
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interlayer-modified two-dimensional layered hexaniobate K4Nb6O17 as an anode material for lithium-ion batteries†
Satheeshkumar
Elumalai‡
 *a,
Selvamani
Vadivel§
b and
Masahiro
Yoshimura
*ac
*a,
Selvamani
Vadivel§
b and
Masahiro
Yoshimura
*ac
aDepartment of Material Science and Engineering, National Cheng Kung University, Tainan 701, Taiwan, Republic of China. E-mail: analyticalsathi@gmail.com; yoshimura@msl.titech.ac.jp
bCSIR-CECRI, Karaikudi 630 006, Tamil Nadu, India
cProfessor Emeritus, Materials and Structures Laboratory, Tokyo Institute of Technology, Tokyo 152-8550, Japan
First published on 2nd March 2021
Abstract
Scalable two-dimensional (2D) layered hexaniobate K4Nb6O17 was synthesized by the solid-state approach, and subsequent interlayer-modified niobate was prepared as an anode for lithium-ion batteries (LIBs) to improve the interfacial charge storage and in turn the electrochemical performance. The modified anode exhibited an excellent lithium storage ability of 480 mA h g−1 at the initial lithiation (0.05 A g−1) and delivered a reversible capacity of 133 mA h g−1 after 750 cycles at 0.5 A g−1.
High-energy, low carbon footprint-based energy storage systems with uninterrupted supply have attracted significant attention in recent years, particularly for applications in advanced secondary lithium-ion batteries (LIBs) and fuel-cell technologies.1–4 Compared to later technology, LIBs are cheaper and safer for operations.5 In LIBs, similar to cathodes, anode materials also play a significant role in enhancing the performance, particularly graphite and silicon-graphite composites, owing to their low lithiation potential as well as volume expansion.6 Besides, layered alkali metal oxide frameworks have shown very fast lithium diffusion, and are promising as anodes for high-rate and high-voltage applications, and therefore directed the attention towards new anode materials.7 Recently, transition metal chalcogenides have shown significant importance owing to high reversible capacities and moderate lithiation potentials with safer lithium shuttling, even at high current densities.7,8 Compared to alloy-type anodes, metal oxides, particularly Ti- and Nb-based mono or mixed bimetallic systems, display several advantages, including reasonable reversible lithiation capacity as well as long-term cycling stability with significantly low volume expansion.4,9
Two-dimensional (2D) materials, particularly multi-layered metal oxides have attracted considerable attention in numerous fields, such as electrochemical sensors, catalysis, capacitors and LIBs. Titanium niobium oxide (TNO) anode materials4 are considered to be alternative high capacity anode materials for the replacement of >1 V operating Li4Ti5O12 (LTO) in LIBs, where the disorderly shared edge and corners of the Ti4+, Nb5+ octahedral site facilitates efficient lithium diffusion and electron transfer via the dual redox.10 Besides, niobium (Nb) is increasingly applied in the composition of LIB materials to meet the current demand for higher performance, longer life, and safer batteries.5 Moreover, it helps to increase the energy density and rate capability upon cycling with improved cycling stability, even at low temperatures.8 Similarly, 2D layered hexaniobate K4Nb6O17 (LH) is another interesting alkali metal niobate material with an asymmetric stacking of Nb6O174− and K+ ions. Two chemically different interfaces of K+ ions are presented in K4Nb6O17 (Fig. S1, ESI†) between the NbO6 octahedral site and are designated as interlayer-I (hydrophilic) and interlayer-II (hydrophobic), respectively.11 The modified interlayer-I can allow improved interfacial charge transfer and/or solid-state diffusion in electrochemical performance.11 It is widely believed that Nb-based layered materials with the ability of lithium accommodation can outperform, and the low intercalation potential further offers a wide operating window.1,9,12
In this study, a simple amino acid, namely glycine (Gly), was chosen as a model of small molecules to emphasize the interlayer modification of LH. Interlayer-I shows crystallographically different intercalation properties that allow an exchange of cations or small molecules (such as H+ and H3O+).13–16 In contrast, interlayer-II remained unchanged and chemically inert. However, the protonation of LH (PLH) is required to achieve the maximum exchange of Gly intercalation in PLH (Gly@PLH).13–16 To prove this, pristine LH was intercalated with aqueous Gly without a protonation step. The resulting X-ray diffraction (XRD) pattern shows that there is no significant expansion of the interlayer-I, even for a longer reaction time (6 days) (Fig. S2, ESI†). However, a significant contraction of the (040) reflection plane was observed due to the preferential replacement of labile K+ in the interlayer-I by the H+ ions present in the aqueous Gly solution, and not by a Gly molecule. It is, therefore, a piece of evidence that Gly molecules were not intercalated into the pristine LH sample, and it revealed that interlayer-I must be protonated to accommodate small organic molecules, such as Gly.16
The XRD patterns of all samples indicated that they were highly crystalline (Fig. 1), and no trace of starting precursors were found in pre/post-intercalation (Fig. S3, ESI†). The (040) reflection plane (corresponding to interlayer-I) in PLH showed an interlayer spacing of d040 = 7.71 Å (at 2θ = 9.78°), whereas pristine LH showed at 2θ = 11.46° with a d040 9.04 Å. Such contraction of the interlayer spacing is obvious because the ionic radius of H+ (as H3O+) is smaller than that of K+. After the Gly intercalation, the PLH sample showed a significant expansion of the interlayer-I spacing to 9.49 Å (at 2θ = 9.32°) from 7.71 Å. Such increment in the interlayer spacing, further suggesting that the Gly molecules were successfully modified between the NbO6 layers of interlayer-I in PLH. The difference in the interlayer spacing of PLH and Gly@PLH was calculated (Fig. 1), and was found to be 1.78 Å, which proved the successful inclusion of Gly into PLH.13–17
 | ||
| Fig. 1 The powder XRD pattern of the as-synthesized pristine-layered hexaniobate K4Nb6O17 (LH), protonated LH (PLH) and Gly intercalated PLH (Gly@PLH). | ||
The disappearance of the reflection plane (red spectrum) at 2θ = 41.2° was due to the contraction of the niobate layer by the exchange of H+ from K+ compared to LH. After the Gly intercalation, a similar peak (2θ = 41.2°) reappeared, suggesting that interlayer-I was occupied by Gly molecules between the niobate layers.18 The peaks (Gly@PLH) at 2θ = 26° and 31° showed a relatively increased intensity compared to PLH; however, these peaks are not corresponding to the Gly powder (Fig. S4, ESI†).
The FTIR spectrum (Fig. S5, ESI†) of PLH showed an emerging new band at 955 cm−1, corresponding to the bridge OH bending vibration (δ-OH), and the disappearance of the IR band at 776 cm−1 revealed the distorted NbO6 layer due to the contraction of the interlayer spacing by the exchange of K+ by H+ (as H3O+).19–21 Raman analysis further (new band appeared at 937 cm−1, Fig. S6, ESI†) confirmed the proton exchange in LH.19–21 After the Gly intercalation, the IR band at 776 cm−1 reappeared, whereas the δ-OH band at 955 cm−1 (for Raman, at 937 cm−1; Fig. S6, ESI†) eventually disappeared, suggesting the successful intercalation of Gly within interlayer-I.17,22–24
The field emission scanning electron microscope (FE-SEM) images show the expansion of layered structures with a reduced lateral size of the nanosheets (3–4 μm) for Gly@PLH (Fig. S7g–i, ESI†), whereas the pristine LH exhibited a closely-stacked layered nanostructure with a lateral size of about 20–30 μm. It should be noted that there was no interlayer expansion observed when the pristine LH (non-protonated LB) was used for Gly intercalation (Fig. S8, ESI†). Thus, it was confirmed that the protonation step was required for the intercalation of Gly.16 These observations were consistent with the XRD results (Fig. S3, ESI†). The morphology of LH was not affected as long as the exchange was carried out in a highly acidic solution (2 M HCl) even for three days (Fig. S8, ESI†). The EDX mapping showed the presence of K, Nb, O, C and N elements in Gly@PLH (Fig. S7, ESI†).
The low (Fig. 2a) and high-resolution transmission electron microscopy (HR-TEM) images (Fig. 2b) showed that few-layered LH (K4Nb6O17) nanosheets were obtained by the interaction with Gly in aqueous solutions. The HR-TEM image (Fig. 2c) and corresponding selected area electron diffraction (SAED) pattern (Fig. 2d) revealed that the layered nanosheets were single crystals, which grew perpendicular to the b-axis of the K4Nb6O17 crystal, which is perpendicular to the basal plane of the plate-like single crystal.13,15 The scanning transmission electron microscopy (STEM)-elemental mapping (EDX) and energy-dispersive X-ray spectroscopy (EDS) analysis demonstrated the presence of K, Nb, O, C and N elements (Fig. 2) and their percentage atomic composition (Fig. S9, ESI†), which confirmed the successful intercalation of Gly into the layered niobates.
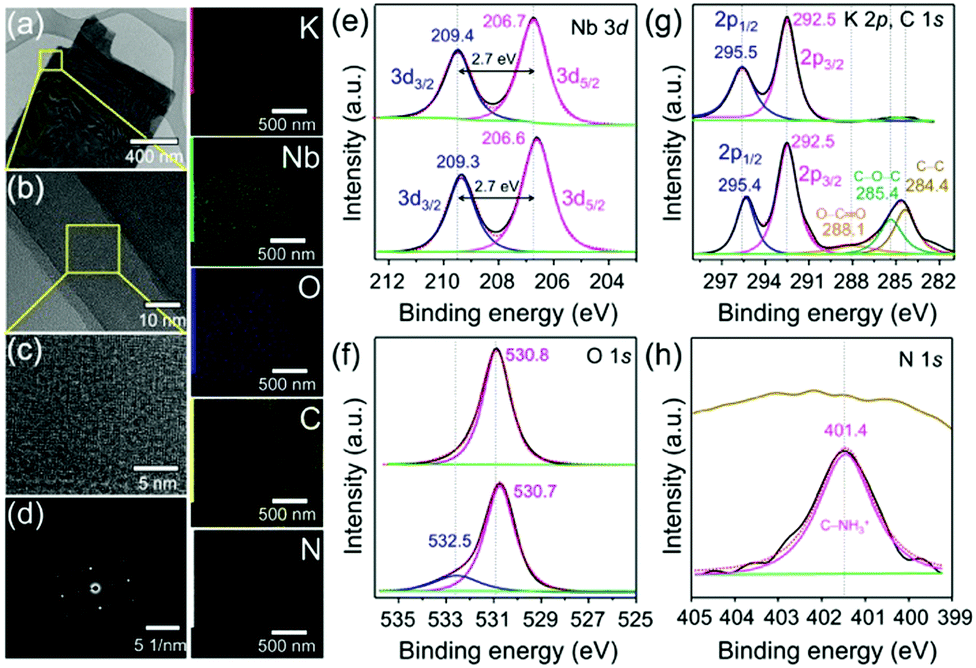 | ||
| Fig. 2 Low-resolution TEM images of Gly@PLH (a) and the corresponding high-resolution TEM images (b and c) and the SAED diffraction pattern for the selected area of (b). The STEM-elemental mapping (EDX) of the TEM image (a) demonstrates the presence of K, Nb, O, C and N elements in Gly@PLH. High-resolution XPS analysis of the deconvoluted core energy level spectra of (e) Nb 3d, (f) O 1s, (g) K 2p, C 1s and (h) N 1s pristine LH and Gly@PLH (top and bottom, respectively). | ||
The X-ray photoelectron spectroscopy (XPS) survey spectrum (Fig. S10, ESI†) showed the presence of Gly in PLH. Fig. 2e presents the peak of Nb 3d3/2 (at 209.4 eV) and Nb 3d5/2 (at 206.7 eV) components, corresponding to the Nd5+ valence state of niobate. Moreover, a small shift in the binding energy (B.E.) of 0.1 eV was observed for Gly@PLH (Fig. 2e, bottom). However, the spin–orbit splitting value of Nb in both samples remained the same at about 2.7 eV, suggesting that the oxidation state of Nb5+ still remained unchanged, and therefore it revealed a non-covalent interaction between Gly and niobate ligands in interlayer-I of PLH.25,26
Fig. 2f (top) shows the O 1s B.E. peak at 530.8 eV, attributed to the O2− (lattice O), whereas Gly@PLH (bottom) shows two chemically distinguished peaks at 530.7 and 532.5 eV, corresponding to O2− (lattice O) and –OH, respectively, which revealed the presence of Gly between the interlayer of Nb2O5. Alternatively, such a high B.E. peak could be endorsed by the location of Gly at nearby the niobium ions.27 It should be noted that a small downward shift (0.1 eV) of lattice O in the Gly@PLH sample revealed that the chemical environment changed in the NbO6 layers.
In the K 2p core-level region (Fig. 2g, top),LH was fitted by the two components at 295.5 and 292.5 eV, corresponding to 2p1/2 and 2p3/2 of potassium. The peak position of K 2p1/2 shifted to lower (Fig. 2g, bottom) due to the chemical changes in interlayer-I, whereas the peak position of K 2p3/2 remained unchanged, even after Gly intercalation. This small 0.1 eV downshifted K 2p1/2 line showed to be different in the chemical environment of K+ ions in the interlayer of Gly@PLH.28–30Fig. 2g (bottom) shows the presence of O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–O–C, and C–C components at 288.1, 285.4 and 284.4 eV, respectively, in the C 1s core-level spectrum of Gly@PLH, which further confirmed the presence of Gly in PLH.
O, C–O–C, and C–C components at 288.1, 285.4 and 284.4 eV, respectively, in the C 1s core-level spectrum of Gly@PLH, which further confirmed the presence of Gly in PLH.
Besides, the N 1s core energy level spectrum (Fig. 2h, bottom) of Gly@PLH shows a peak at 401.4 eV, which corresponds to the protonated C–NH3+.31 According to C 1s and N 1s XPS spectra (Fig. 2g and f; bottom) of the Gly@PLH sample, it was more evident that the Gly molecules were modified in interlayer-I between the NbO6 layers because there was no other source of N atoms in the sample than Gly.
The three consecutive cycles of CV curves were recorded for pristine LH and glycine intercalated LH (Gly@PLH) at a scan rate of 0.1 mV s−1 between 3.0 to 0.01 V (Fig. 3a and b). The first cycle was entirely different from the rest of the cycles, especially while sweeping in the cathodic direction (upon lithiation). A low intense sharp peak was noticed at 0.66 V vs. Li/Li+ corresponding to the reduction of the electrolyte with the simultaneous formation of solid electrolyte interface (SEI), followed by a sharp peak until a lower cut-off voltage related to the lithium intercalation. Interestingly, the CV pattern obtained closely resembled disorderly oriented carbon nano-sheets, which have already been reported earlier.6 During the anodic sweep, the intercalated lithium-ions were deintercalated 0.1–0.2 V. From the second cycle onwards, no significant change has been noticed, which confirms the formation of stable SEI in the initial cycle. A broad peak was noticed in the voltage region between 1.25 and 2.5 V, showing that the Nb underwent a typical lithiation, as observed from the redox peak (Fig. 3a and b). It can be seen from the normalised CV curves, the Gly-intercalated PLH displayed a significantly higher current compared to pristine LH, and this could be related to the expansion of the interlayer distance, as perceived from the XRD (Fig. 1).
 | ||
| Fig. 3 Cyclic voltammograms recorded at the scan rate of 0.1 mV s−1 of (a) LH and (b) Gly@PLH sample. (c) Normalized life-cycle study at a specific current of 0.5 A g−1. (d) Comparison rate performance along with their respective coulombic efficiencies. Selected cycling profiles obtained in the life-cycle study of (e) LH and (f) Gly@PLH samples. | ||
Fig. 3c depicts the comparison of capacity retention as a function of cycle number along with the respective coulombic efficiencies at a specific current of 0.5 A g−1. The Gly@PLH sample exhibited a significantly improved reversible capacity than that of pristine LH, which could be attributed to the elongated inter-slab distance. Interestingly, the discharge capacity gradually increased upon cycling in both cases and could be related to the reversible storage of lithium ions in gel-like SEI, as well as significant widening of the inter-slap distance on prolonged cycling. Initially, Gly@PLH delivered a high reversible capacity of about 96 mA h g−1 and gradually increased to 133 mA h g−1, whereas pristine LH limited the lithium insertion, delivered a delithiation capacity of about 58 mA h g−1 and reached 108 mA h g−1 after 750 cycles. It can be observed from the differential capacity plot that a slight contribution in lithiation also happened in LH, particularly niobate, which could be attributed to the delamination of the layered structure (a small and a broad redox peaks at higher voltage). However, such delamination was not observed in Gly@PLH (owing to the absence of such redox).
It can be seen from the stability graph (Fig. 3c) that the progressive increment of discharge capacity was significantly higher in pristine LH as compared to the sandwiched one, where the intercalated Gly molecule strongly bridged the adjacent layers and retained the structural integrity. Such interactions could also be observed and differentiated from the differential specific capacity plot (Fig. S11, ESI†). The redox peak current (related to Nb5+ to Nb3+) slightly increased in the pristine LH due to the partial delamination of the niobate layer from the bulk upon long cycling. Fig. S12 (ESI†) displays the comparison of the formation cycle lithiated at a specific current of 50 mA g−1, which showed an initial lithiation capacity of about 482 and 385 mA h g−1 for Gly@PLH and LH, respectively. Similarly, the first lithium reversibilities were 219 and 178 mA h g−1 with initial coulombic efficiencies (ICE) of 45.4 and 46.2%, for the aforementioned materials. The high lithiation capacity of Gly@PLH was attributed to the improved interlayer spacing compared to the LH. It can be noticed from ICE that more electrolyte decomposition occurred along with lithiation because of the significant enlargement of inter-slabs.
Fig. 3d and e represent the selected cycles of lithium insertion and extraction recorded profiles at a specific current of 0.5 A g−1. The rate retention study of the material is shown in Fig. 3f, where the cell is subjected to cycling at different specific currents from 0.05 A g−1 to 2.0 A g−1, followed by switching back to a low specific current of 0.5, and 0.25 A g−1. The cells have been cycled for eleven times, and the first cycle was ignored due to the kinetic limitation at each specific currents. It can be noticed from the rate performance test that Gly@PLH displayed an average discharge capacity of 180 mA h g−1 at 0.05 A g−1, which reduced to 142, 107, 87, 64 and 48 mA h g−1 upon increasing the specific current to 0.1, 0.25, 0.5, 1.0 and 2.0 A g−1, respectively. Similarly, the pristine LH showed a reversible capacity of about 130, which reduced to 101, 79, 62, 51 and 40, respectively for the afore-mentioned specific currents. Interestingly, when the specific current was switched back to 1.0, 0.5 and 0.25 A g−1, the Gly@PLH retained its capacity of about 70, 98 and 116 mA h g−1, and the pristine LH showed 57, 74 and 90 mA h g−1, respectively.
In order to study the interfacial properties, electrochemical impedance measurements (EIS) were performed before and after cycling for both cells. The Nyquist plot (Fig. S13, ESI†) showed a partially overlapped two-depressed semi-circle from high to middle, followed by a low-frequency sloping line related to semi-infinite Warburg diffusion (W). After the equilibration time of ageing, the half-cell was subjected to the EIS analysis, which showed bulk electrode resistances (Rs) of about 4.1 and 3.2 Ω for Gly@PLH and LH materials, respectively. Upon the steady-state life cycle study, the resistances slightly varied to 3.6 and 3.5 Ω, where the former decreasing resistance was related to the improved electrode wetting, and the later could be related to thick SEI layer. Similarly, the charge transfer resistance (Rct) expanded interlayer reduced from 133 to 37 Ω, for Gly@PLH and 51 from 177 Ω for LH. From the Nyquist plot, the glycine intercalation significantly improved the electrolyte contact by widening the interlayer spacing, which effectively formed a stable SEI with lower resistance and facilitated a superior lithium diffusion. All the Nyquist plots were fitted with a fitting software, and the respective Randels equivalent circuits are also displayed (Fig. S14, ESI†).
In summary, we have developed an interlayered modified layered hexaniobate of K4Nb6O17 (LH) anode for LIBs. Combining the powder XRD, FTIR and XPS analyses, we have shown that interlayer-I spacings in PLH could accommodate glycine (Gly) molecules; however, this could be achieved only after the protonation of LH. The Gly@PLH anode exhibited a reversible capacity of 133 mA h g−1 after 750 cycles at 0.5 A g−1. We believe that Nb-based materials with a properly modified interlayer have the potential for developing new anode electrode materials for LIB applications.
Author contributions
Conceptualization, S. E., S. V. and M. S.; formal analysis, S. E., S. V. and M. S.; anode materials synthesizing and characterization analysis, S. E.; electrochemical characterization, S. V.; writing–original draft preparation, S. E., S. V. and M. S.; writing–review and editing, S. E., S. V. and M. S.; supervision, S. E. and M. S.; project administration and management, S. E. and M. S.; funding acquisition, M. S. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr Satheeshkumar Elumalai acknowledges support from National Cheng Kung University (grant No. HUA 103-3-3-158), Taiwan. All authors are grateful to Dr Chia-Ying Su and Mr Bo-Yong Wu for their help in this research project.Notes and references
- N. Nitta, F. Wu, J. T. Lee and G. Yushin, Mater. Today, 2015, 18, 252 CrossRef CAS.
- J. B. Goodenough, ACS Catal., 2017, 7, 1132 CrossRef CAS.
- J. B. Goodenough and M. H. Braga, Dalton Trans., 2018, 47, 645 RSC.
- K. J. Griffith, Y. Harada, S. Egusa, R. M. Ribas, R. S. Monteiro, R. B. Von Dreele, A. K. Cheetham, R. J. Cava, C. P. Grey and J. B. Goodenough, Chem. Mater., 2020, 33(1), 4–18, DOI:10.1021/acs.chemmater.0c02955.
- M. H. Braga, N. S. Grundish, A. J. Murchison and J. B. Goodenough, Energy Environ. Sci., 2017, 10, 331 RSC.
- V. Selvamani, N. Phattharasupakun, J. Wutthiprom and M. Sawangphruk, Sustainable Energy Fuels, 2019, 3, 1988 RSC.
- C. P. Kocer, K. J. Griffith, C. P. Grey and A. J. Morris, Chem. Mater., 2020, 32, 3980 CrossRef CAS PubMed.
- Z. Liu, W. Dong, J. Wang, C. Dong, Y. Lin, I. W. Chen and F. Huang, iScience, 2020, 23, 100767 CrossRef CAS PubMed.
- Q. Deng, Y. Fu, C. Zhu and Y. Yu, Small, 2019, 15, e1804884 CrossRef PubMed.
- B. Guo, X. Yu, X.-G. Sun, M. Chi, Z.-A. Qiao, J. Liu, Y.-S. Hu, X.-Q. Yang, J. B. Goodenough and S. Dai, Energy Environ. Sci., 2014, 7, 2220 RSC.
- V. Augustyn, J. Mater. Res., 2017, 32, 2–15 CrossRef CAS.
- L. Yan, X. Rui, G. Chen, W. Xu, G. Zou and H. Luo, Nanoscale, 2016, 8, 8443 RSC.
- R. Abe, M. Hara, J. N. Kondo, K. Domen, K. Shinohara and A. Tanaka, Chem. Mater., 1998, 10, 1647 CrossRef CAS.
- N. Miyamoto, K. Kuroda and M. Ogawa, J. Am. Chem. Soc., 2001, 123, 6949 CrossRef CAS.
- R. Kaito, K. Kuroda and M. Ogawa, J. Phys. Chem. B, 2003, 107, 4043 CrossRef CAS.
- M. A. Bizeto, A. L. Shiguihara and V. R. L. Constantino, J. Mater. Chem., 2009, 19, 2512 RSC.
- C. Chen, M. Boota, P. Urbankowski, B. Anasori, L. Miao, J. Jiang and Y. Gogotsi, J. Mater. Chem. A, 2018, 6, 4617 RSC.
- M. Eguchi, M. S. Angelone, H. P. Yennawar and T. E. Mallouk, J. Phys. Chem. C, 2008, 112, 11280 CrossRef CAS.
- M. A. Bizeto, F. Leroux, A. L. Shiguihara, M. L. A. Temperini, O. Sala and V. R. L. Constantino, J. Phys. Chem. Solids, 2010, 71, 560 CrossRef CAS.
- M. M
![[a with combining cedilla]](https://www.rsc.org/images/entities/char_0061_0327.gif) czka, M. Ptak, A. Majchrowski and J. Hanuza, J. Raman Spectrosc., 2011, 42, 209 CrossRef.
czka, M. Ptak, A. Majchrowski and J. Hanuza, J. Raman Spectrosc., 2011, 42, 209 CrossRef. - Y. Ide and W. Shirae, Inorg. Chem., 2017, 56, 10848–10851 CrossRef CAS PubMed.
- S. Kumar, A. K. Rai, V. B. Singh and S. B. Rai, Spectrochim. Acta, Part A, 2005, 61, 2741 CrossRef PubMed.
- I. A. Mudunkotuwa, A. A. Minshid and V. H. Grassian, Analyst, 2014, 139, 870 RSC.
- E. Satheeshkumar, A. Bandyopadhyay, M. B. Sreedhara, S. K. Pati, C. N. R. Rao and M. Yoshimura, ChemNanoMat, 2017, 3, 172 CrossRef CAS.
- N. Özer, T. Barreto, T. Büyüklimanli and C. M. Lampert, Sol. Energy Mater. Sol. Cells, 1995, 36, 433 CrossRef.
- M. A. B. Gomes, L. O. d. S. Bulhões, S. C. de Castro and A. J. Damião, J. Electrochem. Soc., 2019, 137, 3067 CrossRef.
- Q. Deng, M. Li, J. Wang, P. Zhang, K. Jiang, J. Zhang, Z. Hu and J. Chu, Sci. Rep., 2017, 7, 1883 CrossRef PubMed.
- S. C. Abrahams, P. B. Jamieson and J. L. Bernstein, J. Chem. Phys., 1971, 54, 2355 CrossRef CAS.
- Y. Joseph, G. Ketteler, C. Kuhrs, W. Ranke, W. Weiss and R. Schlögl, Phys. Chem. Chem. Phys., 2001, 3, 4141–4153 RSC.
- R. Gostynski, J. Conradie and E. Erasmus, RSC Adv., 2017, 7, 27718 RSC.
- J. S. Stevens, A. C. de Luca, M. Pelendritis, G. Terenghi, S. Downes and S. L. M. Schroeder, Surf. Interface Anal., 2013, 45, 1238 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00055a |
| ‡ These authors are equaly contributed to this reseach. |
| § The author (S.V.) present address: Department of Chemical and Biomolecular Engineering, Vidyasirimedhi Institute of Science and Technology (VISTEC), Rayong, 21210, Thailand. |
| This journal is © The Royal Society of Chemistry 2021 |