmospheric CO2 capture at ambient temperature and pressure.44 The authors also demonstrated the influence of relative humidity and how the relative humidity tunes the atmospheric CO2 adsorption capacity of an adsorbent by performing the study using seasonal ambient air (Fig. 2E and F). The adsorption kinetics of abundant isotopes of CO2 (12C16O2, 13C16O2, and 12C16O18O) present in the ambient air have also been discussed in detail. Recently, the same group also developed chromogenic nanocomposites (NCs) through the functionalization of silica nano-flowers with phenolphthalein as chromogen for the visual detection and estimation of CO2. The concentration dependent CO2 uptake by the chromogenic NCs was detected by the naked eye through colour change. To increase accuracy, a spectroscopic tool using the developed optical device (CapNanoScope) was established for quantitative measurement of CO2 with good selectivity (Fig. 3).82 Singh et al. reported the preparative strategy to achieve functionalized fibrous silica (KCC-1) using different types of amine through physisorption or covalent attachment for CO2 uptake. They also elucidated the benefits of functionalized fibrous silica (KCC-1) over conventional silica in terms of controllable textural properties for post-functionalized silica, convenient amine loading amount and accessible amine sites.83 Their approach claimed that porous fibrous KCC-1 could be a possible alternative over traditional MCM-41 and SBA-15. Recently, silica nanosheets have been prepared by a wet-chemical technique in water–cyclohexane mixed solvent, and attained a large surface area (1420 m2 g−1) and huge pore volume (3.47 cm3 g−1).84 After surface functionalization using tetraethylenepentamine (TEPA) molecules, they showed a CO2 uptake value of 3.8 mmol g−1 at 75 °C with rapid adsorption kinetics and long-term stability. Despite the extensive usage of silica materials, porous alumina (Al2O3) has also been employed as an alternative candidate for CO2 adsorption. Yong et al. utilized commercial alumina as an adsorbent for CO2 uptake and showed uptake capacity of approximately 0.30 mmol g−1 at 300 °C and 1 bar.85 In this regard, mesoporous γ-Al2O3, with controllable surface area, exclusive pore volume, narrow pore-size distribution and nanocrystallinity was synthesized by a solution-combustion and ball-milling method.86 These γ-Al2O3 also displayed 1.94 mmol g−1 CO2 adsorption performance at 60 °C and 1.5 MPa pressure. Recently, hydroxide modified activated alumina granules have been used for the same and achieved an adsorption capacity of about 146.70 and 130.84 mg g−1 at 20 °C and 6 bar pressure for NaOH and KOH, respectively.87 Ramaprabhu and co-workers prepared graphene based Fe3O4 nanocomposites having CO2 adsorption performance of 60, 35, and 24 mmol g−1 at 11 bar pressure at 25, 50, and 100 °C, respectively.88 Conventional inorganic metal oxides have also been explored as an adsorbent for CO2 uptake over the years. There are numerous reports on other adsorbents for CO2 capture, such as lithium zirconates, strontium titanates, hydrotalcites, calcium oxide, magnesium oxide, etc.89–91 Lithium containing material supports usually exhibit superb performance, but excessive diffusion resistance impedes their application for real-world problems.92 However, the importance of calcium supported materials is well-known for their large abundance, low market price, high adsorption efficiency, rapid kinetics and excellent stability.93 The stoichiometric reaction between calcium oxide (CaO) and CO2 forms calcium carbonate (CaCO3) at high temperature, and recyclability is performed after thermal decarbonation treatment. The tuning of synthetic parameters results in the formation of calcium supported materials with controllable pore architecture, which further activates the CO2 adsorption process. Adsorption performance can be further improved by incorporation of additives, like MgO, NaCl, CaTiO3, etc. within the material network, or reactivation treatment by steam hydration.94 Extraordinary CO2 capture performance with long-term cycling stability was further obtained for the CaO/Ca12Al14O33 matrix by Li et al.95 In this respect, CaO dispersed in the ordered network of SBA-15 exhibited adsorption performance of about 10 mol kg−1 with relatively high sorbent stability.96
 | ||
| Fig. 2 (A and B) FESEM images of dandelion flower-like SiO2 with hierarchical assembly. Inset image shows the architecture of the dandelion flower in figure (B). (C) Panoramic TEM image of flower-like SiO2 and (D) high resolution view of the distinct flower-like SiO2 with elongated spikes. (E) Pictorial diagram showing functionalization strategy of SiO2via covalent attachment of aminosilanes. Reproduced with permission from ref. 81. (F) Pictographic image of clay-based nanocomposites for CO2 fixation from seasonal ambient air. The figure is reproduced with permission from ref. 44. | ||
 | ||
| Fig. 3 Chromogenic silica nanoflowers-based optical device (CapNanoScope) has been developed for the detection and estimation of CO2 in the presence of environmentally relevant gases at ambienttemperature and pressure. The figure is reproduced with permission from ref. 82. | ||
Likewise, layered double hydroxides (LDHs), classified as clay material with exposed anionic or basic sites, can also be employed for CO2 adsorption.97 Costa and co-workers reported Mg–Al–CO3 based layered double hydroxide for CO2 capture from flue gases at elevated temperature up to 200 °C.98 Even after multiple cycles, regenerated adsorbent attained an adsorption capacity up to 98% of its initial intake. Huang et al. studied LiAl2-layered double hydroxides for CO2 fixation under controllable experimental conditions, such as the Li/Al ratio in the synthetic process, annealing temperature, role of K2CO3 as an additive, impact of charge compensating anions and adsorption temperature.99 By optimal doping with K2CO3, CO2 intake performance was significantly increased up to 1.27 and 0.83 mmol g−1 at 60 and 200 °C, respectively. The Li/Al hydrotalcite network can be strategically modified with polyethylenimine (PEI) loading for the same purpose.100 The fabrication of layered double oxide (LDO) grafted with exfoliated reduced graphene oxide (rGO) and layered titanate nanosheets showed a synergistic impact on CO2 adsorption compared to their pristine LDO as well as LDO–rGO composite.101 Their strategy unequivocally corroborates the importance of restacked assembly of rGO nanosheets consisting of titanate counter parts to construct splendid hybrid adsorbents for CO2 capture. In recent time, Xia and co-workers have mechanistically manifested the structural tuning of bulk ZnAl layered double hydroxides (ZnAl-LDHs) to 2D monolayer ZnAl-LDHs (monolayer-LDHs) for low temperature CO2 intake applications.102 The experimental findings have been further computationally supported in terms of electronic activity of porous monolayer-LDHs.
Traditionally, ionic liquids (ILs) have been elucidated as “designer solvents” for CO2 activation. The use of ILs for CO2 fixation can be further extended to the commercial market due to their nano-volatility nature, intrinsic structural modulation and extensive affinity towards CO2.103 To enhance the inherent CO2 capture capacity, amine modified “task-specific” structural tuning has been designed for ionic-liquids.104,105 It is worth mentioning that the anion moiety of ILs has a pivotal impact on CO2 fixation compared to their cationic counter parts.106,107 Wang et al. introduced an equimolar combination of imidazolium-based ionic liquids and a super base for CO2 capture.108 The intake can be further improved using a super base ionic liquid-containing deep eutectic solvent strategy that operates synergistically along with the formation of carbamate and carbonate for IL and EG with CO2, respectively.109 On other hand, utilization of polymeric ionic liquids (PILs) has been considered a lot for bulk-scale capture in the industrial circuit.110 The benefit of PILs over ionic liquid monomers (ILMs) can be attributed to the basis of their macromolecular framework with strong intra-chain bindings, solid-state appearance, controllable meso-to-nano assembly, improved stability as well as durability and easy processibility.111–114 Luo and co-workers studied composite network comprising of PILs with a meso-silica core for broad applications in CCS.115 They also reported meso-SiO2-P[VBTMA][BF4] and SiO2-P[VBTMA][PF6] as adsorbents with CO2 intake values of 0.4025 and 0.3793 mmol g−1, respectively, for simulated flue gas containing 10 vol% CO2 at 30 °C.
Pragmatic scientific interest has also focused on the membrane based CO2 fixation approach due to its low cost, environmental compatibility and user friendly advantages. Monoethanolamine (MEA) based membrane modules are conventionally established for capture purposes.116 For industrial flue gas, where the concentration of CO2 is more than 11%, membrane properties are very important for bulk-mode implementation.117 Fu et al. investigated a thin film composite (TFC) within membrane reactor, modified by a permeable polydimethylsiloxane (PDMS) intermediate layer for effective CO2 capture applications.118 Qiao and co-workers reported a membrane with continuous assembly of polymers (CAP) technology, having high permeance as well as excellent gas separation selectivity.119 Recently, a solid-state facilitated transport membrane has been constructed by CO2-philic amine-compatible poly(vinyl alcohol)-g-poly(oxyethylene methacrylate) (PVA-g-POEM) graft copolymer and diethylenetriamine (DETA) carriers for CO2 fixation.120 Interestingly, a solid-state PVA-g-POEM membrane comprising of DETA, shows excellent stability without any degradation in efficiency for two weeks. Still, membrane based technology is limited in the commercial portfolio because of incompatible material design, complex engineering economics and dynamic mechanics.121 However, recyclability followed by reusability of the adsorbents is still a challenging task for daily users. Additionally, use of organic solvent during the grafting process restricts its implementation due to its high viscosity, low diffusivity and serious toxicity. For an alternative option, supercritical fluid (SCF) could be used instead of organic solvent for the grafting process. The low viscosity and high diffusivity of SCF enables accessibility of the porous framework, which indeed facilitates CO2 adsorption.122
To overcome the difficulties during regeneration of adsorbent, an efficient solid support could be designed with hierarchical porosity, consisting of a narrow and ultra-narrow microporous network. Porous organic polymers (POPs) are a renowned class of covalent bonded network of lighter elements like hydrogen (H), carbon (C), nitrogen (N), oxygen (O), etc.123 The state-of-the-art synthesis approach, particularly for porous aromatic frameworks (PAFs), hypercrosslinked polymers (HCPs), polymers of intrinsic microporosity (PIMs), crystalline triazine-based frameworks (CTFs), and conjugated micro-porous polymers (CMPs), already has been illustrated in the literature.124–126 POPs, having originated from robust monomers are efficient candidates for CO2 capture due to their textural property as well as mechanical stability.126 For high pressure CO2 uptake, controllable textural property is a critical parameter, which can assist condensation of CO2 inside the accessible pores after completion of monolayer coverage, and thus enhances the adsorption capacity. Best performance and selectivity could be achieved for the porous framework with a pore diameter close to 3.3 Å, as it is the kinetic diameter of the CO2 molecule.127Fig. 4 illustrates the distribution of CO2 and N2 gases in a nanoporous azobenzene polymer. HCPs are an interesting class of cross-linked materials with long-range connectivity, having mechanical inertness even after a long cycle.128 For HCPs, extensive capture and storage of CO2 could be facilitated by oxygen rich functional groups through dipole–quadruple interactions. In this context, MOFs can be recommended as an excellent alternative for CO2 capture and storage. MOFs are also known as 3-D coordinated polymers of metal unit and organic linkage, where a definite channel pore controls diffusion rate, thereby indicating selective separation of CO2 from flue gases.129 Yaghi and co-workers designed MOFs with important textural features, like interpenetrated channels, square channels, an amino functionalized network, and open metal site for CO2 capture.130–133 Furukawa et al. succeeded in preparing enormous porous MOF-210 from Zn4O(CO2)6 through expanding organic linkers. Furthermore, MOF-200 was prepared by slightly manipulating the synthetic parameters, and reported as an excellent adsorbent with a CO2 uptake capacity of ∼16.2 mmol g−1 at 25 °C and 50 bar pressure.134 Britt et al. demonstrated the fabrication of Mg-MOF-74 with an open magnesium site rendering a dynamic capacity of about 8.9 wt% with the benefit of regeneration at 80 °C.135 The adsorption performance was also examined for MIL-53, which showed a CO2 uptake capacity of 10.0 mmol g−1 at 30 bar pressure.136 In addition, microstructural shrinkage was encountered due to the interaction between the hydroxyl group and CO2 in MIL-53, while pore accessibility could be regained with increasing CO2 pressure.
 | ||
| Fig. 4 Pictorial presentation of gas distributions of N2 (blue) and CO2 (red) on (A) flat surface and (B) micropore composed of the same functional unit in nanoporous azobenzene polymer. The figure is reproduced with permission from ref. 127. | ||
Conventional zeolites are much more thermally stable than MOFs due to stronger Si–O and Al–O linkage. Unique features such as a cage-like porous network, zeolitic microporosity and mechanical inertness have attracted researchers for their potential applications in CO2 capture. Hedin and co-workers prepared alumina phosphate (AlPO4-17, AlPO4-18, AlPO4-53, and AlPO4-25) by a hydrothermal crystallization technique followed by annealing, demonstrating remarkable CO2 uptake capacity and selectivity.137 Furthermore, relative low water uptake was usually addressed, revealing the requirement for a lower energy cost during the regeneration process. In this regard, selective adsorption of CO2 from the binary mixture of CO2/CH4 was also achieved for zeolite 5A.138 Yang et al. designed cation-exchanged zeolitic chalcogenides (M@RWY) using a sequential ion-exchange approach particularly for Cs+, Rb+ and K+. Specifically, a CO2 adsorption efficiency of about 6.3 mmol g−1 was found by K+-exchanged chalcogenides (K@RWY) with isosteric heat of 35–41 kJ mol−1.139 This investigation proposes K+-exchanged chalcogenides (K@RWY) as an efficient candidate for CO2 capture with excellent reusability and a water tolerant nature. Zeolitic imidazolate frameworks (ZIFs) are precisely associated with the metal (M) and imidazole (Im) structural network with conventional zeolite-like topological features. Most importantly, the 145° bond angle is accounted for in the M–Im–M linkage, which is similar to the O–Si–O bond angle for the zeolitic framework. ZIFs have been known for their inimitable features, like mechanical inertness, porous architecture, controllable pore-size distribution and organic building blocks. In addition, the hydrophobic nature makes them stable under the influence of moisture, which is the most prominent feature despite having a resemblance to conventional MOFs and zeolites.140,141 For conventional ZIFs, CO2 uptake capacity was reported at about 3.12 mmol g−1 at 0 °C.142 Despite having identical topological characteristics, the higher surface area increases the number of accessible sites for van der Waals interactions with CO2, displaying higher adsorption performance. Besides the BET surface area, pore diameter and volume also have an intrinsic role in regulating gas uptake capacity. Chen et al. prepared rho-ZMOF and sod-ZMOF using 4,5-imidazoledicarboxylic acid and the best performance was obtained for K+-exchanged sod-ZMOF.143 In this context, the larger pore volume of rho-ZMOF allowed 25% more CO2 uptake than sod-ZMOF at high pressure, indicating that free space throughout the zeolitic network could be the predominant factor for rho-ZMOF. Aguado and co-workers described the aqueous based preparative approach of ZIF-93 using a stoichiometric amount of metal and ligand in the presence of ammonia at room temperature, obtaining a useful yield of about 80%.144 The advantages of this method are the use of eco-friendly solvent and low temperature, together with great activity towards CO2 uptake (∼8.0 and 1.6 mmol g−1 at 30 and 1 bar, respectively). To enhance the further permeability factor for CO2, a membrane was constructed from the composite of polymer (Matrimid 5218) and ZIF-8, which enabled free space to accommodate higher CO2 loading.145 For industrial implementation of technology, a super hydrophobic adsorbent with long-term stability is demanded by the day-to-day commercial market. Gao et al. introduced a pre-seeding strategy followed by a two-step crystallization technique to fabricate a composite network with commercial zeolite-5A core and ZIF-8 shell.146 The 5A@ZIF-8 composite can be used to achieve the dynamic hydrophobic character, high CO2 uptake capacity and high stability, even after several adsorption–desorption cycles.
For commercial purposes, zeolitic adsorbents could be employed due to their moisture sensitivity and optimized porosity. Again, expensive imidazole derivatives restrict implementation of ZIFs as adsorbents for real world problems. However, porous carbons have been claimed as potential adsorbents with the benefit of accessible surface sites, low cost, reduced hydrophilic nature and simple regeneration technique.147 By virtue of the precursor source, synthesis of carbonaceous materials has been reported from bio-mass, polymers, ionic-liquids, organic frameworks and carbohydrates.148 For the carbon derived adsorbents, distribution of micropores and density of surface functional groups are considered as key parameters for the adsorption. Initially, CO2 could be polarized over surface functional groups, followed by adsorption throughout the micro-porous network. Hierarchical porosity with interconnected network and controllable pore volume further triggers the diffusion dynamics, and subsequently enhances CO2 adsorption capacity for adsorbents. Gogotsi et al. demonstrated the correlation between textural properties and CO2 uptake capacity relative to pressure, and established that CO2 uptake was maximum for the pore size of about 0.5 nm at 0.1 bar, while pores around 0.8 nm were more significant at 1 bar.149 Despite having large surface area, the low porosity and large crystallite size restricted the diffusion of gases within the micro-porous carbonaceous network. To overcome the issue, numerous strategies, such as controllable nano-scale dimensions, introduction of mesoporosity, hierarchical connectivity and narrow pore size distribution, have been taken into consideration during the fabrication of such adsorbents.150–154 In this regard, improved adsorption kinetics were attained for sheet-like morphology due to the greater exposed surface site and shorter diffusion trajectory in comparison with spherical nanostructures.155 Kowalewski and co-workers synthesized nitrogen doped nano-carbon by pyrolysis of the block-copolymer containing polyacrylonitrile (PAN) and employed it for the selective capture of CO2.156 This study directly depicts the influence of nitrogen content and surface area on the adsorption capacity of CO2. Although CO2 capture has gained a lot of interest, storage in nano-pores could partially solve the carbon emission problem too, because the physisorbed CO2 could be released after several years underground. Again, the high cost associated with CO2 transportation and injection to geologically suitable storage sites, could make such a process energy-intensive and costly.
3. Conversion of CO2
To circumvent the aforementioned challenges, it is inevitable to convert the captured CO2 to value-added products through hydrogenation, cycloaddition, photocatalysis and electrocatalysis reactions.157 For simultaneous CO2 capture and conversion, materials should be designed with hierarchical porosity (interconnected macro-, meso- and micropores) for smooth diffusion of reactants to the catalytic sites. Therefore, a dual distribution of pores is required for catalytic applications; because micropores/ultra-micropores are good for CO2 storage but could be a hindrance in catalysis. It should be pointed out that CO2 is suitable as a C1 feedstock and has broad scope for serving as C1 to produce important products e.g., fuel, commodity chemicals, agrochemicals and valuable materials.158 Besides the aforesaid advantages, CO2 is a cheap and readily abundant resource. Thus, CO2 conversion is potentially meritorious to mitigate the adverse effect of green-house gas emissions. In Fig. 5, we have illustrated some important products, which can be derived from CO2. It can be stated that porous materials generally capture and store up to ∼10–40 wt% CO2 in their framework and the concentration of CO2 inside the pores may be tens to hundreds of times higher than that of gaseous CO2 in the ambient atmosphere, which is significant when CO2 is considered as a substrate. However, owing to the high chemical stability of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond (bond enthalpy +805 kJ mol−1), CO2 transformation would be challenging; hence, it requires high energy input to break the bond. To resolve this issue, those materials having high surface area, large adsorption capacity of CO2 and stable catalytic sites, would be significant for the capture and conversion of CO2 at atmospheric pressure and at room temperature.
O bond (bond enthalpy +805 kJ mol−1), CO2 transformation would be challenging; hence, it requires high energy input to break the bond. To resolve this issue, those materials having high surface area, large adsorption capacity of CO2 and stable catalytic sites, would be significant for the capture and conversion of CO2 at atmospheric pressure and at room temperature.
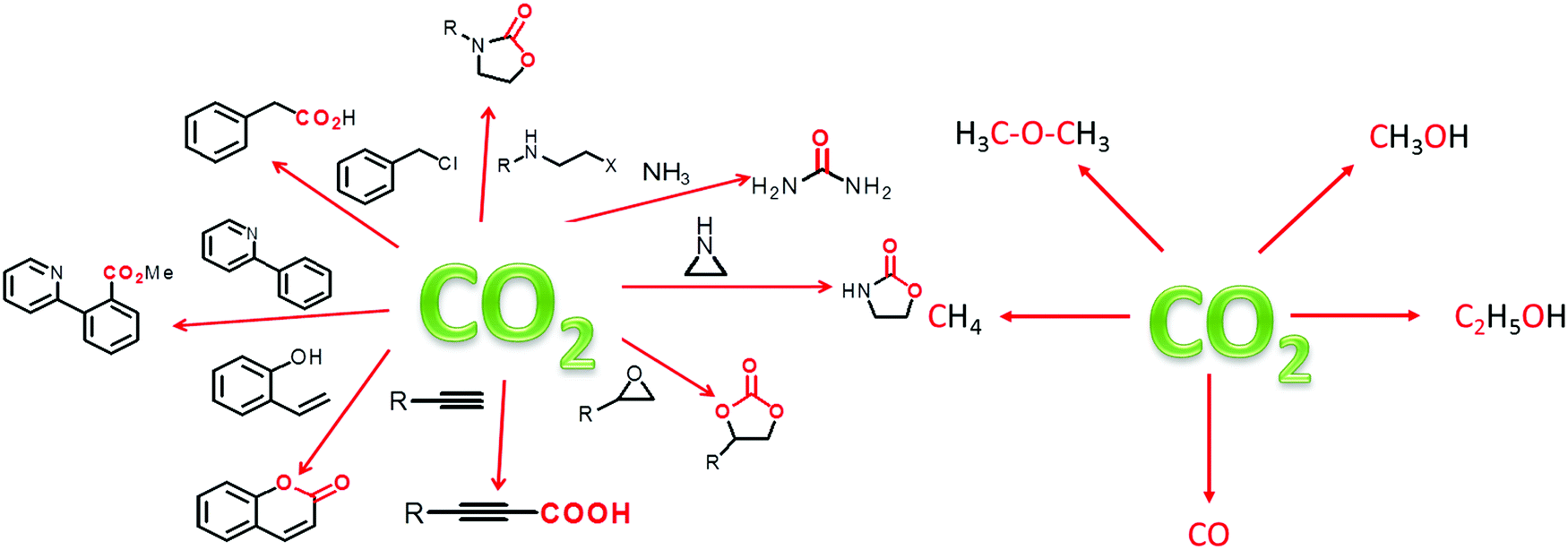 | ||
| Fig. 5 Schematic representation for the formation of different value added products obtained during CO2 conversion: (left) CO2 addition products and (right) CO2 reduction products. | ||
3.1. Metal oxides-based catalyst for CO2 conversion
Microstructural engineering of mesoporous metal oxide endows tremendous industrial awareness, particularly for the catalytic conversion of CO2. It can be believed that the oddity of formulation and defect design provides a diverse range of properties. Another potential outlook is to control the textural property of catalysts, which includes surface area, pore size, pore geometry and pore-size distribution. Further, d-electrons confined in the nano-domain along with long-range porosity and dynamic redox activity make mesoporous transition metal oxides much more interesting among traditional non-siliceous oxide materials.159 Apart from this, the influence of surface defects is regarded as a key issue behind the mechanical, optical, thermal, electrical and magnetic behavior of metal oxides.160–164 The prudent choice of metal oxides is due to their simple preparative strategy, earth abundance, low-cost and tuneable shape or size.165 Conventionally, a catalytic framework is an integrated system of active site and supporting material. Metal centres are considered as the active site, where metal oxides (such as SiO2, Al2O3, MgO, ZrO2, ZnO, CeO2, etc.) have been employed as the support system.166,167 The synergy between active site and support system results from the active site dispersion, distribution of defects, particle size, morphological impact and mechanical properties.168Cu/ZnO/Al2O3 is a well-known catalyst for methanol production under low pressure CO2/CO/H2. However, an excessive amount of CO reduces methanol production and also poisons the platinum (Pt) catalyst in fuel cells.169 Alumina is usually used for its high temperature stability and surface area support. For better activity, Al2O3 is replaced with ZrO2 which enables a weak hydrophilic interaction, thereby restricting the poisonous effect of water for the catalytic active center compared to Al2O3. Furthermore, ZrO2 delivers better Cu dispersion which provides a high CO2 adsorption rate and selectivity for methanol.170 Again, the ZrO2 incorporation can influence surface basicity, hydrogen dissociation, active intermediate formation and a mechanistic portfolio for methanol production. Amenomiya et al. compared the catalytic activity of Cu loaded on different supports, namely Cu/SiO2, Cu/Al2O3, Cu/TiO2, Cu/ZrO2 and Cu/ZnO. Among the different catalysts, the best catalytic performance was observed for Cu/ZrO2 in terms of efficiency of CO2 reduction and selectivity of methanol generation. The 40% CuO loaded in ZrO2 results in 4% conversion as well as 81% selectivity at 220 °C and 5 MPa.171 Notably, the monoclinic crystal structure of ZrO2 has been considered as more effective than tetragonal for such applications.172 Satokawa and co-workers achieved more selectivity towards methanol utilizing amorphous ZrO2 by suppressing the decomposition of methanol (Fig. 6).173 In2O3/ZrO2 can be used as a splendid catalyst for CO2 hydrogenation with 100% methanol selectivity under all tested experimental conditions (200–300 °C, 5 MPa, 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1) and 1000 h stability at 300 °C under streaming conditions. Here, ZrO2 helps to protect the In2O3 phase from high-temperature annealing, and is responsible for the uniform distribution of oxygen defects.174 Likewise, ZnO–ZrO2 can be treated as a catalyst to attain 10% CO2 conversion efficiency, with selectivity up to 91% for methanol.175 However, the addition of ZnO within the Cu/ZrO2 system increases adsorption performance of CO/H2 and therefore the yield of methanol is improved to a certain extent. Despite the impressive role of ZnO, the catalytic role of other supportive metal oxides, such as WO3, Cr2O3, MoO3 and so on has also been checked.176,177 Analogous to Cu/ZnO/ZrO2, several other ternary catalytic frameworks, e.g.; Ag/ZnO/ZrO2, Au/ZnO/ZrO2 and Au/CeO2/ZrO2 have been employed for the same.178,179 Interestingly, Cu/ZnO/ZrO2/Al2O3 provides an enormous yield of methanol, which is 11% higher than Cu/ZnO/Al2O3 and 80% higher than commercial catalysts.
000 h−1) and 1000 h stability at 300 °C under streaming conditions. Here, ZrO2 helps to protect the In2O3 phase from high-temperature annealing, and is responsible for the uniform distribution of oxygen defects.174 Likewise, ZnO–ZrO2 can be treated as a catalyst to attain 10% CO2 conversion efficiency, with selectivity up to 91% for methanol.175 However, the addition of ZnO within the Cu/ZrO2 system increases adsorption performance of CO/H2 and therefore the yield of methanol is improved to a certain extent. Despite the impressive role of ZnO, the catalytic role of other supportive metal oxides, such as WO3, Cr2O3, MoO3 and so on has also been checked.176,177 Analogous to Cu/ZnO/ZrO2, several other ternary catalytic frameworks, e.g.; Ag/ZnO/ZrO2, Au/ZnO/ZrO2 and Au/CeO2/ZrO2 have been employed for the same.178,179 Interestingly, Cu/ZnO/ZrO2/Al2O3 provides an enormous yield of methanol, which is 11% higher than Cu/ZnO/Al2O3 and 80% higher than commercial catalysts.
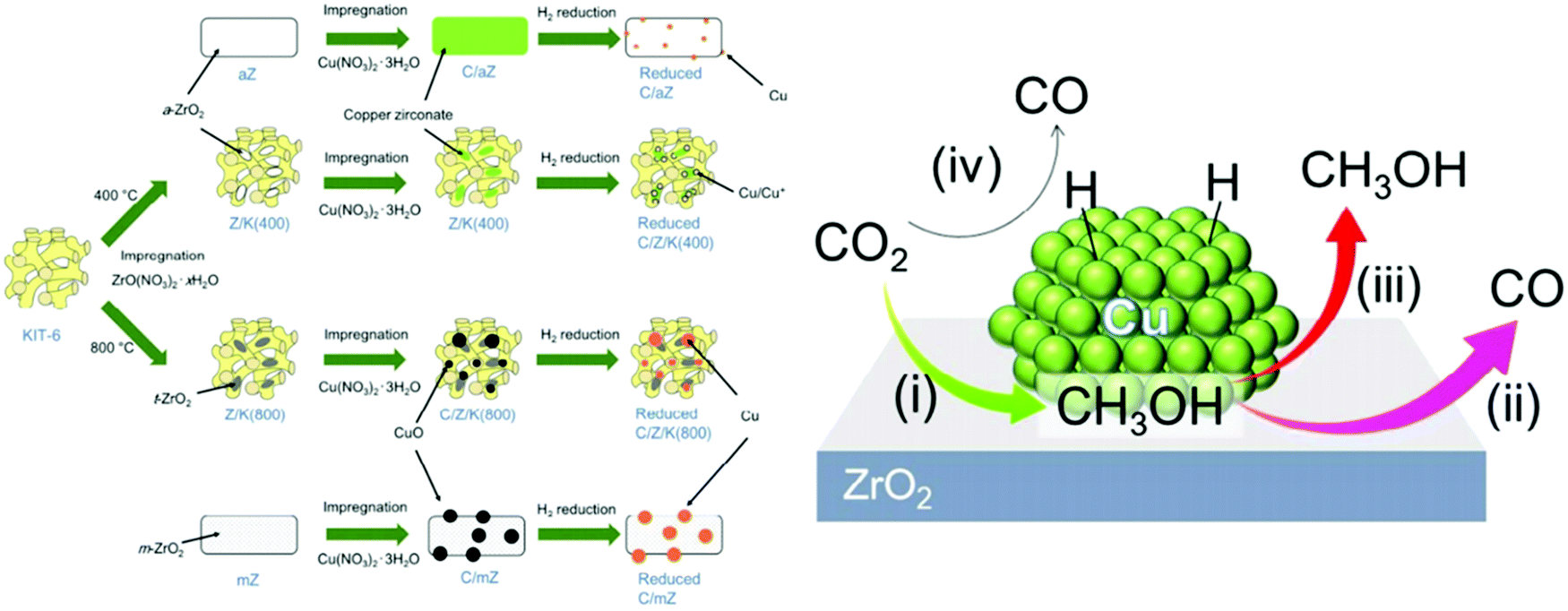 | ||
| Fig. 6 (left) Pictographic image of the catalyst fabrication of C/aZ, C/Z/K(400), C/Z/K(800) and C/mZ (from the top). (right) Proposed mechanistic view of the methanol hydrogenation process from CO2 using Cu nanoparticles embedded ZrO2. (i) CO2 hydrogenation to methanol at the interface between Cu and ZrO2. (ii) Methanol decomposition into CO is observed for strongly adsorbed methanol from the ZrO2 surface. (iii) Desorption of methanol is easy for weakly adsorbed methanol from the ZrO2 surface that further suppresses methanol decomposition. (iv) Conversion of CO2 into CO takes place on the metallic Cu surface via the RWGS reaction. The figures are reproduced with permission from ref. 173. | ||
Incorporation of ZrO2 (5 atomic%) plays a crucial role in reducing reversible adsorption of water and CO, which in turn increases the methanol content from both a yield and selectivity point of view.180 The deliberate amount of ZrO2 addition during the preparation of the catalytic framework may be the strategy for optimum surface area, distribution of active sites and high-temperature stability.181 Saito et al. described the utility of Cu/ZnO/ZrO2/Al2O3/Ga2O3 as a multicomponent catalytic composite that holds extraordinary performance for the catalytic reduction of CO2 to methanol.182 In recent times, the catalytic reduction of CO2 to ethanol is regarded as the most challenging task when considering the selectivity and yield. Numerous studies have demonstrated that selective production of ethanol can be obtained using a metal oxide supported catalytic network, like Pt/Co3O4, Pd2CuNPs/P25, CoAlOx, etc.183,184 Nobel metal-based catalysts are well-known for their outstanding ability in the C–C coupling reaction, which is an essential requirement for ethanol formation. However, the high cost and tendency towards formation of higher chain alcohol, restricts the implementation of such a catalyst in routine work. Wang et al. optimized the synthetic parameters by adjusting the composition of Co–CoOx, and pre-reduction temperature regarding the preparation of CoAlOx catalyst for the same purpose.185 At a pre-reduction temperature of 600 °C, the yield of ethanol production is about 0.444 mmol g−1 h−1 with 92.1% selectivity at 140 °C. The impressive catalytic activity can be ascribed to the co-existence of a Co–CoO elemental phase within the Co–Al double layer hydroxide, which indeed triggers the formation of acetate from formate via the insertion reaction. However, poor selectivity and a lower yield of ethanol was encountered at the pre-reduction temperature of 400 °C. This basically indicated the lower hydrogenation tendency to attain the intermediate step of acetate, which might assist the greater selectivity for methanol production.
Likewise, the production of dimethyl ether directly through the catalytic reduction of CO2 provides one step forward to valuable petrochemicals. One-step conversion of CO2 to dimethyl ether requires an efficient dehydration agent along with a hydrogenation catalyst, promoter and support network.186 In this regard, efficient acidic catalysts, such as HZSM-5, NaZSM-5, γ-Al2O3, etc. have been employed for the dehydration of methanol.187–193 Wang and co-workers reported a physical mixture of CuO–ZnO–Al2O3–ZrO2/HZSM-5 for the same issue, and achieved a convenient yield of dimethyl ether via catalytic reduction of CO2.194 The framework of the Cu–Fe–Zr/HZSM-5 composite has been fabricated for the synthesis of dimethyl ether from CO2 and H2.195 The importance of ZrO2 doping within the CuO–Fe2O3 catalyst might be the cause of the enhancement in surface area, resulting in enhancement of hydrocarbon selectivity. The excellent conversion efficiency (28.4%) and selectivity (64.5%) for dimethyl ether has been attained by 1.0% ZrO2 addition at 260 °C and 3.0 MPa with gas hourly space velocity of 1500 mL gcat−1 h−1. The optimized doping amount for ZrO2 is not only important for the improvement of surface area, but also it influences the reducing feature of the CuO counter-part through deploying outer-shell electron density of Cu+2. Mesoporous ordered bi-functional Cu/γ-Al2O3 catalyst can be used for the same purpose in the presence of promoters, such as gallium or zinc oxide.196 Phase transformation of promoters directly controls the oxidation states of Cu NPs as well as the degree of aggregation. Furthermore, distribution of acidic sites on mesoporous γ-Al2O3 influences the selectivity and rate of dimethyl ether production. The partial appearance of spinel type CuAl2O4 on the catalytic surface might be an active site for methanol dehydration. Notably, Cu–Zn–Al/HZSM-5 possesses 97% efficient CO2 conversion rate at 280 °C and an elevated reaction pressure of about 36 MPa.197 Similarly, Silva et al. showed the benefit of Al2O3 as the catalytic support over Nb2O5 for Cu–ZnO catalyst for catalytic reduction of CO2.198 Recently, Suh and co-workers reported the bifunctional activity of Al2O3/Cu/ZnO (shown in Fig. 7) for direct dimethyl ether production from synthesis gas with two to three times more yield than a conventional catalytic framework.199 This synthetic approach provides more accessible Cu atoms throughout the acidic network of Al2O3, attributing to the extraordinary performance for the catalytic reduction of CO2. Furthermore, the same research group demonstrated a sequential precipitation method for the preparation of Al2O3/Cu/ZnO for single step conversion of CO2 to dimethyl ether.200 The best performance has been attained for 80% loading of the aluminium component, which displays a selectivity of 75% for dimethyl ether and 25% CO2 conversion at 300 °C and 50 bar. To enhance the further selectivity towards dimethyl ether, a dual-bed reactor system approach can be employed with the primary layer of Al2O3/Cu/ZnO having 80% loading of the aluminium component, and the secondary layer of zeolite ferrierite. This strategy boosts the selectivity up to 90% for dimethyl ether with little improvement in CO2 conversion performance.
 | ||
| Fig. 7 Catalyst structure for syngas-to-DME conversion. The as-prepared sample is composed of a (Cu,Zn)2(OH)2CO3 network with amorphous Al(OH)3 as either a shell (surrounding yellow layer) or small aggregates (yellow sphere). The as-prepared sample is transformed into Al2O3/CuO/ZnO after calcination followed by formation of Al2O3/Cu/ZnO via the reduction method. The figures are reproduced with permission from ref. 199. | ||
For the conversion of CO2 to lower olefins (ethylene and propylene), In2O3–ZrO2/SAPO-34 has been found to be an efficient catalyst with 80–90% selectivity at 15 bar and 400 °C.201 The stability of the catalyst was obtained over 50 h without any prominent decay in performance. The influence of ZrO2 addition was further supported with experimental and computational findings by Sun and co-workers.202 In comparison with pure In2O3, incorporation of a zirconium component can accelerate the formation of a greater number of oxygen vacancies, which originate due to the appearance of In1−xZrxOy on the surface of metal oxide. The oxygen defect site further triggers the CO2 chemisorption and stabilizes the intermediate that suppresses the CO production followed by reduction in RWGS processes. Optimization of synthetic parameters for the preparation of the catalytic framework with the above elemental composition further assists the enhancement of the stability even up to 150 h.203 The catalytic network of In1−xZrxOy and catalytic support of SAPO-34 zeolite have been required for molecular activation of CO2 and C–C coupling purposes, respectively. In addition, the ZnZrO/SAPO catalyst can be used to attain 80% selectivity for the production of lower olefins.204 High selectivity over the core–shell microstructure of CuZnZr@(Zn-)SAPO-34 can also be obtained for methanol to olefin conversion.205 Interestingly, incorporation of Zn within SAPO-34 reduces the acidity of the composite framework, which impedes the secondary hydrogenation reaction. Liu et al. performed selective conversion of CO2 to lower olefin using ZnGa2O4/SAPO-34 as the bifunctional catalyst.206 Here, development of oxygen vacancies over the ZnGa2O4 network are responsible for the formation of formate and methoxide-like intermediate species that are ultimately transformed into methanol. The ZnGa2O4/SAPO-34 catalyst maintains selectivity of undesirable CH4 and CO at less than 1% and 50%, respectively, indicating the prudent catalytic conversion rate of CO2 with rational selectivity towards lower olefins. Fabrication of an OX-ZEO (oxide-zeolite) catalyst with elemental composition of ZnCrOx/MSAPO has been reported as a superlative catalyst for light olefins with 110 h stability without any loss of activity.207 The activation of CO and H2 can be processed over the surface of partially reduced ZnCrOx, whereas a hierarchical mesoporous network of SAPO induces C–C coupling. The topological features of oxide and oxide/zeolite composition have a significant impact on CO conversion, while olefin selectivity can be tuned by the pore architecture and acidity of zeolite. Recently, Ye and co-workers derived a Fe0.30Co0.15Zr0.30K0.10O1.63 catalyst with enriched oxygen defects rendering 33.24% more light olefin space time yields (STY) than Fe0.60Co0.30K0.10O1.40 at 310 °C and 2.0 MPa.208 The promising yield can be attributed to the distribution of oxygen defects throughout the framework of partially reduced Fe0.30Co0.15Zr0.30K0.10O1.63, thus facilitating adsorption and subsequent activation of CO2 that eventually enables the formation of CO as an intermediate for the generation of light olefins. The use of HZSM-5 instead of SAPO-34 has been proven as an interesting move for the production C5+ olefins and gasoline-range hydrocarbons. The catalytic composition with reducible In2O3 in the presence of HZSM-5 can be employed to acquire a high octane number towards 78.6% selectivity of gasoline-range hydrocarbons with non-desirable 1% CH4.209 The close proximity of In2O3 and HZSM-5 can be promising to inhibit the RWGS process, which thus endows high yields for gasoline-range hydrocarbon. Furthermore, ZnAlOx/HZSM-5 was employed for CO2 hydrogenation to aromatics with 73.9% selectivity and 9.1% CO2 conversion by Liu and co-workers.210 The presence of Zn2+ is responsible for the CO2 activation and the acid site throughout ZnAlOx/HZSM-5 assists aromatization of intermediate species. Cui et al. have recently elucidated the catalytic framework consisting of sodium modified spinel ZnFeOx and HZSM-5, for direct conversion of aromatics from CO2 and H2.211 The aromatic selectivity reached up to 75.6% with 41.2% CO2 conversion, demonstrating the importance of elemental composition, particularly the existence of sodium within the catalyst. The enormous aromatic yield and convenient stability have been attained owing to the optimized amount of sodium, the density of the acidic network and hierarchical porosity of the catalyst.
Linear organic carbonates are well-known for their widespread application as alkylating, as well as carbonylating, agents for polycarbonate species and oxygenated fuel additives. The clean production of linear organic carbonates is preferable to avoid toxic ingredients from a green and sustainable point of view. Choi et al. demonstrated an inimitable strategy to prepare dimethyl carbonate (DMC) from methanol and CO2 in the presence of dialkyltin methoxide as the catalyst. They thoroughly investigated the intermediate structure by X-ray crystallography and further performed thermolysis of the intermediate to prepare DMC.212 Mikkola and co-workers reported ZrO2 doped with KCl promoted by magnesium turnings for the synthesis of DMC.213 Similarly, polyethylene glycol enfolded potassium bromide can be employed as a splendid catalyst for the manufacture of DMC.214 Recently, the synthesis of DMC has been computationally studied using DFT modelling from CO2 and dimethoxytin(IV) complexes.215 Preparation of diethyl carbonate (DEC) was extensively investigated through the carboxylation of ethanol in the presence of different catalysts. Darbha et al. introduced ceria as a catalyst along with 2-cyanopyridine for trapping water molecules that shift the equilibrium to increase the yield of product.216 Apart from that, a bimetallic Cu–Ni alloy supported by activated carbon, was also reported as a catalytic network for the same purpose.217 In recent times, synthesis of DEC has also been successfully performed from CO2 and ethanol over Ni–Cu@Na3PW12O40 catalytic surfaces with good yield under optimal conditions.218
A large study has focused on the preparation of cyclic carbonate from epoxide using CO2 as a source of carbonyl groups. Pd NPs embedded within nanorod-like mesoporous TiO2 as the heterogeneous catalyst were developed to prepare cyclic carbonate through CO2 fixation at ambient pressure and room temperature. They also demonstrated the influence of Pd NPs as an electron donor, which played a deliberate role in activating the CO2 molecules, and also the formation of oxyanion species at an intermediate reaction step.219 Recently, Mahalingam and co-workers designed Au NPs incorporated parallelepiped α-Fe2O3 nanostructures for the same purpose.220 The synthetic strategy was further generalized for Ag and Cu integrated hematite architectures and the percentage of epoxide conversion was reasonably studied for all the samples. The catalytic trend was found to be as follows: Cu/α-Fe2O3 < Ag/α-Fe2O3 < Au/α-Fe2O3. The reusability of the best catalyst was checked over several cycles, showing remarkable catalytic performance (above 80%) even after 6 cycles (Fig. 8A).220 Bondarenko et al. reported a solvent-free approach for epoxide to cyclic carbonate conversion using ZnCl2/Al2O3. Interestingly, this catalyst was also utilized for the synthesis of sulphur-substituted cyclic carbonate products from the reaction between epoxide and carbon disulphide (CS2).221 Carboxylic acid grafted SBA-15 was reported as a splendid catalyst for the synthesis of a wide range of cyclic carbonates from epoxide, at atmospheric pressure and room temperature. A post-synthetic focalization technique was employed for the high loading of carboxylic acid over ordered silica along with exclusive pore-wall features, high surface area and thermal stability.222 Prasad et al. succeeded in fabricating hollow marigold CuCo2O4 spinel microspheres by solvothermal method and carried out solvent-free epoxide to cyclic carbonate conversion. They also investigated the reusability of the catalytic system (CuCo2O4 and tetrabutylammonium iodide combination) in up to five consecutive cycles without any loss in performance (Fig. 8B).223 Numerous other materials, like Si/SiC@C hollow-nanospheres, TiPO4 flowers, and aluminum porphyrin complexes, can also be used as a catalyst to achieve high conversion efficiency for the chemical fixation of CO2 under milder conditions.224–226 Although zeolites,227 titanosilicates,228 and metal oxides229 were reported as CO2 fixation catalyst, but they mostly required elevated temperature, pressure and extensive purification steps. Additionally, low product yield could enhance the energy/cost due to increased number of recycling steps. Therefore, it is challenging to develop efficient catalysts to solve the aforesaid issues. To address this concern, we have chosen various functional nanoporous materials viz. MOFs, POPs, COFs as heterogeneous supports for the activation of CO2 with/without using noble metal catalysts, and underlined their recent developments in this article. Moreover, we have illustrated some of the recent progress on such ionic frameworks for CO2 fixation reactions.
 | ||
| Fig. 8 Pictorial image of the proposed mechanism for the epoxide to cyclic carbonate conversion. The figures are reproduced with permission from (A) ref. 220 and (B) ref. 223. | ||
3.2. MOFs as catalysts for the conversion of CO2
Metal–organic frameworks (MOFs) are hybrid materials with coordinatively unsaturated metal sites and chelated with organic ligands, have outstanding capability towards chemical interaction with CO2, which is highly inspiring to the scientists for applying MOF-based catalysts in favor of CO2 fixation and conversion purposes.230,231 The catalytic potential of MOFs can be readily controlled by judicious selection of metal-ions/organic ligands in order to tailor their surface property. Another important parameter is the stability of MOFs during catalysis. Metal ions with high coordination number together with high hydrophobicity MOF crystals could be suitable for this purpose. Darensbourg232 demonstrated that MOFs can capture and store CO2, while releasing the CO2 (by heating) for chemical fixation. He anticipated that MOFs with metallosalen as the catalytic sites can easily convert CO2 and epoxides to polycarbonate plastics. In this context, it is pertinent to mention that CO2 conversion to carbonates is highly atom economic, and a most elegant artificial CO2 fixation reaction. Gupta et al. have recently shown that zinc-based MOF can be used for CO2 fixation through catalytic conversion of epoxide to cyclic carbonate.233 They also found exceptional thermal stability and prudent selectivity for CO2 adsorption, which can be accredited to their high surface area and microporous nature. The splendid performance of the catalyst was thoroughly studied and explained on the basis of an abundance of unsaturated sites and Lewis acid–base interactions. The utility of MOF with ultra-high concentrations of Zn was further investigated for CO2 conversion by Jiang and co-workers.234 They artistically not only attempted to incorporate a single atom within the porous network, but also introduced the photothermal effect in CO2 cycloaddition trajectory. For a similar purpose, a 3D Zn-MOF with profuse Lewis active sites was designed and implemented as the catalyst for cyclic carbonate conversion in solvent-free conditions at ambient pressure (Fig. 9).235 Li et al. prepared zinc-based 3D-MOFs with abundant active centers for solvent-free CO2 fixation in the presence of TBAB, and found high TOF values, and excellent stability and recyclability of catalyst up to seven cycles for epoxide to cyclic carbonate conversion.235 On the other hand, Peppel et al.236 reported the cycloaddition reaction between CO2 and epoxides, to carbonates at very high pressure and high temperature, in the presence of Lewis acid as the catalyst and Lewis base as the cocatalyst. Mechanistically, it can be said that a typical acid catalyst (e.g., metal or proton) initially coordinates to the epoxide and activates it towards neucleophilic attack by the co-catalyst (commonly used tetraalkylammonium halide) to form halo-alkoxide. The intermediate halo-alkoxide then reacts with CO2 to yield cyclic carbonates with regeneration of the cocatalyst. The rate law for cycloaddition of CO2 to epoxide has been postulated by North and Pasquale using homogeneous Al(salen) complex as the catalyst.237 With a change in concentration of catalyst, co-catalyst, and CO2, they found that the reaction is first-order for epoxide, CO2, and catalyst concentration, while second-order with respect to co-catalyst concentration. The second-order dependence of the rate on the tetrabutylammonium bromide cocatalyst concentration implies that two separate molecules of tetrabutylammonium bromide are involved in the catalytic cycle before the rate-determining step.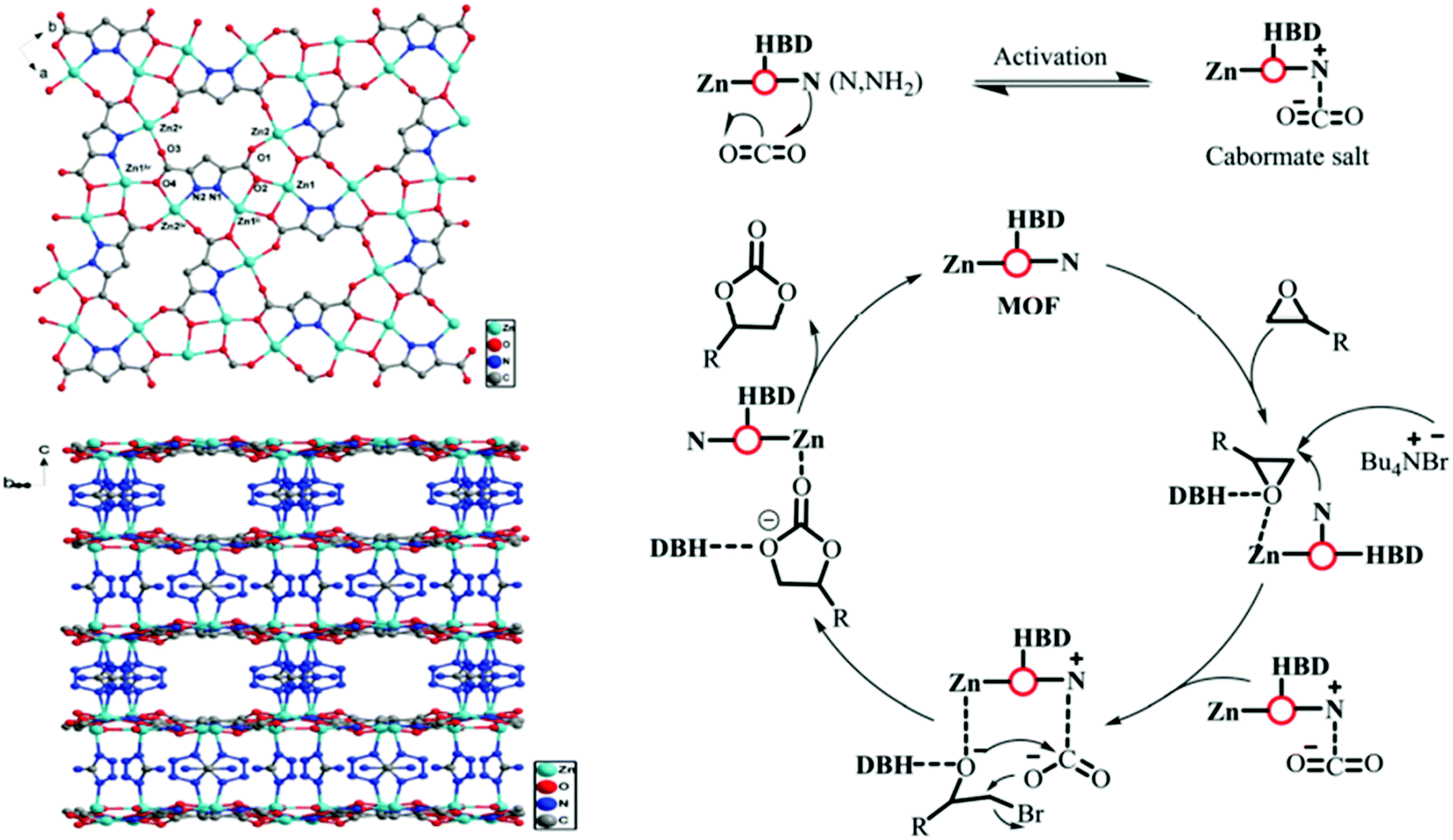 | ||
| Fig. 9 (left) View down the c axis, showing the 2D sheet structure built of Zn centers connected by the PZDC2− anion, and corresponding view of the 3D framework of Zn(PZDC)(ATZ). (right) Possible reaction mechanism for CO2 coupling over Zn(PZDC)(ATZ)/Bu4NBr catalysts. HBD stands for NH2 and water in the Zn(PZDC)(ATZ) catalyst. Figures are reproduced with permission from ref. 191. | ||
Lin and co-workers fabricated Cu(I) sites on Zr12 cluster-based MOFs for ethanol production through selective hydrogenation of CO2.238 Interestingly, the selective hydrogenation strategy can also be instigated by Zr-based MOFs with a uniform distribution of Ni components, particularly for the catalytic conversion of CO2 into CH4.239 Cyclization of propargylic alcohols using CO2 is of great importance to the industrial circuit. Zhao and co-workers proposed noble-metal-free MOFs for solvent-free cyclization of propargylic alcohols and CO2 under mild conditions with large of turnover number (TON) yield.240 More recently, a series of ruthenium complexes immobilized on azolium-based MOFs have been reported for CO2 to formic acid conversion with convenient yield and superior recyclability.241 Tshuma et al. also prepared efficient lanthanum (La)-based MOFs for catalytic hydrogenation of CO2 to formate under optimized reaction conditions.242 Several homogeneous catalysts containing porphyrin/salen sites show high activity under harsh reaction conditions, which may be insignificant for an economical process.243 Nevertheless, MOF-based catalysts hold inherent advantages of having vacant/partially coordinated metal ions that can be readily available as the Lewis acid site for the activation of epoxides. Additionally, MOFs having organic ligands can also be useful as the Lewis base. Consequently, we found that MOFs have dual advantages for serving as the bifunctional catalyst in this reaction.
Although in the previous section we have discussed important applications of MOF-based catalysts for CO2 conversion to cyclic carbonates, ZIF is limited in this application. If the linkers of MOFs are catalytically inactive or the metal centre is coordinatively saturated, the catalysis takes place through the defect sites of the MOFs surface. ZIF-8 is exemplary in this regard.244 In this work, ZIF-8 was used as the catalyst for CO2 cycloaddition to produce cyclopropene carbonate at 70–80 °C for 4 h and 7 bar CO2 pressure without any solvent or cocatalyst. The reaction gives low to moderate yield of product (44%). Later, ZIF-8 was functionalized with ethylene diamine, which improves the product yield to 73%. It has been found that the ZIF-8 topology is very important for this reaction, as it possesses both Lewis acidic and basic properties, which in turn outperform other catalysts containing single active sites e.g., titanosilicate (acidic) or NH3 adsorbed zeolites. In ZIF-8, catalysis takes place at the defect sites because styrene oxide is too large to penetrate inside the ZIF-8 pores. The recyclability of ZIF-8 for this reaction is moderate as evidenced by no significant loss of product. However, ∼15% basic sites on the ZIF-8 structure are lost due to its repeated use. When ZIF-68, having larger pores (10.3 and 7.8 Å), is used, a cycloaddition reaction takes place without any hindrance.245 The exclusive benefit for synergistic CO2 fixation can be further extended into simultaneous capture and conversion into value added chemicals for several porous materials under mild conditions.246
Another Zn-based framework MOF-5 (Zn4O),247 having coordinatively saturated metal centers, cannot catalyze a CO2 cycloaddition reaction in the absence of any co-catalyst. In contrast to ZIF, MOF-5 has no basic site in the framework; hence, it requires tetraalkylammonium halide as co-catalyst at 50 °C and 60 bar pressure, producing nearly 100% propylene carbonate. Nevertheless, MOF-5 is devoid of any catalytic functionality; therefore, it is primarily the defect sites that are responsible for catalysis. Carrasco et al. reported one-step microwave synthesis of metalated PCN-222 for cycloaddition of CO2 with epoxides and aziridines at atmospheric pressure under mild conditions (Fig. 10). In addition, cobalt as a catalytic site displays remarkable performance compared to others such as Ni, Cu, Zn.248 Again, ZIF-78 hexagonal prisms having the same length and thickness of 5 μm, were used in the synthesis of propylene carbonate (PC) from CO2 and propylene oxide (PO).249 Owing to dual functional sites of ZIF-78, i.e., Lewis acidic (Zn) and basic (–NH of imidazole linker) sites, the reaction takes place without the addition of any co-catalyst or solvent at 150 °C, 10 kg cm−2 pressure and 15 h of reaction time. The yield of the product PC is 54%. Schematic representation for the ZIF-78 catalyzed cycloaddition of CO2 to epoxide is shown in Fig. 11.
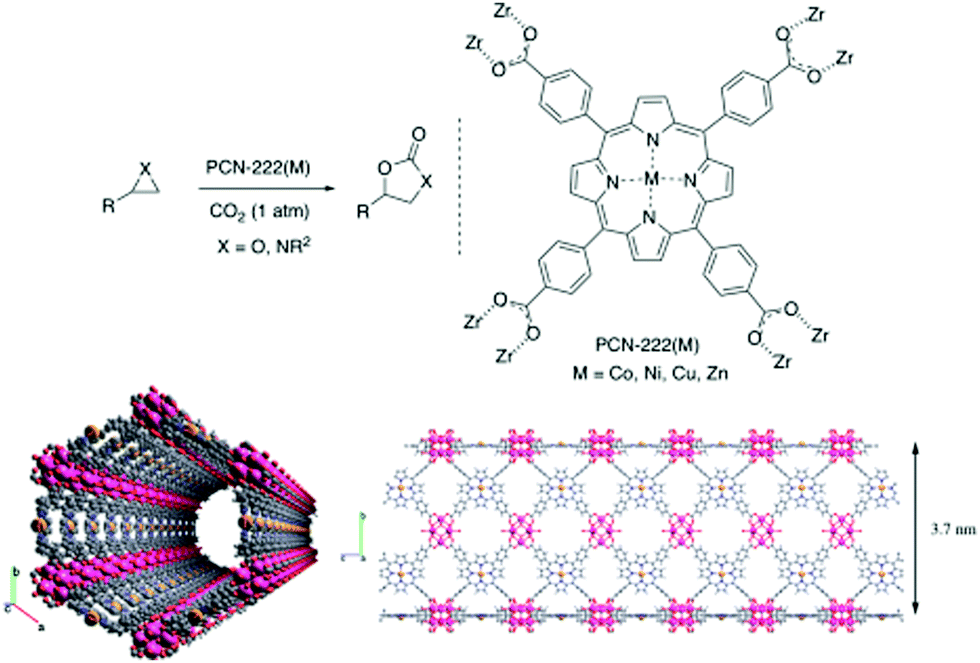 | ||
| Fig. 10 (top) Schematic presentation of cycloaddition reaction of CO2 with epoxide and aziridines using PCN-222(M). (bottom) Two different projections of PCN-222(Co) channels along different axis. Figures are reproduced with permission from ref. 248. | ||
 | ||
| Fig. 11 ZIF-78 catalyzed cycloaddition of CO2 to epoxides, reproduced with permission from ref. 249. | ||
In 2016, Park and co-workers258 investigated the catalytic role of high surface area (>1400 m2 g−1) CZ-ZIF with sodalite topology, consisting of bimetallic Co, Zn as the active sites and 2-methylimidazole as the ligand sites, for the cycloaddition of CO2 to epoxide. CZ-ZIF displayed high catalytic efficacy to five-membered cyclic carbonates using CO2 as the C1 source under solvent- and co-catalyst-free conditions with good selectivity, which is better than ZIF-67, and the framework stability can be comparable to ZIF-8. In other research, Park and co-workers259 prepared amine functionalized ZIF-90 (F-ZIF-90) by post-functionalization of ZIF-90 with hydrazine, having reduced effective pore volume and surface area. This F-ZIF-90 was studied for the catalytic conversion of allyl glycidyl ether (AGE) and CO2 to cyclic carbonate at 120 °C for 6 h with 1.17 MPa initial pressure of CO2. The yield and selectivity of carbonate can be increased by >50% after post-functionalization of ZIF-90 with amines. In recent times, Zhong et al. synthesized MOFs with a zeolitic microstructure and reported admirable heterogeneous catalytic activity through a super large cage.260 They also obtained high yields, unique selectivity, long durability and outstanding recyclability even after five cycles of catalytic performance. Zhang et al. studied the catalytic efficiency of zeolite-like metal organic frameworks (ZMOF) for CO2 cycloaddition and established the superiority of rho-ZMOF over sod-ZMOF with excellent selectivity and conversion efficiency.261 In Table 1, we compare the activity of MOFs and ZIFs as catalysts for the cycloaddition of CO2 to epoxide.
|
|
|||||
|---|---|---|---|---|---|
| Entry | MOF/ZIF catalyst | Substrate | Reaction conditions | Yield, % | Ref. |
| 1 | MOF-505 | Propylene oxide | 25 °C, 1 atm, 48 h | 48 | 250 |
| 2 | BIT-103 | Propylene oxide | 160 °C, 30 atm. | 95.2 | 251 |
| 3 | ZIF-8-NH2 | Epochlorohydrin | 80 °C, 6.9 atm., 4 h | 73.1 | 244 |
| 4 | ZIF-68 | Styrene oxide | 120 °C, 9.9 atm., 12 h | 93.3 | 245 |
| 5 | Cr-MIL-101 | Styrene oxide | 25 °C, 8 atm., 48 h | 95 | 252 |
| 6 | ZIF-22 | Epochlorohydrin | 120 °C, 11.8 atm., 2 h | 99 | 253 |
| 7 | PCN-222(Co) | 1,2-Epoxypentane | 25 °C, 1 atm., 12 h | 99 | 248 |
| 8 | MIL-101-P(n-Bu)3Br. | Propylene oxide | 80 °C, 19.7 atm., 8 h | 99.1 | 254 |
| 9 | MIL-68-NH2 | Styrene oxide | 150 °C, 7.9 atm., 8 h | 74 | 255 |
| 10 | Ni-TCPE1 | Styrene oxide | 100 °C, 9.9 atm., 12 h | >99 | 256 |
| 11 | [Cu4[(C57H32N12)(COO)8]]n | 2-Ethyloxirane | 25 °C, 1 atm., 48 h | 83 | 257 |
| 12 | MIL-101-P(n-Bu)3Br | Propylene oxide | 80 °C, 19.7 atm., 8 h | 99.1 | 254 |
| 13 | ZIF-8 | Epochlorohydrin | 80 °C, 6.9 atm., 4 h | 43.7 | 244 |
| 14 | MOF-5 | Propylene oxide | 80 °C, 19.7 atm., 8 h | 2.5 | 247 |

Another impressive strategy is photocatalysis of CO2 to form fuels, such as formic acid, methanol and methane. As photocatalysis requires only solar energy as the abundant and renewable energy source, and water as the proton source. Therefore, this would be a very attractive process for carbon conversion purposes because the other approach, like Fischer–Tropsch process is energy intensive, expensive and contributes further towards CO2 emissions.262 Although inorganic semiconductors e.g., TiO2, ZnO, CdS, ZnS are readily available, non-toxic and stable, the catalytic performance of these materials is limited due to the uncontrolled recombination of photo-generated electrons and holes, low uptake of CO2 and larger absorption cross-section of UV light.263 Consequently, a great effort has been made to utilize MOFs as the photocatalysts for CO2 conversion by tailoring their band gap, band edge, organic ligands, etc. For the successful synthesis of MOF-based photocatalysis towards CO2 conversion, it can be said that the lowest unoccupied crystal orbital (LUCO) must be above half the redox potential of the CO2 reduction reaction, depending on which product is formed. For example, the redox potential for the conversion of CO2 to formic acid is −3.42 eV, but for methanol and methane it is −3.65 and −3.79 eV, respectively. Again, the electronic properties of MOFs can be monitored through the energy level of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of the ligand. For long lived charge separation, the empty d orbital of the metal should overlap with the LUMO of the ligand to facilitate efficient ligand to metal charge transfer (LMCT). Therefore, the above-mentioned characteristics of MOF clearly signify their true requirement for CO2 conversion. In Table 2, we summarize several important MOF materials that have been utilized as photocatalysts for CO2 conversion reactions.
| Entry | MOF | Light | Proton donor | Product | Yield, μmol g−1 h−1 | Ref. |
|---|---|---|---|---|---|---|
| 1 | Co-ZIF-9 | Visible | TEOA | CO, H2 | — | 264 |
| 2 | PCN-222 | Visible | TEOA | Formate | 60 | 265 |
| 3 | Ru-MOF | Visible | TEOA | Formate | 77.2 | 266 |
| 4 | MIL-101 (Fe) | Visible | TEOA | Formate | 442.5 | 267 |
| 5 | NH2-MIL-125(Ti) | Visible | TEOA | Formate | 16.3 | 268 |
| 6 | MIL 53 (Fe) | Visible | TEOA | Formate | 222.75 | 267 |
| 7 | Re doped UiO-67 | UV | TEA | CO | — | 269 |
3.3. Polymer based catalysts for fixation of CO2
Analogous to MOFs, POPs containing metallosalen,270,271 metalloporphyrin272 have also been investigated as CO2 fixation catalysts. Deng and co-workers226 have discovered a process utilizing a Co/Al-salen-based conjugated microporous polymer (CMP), where the CMP framework can take CO2 and funnel it to the catalytically active Co/Al site, which is then utilized for the conversion of CO2 to epoxide. It is significant to mention that a high BET surface area (965 m2 g−1) of Co-CMP compared to metallosalen (Al-CMP; 798 m2 g−1) and CMP (772 m2 g−1), triggers good CO2 uptake (2.0 mmol g−1) at atmospheric pressure, which is higher comparable to Al-CMP (1.7 mmol g−1), MC-BIF-3H (1.6 mmol g−1) and MC-BIF-5H (0.23 mmol g−1).273 The authors showed 77.1% yield of cyclic carbonate from homogeneous salen–Co-OAc, which increases to 81.5% at room temperature and atmospheric pressure with Co-CMP in the presence of TBAB as the cocatalyst. So, the utility of Co-CMP is enormous for enhancing CO2 concentration near the catalytic centers of the pores, justifying the high CO2 uptake capacity of Co-CMP compared with salen–Co-OAc. Another interesting finding has been observed for porphyrin-based POP which was further modified by metalation with Zn and Co to the corresponding metalloporphyrin-based organic polymer, M(Por)OP, having dense metalloporphyrin sites in the pore wall. The resulting M(Por)OP shows excellent catalytic performance toward the cycloaddition of CO2 with propylene oxide (PO) to give cyclic propylene carbonate (PC) remarkable selectivity without polycarbonate and other by-products. The reaction proceeds by adding the catalyst, M(Por)OP (M, Zn, Co), co-catalyst (KI, TPPB, TBAB, or DMAP), PO and dichloromethane solvent in a 25 mL stainless autoclave, then adding CO2 at 120 °C for 2.5 h. The effect of the cocatalyst in this reaction is enormous, as without its addition, the activity of M(Por)OP was remarkably reduced, yielding a PC of 12% from Co(Por)OP and 20% from Zn(Por)OP, respectively. In Fig. 12, we provide a pictorial representation for the conversion of CO2 to epoxide, catalyzed by metallosalen and metalloporphyrin units of POPs. Graphitic carbon nitride modified POPs comprising uniform Ag-dispersion were reported as proficient catalysts for carboxylative cyclization of propargyl alcohols using CO2 under mild experimental conditions.274 Phenanthroline-based POPs with high surface area and porosity can be employed for Ir catalyzed CO2 hydrogenation for formate production.275 Ru porphyrin-based POPs were also prepared for the efficient hydrosilylative reduction of CO2 for formate synthesis (Fig. 13).276 Recently, Shao et al. synthesized ultrafine Pd NPs-embedded POPs for the chemical transformation of CO2 into formate under mild condition.277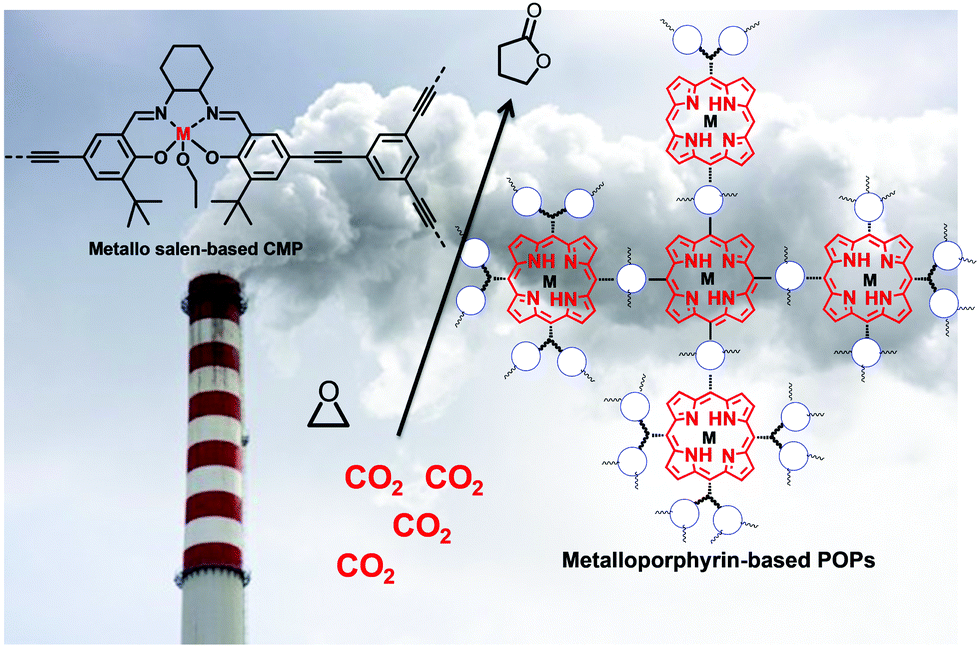 | ||
| Fig. 12 Co and Al-based CMP as the catalyst for chemical fixation of CO2, (M = Co, Al). | ||
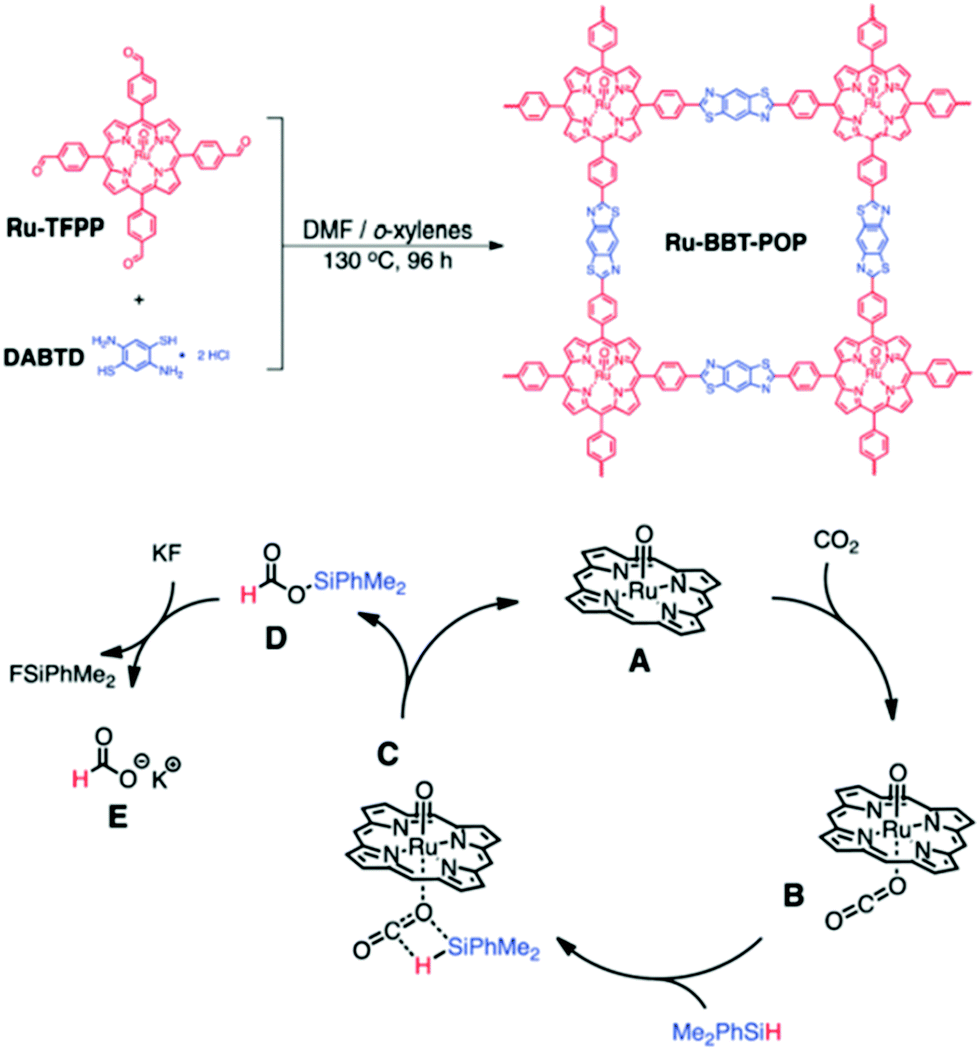 | ||
| Fig. 13 (top) Synthesis of Ru–BBT–POP. (bottom) Proposed mechanism for the hydrosilylative reduction of CO2 to potassium formate. Figures are reproduced with permission from ref. 276. | ||
Similar to the metal-based catalytic sites, metal free cationic POPs containing imidazole278 and bipyridine279 sites are rationally developed as CO2 fixation catalysts. The utility of cationic polymers for cycloaddition reactions is broad as it accommodates the counter-anion, cocatalyst within a single framework, providing benefits both from economic and environmental perspectives. Nitrogen-rich POPs were used as heterogeneous catalysts for catalytic conversion of CO2 in the absence of solvents, metals and additives.280 Furthermore, the synthetic design with imidazolium-based ionic liquids on click-based POPs can tune the textural property (surface area and porosity), selectivity towards CO2 over N2, CO2 sorption capacity and catalytic performance, particularly for epoxide to cyclic carbonate conversion. In 2016, Guo et al.278 prepared a covalent organic polymer (PAD-3) containing both imidazole and amine sites using azobisisobutyronitrile (AIBN) initiated radical polymerization between amino-functionalized imidazolium bromide and divinyl benzene, giving rise to an ionic copolymer catalyst having small nanopores (3.0–3.2 nm), moderate porosity (80–392 m2 g−1) and high ionic density with superior basicity (Fig. 14).
 | ||
| Fig. 14 (A) Synthesis of cationic nanoporous polymers and (B) their application as CO2 fixation catalysts, reproduced with permission from ref. 278. | ||
It is known to all that catalysis in halogen free conditions has much importance for the environment. The catalyst shows promising advantages in CO2 fixation to cyclic carbonates in the absence of any solvent or additives and has high activity and stability compare to various control catalysts, including halogen-containing analogues. Furthermore, high yields were obtained over a wide scope of substrates even under atmospheric pressure and less than 100 °C. Later, the effect of amine functionality on promoting the CO2 addition activity was probed on the basis of in situ Fourier transform infrared (FT-IR) spectroscopy and density functional theory (DFT) calculations of the reaction intermediates. Again, Coskun and co-workers281 prepared another cationic polymer comprising of sterically confined N-heterocyclic carbine, which is stable in air and contains super basic characteristics as well as having a strong ability to activate CO2 (Fig. 15). In this way, a tetrahedral core comprising of tetraphenyl methane incorporating 2,6-diisopropyl aniline moieties at the terminal position could successfully make a 3D nanoporous polymer, NP-NHC. Owing to the super basic nature of NP-NHC, it undergoes chemical fixation of CO2 to carbonates at room temperature and ambient pressure, with 97% efficiency for the formation of epoxides. Interestingly, it undergoes substrate selectivity due to the availability of sterically hindered imidazolium catalytic sites, which provide a unique opportunity in tailoring a metal free approach for simultaneous capture and conversion of CO2. 4,4′-Bipyridine containing cationic frameworks having a tetraphenyl core have also been synthesized by Coskun and coworkers.235 By virtue of large cationic charge in the backbone, different counter anions (Cl−, BF4−, PF6−) can be incorporated in the polymer by simply varying the counter anions, both gas sorption and catalytic properties of PCPs can easily be tuned for metal-free capture and conversion of CO2 into cyclic carbonates with excellent yield.
 | ||
| Fig. 15 Schematic representation of the preparation of porous cationic frameworks with different counter anions, reproduced with permission from ref. 235. | ||
Apart from amorphous POPs, crystalline COFs remain almost unexplored as CO2 conversion catalysts.282,283 However, few functional COFs having N-rich porphyrin and/or imine sites along with aromatic –OH groups, show metal-free fixation of CO2 to epoxide and oxazolidinones formation. Bhanage and co-workers prepared chemically stable bifunctional COFs (2,3-DhaTph and 2,3-DmaTph) containing two incompatible catalytic sites, viz. acidic (catechol) and basic (porphyrin).282 The cooperative effect originated from the presence of hydrogen bond donor (HBD) in 2,3-DhaTph and 2,3-DmaTph catalysts accelerates the cycloaddition reaction of CO2 with epoxides by promoting the ring opening of epoxide through nucleophilic attack and stabilizing the oxyanion intermediate, which was also demonstrated by computational analysis.284 It was observed that 2,3-DhaTph and 2,3-DmaTph catalyzed synthesis of cyclic carbonates (yield 98%) requires 1 atm. CO2 pressure, 110 °C temperature and 12 h time with a ratio of styrene oxide:cocatalyst = 200:1. In another work, porphyrin in DhaTph and DmaTph was replaced by triazine units and the resulting COFs prepared by Liu and co-workers283 are named as COF-JLU6 and COF-JLU7, which also possess a similar functional group effect for the activation of CO2 to carbonates (yield >90%) under 0.1 MPa CO2, 40 °C and 48 h of reaction time with styrene oxide:cocatalyst = 20:1. Additionally, COF-JLU6 and COF-JLU7 have high BET specific surface area up to 1390 m2 g−1 and large pore volume of 1.78 cm3 g−1. Among these COFs, COF-JLU7 displays CO2 uptake of 3.5 mmol g−1 at 273 K and 1 bar.
3.4. Metal NPs as active sites for CO2 conversion
In addition to the metal complex supported by MOFs and POPs, metal NPs mediated CO2 fixation reactions have also attracted a lot of interest for carbon removal applications.285 Recently, progress has been made in CO2 transformation to methanol, formic acid, carbonates, urea, etc. It is known to all that metal NPs demonstrate poor catalytic performance because of their agglomeration in solution due to their high surface-to-volume ratio. This can be prevented through immobilizing them on solid microporous and mesoporous supports. In this regard, a wide range of NPs, e.g., Ag, Fe, Zn, Pd, Cu, Ni have been loaded over the porous frameworks of the support and subsequently proven to be stabilized during the catalytic process. Therefore, depending on the nanostructure and the type of NPs, several substrates can be activated and bonded with CO2. For instance, Ni NPs at the surface of bimetallic CuNi NPs can adsorb and strongly activate CO2, which facilitates a prompt reaction with electron-rich substrates and finally forms CO2-addition products.286 Furthermore, CuPt bimetallic nanoclusters on a TiO2 support can also strongly bind CO2 intermediates, which in turn increase the CH4 formation rate.287 The enhanced hydrogenation activity of CO2 to methanol using Ga-doped Cu–ZnO supported catalysts was observed possibly due to the formation of very reactive CuZn species.288 Again, Pt NPs deposited on Co can be very effective in CO2 methanation reactions because of the formation of PtCo bimetallic species. It can be said that metallic NPs (Ag/Cu) have great potential for the activation of alkyne to electron-rich acetylides during the synthesis of carboxylates, where CO2 is used as the carboxylating agent.289 Ag NPs show good efficiency for the carboxylation of terminal alkynes.290 Likewise, CeO2 NPs are utilized for the synthesis of urea from amines and CO2. At high CO2 pressure, Pd NPs produced oxazolidinones from propargylamines, aryl halides and CO2291 Furthermore, Chen and co-workers292 showed that Ag NPs trapped in MOF (MIL-101) can also act as good heterogeneous catalysts for the carboxylation of terminal alkynes with CO2, suggesting Ag NPs and its salts are efficient catalysts for this reaction. Fig. 16 illustrates the carboxylation of terminal alkynes with CO2 to propiolic acid using the highly efficient Ag-MIL-101 catalyst.246
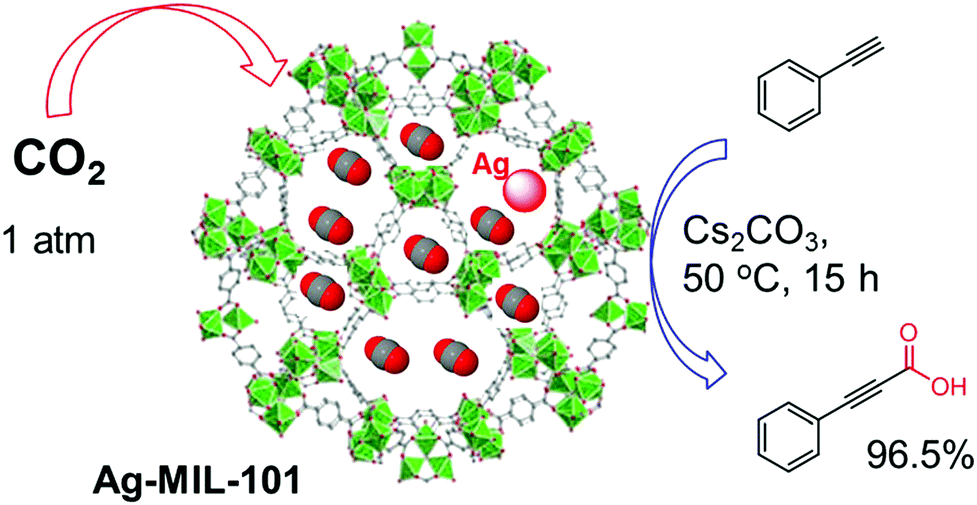 | ||
| Fig. 16 Carboxylation of terminal alkynes with CO2 using Ag-MIL-101 catalyst, reproduced with permission from ref. 246. | ||
Apart from NPs stabilized by MOFs, Yang et al.293 demonstrated the stabilization and activation of NPs utilizing mesoporous poly(triphenylphosphine) with azo functionality, (poly(PPh3)-azo), which was synthesized by oxidative polymerization of P(m-NH2Ph)3 under ambient conditions (Fig. 17). They showed that the azo and PPh3 moieties in the polymer are essential for coordination with metal ions (e.g., Ru3+) and also to reduce Ag+ to the metallic form. The resultant metalated poly(PPh3)-azo (e.g., poly(PPh3)-azo-Ag or -Ru) was found to be a highly efficient catalyst for CO2 transformation. Poly(PPh3)-azo-Ru exhibited good activity for the methylation of amines with CO2. Interestingly, the authors demonstrated that the high performance of the catalyst could be because of cooperative effects between the polymer frameworks and the metal species. Bimetallic Ni-Pd/SBA-15 alloy was prepared for selective hydrogenation of CO2 to CH4.294 They also established that a 3:1 ratio of Ni:Pd was the best combination, with convenient 0.93 mol of CH4 yield per mol of CO2 at 430 °C. Bustinza et al. successfully designed bimetallic Ru–Ni based alumina coated monoliths using a consecutive impregnation method for CO2 methanation with low pressure drop at high space velocities.295 Recently, Chaudret and co-workers reported a surface engineering technique to fabricate an Fe–Ni catalytic network for selective magnetically induced CO2 hydrogenation towards CH4 under mild conditions (Fig. 18).296 Fascinatingly, the Fe–Ni based bimetallic cluster also acted as a superior heating agent under a low magnetic field. Interestingly, bimetallic Au–Cu embedded Cu submicron arrays demonstrated the capability of ethanol formation through CO2 reduction.297 Tsang and co-workers demonstrated a Rh–In catalyst for industrially scalable methanol production, and also showed excellent efficiency to inhibit reverse water gas shift under controlled H2-deficient flow conditions.298
 | ||
| Fig. 17 CO2 transformation with mesoporous poly(PPh3)-azo-Ag or -Ru, reproduced with permission from ref. 293. | ||
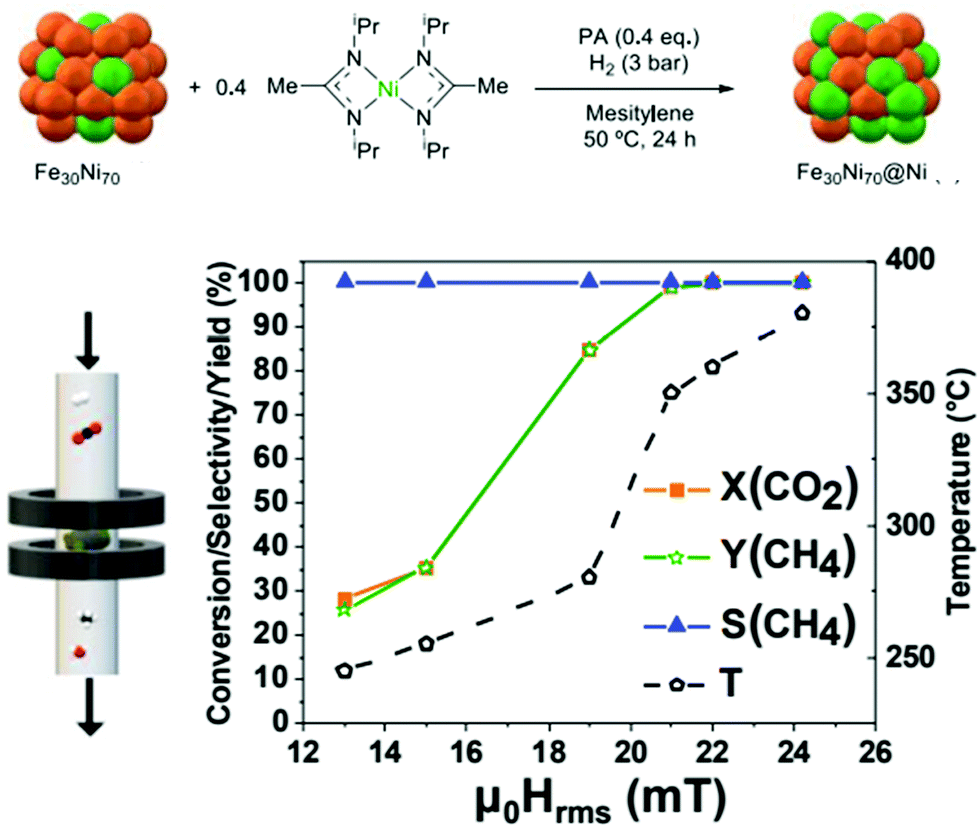 | ||
| Fig. 18 (top) Synthesis of Fe30Ni70@Ni NPs by decomposition of Ni[iPrNC(CH3)NiPr]2 under a H2 atmosphere in the presence of Fe30Ni70. (bottom) Magnetically induced CO2 methanation in a down-flow reactor using Fe30Ni70@Ni NPs, reproduced with permission from ref. 296. | ||
Furthermore, photochemical and electrochemical pathways for CO2 fixation reactions with metal NPs are equally efficient, economic and demanding for sustainable chemistry research. In this regard, several inorganic nanomaterials, TiO2, ZnO, CdS, ZnS, SrTiO3, etc. when suspended in water with a rich CO2 environment, can reduce CO2 to formaldehyde, formic acid, methanol and methane.299 For the photocatalytic reduction of CO2, Li and co-workers300 reported the increase in CH4 selectivity from the Cu NPs deposited on the TiO2 surface, which could be attributed to the presence of surface defect sites for the stabilization of electrons and holes. Additionally, graphene oxide (GO) shows favorable band-gap and a low electron–hole recombination rate, which favors the activity of metal NPs for selective photoreduction of CO2 to CH4.301 Electrochemically, CO2 can also be converted to CH4 in the presence of Cu–Pd nanoalloy deposited on an organic polymer. A synergy between Cu–CO and the in situ generated Pd–H, might be crucial for achieving high CH4 selectivity.302 Additionally, Cu NPs deposited on graphene nanosheets are also effective in the electrochemical reduction of CO2. Furthermore, Bi NPs on ionic liquid303 and Ag NPs on carbon nanotubes304 can also promote the electrochemical reduction of CO2 to CO. Again, Rybchenko et al. observed high Faradaic efficiency for methanol synthesis from pyridine as a simple organic precursor under high CO2 pressure over a Pt-based electrode.305 Song et al. proposed a one-pot silver-catalysed method for the synthesis of 2-oxazolidinones using propargylic alcohols, CO2, and 2-aminoethanols.306 The scope of application was further extended to synthesize cyclic urea using diamines, CO2 at low pressure (∼0.3 MPa) in the presence of CeO2 as the catalyst.307 Sun et al. also employed CO2 for the preparation of 4-aryl-2-quinolinones using N-tosylhydrazones, 2-iodoanilines at atmospheric pressure.308 The synthetic protocol was performed in the presence of palladium as the catalyst with the instantaneous development of two C–C, one CC and one C–N bond formations in amide. All these investigations actually demonstrate the efficacy of metal NPs based catalyst for CO2 transformation reactions.
4. Outlook and future directions
Porous networks have been considered as an ideal candidate for CO2 fixation under ambient conditions owing to their controllable microstructure, optimized distribution of active centers and surface area with hierarchical porosity and surface acidity. In addition, surface functionalization with amine groups together with the presence of heteroatoms has been established to have significant influence on CO2 adsorption. However, direct use of atmospheric CO2 is still a challenging issue due to the low concentration in ambient air, which is around 400 ppm. Interestingly, the low energy strategy using MEA and their derivatives were chosen for ultimate CO2 fixation from atmospheric resources. The acid assisted recycling technique to release the absorbed CO2 for long-term use was reported and established as a novel CO2 generator. Likewise, the unique strategy of CO2 fixation using oxazolidinone derivatives to manufacture valuable organic heterocyclic compounds, well-known intermediates in the pharmaceutical industry, has also been demonstrated. Recently, significant progress has been made in the preparation of organic heterocyclic compounds using CO2 under ambient conditions. Again, metal-free CO2 reduction has captured a lot of attention due to the simple synthetic approach involving cheap earth-profuse reagents under mild conditions. Furthermore, low energy of activation and low toxicity of reagents motivated a metal-free CO2 incorporation mechanism in heterocyclic compounds.309,310 A unique strategy to reduce carbon dioxide to methoxyborane in the presence of several hydroboranes was reported employing a N-heterocyclic carbene based homogeneous catalyst under ambient conditions. The efficiency of the mechanistic pathway was further supported by the turnover number calculation.311 Traditionally, ionic liquids (ILs) have been elucidated as “designer solvents” for CO2 activation.312,313 Still, the potential requirement is very high in the commercial market for inexpensive catalysts to be used for CO2 activation together with organic molecules under ambient conditions.The understanding of a sustainable synthetic strategy is extremely important in order to eliminate hazardous substances, restricting global warming, reducing human contact to hazardous chemicals, utilizing green reagents, recycling organic solvents, sensible use of natural wastes and carbon-free energy redeemable processes. The preparative approach can be improved by greener routes for numerous classes of materials; including polymers, ceramics, MOFs, nanostructured compounds, etc.314 Several bottom-up processing techniques are already available in the industrial arena, such as sol–gel, solvothermal, solid-state decomposition, electro-deposition, sono-chemical methods etc. The key benefit of these methods is the adequate textural precision with controlled metallurgical defects. However, the high cost and low yield of products can be considered as major limitations for these methods in the commercial zone. On the other side, a top-down strategy, particularly a mechanical (ball milling) technique, can be prescribed as the most conventional technique with high productivity and cheap budget. However, mechanical techniques exhibit random structural orientation, and haphazard size and shape for synthetic products. Even after the diverse advantages of the top-down strategy, the bottom-up method is more frequently used and considered to be a more suitable route for the manufacture of nanostructured materials with controllable microstructure and porosity.315–317
Catalytic conversion of CO2 endows several potential benefits from a sustainable and clean environment context. However, the blue print of catalytic transformation pathways has several critical bottlenecks that need to be overcome for the successful implementation of this technology. It is extremely important to avoid the use of hazardous solvents and reagents during fabrication of catalysts. In addition, recycling catalysts with nominal release of environmental pollutants has been pointed out as a pivotal factor for long-term applications. Catalytic reduction of CO2 into different valuable products requires a mechanical set-up with a continuous H2 flow facility. Surprisingly, the bulk-scale H2 production is still dependent upon fossil fuel consumption, steam reforming, partial oil oxidation and much more. Therefore, generation of H2 with zero carbon emissions is regarded as the most convenient mode of the green revolution. In this prospect, water splitting is believed to be the most conventional way for large scale H2 production in modern times. Biomass conversion is also considered as an alternative strategy for the same issue. The in situ H2 production via water splitting during catalytic reduction of CO2 into valuable chemicals is believed to be an energy saving mode evoked recently in the commercial arena. In this context, the fabrication of electrocatalysts has emerged as a promising technology for the electrocatalytic reduction of CO2, since H2 is generated via in situ water splitting. Catalytic reduction of CO2 into formic acid is still an industrially challenging task due to the requirement of precious metal complexes as the catalyst, and toxic phosphine compounds as ligands together with harsh reaction conditions. Apart from that, several bottlenecks have been reported, such as formic acid stability, selectivity of solid catalyst and yield of product. Hence, the selection of a catalyst is not only a crucial feature but also the most tricky aspect for the successful implementation in a large scale production house. Although zeolites are advantageous from the thermal stability and surface acidic site point of view, restricted diffusion throughout the zeolitic network limits their potential application for long-term use. To overcome these difficulties, MOFs, COFs, ZIFs and POPs have successfully been employed due to their crystalline network together with pore opening. However, stability and regeneration problems restrict their industrial application. Though the carbonaceous materials are well-known for their high surface area and low-cost resources, poor selectivity and low capacity confine their application in CO2 fixation. Presently, there is no entirely established strategy available to combat with the fixation issue. Encouragingly, mesoporous oxides can be an alternative option due to their massive abundance, low cost, hierarchical porosity, mechanical strength and simple synthetic route. Functionalized, naturally occurring clay materials also have huge scope under the same scenario. Thus, the uniform distribution of metal nanoparticles throughout the porous microstructural framework with controllable shape and size may be a unique alternative for the long-standing CO2 fixation issue.
5. Conclusion
The day by day rise of the human population intensifies industrialization and technological empowerment that inflates CO2 emissions in the atmosphere. Furthermore, deforestation and insensitive fuel consumption compound the situation. To reduce greenhouse gas emissions and combat global warming, strategic CO2 fixation into valuable chemicals has emerged as an attractive option for a green economy. In this review, we have discussed the present status of CO2 fixation, and elucidated different catalytic networks employed for the synthesis of value-added chemicals. The importance of catalytic frameworks is attributed to the basis of composition, porosity, surface area, thermal stability and architecture. In addition, catalytic performance of different materials is described by means of their capacity, selectivity, recyclability and yield of product. The preparative approaches to design a catalytic network have been summarized and presented in brief to support the reader as well as facilitate the researcher work in this advanced field. The advantages of multicomponent catalysts are explored step-wise and we briefly illustrate their role in the conversion of CO2 into selective chemicals. From the commercial aspect, design and preparative cost of the catalyst play the crucial role in the ultimate production of value-added chemicals. In organic–inorganic hybrid materials, metal or metal oxide NPs in the presence of a porous counterpart exhibit synergetic influence that may help in capturing CO2 and then transform it into active intermediates, followed by product formation under ambient conditions. The fabrication of a catalyst is governed in such a way that the selective capture of CO2 can be possible even in the presence of moisture, CO, O2 and NOx in flue gas. An ionic liquid derived catalytic framework also has unique features, such as high thermal stability, delocalized charge distribution and volatility, which in turn may come up with their own benefits during the catalytic reaction performed even at high temperature and pressure. It is worth stating that scalable synthesis from earth abundant elements is an essential requirement to develop a catalyst for large scale implementation. Utilization of natural resources, ranging from biomass, rice husk, fly ash and many more, to prepare a porous catalytic network will overcome such a concern and may be an appropriate alternative. Ultimately, sustainable living demands CO2 fixation with zero carbon output that encourages low-cost catalyst fabrication with high yield and selectivity.Abbreviations
| MOFs | Metal organic frameworks |
| ZIFs | Zeolitic imidazolate frameworks |
| NPs | Nanoparticles |
| POPs | Porous organic polymers |
| CCS | Carbon capture and storage |
| CCU | Carbon capture and utilization |
| GHSV | Gas hourly space velocity |
| RWGS | Reverse water gas shift |
| DME | Dimethylether |
| LIB | Lithium-ion battery |
| DAC | Direct air capture |
| COFs | Covalent organic frameworks |
| MEA | Monoethanolamine |
| MDEA | Methyldiethanolamine |
| DETA | Diethylenetriamine |
| TETA | Triethylenetetramine |
| AEEA | Aminoethylethanolamine |
| PEI | Polyethylenimine |
| APS | (3-Aminopropyl)triethoxysilane |
| 2N-APS | N-[(3-Trimethoxysilyl)propyl]ethylenediamine |
| 3N-APS | N-[(3-Trimethoxysilyl)propyl]diethylenetriamine |
| TEPA | Tetraethylenepentamine |
| SCF | Supercritical fluid |
| PAFs | Porous aromatic frameworks |
| HCPs | Hypercrosslinked polymers |
| PIMs | Polymers of intrinsic microporosity |
| CTFs | Crystalline triazine-based frameworks |
| CMPS | Conjugated micro-porous polymers |
| PAN | Polyacrylonitrile |
| STY | Space time yields |
| AGE | Allyl glycidyl ether |
| ZMOF | Zeolite-like metal organic frameworks |
| LUCO | Lowest unoccupied crystal orbital |
| HOMO | Highest occupied molecular orbital |
| LUMO | Lowest unoccupied molecular orbital |
| LMCT | Ligand to metal charge transfer |
| CMP | Conjugated microporous polymer |
| PO | Propylene oxide |
| PC | Propylene carbonate |
| AIBN | Azobisisobutyronitrile |
| FTIR | Fourier transform infrared |
| DFT | Density functional theory |
| HBD | Hydrogen bond donor |
| GO | Graphene oxide |
| rGO | Reduced graphene oxide |
| ILs | Ionic liquids |
| M | Metal |
| Im | Imidazole |
| CS2 | Carbon disulphide |
| TON | Turnover number |
| La | Lanthanum |
| DMC | Dimethyl carbonate |
| DEC | Diethyl carbonate |
| AC | Activated carbon |
| EG | Ethylene glycol |
| PILs | Polymeric ionic liquids |
| ILMs | Ionic liquid monomers |
| TFC | Thin film composite |
| PDMS | Polydimethylsiloxane |
| PVA-g-POEM | Poly(vinyl alcohol)-g-poly(oxyethylene methacrylate) |
| LDO | Layered double oxide |
| ZnAl-LDHs | ZnAl layered double hydroxides |
| CAP | Continuous assembly of polymers |
| MUF | Massey university framework |
| UTSA | University of Texas at San Antonio |
| BBL | Benzimidazobenzophenanthroline |
| DEA | Diethanolamine |
| SBA-15 | Santa Barbara amorphous type material |
| MCM-41 | Mobil composition of matter no. 41 |
| KCC-1 | KAUST catalysis Center |
| BIT | Beijing Institute of Technology |
| SAPO | Silicoaluminophosphate |
| OX-ZEO | Oxide-zeolite |
| TOF | Turn over frequency |
| PZDC | Pyrazoledicarboxylic acid |
| ATZ | Aminoterazole |
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We acknowledge the Technical Research Centre project (AI/1/64/SNB/2014) funded by the Department of Science and Technology (DST), Government of India at S. N. Bose National Centre for Basic Sciences, Kolkata, India. All the authors also acknowledge S. N. Bose National Centre for Basic Sciences, Kolkata, India.References
- A. Busch, S. Alles, Y. Gensterblum, D. Prinz, D. N. Dewhurst, M. D. Raven, H. Stanjek and B. M. Krooss, Int. J. Greenhouse Gas Control, 2008, 2, 297–308 CrossRef CAS.
- J. Rogelj, W. Hare, J. Lowe, D. P. Van Vuuren, K. Riahi, B. Matthews, T. Hanaoka, K. Jiang and M. Meinshausen, Nat. Clim. Change, 2011, 1, 413–418 CrossRef.
- M. Höök and X. Tang, Energy Policy, 2013, 52, 797–809 CrossRef.
- Y. Jiang, P. Tan, S. C. Qi, X. Q. Liu, J. H. Yan, F. Fan and L. B. Sun, Angew. Chem., Int. Ed., 2019, 58, 6600–6604 CrossRef CAS PubMed.
- J. Rogelj, M. Den Elzen, N. Höhne, T. Fransen, H. Fekete, H. Winkler, R. Schaeffer, F. Sha, K. Riahi and M. Meinshausen, Nature, 2016, 534, 631–639 CrossRef CAS PubMed.
- L. Cheng, Y. Jiang, S. C. Qi, W. Liu, S. F. Shan, P. Tan, X. Q. Liu and L. B. Sun, Chem. Mater., 2018, 30, 3429–3437 CrossRef CAS.
- K. Kole, S. Das, A. Samanta and S. Jana, Ind. Eng. Chem. Res., 2020, 59, 21393–21402 CrossRef CAS.
- S. J. Davis, K. Caldeira and H. D. Matthews, Science, 2010, 329, 1330–1333 CrossRef CAS PubMed.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- O. Jacquet, X. Frogneux, C. D. N. Gomesa and T. Cantat, Chem. Sci., 2013, 4, 2127–2131 RSC.
- M. Mikkelsen, M. Jørgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 RSC.
- G. Kupgan, L. J. Abbott, K. E. Hart and C. M. Colina, Chem. Rev., 2018, 118, 5488–5538 CrossRef CAS PubMed.
- J. Yu, L.-H. Xie, J.-R. Li, Y. Ma, J. M. Seminario and P. B. Balbuena, Chem. Rev., 2017, 117, 9674–9754 CrossRef CAS.
- H. Balat and C. Öz, Energy Explor. Exploit., 2007, 25, 357–392 CrossRef CAS.
- A. Modak and S. Jana, Advances in Porous Adsorbents for CO2 Capture and Storage, Carbon Dioxide Chemistry, Capture and Oil Recovery, InTechOpen, 2018, pp. 165–183 Search PubMed.
- S. H. Jun, J. Yang, H. Jeon, H. S. Kim, S. P. Pack, E. Jin and J. Kim, Environ. Sci. Technol., 2020, 54, 1223–1231 CrossRef CAS.
- A. Rafiee, K. R. Khalilpour, D. Milani and M. Panahi, J. Environ. Chem. Eng., 2018, 6, 5771–5794 CrossRef CAS.
- M. D. Garbaa, M. Usman, S. Khan, F. Shehzad, A. Galadima, M. F. Ehsane, A. S. Ghanem and M. Humayun, J. Environ. Chem. Eng., 2021, 9, 104756 CrossRef.
- M. He, Y. Sun and B. Han, Angew. Chem., Int. Ed., 2013, 52, 9620–9633 CrossRef CAS.
- S. Zeng, X. Zhang, L. Bai, X. Zhang, H. Wang, J. Wang, D. Bao, M. Li, X. Liu and S. Zhang, Chem. Rev., 2017, 117, 9625–9673 CrossRef CAS PubMed.
- A. S. Bhown and B. C. Freeman, Environ. Sci. Technol., 2011, 45, 8624–8632 CrossRef CAS PubMed.
- J. Shang, X. Guo, Z. Li and Y. Deng, Green Chem., 2016, 18, 3082–3088 RSC.
- W.-G. Cui, X.-Y. Zhuang, Y.-T. Li, H. Zhang, J.-J. Dai, L. Zhou, Z. Hu and T.-L. Hu, Appl. Catal., B, 2021, 287, 119959 CrossRef CAS.
- Y. Yan, Y. Dai, H. He, Y. Yu and Y. Yang, Appl. Catal., B, 2016, 196, 108–116 CrossRef CAS.
- L. Pastor-Pérez, F. Baibars, E. Le Sache, H. Arellano-Garcia, S. Gu and T. Reina, J. CO2 Util., 2017, 21, 423–428 CrossRef.
- G. Olah, A. Goeppert and G. K. Prakash, Beyond Oil and Gas: The Methanol Economy, Wiley-VCH, 2011, pp. 1–350 Search PubMed.
- S. Tada, S. Kayamori, T. Honma, H. Kamei, A. Nariyuki, K. Kon, T. Toyao, K. I. Shimizu and S. Satokawa, ACS Catal., 2018, 8, 7809–7819 CrossRef CAS.
- P. Bhanja, A. Modak and A. Bhaumik, Chem. – Eur. J., 2018, 24, 14189–14197 CrossRef PubMed.
- K. Saravanan, H. Ham, N. Tsubaki and J. W. Bae, Appl. Catal., B, 2017, 217, 494–522 CrossRef CAS.
- L. Guo, J. Sun, Q. Ge and N. Tsubaki, J. Mater. Chem. A, 2018, 6, 23244–23262 RSC.
- T. Biswas and V. Mahalingam, New J. Chem., 2017, 41, 14839–14842 RSC.
- A.-A. G. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951–976 CrossRef CAS PubMed.
- A. J. Wills, Y. K. Ghosh and S. Balasubramanian, J. Org. Chem., 2002, 67, 6646–6652 CrossRef CAS PubMed.
- J. P. Mayer, G. S. Lewis, M. J. Cuetius and J. Zhang, Tetrahedron Lett., 1997, 38, 8445–8448 CrossRef CAS.
- I. Vauthey, F. Valot, C. Gozzi, F. Fache and M. Lemaine, Tetrahedron Lett., 2000, 41, 6347–6350 CrossRef CAS.
- W. J. Blank, Certain hydroxyalkylcarbamates, polymers and uses thereof, US Pat., 4820830, 1989 Search PubMed.
- R. Nomura, Y. Hasegawa, M. Ishimoto, T. Toyosaki and H. Matsuda, J. Org. Chem., 1992, 57, 7339–7342 CrossRef CAS.
- M. Tamura, M. Honda, Y. Nakagawa and K. Tomishige, J. Chem. Technol. Biotechnol., 2014, 89, 19–33 CrossRef CAS.
- A. Modak and S. Jana, Microporous Mesoporous Mater., 2019, 276, 107–132 CrossRef CAS.
- X. Zhang, W. Li and A. Lu, New Carbon Mater., 2015, 30, 481–501 CrossRef CAS.
- K. S. Lackner, H. Ziock and P. Grimes, Carbon Dioxide Extraction from Air: Is It an Option? Proceedings of the 24th Annual Technical Conference on Coal Utilization & Fuel Systems, Clearwater, FL, 1999, pp. 885–896.
- F. S. Zeman and K. S. Lackner, World Resour. Rev., 2004, 16, 157–172 Search PubMed.
- X. Shi, H. Xiao, H. Azarabadi, J. Song, X. Wu, X. Chen and K. S. Lackner, Angew. Chem., Int. Ed., 2020, 59, 6984–7006 CrossRef CAS PubMed.
- S. Das, C. Ghosh and S. Jana, J. Mater. Chem. A, 2016, 4, 7632–7640 RSC.
- E. S. Sanz-Pérez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef PubMed.
- D. Wu, Q. Yang, C. Zhong, D. Liu, H. Huang, W. Zhang and G. Maurin, Langmuir, 2012, 28, 12094–12099 CrossRef CAS PubMed.
- D. Iruretagoyena, X. W. Huang, M. S. P. Shaffer and D. Chadwick, Ind. Eng. Chem. Res., 2015, 54, 11610–11618 CrossRef CAS.
- S. Das, A. Maity, M. Pradhan and S. Jana, Anal. Chem., 2016, 88, 2205–2211 CrossRef CAS PubMed.
- S. Jana, S. Das, C. Ghosh, A. Maity and M. Pradhan, Sci. Rep., 2015, 5, 8711 CrossRef CAS PubMed.
- M. Yuan, R. Yang, S. Wei, X. Hu, D. Xu, J. Yang and Z. Dong, J. Colloid Interface Sci., 2019, 538, 720–730 CrossRef CAS.
- O. T. Qazvini and S. G. Telfer, J. Mater. Chem. A, 2020, 8, 12028–12034 RSC.
- Y. Liu, V. C. Kravtsov and M. Eddaoudi, Angew. Chem., Int. Ed., 2008, 47, 8446–8449 CrossRef CAS.
- S. Xiong, Y. Gong, H. Wang, H. Wang, Q. Liu, M. Gu, X. Wang, B. Chen and Z. Wang, Chem. Commun., 2014, 50, 12101–12104 RSC.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O’Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef PubMed.
- H. J. Noh, Y. K. Im, S. Y. Yu, J. M. Seo, J. Mahmood, T. Yildirim and J. B. Baek, Nat. Commun., 2020, 11, 1–8 Search PubMed.
- R. Balasubramanian and S. Chowdhury, J. Mater. Chem. A, 2015, 3, 21968–21989 RSC.
- A. B. Rao and E. S. Rubin, Ind. Eng. Chem. Res., 2006, 45, 2421–2429 CrossRef CAS.
- A. B. Rao and E. S. Rubin, Environ. Sci. Technol., 2002, 36, 4467–4475 CrossRef CAS.
- M. R. M. Abu-Zahra, L. H. J. Schneiders, J. P. M. Niederer, P. H. M. Feron and G. F. Versteeg, Int. J. Greenhouse Gas Control, 2007, 1, 37–46 CrossRef CAS.
- C. Nwaoha, T. Supap, R. Idem, C. Saiwan, P. Tontiwachwuthikul, M. J. AL-Marri and A. Benamor, Petroleum, 2017, 3, 10–36 CrossRef.
- S. Mudhasakul, H. M. Ku and P. L. Douglas, Int. J. Greenhouse Gas Control, 2013, 15, 134–141 CrossRef CAS.
- S. Tian, Y. Hou, W. Wu, S. Ren and J. Qian, J. Taiwan Inst. Chem. Eng., 2015, 49, 95–99 CrossRef CAS.
- M. Yuana, G. Gaoa, X. Hub, X. Luoa, Y. Huanga, B. Jina and Z. Lianga, Ind. Eng. Chem. Res., 2018, 57, 6189–6200 CrossRef.
- S. Y. Choi, S. C. Nam, Y. I. Yoon, K. T. Park and S. J. Park, Ind. Eng. Chem. Res., 2014, 53, 14451–14461 CrossRef CAS.
- M. Caplow, J. Am. Chem. Soc., 1968, 90, 6795–6803 CrossRef CAS.
- P. V. Danckwerts, Chem. Eng. Sci., 1979, 34, 443–446 CrossRef CAS.
- P. M. M. Blauwhoff, G. F. Versteeg and W. P. M. Van Swaaij, Chem. Eng. Sci., 1983, 38, 1411–1429 CrossRef CAS.
- H. B. Xie, Y. Zhou, Y. Zhang and J. K. Johnson, J. Phys. Chem. A, 2010, 114, 11844–11852 CrossRef CAS PubMed.
- B. Lv, B. Guo, Z. Zhou and G. Jing, Environ. Sci. Technol., 2015, 49, 10728–10735 CrossRef CAS PubMed.
- B. Arstad, R. Blom and O. Swang, J. Phys. Chem. A, 2007, 111, 1222–1228 CrossRef CAS.
- B. A. Oyenekan and G. T. Rochelle, AIChE J., 2007, 53, 3144–3154 CrossRef CAS.
- Y. Belmabkhout and A. Sayari, Energy Fuels, 2010, 24, 5273–5280 CrossRef CAS.
- X. Xu, C. Song, J. M. Andresen, B. G. Miller and A. W. Scaroni, Energy Fuels, 2002, 16, 1463–1469 CrossRef CAS.
- X. Xu, C. Song, B. G. Miller and A. W. Scaroni, Ind. Eng. Chem. Res., 2005, 44, 8113–8119 CrossRef CAS.
- D. Wang, C. Sentorun-Shalaby, X. Ma and C. Song, Energy Fuels, 2011, 25, 456–458 CrossRef CAS.
- X. Ma, X. Wang and C. Song, J. Am. Chem. Soc., 2009, 131, 5777–5783 CrossRef CAS PubMed.
- D. M. D’Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef.
- O. Leal, C. Bolívar, C. Ovalles, J. J. García and Y. Espidel, Inorg. Chim. Acta, 1995, 240, 183–189 CrossRef CAS.
- L. A. Darunte, A. D. Oetomo, K. S. Walton, D. S. Sholl and C. W. Jones, ACS Sustainable Chem. Eng., 2016, 4, 5761–5768 CrossRef CAS.
- F.-Y. Chang, K.-J. Chao, H.-H. Cheng and C.-S. Tan, Sep. Purif. Technol., 2009, 70, 87–95 CrossRef CAS.
- S. Das, A. Samanta and S. Jana, Chem. Eng. J., 2019, 374, 1118–1126 CrossRef CAS.
- K. Kole, A. Halder, S. Singh, A. Samanta, S. Das, A. K. Kundu, D. Bhattacharyya, S. K. Pal and S. Jana, ACS Appl. Nano Mater., 2020, 3, 4321–4328 CrossRef CAS.
- B. Singh and V. Polshettiwar, J. Mater. Chem. A, 2016, 4, 7005–7019 RSC.
- B. Singh and V. Polshettiwar, Nanoscale, 2019, 11, 5365–5376 RSC.
- Z. Yong, V. Mata and A. E. Rodrigues, J. Chem. Eng. Data, 2000, 45, 1093–1095 CrossRef CAS.
- F. Granados-Correa, J. Bonifacio-Martínez, H. Hernandez-Mendoza and S. Bulbulian, J. Air Waste Manage. Assoc., 2016, 66, 643–654 CrossRef CAS.
- N. K. Mohammad, A. Ghaemi and K. Tahvildari, Int. J. Greenhouse Gas Control, 2019, 88, 24–37 CrossRef CAS.
- A. K. Mishra and S. Ramaprabhu, J. Appl. Phys., 2014, 116, 064306 CrossRef.
- J. Baniecki, M. Ishii, K. Kurihara, K. Yamanaka, T. Yano, K. Shinozaki, T. Imada and Y. Kobayashi, J. Appl. Phys., 2009, 106, 054109 CrossRef.
- G. Pacchioni, J. M. Ricart and F. Illas, J. Am. Chem. Soc., 1994, 116, 10152–10158 CrossRef CAS.
- Q. Wang, J. Luo, Z. Zhong and A. Borgna, Energy Environ. Sci., 2011, 4, 42–55 RSC.
- K. Nakagawa, M. Kato, S. Yoshikawa and K. Essaki, J. Mater. Sci. Lett., 2002, 21, 485–487 CrossRef.
- V. Manovic and E. J. Anthony, Energy Fuels, 2008, 22, 1851–1857 CrossRef CAS.
- V. Manovic and E. J. Anthony, Environ. Sci. Technol., 2007, 41, 1420–1425 CrossRef CAS PubMed.
- Z. S. Li, N. S. Cai and Y. Y. Huang, Ind. Eng. Chem. Res., 2006, 45, 1911–1917 CrossRef CAS.
- C. H. Huang, K. P. Chang, C. T. Yu, P. C. Chiang and C. F. Wang, Chem. Eng. J., 2010, 161, 129–135 CrossRef CAS.
- A. de Roy, C. Forano, K. E. Malki and J. P. Besse, Anionic Clays: Trends in Pillaring Chemistry, Expanded Clays and Other Microporous Solids, van Nostrand Reinhold, New York, 2002, ch. 7 Search PubMed.
- M. K. Ram Reddy, Z. P. Xu, G. Q. Lu and J. C. Diniz da Costa, Ind. Eng. Chem. Res., 2006, 45, 7504–7509 CrossRef CAS.
- L. Huang, J. Wang, Y. Gao, Y. Qiao, Q. Zheng, Z. Guo, Y. Zhao, D. O'Hare and Q. Wang, J. Mater. Chem. A, 2014, 2, 18454–18462 RSC.
- S. P. Chen, X. Sun, X. Luo and Z. Liang, Ind. Eng. Chem. Res., 2019, 58, 1177–1189 CrossRef CAS.
- H. B. Mohd Sidek, Y. K. Jo, I. Y. Kim and S. J. Hwang, J. Phys. Chem. C, 2016, 120, 23421–23429 CrossRef CAS.
- L. Zhang, Y. Meng, G. Pan and S. Xia, Inorg. Chem., 2020, 59, 17722–17731 CrossRef CAS PubMed.
- S. Zeng, X. Zhang, L. Bai, X. Zhang, H. Wang, J. Wang, D. Bao, M. Li, X. Liu and S. Zhang, Chem. Rev., 2017, 117, 9625–9673 CrossRef CAS PubMed.
- E. D. Bates, R. D. Mayton, I. Ntai and J. H. Davis, J. Am. Chem. Soc., 2002, 124, 926–927 CrossRef CAS PubMed.
- M. J. Muldoon, S. N. Aki, J. L. Anderson, J. K. Dixon and J. F. Brennecke, J. Phys. Chem. B, 2007, 111, 9001–9009 CrossRef CAS PubMed.
- J. L. Anthony, J. L. Anderson, E. J. Maginn and J. F. Brennecke, J. Phys. Chem. B, 2005, 109, 6366–6374 CrossRef CAS PubMed.
- C. Cadena, J. L. Anthony, J. K. Shah, T. I. Morrow, J. F. Brennecke and E. J. Maginn, J. Am. Chem. Soc., 2004, 126, 5300–5308 CrossRef CAS PubMed.
- C. Wang, H. Luo, X. Luo, H. Li and S. Dai, Green Chem., 2010, 12, 2019–2023 RSC.
- H. Yan, L. Zhao, Y. Bai, F. Li, H. Dong, H. Wang, X. Zhang and S. Zeng, ACS Sustainable Chem. Eng., 2020, 8, 2523–2530 CrossRef CAS.
- S. Zulfiqar, M. I. Sarwar and D. Mecerreyes, Polym. Chem., 2015, 6, 6435–6451 RSC.
- D. Mecerreyes, Prog. Polym. Sci., 2011, 36, 1629–1648 CrossRef CAS.
- J. Tang, H. Tang, W. Sun, M. Radosz and Y. Shen, Polymer, 2005, 46, 12460–12467 CrossRef CAS.
- E. I. Privalova, E. Karjalainen, M. Nurmi, P. Mäki-Arvela, K. Eränen, H. Tenhu, D. Y. Murzin and J.-P. Mikkola, ChemSusChem, 2013, 6, 1500–1509 CrossRef CAS PubMed.
- A. Wilke, J. Yuan, M. Antonietti and J. Weber, ACS Macro Lett., 2012, 1, 1028–1031 CrossRef CAS.
- H. Cheng, P. Wang, J. Luo, J. Fransaer, D. E. De Vos and Z.-H. Luo, Ind. Eng. Chem. Res., 2015, 54, 3107–3115 CrossRef CAS.
- J. Husebye, A. L. Brunsvold, S. Roussanaly and X. Zhang, Energy Procedia, 2012, 23, 381–390 CrossRef.
- S. Roussanaly, R. Anantharaman, K. Lindqvist and B. Hagen, Sustainable Energy Fuels, 2018, 2, 1225–1243 RSC.
- Q. Fu, A. Halim, J. Kim, J. M. Scofield, P. A. Gurr, S. E. Kentish and G. G. Qiao, J. Mater. Chem. A, 2013, 1, 13769–13778 RSC.
- Q. Fu, J. Kim, P. A. Gurr, J. M. Scofield, S. E. Kentish and G. G. Qiao, Energy Environ. Sci., 2016, 9, 434–440 RSC.
- D. H. Kim, M. S. Park, N. U. Kim, D. Y. Ryu and J. H. Kim, Macromolecules, 2018, 51, 5646–5655 CrossRef CAS.
- R. Khalilpour, K. Mumford, H. Zhai, A. Abbas, G. Stevens and E. S. Rubin, J. Cleaner Prod., 2015, 103, 286–300 CrossRef CAS.
- C. H. Yu, C. H. Huang and C. S. Tan, Aerosol Air Qual. Res., 2012, 12, 745–769 CrossRef CAS.
- A. Modak, J. Mondal and A. Bhaumik, ChemCatChem, 2013, 5, 1749–1753 CrossRef CAS.
- S. Das, P. Heasman, T. Ben and S. L. Qiu, Chem. Rev., 2017, 117, 1515–1563 CrossRef CAS PubMed.
- L. Zou, Y. Sun, S. Che, X. Yang, X. Wang, M. Bosch, Q. Wang, H. Li, M. Smith, S. Yuan, Z. Perry and H. C. Zhou, Adv. Mater., 2017, 29, 1700229 CrossRef PubMed.
- Y. Xu, S. Jin, H. Xu, A. Nagai and D. Jiang, Chem. Soc. Rev., 2013, 42, 8012–8031 RSC.
- O. Buyukcakir, S. H. Je, J. Park, H. A. Patel, Y. Jung, C. T. Yavuz and A. Coskun, Chem. – Eur. J., 2015, 21, 15320–15327 CrossRef CAS PubMed.
- L. X. Tan and B. Tan, Chem. Soc. Rev., 2017, 46, 3322–3356 RSC.
- A. E. Baumann, D. A. Burns, B. Liu and V. S. Thoi, Commun. Chem., 2019, 2, 86 CrossRef.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS PubMed.
- A. R. Millward and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 17998–17999 CrossRef CAS.
- N. Getachew, Y. Chebude, I. Díaz and M. Sánchez-Sánchez, J. Porous Mater., 2014, 21, 769–773 CrossRef CAS.
- H. Li, M. Eddaoudi, M. O’Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS.
- H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. O. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424–428 CrossRef CAS PubMed.
- D. Britt, H. Furukawa, B. Wang, T. G. Glover and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20637–20640 CrossRef CAS PubMed.
- S. S. Bourrelly, P. L. Llewellyn, C. Serre, F. Millange, T. Loiseau and G. Férey, J. Am. Chem. Soc., 2005, 127, 13519–13521 CrossRef CAS.
- Q. Liu, N. C. O. Cheung, A. E. Garcia-Bennett and N. Hedin, ChemSusChem, 2011, 4, 91–97 CrossRef CAS PubMed.
- M. Mofarahi and F. Gholipour, Microporous Mesoporous Mater., 2014, 200, 1–10 CrossRef CAS.
- H. Yang, M. Luo, X. Chen, X. Zhao, J. Lin, D. Hu, D. Li, X. Bu, P. Feng and T. Wu, Inorg. Chem., 2017, 56, 14999–15005 CrossRef CAS PubMed.
- S. A. Moggach, T. D. Bennett and A. K. Cheetham, Angew. Chem., Int. Ed., 2009, 48, 7087–7089 CrossRef CAS.
- Y.-Q. Tian, Z.-X. Chen, L.-H. Weng, H.-B. Guo, S. Gao and D. Y. Zhao, Inorg. Chem., 2004, 43, 4631–4635 CrossRef CAS.
- A. Phan, C. J. Doonan, F. J. Uribe-romo, C. B. Knobler, M. O’Keeffe and O. M. Yaghi, Acc. Chem. Res., 2010, 43, 58–67 CrossRef CAS PubMed.
- C. Chen, J. Kim, D. A. Yang and W. S. Ahn, Chem. Eng. J., 2011, 168, 1134–1139 CrossRef CAS.
- E. V. Ramos-Fernandez, A. Grau-Atienza, D. Farrusseng and S. Aguado, J. Mater. Chem. A, 2018, 6, 5598–5602 RSC.
- Q. Song, S. K. Nataraj, M. V. Roussenova, J. C. Tan, D. J. Hughes, W. Li, P. Bourgoin, M. A. Alam, A. K. Cheetham, S. A. Al-Muhtaseb and E. Sivaniah, Energy Environ. Sci., 2012, 5, 8359–8369 RSC.
- F. Gao, Y. Li, Z. Bian, J. Hu and H. Liu, J. Mater. Chem. A, 2015, 3, 8091–8097 RSC.
- N. P. Wickramaratne and M. Jaroniec, ACS Appl. Mater. Interfaces, 2013, 5, 1849–1855 CrossRef CAS PubMed.
- K. X. Yao, Y. L. Chen, Y. Lu, Y. F. Zhao and Y. Ding, Carbon, 2017, 122, 258–265 CrossRef CAS.
- V. Presser, J. McDonough, S.-H. Yeon and Y. Gogotsi, Energy Environ. Sci., 2011, 4, 3059–3066 RSC.
- G. P. Hao, W. C. Li, S. Wang, G. H. Wang, L. Qi and A. H. Lu, Carbon, 2011, 49, 3762–3772 CrossRef CAS.
- G.-P. Hao, W.-C. Li, D. Qian, G.-H. Wang, W.-P. Zhang, T. Zhang, A.-Q. Wang, F. Schuth, H.-J. Bongard and A.-H. Lu, J. Am. Chem. Soc., 2011, 133, 11378–11388 CrossRef CAS PubMed.
- G. P. Hao, Z. Y. Jin, Q. Sun, X. Q. Zhang, J. T. Zhang and A. H. Lu, Energy Environ. Sci., 2013, 6, 3740–3747 RSC.
- J. Yu, M. Guo, F. Muhammad, A. Wang, F. Zhang, Q. Li and G. Zhu, Carbon, 2014, 69, 502–514 CrossRef CAS.
- J. Yu, M. Guo, F. Muhammad, A. Wang, G. Yu, H. Ma and G. Zhu, Microporous Mesoporous Mater., 2014, 190, 117–127 CrossRef CAS.
- Z. Y. Jin, Y. Y. Xu, Q. Sun and A. H. Lu, Small, 2015, 11, 5151–5156 CrossRef CAS PubMed.
- M. Zhong, S. Natesakhawat, J. P. Baltrus, D. Luebke, H. Nulwala, K. Matyjaszewski and T. Kowalewski, Chem. Commun., 2012, 48, 11516–11518 RSC.
- M. S. Duyara, M. A. ArellanoTreviñob and R. J. Farrautoa, Appl. Catal., B, 2015, 168–169, 370–376 CrossRef.
- X. Gao, B. Yu, Z. Yang, Y. Zhao, H. Zhang, L. Hao, B. Han and Z. Liu, ACS Catal., 2015, 5, 6648–6652 CrossRef CAS.
- Y. Ren, Z. Ma and P. G. Bruce, Chem. Soc. Rev., 2012, 41, 4909–4927 RSC.
- R. J. D. Tilley, Defects in Solids, John Wiley & Sons, Inc., 2008 Search PubMed.
- A. M. Stoneham, Theory of Defects in Solids: Electronic Structure of Defects in Insulators and Semiconductors, Oxford University Press, 2001 Search PubMed.
- A. Wold and K. Dwight, Solid State Chemistry: Synthesis, Structure, and Properties of Selected Oxides and Sulfides, Springer Science & Business Media, 1993 Search PubMed.
- D. M. Smyth, The Defect Chemistry of Metal Oxides, Oxford University Press, 2000 Search PubMed.
- J. Jupille and G. Thornton, Defects at Oxide Surfaces, Springer Series in Surface Sciences, 2015 Search PubMed.
- J. Jia, C. Qian, Y. Dong, Y. F. Li, H. Wang, M. Ghoussoub, K. T. Butler, A. Walsh and G. A. Ozin, Chem. Soc. Rev., 2017, 46, 4631–4644 RSC.
- R. E. Owen, P. Plucinski, D. Mattia, L. Torrente-Murciano, V. P. Ting and M. D. Jones, J. CO2 Util., 2016, 16, 97–103 CrossRef CAS.
- K. Chang, H. Zhang, M.-J. Cheng and Q. Lu, ACS Catal., 2020, 10, 613–631 CrossRef CAS.
- K. Z. Li and J. G. Chen, ACS Catal., 2019, 9, 7840–7861 CrossRef CAS.
- K. Ahmad and S. Upadhyayula, Sustainable Energy Fuels, 2019, 3, 2509–2520 RSC.
- Y. Wang, S. Kattel, W. Gao, K. Li, P. Liu, J. G. Chen and H. Wang, Nat. Commun., 2019, 10, 1166 CrossRef PubMed.
- Y. Amenomiya, Appl. Catal., 1987, 30, 57–68 CrossRef CAS.
- K. T. Jung and A. T. Bell, Catal. Lett., 2002, 80, 63–68 CrossRef CAS.
- S. Tada, A. Katagiri, K. Kiyota, T. Honma, H. Kamei, A. Nariyuki, S. Uchida and S. Satokawa, J. Phys. Chem. C, 2018, 122, 5430–5442 CrossRef CAS.
- O. Martin, A. J. Martín, C. Mondelli, S. Mitchell, T. F. Segawa, R. Hauert, C. Drouilly, D. Curulla-Ferré and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2016, 55, 6261–6265 CrossRef CAS PubMed.
- J. Wang, G. Li, Z. Li, C. Tang, Z. Feng, H. An, H. Liu, T. Liu and C. Li, Sci. Adv., 2017, 3, e1701290 CrossRef.
- G. Wang, D. Mao, X. Guo and J. Yu, Int. J. Hydrogen Energy, 2019, 44, 4197–4207 CrossRef CAS.
- G. Wang, D. Mao, X. Guo and J. Yu, Appl. Surf. Sci., 2018, 456, 403–409 CrossRef CAS.
- Z. Shi, Q. Tan and D. Wu, Mater. Chem. Phys., 2018, 219, 263–272 CrossRef CAS.
- J. Słoczynski, R. Grabowski, A. Kozłowska, P. Olszewski, J. Stoch, J. Skrzypek and M. Lachowska, Appl. Catal., A, 2004, 278, 11–23 CrossRef.
- X. An, J. Li, Y. Zuo, Q. Zhang, D. Wang and J. Wang, Catal. Lett., 2007, 118, 264–269 CrossRef CAS.
- P. Gao, F. Li, F. K. Xiao, N. Zhao, N. N. Sun, W. Wei, L. S. Zhong and Y. H. Sun, Catal. Sci. Technol., 2012, 2, 1447–1454 RSC.
- M. Saito, T. Fujitani, M. Takeuchi and T. Watanabe, Appl. Catal., A, 1996, 138, 311–318 CrossRef CAS.
- S. Bai, Q. Shao, P. Wang, Q. Dai, X. Wang and X. Huang, J. Am. Chem. Soc., 2017, 139, 6827–6830 CrossRef CAS PubMed.
- Z. He, Q. Qian, J. Ma, Q. Meng, H. Zhou, J. Song, Z. Liu and B. Han, Angew. Chem., Int. Ed., 2016, 55, 737–741 CrossRef CAS PubMed.
- L. Wang, L. Wang, J. Zhang, X. Liu, H. Wang, W. Zhang, Q. Yang, J. Ma, X. Dong, S. J. Yoo, J. G. Kim, X. Meng and F. S. Xiao, Angew. Chem., Int. Ed., 2018, 57, 6104–6108 CrossRef CAS PubMed.
- R.-P. Ye, J. Ding, W. Gong, M. D. Argyle, Q. Zhong, Y. Wang, C. K. Russell, Z. Xu, A. G. Russell, Q. Li, M. Fan and Y.-G. Yao, Nat. Commun., 2019, 10, 5698 CrossRef CAS PubMed.
- Z. Wang, J. Wang, J. Diao and Y. Jin, Chem. Eng. Technol., 2001, 24, 507–511 CrossRef CAS.
- Z. L. Wang, J. Diao, J. F. Wang, Y. Jin and X. D. Peng, Chin. J. Chem. Eng., 2001, 9, 412–416 CAS.
- F. Xie, H. S. Li, X. L. Zhao, F. Ren, D. Z. Wang, J. F. Wang and J. L. Liu, Chin. J. Catal., 2004, 25, 403–408 CAS.
- J. Erena, R. Garona, J. M. Arandes, A. T. Aguayo and J. Bilbao, Int. J. Chem. React. Eng., 2005, 3, 1–15 Search PubMed.
- J. Erena, R. Garona, J. M. Arandes, A. T. Aguayo and J. Bilbao, Catal. Today, 2005, 107–108, 467–473 CrossRef CAS.
- A. T. Aguayo, J. Erena, I. Sierra, M. Olazar and J. Bilbao, Catal. Today, 2005, 106, 265–270 CrossRef CAS.
- A. T. Aguayo, J. Erena, D. Mier, J. M. Arandes, M. Olazar and J. Bilbao, Ind. Eng. Chem. Res., 2007, 46, 5522–5530 CrossRef CAS.
- X. An, Y. Z. Zuo, Q. Zhang, D. Z. Wang and J. F. Wang, Ind. Eng. Chem. Res., 2008, 47, 6547–6554 CrossRef CAS.
- R. W. Liu, Z. Z. Qin, H. B. Ji and T. M. Su, Ind. Eng. Chem. Res., 2013, 52, 16648–16655 CrossRef CAS.
- H. Ham, S. W. Baek, C.-H. Shin and J. W. Bae, ACS Catal., 2018, 9, 679–690 CrossRef.
- A. Bansode and A. Urakawa, J. Catal., 2014, 309, 66–70 CrossRef CAS.
- R. J. da Silva, A. F. Pimentel, R. S. Monteiro and C. J. A. Mota, J. CO2 Util., 2016, 15, 83–88 CrossRef CAS.
- C. Jeong, H. Ham, J. W. Bae, D. Kang, C. Shin, J. H. Baik and Y. Suh, ChemCatChem, 2017, 9, 4484–4489 CrossRef CAS.
- C. Jeong, J. Kim, J. Kim, S. Lee, J. W. Bae and Y. Suh, Catalysts, 2019, 9, 524 CrossRef.
- J. Gao, C. Jia and B. Liu, Catal. Sci. Technol., 2017, 7, 5602–5607 RSC.
- S. Dang, P. Gao, Z. Liu, X. Chen, C. Yang, H. Wang, L. Zhong, S. Li and Y. Sun, J. Catal., 2018, 364, 382–393 CrossRef CAS.
- P. Gao, S. Dang, S. Li, X. Bu, Z. Liu, M. Qiu, C. Yang, H. Wang, L. Zhong, Y. Han, Q. Liu, W. Wei and Y. Sun, ACS Catal., 2017, 8, 571–578 CrossRef.
- Z. Li, J. Wang, Y. Qu, H. Liu, C. Tang, S. Miao, Z. Feng, H. An and C. Li, ACS Catal., 2017, 7, 8544–8548 CrossRef CAS.
- J. Chen, X. Wang, D. Wu, J. Zhang, Q. Ma, X. Gao, X. Lai, H. Xia, S. Fan and T. Zhao, Fuel, 2019, 239, 44–52 CrossRef CAS.
- X. Liu, M. Wang, C. Zhou, W. Zhou, K. Cheng, J. Kang, Q. Zhang, W. Deng and Y. Wang, Chem. Commun., 2018, 54, 140–143 RSC.
- F. Jiao, J. Li, X. Pan, J. Xiao, H. Li, H. Ma, M. Wei, Y. Pan, Z. Zhou, M. Li, S. Miao, J. Li, Y. Zhu, D. Xiao, T. He, J. Yang, F. Qi, Q. Fu and X. Bao, Science, 2016, 351, 1065–1068 CrossRef CAS PubMed.
- J. Ding, L. Huang, W. Gong, M. Fan, Q. Zhong, A. G. Russell, H. Gua, H. Zhang, Y. Zhang and R. Ye, J. Catal., 2019, 377, 224–232 CrossRef CAS.
- P. Gao, S. Li, X. Bu, S. Dang, Z. Liu, H. Wang, L. Zhong, M. Qiu, C. Yang, J. Cai, W. Wei and Y. Sun, Nat. Chem., 2017, 9, 1019–1024 CrossRef CAS PubMed.
- Y. Ni, Z. Chen, Y. Fu, Y. Liu, W. Zhu and Z. Liu, Nat. Commun., 2018, 9, 3457 CrossRef PubMed.
- X. Cui, P. Gao, S. Li, C. Yang, Z. Liu, H. Wang, L. Zhong and Y. Sun, ACS Catal., 2019, 9, 3866–3876 CrossRef CAS.
- J. C. Choi, T. Sakakura and T. Sako, J. Am. Chem. Soc., 1999, 121, 3793–3794 CrossRef CAS.
- V. Eta, P. Maki-Arvela, A. R. Leino, K. Kordás, T. Salmi, D. Y. Murzin and J. P. Mikkola, Ind. Eng. Chem. Res., 2010, 49, 9609–9617 CrossRef CAS.
- S. Kumar and S. L. Jain, Ind. Eng. Chem. Res., 2014, 53, 15798–15801 CrossRef CAS.
- M. Poor Kalhor, H. Chermette and D. Ballivet-Tkatchenko, Ind. Eng. Chem. Res., 2020, 59, 6867–6873 CrossRef CAS.
- G. G. Giram, V. V. Bokade and S. Darbha, New J. Chem., 2018, 42, 17546–17552 RSC.
- O. Arbeláez, A. Orrego, F. Bustamante and A. L. Villa, Top. Catal., 2012, 55, 668–672 CrossRef.
- M. Zhang, Chem. Pap., 2020, 74, 4493–4505 CrossRef CAS.
- R. Khatun, P. Bhanja, P. Mondal, A. Bhaumik, D. Das and S. M. Islam, New J. Chem., 2017, 41, 12937–12946 RSC.
- G. Tudu, S. Ghosh, T. Biswas and V. Mahalingam, New J. Chem., 2020, 44, 11887–11894 RSC.
- G. N. Bondarenko, E. G. Dvurechenskaya, O. G. Ganina, F. Alonso and I. P. Beletskaya, Appl. Catal., B, 2019, 254, 380–390 CrossRef CAS.
- R. Khatun, P. Bhanja, R. A. Molla, S. Ghosh, A. Bhaumik and S. M. Islam, Mol. Catal., 2017, 434, 25–31 CrossRef CAS.
- D. Prasad, K. N. Patil, J. T. Bhanushali, B. M. Nagaraja and A. H. Jadhav, Catal. Sci. Technol., 2019, 9, 4393–4412 RSC.
- M. Liu, Y. Luo, L. Xu, L. Sun and H. Du, Dalton Trans., 2016, 45, 2369–2373 RSC.
- A. H. Chowdhury, I. H. Chowdhury and S. M. Islam, Ind. Eng. Chem. Res., 2019, 58, 11779–11786 CrossRef CAS.
- Y. Qin, H. Guo, X. Sheng, X. Wang and F. Wang, Green Chem., 2015, 17, 2853–2858 RSC.
- E. J. Doskocil, S. V. Bordawekar, B. G. Kaye and R. J. Davis, J. Phys. Chem. B, 1999, 103, 6277–6282 CrossRef CAS.
- R. Srivastava, D. Srinivas and P. Ratnasamy, J. Catal., 2005, 233, 1–15 CrossRef CAS.
- H. Yasuda, L. N. He and T. Sakakura, J. Catal., 2002, 209, 547–550 CrossRef CAS.
- W. G. Cui and T. L. Hu, Small, 2020, 16, 2003971 Search PubMed.
- W. G. Cui, G. Y. Zhang, T. L. Hu and X. H. Bu, Coord. Chem. Rev., 2019, 387, 79–120 CrossRef CAS.
- D. J. Darensbourg, Chem. Rev., 2007, 107, 2388–2410 CrossRef CAS PubMed.
- V. Gupta and S. K. Mandal, Inorg. Chem., 2020, 59, 4273–4281 CrossRef CAS PubMed.
- Q. Yang, C. C. Yang, C. H. Lin and H. L. Jiang, Angew. Chem., Int. Ed., 2019, 58, 3511–3515 CrossRef CAS PubMed.
- Y. Li, X. Zhang, P. Xu, Z. Jiang and J. Sun, Inorg. Chem. Front., 2019, 6, 317–325 RSC.
- W. J. Peppel, Ind. Eng. Chem., 1958, 50, 767–770 CrossRef CAS.
- M. North and R. Pasquale, Angew. Chem., Int. Ed., 2009, 48, 2946–2948 CrossRef CAS PubMed.
- B. An, Z. Li, Y. Song, J. Zhang, L. Zeng, C. Wang and W. Lin, Nat. Catal., 2019, 2, 709–717 CrossRef CAS.
- L. Zeng, Y. Wang, Z. Li, Y. Song, J. Zhang, J. Wang, X. He, C. Wang and W. Lin, ACS Appl. Mater. Interfaces, 2020, 12, 17436–17442 CrossRef CAS PubMed.
- S. L. Hou, J. Dong, X. L. Jiang, Z. H. Jiao and B. Zhao, Angew. Chem., Int. Ed., 2019, 58, 577–581 CrossRef CAS PubMed.
- C. Wu, F. Irshad, M. Luo, Y. Zhao, X. Ma and S. Wang, ChemCatChem, 2019, 11, 1256–1263 CrossRef CAS.
- P. Tshuma, B. C. E. Makhubela, L. Ohrstrom, S. A. Bourne, N. Chatterjee, I. N. Beas, J. Darkwa and G. Mehlana, RSC Adv., 2020, 10, 3593–3605 RSC.
- M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514–1539 RSC.
- C. M. Miralda, E. E. Macias, M. Zhu, P. Ratnasamy and M. A. Carreon, ACS Catal., 2012, 2, 180–183 CrossRef CAS.
- L. Yang, L. Yu, G. Diao, M. Sun, G. Cheng and S. Chen, J. Mol. Catal. A: Chem., 2014, 392, 278–283 CrossRef CAS.
- Y. Zhang and D. S. W. Lim, ChemSusChem, 2015, 8, 2606–2608 CrossRef CAS PubMed.
- J. L. Song, Z. F. Zhang, S. Q. Hu, T. B. Wu, T. Jiang and B. X. Han, Green Chem., 2009, 11, 1031–1036 RSC.
- S. Carrasco, A. Sanz-Marco and B. Martín-Matute, Organometallics, 2019, 38, 3429–3435 CrossRef CAS.
- Y. F. Lin, K. W. Huang, B. T. Ko and K. Y. A. Lin, J. CO2 Util., 2017, 22, 178–183 CrossRef CAS.
- K. W. Frese, J. Electrochem. Soc., 1991, 138, 3338–3344 CrossRef CAS.
- X. Huang, Y. Chen, Z. Lin, X. Ren, Y. Song, Z. Xu, X. Dong, X. Li, C. Hu and B. Wang, Chem. Commun., 2014, 50, 2624–2627 RSC.
- O. V. Zalomaeva, A. M. Chibiryaev, K. A. Kovalenko, O. A. Kholdeeva, B. S. Balzhinimaev and V. P. Fedin, J. Catal., 2013, 298, 179–185 CrossRef CAS.
- G. Y. Hwang, R. Roshan, H. S. Ryu, H. M. Jeong, S. Ravi, M. I. Kim and D. W. Park, J. CO2 Util., 2016, 15, 123–130 CrossRef CAS.
- D. Ma, B. Li, K. Liu, X. Zhang, W. Zou, Y. Yang, G. Li, Z. Shi and S. Feng, J. Mater. Chem. A, 2015, 3, 23136–23142 RSC.
- T. Lescouet, C. Chizallet and D. Farrusseng, ChemCatChem, 2012, 4, 1725–1728 CrossRef CAS.
- Z. Zhou, C. He, J. Xiu, L. Yang and C. Duan, J. Am. Chem. Soc., 2015, 137, 15066–15069 CrossRef CAS.
- P. Z. Li, X. J. Wang, J. Liu, J. S. Lim, R. Zou and Y. Zhao, J. Am. Chem. Soc., 2016, 138, 2142–2145 CrossRef CAS.
- R. R. Kuruppathparambil, R. Babu, H. M. Jeong, G. Y. Hwang, G. S. Jeong, M. Kim, D. W. Kim and D. W. Park, Green Chem., 2016, 18, 6349–6356 RSC.
- T. Jose, Y. Hwang, D. W. Kim, M. Kim and D. W. Park, Catal. Today, 2015, 245, 61–67 CrossRef CAS.
- K. Zhong, R. F. Yang, W. Y. Zhang, Y. T. Yan, X. J. Gou, W. H. Huang and Y. Y. Wang, Inorg. Chem., 2020, 59, 3912–3918 CrossRef CAS.
- S. Zhang, M. S. Jang, J. Lee, P. Puthiaraj and W. S. Ahn, ACS Sustainable Chem. Eng., 2020, 8, 7078–7086 CrossRef CAS.
- J. Kothandaraman, A. Goeppert, M. Czaun, G. A. Olah and G. K. S. Prakash, J. Am. Chem. Soc., 2016, 138, 778–781 CrossRef CAS.
- W. Tu, Y. Zhou and Z. Zou, Adv. Mater., 2014, 26, 4607–4626 CrossRef CAS.
- S. Wang, W. Yao, J. Lin, Z. Ding and X. Wang, Angew. Chem., Int. Ed., 2014, 126, 1052–1056 CrossRef.
- H. Q. Xu, J. Hu, D. Wang, Z. Li, Q. Zhang, Y. Luo, S. H. Yu and H. L. Jiang, J. Am. Chem. Soc., 2015, 137, 13440–13443 CrossRef CAS PubMed.
- S. Zhang, L. Li, S. Zhao, Z. Sun, M. Hong and J. Luo, J. Mater. Chem. A, 2015, 3, 15764–15768 RSC.
- D. Wang, R. Huang, W. Liu, D. Sun and Z. Li, ACS Catal., 2014, 4, 4254–4260 CrossRef CAS.
- Y. Fu, D. Sun, Y. Chen, R. Huang, Z. Ding, X. Fu and Z. Li, Angew. Chem., Int. Ed., 2012, 51, 3364–3367 CrossRef CAS PubMed.
- C. Wang, Z. Xie, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 13445–13454 CrossRef CAS PubMed.
- Y. Xie, T. T. Wang, X. H. Liu, K. Zou and W. Q. Deng, Nat. Commun., 2013, 4, 1960 CrossRef.
- M. H. Alkordi, J. Weselinski, V. D'Elia, S. Barman, A. Cadiau, M. N. Hedhili, A. J. Cairns, R. G. AbdulHalim, J. M. Basset and M. Eddaoudi, J. Mater. Chem. A, 2016, 4, 7453–7460 RSC.
- A. Chen, Y. Zhang, J. Chen, L. Chen and Y. Yu, J. Mater. Chem. A, 2015, 3, 9807–9816 RSC.
- S. Zheng, T. Wu, J. Zhang, M. Chow, R. A. Nieto, P. Feng and X. Bu, Angew. Chem., Int. Ed., 2010, 122, 5490–5494 CrossRef.
- C. Du, X. Lan, G. An, Q. Li and G. Bai, ACS Sustainable Chem. Eng., 2020, 8, 7051–7058 CrossRef CAS.
- G. H. Gunasekar and S. Yoon, J. Mater. Chem. A, 2019, 7, 14019–14026 RSC.
- G. M. Eder, D. A. Pyles, E. R. Wolfson and P. L. McGrier, Chem. Commun., 2019, 55, 7195–7198 RSC.
- X. Shao, X. Miao, X. Yu, W. Wang and X. Ji, RSC Adv., 2020, 10, 9414–9419 RSC.
- Z. Guo, X. Cai, J. Xie, X. Wang, Y. Zhou and J. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 12812–12821 CrossRef CAS PubMed.
- O. Buyukcakir, S. H. Je, D. S. Choi, S. N. Talapaneni, Y. Seo, Y. Jung, K. Polychronopoulou and A. Coskun, Chem. Commun., 2016, 52, 934–937 RSC.
- C. Cui, R. Sa, Z. Hong, H. Zhong and R. Wang, ChemSusChem, 2020, 13, 180–187 CrossRef CAS.
- S. N. Talapaneni, O. S. H. Buyukcakir, S. H. Je, S. Y. Srinivasan, Y. Seo, K. Polychronopoulou and A. Coskun, Chem. Mater., 2015, 27, 6818–6826 CrossRef CAS.
- V. Saptal, D. B. Shinde, R. Banerjee and B. M. Bhanage, Catal. Sci. Technol., 2016, 6, 6152–6158 RSC.
- Y. Zhi, P. Shao, X. Feng, H. Xia, Y. Zhang, Z. Shi, Y. Mu and X. Liu, J. Mater. Chem. A, 2018, 6, 374–382 RSC.
- M. Liu, K. Gao, L. Liang, F. Wang, L. Shi, L. Sheng and J. Sun, Phys. Chem. Chem. Phys., 2015, 17, 5959–5965 RSC.
- Y. Tsuji and T. Fujihara, Chem. Commun., 2012, 48, 9956–9964 RSC.
- N. Austin, B. Butina and G. Mpourmpakis, Prog. Nat. Sci.: Mater. Int., 2016, 26, 487–492 CrossRef CAS.
- S. H. Lee, S. Jeong, W. D. Kim, S. Lee, K. Lee, W. K. Bae, J. H. Moon, S. Lee and D. C. Lee, Nanoscale, 2016, 8, 10043–10048 RSC.
- M. M. J. Li, Z. Y. Zeng, F. L. Liao, X. L. Hong and S. C. E. Tsang, J. Catal., 2016, 343, 157–167 CrossRef CAS.
- X. Zhang, W. Z. Zhang, X. Ren, L. L. Zhang and X. B. Lu, Org. Lett., 2011, 13, 2402–2405 CrossRef CAS PubMed.
- D. Y. Yu, M. X. Tan and Y. G. Zhang, Adv. Synth. Catal., 2012, 354, 969–974 CrossRef CAS.
- P. G. Dominguez, L. Fehr, G. Rusconi and C. Nevado, Chem. Sci., 2016, 7, 3914–3918 RSC.
- X. H. Liu, J. G. Ma, Z. Niu, G. M. Yang and P. Cheng, Angew. Chem., Int. Ed., 2015, 54, 988–991 CrossRef CAS.
- Z. Yang, B. Yu, H. Zhang, Y. Zhao, Y. Chen, Z. Ma, G. Ji, X. Gao, B. Han and Z. Liu, ACS Catal., 2016, 6, 1268–1273 CrossRef CAS.
- Y. Li, H. Zhang, L. Zhang and H. Zhang, Int. J. Hydrogen Energy, 2019, 44, 13354–13363 CrossRef CAS.
- A. Bustinza, M. Frías, Y. Liu and E. García-Bordejé, Catal. Sci. Technol., 2020, 10, 4061–4071 RSC.
- D. D. Masi, J. M. Asensio, P. F. Fazzini, L. M. Lacroix and B. Chaudret, Angew. Chem., Int. Ed., 2020, 59, 6187–6191 CrossRef PubMed.
- S. Shen, X. Peng, L. Song, Y. Qiu, C. Li, L. Zhuo, J. He, J. Ren, X. Liu and J. Luo, Small, 2019, 15, 1970193 CrossRef.
- M. M. J. Li, H. Zou, J. Zheng, T. S. Wu, T. S. Chan, Y. L. Soo, X. P. Wu, X. Q. Gong, T. Chen, K. Roy, G. Held and S. C. E. Tsang, Angew. Chem., Int. Ed., 2020, 132, 16173–16180 CrossRef.
- K. Mori, H. Yamashita and M. Anpo, RSC Adv., 2012, 2, 3165–3172 RSC.
- L. J. Liu, F. Gao, H. L. Zhao and Y. Li, Appl. Catal., B, 2013, 134, 349–358 CrossRef.
- I. Shown, H. C. Hsu, Y. C. Chang, C. H. Lin, P. K. Roy, A. Ganguly, C. H. Wang, J. K. Chang, C. I. Wu, L. C. Chen and K. H. Chen, Nano Lett., 2014, 14, 6097–6103 CrossRef CAS PubMed.
- S. Zhang, P. Kang, M. Bakir, A. M. Lapides, C. J. Dares and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 15809–15814 CrossRef CAS PubMed.
- J. Medina-Ramos, J. L. DiMeglio and J. Rosenthal, J. Am. Chem. Soc., 2014, 136, 8361–8367 CrossRef CAS.
- S. Ma, R. Luo, J. I. Gold, A. Z. Yu, B. Kim and P. J. A. Kenis, J. Mater. Chem. A, 2016, 4, 8573–8578 RSC.
- S. I. Rybchenko, D. Touhami, J. D. Wadhawan and S. K. Haywood, ChemSusChem, 2016, 9, 1660–1669 CrossRef CAS PubMed.
- Q. W. Song, Z. H. Zhou, M. Y. Wang, K. Zhang, P. Liu, J. Y. Xun and L. N. He, ChemSusChem, 2016, 9, 2054–2058 CrossRef CAS PubMed.
- M. Tamura, K. Noro, M. Honda, Y. Nakagawa and K. Tomishige, Green Chem., 2013, 15, 1567–1577 RSC.
- S. Sun, W. M. Hu, N. Gu and J. Cheng, Chem. – Eur. J., 2016, 22, 18729–18732 CrossRef CAS.
- P. Sreejyothi and S. K. Mandal, Chem. Sci., 2020, 11, 10571–10593 RSC.
- P. K. Hota, S. C. Sau and S. K. Mandal, ACS Catal., 2018, 8, 11999–12003 CrossRef CAS.
- S. C. Sau, R. Bhattacharjee, P. K. Vardhanapu, G. Vijaykumar, A. Datta and S. K. Mandal, Angew. Chem., Int. Ed., 2016, 55, 15147–15151 CrossRef CAS PubMed.
- E. D. Bates, R. D. Mayton, I. Ntai and J. H. Davis, J. Am. Chem. Soc., 2002, 124, 926–927 CrossRef CAS PubMed.
- C. M. Wang, H. M. Luo, D. E. Jiang, H. R. Li and S. Dai, Angew. Chem., Int. Ed., 2010, 49, 5978–5981 CrossRef CAS.
- H. H. Duan, D. S. Wang and Y. D. Li, Chem. Soc. Rev., 2015, 44, 5778–5792 RSC.
- C. Dhand, N. Dwivedi, X. J. Loh, A. N. J. Ying, N. K. Verma, R. W. Beuerman, R. Lakshminarayanan and S. Ramakrishna, RSC Adv., 2015, 5, 105003 RSC.
- D. V. Talapin and E. V. Shevchenko, Chem. Rev., 2016, 116, 10343–10345 CrossRef CAS PubMed.
- A. Stolle, T. Szuppa, S. E. S. Leonhardt and B. Ondruschka, Chem. Soc. Rev., 2011, 40, 2317–2329 RSC.
| This journal is © The Royal Society of Chemistry 2021 |