
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel 3D-networked melamine–naphthalene–polyamic acid nanofillers doped in vinyl ester resin for higher flame retardancy
Ravi Kumar
Cheedarala
 a,
M. N.
Prabhakar
a,
Beom Gon
Cho
b,
Young Bin
Park
b and
Jung
Il Song
*a
a,
M. N.
Prabhakar
a,
Beom Gon
Cho
b,
Young Bin
Park
b and
Jung
Il Song
*a
aResearch Institute of Mechatronics Engineering, Department of Mechanical Engineering, Changwon National University, Changwon, Gyeongnam 51140, Republic of Korea. E-mail: jisong@changwon.ac.kr
bDepartment of Mechanical Engineering, Ulsan National Institute of Science and Technology (UNIST), UNIST-gil 50, Eonyang-eup, Ulju-gun, Ulsan 689-798, Republic of Korea
First published on 11th May 2021
Abstract
In this study, melamine–naphthalene covalent polyamic acid and its metal carboxylate salts were synthesized as building blocks of nanofillers (Nt–Mn–PA–COOM, M = H, Li, Na, and K). The Nt–Mn–PA–COOM nanofillers were added as a dopant to vinyl ester resin at 1–5 wt% and their flame retardancy and thermal properties were evaluated. The flame retardancies of 5 wt% of Nt–Mn–PA–COOK–VE and Nt–Mn–PA–COONa–VE were found to improve the heat release rate by up to 95% and 90%, respectively, in comparison with the pristine vinyl ester (VE) composite. These results demonstrated significant progress in the fireproofing performance of VE composites. The burning times of Nt–Mn–PA–COONa–VE and Nt–Mn–PA–COOK–VE composites were shown to be higher 10.6 min, and 12.1 min, respectively, when compared with the pure VE composite (6.28 min). Also, the burning rates of Nt–Mn–PA–COONa–VE and Nt–Mn–PA–COOK–VE concerning flame retardancies were decreased from 11.92 min to 7.07 and 6.19 min, respectively. The delay in burning time, and the reduction of burning rate clearly refer to the enhancement of flame retardancy behavior of the Nt–Mn–PA–COOM–VE composites. It was found that 5 wt% of nanofiller dopants Nt–Mn–PA–COOM–VE (M = Na, and K) in VE was the ideal loading for considering the integrated perspective for flame retardancy applications. The excellent flame retardancy and thermal performance of the Nt–Mn–PA–COOM–VE (M = Na, and K) composites made them promising candidates for developing novel flame retardancy nanofiller dopants.
1. Introduction
Char-forming flame retardant (FR) fillers react to form a carbonaceous layer on the surface of the host polymer material. The carbonaceous layer insulates the polymer, thereby decelerating pyrolysis, and creates a boundary that delays the release of surplus gases for fuel combustion. The FR mechanism involves endothermic degradation, thermal shielding, dilution of the gas phase, and gas-phase radical quenching. These four important characteristic features have played a key role in enhancing the flame retardancy of materials. In particular, vinyl ester resin is a widely popular thermoset polymer, produced by the esterification of epoxy bis-phenol A with acrylic acid,1–4 used in laminating processes. Vinyl ester is typically activated with methyl ethyl ketone peroxide. It displays greater endurance, better physical and mechanical properties, enhanced heat and corrosion resistance, good adhesion, and rapid curing over polyester and epoxy resins.5–10 Consequently, they are extensively used in composite pipes, printed circuit board coatings, adhesives, and various other industrial fields.11–16 In homebuilt airplanes, especially, the Glasair kit planes extensively use vinyl ester fiberglass-reinforced structures. Vinyl ester resin (VER) is commonly used in the maritime industry because it exhibits high corrosion resistance and hydrophobicity to water. VER is extensively used to manufacture fiberglass-reinforced plastic (FRP) tanks and vessels as per BS4994. Owing to the abundant advantages of VER laminates in both industrial and commercial domains, scientists and engineers often examine the intumescent flame retardant properties of VERs. While VER displays many anticipated properties, such as resiliency, flexibility, and low cost, its intrinsic flammability cannot be ignored. Customized VERs burn easily when they are exposed to fire owing to their inherent chemical structures. This hinders their applications in some areas with high flame-retardant requirements. Thus, improving the flame retardancy of VERs is of great significance for expanding their applications.17–23 Recently, polyimides have been used as nanofillers to improve the flame-retardant performance of polymer materials owing to their high reactivity, good thermal properties, high flame-retardant efficiency, and relatively environmentally friendly features.24–28Aromatic polyimides exhibit inherent fire-resistant properties owing to strong imidic linkages and π–π stacking between each monomeric block. These advantages are anticipated to increase their thermo-oxidative stability and flame-retardant properties, which are crucial for industrial applications. For instance, A. B. Morgana et al. used inorganic materials, such as 40 wt% of alumina, 10 wt% of fumed silica, and 5 wt% of carbon nanofibers to improve the thermal strength and flammability performance of polyimides but their heat release rate (HRR) was poor in comparison with the authentic polyimide.29 J. Liu et al. used 20 wt% SiO2 in BTDA–ODA polyimide, which increased its limiting oxygen index (LOI) value by up to 37%.30 In another study, S. Koytepe et al. investigated zinc borate nanoparticles incorporated in a polyimide matrix for the enhanced thermal stability of polyimides.31
According to previous reports, when different flame-retardant groups were introduced in a molecular skeleton, a synergistic fire-retardant effect was produced. Therefore, some fire retardants containing phosphaphenanthrene and other flame-retardant components, such as triazine, silsesquioxane, and triazine-trione, were successively developed structurally to further enhance the flame retardancy of resin systems.30 Additionally, Bai et al. synthesized a star-shaped flame retardant [2,4,6-tris(p-formylphenoxy)-1,3,5-triazine], featuring phosphaphenanthrene and triazine groups, and prepared a series of fire-retardant UPR composites with various amounts of TRIPOD–DOPO.31 The test results showed that the introduction of TRIPOD–DOPO improved the flame retardancy and thermal stability of UPR. Moreover, their groups also synthesized a liquid monomer [DOPO–vinyltrimethoxysilane (VTS)] containing phosphorus and silicon and prepared flame-retardant UPR–SiO2 hybrid materials by introducing DOPO–VTS and methacryloxypropyl trimethoxy silane (KH570) into UPR. When the content of DOPO–VTS was 20%, the LOI value of UPR increased from 21.0 to 27.0%.32 Traditionally, flame retardancy (FR) polymeric materials have been used earlier such as halogenated FR (i.e. tetrabromobisphenol A, hexabromocyclododecane, polybrominated bisphenyls (PBBs) and polybrominated diphenyl ethers (PBDEs)) have been used at an estimated volume of 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons per year globally. However, halogenated polymers produce huge amounts of toxic, corrosive, and halogenated gases during the combustion process. Owing to the gigantic evolution of toxic pollutants released into the environment, a robust substitute is required, which is environmentally benign, to protect the planet.
000 tons per year globally. However, halogenated polymers produce huge amounts of toxic, corrosive, and halogenated gases during the combustion process. Owing to the gigantic evolution of toxic pollutants released into the environment, a robust substitute is required, which is environmentally benign, to protect the planet.
This research work aims to explain the advances in both inorganic and organic materials to generate novel, robust, and multifunctional materials with excellent flame retardant characteristics. To the best of our knowledge, we developed novel organic fillers based on naphthalene dicarboxylic acid salts (i.e. M = H, Li, Na, and K) and melamine as a core for the first time. The inorganic–organic hybrid nanofillers exhibited versatile characteristics owing to their physical and chemical properties. These features include strong binding property by chelation with host polymers, and they exhibit higher thermophysical properties over a wide range of applied temperatures. Additionally, they possess a variety of novel factors provided by architecturally diverse metal ions, such as Li, Na, and K. Polyamic acid-based fillers, especially 3D networked naphthalenedicarboxylic acid coupled melamine–polyamic acid salts as nanofillers have been attracting attention as potential substituents for conventional FR materials owing to their excellent thermal stability and low flammability.33 However, to the best of our knowledge, this is the first report in the literature using Nt–Mn–PA–COO–M (i.e. M = H, Li, Na, and K) for FR applications. Their high thermal stability and thermo-chemo-physical properties are promising candidates for non-halogen FR materials.
Herein, we demonstrated a simple synthetic protocol using naphthalene dianhydride as the peripheral moiety and melamine as the backbone network of the novel dianhydride–melamine filler for the FR 3D networked hybrid filler. First, we aim to design and synthesize naphthalene–melamine–polyamic acid (Nt–Mn–PA–COOH) using the solution mediated amide coupling method. Second, we prepared alkali metal carboxylic salts (Nt–Mn–PA–COO–M, i.e. M = Li, Na, and K) to investigate the efficiency of their FR properties within VER. The Nt–Mn—PA–COO—M/VER polymer composite is prepared by dispersing 5 wt% of Nt–Mn–PACOOM in VER. During the intumescent flame retardancy test, the environmental oxygen cannot penetrate the 3D-networked Nt–Mn–PACOOM polymeric system owing to the strong interfacial interaction caused by π–π stacking in naphthalene rings, which can prevent elemental oxygen from entering the host polymers. Systematically, when the size of the dopant metal atom (i.e. Li(3) > Na(11) > K(20)) is increased in the host polymer then a huge space is occupied within the VER polymer network and automatically the burning property will be delayed due to there being no space available for oxygen gas to enter. Also, the experimental results of the HRR strongly support our hypothesis before and after the insertion of metal ions. Besides, the proposed Nt–Mn–PA–COOM–VER composites show robust thermal stability even at 600 °C. To measure the FR of the composites, LOI measurements and UL-94 VTM tests are essential. The novel Nt–Mn–PA–COOM–VER composites showed better thermal stability, even at 600 °C, of up to 96.5% in comparison with the pristine VER polymer composite (Scheme 1).
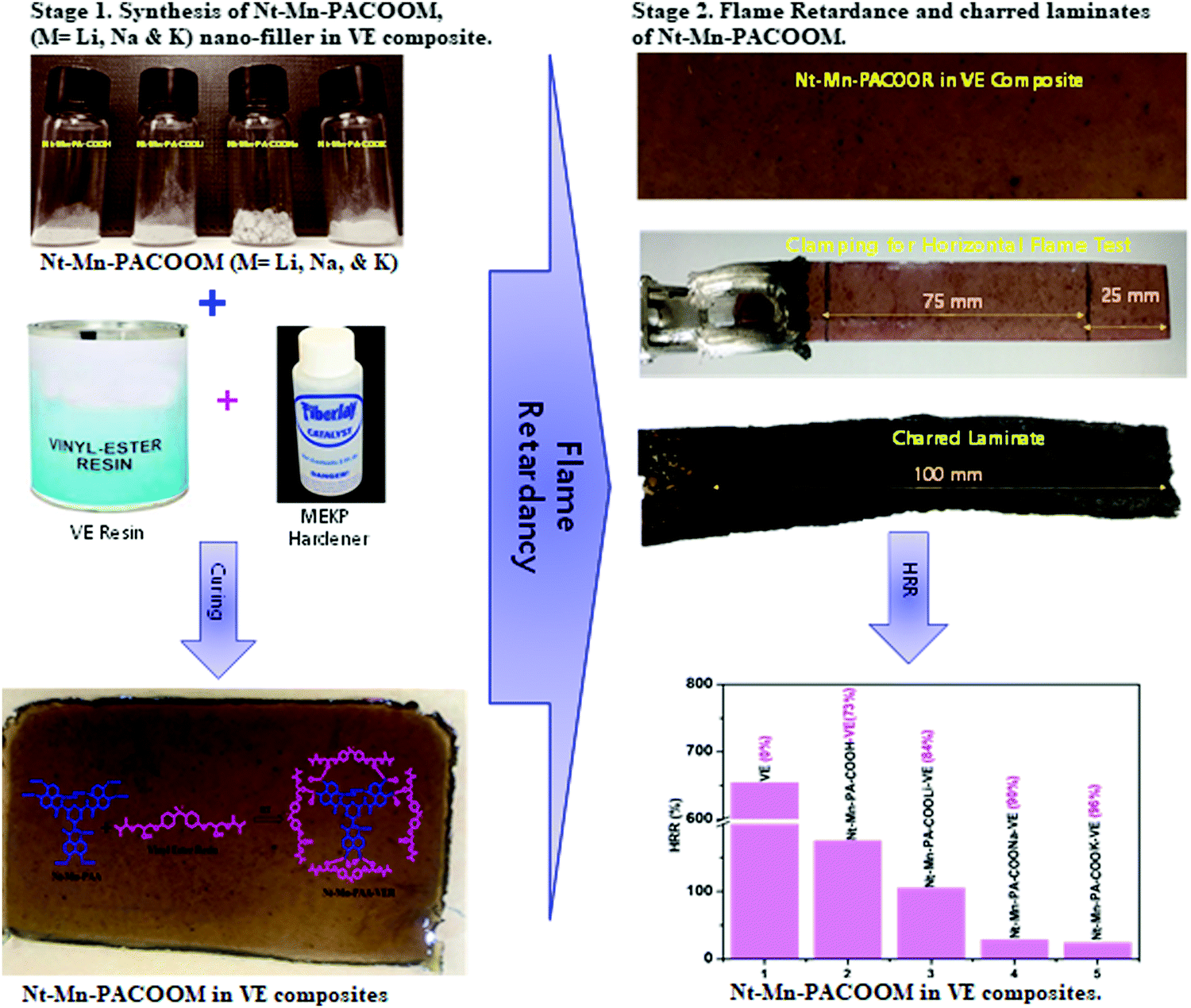 | ||
| Scheme 1 Schematic representation of the synthesis of Nt–Mn–PACOOM in VE composite and the flame retardant properties. | ||
2. Experiments
2.1. Materials
All chemicals were of commercially available grade and used without further purification. All solvents were dried using a 4 Å molecular sieve before use. Vinyl ester (specific gravity = 1.03 and viscosity = 150 cps) was supplied by CCP composites, Korea. Methyl ethyl ketone peroxide (MEKP) (hardener), cobalt naphthalate (accelerator), and Frekote 700-NC (releasing agent) were purchased from CCP composites, Korea. Melamine (Mn), naphthalene dianhydride (Ntda), and DMF were purchased from Aldrich, Korea.2.2. Instrumentation
Chemical composition analysis has been investigated using an X-ray spectrometer (EDX) with a Rönteck flash detector analyzer associated with scanning electron microscopy (SEM, Leo-Evo 40xVP). Incident electron beam energies from 10 to 30 keV were used. In all cases, the beam was normally incident to the sample surface and the measurement time was 100 s. All EDX spectra were corrected using ZAF correction, which considered the influence of the matrix material on the obtained spectra.2.3. Characterization
Fourier transform infrared (FT-IR) spectra were recorded using a Nicolet 6700 FT-IR spectrometer (Thermo Electron Scientific Instruments). The test was performed using the KBr pellet pressing method in the scanning range of 400–4000 cm−1. Cone calorimetry measurements were performed using an FTT0007cone calorimeter (FTT, United Kingdom) according to the ISO5660 standard. The external heat flux was 50 kW m−2 and the dimensions of all casting samples were 100 × 100 × 3 mm3. The morphologies of char residues after the cone calorimetry tests were analyzed via TESCAN VEGA3 tungsten filament SEM (TESCAN, Czech) under an acceleration voltage of 20 kV. TGA was performed under nitrogen with a Netzsch STA449F3 instrument (TA Co.). The heating rate was 10 °C min−1 and the testing temperature range was 30–700 °C.2.4. Preparation of Nt–Mn–PA–COOH
A mixture of melamine (1.26 g, 10 mmol) and naphthalene dianhydride (Ntda) (4.02 g, 15 mmol) was refluxed in DMF (30 mL) for 15 h under a nitrogen atmosphere at 150 °C. After completion of the reaction, the solid was separated, washed using ethanol, and dried at 75 °C in a vacuum oven for 5 h to obtain Nt–Mn–PA–COOH as a white solid in 87% yield.2.5. Preparation of Nt–Mn–PA–COOM (M = Li, Na, and K) (general procedure)
A mixture of Nt–Mn–PA–COOH (2 g, 20 mmol) and base (i.e., LiOH, NaOH, and KOH) (40 mmol) was stirred in pure ethanol (50 ml) for 2 h at 60 °C. The product was filtered, washed with ethanol, and dried to obtain the corresponding metalized Nt–Mn–PA–COOM (i.e., M = Li, Na, and K) in quantitative yield.2.6. Preparation of Nt–Mn–PA–COOM–VER composite (general procedure)
A total of 1 g, 2 g, 3 g, 4 g, and 5 g of Nt–Mn–PA–COOM (M = Li, Na, and K) was mixed with VER (100 g) individually at RT. After the formation of a homogeneous solution in 1 h, simultaneously, MEKP hardener (1 g) and accelerator (1 g) were added. After stirring for 1 h, the polymer mixtures were transferred into suitable molds (30 cm × 30 cm × 1 cm) and air-dried for 12 h. The four fabricated samples (i.e. Nt–Mn–PA–COOH–VE, 1–5 wt%, Nt–Mn–PA–COOLi–VE, 1–5 wt%, Nt–Mn–PA–COONa–VE, 1–5 wt%, and Nt–Mn–PA–COOK–VE, 1–5 wt%) were cut into dimensions of 20 cm × 1 cm and used for the horizontal flame tests.2.7. Horizontal burning test
The horizontal burning test was used to evaluate the effect of the fire retardants on the flammability of thermoplastic starch biopolymer (TPS) composites according to UL-94 standards (Araoetal, 2014). In brief, the sample was held horizontally and a flame produced by an organic solvent (butane starch) was applied to one end of the sample for 30 s. The height and angle of the flame in the vertical direction were 10 mm and 45°, respectively. The time taken for the flame to reach the second reference mark (80 mm from the end) from the first reference mark (20 mm from the end) was measured. Prior to the test, the specimens were dried at 80 °C for 24 h and the tests were conducted, simultaneously, at least three times for each composition.2.8. Thermogravimetric analysis (TGA)
The thermal stability and decomposition behavior of the constituents of the composites were analyzed using a thermogravimetric analyzer (Model: TGAQ600/SDT, TA instruments, USA) at a heating rate of 10 °C min−1 under both inert (N2) and air atmospheres. The scanning scope ranged from 30–700 °C. The residue was evaluated as the specimen's residual weight at 700 °C.3. Results and discussions
3.1. Synthesis of 3D networked Nt–Mn–PA–COOM nanofillers and Nt–Mn–PA–COOM–VE composites
In this work, we synthesized highly flame-retardant nanofillers which were prepared using inorganic–organic 3D-networked polyamic acid nanofillers with increasing sizes of metal ions (i.e. Li, Na, and K). A mixture of naphthalene dianhydride and melamine was reacted in dry DMF at room temperature and produced the corresponding naphthalene–melamine–polyamic acid (Nt–Mn–PA–COOH) in good yield, as shown in Fig. 1. Then, the Nt–Mn–PA–COOH was reacted with different alkali metal hydroxides M–OH (i.e. M = Li, Na, and K) in ethanol, and produced the corresponding carboxylic acid salts of Nt–Mn–PA–COOM (i.e. M = Li, Na, and K) in quantitative yields (Stage 1).32 Consequently, for the synthesis of Nt–Mn–PA–COOM–VE composites, first, Nt–Mn–PA–COOM (i.e. M = Li, Na, and K) was mixed with VER, MEKP, hardener, and accelerator, respectively. After stirring for 1 h, the polymer mixture was transferred into a suitable mold and air-dried for 12 h. Four samples with different wt% were prepared (i.e. Nt–Mn–PA–COOH–VE, 1–5 wt%, Nt–Mn–PA–COOLi–VE, 1–5 wt%, Nt–Mn–PA–COONa–VE, 1–5 wt%, and Nt–Mn–PA–COOK–VE, 1–5 wt%) respectively. The Nt–Mn–PA–COOM–VER composites were cut into dimensions of 20 cm × 1 cm for the horizontal flame tests (Stage 2).33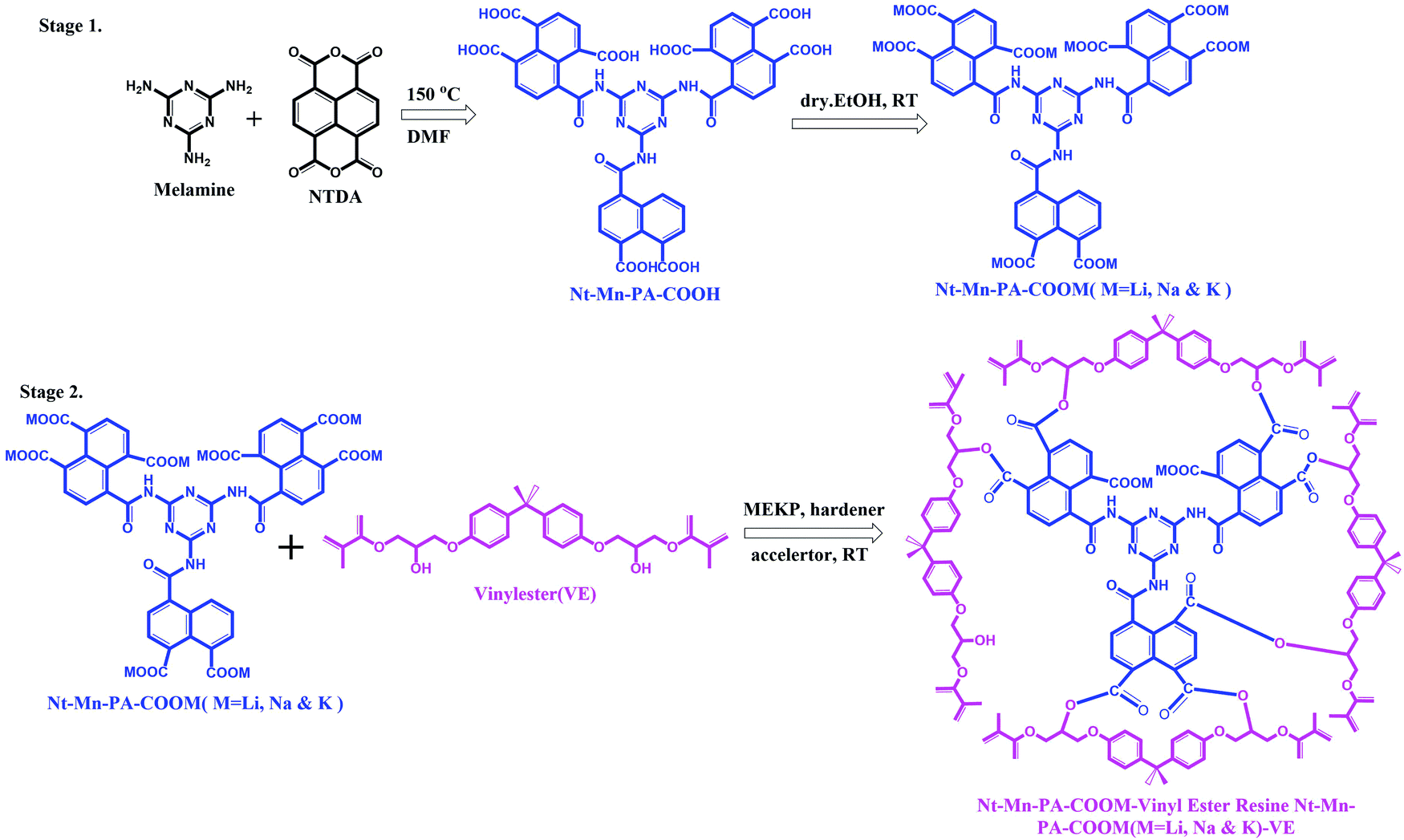 | ||
| Fig. 1 Syntheses of 3D-networked inorganic–organic polyamic acid and its Li, Na, and K metal salt nanofillers (Stage 1) and Nt–Mn–PA–COOM–VE composite (Stage 2). | ||
3.2. Structural characterization of Nt–Mn–PA–COOR–VE composites
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N in Nt–Mn–COONa.34–38 However, in the case of the Nt–Mn–COOK network, peaks were observed at 3467 cm−1 for –OK, 1596 cm−1 for CO–NH–, and 1416 cm−1 and 1342 cm−1 for CN of the triazine. Vibrational peaks were observed at ∼773 cm−1 for –CH (wag) and ∼612 cm−1 for –C–C– (stretch) in all three spectra due to the structural stability of the triazine ring, which was interconnected with the naphthalene ring systems. The absorbance of amide in Nt–Mn–COOH shifted from 1651 cm−1 to ∼1585 cm−1 to 1596 cm−1, indicating that the amidation reaction occurred between melamine and naphthalene dianhydride. Thus, the results of FT-IR analysis proved the formation of Nt–Mn–PA–COOM.38
N in Nt–Mn–COONa.34–38 However, in the case of the Nt–Mn–COOK network, peaks were observed at 3467 cm−1 for –OK, 1596 cm−1 for CO–NH–, and 1416 cm−1 and 1342 cm−1 for CN of the triazine. Vibrational peaks were observed at ∼773 cm−1 for –CH (wag) and ∼612 cm−1 for –C–C– (stretch) in all three spectra due to the structural stability of the triazine ring, which was interconnected with the naphthalene ring systems. The absorbance of amide in Nt–Mn–COOH shifted from 1651 cm−1 to ∼1585 cm−1 to 1596 cm−1, indicating that the amidation reaction occurred between melamine and naphthalene dianhydride. Thus, the results of FT-IR analysis proved the formation of Nt–Mn–PA–COOM.38
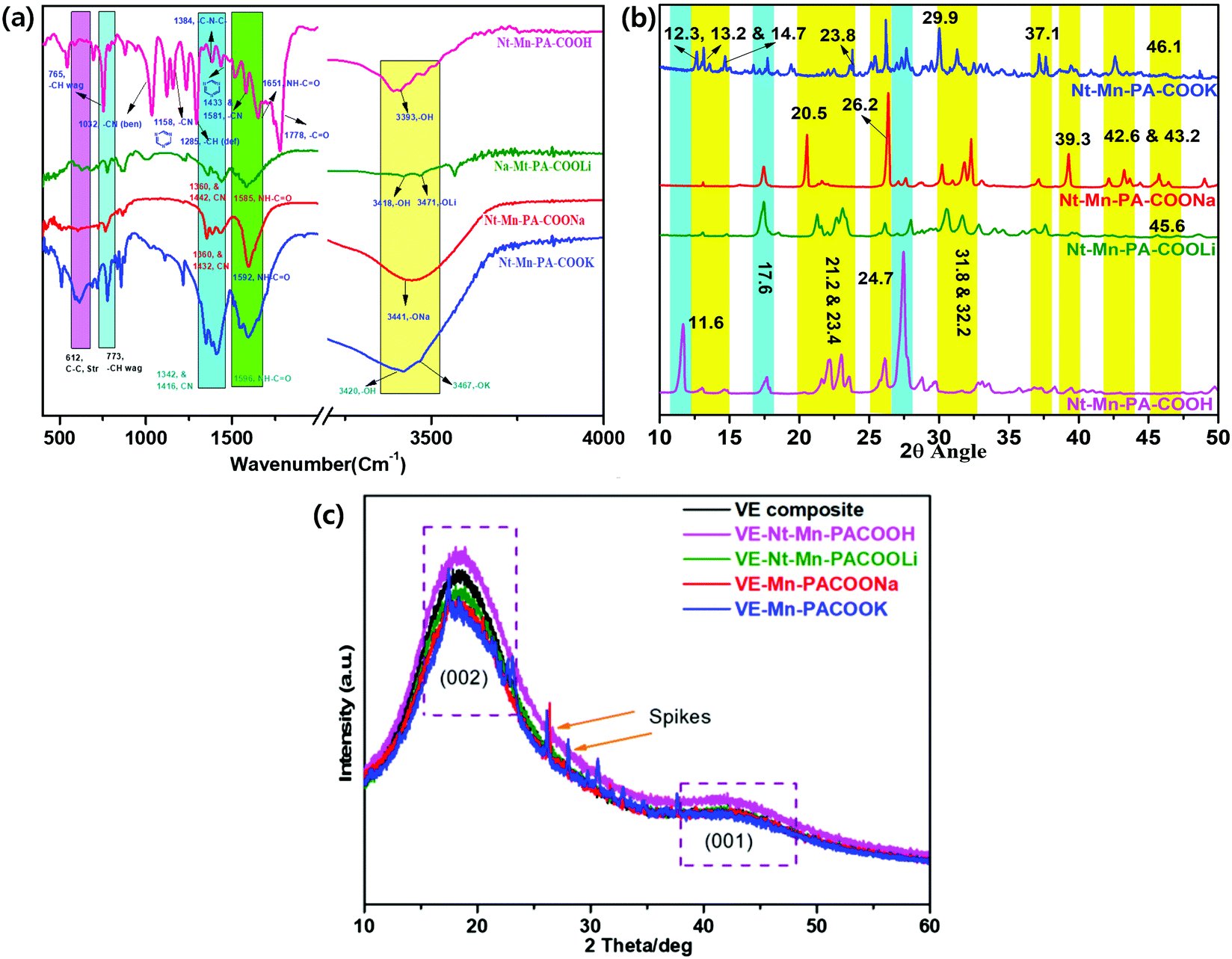 | ||
| Fig. 2 (a) FT-IR, (b) powder XRD analyses of Nt–Mn–PACOOM ionic salts, and (c) XRD analyses of 3D-networked Nt–Mn–PACOOM–VER composites. | ||
XRD analysis of the Nt–Mn–PA–COOM–VE composites (where M = H, Li, Na and K) was determined the properties of the crystalline nature. Fig. 2c shows the XRD patterns obtained for the pristine VE panel and VE panels doped with Nt–Mn–PA–COOM. The XRD pattern of pristine VE demonstrated the typical amorphous structure that possessed (002) and (001) broad diffraction peaks located at the 2θ angles of 22° and 44°, respectively.39 Compared with the pristine VE composite, the Nt–Mn–PACOOM–VE panels had no significant impact on the crystal structure except for sharp peaks. XRD analysis of Nt–Mn–PA–COONa and laminated Nt–Mn–PA–COOK–VE demonstrated some sharper peaks superimposed on the broad peaks, which indicates the presence of crystalline components in the amorphous membrane. The sharper peaks were observed especially for Nt–Mn–PA–COONa–VE and Nt–Mn–PA–COOK–VE owing to the intercalation of Na+ and K+ ions. The ion sizes of Na+ and K+ are 11 and 19 in comparison to pristine VEs, Nt–Mn–PA–COOH–VE and Nt–Mn–PA–COOLi–VE composites, respectively. The broad peaks observed in Nt–Mn–PA–COONa–VE, and Nt–Mn–PA–COOK–VE changed into sharp peaks and the degree of crystallinity increased due to the formation of Na+ and K+ ionic clusters at carboxylic moieties, which enhanced the flame retardancy property while inhibiting the penetration of oxygen gas.41,42 However, the sharp peaks suggested that the amount of crystalline components is significantly high comparison with that in the amorphous parts.
Confirmation of metal ions in Nt–Mn–PA–COOM in VE composites. Fig. 3 shows the energy-dispersive X-ray (EDX) SEM analyses, which revealed the presence of metals ions such as Li, Na, and K within the Nt–Mn–PA–COOM fillers. The EDX analysis showed the dopant nanofillers such as Nt–Mn–PA–COOM (i.e., M = Li, Na, and K), in which the metal ions are varied by size, established the self-assembly of Nt–Mn–PA–COOM nanofillers simultaneously absorbed on a monolayer film of the polymer via physical and chemical arrangements.43 Controlling more than a single type of nanofiller in exact positions could provide an opportunity to utilize the exceptional properties of each type of nanofiller. Thus, the methodology established in this study can be a good example of how the diverse types of functional nanometer-sized structure blocks can be organized in specific arrangements via physical and chemical self-assembly procedures on organized patterns, as shown in Fig. 3a–d and Table 1. The surface morphology of Nt–Mn–PA–COOH was plain and smooth with no visible deposits, except for some micro holes. However, the Nt–Mn–PA–COOLi composite exhibited a rough surface morphology with depositions due to the presence of Li+ ions. Nevertheless, the surface morphologies were showed similar structural patterns due to the closest ionic sizes of H and Li-ions. Instead, noteworthy changes in the surface morphologies were found for the Nt–Mn–PA–COONa and Nt–Mn–PA–COOK composites, as shown in Fig. 3c and d. Na and K metal ion aggregations were found on the surface, which further supports the anchoring with the base VER composite. The EDS spectra reveal the amount of deposition of Na+ and K+ ions within the polymer network by wt% and at%. In particular, the amount of wt% and at% showed the rigid morphologies on the surface by coordinating the covalent bond with the host VER polymer network (Table 1).
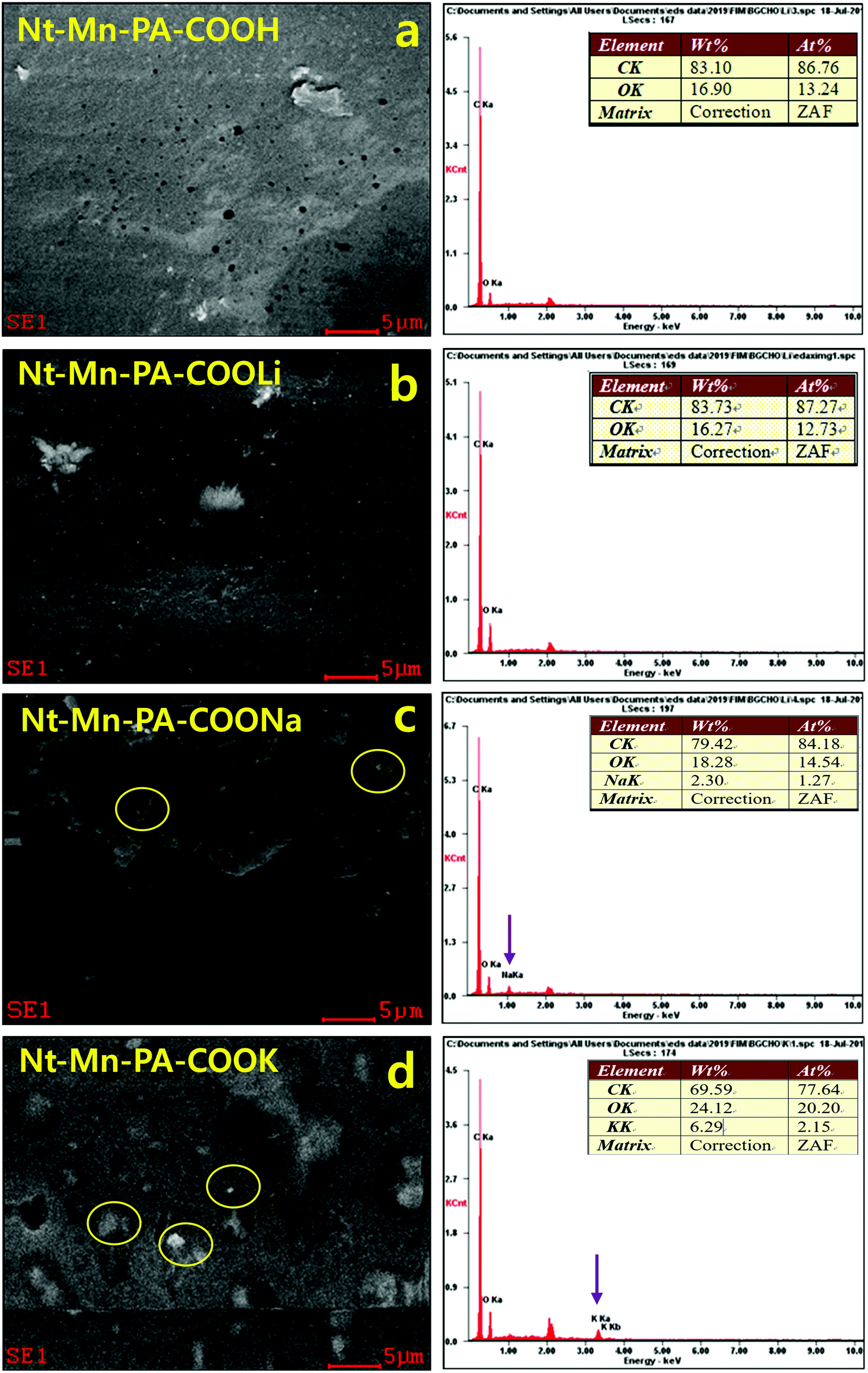 | ||
| Fig. 3 EDX analyses of 3D-networked Nt–Mn–PACOOM in VE. | ||
| Composites | wt% | at% | ||||||
|---|---|---|---|---|---|---|---|---|
| C | O | Na | K | C | O | Na | K | |
| Nt–Mn–PA–COOH–VE | 83.10 | 16.90 | — | — | 86.76 | 13.24 | — | — |
| Nt–Mn–PA–COOLi–VE | 83.73 | 16.27 | — | — | 87.27 | 12.73 | — | — |
| Nt–Mn–PA–COONa–VE | 79.42 | 18.28 | 2.30 | — | 84.18 | 14.54 | 1.27 | — |
| Nt–Mn–PA–COOK–VE | 69.59 | 24.12 | — | 6.29 | 77.64 | 20.20 | — | 2.15 |
3.3. DSC, and TGA analyses
DSC measurements demonstrate the chemical changes within the polymer constituents when subjected to heat. Fig. 4a shows the DSC curves of pristine VE and the composites enhanced with Nt–Mn–PA–COOM. In comparison with pristine VE, the melting endothermic peak of the composites was shifted to a higher temperature, which indicated that the filling of the Nt–Mn–PA–COOM nanofillers improved the melting temperature (Tm) of the VE matrix. Nt–Mn–PA–COOM consists of 3D-networked melamine backbone–naphthalene carboxylic acid moieties.43 Additionally, the VE molecular chain became more stable after addition of the Nt–Mn–PA–COOM nanofillers. Thus, Nt–Mn–PA–COOM nanofillers are helpful factors in hindering the melting of the composites. For VE, which only has nonpolar groups in the molecular chains, the Nt–Mn–PA–COOH and Nt–Mn–PA–COOLi nanofillers demonstrated the same effect on its melting temperature, which resulted in slightly different Tm values for the experimental and control groups. Furthermore, the addition of Nt–Mn–PA–COONa and Nt–Mn–PACOOK nanofillers improved the crystallinity of the VE composite, thereby indicating that the Nt–Mn–PA–COONa and Nt–Mn–PA–COOK nanofillers had a higher nucleation function due to the high aspect ratios and appropriate particle sizes. Moreover, inducing the formation of a trans crystalline nature (TC) can improve the crystallinity of the VE composite polymer. By increasing the size of the nanofiller ions (i.e., H, Li, Na, and K) in the host polymer, the number of hydrogen bonds between the VE and the nanofillers was increased due to the formation of chemical bonds between CO in the VE polymer chains, along with free –OH, and –NH in Nt–Mn–PA–COOM. Due to formation of strong covalent hydrogen bonds between the dopant nanofiller and the guest VE polymer, the nucleation density was increased, which is reflected in the crystallinity of the polymer network being stable even at higher temperatures such as 284–507 °C. The results were attributed to the excellent interfacial compatibility between Nt–Mn–PA–COOM and the VE matrix by the formation of metal ion–lone pairs of carbonyls through strong interactions by chelation, evidenced by the FT-IR analyses, as shown in Fig. 2a.
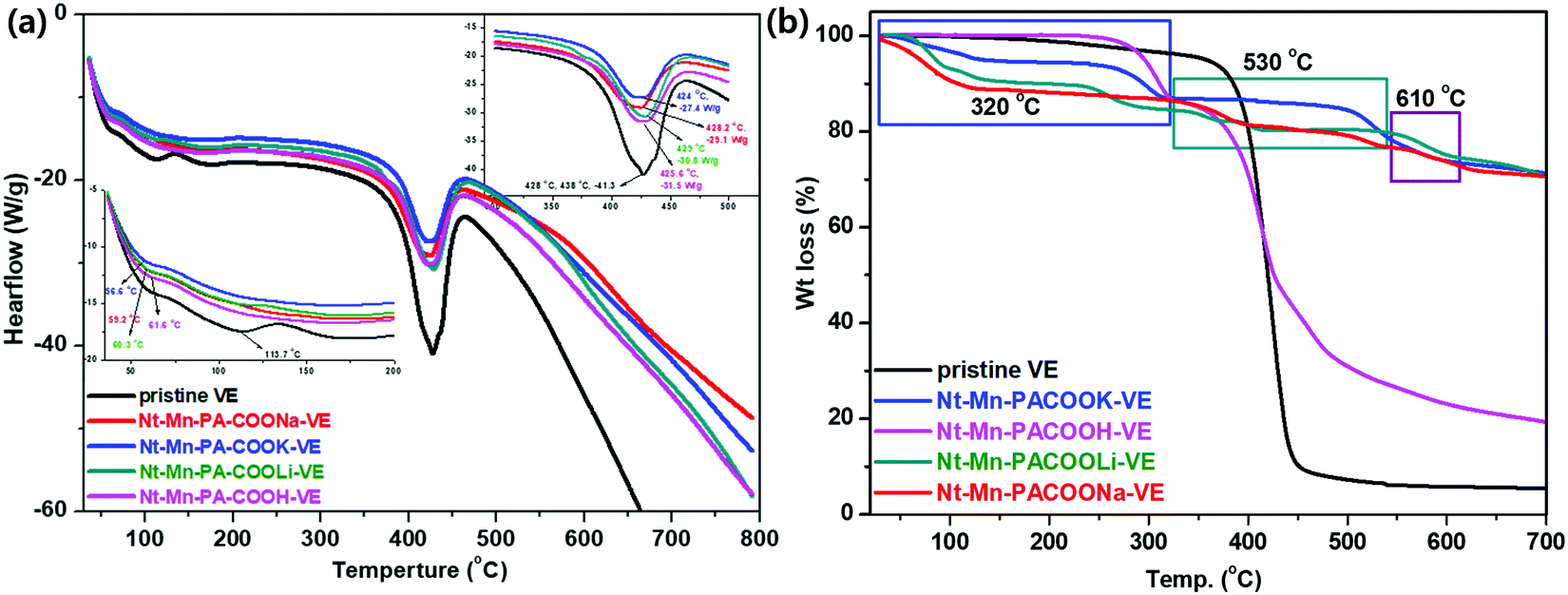 | ||
| Fig. 4 (a) DSC and (b) TGA analyses of 3D-networked Nt–Mn–PA–COOM–VE. | ||
Fig. 4b shows the TGA analysis of the Nt–Mn–PA–COOM–VE composites. The characteristic features of individual composites are illustrated in comparison with the pure VE composite. The thermogram of VE differs from that of the Nt–Mn–PA–COOM composites (i.e., M = Li, Na, and K) due to the availability of the nanofiller with carboxylate metal salts. Heat degradation for VE started at 390 °C (Tonset) to 481 °C (Tmax) and char residue at 700 °C for losses in wt% of 10, 30, and 94.5, respectively, owing to decomposition by the evolution of CO2, and aliphatic carbon chains. The Nt–Mn–PA–COOM–VE composites demonstrated independent decompositions for Nt–Mn–PA–COOH–VE, Nt–Mn–PA–COOLi–VE, Nt–Mn–PA–COONa–VE, and Nt–Mn–PA–COOK–VE, respectively, as shown in Table 2.
| Thermogravimetric data in nitrogen | Horizontal burning test data in open air | |||||
|---|---|---|---|---|---|---|
| Composite | T (onset) 10% [°C] | T (max) 30% [°C] | Residue at 700 °C [%] | Burning time (min) | Burning rate mm min−1 | After glow |
| VE | 390 | 481 | 5.5 | 6.2 | 11.9 | Yes |
| Nt–Mn–PA–COOH–VE | 283 | 450 | 19 | 7.7 | 9.7 | Yes |
| Nt–Mn–PA–COOLi–VE | 243 | 545 | 70 | 9.3 | 8.0 | Yes |
| Nt–Mn–PA–COONa–VE | 360 | 535 | 70 | 10.6 | 7.0 | Yes |
| Nt–Mn–PA–COOK–VE | 284 | 507 | 70 | 12.1 | 6.1 | Yes |
At the first stage between 50 °C and 322 °C, a reduced weight loss of 10% was recorded due to the removal of CO2 from the carboxylate chains. The second decomposition stage occurred from 428 °C and involved a weight loss of 51%, which ended with breaking of the naphthalene rings of Nt–Mn–PA–COOM. From 450 to 700 °C, the third stage of decomposition involved a weight loss of 30% from Nt–Mn–PA–COOH due to the decomposition of the triazine ring. This change might occur due to decomposition of the metal ion carboxylated chains in Nt–Mn–PA–COOM, such as COOH, COOLi, COONa, and COOK groups.
Next, the TGA thermograms of Nt–Mn–PA–COOM (i.e. Li, Na, and K) were found to be similar to each other due to the formation of salts. They underwent a three-stage decomposition. The first stage involved the loss of water and carboxylic acid salts, the second corresponded to the decomposition of aromatic and triazine rings, and the third stage involved the vaporization and elimination of volatile fragments. With the first stage between 50 °C and 450 °C, almost 10% preliminary weight loss occurred. The second stage had a 5% loss in weight just above 535 °C, which involved the aromatic naphthalene ring of Nt–Mn–PA–COOM. The third stage of decomposition, which ends at around 700 °C, involved a weight loss of approximately 29% from the triazine skeleton. Eventually, after the formation of Li, Na, and K salts of carboxylic acids, the structure of Nt–Mn–PA–COOM was thermally stable even at high temperatures. The TGA results suggested that after doping the Nt–Mn–COOM nanofillers, the thermal stability of the composites was significantly improved because the size of the metal ions in the carboxylic acids (i.e., H, Li, Na, and K) gradually increased, which hindered the burning intensity.
3.4. Cone colorimetry tests
Cone colorimetry measurements were performed to further study the fire-retardant performance of the as-prepared Nt–Mn–PA–COOM–VE composites. Characteristic burning parameters include the peak heat release rate (HRR). Fig. 5 shows the HRR curves of the flame-retardant Nt–Mn–PA–COOM–VE (R = H, Li, Na, and K) composites, respectively. In comparison with those of the flame-retardant VE composites, the curve of the neat VE composite presented the sharpest changes among all the measured samples. The HRR values reached were high compared with the pristine VE. This trend indicated that the ignited neat VER underwent violent burning, accompanied with a large heat release. Importantly, as the size of the metal ions as the dopant in the Nt–Mn–PA–COOH composite was increased, the HRR was gradually reduced due to the formation of chemical bonds between CO in the VE polymer chains, and free –OH and –NH in Nt–Mn–PA–COOM, which further improved the nucleation density of the HRR in the VE composite.
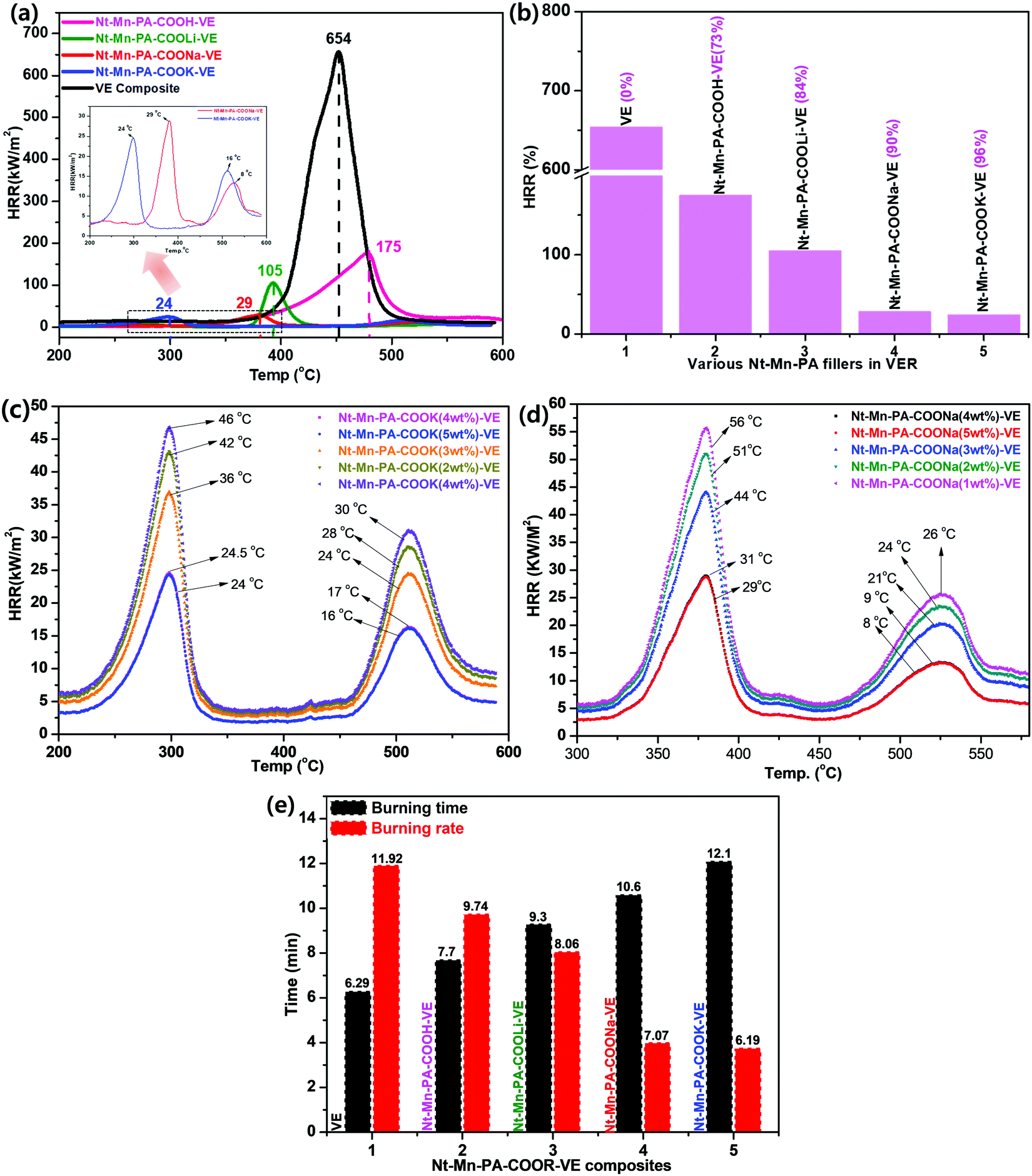 | ||
| Fig. 5 Heat release rate (HRR) analyses in 3D-networked Nt–Mn–PACOOM–VE (a and b), HRR of various wt% of nanofillers of Nt–Mn–PA–COOK–VE (c) and Nt–Mn–COONa–VE (d), and horizontal burning tests for 3D-networked Nt–Mn–PACOOM–VE composites (e). | ||
Fig. 5a shows that the HRR values of Nt–Mn–PA–COOK–VE and Nt–Mn–PA–COONa–VE were significantly reduced compared with the HRR values of pristine VE, and the Nt–Mn–PA–COOH–VE and Nt–Mn–PA–COOLi–VE composites. Besides, the HRR values of Nt–Mn–PA–COOK, and Nt–Mn–PA–COONa were lowered by approximately 27 and 25 times when compared with the HRR value of the pure VER composite due to the incorporation of Na and K metal ions, which made strong ionic chelation between the metal ions and the host VER polymer. In particular, micro calorimeter analysis of the Nt–Mn–PA–COOM–VE composites for the HRR were conducted for the flame retardancy of the as-prepared pure VE and Nt–Mn–PA–COOM–VE composites. A superior flame retardancy effect was observed in comparison with the pure VE composite. We tested three sets of composites for each experiment, for obtaining concurrent results, and produced the average results. The HRRs of VE and Nt–Mn–PA–COOM–VE (i.e., R = H, Li, Na, and K) composites were observed at 654 kW m−2, 175 kW m−2, 105 kW m−2, 29 kW m−2, and 24 kW m−2, respectively. The Nt–Mn–PA–COOK, and Nt–Mn–PA–COONa composites exhibited lower HRRs, and higher thermal stabilities compared with Nt–Mn–PA–COOLi, Nt–Mn–PA–COOH, and the pure VER composite. Fig. 5b shows HRRs of 96%, 90%, 84%, and 73% for Nt–Mn–PA–COOK, Nt–Mn–PA–COONa, Nt–Mn–PA–COOLi, and Nt–Mn–PA–COOH, respectively, as compared with pure VE. These results concluded that after doping with the nanofillers, the stability of the composites is increased and they exhibit a lower HRR.43
Next, we investigated the Nt–Mn–PA–COOK–VE and Nt–Mn–COONa–VE nanofiller dopant quantities until saturated HRR data were obtained using various wt% loadings. We tested the nanofiller dopants from 1 wt% to 5 wt%. The saturation point was observed at 4 wt% of Nt–Mn–PA–COOK–VE and Nt–Mn–COONa–VE. Next, we observed two different HRR curves obtained from Nt–Mn–PA–COOK (Fig. 5c) and Nt–Mn–COONa (Fig. 5d). Our nanofiller exhibited a 3D network structure with π–π stacking within the polymer network. In general, the HRR analysis exhibits a single decomposition curve but in our case we found two different curves coming from 250 °C to 350 °C, and from 460 °C to 560 °C, respectively, due to the decomposition of pristine VE and Nt–Mn–PA–COOM nanofillers. This kind of trend in decomposition is unique and significant.44
3.5. Horizontal burning test
During fire accidents, the material with a maximum tolerance to fire is called a flame retardant material. To estimate the flame retardancy properties of the novel Nt–Mn–PA–COOM–VE composites, a horizontal burning test was performed and the burning time and rate results are shown in Table 2. The Nt–Mn–PA–COOM–VE composites exhibited a high burning time and low burning rate due to the lack of flammability by the occupying metal ions, which are strongly anchored to the host polymer by metal chelation in ionic clusters, as shown in Fig. 5f. These ionic clusters strongly prevent oxygen from entering the polymer chains. The burning time and burning rate of all the polymer composites are shown in increasing order from 6.2 min to 12.1 min. The burning rate gradually decreased from 11.9 to 6.1 mm min−1 for VE and Nt–Mn–PA–COOM–VE (i.e., R = H, Li, Na, and K), respectively. The results showed that the novel Nt–Mn–PA–COOM as nanofillers exhibited significant flame retardancy properties.3.6. Morphologies of char residues
To investigate the doping concentration of Nt–Mn–COOM (i.e., R = H, Li, Na, and K) in the VE composite, the morphologies of Nt–Mn–COOM were studied and the results are shown in Fig. 6. The dispersions could be observed as white beads with a diameter of 500 nm. The metal size increased with the increasing atomic number of the metal. Fire resistance increased remarkably with the content of Nt–Mn–PA–COOM because the metal ions were strongly coordinated with the base VER organic matrices and formed a shield on the surface of the burning polymer to retard its further combustion. In particular, after the flame tests, the surface morphologies of the Nt–Mn–PA–COOM composites were revealed. Fig. 6a shows that cured Nt–Mn–PA–COOH (5 wt%) barely had any char residue after the flame test, whereas the amount of carbon residue in the flame-retardant VE thermosets evidently increased with the increasing metal size in Nt–Mn–PA–COOM (i.e., M = Li, Na, and K) due to the catalytic char formation effect of the metal ions contained as dopants on VE.40Fig. 6b shows the lattice-like porous network morphology of Nt–Mn–PA–COOLi with irregular holes due to the sizes of Li+ ion and H+ ion, which are more or less similar in the Nt–Mn–PA–COOH and Nt–Mn–PA–COOLi polymers. However, as the metal size increased in Fig. 6c and d the pores were filled by the Na and K because of the increasing size of the metal. In the order of Nt–Mn–PA–COOH < Nt–Mn–PA–COOLi < Nt–Mn–PA–COONa < Nt–Mn–PA–COOK–VE, the composites displayed higher flame retardancy. Additionally, the surface morphology gradually changed with the disappearance of pores, which were occupied by the metal ions, and a compact char-layer surface was formed. Such a compact structure is beneficial for isolating heat and hindering pyrolysis gas transport for the penetration of oxygen gas during the flame tests, thereby protecting the underlying matrix from further combustion.45 The changes in the char residue morphologies were consistent with the good flame retardancy of the Nt–Mn–PA–COOM–VEs, as discussed in the sections on the HRR tests.46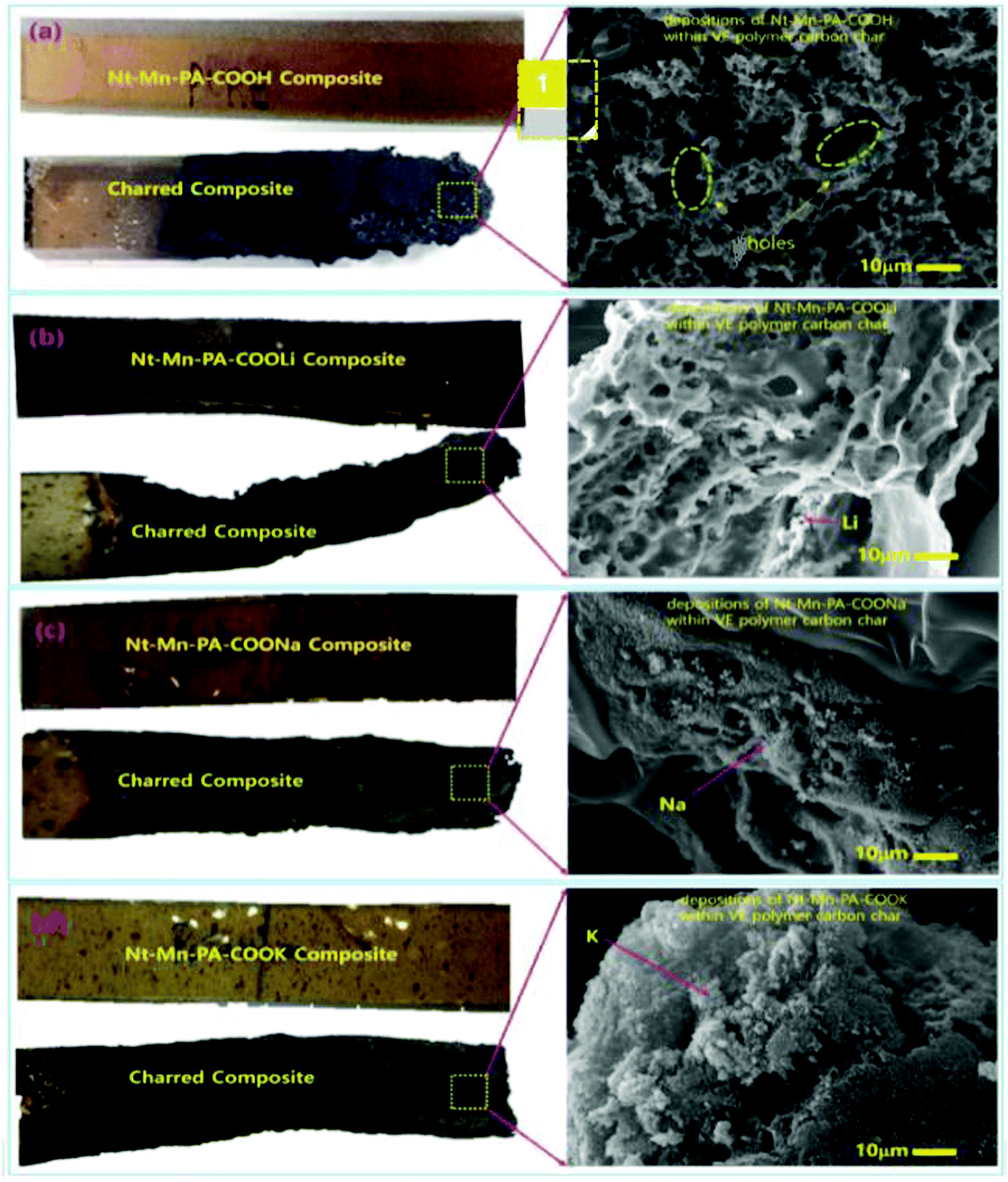 | ||
| Fig. 6 Digital images and magnified charred SEM images of Nt–Mn–PA–COOM composites (i.e., M = Li, Na, and K) before and after the flame tests. (a) Porous surface morphology with irregular pores; (b) Li metal depositions with the polymer network; (c) dense Na; and (d) K metal depositions were observed on surface. | ||
3.7. Mechanism of flame retardancy
The pyrolysis and combustion of polymer composites occur in numerous stages. The polymeric substrate heated by an external heat source is pyrolyzed with the generation of combustible fuel. Usually, only a part of this fuel is fully combusted in the flame by combining with a stoichiometric quantity of atmospheric oxygen. The other part remains and can be combusted by more forcing means, such as in the presence of a catalyst and by an excess of oxygen. Some of the released heat is fed back to the sample to continue the burning cycle of pyrolysis. The other part is lost to the environment. The polymer was decomposed into volatiles and combustibles by the pyrolysis process. The amount of gaseous products controls the flammability of the substrate. A flame-retardant reaction occurs when a condensed phase chemical mechanism alters the pyrolysis path of the substrate and substantially reduces the amount of gaseous combustibles, usually by favoring the formation of carbonaceous char and water. When the combustion is decreased with an increase in the amount of flame-retarding agent, the excess heat is released. In the gas-phase chemical mechanism, the amount of leftover combustible matter is constant but the heat released in the combustion decreases with an increase in the amount of flame-retarding agent. The flame-retarding moiety must be volatile and reach the flame in the gaseous form. Otherwise, it decays and provides the active fraction of its molecule to the gaseous phase. The char residue left over in the substrate has a lesser amount of active agent. In addition, the presence of the gas-phase active agent should not affect the configuration of the volatiles reaching the flame.47With the abovementioned results, the possible flame-retardant and smoke suppressant mechanisms are proposed in Fig. 7. The early decomposition of Nt–Mn–PA–COOM–VE instigated the generation of CO2 through the combustion of polymeric chains by degradation into monomers, including dehydration of the VE matrix as a protective char, as shown in Fig. 7a.48 During the combustion process, a protective char layer was formed and thermal decomposition of Nt–Mn–PA–COOM–VE into a naphthalene–melamine (Nt–Mn) skeletal structure was observed. In addition, Li, Na, and K ions can help to accelerate the decomposition of Nt–Mn–PA–COOM–VE and improve the char layer formation rate. An early formed intumescent char can act as a barrier that prevents heat and O2 from being transmitted into the polymer matrix, thereby hindering the combustible gases that are fed back into the flame zone and subsequently suppressing the release of smoke and toxic gases, as shown in Fig. 7b and c. Subsequently, Li, Na, and K ions not only played a role in the oxidation of CO, but also exhibited a good synergic effect in Nt–Mn–PA–COOM for improving the formation amount, intumescent degree, and compactness of the char.49,50
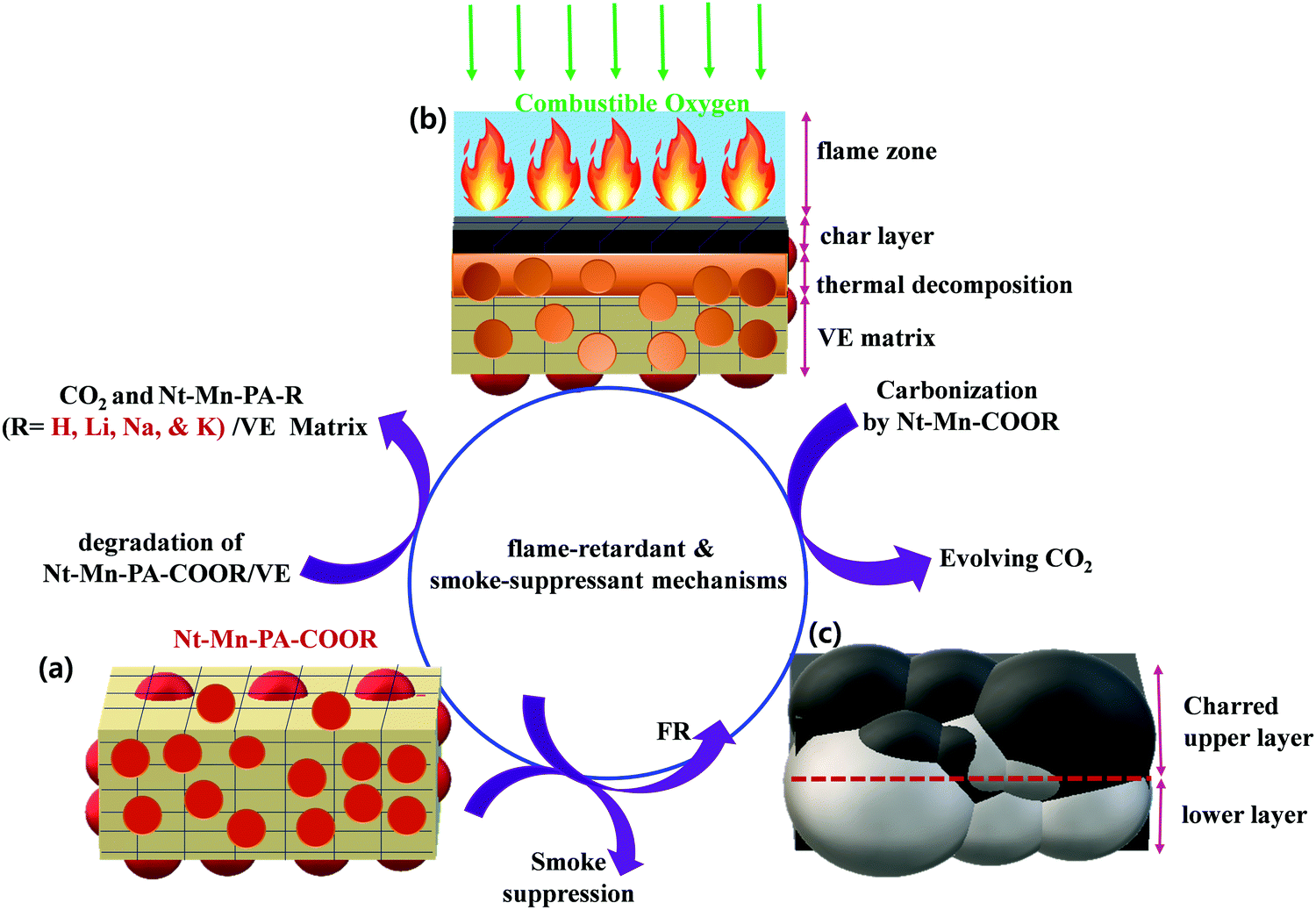 | ||
| Fig. 7 Flame retardancy mechanism using 3D-networked Nt–Mn–PACOOM–VE composites. | ||
4. Conclusion
In this study, we prepared a series of Nt–Mn–PA–COOM nanofillers doped in VE at various wt% ratios. The introduction of Nt–Mn–PA–COOM in VE exhibited excellent flame retardancy properties owing to the metal chelation effect with the base polymer chains. Mainly, the Nt–Mn–PA–COOM nanofillers were chemically bonded with the core VE polymer via the esterification reaction, thereby enhancing the endurance of the polymer chains. When the dopant metal ion size was increased, the flame retardancy property also increased. These significant results were obtained due to the chelation of metal ions with the host polymer, thereby forming metal ion clusters with strong chelation. These metal ion clusters strongly inhibited the penetration of elemental oxygen gas during burning. The HRR and horizontal tests for the novel nanofiller composites demonstrated superior flame retardancy properties with respect to the pure VE composite. All results showed that the comprehensive properties of flame retardant VE were best when the amount of Nt–MN–PA–COOK was 5 wt%. The novel Nt–Mn–PA–COOM nanofiller exhibited the maximum flame retardancy property of 96% (K), 90% (Na), 84% (Li), and 73% (H) when compared with pristine VER. Our work can be validated for the flame retardancy property of effective nanofillers for commercial applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) No. 2017R1A2B2011730, No. 2018R1A6A1A03024509 and 2019R1A2C1011113.References
- S. Jaiswal and B. Gaur, New trends in vinyl ester resins, Rev. Chem. Eng., 2014, 30, 567 Search PubMed.
- E. Can, E. Kınac and G. R. Palmese, Eur. Polym. J., 2015, 72, 129 Search PubMed.
- M. Sultania, J. S. P. Rai and D. Srivastava, Eur. Polym. J., 2010, 46, 2019 Search PubMed.
- M. Sultania, S. B. Yadaw, J. S. P. Rai and D. Srivastava, Mater. Sci. Eng., A, 2010, 527, 4560 Search PubMed.
- J. S. Arrieta, E. Richaud, B. Fayolle and F. Nizeyimana, Polym. Degrad. Stab., 2016, 129, 142 Search PubMed.
- A. C. Garay, L. T. Paese, J. A. Souza and S. C. Amico, Rev. Mater., 2015, 20, 64 Search PubMed.
- P. Li, X. Yang, Y. Yu and D. Yu, J. Appl. Polym. Sci., 2010, 92, 1124 Search PubMed.
- S. Huo, V. S. Chevali and C. A. Ulven, J. Appl. Polym. Sci., 2013, 128, 3490 Search PubMed.
- N. Agarwal, A. Singh, I. K. Varma and V. Choudhary, J. Appl. Polym. Sci., 2008, 108, 1942 Search PubMed.
- S. Grishchuk, N. Castellà, A. A. Apostolov and J. KargerKocsis, J. Compos. Mater., 2011, 46, 941 Search PubMed.
- A. M. Atta, S. M. El-Saeed and R. K. Farag, React. Funct. Polym., 2006, 66, 1596 Search PubMed.
- A. N. da Silva, G. S. Mori and J. R. M. d’Almeida, Mater. Res., 2015, 18, 121 Search PubMed.
- M. Shamsuddoha, L. P. Djukic, M. M. Islam, T. Aravinthan and A. Manalo, J. Compos. Mater., 2016, 51, 1605 Search PubMed.
- H. W. Cui, J. T. Jiu, S. Nagao, T. Sugahara, K. Suganuma, H. Uchida and K. A. Schroder, RSC Adv., 2014, 4, 15914 Search PubMed.
- B. K. Kandola, J. R. Ebdon and C. Zhou, React. Funct. Polym., 2018, 129, 111 Search PubMed.
- U. Javaid, Z. M. Khan, M. B. Khan, M. Bassyouni, S. M. S. AbdelHamid, M. H. Abdel-Aziz and S. W. ul Hasan, Composites, Part B, 2016, 91, 257 Search PubMed.
- L. Ferry, J. M. L. Cuesta, C. Chivas, G. M. W. Hoy and H. Dvir, Polym. Degrad. Stab., 2001, 74, 449 Search PubMed.
- H. S. Patel and B. K. Patel, Int. J. Polym. Mater., 2009, 58, 312 Search PubMed.
- X. S. Chen, Z. Z. Yu, W. Liu and S. Zhang, Polym. Degrad. Stab., 2009, 94, 1520 Search PubMed.
- C. Kaynak and N. A. Isitman, Polym. Degrad. Stab., 2011, 96, 798 Search PubMed.
- R. Kheyrabadi, H. Rahmani and S. H. M. Najafi, Polymer, 2017, 30, 202 Search PubMed.
- S. Y. Lu and I. Hamerton, Prog. Polym. Sci., 2002, 27, 1661 Search PubMed.
- L. Zang, S. Wagner, M. Ciesielski, P. Müller and M. Döring, Polym. Adv. Technol., 2011, 22, 1182 Search PubMed.
- Z. Bai, L. Song, Y. Hu and R. K. K. Yuen, Ind. Eng. Chem. Res., 2013, 52, 12855 Search PubMed.
- L. P. Gao, D. Y. Wang, Y. Z. Wang, J. S. Wang and B. Yang, Polym. Degrad. Stab., 2008, 93, 1308 Search PubMed.
- S. Yang, J. Wang, S. Huo, M. Wang and L. Cheng, Ind. Eng. Chem. Res., 2015, 54, 7777 Search PubMed.
- S. Yang, J. Wang, S. Huo, J. Wang and Y. Tang, Polym. Degrad. Stab., 2016, 126, 9 Search PubMed.
- S. Yang, J. Wang, S. Huo, M. Wang, J. Wang and B. Zhang, Polym. Degrad. Stab., 2016, 128, 89 Search PubMed.
- Ç. E. Emrah and A. Güngör, Polym. Degrad. Stab., 2013, 98, 927–933 Search PubMed.
- J. Liu, Y. Gao, F. Wang, D. Li and J. Xu, J. Mater. Sci., 2002, 37, 3085–3088 Search PubMed.
- S. Vural and T. Seçkin, Mater. Res. Bull., 2009, 44, 369–376 Search PubMed.
- Z. Li, J. Zhou, R. Xu, S. Liu, Y. Wang, P. Li, W. Wu and M. Wu, Chem. Eng. J., 2016, 287, 516–522 Search PubMed.
- R. K. Cheedrala, S. Vijaya and J. W. Park, Synth. Commun., 2009, 39, 1966–1980 Search PubMed.
- R. K. Cheedrala, G. H. Kim, S. Cho, J. H. Lee, J. Kim, H. K. Song, J. Y. Kim and C. Yang, J. Mater. Chem., 2011, 21, 843–850 Search PubMed.
- R. K. Cheedarala, E. J. Park, K. Kong and Y. B. Park, Int. J. Heat Mass Transfer, 2016, 100, 396–406 Search PubMed.
- R. K. Cheedarala, L. C. Duy and K. K. Ahn, Nano Energy, 2018, 44, 430–437 Search PubMed.
- R. K. Cheedarala, A. N. Parvez and K. K. Ahn, Nano Energy, 2018, 53, 362–372 Search PubMed.
- R. K. Cheedarala, M. Shahriar, J. H. Ahn, J. Y. Hwang and K. K. Ahn, Nano Energy, 2019, 65, 104017 Search PubMed.
- D. B. Thomas and H. Satoshi, Nat. Rev. Mater., 2018, 3, 431–440 Search PubMed.
- Y. Arash, W. H. H. Günther, T. M. Scott and A. G. Olivia, PLoS One, 2020, 15, e0234774 Search PubMed.
- A. Vahid and A. Alireza, RSC Adv., 2016, 6, 22331–22340 Search PubMed.
- R. K. Cheedarala and J. I. Song, RSC Adv., 2019, 9, 31735–31746 Search PubMed.
- R. K. Cheedarala, C. D. Kee and I. K. Oh, Adv. Funct. Mater., 2013, 23, 6007–6018 Search PubMed.
- R. K. Cheedarala, J. J. Jeon, C. D. Kee and I. K. Oh, Adv. Funct. Mater., 2014, 24, 6005–6015 Search PubMed.
- M. N. Prabhakar, R. S. Attaur and J. I. Song, Carbohydr. Polym., 2017, 168, 201–211 Search PubMed.
- H. Duan, S. Ji, T. Yin, X. Tao, Y. Shen and H. Ma, J. Appl. Polym. Sci., 2019, 47997 Search PubMed.
- S. Yang, J. Wang, S. Huo, L. Cheng and M. Wang, Polym. Degrad. Stab., 2015, 115, 63 Search PubMed.
- M. Gao, H. Wang, Y. Wang, T. Shen and W. Wu, J. Vinyl Addit. Technol., 2016, 22, 350 Search PubMed.
- R. K. Cheedarala and J. I. Song, Int. J. Heat Mass Trasfer, 2020, 162, 120391 Search PubMed.
- C. Venkata, R. K. Cheedarala and J. I. Song, J. Appl. Polym. Sci., 2020 DOI:10.1002/app.50227.
| This journal is © The Royal Society of Chemistry 2021 |