
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAtomic layer deposition of nano-scale molybdenum sulfide within a metal–organic framework for highly efficient hydrodesulfurization†
Lei
Yu
ab,
Wen-Gang
Cui
ab,
Qiang
Zhang
ab,
Zhuo-Fei
Li
ab,
Yan
Shen
ab and
Tong-Liang
Hu
 *abc
*abc
aSchool of Materials Science and Engineering, Tianjin Key Laboratory of Metal and Molecule-Based Material Chemistry, National Institute for Advanced Materials, Nankai University, Tianjin 300350, China. E-mail: tlhu@nankai.edu.cn
bTianjin Key Lab for Rare Earth Materials and Applications, Nankai University, Tianjin 300350, China
cState Key Laboratory of Coordination Chemistry, Nanjing University, Nanjing 210023, China
First published on 13th January 2021
Abstract
Molybdenum sulfide (MoS2) exhibits extremely encouraging performance in applications of hydrodesulfurization (HDS). However, due to its low active sites, incomplete sulfidation and accounting for more than 70% of the quality of the supported catalyst for alumina, without playing a catalytic role, the γ-Al2O3 supported catalyst has low reactivity and high cost. Here, we selectively functionalized MoS2 on the nodes of a metal–organic framework (MOF, NU-1000) through atomic layer deposition (ALD) and explored its catalytic performance in the HDS of dibenzothiophene. The AIM-60 (AIM = ALD in a metal–organic framework) catalyst obtained was characterized by X-ray diffraction (XRD), N2 adsorption–desorption, thermogravimetric analysis (TG), X-ray photoelectron spectroscopy (XPS), Fourier transform infrared spectroscopy (FT-IR), and scanning and transmission electron microscopy (SEM and TEM). The conversion rate of the AIM-60 material is 86.7% at 250 °C and 3 MPa, which is much better than that of traditional MoS2/γ-Al2O3 (8.6%) with the same loading. In addition, the AIM-60 catalyst shows a preference for the direct desulfurization (DDS) pathway, and it can directly convert dibenzothiophene to biphenyl, with excellent stability in real-life tests. The results indicate that AIM-60 with high activity and high stability represents a new class of HDS catalyst.
Introduction
Over the years, the burning of sulfur compounds has accelerated environmental degradation and had a serious impact on human life.1,2 Desulfurization has become one of the major problems for the petroleum industry, especially in the context of strict legislation and increasing environmental awareness.3 In China, the United States and other countries and regions, the sulfur content in fuel is strictly limited to less than 10 μg g−1.4–7 Although some methods, such as oxidative desulfurization, adsorption desulfurization, and biodesulfurization have been reported in the literature, hydrodesulfurization (HDS) is still the most important method in the petroleum industry.8–10 Nevertheless, the key point is the development of an efficient hydrodesulfurization catalyst. In terms of HDS catalysts, Co–Mo–S or Ni–W–S loaded on γ-alumina is one of the most common catalytic systems, in which, generally, cobalt or nickel act as an auxiliary agent.11–15 At the same time, alumina accounts for more than 70% of the quality of the supported catalyst, without playing a catalytic role.16 Therefore, in the search for new methods to achieve good dispersion of active metals, we turned to metal–organic framework materials (MOFs),17–20 as a novel catalytic material consisting of metal–inorganic nodes and organic linkers, which have aroused great interest in other applications.21–23 A small portion of them exhibit unique thermal stability and relatively large pore size, which are critical for catalytic and energy conversion applications.24,25 There is increasing interest in the use of atomic layer deposition (ALD) to incorporate single-site metal clusters into MOF.26–29 Of particular interest is the introduction of metal atoms into nanomaterials at the atomic scale to prepare materials with a uniform distribution of active sites. However, due to the instability of most MOFs in a hydrogen sulfide atmosphere, the fixation of metal sulfides on MOFs is rare.30,31 In recent years, zirconium-based MOF NU-1000 has attracted a lot of attention because of its strong resistance to hydrogen sulfide.32 NU-1000 is especially suitable for use as a carrier for metal sulfide deposition for the following reasons: (1) the available hydroxy/water ligands on the Zr6 node in NU-1000 can be combined with an ALD precursor,33–36 (2) large mesoporous channels can satisfy the diffusion of ALD precursors and reaction substrates and promote the effective binding of catalysts and substrates,37 (3) high stability under the reaction conditions required for desulfurization applications,32 (4) with high specific surface area, more active centers could be obtained, and the adjustability of the structure may provide synergistic catalytic activities.In order to develop a high-performance hydrodesulfurization catalyst, herein we incorporate a highly stable and dispersed molybdenum disulfide within NU-1000 by the ALD method. The HDS of dibenzothiophene (DBT) was selected as the model reaction to prove the special properties of as-prepared AIM-X (X = 30, 40, 50, 60, 70, 80, 90, 100), where X represents the number of ALD cycles. The physicochemical properties and hydrodesulfurization activity of these catalysts were thoroughly studied. The conversion rate of AIM-60 is 86.7% at 250 °C and 3 MPa, which is much higher than that of MoS2/γ-Al2O3, and AIM-60 prefers the direct desulfurization path. In addition, the conversion rate remains basically unchanged after 20 h of circulation.
Experimental section
Materials
All reagents are analytically pure and were used directly without further purification. Zirconium tetrachloride (ZrCl4), dibenzothiophene (C12H8S), 4,6-dimethyldibenzothiophene (C14H12S), hexacarbonyl molybdenum (Mo(CO)6), benzoic acid (C6H5COOH), 1,3,6,8-terakis(p-benzoic acid)-pyrene (H4TBAPy), ammonium molybdate ((NH4)6Mo7O24·4H2O), acetone (CH3COCH3), hydrochloric acid (HCl), hexane (CH3(CH2)4CH3) (chromatographic pure) and ethanol (C2H5OH) were purchased from Sigma-Aldrich. The gases employed were N2, 2% H2S in N2, and 10% H2 in Ar.Synthesis of NU-1000
NU-1000 was synthesized and activated according to previous work with slight modification.34,36 In short, an autoclave (100 mL) was charged with ZrCl4 (420 mg, 1.80 mmol), benzoic acid (16.2 g, 132.6 mmol) and 48 mL of N,N-dimethylformamide (DMF), which was sonicated for 30 min. The clear solution was subsequently incubated in an 80 °C oven for 1 h. After cooling down to room temperature, H4TBAPy (240 mg, 0.36 mmol) was added to this solution and the yellow suspension was sonicated for 20 min, then placed in a 120 °C oven for 48 h. Single yellow crystals were isolated via centrifugation (9000 rpm, 5 min) and washed with DMF twice. Then they were mixed with 72 mL of DMF and 3 mL of 8 M HCl. This mixture was heated in a 100 °C oven for 24 h. After cooling down to room temperature, the sample was washed twice with DMF to remove HCl impurity and washed with acetone twice and then soaked in acetone overnight. Before using, NU-1000 was filtered and activated at 80 °C under vacuum for 12 h.Synthesis of AIM-X
ALD of MoS2 was conducted on an ALD instrument (Fig. S1, ESI†). In a typical procedure, activated NU-1000 (about 80 mg) was allowed to equilibrate for 2 h in the ALD sample chamber at 170 °C with 25 mL min−1 N2 flow. Samples were prepared with 30, 40, 50, 60, 70, 80, 90, 100 ALD cycles, where each ALD cycle consisted of multiple Mo precursor pulses, followed by multiple H2S pulses. The prepared samples were recorded as AIM-30, AIM-40, AIM-50, AIM-60, AIM-70, AIM-80, AIM-90, and AIM-100, respectively.Synthesis of MoS2-Al
MoS2-Al was prepared in much the same way as AIM-60, except for using γ-Al2O3 instead of NU-1000.Synthesis of MoS2/γ-Al2O3
For comparison, MoS2/γ-Al2O3 was also prepared by the incipient wetness coimpregnation method, according to the previous literature.38 Typically, ammonium molybdate (50.8 mg) was added to deionized water (6.50 mL) and stirred until it was dissolved. γ-Al2O3 (5.00 g) was quickly added into the above solution, evenly stirred, sealed with plastic wrap, and sonicated for 15 min. The solution was kept at room temperature for 12 h, then dried in a 110 °C oven for 10 h, and finally calcined in a muffle furnace at 500 °C for 2 h. The obtained sample was first reduced in 10% H2 in Ar and then presulfided in 2% H2S in N2, as shown in Fig. S2 (ESI†). The final sample obtained was labeled MoS2/γ-Al2O3.Synthesis of MoS2/γ-Al2O3 (H)
MoS2/γ-Al2O3 (H) was prepared in much the same way as MoS2/γ-Al2O3, except it was presulfided in 2% H2S in N2 at 360 °C for 4 h.Hydrodesulfurization of dibenzothiophene
The HDS of DBT was carried out in a 100 mL high-pressure reactor. Typically, AIM-X catalyst (0.10 g) was loaded in the reactor. A 15 mL n-hexane solution with 0.58 wt% DBT (sulfur content of 1000 μg g−1) was poured into the reactor. The HDS reaction was performed under the follow conditions: pressure of 3.0 MPa, temperature of 250 °C, stirring rate of 600 rpm, reaction time of 4 h. A final pressure range between 5.5 and 6.5 MPa inside the reactor was reached. The DBT contents before and after the reaction were measured on a Shimadzu GC-2014C gas chromatograph, equipped with a DB-WAX capillary column (length 30 m, diameter 0.25 mm) and an auto injector. The DBT conversion was calculated according to the following equation:where Δc is the DBT conversion, and c0 and c1 are DBT concentration before and after the reaction, respectively.
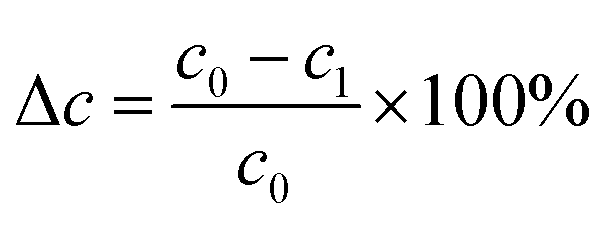
According to the literature,3,39,40 the HDS of DBT is considered to follow a quasi-first-order kinetic model, and the reaction rate constant (kHDS, mol g−1 h−1) could be calculated with the following equation:
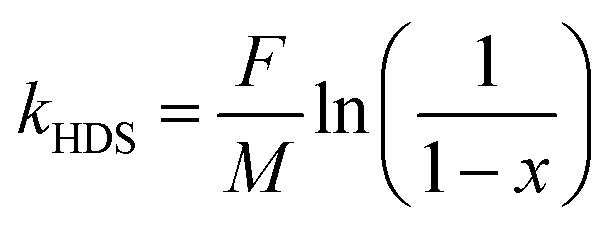
The HDS reaction was performed in succession using the same catalyst to evaluate the recyclability of the AIM-X catalyst. After each HDS reaction, the AIM-X catalyst was isolated via centrifugation (9000 rpm, 5 min), washed twice with ethanol, and dried in a vacuum oven at 60 °C for 3 h.
Method and instruments
Powder X-ray diffraction (PXRD) patterns were obtained on an X-ray diffractometer (Rigaku MiniFlex600) with Cu Kα radiation. Nitrogen sorption isotherms were collected on a Micrometrics ASAP 2020M volumetric gas adsorption analyzer at 77 K. Before measurement, samples were degassed at 150 °C for 10 h. The surface morphology of the samples was analyzed by scanning electron microscopy (SEM) (FEI Nanosem 430, Hitachi S-4800), energy dispersion spectra and field emission transmission electron microscopy (TEM) (FEI-Tecnai, G2F-20). The elemental contents in the samples were collected on an ICP-OES spectrometer. Thermogravimetric analysis (TGA) data were collected by a thermogravimetric analyzer (TG-8121), at a test temperature is 30–600 °C with a heating rate of 10 °C min−1 under Ar. The surface composition and chemical status were studied using X-ray photoelectron spectroscopy (XPS) employing a ThermoFisher EscaLab 250Xi spectrometer with Al Kα (1486.6 eV). The calibration of the spectra binding energy was performed with the C1s peak of the aliphatic carbons at 284.8 eV. Fourier transform infrared (FT-IR) spectra were measured on a Bruker TENSOR 27 FT-IR spectrometer at 273 K and the spectra were collected in the spectral window of 1000–4000 cm−1 at a resolution of 1 cm−1.Results and discussion
Synthesis and characterization of AIM-X
Using Mo(CO)6 and H2S as molybdenum source and sulfur source, respectively, the synthesized NU-1000 was functionalized by ALD. One ALD MoS2 cycle consists of four steps: Mo(CO)6 pulse, N2 purging, H2S pulse, and N2 purging (Fig. S3, ESI†). Due to the fact that the AIM-60 catalyst had the highest catalytic performance in the HDS experiment of DBT, a series of characterizations was carried out with AIM-60 as the representative. The synthesized NU-1000 and AIM-60 were firstly characterized by PXRD. As shown in Fig. 1, the PXRD pattern of AIM-60 confirms that NU-1000 maintains its crystallinity after molybdenum sulfide functionalization. Due to the low Mo loading in the AIM-60 catalyst, there is no characteristic peak of molybdenum species in the PXRD pattern. For comparison, we also synthesized γ-Al2O3 supported catalysts according to the literature (Fig. S4, ESI†).38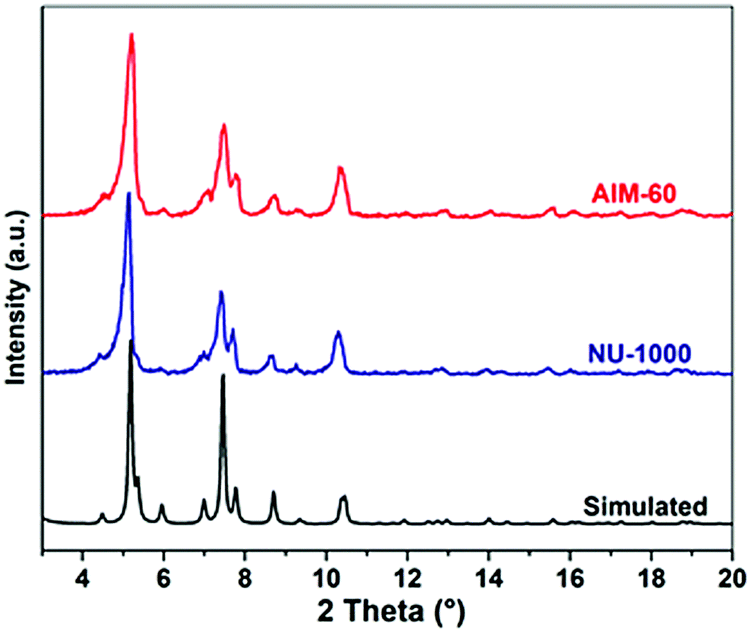 | ||
| Fig. 1 PXRD patterns of AIM-60, NU-1000, and the simulated pattern of NU-1000 from crystallographic data. | ||
The pore structure of the synthesized AIM-60 was analyzed using a nitrogen physical adsorption and desorption isotherm. As shown in Fig. 2a, the isotherm of as-synthesized NU-1000 has obvious H1-type hysteresis loops in the relative pressure range of 0.2–0.3, indicating the existence of an obvious mesoporous structure. After incorporation of MoS2, the hysteresis loop moved to a smaller pressure range, indicating that the diameter of the mesopore of NU-1000 decreased and the adsorption amount also decreased significantly. When introducing MoS2 into Al2O3 by the impregnation method, its specific surface area was also significantly reduced. From the pore size distribution in Fig. 2b, the most probable pore size distribution of the investigated samples decreases in the order of NU-1000 > AIM-60, and these declines are attributed to deposition of active metals. Details of the changes in pore diameter distribution, specific surface area and ICP-OES results are summarized in Table 1. By calculation from the ICP-OES data, the S/Mo atomic ratio of the synthesized AIM-60 is close to 2, which is proof of the formation of MoS2. The specific surface area and the pore diameter of AIM-60 are much higher than those of MoS2/γ-Al2O3 with the same loading, indicating that AIM-60 has a better adsorption effect and thus could improve its HDS catalytic activity. Fig. S5 (ESI†) shows the isotherms of AIM-30, AIM-60 and AIM-90 in nitrogen adsorption and desorption. According to the IUPAC classification, all of the isotherms exhibit type IV characteristic. There are significant hysteresis loops in all the isotherms of AIM-30, AIM-60 and AIM-90, indicating a mesoporous nature. The ICP-OES results for AIM-X are shown in Fig. S6 (ESI†).
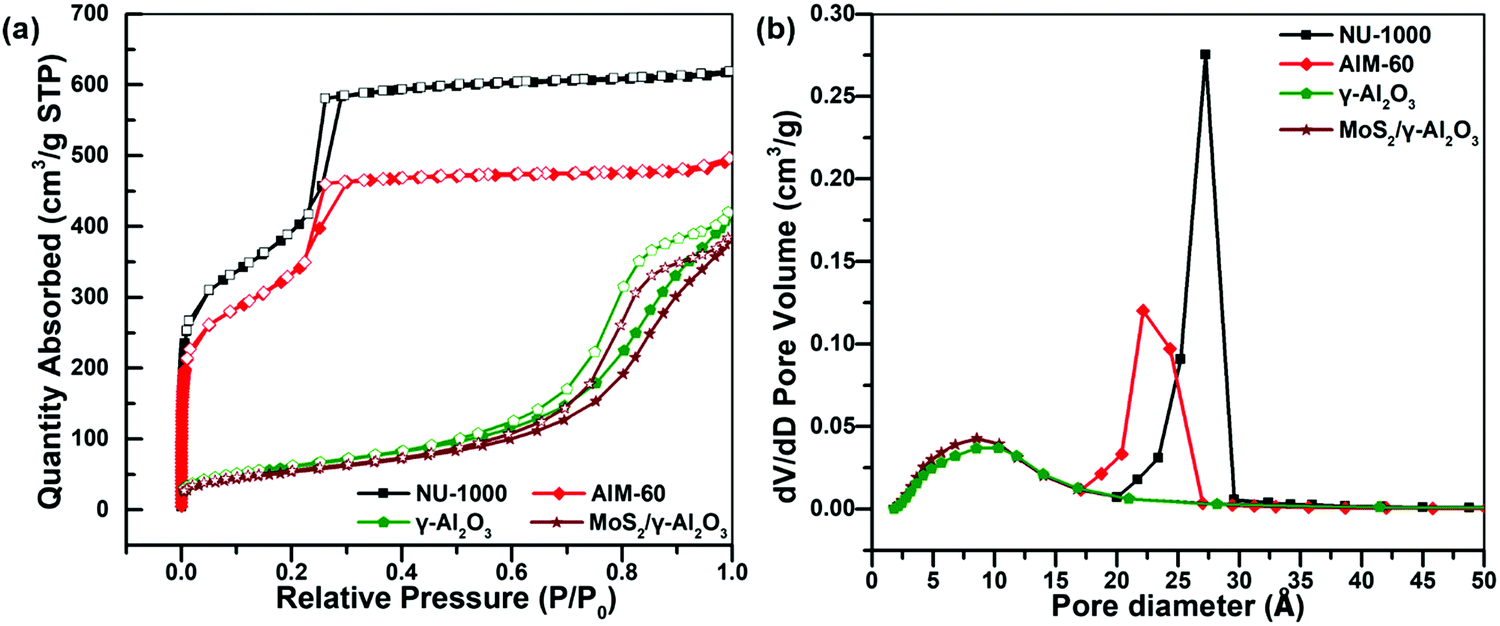 | ||
| Fig. 2 (a) N2 physical adsorption–desorption isotherms of the synthesized samples at 77 K. (b) Pore size distribution of the synthesized samples. | ||
Through infrared spectrum analysis, the binding sites of MoS2 and NU-1000 in the catalyst were revealed, and the obtained spectrum is shown in Fig. S7 (ESI†). The characteristic bands detected at 3673 and 3655 cm−1 are attributed to node bridges in the combination of μ3-OH, terminal OH and terminal OH2 groups, consistent with previous reports on NU-1000.33–36 Compared with NU-1000, the peak value of AIM-60 at 3674 cm−1 was significantly reduced. These data seem to confirm the hypothesis that the growth of molybdenum sulfide initially occurred by reacting with the hydroxide and water ligands of the Zr6 node in NU-1000. In addition, according to the pore size distribution (Fig. 2b), the pore size is reduced from 27 to 24 Å, which indicates the growth of molybdenum sulfide in NU-1000.
In order to study the morphology and particle size distribution of NU-1000, AIM-60, γ-Al2O3 and MoS2/γ-Al2O3, we conducted SEM analysis and the results obtained are shown in Fig. 3. The results clearly show that NU-1000 has clear spindle form and γ-Al2O3 is a block-shaped crystal. After adding a relatively low MoS2 content, the surface of the AIM-60 and MoS2/γ-Al2O3 samples became rough, and the particle size did not change significantly. Generally, a larger surface area can achieve better dispersibility. The particle size distribution of the AIM-60 catalyst was more uniform than that of the corresponding MoS2/γ-Al2O3. And the N2 physical adsorption analysis proved that AIM-60 has a significantly larger surface area than MoS2/γ-Al2O3. In addition, the SEM-EDS curve in Fig. S8 (ESI†) clearly shows that NU-1000 was successfully modified by MoS2. A typical TEM image of AIM-60 is shown in Fig. 3e, and the corresponding EDX spectra demonstrate that AIM-60 contains uniformly distributed elements of Zr, Mo, and S (Fig. 3f–h). The overlap between Mo and S indicates the formation of a metallic sulfide, rather than a separate mixture of Mo and S. As shown in the inset of Fig. 3i, the atomic ratio of Mo/S is calculated to be approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, which is related to the stoichiometric composition of MoS2.
:2, which is related to the stoichiometric composition of MoS2.
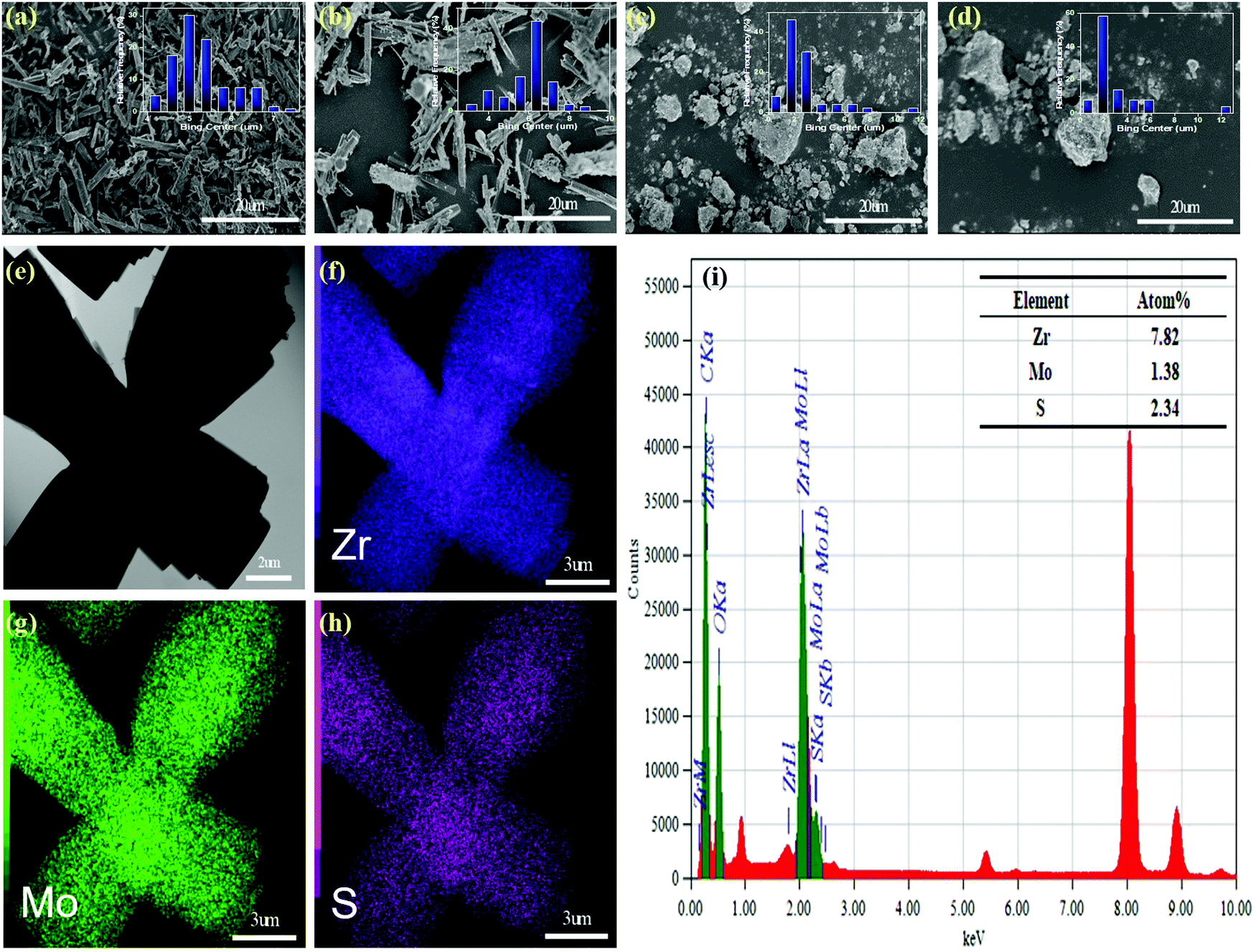 | ||
| Fig. 3 SEM images of the as-prepared NU-1000 (a), AIM-60(b), γ-Al2O3(c) and MoS2/γ-Al2O3. (d) TEM image of the as-prepared AIM-60 (e), and the corresponding elemental mappings of (f) Zr, (g) Mo, and (h) S, EDX spectra (i), and atomic percentage (inset). | ||
In order to investigate the thermal stability of the synthesized catalysts, TGA was carried out in an Ar atmosphere. As shown in Fig. 4a, the TG curves of AIM-60 and NU-1000 are similar, and they decompose above 400 °C, showing that NU-1000 has excellent thermal stability, and it can maintain a relatively complete skeleton structure at 400 °C. However, when the temperature exceeded 400 °C, the mass of the catalyst began to decline rapidly, which may be caused by the collapse of the skeleton of NU-1000 and the destruction of the organic ligands. Significantly, AIM-60 had a residual ash mass of 39.17% at 600 °C, while NU-1000 had a residual ash mass of 37.20%. The difference in mass fraction might be caused by MoS2 deposition in AIM-60 relative to NU-1000. After thermogravimetric analysis, as shown in Fig. 4b, the PXRD patterns of AIM-60 and NU-1000 showed the characteristic peaks of ZrO2 at 30.1°, 34.9°, 50.2°, 59.7°, 62.6° and 73.9° (ZrO2, PDF#49-1642).
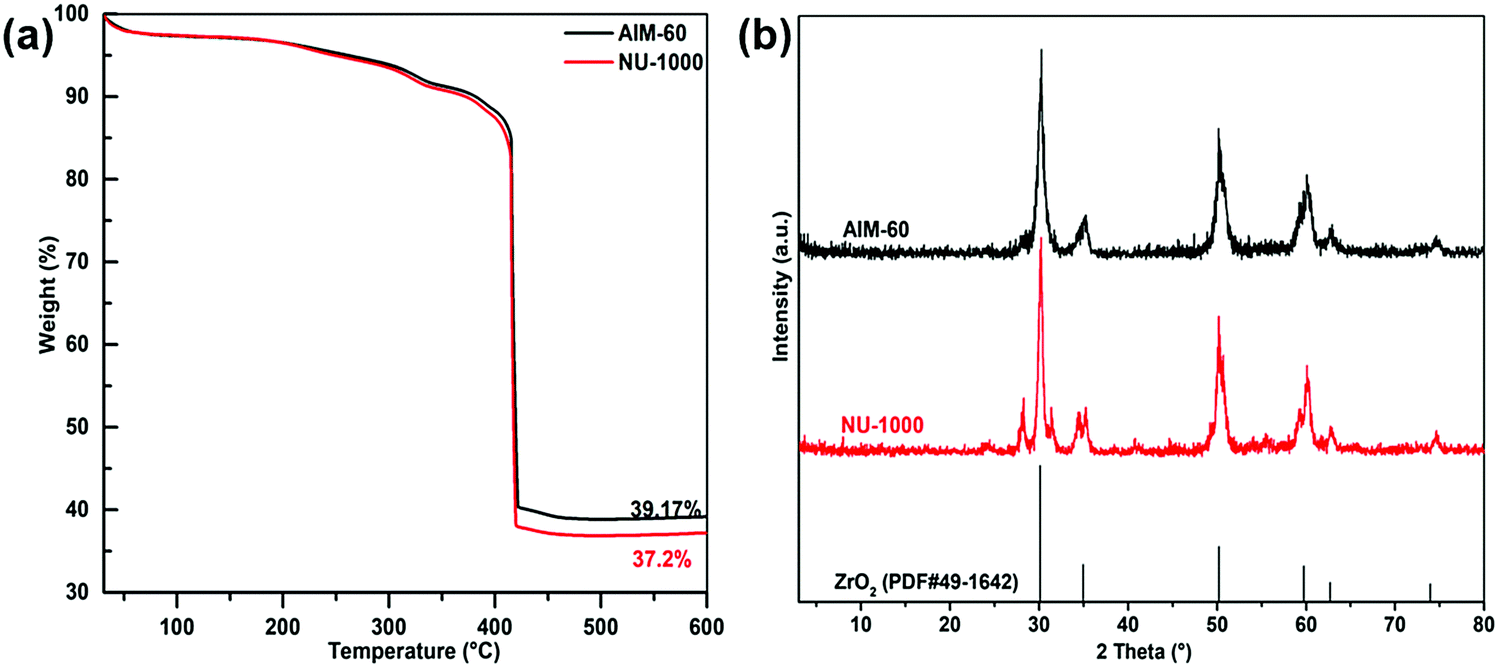 | ||
| Fig. 4 (a) Thermogravimetric curves for NU-1000 (red) and AIM-60 (black) in Ar. (b) PXRD patterns of AIM-60 and NU-1000 after thermogravimetric treatment. | ||
In order to better understand the chemical state, sulfurization degree (SD) and surface composition of the active metal of the catalysts, XPS analysis of AIM-60 and MoS2/γ-Al2O3 was conducted. As shown in Fig. 5, for S2p, peaks at around 162.28 eV and 163.43 eV can be assigned to S2− and some terminal S22−, peaks at around 164.58 eV and 165.88 eV can be attributed to the bridging S22−, and the peak of SO42− is located near 169.3 eV. During the experiment, S2− was inevitably oxidized to form S22− and SO42− due to contact with air.41–43 By Gaussian fitting, the Mo 3d spectrum can be decomposed into two double peaks of the spin orbit, namely Mo 3d5/2 and Mo 3d3/2. The binding energies of Mo 3d5/2 and Mo 3d3/2 in the MoS2 (Mo4+) phase are 228.0 eV and 232.0 eV, respectively. The binding energies of Mo 3d5/2 and Mo 3d3/2 of MoOxSy (Mo5+) mesophase are 230.9 eV and 234.2 eV, respectively. The binding energies of Mo 3d5/2 and Mo 3d3/2 in MoO3 (Mo6+) phase are 232.8 eV and 235.8 eV, respectively. The SD of a Mo-based sulfide catalyst is defined as the atomic ratio of Mo4+ to the sum of Mo4+, Mo5+ and Mo6+, wherein Mo4+, Mo5+ and Mo6+ represent MoS2 (Mo4+), MoOxSy (Mo5+) and MoO3 (Mo6+) phases, respectively.44,45 The XPS fitting results in Table 2 show that the Mo SDs of AIM-60 and MoS2/γ-Al2O3 are 40.82% and 7.22%, respectively, suggesting that AIM-60 should show higher HDS activity.
 | ||
| Fig. 5 XPS spectra of AIM-60 and MoS2/γ-Al2O3 catalysts: S 2p spectra deconvolution of AIM-60 (a) and MoS2/γ-Al2O3 (c), Mo 3d spectra deconvolution of AIM-60 (b) and MoS2/γ-Al2O3 (d). | ||
Catalytic performances of AIM-X catalysts
Studies have shown that among the different S-containing molecules in feedstocks, dibenzothiophene is considered a very difficult molecule to desulfurize.3,40 Therefore, the hydrodesulfurization reaction of dibenzothiophene is often used as a model reaction when evaluating the catalytic performance of hydrodesulfurization catalysts. Considering that the addition of an appropriate amount of MoS2 can regulate the physical and chemical properties of the catalyst, we prepared an AIM-X series of catalysts with different deposition cycles through ALD. Here, the HDS activities of the investigated AIM-X series catalysts and MoS2/γ-Al2O3 were evaluated at 180, 200, 230 and 250 °C and the DBT conversions are shown in Fig. 6.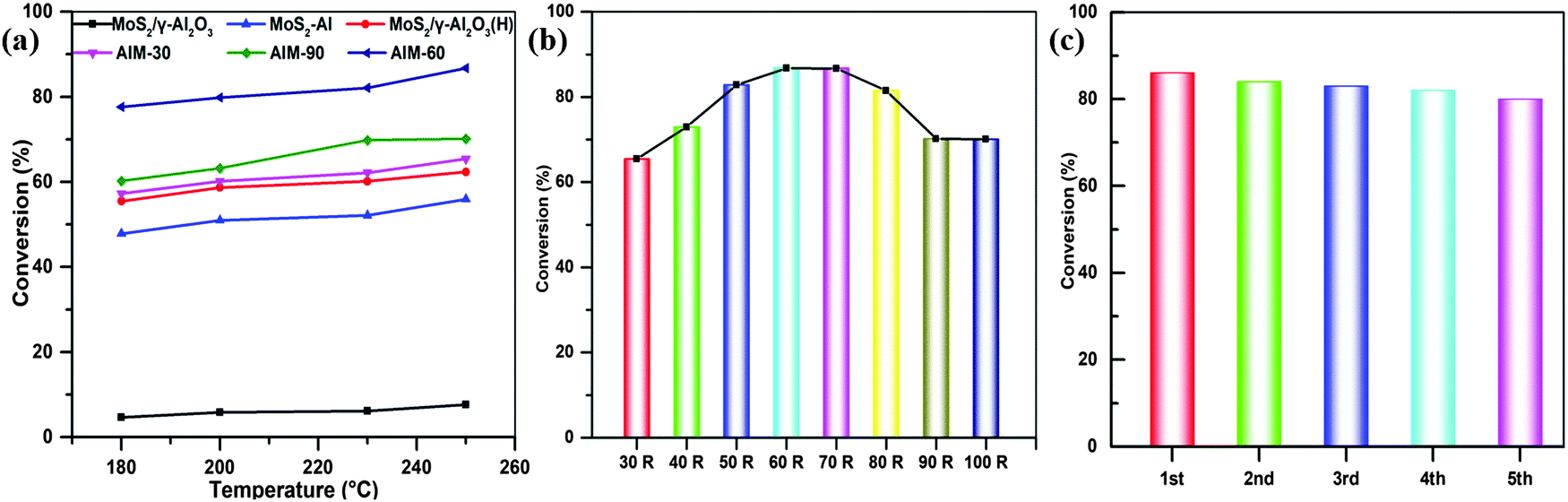 | ||
| Fig. 6 (a) Dibenzothiophene conversions over the investigated catalysts at different reaction temperatures. (b) HDS activity comparison between different AIM-X catalysts at 250 °C. (c) Reusability of the AIM-60 catalyst at 250 °C. | ||
As shown in Fig. 6a, the conversions of DBT clearly show that the activities of the investigated catalysts increased with an increase in temperature. At all temperatures investigated, the conversions of the catalysts are in the order of MoS2/γ-Al2O3 < MoS2-Al < MoS2/γ-Al2O3 (H) < AIM-30 < AIM-90 < AIM-60. The DBT conversions of the AIM-X catalysts increased from 65.40% for AIM-30 to 86.74% for AIM-60 and then decreased to 70.03% for AIM-100 as the number of ALD cycles increased from 30 to 100 (Fig. 6b). In order to test the stability of the catalyst, AIM-60 was recycled after the reaction and the HDS reaction of DBT was performed under the same conditions. As shown in Fig. 6c, the conversion rate was still higher than 80% after 5 cycles. The PXRD of the AIM-X samples remained intact after the hydrodesulfurization reaction (Fig. S9, ESI†), indicating the high stability of the AIM-X catalysts.
Based on the results above, the high catalytic efficiency of AIM-60 is attributed to its unique physicochemical properties. The N2 physical adsorption analysis of the catalyst shows that the specific surface area and pore size of AIM-60 are much larger than those of MoS2/γ-Al2O3, which results in a better and more uniform distribution of active nanometals. In addition, XPS analysis shows that the AIM-60 catalyst provided a higher degree of sulfidation than MoS2/γ-Al2O3 with the same loading, which means that it has active sites with higher adsorption affinity, which in turn makes the DBT more effectively desulfurized. Consequently, a better desulfurization effect can be achieved on the active site of the catalyst AIM-60. Compared with a conventional catalyst supported on γ-Al2O3, all these factors improve the HDS catalytic efficiency of AIM-60. It is worth noting that the catalyst has an excellent cycle stability, with negligible loss of catalytic activity.
HDS reaction mechanisms
Based on previous research results,16,46 there are two possible pathways for the HDS of DBT, as shown in Fig. 7. The first is a direct desulfurization (DDS) pathway and the second is a hydrodesulfurization (HYD) pathway. Intermediates, such as tetrahydrodibenzothiophene (THDBT), cyclohexyl benzene (CHB) and bicyclohexyl (BCH), are considered to be products of the HYD pathway, and biphenyl (BP) is considered to be the product of the DDS pathway. At the identical conversion (10%) of DBT, the HDS product distributions of AIM-60 and MoS2/γ-Al2O3 were compared, and the results are listed in Table 3. The kHDS of AIM-60 is much higher than that of MoS2/γ-Al2O3, indicating that AIM-60 has higher catalytic activity. In order to compare the contribution of DDS and HYD to the overall HDS of the two catalysts, it is appropriate to use the yield ratio of BP/(1-BP) to indicate the selectivity of the DDS/HYD route. As shown in Table 3, the DDS/HYD ratio of AIM-60 is 6.87, much higher than 1, indicating that AIM-60 is more inclined to the DDS pathway, while the ratio of MoS2/γ-Al2O3 is 0.96. | ||
| Fig. 7 HDS of DBT reaction pathways over catalysts. | ||
| Samples | k HDS (10−5 mol g−1 h−1) | Product selectivity (%) | ||
|---|---|---|---|---|
| BP | CHB | DDS/HYD | ||
| a Calculated with DBT conversion at about 10%. | ||||
| AIM-60 | 5.72 | 87.3 | 12.7 | 6.87 |
| MoS2/γ-Al2O3 | 1.86 | 48.9 | 51.1 | 0.96 |
| MoS2-Al | 4.76 | 39.3 | 60.7 | 0.65 |
Conclusions
In summary, we have developed a method to prepare a hydrodesulfurization catalyst by loading MoS2 in NU-1000 through ALD, using molybdenum hexacarbonyl and hydrogen sulfide as the molybdenum source and sulfur source, respectively. The synthesized AIM-X composites exhibited excellent HDS activity and the conversions of the AIM-X series of catalysts were much higher than that of MoS2/γ-Al2O3. The conversion rate of AIM-60 reached its highest, 86.74%, at 250 °C and 3 MPa. Due to the large number of sources of transition metal precursors, we believe that ALD can be used to deposit other metal sulfides in other MOFs for other catalytic processes, such as dehydrogenation or deoxygenation. The AIM-X series catalysts provide a new prospect for the development of ecologically friendly HDS processes, which can reduce hydrogen consumption during desulfurization and maintain excellent desulfurization activity and high stability.Author contributions
L. Yu and Q. Zhang performed the experiments; L. Yu contributed to write the manuscript; L. Yu and W. G. Cui analyzed the data; Y. Shen, Z. F. Li, and W. G. Cui contributed to revising the manuscript; and T. L. Hu conceived the study, designed and supervised the overall project, and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21673120 and 21421001), the Natural Science Foundation of Tianjin (20JCYBJC01330) and the Fundamental Research Funds for the Central Universities, Nankai University (63196005).References
- W. Zhou, L. Yang, L. Liu, Z. Chen, A. Zhou, Y. Zhang, X. He, F. Shi and Z. Zhao, Appl. Catal., B, 2020, 268, 118428 CrossRef CAS.
- X. Chen, M. Zhang, Y. Wei, H. Li, J. Liu, Q. Zhang, W. Zhu and H. Li, Inorg. Chem. Front., 2018, 5, 2478–2485 RSC.
- A. López-Benítez, G. Berhault and A. Guevara-Lara, Appl. Catal., B, 2017, 213, 28–41 CrossRef.
- G. Yang, J. Han, Z. Qiu, X. Chen, Z. Feng and J. Yu, Inorg. Chem. Front., 2020, 7, 1975–1980 RSC.
- A. N. Varakin, A. V. Mozhaev, A. A. Pimerzin and P. A. Nikulshin, Appl. Catal., B, 2018, 238, 498–508 CrossRef CAS.
- H. Li, J. Liu, J. Li, Y. Hu, W. Wang, D. Yuan, Y. Wang, T. Yang, L. Li, H. Sun, S. Ren, X. Zhu, Q. Guo, X. Wen, Y. Li and B. Shen, ACS Catal., 2017, 7, 4805–4816 CrossRef CAS.
- G. Ye, H. Qi, W. Zhou, W. Xu and Y. Sun, Inorg. Chem. Front., 2019, 6, 1267–1274 RSC.
- Z. Cao, X. Zhang, R. Guo, S. Ding, P. Zheng, J. Fan, J. Mei, C. Xu and A. Duan, Chem. Eng. J., 2020, 400, 125886 CrossRef CAS.
- Y. Pan, M. Chen, M. Hu, M. Tian, Y. Zhang and D. Long, Appl. Catal., B, 2020, 262, 118266 CrossRef CAS.
- A. A. Toutov, M. Salata, A. Fedorov, Y. F. Yang, Y. Liang, R. Cariou, K. N. Betz, E. P. A. Couzijn, J. W. Shabaker, K. N. Houk and R. H. Grubbs, Nat. Energy, 2017, 2, 17008 CrossRef CAS.
- A. Alsalme, N. Alzaqri, A. Alsaleh, M. R. H. Siddiqui, A. Alotaibi, E. F. Kozhevnikova and I. V. Kozhevnikov, Appl. Catal., B, 2016, 182, 102–108 CrossRef CAS.
- R. Singh, D. Kunzru and S. Sivakumar, Appl. Catal., B, 2016, 185, 163–173 CrossRef CAS.
- A. Bodin, A. L. N. Christoffersen, C. F. Elkjaer, M. Brorson, J. Kibsgaard, S. Helveg and I. Chorkendorff, Nano Lett., 2018, 18, 3454–3460 CrossRef CAS.
- X. Liu, S. Ding, Q. Wei, Y. Zhou, P. Zhang and Z. Xu, Fuel, 2021, 285, 119039 CrossRef CAS.
- S. Shan, P. Yuan, W. Han, G. Shi and X. Bao, J. Catal., 2015, 330, 288–301 CrossRef CAS.
- C. Larabi, P. K. Nielsen, S. Helveg, C. Thieuleux, F. B. Johansson, M. Brorson and E. A. Quadrelli, ACS Catal., 2012, 2, 695–700 CrossRef CAS.
- W. G. Cui and T. L. Hu, Small, 2020, 16, 2003971 CrossRef.
- Y. Shen, L. W. Bao, F. Z. Sun and T. L. Hu, Mater. Chem. Front, 2019, 3, 2363–2373 RSC.
- W. G. Cui, T. L. Hu and X. H. Bu, Adv. Mater., 2020, 32, 1806445 CrossRef CAS.
- M. H. Yu, B. Space, D. Franz, W. Zhou, C. He, L. Li, R. Krishna, Z. Chang, W. Li, T. L. Hu and X. H. Bu, J. Am. Chem. Soc., 2019, 141, 17703–17712 CrossRef CAS.
- Y. L. Peng, J. Liu, H. F. Zhang, D. Luo and D. Li, Inorg. Chem. Front., 2018, 5, 1563–1569 RSC.
- Y. Wang, Y. Wang, L. Zhang, C. S. Liu and H. Pang, Inorg. Chem. Front., 2019, 6, 2514–2520 RSC.
- T. L. Hu, H. Wang, B. Li, R. Krishna, H. Wu, W. Zhou, Y. Zhao, Y. Han, X. Wang, W. Zhu, Z. Yao, S. Xiang and B. Chen, Nat. Commun., 2015, 6, 7328 CrossRef CAS.
- W. G. Cui, G. Y. Zhang, T. L. Hu and X. H. Bu, Coord. Chem. Rev., 2019, 387, 79–120 CrossRef CAS.
- Z. Li, A. W. Peters, V. Bernales, M. A. Ortuno, N. M. Schweitzer, M. R. DeStefano, L. C. Gallington, A. E. Platero-Prats, K. W. Chapman, C. J. Cramer, L. Gagliardi, J. T. Hupp and O. K. Farha, ACS Cent. Sci., 2017, 3, 31–38 CrossRef CAS.
- Z. Li, N. M. Schweitzer, A. B. League, V. Bernales, A. W. Peters, A. B. Getsoian, T. C. Wang, J. T. Miller, A. Vjunov, J. L. Fulton, J. A. Lercher, C. J. Cramer, L. Gagliardi, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2016, 138, 1977–1982 CrossRef CAS.
- L. C. Gallington, I. S. Kim, W. G. Liu, A. A. Yakovenko, A. E. Platero-Prats, Z. Li, T. C. Wang, J. T. Hupp, O. K. Farha, D. G. Truhlar, A. B. F. Martinson and K. W. Chapman, J. Am. Chem. Soc., 2016, 138, 13513–13516 CrossRef CAS.
- H. F. Barton, A. K. Davis and G. N. Parsons, ACS Appl. Mater. Interfaces, 2020, 12, 14690–14701 CrossRef CAS.
- Z. Li, A. W. Peters, J. Liu, X. Zhang, N. M. Schweitzer, J. T. Hupp and O. K. Farha, Inorg. Chem. Front., 2017, 4, 820–824 RSC.
- S. Relijic, A. Broto-Ribas, C. Cuadrado-Collados, E. O. Jardim, D. Maspoch, I. Imaz and J. Silvestre-Albero, Chem. – Eur. J., 2020, 26, 1–11 CrossRef.
- W. Zhang, J. Wang, L. Su, H. Chen, L. Zhang, L. Lin, X. Chen, J. Song and H. Yang, Sci. China: Chem., 2020, 63, 1315–1322 CrossRef CAS.
- A. W. Peters, Z. Li, O. K. Farha and J. T. Hupp, ACS Nano, 2015, 9, 8484–8490 CrossRef CAS.
- A. E. Platero-Prats, A. B. League, V. Bernales, J. Ye, L. C. Gallington, A. Vjunov, N. M. Schweitzer, Z. Li, J. Zheng, B. L. Mehdi, A. J. Stevens, A. Dohnalkova, M. Balasubramanian, O. K. Farha, J. T. Hupp, N. D. Browning, J. L. Fulton, D. M. Camaioni, J. A. Lercher, D. G. Truhlar, L. Gagliardi, C. J. Cramer and K. W. Chapman, J. Am. Chem. Soc., 2017, 139, 10410–10418 CrossRef CAS.
- W. G. Liu and D. G. Truhlar, Chem. Mater., 2017, 29, 8073–8081 CrossRef CAS.
- A. E. Platero-Prats, Z. Li, L. C. Gallington, A. W. Peters, J. T. Hupp, O. K. Farha and K. W. Chapman, Faraday Discuss., 2017, 201, 337–350 RSC.
- P. C. Lemaire, D. T. Lee, J. Zhao and G. N. Parsons, ACS Appl. Mater. Interfaces, 2017, 9, 22042–22054 CrossRef CAS.
- H. Chen, P. Liao, M. L. Mendonca and R. Q. Snurr, J. Phys. Chem. C, 2018, 122, 12362–12368 CrossRef CAS.
- H. Sun, H. Sun, X. Zhang, Q. Yu, P. Zeng, Q. Guo, D. Wang, G. Wen, W. Zhang, S. He and B. Shen, ACS Catal., 2019, 9, 6613–6623 CrossRef CAS.
- X. Wang, C. Xiao, J. Mei, M. H. Alabsi, Y. Shi, Z. Zhao, A. Duan, K. W. Huang and C. Xu, ACS Appl. Mater. Interfaces, 2020, 12, 40404–40414 CrossRef CAS.
- X. Wang, J. Mei, Z. Zhao, P. Zheng, Z. Chen, D. Gao, J. Fu, J. Fan, A. Duan and C. Xu, ACS Catal., 2018, 8, 1891–1902 CrossRef CAS.
- Z. Liu, W. Han, D. Hu, H. Nie, Z. Wang, S. Sun, Z. Deng and Q. Yang, Catal. Sci. Technol., 2020, 10, 5218–5230 RSC.
- W. Zhou, A. Zhou, Y. Zhang, C. Zhang, Z. Chen, L. Liu, Y. Zhou, Q. Wei and X. Tao, J. Catal., 2019, 374, 345–359 CrossRef CAS.
- T. A. Saleh and S. A. AL-Hammadi, Chem. Eng. J., 2021, 406, 125167 CrossRef CAS.
- X. Wang, P. Du, K. Chi, A. Duan, C. Xu, Z. Zhao, Z. Chen and H. Zhang, Catal. Today, 2017, 291, 146–152 CrossRef CAS.
- X. Wang, H. Fang, Z. Zhao, A. Duan, C. Xu, Z. Chen, M. Zhang, P. Du, S. Song, P. Zheng and K. Chi, RSC Adv., 2015, 5, 99706–99711 RSC.
- K. Yang, X. Chen, J. Qi, Z. Bai, L. Zhang and C. Liang, J. Catal., 2019, 369, 363–371 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00955e |
| This journal is © The Royal Society of Chemistry 2021 |