
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEugenol micro-emulsion reinforced with silver nanocomposite electrospun mats for wound dressing strategies†
Lakshimipriya
Sethuram
,
John
Thomas
,
Amitava
Mukherjee
 and
Natarajan
Chandrasekaran
*
and
Natarajan
Chandrasekaran
*
Centre for Nanobiotechnology, VIT University, Vellore, Tamilnadu, India. E-mail: nchandrasekaran@vit.ac.in; Fax: +91 416 2243092; Tel: +91 416 2202624
First published on 4th March 2021
Abstract
Wound management is a complex process that involves the application of tissue scaffolds/nanocomposites for wound healing treatment. An ideal wound dressing possesses excellent mechanical properties, good biocompatibility and effective antibacterial activity. The present study optimized silver nanoparticles (AgNPs) using a eugenol microemulsion (EuME) incorporated with polyvinyl alcohol (synthetic polymer) to fabricate efficacious nanofibers via an electrospinning technique by optimizing polymer concentration, applied voltage, needle tip–collector distance and flow rate adjusted to 9%, 25 kV, 15 cm and 0.5 ml h−1 respectively. The results of SEM showed a homogeneous distribution of well-oriented electrospun nanofibers (ESNs) with a uniform pore diameter of 404.1 nm and elemental silver composition of 13.93%. The hydrogen bonding and physical interaction between the nanofibers were observed by FTIR. The thermal stability and specific functionality of nanofibers were investigated by TGA/DTA analysis. The synthesized eugenol microemulsion-silver nanoparticle nanofibers (EuME–AgNP–NFs) were compared with silver bandaid-suspended nanoparticle nanofibers (SBD–AgNP–NFs) to monitor the efficiency and toxicity in the biological system. The eugenol microemulsion-silver nanoparticles (EuME–AgNPs) exhibited the highest antibacterial efficacy against Staphylococcus aureus. The silver release behaviour of EuME–AgNP–NFs showed sustained and controlled release of silver ions in the simulated wound fluid system. The interaction of EuME–AgNPs with lymphocytes and erythrocytes revealed the maximum rate of cell viability (69.81%) and minimum rate of red blood cell breakdown (19.44%) in the human biological system when compared with silver bandaid–silver nanoparticles (SBD–AgNPs). The fabricated EuME–AgNPs–NFs with effective antibacterial activity and sustained release of silver ions provide a suitable environment for wound healing and could be used for cut wounds in clinical practice.
1. Introduction
A wound infection is an excavation or localized defect of the skin in which pathogenic microorganisms invade the viable tissues of the wound, causing tissue damage and inflammation. The microbial factors such as depth, size, site and type of the wound, and the level and state of exogeneous contamination are important parameters for the causes of wound infections.1 There are various types of wound infections such as surgical wounds, acute tissue wounds, cut wounds, bite wounds, burn wounds, diabetic wounds and pressure ulcer wounds. The survey report (2002) by Nosocomial Infection National Surveillance Service (NINSS) reveals that 30% of individuals have been affected with bacterial wound infections worldwide. The health care costs have increased to nearly £1 billion pounds per annum.2 The pathogenic microorganisms present on the surface of wound infections are polymicrobial involving both anaerobic and aerobic microbial strains such as Pseudomonas aeruginosa, Staphylococcus aureus and hemolytic streptococci.3,4 The Gram positive bacterium Staphylococcus aureus is found to be predominant in all types of acute and chronic wound infections and methicillin-resistant Staphylococcus aureus (MRSA) is still increasing at a fast rate and gaining antimicrobial resistance to various synthetic and natural antibiotics resulting in urgent demand for new therapeutics.5,6Wound dressings play an important role in the treatment of wound infections. There are different types of wound dressings namely gauze, foams, alginates, hydrogels and hydrocolloids. A good wound dressing should possess excellent tensile strength, promote effective wound healing, prevent bacterial infections and provide moist environment and permeability for water and gaseous exchange.7 Electrospun nanofibers (ESNs) are nanostructural networks that possess extraordinary characteristic features such as high porosity, better tensile strength and good flexibility with defined structures. ESNs reinforced with silver nanoparticles (AgNPs) can be used as nanocomposites, 3D scaffolds and as wound dressings in various biomedical applications.8,9 Electrospinning is a fabrication technique that uses electrostatic forces to produce multifunctional homogeneous polymeric ESNs with excellent mechanical properties and greater encapsulation efficiency.10 Nanofibrous mats can be loaded with different kinds of therapeutic or bioactive agents such as vitamins, growth factors (GFs) and antibiotics, anti-inflammatory, analgesics and antimicrobial compounds which may be used for treatment of cut wounds, burn wounds, diabetic wounds and other pressure ulcer wounds.11 The electrospinning technique has the ability to incorporate medicinal derivatives and plant extracts like essential oils for effective wound healing applications. Material availiability, cost-effectiveness and traditional therapeutic properties are the main advantages of using essential oils in nanofibrous mats. Sodium alginate combined with lavender oil produces bioactive wound dressings using electrospinning and the efficiency has been tested for treatment of skin burns. Lavender oil–alginate based nanofibrous mats are promising wound dressings for skin burn treatment.12 The combination of conventional antimicrobial agents with essential oils shows enhanced antibacterial properties in vitro. For example, by combining hydrophobic peppermint oil, hydrophilic chlorhexidine digluconate (CHG) and amphiphilic octenidine.2HCl (OCT) to synthesize silica nanocapsules an enhanced disinfection effect can be seen on Bacillus subtilis and Escherichia coli K-12. The combinations of these functional agents provide a smooth platform for the optimization of multifunctional nanomaterials.13 Generally essential oils containing chemical derivatives such as phenolic compounds and volatile terpenoids show antibacterial properties against a wide range of microbial pathogens. The antimicrobial properties of nanofibers loaded with three different essential oils such as cinnamon bark oil, tea tree oil and clove oil were studied qualitatively and quantitatively using three various strains (Staphylococcus aureus, Escherichia coli and Pseudomonas aeruginosa). The nanofibers containing cinnamon essential oil show excellent antimicrobial activity against the selected pathogens.14 The presence of the eugenol compound in cinnamon bark essential oil provides antimicrobial effect against the microorganisms. According to some authors the antimicrobial activity of eucalyptus oil incorporated with cyclodextrin complexes mats shows a homogeneous and continuous microstructure in order to produce zein ultrafine nanofibrous mats. The combination of eucalyptus oil with β-cyclodextrin complexes exhibits an excellent antimicrobial effect against Gram-negative and Gram-positive bacteria.15
Silver is a well-known therapeutic antimicrobial agent for treating bacterial infections. The hindering action of antimicrobial silver is attributed to interaction with thiol groups of the respiratory enzymes inhibiting the process of replication in bacteria.16 The prominent application of AgNPs eliminate microorganisms by preventing cellular respiration and replication in both prokaryotic and eukaryotic cells. The chemically synthesized AgNPs is toxic to the human system and causes adverse effects. To overcome this, the formulation of green synthesized AgNPs using bioactive agents like plant extract derivatives or essential oils is required for wound healing applications. Polyvinyl alcohol (PVA) is a nontoxic, aqueous soluble synthetic polymer with greater mechanical properties and stability. PVA mats exhibit a larger surface area to volume ratio and smaller pore size compared to conventional nonwoven fabrics. Electrospun PVA nanofibers possess higher swelling capacity with flexibility17,18 and prove to be a promising base matrix employed by pharmaceuticals showing greater benefits and prevalence in drug delivery paradigms with less adverse effects. PVA capped AgNPs incorporated with a chitosan-agarose matrix (CAM) exhibit a predominant antibacterial activity with controlled and sustained release of silver ions in the biological system. PVA-AgNPs incorporated with CAM accelerated the wound healing potential in Wistar albino rat models and were found to be comparatively more effective than other synthetic polymer capped AgNPs which are reported in a previous study.19
Eugenol is an antimicrobial phenolic component of cinnamon essential oil known for its anti-inflammatory, antioxidant, anticarcinogenic and analgesic properties.20,21 Eugenol is highly volatile and insoluble in water, gets affected by enzymatic and chemical degradation, and undergoes thermal decomposition and volatilization. To overcome the disadvantages, formulation of emulsion systems using eugenol essential oil could be an alternative approach to address the problem of viscosity, volatility and degradation. Microemulsions have low viscosity, are transparent, possess isotropic dispersion and are thermodynamically stable. These require only low-energy systems such as slow magnetic stirring or vortexing to formulate emulsified particles ranging between 5 and 100 nm. These emulsions can be easily synthesized compared to nanoemulsions and conventional emulsions.
An emulsion electrospinning could be an effective and alternative approach to successfully impregnate both hydrophobic and hydrophilic bioactive agents such as metals, essential oils, emulsions, drugs, proteins and enzymes into biocompatible polymeric ESNs. Synthetic polymers incorporated with nanoparticles were made into biocompatible ESNs using emulsion electrospinning. Bioactive compound based nanoparticles incorporated into ESNs exhibit greater tensile strength and stability with dominant antibacterial efficacy against Gram positive and Gram negative strains.22–25 The ESNs encapsulating cyclodextrin complexes of bioactive compounds like eugenol possess good nanoscale porosity, sustained/controlled release with greater stability and show antibacterial/antioxidant properties for biomedical applications.26 The electrospun inclusion complex (IC) of eugenol and cyclodextrin shows excellent antioxidant activity compared to the pure forms of eugenol. The nanofibers of eugenol exhibit accelerated antibacterial efficacy and flexibility. The thermal properties and the dissolving nature of ESNs incorporated with eugenol and cyclodextrins are also enhanced.27 The ESNs loaded with eugenol and polymeric PVA matrix reveal high porosity in the range of 60%–90%. The effective antibacterial efficacy of eugenol was demonstrated against Staphylococcus aureus and Pseudomonas aeruginosa. The in vitro cytotoxic activity of eugenol shows that human dermal fibroblast cells remained viable for 7 days, even after direct interaction with the produced ESNs.11
The dissolution behaviour of AgNPs is closely related to the toxicity of AgNPs. A few reports suggest that the release of Ag+ ions plays a critical role in acute toxicity of living cells.28 The toxicity of AgNPs in bacteria appears to be strongly driven by release of the silver ions. The generation of reactive oxygen species (ROS) from Ag+ ions upon the exposure of bacterial cells is a major contributor to toxicity.29 Additively, Sotiriou and Pratsinis examined the antibactericidal activity of nanosilver particles and silver ions and thus reported that bactericidal activity against the Gram negative Escherichia coli bacteria is strongly dominated by the Ag+ ions rather than the AgNPs.30 The mechanism of Ag+ ion release takes place in three different ways such as diffusion in swelling of the polymer matrix, polymeric degradation, or a combination of both. Controlled drug release is important for longstanding antimicrobial activity. The incorporation of AgNPs into the polymeric matrix was fabricated to form biodegradable ESNs and release behaviour of nanofibers was reported.8,9 There are few reports on the effect of shape and size of AgNPs and the effect of fiber morphology on the release behaviour of AgNPs.
The present study aimed to synthesize AgNPs using a eugenol microemulsion (EuME) termed as eugenol microemulsion–silver nanoparticles (EuME–AgNPs). Similarly, AgNPs using silver bandaid suspensions (SBD) were termed as silver bandaid–silver nanoparticles (SBD–AgNPs). The physicochemical properties of EuME–AgNPs and SBD–AgNPs were characterized and evaluated. The EuME–AgNPs and SBD–AgNPs were incorporated with a polymeric PVA matrix to produce eugenol microemulsion–silver nanoparticle–nanofibers (EuME–AgNP–NFs) and silver bandaid-suspended nanoparticle nanofibers (SBD–AgNP–NFs) respectively using the electrospinning technique to prevent the loss of bioactivity and structural integrity. The properties of EuME–AgNP–NFs and SBD–AgNP–NFs were studied. Furthermore, the study will be subjected to ensuring antimicrobial efficacy (Minimum Inhibitory Concentration) of AgNPs tested on Staphylococcus aureus. The silver ion release behaviour of EuME–AgNP–NFs and SBD–AgNP–NFs in the prepared simulated wound fluid (SWF) system was investigated. The cytotoxic response of EuME–AgNPs and SBD–AgNPs in the lymphocytes or white blood cells (WBCs) (rate of cell viability) and erythrocytes or red blood cells (RBCs) (rate of red blood cells breakdown) was evaluated as well.
2. Experimental section
2.1. Materials
Eugenol essential oil (Reagent Plus, 99%), Tween 20 (polyoxyethylene (20) sorbitan monolaurate), Tween 80 (polyethylene glycol sorbitan monooleate, highly viscous) Brij 93 (polyethylene glycol oleyl ether) (mol wt – 356.58 g mol−1), Span 80 (sorbitan monooleate) (mol wt – 428.62 g mol−1) silver nitrate (AgNO3) (ACS reagent, ≥99.0%), polyvinyl alcohol (PVA) (Mw 89![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–98000, 99% hydrolyzed), potassium chloride (KCl) (≥99.0%), sodium chloride (NaCl) (≥99.5%), calcium chloride (CaCl2) (≤7.0 mm, ≥93.0%), sodium bicarbonate (NaHCO3) (≥99.5%) and bovine serum albumin (BSA) were obtained from Sigma-Aldrich (India). Nutrient broth (NB), Nutrient Agar (NA) and Mueller Hinton Broth (MHB) were procured from Himedia Private Laboratories (India). Deionized ultrapurified water was purchased from the Biowater CascadaTM System (USA). The reagents used for the study were of analytical gradient.
000–98000, 99% hydrolyzed), potassium chloride (KCl) (≥99.0%), sodium chloride (NaCl) (≥99.5%), calcium chloride (CaCl2) (≤7.0 mm, ≥93.0%), sodium bicarbonate (NaHCO3) (≥99.5%) and bovine serum albumin (BSA) were obtained from Sigma-Aldrich (India). Nutrient broth (NB), Nutrient Agar (NA) and Mueller Hinton Broth (MHB) were procured from Himedia Private Laboratories (India). Deionized ultrapurified water was purchased from the Biowater CascadaTM System (USA). The reagents used for the study were of analytical gradient.
2.2. Formulation of eugenol microemulsion (EuME)
Primarily, microemulsions was optimized using eugenol oil and four different surfactants namely hydrophilic non-ionic surfactants with Tween 20/Tween 80 (HLB value of 16.7/15.0) and Brij 93/Span 80 (HLB value of 4.0/4.3). An oil in water (O/W) type of emulsification was used to prepare microemulsions. Pseudo-ternary diagrams were constructed in order to choose the best surfactant for optimization. 6% v/v of eugenol oil was kept constant for all formulations. Various formulations of emulsions were prepared by mixing the oil:surfactant in different ratios ranging from 1:1 to 1:9 respectively. Deionized water was added drop by drop to the organic phase system (oil:surfactant). The process of microemulsion was carried under magnetic stirring using a SpinIt 4010 (Tarsons Products Pvt. Ltd, India) with a stirring speed of 400 rpm at room temperature. The visual appearance of microemulsions was observed. The hydrodynamic droplet size and polydispersity index (pDI) was determined by dynamic light scattering (DLS) technique using a Horiba Scientific nanoparticle analyzer type model ((SZ-100), Japan) operated at room temperature and the effect of stability parameters was studied with regard to different time intervals for the formulated microemulsions. All prepared EuMEs were stored for 24 hours for equilibration. As an end point, the EuME with minimum surfactant concentration mixing ratio, less toxicity and longstanding kinetic stability was chosen for further characterization studies.
2.3. Preparation of silver nanoparticles (AgNPs)
2.4. Characterization of EuME–AgNPs and SBD–AgNPs
2.5. Preparation of EuME, EuME–AgNP and SBD–AgNP electrospinning solutions
Primarily, a PVA solution of 10% w/v was prepared by dissolving PVA powder in a defined mixture of 2% acetic acid and deionized water under constant magnetic stirring at 120 °C for 3 hours. After obtaining a visible transparent solution, three types of stock solutions were prepared. First, to the formulated PVA (base matrix) solution, various percentages of (0, 3%, 6%, 9% v/v) of EuME were added and stirred (300 rpm) using a magnetic stirrer at room temperature for 3 hours, to get uniformly blended homogeneous solutions for electrospinning. Secondly, to the prepared PVA (base matrix) transparent solution, various percentages of (0, 3%, 6%, 9% v/v) EuME–AgNPs were added and stirred (300 rpm) using a magnetic stirrer at room temperature for 3 hours, to get homogeneous samples for fabrication. To compare the efficiency of EuME–AgNPs using a conventional AgNP matrix, SBD–AgNPs were included for the study. Third, various percentage ratios of (0, 3%, 6%, 9% v/v) SBD–AgNPs were incorporated into the PVA (base matrix) solution and stirred (300 rpm) using a magnetic stirrer at room temperature for 3 hours and standardized for the fabrication of nanofibers.2.6. Fabrication of nanofibers using electrospinning
The blended electrospinning solutions (EuME, EuME–AgNPs, SBD–AgNPs) were filled in a 2 ml syringe bearing a metal capillary needle of diameter 9.12 (mm). The nanofibers were produced using the electrospinning technique (ESPIN-NANO model V1-VH) which is set up using the following conditions. The filled syringe was mounted on a syringe pump which permits adjustment and controls the flow rate of solutions. The syringe capillary was connected to the positive lead of a high potential supply and a rotating drum collector was grounded and wrapped with aluminium foil mounted on polypropylene blocks which act as a nanofiber collector. The electrospinning parameters have been optimized under the mentioned experimental conditions adjusted between the flow rate (0.1–1 ml h−1), applied voltage supply (15–25 kV), needle tip to collector distance (15–30 cm) and speed of rotating drum collector (100–300 rpm). The temperature (25 °C) and humidity conditions were kept constant for all the sample solutions. Then, the collected nanofibers after electrospinning were placed in vacuum dried conditions overnight at room temperature to remove the residual solvents present in ESNs. In order to facilitate the structural and functional stability of ESNs in the aqueous media, the nanofibrous mats were incubated under glutaraldehyde vapor conditions for approximately 10 hours following by washing with the 90% aqueous methanol for 2 hours. The characterization of ESNs was performed followed by various in vitro and in vivo experiments.2.7. Characterization of electrospun nanofibers (ESNs)
 | (1) |
2.8. Determination of minimum inhibitory concentration (MIC) – (EuME, EuME–AgNP, SBD–AgNPs)
The antibacterial efficacy of (EuME, EuME–AgNPs, SBD–AgNPs) colloidal AgNP solutions were determined against Staphylococcus aureus (Gram positive bacteria) obtained through American Type (ATCC-25923) Culture Collection. The clinical breakpoint table (EUCAST) (MIC breakpoints monitored for Gentamicin at (R < 18 mm) – Gram positive strain Staphylococcus aureus) was used as key reference for optimization. The chemical aliquots were proportionally diluted from the prepared main stock solutions. The working concentrations of colloidal AgNP solutions were interacted with bacterial suspension containing 106 to 107 (CFU) colony forming units. Triplicate values were taken for analyzing statistical observations.MIC is regarded to be the lowest minimum inhibitory concentration of the colloidal solution (may be a nanoparticle, nanoemulsion, nanocolloid or any antimicrobial drug/agent) that prevents visible growth of microorganism after 12–24 hours incubation at a certain temperature (37 °C). The major use of the MIC is to mainly confirm the resistance and as an effective research tool for studying the breakpoints and activity of antimicrobial agents.31 The grown bacterial cells after overnight incubation in NB were diluted in major ratios of 1:100 MHB (bacterial culture suspension – OD maintained at 0.1) fixed at an optimized cell density of exactly ∼1 × 108 CFU mL−1.Different dilutions of EuME, EuME–AgNPs and SBD suspensions were interacted with MHB media and gentamicin which acts as negative and positive controls respectively. The colloidal solutions were incubated for a time period of 24 hours at 37 °C. Later, resazurin dye of 10 μl was added into all wells and incubated for 2 hours to observe the colour change. Based on the observations, the value of MIC was calculated.32–34
2.9. In vitro silver ion release behaviour
Although wide varieties of drugs/agents have been incorporated into the electrospun fibers so far,35 the sustained/controlled release of hydrophilic synthetic polymers like PVA now remains a major challenge due to their higher solubility in release media such as in simulated wound fluid (SWF). The compatibility of synthetic polymers associated with particles/emulsions creates a strong binding capacity and results in effective encapsulation of nanofibers.To investigate and compare the ion release behaviour of silver on SWF, the process of dialysis was conducted using beaker method at room temperature (37 °C) with known concentrations of AgNP encapsulated nanofibers (0.35 g L−1 Ag). The nanofibers were cut into 1 × 1 cm pieces dissolved in freshly formulated solutions of SWF adjusted with a optimial neutral pH value of 7.4. The chemical composition of SWF was formulated using 3.3604 g of sodium bicarbonate, 5.844 g of sodium chloride, 0.2775 g of calcium chloride, 0.2982 g of potassium chloride and 33.00 g of bovine serum albumin (BSA) was dispersed in 1000 ml of deionized water.33 The process of dialysis was carried under slow magnetic stirring at ambient temperature and readings were recorded at appropriate time intervals.36 The dispersed fluid was replaced by freshly prepared wound fluids at defined time intervals (0, 1, 2, 3, 4, 5, 6, 12, 18 and 24 hours) and the percentage of silver release was measured using a UV-spectrophotometer at 400 nm. The release experiments were conducted in triplicate and evaluated for statistical significance.
2.10. Biosafety studies
 | (2) |
2.11. Statistical analysis
All experimental results were expressed in mean ± SD (standard deviation) of three independent variables. The statistical analysis was evaluated using GraphPad Prism software using a one-way ANOVA test (Version 5; GraphPad Software, La Jolla, CA, USA). The probability values <0.05 were considered statistically significant for all experimental observations.3. Results and discussion
3.1. Formation of microemulsion system
Microemulsions are transparent, thermodynamically stable dispersions of hydrophilic and lipophilic phases. These microemulsions as potential drug delivery systems are applicable for parenteral, topical and oral administrations and support the delivery of biomolecules/bioactive agents as drugs in patients via various routes, and this has gained more attention in biomedical research.42 The present study aimed at optimizing the eugenol microemulsion (EuME) using eugenol (essential oil), Tween 80 (surfactant) and ultrapure deionized water. The formulated microemulsions exhibit definite droplet size, longer shelf-life, thermodynamic stability, lower energy of preparation, higher surface area, optical translucency with greater solubilization efficiency and drug mobility.43Preliminary optimization of spontaneous emulsion system was prepared using eugenol (essential oil) with four different types of surfactants such as Tween 20/Tween 80/Brij 93/Span 80. The surfactants were interacted in order to select the best optimized surfactant for microemulsion formulation. The hydrophilic non-ionic surfactant Tween 80 when mixed with eugenol oil forms a micelle layer supporting the formation of microemulsion system (oil-in-water emulsion system) as shown in Fig. S1 (ESI†). Tween 80 is especially designed to be a non-toxic, biocompatible, and environmentally friendly non-ionic surfactant which is commercially inexpensive.44 Fig. S1(b) (ESI†) shows the visual appearance of a spontaneous emulsion system using Tween 80 and demonstrates a grey transparent colour at 1:5 ratio [100 μl (oil):500 μl (Tween 80)]. The results show that a clear and transparent emulsion forms with Tween 80 formulation at 1:5 when compared with Tween 20 formulation [Fig. S1(a), ESI†] where the emulsion forms later at 1:6 ratio [100 μl (oil):600 μl (Tween 20)]. The distribution of droplet size while using Tween 80 at 1:5 was found to be 28.4 nm with a polydispersity index of 0.457. Interaction of eugenol with low HLB (Hydrophilic Lipophilic Balance) value surfactants such as Brij 93 and span 80 gives a milky appearance as shown in Fig. S1(c) and (d) (ESI†). The results show that Brij 93 and span 80 do not interact with eugenol oil and there is no formation of a micelle layer to produce emulsions. The experimental results clearly state that Tween 80 exhibits the best optimium surfactant characteristics with the minimum surfactant concentration ratio at 1:5.
The pseudo-ternary diagram yields a greater insight into phase behaviour with varying compositions of predefined mixtures. The phase diagrams were obtained using eugenol (essential oil) as the oil phase, Tween 80 as non-ionic surfactant of HLB value (15.0) and ultra pure water as the aqueous phase system as shown in Fig. 1.
 | ||
| Fig. 1 Pseudo-ternary (phase) diagrams of eugenol oil with Tween 80(surfactant). | ||
The pseudo-ternary phase systems showed a wider range of clear and transparent emulsions. Fig. 1 demonstrates that Tween 80 was found to be more suitable for the formation of a microemulsion formulation with eugenol oil. Thus, the oil in water type of emulsification using eugenol oil and Tween 80 forms the bigger area of clear and transparent emulsions when compared with other non-ionic surfactants.
Microemulsions were optimized using eugenol oil, Tween 80 and ultrapure deionized water. The eugenol microemulsion was formulated by varying ratios of oil:surfactant ranging from 1:1 to 1:9. Clear and transparent emulsions were observed from 1:5 to 1:9. The homogeneous droplet size distribution of EuME was observed in the ratios of 1:5, 1:6, 1:7, 1:8 and 1:9 which were found to be 28.4 nm, 20.8 nm, 19.2 nm, 17.3 nm and 15.0 nm with pDI of 0.457, 0.228, 0.207, 0.191 and 0.187 respectively. The values of the zeta potentials of 1:5 to 1:9 were measured to be −1.4 mV, −1.2 mV, −1.1 mV, −1.1 mV and −0.9 mV respectively. The size distribution images of EuME using Tween 80 are shown in Fig. S2 (ESI†). The results demonstrate that “As surfactant concentration increases, gradual decrease in droplet size is observed”. The microemulsions possessing low surfactant concentrations with a broader spectrum of compositions could be more suitable for enzymatic reactions and drug delivery systems.45 Based on these reports, while comparing the Tween 80 emulsion formulation and Tween 20 (data not shown) formulation, eugenol with Tween 80 determined to be more suitable for further characterization. The present study formulated the droplet size of microemulsion in the medium range of 25–30 nm with pDI less than 0.500 which could be an ideal fit for AgNP formulation using a microemulsion system.
The eugenol microemulsions were subjected to centrifugation, heating–cooling cycles and freeze–thaw cycles to monitor the thermodynamic stability. The experimental results show that creaming and phase separation was observed at a 1:5 ratio of eugenol oil with Tween 20 formulation, whereas eugenol oil with Tween 80 formulation observed at 1:5 was resistant to temperature variations (heating–cooling cycles and freeze–thaw cycles). Kinetic stability of eugenol microemulsions was observed by storing the samples for a prolonged period of time for nearly 8 months as shown in Fig. 2.
 | ||
| Fig. 2 Kinetic stability of formulated EuME using Tween 80 and Tween 20 as surfactants. | ||
Fig. 2 shows the kinetic stability of eugenol oil with Tween 80 formulation that exhibits excellent stability for an approximately 8 month period which was predominantly evident by calculating the average droplet size and pDI during different time periods. However, eugenol oil with Tween 20 formulation was observed to have a layer of phase separation in the ratio of 1:5 itself after 24 hours. The results confirm that eugenol oil with Tween 80 formulation is found to be a suitable combination for further synthesis and characterization of silver nanomaterials.
3.2. Preparation and characterization of silver nanoparticles (AgNPs)
The formation of AgNPs occurs due to reduction (decreased state) of Ag+ (silver ions) into Ag0 (metallic silver) resulting in the formation of a yellowish brown colour which confirms the presence of AgNPs. The AgNPs were synthesized using varying concentrations of formulated EuME (1%, 3%, 5%, 7% & 9%) which acts as a reducing and stabilizing agent and 7 mL of 1 mM AgNO3 solution was added dropwise to the reducing agent. The solution was incubated for 40 hours in the magnetic stirrer at 400 rpm resulting in the formation of stable AgNPs. The SPR peak of EuME–AgNPs was observed to have a maximum absorbance at a wavelength of 412 nm which confirmed the presence of AgNPs as shown in Table 1. The conversion efficiency of the synthesized AgNPs reveals that the experimental yield (>99%) observed was nearly equivalent to the theoretical yield (96%) respectively. The eugenol compounds present in AgNPs possess a wide variety of the functional entities with greater affinity for metals which aids in stabilization of nanoparticles.46 The phenolic ring present in the eugenol structure acts as reducing agent in the nanoparticle system. The distribution of particle size of EuME–AgNPs was determined by DLS which measures the hydrodynamic radii of nanoparticles. The mean hydrodynamic particle size of EuME–AgNPs was observed to be 38.6 nm with a polydispersity index (pDI) of 0.341 and zeta potential of −1.4 mV as shown in Table 1. In the above case, a pDI less than 0.4 indicates a perfect distribution of nanoparticles in the chemical system. The particle size of EuME–AgNPs in TEM was observed as 25.1 ± 0.2 which was slightly less than the hydrodynamic diameter. The optimium pH of EuME–AgNPs is 7.4 with the mass concentration of 25.35 mg L−1 measured by AAS analysis. A few reports show that the particle size of eugenol based AgNPs using 1 mM volumetric composition of AgNO3 results in 20–30 nm sized and homogeneous nanoparticles. The eugenol based AgNPs possess excellent antibacterial activity against Staphylococcus aureus, Escherichia coli and Candida albicans.24 The yield efficiency and thermodynamic stability of EuME–AgNPs in the present study is higher when compared with the other studies in the literature.46| AgNP suspensions | SPR peak | Hydrodynamic diameter (nm) | pDI | TEM | pH | Mass concentration (mg L−1) | MIC value (In terms of %) | Zeta potential (mV) |
|---|---|---|---|---|---|---|---|---|
| EuME–AgNPs | 412 | 38.6 | 0.341 | 25.1 ± 0.2 | 7.4 | 25.35 | 0.2 | −1.4 |
| SBD–AgNPs | 403 | 32.2 | 0.306 | 20.1 ± 0.5 | 7.0 | 23.15 | 0.4 | −4.3 |
The SBD–AgNPs prepared using conventional non-adhesive bilayer pads were characterized to evaluate the maximum absorbance level and were measured to have a wavelength at 403 nm. The particle size of SBD–AgNPs was determined by DLS which measures the hydrodynamic radii of nanoparticles. The mean hydrodynamic particle size of SBD–AgNPs was observed to be 32.2 nm with the polydispersity index (pDI) of 0.306 and zeta potential of −4.3 mV as shown in Table 1. The particle size of SBD–AgNPs in TEM was observed to be 20.1 ± 0.5 which was slightly smaller than the measurement of the hydrodynamic diameter. The optimium pH of SBD–AgNPs is 7.0 with a mass concentration of 23.15 mg L−1 measured by AAS analysis. Fig. 3 shows micrographs of EuME–AgNPs and SBD–AgNPs measured by TEM. The uniform dispersion of EuME–AgNPs shown in Fig. 3(a) provides evidence of binding between the oil's phytochemicals and is mainly responsible for the steric hindrance in nanoparticles. The TEM micrograph of SBD–AgNPs shows different shapes (namely oval, circular, triangular, hexagon, and pentagon) of particles which may be due to the combination of other polymeric mixtures namely polyamides and metals which may interrupt the homogeneous distribution of nanoparticles as shown in Fig. 3(b).
 | ||
| Fig. 3 TEM images of AgNPs (a) EuME–AgNPs (b) SBD–AgNPs and kinetic stability of EuME–AgNPs and SBD–AgNPs with respect to incubation time (days). | ||
The thermodynamic studies of EuME–AgNPs and SBD–AgNPs were evaluated under different condition parameters such as heating–cooling cycle, freeze–thaw conditions and kinetic stability of nanoparticles. The observed results showed that EuME–AgNPs at various concentrations were found to be stable under temperature variations and were capable of surviving heating–cooling and freeze–thaw conditions. However, SBD–AgNPs at varying concentrations could not sustain and withstand the temperature variations and incongruity. The kinetic stability parameter of EuME–AgNPs and SBD–AgNPs was investigated with respect to a prolonged period of time as shown in Fig. 3. EuME–AgNPs were shown to have effective kinetic stability for more than 8 months by evaluating their particle size with respect to intervals of time as shown in Fig. 3(c). The particle size of EuME–AgNPs was also found to be stable with respect to time duration in months whereas the kinetic stability of SBD–AgNPs was found not to be consistently stable and the particle size of SBD–AgNPs varied (increase and decrease) with respect to time in months as shown in Fig. 3(d).
3.3. Effect of spinning parameters on morphological structure
The biocompatible nanofibers possess effective humidity, huge porosity, rate of oxygen exchange and significant antibacterial activity. Nanofibers act as a promising matrix with numerous advantages such as higher surface area, and small diameter with narrow fibrous distribution. In brief, the nanofibers collectively range between micrometers and nanometers. Nanofibers utilize excipients to deliver therapeutic agents with higher efficiency and lower adverse effects to the specific site of infection.47 The blended electrospinning solutions of EuME (3%, 6% & 9%), EuME–AgNPs (3%, 6% & 9%) and SBD–AgNPs (3%, 6% & 9%) were fabricated using various parameters such as effect of concentration, effect of applied voltage, effect of needle tip to collector distance and effect of flowrate. The eugenol oil was formulated into a eugenol microemulsion (EuME) in order to reduce the surface tension and viscosity of the essential oil. After that, the EuME was fabricated into biocompatible nanofibers. So, the nanofibers containing eugenol essential oil do not evaporate during electrospinning because the eugenol interacts with the surfactant to form a micelle and gets encapsulated to form microemulsions. Emulsion electrospinning is an effective technique to incorporate bioactive agents/ingredients which can be used for effective wound healing applications.23,24 Among the different concentrations of EuME–AgNPs, only 9% of EuME–AgNPs exhibited homogeneous and smooth distribution of nanofibers which suggested effective encapsulation of the eugenol compound with a silver complex on a PVA based matrix and morphological examination using SEM is shown in Fig. 4. | ||
| Fig. 4 SEM images of EuME–AgNP–NFs at different concentrations (a) 3% of EuME–AgNPs–NFs (c) 6% of EuME–AgNPs–NFs (e) 9% of EuME–AgNPs–NFs and elemental analysis of EuME–AgNPs–NFs (b) presence of elemental silver in 3% EuME–AgNPs–NFs (d) presence of elemental silver in 6% EuME–AgNPs–NFs (f) presence of elemental silver in 9% EuME–AgNPs–NFs. | ||
Nanofibers using various synthetic and biopolymers incorporated with wound healing components serve as an excellent scaffold in tissue engineering systems. The electrospinning parameters such as concentration, flow rate, applied voltage and needle tip to collector distances have been optimized for formulating EuME–AgNPs–NFs and SBD–AgNPs–NFs. Among the varying concentrations (3%, 6% & 9%) of EuME–AgNPs–NFs, the concentration ratio of 9% EuME–AgNPs–NFs shows high encapsulation efficiency with homogeneous and bead-free distribution of nanofibers. The optimized applied voltage, needle tip to collector distance and flow rate of 9% EuME–AgNPs–NFs was fixed at 25 kV, 15 cm and 0.5 ml h−1 respectively to produce a broader and smooth distribution of the nanofiber matrix. The morphological observations of EuME–AgNPs–NFs were studied as shown in Fig. 5(a–c) at different magnifications which shows that eugenol components possess a homogeneous and broader distribution of bead-free nanofibers conjugated with the AgNP complex which may be used as effective nanofibrous membranes for antibacterial and cut wound infections.
Similarly, SBD–AgNPs were fabricated into nanofibers termed SBD–AgNPs–NFs. Varying concentrations (3%, 6% & 9%) of SBD–AgNPs–NFs were prepared and fabricated using various parameters such as applied voltage, needle tip to collector distance and flow rate fixed at 25 kV, 15 cm and 0.5 ml h−1 respectively as shown in Fig. 5(d–f) at different magnifications. Silver bandaids were functionalized with polyamide and fabricated into nanofibers. Silver ions (Ag+) were photoreduced to silver nanoparticles (AgNPs). Upon electrospinning of SBD–AgNPs with PVA, polyamides or amidoxime groups get distributed on the fiber surface which shows a heterogeneous distribution of nanofibers on the surface morphology.
 | ||
| Fig. 5 SEM observations of electrospun AgNP nanofibers (a–c) EuME–AgNPs–NFs at different magnifications (10.00k×, 40.00k×, 50.00k×), (d–f) SBD–AgNP–NFs at different magnifications (10.00k×, 40.00k×, 50.00k×). | ||
3.4. Characterization of synthesized AgNP nanofibers
The IR spectrum provides information about molecular changes of organic molecules present on the surface of nanoparticles. An FTIR measurement identifies qualitative results and the relative amount of quantitative observations. FTIR is used to analyze the chemical compositions of a wide variety of polymers, biological samples, minerals, and adhesives, organic and inorganic chemicals. It identifies possible biomolecules required for efficient encapsulation and stability of metallic nanoparticles. The FTIR spectrum provides information about the potential biomolecules present in the sample which is mainly responsible for capping efficiency of the metallic AgNPs. Fig. 6(a–c) shows the FTIR spectrum of synthesized ESNs. The strong band at 3473 cm−1 was functionally associated with O–H stretching of the PVA matrix. The peak between 1270 cm−1 and 1462 cm−1 which is exactly at 1390 cm−1 is clearly indicative of C–O–H stretching that confirms the presence of the (10%) PVA matrix as expressed in Fig. 6(a). The results of 10% PVA matrix showed similar observations in the functional groups as explained by Abd El-aziz.49 Clearly, the hydroxyl bond between 3000 and 3700 cm−1 reveals occurrence of O–H stretching in EuME–AgNPs–NFs, a strong bond vibration at 1307 cm−1 possess C–H deformation of eugenol-methyl, bond at 1641 cm−1 attributes to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C aromatic bond stretching, 1732 cm−1 shows CO stretching of clear formation of carboxylic acid, 1249 cm−1 shows C–O stretching of stronger formation of phenolic hydroxyl as shown in Fig. 6(b). The major components of eugenol based nanomaterials are a benzene ring, phenolic hydroxyl group, double bond formation and also other groups. The functional groups present in EuME–AgNPs–NFs were found to be similar as explained by Mouro and Gao.11,50 Polyamides were the main components of the silver bandaid based nanofibers. A stronger vibration at 1714 cm−1 shows CO stretching of carboxylic acid which quantifies the presence of polyamide structures. Similarly, the vibration at 1217 cm−1 shows CH2 stretching of the α form of amides, at 785 cm−1 occurring with the CO–NH stretching vibration in amide planes as shown in Fig. 6(c). The results of SBD–AgNPs–NFs (i.e., presence of polyamides) were similar to the results observed by Rotter and Zimudzi.51,52
C aromatic bond stretching, 1732 cm−1 shows CO stretching of clear formation of carboxylic acid, 1249 cm−1 shows C–O stretching of stronger formation of phenolic hydroxyl as shown in Fig. 6(b). The major components of eugenol based nanomaterials are a benzene ring, phenolic hydroxyl group, double bond formation and also other groups. The functional groups present in EuME–AgNPs–NFs were found to be similar as explained by Mouro and Gao.11,50 Polyamides were the main components of the silver bandaid based nanofibers. A stronger vibration at 1714 cm−1 shows CO stretching of carboxylic acid which quantifies the presence of polyamide structures. Similarly, the vibration at 1217 cm−1 shows CH2 stretching of the α form of amides, at 785 cm−1 occurring with the CO–NH stretching vibration in amide planes as shown in Fig. 6(c). The results of SBD–AgNPs–NFs (i.e., presence of polyamides) were similar to the results observed by Rotter and Zimudzi.51,52
 | ||
| Fig. 6 FTIR spectrum analysis of ESNs (a) only 10% PVA matrix (b) EuME–AgNPs–NFs (c) SBD–AgNPs–NFs; XRD observation patterns of ESNs (d) only 10% PVA matrix (e) EuME–AgNPs–NFs (f) SBD–AgNPs–NFs; TGA/DTA traces of synthesized ESNs (g) only 10% PVA matrix (h) EuME–AgNPs–NFs (i) SBD–AgNPs–NFs. | ||
X-ray diffraction (XRD) patterns have been a useful tool for analyzing the structure as well as crystallization of the polymers, biomaterials and nanocomposites in the biomedical engineering. It also measures the average spacings between layers or rows of atoms, and determines the orientation of a single crystal or grain. The size, shape and internal stress of smaller crystalline regions can also be elucidated. Fig. 6(d–f) determines XRD patterns of the pure 10% PVA matrix, EuME–AgNPs–NFs and SBD–AgNPs–NFs respectively. The experimentally observed maximum diffraction peak was found at 2θ = 20° which corresponds to a d spacing of 4.4801 Å indicating the appearance of a distinctive semicrystalline structure in the 10% pure PVA matrix as shown in Fig. 6(d). The XRD diffraction patterns of PVA/EuME–AgNPs–NFs indicate that eugenol molecules were uniformly distributed along with the PVA matrix showing salient broad diffraction peaks centered at 2θ ∼ 19° and 20°, 2θ ∼ 39°, 44°, 65°, 78° and 83° in the eugenol nanofibers along with slightly intensified diffraction peaks at 2θ ∼ 22° and 23° were observed determining that channel type crystals of EuME–AgNPs–NFs were elucidated along with the PVA matrix system as shown in Fig. 6(e). The XRD patterns of EuME–AgNPs–NFs reveal that channel type diffraction crystals were encapsulated in the PVA base matrix. The synthesized eugenol based nanofibers were not crystalline in nature confirming the amorphous structure. These results were found to be similar to a thermally stable eugenol and cyclodextrin inclusion complex.26 The XRD patterns of PVA/SBD–AgNPs–NFs indicate that silver bandaid nanofibers were homogeneously distributed along with the 10% PVA matrix fabricated nanofibers. SBD shows salient broad and intensified diffraction peaks centered at 2θ ∼ 65° and 80° that was noticed in the diffraction crystals which was encapsulated along with the PVA matrix. Slight diffraction peaks of SBD–AgNPs–NFs were observed at 2θ ∼ 42° and 44° by elucidating the diffraction variation peaks as shown in Fig. 6(f). In brief, XRD patterns reveal that EuME–AgNPs–NFs and SBD–AgNPs–NFs (channel type crystals) were equally dispersed along with the PVA base matrix.
Thermogravimetric analysis (TGA) is helpful for analyzing the purity and chemical composition of nanomaterials, ignition and drying temperatures of nanomaterials and the temperature of chemical compounds. Differential thermal analysis (DTA) analyzes the temperature of transitions, melting points and reactions of substances. The thermal stabilities of the pure 10% PVA matrix, EuME–AgNPs–NFs and SBD–AgNPs–NFs were investigated using a thermogravimetric analyzer as shown in Fig. 6(g–i). The TGA analysis of EuME–AgNPs–NFs and SBD–AgNPs–NFs was done for comparison. The TGA curve of the pure 10% PVA matrix shows that the amount of weight loss was 69.19% which was nearly 1.081 mg lying in the temperature range between 100 °C and 350 °C. There was a medium amount of crystallization and decomposition of the sample and the DTA profile of the 10% PVA matrix was found to be nearly 289.65 °C which is below 300 °C as shown in Fig. 6(g). The TGA curve of EuME–AgNPs–NFs shows that the amount of weight loss was 80.87% which was nearly 1.389 mg lying in temperature range between 100 °C and 420 °C. The results show that there was greater crystallization and decomposition of the sample and the DTA profile of EuME–AgNPs–NFs was found to be nearly 278.39 °C which is below 300 °C as shown in Fig. 6(h). The combination of eugenol with the other inclusion complex possesses high encapsulation efficacy resulting in higher stability and greater hydrophobic interaction.26 The TGA graph of SBD–AgNPs–NFs shows that the amount of weight loss was 67.23% which was nearly 1.499 mg lying in the temperature range between 100 °C and 320 °C. The results reveal that there was incomplete crystallization and decomposition of the sample and the DTA profile of SBD–AgNPs–NFs was found to be nearly at 79.42 °C and also the decomposition at 282.86 °C as shown in Fig. 6(i). In brief, the TGA results determine that the PVA matrix has been homogeneously encapsulated with EuME–AgNPs–NFs and SBD–AgNPs–NFs. PVA/EuME–AgNPs–NFs can withstand temperature till 420 °C and possess sharp bended distribution of curve leading to consistent saturation when compared with PVA/SBD–AgNPs–NFs, where the temperature was only at 300 °C leading to blend-ended distribution of curve. These results prove that EuME–AgNPs–NFs showed sustained thermal decomposition and could withstand the temperature for a longer time with a sharp curve distribution when compared with SBD–AgNPs–NFs (Fig. 6).
 | ||
| Fig. 7 Porosity of electrospun nanofibers (only 10% PVA matrix, EuME–AgNPs–NFs, SBD–AgNPs–NFs). | ||
The porosity of the treated ESNs (only 10% PVA matrix, EuME–AgNPs–NFs and SBD–AgNPs–NFs) is shown in Fig. 7. The percentage of porosity of the 10% PVA matrix, EuME–AgNPs–NFs and SBD–AgNPs–NFs was found to be 90.9%, 86.9% and 74% respectively. The porosities of treated ESNs ranged between 60 and 90% and further in vitro studies were performed. High porosity is essential for the cell growth and cell infiltration. It is also required for nutrient and oxygen exchange for the living cells. Considering Fig. 7, it can be concluded that eugenol embedded ESNs are relatively porous and highly efficient for encapsulation and nutrient exchange compared to the SBD impregnated ESNs.
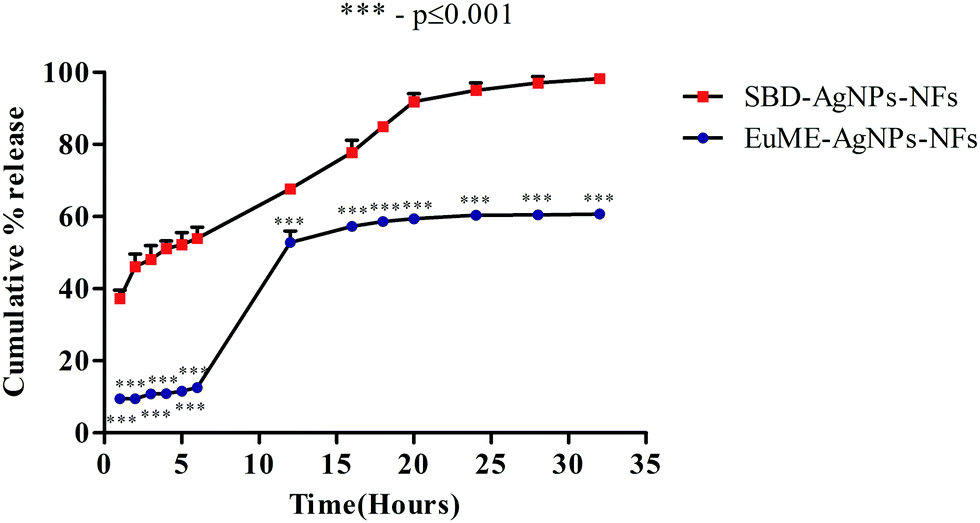 | ||
| Fig. 8 Silver ion release behaviour of EuME–AgNPs–NFs and SBD–AgNP–NFs. | ||
3.5. Evaluation of minimum inhibitory concentration (MIC)
Natural components like eugenol possess attractive properties that could rapidly reduce the usage of commercial/conventional antibiotics. Studies have reported that eugenol exhibits potentially beneficial properties such as antioxidant, antimicrobial, anti-inflammatory and anticarminative activities.24,53 The mechanism of the antimicrobial action of eugenol is determined by the influence of free hydroxyl groups present in the respective molecule. The antimicrobial effect of EuME, EuME–AgNPs and SBD–AgNPs was completely concentration and dose dependent and they were found to be competent against the microbial strains of Gram-positive Staphylococcus aureus.54 The MIC of EuME–AgNPs and SBD–AgNPs was determined against Staphylococcus aureus using a broth microdilution process. The EuME, EuME–AgNPs and SBD–AgNPs exhibited MIC values of 0.25%, 0.2% and 0.4% respectively. The experimental results show that the minimum concentration needed for the growth inhibition of Staphylococcus aureus of EuME–AgNPs was 0.2% whereas SBD–AgNPs require 0.4% of the sample for growth inhibition. Thus, the efficiency of EuME–AgNPs was higher and more effective when compared with SBD–AgNPs.A few reports on in vitro studies show that eugenol exhibits effective antibacterial activity against microbial pathogens such as Staphylococcus aureus, Escherichia coli, Bacillus cereus and Yersinia enterocolitica. The MIC values of as Staphylococcus aureus, Escherichia coli, Bacillus cereus and Yersinia enterocolitica range between 0.10% and 0.25% respectively.11,24 A few literature studies reported that eugenol helps in eradicating established biofilms of methicillin sensitive Staphylococcus aureus (MSSA) and methicillin resistant Staphylococcus aureus (MRSA) clinical strains55 by damaging the cell membrane thereby causing leakage of cell contents. It also decreases the expression of enterotoxin genes and related biofilms. The three essential oils, teatree, clove and cinnamon bark, have shown excellent antibacterial effect against Staphylococcus aureus, Escherichia coli and Pseudomonas aeruginosa and cinnamon essential oil containing the eugenol compound was not cytotoxic to the biological system and possessed anti-UV properties.13,14 Other essential oils such as eucalyptus oil containing zein show antibacterial activity against Gram-positive and Gram-negative strains. The growth reduction of Staphylococcus aureus was 24.3% and of L.monocytogenes was 28.5%.15 The present results show that the growth inhibition of Staphylococcus aureus was even faster when interacting with EuME–AgNPs. The synthesized eugenol based AgNPs have to be tested against the multidrug resistant and susceptible strain which has to be included in our future research outcomes. The eugenol based silver nanocomposites could be a promising strategy in various anti-bacterial and anti-inflammatory settings of wound healing applications.
3.6. Silver ion release behaviour of EuME–AgNPs–NFs and SBD–AgNPs–NFs
The release behaviour of different chemical components of silver ions (Ag+) released from wound dressings has multi-dimensional effects on the human body. Wound dressings containing silver are present in different forms such as (a) inorganic compounds like silver phosphate, silver sulfate, silver chloride and as silver oxides (b) metallic silver (Ag0), a nanocrystalline particle (c) organic complexes like silver alginate, silver based zinc allantoinate and silver carboxymethyl cellulose.56 The sudden release of Ag+ ions from AgNPs induces the formation of chronic toxicity and reactive oxygen species (ROS) which greatly affects the cytotoxicity level of AgNPs/AgNPs-ESNs.57 Though there are numerous reports on the release behaviour of natural and synthetic nanofibers using AgNPs, the effect of the shape, size of AgNPs and fibrin morphology of ESNs on the release behaviour for understanding the release studies of AgNP nanofibers on the biological system still remains a challenging task. To overcome these limitations, the present study focuses on the comparison of EuME–AgNPs–NFs and SBD–AgNPs–NFs which will be evaluated based on the release behaviour of Ag+ ions from AgNPs with respect to time period and the effect of structural morphology of nanofibers on the dissolution system will be analyzed.Dissolution studies of formulated nanofibers were analyzed by monitoring the percentage of cumulative release with respect to the time period. Release of ionic silver into a normal saline/phosphate buffer saline (PBS) may be easier for understanding the interaction of normal saline based cleansers with wound dressings. There are few reports monitoring the release behaviour of nanofibers in more complex media such as in inorganic and organic mixtures which pose a major problem in real wound exudates. To examine the interactions between SWF and synthesized nanofibers, the time of Ag+ ion release was analyzed and SPR peak ranges between 400 and 430 nm. As shown in Fig. 8, cumulative release of EuME–AgNPs–NFs was calculated with respect to time intervals during 1, 2, 3, 4, 5, 6, 12, 16, 18, 20, 24, 28 and 32 hours showing a sustained percentage release of Ag+ ions when compared with SBD–AgNPs–NFs. The observed cumulative release of EuME–AgNPs–NFs and SBD–AgNPs–NFs was monitored during the respective time intervals as shown in Fig. 8. The percentage of cumulative release of EuME–AgNPs–NFs and SBD–AgNPs–NFs during the 32nd hour was 60% and 98% respectively. The obtained results imply that EuME–AgNPs–NFs show sustained release of silver ions in the simulated medium when compared with the SBD–AgNPs–NFs which show a sudden and rapid release of silver ions in the simulated medium. The sustained release of silver ions from EuME–AgNPs–NFs in SWF solution shows a potentially stable mode of release in the prepared biological system whereas the ion release behaviour of silver ions released from the SBD–AgNPs–NFs might be due to the cytotoxic presence of silver ions in the SBD suspension. The silver ion release was predominantly controlled by a diffusion mechanism referred to as Fickian diffusion.
The results of the cumulative release of EuME–AgNPs–NFs clearly depict that sustained release behaviour of ionic silver might be related to the activity of compound eugenol being embedded and incorporated along with the silver composite system to enhance the antimicrobial potential. The sustained release of silver ions has a greater impact on the cytotoxicity behaviour resulting in higher antimicrobial efficacy and wound healing potential in cut wounds.
Statistical analysis reveals that EuME–AgNPs–NFs show greater (***) significant difference (p value ≤ 0.001) when compared with SBD–AgNPs–NFs as shown in Fig. 8. The sustained release behaviour of EuME–AgNPs–NFs is significantly different when compared with the SBD–AgNPs–NFs which exhibit a sudden release of silver ions in the simulated wound system. A few reports shows that the in vitro dissolution behaviour of eugenol based compounds relates to good solubility properties in the organic solvent. The sustained mode of release of EuME–AgNPs–NFs may be due to diffusion properties of eugenol which gets dispersed into the polymeric matrix and is known as the dominant mechanism. The process of diffusion takes place in three different stages: the first stage is the impregnation of the eugenol microemulsion into silver nanoparticles, the second stage is the change of polymeric suspension into a base matrix and the third step is the trans-diffusion of the eugenol compound from the synthesized scaffold matrix. The release mechanism of silver ions present in eugenol compounds was found to be similar to that of other literature studies.58 The experimental results prove that the EuME–AgNPs–NFs exhibit a sustained mode of silver ion release in the prepared SWF solution whereas SBD–AgNPs–NFs exhibit a sudden and rapid release of silver ions in the simulated wound system.
The impregnation of nanoparticles and polymers plays an important role in the development of controlled and sustained release of ESNs. The characteristic of the drugs released from nanofibers is solely dependent on the distribution of the major component present within the shell or core phase and the structural morphology of the nanofibers. Another important factor related to the sustained release from the nanofibers is the compatibility of the compound and the polymer. The compatibility mainly refers to physical interaction that occurs between polymer chains and the component molecules. The compatibility is directly proportional to the solubility of the drug/compound in the polymeric solvent matrix.59 The reports were found to be in correlation with the present work showing that EuME–AgNPs were uniformly distributed and impregnated in the PVA polymeric matrix leading to a sustained release of the silver ions present in the eugenol compound whereas with the SBD–AgNPs–NFs, the suspensions were unevenly and heterogeneously distributed in the polymeric matrix leading to uncontrolled and sudden release of the silver ions present in the SBD suspension.
Fig. 3(a and b) show the shape of EuME–AgNPs and SBD–AgNPs evaluated by transmission electron microscopy (TEM). The morphology of EuME–AgNPs obtained was homogeneously spherical in shape and uniformly dispersed in the polymeric matrix whereas, the morphological shape of SBD–AgNPs obtained was of a heterogeneous shape distribution such as triangular, oval, hexagonal and mostly pentagonal. The sustained release behaviour of ionic silver from EuME–AgNPs–NFs might be due to the homogeneous shape distribution of AgNPs in the nanofiber matrix. Likewise, the uncontrolled and sudden release of ionic silver from SBD–AgNPs–NFs might be due to the heterogeneous shape distribution of SBD suspended in the nanofiber matrix. The mode of release was nearly proportional to the surface area of synthesized AgNPs. The results obtained were found to be similar to other literature studies.60 The reports reveal that the nanoparticle morphology is solely dependent on the release behaviour of Ag+ ions. The nanoparticles with different shape morphologies affects the dissolution behaviour of silver ions in the biological system. The experimental results show that the release behaviour (sustained or sudden release of silver ions) is also dependent on the shape and structure of nanoparticles.
Fig. 5 showing the morphological view of nanofibers (EuME–AgNPs–NFs and SBD–AgNPs–NFs). The effect of fiber morphology on Ag+ ion release is well explained. The structure of EuME–AgNPs–NFs was uniformly distributed and of equal fiber shape dispersion in the polymeric matrix. The sustained release of silver ions from EuME–AgNPs–NFs in SWF may be due to the homogeneous shape distribution of nanofibers in the PVA matrix. Likewise, the structural morphology of SBD–AgNPs–NFs was elucidated as shown in Fig. 5. The fiber distribution of SBD–AgNPs–NFs was heterogeneous with uneven and narrow dispersion of fibers in the polymeric matrix. The fiber morphology has an effect on the Ag+ ion release in the biological system.61 In brief, nanofibers loaded with AgNPs have higher efficiency of loading capacity with tunable dissolution mechanisms. Also, sustained release of nanofibers reduces dose specific silver ion toxicity and thereby increases antibacterial activity. The effect of fiber morphology is an important consideration in terms of Ag+ ion release in the biological system. Therefore, the smooth and broader distribution of nanofibers like EuME–AgNPs–NFs results in sustained and controlled release of silver ions whereas, narrow distribution of nanofibers like SBD–AgNPs–NFs results in a sudden and rapid release of silver ions in the simulated wound fluid system.
3.7. Evaluation of cytotoxicity
 | ||
| Fig. 9 Percentage of WBC viability with EuME–AgNPs and SBD–AgNPs. | ||
The reason may be due to the effect of toxicity level which was higher in SBD–AgNPs when compared with EuME–AgNPs. These EuME–AgNPs possess the minimum cytotoxicity gradient (p ≤ 0.001) which may be related to the encapsulation of the eugenol compound with the OH group that is embedded on the surface of AgNPs by providing anti-angiogenic efficiency and greater stability. The statistical observations reveal that EuME–AgNPs possess a greater significant level of difference (***) which is denoted by a p value ≤ 0.001 compared to statistically observed values of SBD–AgNPs and 1 mM AgNO3. The reason might be related to the percentage of cell viability of EuME–AgNPs on the human blood lymphocytes. Though the fabricated SBD sheets conjugated on the polyethylene polymers possess anti-inflammatory activity, the cytotoxic level is higher when compared with the eugenol encapsulated nanofibers. The findings clearly depict that EuME–AgNPs possess a higher rate of cell viability on the human blood lymphocytic cells when compared with the SBD–AgNPs which exhibit a lower rate of cell viability and greater level of cytotoxicity.
 | ||
| Fig. 10 Rate of RBC breakdown with EuME–AgNPs and SBD–AgNPs. | ||
The reason may be due to the higher toxicity level of SBD–AgNPs which show the maximum rate of RBCs breakdown when compared with the EuME–AgNPs which show the minimum rate of RBC breakdown with a lower cytotoxic response. The statistical observations show that the rate of RBC breakdown of EuME–AgNPs shows a greater significant difference (***) which is denoted by a p value ≤ 0.001 compared to the statistically observed differences of SBD–AgNPs and 1 mM AgNO3. The results might be related to fewer cytotoxic properties of the EuME–AgNPs which show the minimum rate of RBC breakdown. Although SBD possesses anti-inflammatory properties and anti-bacterial efficiency, the cytotoxic effect is found to be higher with an increase in the rate of breakdown of red blood cells. The findings conclude that EuME–AgNPs have a minimum cytotoxic effect in the RBC breakdown and provide authentic evidence for choosing EuME–AgNPs as antimicrobial wound dressings for cut wound applications.
4. Conclusion
Electrospinning techniques show great promise for fabricating sustained release composite (nanofiber) materials with a defined release behaviour that depends on the nanoparticle loading capacity, surface chemistry and antibacterial efficacy. Using these perspectives, EuME–AgNPs were synthesized using a green synthesis (eugenol essential oil) and fabricated using an electrospinning techique. These EuME–AgNPs–NFs were found to be comparatively more efficient than SBD–AgNPs–NFs in terms of the structural morphology, defined chemical characteristics, homogeneous and broader distribution with a PVA polymeric matrix, antibacterial efficiency with sustained and controlled mode of release in SWF and most importantly minimum toxicity in the case of rate of cell viability (WBCs) and rate of cell hemolysis (RBCs). The eugenol based nanoscaffolds (EuME–AgNPs–NFs) exhibit a sustained and controlled mode of release in simulated wound systems with minimum cytotoxicity level in the biological system. The EuME–AgNPs–NFs (nanofibers using eugenol) could be the best alternative as potential scaffolds for treating cut wounds in biomedical engineering.Conflicts of interest
All authors declare no professional or personal conflicts of interest.Acknowledgements
The authors are thankful to the Council of Scientific and Industrial Research (CSIR-SRF) (File No. 09/844(0099)/2020 EMR-1) for funding this work and VIT University for providing the lab facilities to carry out the research work.References
- P. Bowler, B. Duerden and D. G. Armstrong, Clin. Microbiol. Rev., 2001, 14, 244–269 CrossRef CAS.
- J. Wilson, V. Ward, R. Coello, A. Charlett and A. Pearson, J. Hosp. Infect., 2002, 52, 114–121 CrossRef CAS PubMed.
- J. V. Vayalumkal and T. Jadavji, Paediatr Drugs, 2006, 8, 99–111 CrossRef PubMed.
- D. C. Daltrey, B. Rhodes and J. Chattwood, J. Clin. Pathol., 1981, 34, 701–705 CrossRef CAS.
- J. Hutchinson and M. McGuckin, Am. J. Infect. Control, 1990, 18, 257–268 CrossRef CAS.
- R. L. Nichols and J. W. Smith, Clin. Infect. Dis., 1994, 18, S280–S286 CrossRef.
- V. Jones, J. E. Grey and K. G. Harding, BMJ, 2006, 332, 777–780 CrossRef.
- L. Du, H. Xu, T. Li, Y. Zhang and F. Zou, Fiber Polym., 2016, 17, 1995–2005 CrossRef CAS.
- J. Y. Chun, H. K. Kang, L. Jeong, Y. O. Kang, J.-E. Oh, I.-S. Yeo, S. Y. Jung, W. H. Park and B.-M. Min, Colloids Surf., B, 2010, 78, 334–342 CrossRef CAS PubMed.
- A. Greiner and J. H. Wendorff, Angew. Chem., Int. Ed., 2007, 46, 5670–5703 CrossRef CAS PubMed.
- C. Mouro, M. Simões and I. C. Gouveia, Adv. Polym. Technol., 2019, 2019(1), 1–11 CrossRef.
- H. Hajiali, M. Summa, D. Russo, A. Armirotti, V. Brunetti, R. Bertorelli, A. Athanassiou and E. Mele, J. Mater. Chem. B, 2016, 4, 1686–1695 RSC.
- S. Jiang, B. C. Ma, J. Reinholz, Q. Li, J. Wang, K. A. Zhang, K. Landfester and D. Crespy, ACS Appl. Mater. Interfaces, 2016, 8, 29915–29922 CrossRef CAS.
- J. Y. Lee, D. Yoon, B. C. Son, A. I. Rezk, C. H. Park and C. S. Kim, J. Nanosci. Nanotechnol., 2020, 20, 5376–5380 CrossRef CAS.
- M. D. Antunes, G. da Silva Dannenberg, Â. M. Fiorentini, V. Z. Pinto, L.-T. Lim, E. da Rosa Zavareze and A. R. G. Dias, Int. J. Biol. Macromol., 2017, 104, 874–882 CrossRef CAS PubMed.
- J. Richard, J. Burns, 2002, 1, 11–19 Search PubMed.
- B. Ding, H. Y. Kim, S. C. Lee, C. L. Shao, D. R. Lee, S. J. Park, G. B. Kwag and K. J. Choi, J. Polym. Sci., Part B: Polym. Phys., 2002, 40, 1261–1268 CrossRef CAS.
- C. Zhang, X. Yuan, L. Wu, Y. Han and J. Sheng, Eur. Polym. J., 2005, 41, 423–432 CrossRef CAS.
- L. Sethuram, J. Thomas, A. Mukherjee and N. Chandrasekaran, RSC Adv., 2019, 9, 35677–35694 RSC.
- M. C. Raja, V. Srinivasan, S. Selvaraj and S. Mahapatra, Pharm. Anal. Acta, 2015, 6, 367 Search PubMed.
- X.-J. Kong, X.-W. Liu, J.-Y. Li and Y.-J. Yang, Curr. Opin. Complement Alternat. Med., 2014, 1, 8–11 Search PubMed.
- X. Wang, Y. Yuan, X. Huang and T. Yue, J. Appl. Polym. Sci., 2015, 132, 41811 Search PubMed.
- C. Wang and M. Wang, 2012.
- V. Tekin, O. Kozgus Guldu, E. Dervis, A. Yurt Kilcar, E. Uygur and F. Z. Biber Muftuler, Appl. Organomet. Chem., 2019, 33, e4969 CrossRef.
- N. Cai, C. Han, X. Luo, G. Chen, Q. Dai and F. Yu, Macromol. Mater. Eng., 2017, 302, 1600364 CrossRef.
- F. Kayaci, Y. Ertas and T. Uyar, J. Agric. Food Chem., 2013, 61, 8156–8165 CrossRef CAS PubMed.
- A. Celebioglu, Z. I. Yildiz and T. Uyar, J. Agric. Food Chem., 2018, 66, 457–466 CrossRef CAS PubMed.
- W. S. Lee, E. Kim, H.-J. Cho, T. Kang, B. Kim, M. Y. Kim, Y. S. Kim, N. W. Song, J.-S. Lee and J. Jeong, Nanomaterials, 2018, 8, 652 CrossRef PubMed.
- C. Greulich, J. Diendorf, T. Simon, G. Eggeler, M. Epple and M. Köller, Acta Biomater., 2011, 7, 347–354 CrossRef CAS PubMed.
- G. A. Sotiriou and S. E. Pratsinis, Environ. Sci. Technol., 2010, 44, 5649–5654 CrossRef CAS PubMed.
- T.-N. Khieu, M.-J. Liu, S. Nimaichand, N.-T. Quach, S. Chu-Ky, Q.-T. Phi, T.-T. Vu, T.-D. Nguyen, Z. Xiong and D. M. Prabhu, Front. Microbiol., 2015, 6, 574 Search PubMed.
- P. Manivasagan, J. Venkatesan, K. Sivakumar and S.-K. Kim, Crit. Rev. Microbiol., 2016, 42, 209–221 CrossRef CAS.
- C. Bradford, R. Freeman and S. L. Percival, J. Am. Coll. Clin. Wound Spec., 2009, 1, 117–120 Search PubMed.
- A. A. Catherine, H. Deepika and P. S. Negi, J. Essent. Oil Res., 2012, 24, 481–486 CrossRef CAS.
- M. Zamani, M. P. Prabhakaran and S. Ramakrishna, Int. J. Nanomed., 2013, 8, 2997 Search PubMed.
- K. Loza, J. Diendorf, C. Sengstock, L. Ruiz-Gonzalez, J. Gonzalez-Calbet, M. Vallet-Regi, M. Köller and M. Epple, J. Mater. Chem. B, 2014, 2, 1634–1643 RSC.
- C. Vergallo, E. Panzarini, D. Izzo, E. Carata, S. Mariano, A. Buccolieri, A. Serra, D. Manno and L. Dini, 2014.
- A. Zhornik, L. Baranova, I. Volotovski, S. Chizhik, E. Drozd, M. Sudas, Q. B. Ngo, H. C. Nguyen, T. H. Huynh and T. H. Dao, Adv. Nat. Sci., 2015, 6, 025003 Search PubMed.
- L. Q. Chen, B. Kang and J. Ling, J. Nanopart. Res., 2013, 15, 1–9 Search PubMed.
- M. A. Dobrovolskaia, J. D. Clogston, B. W. Neun, J. B. Hall, A. K. Patri and S. E. McNeil, Nano Lett., 2008, 8, 2180–2187 CrossRef CAS.
- Y.-S. Lin and C. L. Haynes, J. Am. Chem. Soc., 2010, 132, 4834–4842 CrossRef CAS PubMed.
- G. Ktistis and I. Niopas, J. Pharm. Pharmacol., 1998, 50, 413–418 CrossRef CAS PubMed.
- V. Ghosh, S. Saranya, A. Mukherjee and N. Chandrasekaran, Colloids Surf., B, 2013, 105, 152–157 CrossRef CAS PubMed.
- B. A. Kerwin, J. Pharm. Sci., 2008, 97, 2924–2935 CrossRef CAS PubMed.
- C. Prieto and L. Calvo, J. Appl. Chem., 2013, 930356 Search PubMed.
- K. Vijayaraghavan, S. K. Nalini, N. U. Prakash and D. Madhankumar, Mater. Lett., 2012, 75, 33–35 CrossRef CAS.
- Y. Liu, S. Zhou, Y. Gao and Y. Zhai, Asian J. Pharm. Sci., 2019, 14, 130–143 CrossRef PubMed.
- M. T. Ahmadi and R. Ismail, Arch. Med. Vet., 2018, 4, 14 Search PubMed.
- A. Abd El-aziz, A. El-Maghraby and N. A. Taha, Arab. J. Chem., 2017, 10, 1052–1060 CrossRef CAS.
- H. Gao, H. Yang and C. Wang, Results Phys., 2017, 7, 3130–3136 CrossRef.
- G. Rotter and H. Ishida, J. Polym. Sci., Part B: Polym. Phys., 1992, 30, 489–495 CrossRef CAS.
- T. J. Zimudzi, K. E. Feldman, J. F. Sturnfield, A. Roy, M. A. Hickner and C. M. Stafford, Macromolecules, 2018, 51, 6623–6629 CrossRef CAS PubMed.
- A. O. Gill and R. A. Holley, Appl. Environ. Microbiol., 2004, 70, 5750–5755 CrossRef CAS PubMed.
- E. Pazos-Ortiz, J. H. Roque-Ruiz, E. A. Hinojos-Márquez, J. López-Esparza, A. Donohué-Cornejo, J. C. Cuevas-González, L. F. Espinosa-Cristóbal and S. Y. Reyes-López, J. Nanomater., 2017, 4752314 Search PubMed.
- M. K. Yadav, S.-W. Chae, G. J. Im, J.-W. Chung and J.-J. Song, PLoS One, 2015, 10, e0119564 CrossRef.
- A. Lansdown and A. Williams, Adv. Wound Care, 2004, 13, 131–136 CrossRef CAS.
- S. Pillai, R. Behra, H. Nestler, M. J.-F. Suter, L. Sigg and K. Schirmer, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 3490–3495 CrossRef CAS PubMed.
- S. A. Agnihotri, N. N. Mallikarjuna and T. M. Aminabhavi, J. Controlled Release, 2004, 100, 5–28 CrossRef CAS PubMed.
- J. Zeng, L. Yang, Q. Liang, X. Zhang, H. Guan, X. Xu, X. Chen and X. Jing, J. Controlled Release, 2005, 105, 43–51 CrossRef CAS PubMed.
- J. Helmlinger, C. Sengstock, C. Groß-Heitfeld, C. Mayer, T. Schildhauer, M. Köller and M. Epple, RSC Adv., 2016, 6, 18490–18501 RSC.
- S. Calamak, E. A. Aksoy, N. Ertas, C. Erdogdu, M. Sagıroglu and K. Ulubayram, Eur. Polym. J., 2015, 67, 99–112 CrossRef CAS.
- C. Liao, Y. Li and S. C. Tjong, Int. J. Mol. Sci., 2019, 20, 449 CrossRef PubMed.
- Z.-M. Xiu, Q.-B. Zhang, H. L. Puppala, V. L. Colvin and P. J. Alvarez, Nano Lett., 2012, 12, 4271–4275 CrossRef CAS PubMed.
- B. Reidy, A. Haase, A. Luch, K. A. Dawson and I. Lynch, Materials, 2013, 6, 2295–2350 CrossRef CAS PubMed.
- Y. J. Lee, J. Kim, J. Oh, S. Bae, S. Lee, I. S. Hong and S. H. Kim, Environ. Toxicol. Chem., 2012, 31, 155–159 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00103e |
| This journal is © The Royal Society of Chemistry 2021 |