
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInverse vulcanization of octenyl succinate-modified corn starch as a route to biopolymer–sulfur composites†
Moira K.
Lauer
a,
Andrew G.
Tennyson
 ab and
Rhett C.
Smith
*a
ab and
Rhett C.
Smith
*a
aDepartment of Chemistry, Clemson University, Clemson, South Carolina 29634, USA. E-mail: rhett@clemson.edu
bDepartment of Materials Science and Engineering, Clemson University, Clemson, South Carolina 29634, USA
First published on 12th March 2021
Abstract
Herein we report a route to sulfur–starch composites by the modification of corn starch with octenyl succinic anhydride (OSA, degree of substitution = 2.6%) and its subsequent reaction with elemental sulfur through an inverse vulcanization process to generate OSSx (where x = wt% sulfur, either 90 or 95). This work represents an expansion into a previously untapped biomass source for the preparation of recyclable thermoplastic materials by this process. Composites OSSx are comprized of 83–89% by mass of extractable sulfur, and have reasonable thermal stability (Td = 214–216 °C) and Tm (DSC) of 118 °C. The starch modification strategy employed herein allowed for lower degree of substitution of the starch than was feasible for other bioploymers, leading to materials with high strength despite relatively low crosslink density relative to that in previous biopolymer–sulfur composites. The low crosslink density resulted in relatively long polysulfur catenates, thus producing materials with impressive flexural strengths (5.3–5.4 MPa) and highlighting the potential for biomass–sulfur materials to exhibit a range of mechanical properties depending on the biopolymer scaffold and modification strategy.
Introduction
Inverse vulcanization has recently gained attention as a way to valorise waste sulfur from petroleum refining by its reaction with a wide range of substrates to give high sulfur-content materials (HSMs).1,2 Inverse vulcanization is a process involving reaction of a majority feed ratio of sulfur with an olefin-bearing comonomer. Theoretically this process can be up to 100% atom economical, as mechanistically inverse vulcanization proceeds via the radical pathway shown in Scheme 1. Through this process materials have been developed for a multitude of applications including IR transparent lenses, cathodes for lithium–sulfur batteries, and materials for oil spill remediation and mercury capture.4–20 More recently, novel strategies have been implemented for inverse vulcanization at reduced temperatures as well as for processing/recycling23–26 HSMs prepared by inverse vulcanization.27–31 Similar HSMs can be prepared from the reaction of aryl halides or anisole derivatives with sulfur as well, although polymerization of these monomers proceeds via different mechanisms than simple inverse vulcanization.32–35 | ||
| Scheme 1 Simplified schematic for the inverse vulcanization process. | ||
In addition to the aforementioned applications of HSMs, our group has recently reported numerous high-strength composite materials prepared by the reaction of sulfur with bio-derived monomers including fatty acids,36–39 triglycerides,42 terpenoids,21,43 amino acid derivatives,44 lignin derivatives,22,35,45,46 cellulose derivatives,3,21 and raw lignocellulosic biomass sources.40,41 In terms of commercialization of biopolymer-derived materials, starch-derived films and composites have recently gained tremendous interest because starch is remarkably simple to solubilize, derivatize and process compared to cellulose. We recently reported a comprehensive review on the merits and progress in application of starch as a structural element for packaging applications.47 Given the desirable properties of starch and its chemical similarity to cellulose, we hypothesized that starch may be an attractive target as a comonomer with sulfur in the quest to expand the repertoire of material properties and sustainably-sourced polymers/composites that can be conveniently prepared. We further hypothesized that the desirable processability and sulfur-miscibility of starch would allow durable composites to be prepared from starch requiring a lower degree of substitution than is required when cellulose is employed as the biopolymer element, thus limiting the amount of non-bio-derived material in the composites.
Starch is ubiquitous and easily produced worldwide, making it a resilient candidate for exploration as a component of sustainable surrogates for environmentally deleterious polymers that plague society. Corn starch is of particular interest as corn is one of the most-produced crops globally and corn starch is typified by smaller and more uniform particle sizes relative to other starch sources.48,49 In 2020, more than 15 billion bushels of corn were produced in the U.S. alone, approximately 70% of which is comprised of starch.47,50 In 2015, approximately 50% of all corn was utilized for animal feed, 30% for bioethanol production, 6% to produce sweeteners, and 11% for export.51 Current trends in food consumption and the demand for more health-conscious options have led to commensurate decrease in the demand for corn-based sweeteners and an increase in the demand for grass-fed, rather than grain-fed cattle.52,53 These trends, coupled with a nearly 50% reduction in the price of corn since 2013 yet constant production volume (Fig. 1) posture corn starch as an untapped potential resource in the development of more value-added and sustainable technologies.
 | ||
| Fig. 1 Trends in the yearly average corn price per bushel (black) and the yearly U.S. corn production (blue) showing relatively steady production but a significant drop in price due to reduced consumer demand. | ||
In the current study, we have thus modified commercial corn starch with octenylsuccinic anhydride (OSA) in order to equip starch with the olefins required for reaction with sulfur through inverse vulcanization. The thermal and mechanical properties of the resultant composites (OSSx, where x = wt% sulfur, either 90 or 95 wt%, Scheme 2) were analysed for comparison to analogous properties of other biomass–sulfur composites.
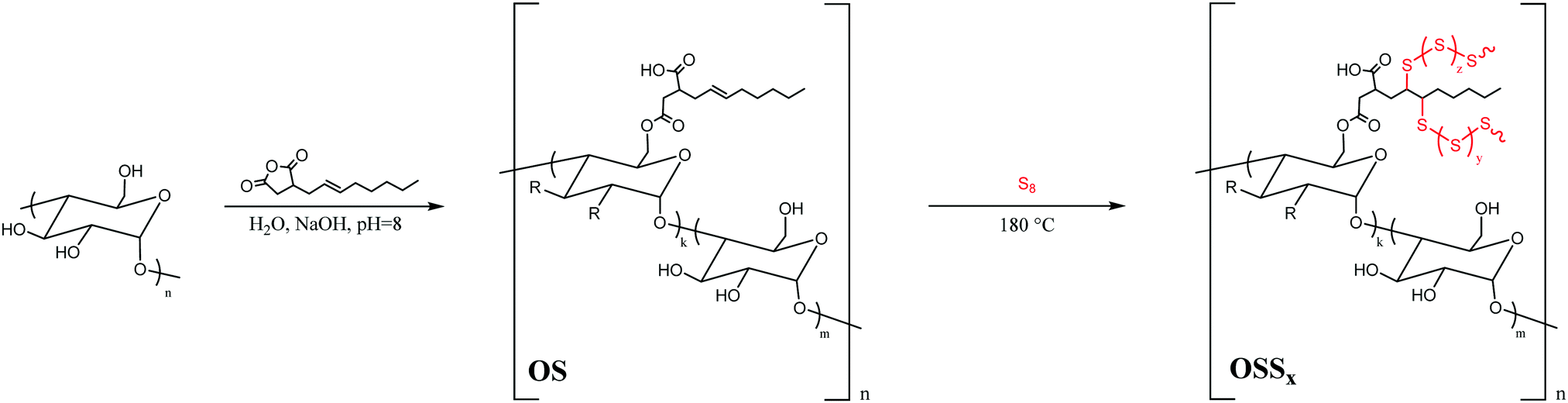 | ||
| Scheme 2 The reaction of starch, shown as poly((1 → 4)α-D-glucopyranose) for simplicity, with octenylsuccinic anhydride (OSA) to yield OS where k = 0.0263 and m = 0.9737 and the subsequent reaction of olefinic OS with sulfur which can occur above 159 °C when cyclic S8 homologates to form radical polymeric sulfur to generate OSSx, (where x = wt% sulfur, either 90 or 95 wt%). | ||
Results and discussion
Starch modification and composite synthesis
Modification of corn starch was carried out by its reaction with octenylsuccinic anhydride (OSA) via a standard literature procedure.54 This particular modification protocol was selected for its favourable incorporation of green chemistry principles: the reaction is carried out at room temperature in aqueous media (full details can be found in the ESI†), to yield OSA-modified starch OS (Scheme 2). Olefination with OSA is also well-precedented to give a product with limited change in crystallinity upon functionalization.55 In our previous work, we have noted that retention of biopolymer crystallinity is a potential contributing factor in compatibilizing sulfur and biopolymer comonomers as well as for imbuing strength to the resultant cellulose–sulfur composites.3,21 The incorporation of hydrophobic alkyl chains should also improve miscibility/compatibilization with sulfur for more facile reaction than has been observed in some cellulose–sulfur systems.56 Modification with OSA also provides another site for modification, a carboxylic acid side chain, which could be further functionalized to incorporate additional olefins to increase crosslink density, although this avenue is not described in the current work.Successful modification of the corn starch was qualitatively confirmed by ATR-FTIR spectroscopic analysis, which revealed carbonyl stretches at 1566 and 1639 cm−1 attributable to ester and carboxylic acid functionalities, while the C–H stretching of the olefin functionality was too weak to observe (ESI,† Fig. S1). The degree of substitution (DS, the number of olefins per anhydroglucose unit) in OS was determined to be 0.0263 ± 0.0005 by acid–base titration. Modification of starch with OSA resulted in a drastic change in hydrophilicity, a property that was readily visualized by qualitative observation of the significantly higher contact angle with water for OS than for native corn starch (Fig. 2A). This observation is consistent with reported quantitative studies that demonstrate significant increases in water contact angle with OSA-modified starch films having DS < 0.03.57
 | ||
| Fig. 2 (A) The increase in water contact angle from an unmodified corn starch film (left, after 0.1 s exposure; eventually water is absorbed by starch) to an OS film (right) provides a visual, qualitative demonstration of increased hydrophobicity upon modification. The water was dyed with food coloring to improve image quality and had no effect on the interaction between starch and water. (B) Digital images of cuboids of OSS95 (left) and OSS90 (right) measuring approximately 15 × 8 × 1.5 mm. | ||
The thermal properties of OS were also noticeably different from those of native corn starch. The decomposition temperature (Td) as assessed by thermogravimetric analysis (TGA) decreased by approximately 25 °C upon modification (293.5 ± 0.5 for OSversus 268.4 ± 0.4 °C for corn starch; Table 1 and ESI,† Fig. S2 and Table S1). The glass transition temperature (Tg) determined from differential scanning calorimetry (DSC) decreased by approximately 9 °C upon modification (98.4 and 89.6 °C for corn starch and OS, respectively, ESI,† Fig. S3). As anticipated, the crystallinity showed a decreased of only 5%, from 20.9% for native corn starch to 15.8% for OS, as determined by powder X-ray diffraction (PXRD, ESI,† Fig. S4).
| T d (°C) | Cold xtl.b (°C) | T m (°C) | S8 melt integratione (J g−1) | Char yieldf (%) | |
|---|---|---|---|---|---|
| a The average temperature at which 5% mass loss was observed. b Taken from the third heating cycle. c Defined as the midpoint of the step in the heat of the DSC thermogram. d The melting temperature for S8 determined by the DSC thermogram. e Taken as an average of the values from heats 3–5 by triplicate analyses. f Residual mass at 800 °C. g Values based on quadruplicate analysis due to the poor thermal conductivity of sulfur and the large dependence on the sample mass (varied from 5–6.5 mg across four trials). h Normalized to eliminate the impact of adsorbed water. | |||||
| Sulfurc | 219.0 ± 2.4g | — | 119 | −45.4 ± 1.3 | −0.2 ± 0.3 |
| OSS95 | 215.6 ± 0.8 | 22.6 | 118 | −40.2 ± 1.1 | 0.6 ± 0.0 |
| OSS90 | 213.8 ± 0.2 | 24.6 | 118 | −37.5 ± 1.2 | 1.6 ± 0.3 |
| OS | 268.4 ± 0.4 | — | — | — | 11.6 ± 0.1h |
Synthesis of OS–sulfur composites (OSSx, where x = wt% sulfur, either 90 or 95 wt%, Scheme 2) was undertaken under an inert atmosphere of N2 at 180 °C. In this process, each of the modified monomers provided one olefin for potential reaction via inverse vulcanization (Scheme 1). Although OS was significantly more miscible with sulfur than native corn starch, the homogenization of OS granules with sulfur still proved challenging and required constant efficient stirring due to phase separation at the reaction interface. Unsurprisingly, homogenization with a higher ratio of OS (i.e.OSS90) required longer reaction times. To eliminate any heating-duration effects, however, both samples were heated for 440 minutes at 180 °C, the conditions required to homogenize the OSS90 reaction mixture. The resultant materials were allowed to cool before remelting and pouring into appropriate moulds to shape the material into rectangular prisms or cylinders for flexural and compressional strength analysis, respectively. Both materials were golden-brown in colour upon cooling, with OSS95 taking on a slightly darker colour (Fig. 2B). All subsequent thermal, compositional, and mechanical analyses were conducted after samples were cured for 15 days at room temperature to ensure that any metastable polymeric sulfur that was not sufficiently stabilized by the crosslinked network would relax to the stable S8 allotrope. The mechanical properties of HSMs can vary wildly within the first few days of their preparation, so this pre-characterization curing is essential if the data are to be used to draw conclusions relevant to eventual practical applications.
Elemental analysis confirmed that OSS95 and OSS90 contained 95 and 90 wt% of sulfur, respectively. Although IR spectroscopy was not sensitive enough to assess the consumption of olefins, indirect evidence for successful sulfur crosslinking was abundant. Fractionation of the composites in CS2 revealed that only 82 and 86% of OSS90 and OSS95, respectively, were comprised of CS2-soluble S8, indicating that a surprisingly large proportion of sulfur – 8.4 and 10 wt% of monomer feed sulfur for OSS95 and OSS90 respectively – was crosslinked. These crosslinkable sulfur values compare to only up to 7.4 wt% of monomer feed sulfur that were stabilized as crosslinking catenates in analogous cellulose–sulfur composites (PCSx) despite the fact that there were only ∼20% the available crosslinkable sites in OSSx composites (Table 2) compared to the number available in PCSx.58 Sulfur ranks (RS, average number of sulfur atoms per crosslinking polysulfur chain) were calculated to be 170 and 69 for OSS95 and OSS90, respectively (calculations and raw data provided in the ESI† and accompanying Table S2 and eqn (S2)). The RS in OSSx composites are towards the high end of the range reported for previously-reported biopolymer–sulfur composites prepared by inverse vulcanization of methylpropene-derivatized cellulose (PCSx, RS = 24–58),58 geraniol-derivatized cellulose (GCS90, RS = 22),59 allyl lignin (LSx, RS = 49–96),46 or allylated lignocellulose biomass (APSx, RS = 20–21).40,41 A comparison of several properties for OSSx and these related biopolymer–sulfur composites is provided in Table 2.
| Crosslinkable sitesa (μmol olefin per g composite) | CS2 solubleb (%) | Sulfur rankc | Flexural strength (MPa) | Flexural modulus (MPa) | Compressional strength (MPa) | |
|---|---|---|---|---|---|---|
| a Determined by the extent of modification and incorporation of organic material into the composite. b The wt% of material that was soluble in CS2 upon fractionation. c The average number of sulfur atoms per crosslink. d No data were reported in the associated manuscript. e Data from the ref. 3. f Data from the reference. g Data from the ref. 21 and 22 h Data from the ref. 40 and 41 | ||||||
| OSS95 | 7.84 | 86 | 170 | 5.4 | 690 | 11.9 ± 3.2 |
| OSS90 | 15.7 | 82 | 69 | 5.3 | 660 | 10.9 ± 1.9 |
| PCS95 | 35.6 | 88 | 58 | NDd | NDd | NDd |
| PCS90 | 71.2 | 84 | 24 | NDd | NDd | NDd |
| GCS90 | 141 | 71 | 22 | >4.9 | 950 | NDd |
| LS95 | 45 | 87 | 49 | ND | ND | NDd |
| LS90 | 73.5 | 84 | 96 | 1.7 | 60 | NDd |
| APS95 | 95.0 | 89 | 20 | 4.8 | 690 | 35.7 ± 1.8 |
| APS90 | 190 | 77 | 21 | 6.7 | 1490 | 24.1 ± 5.7 |
Thermal analysis provides an additional method to quantify the amount of free S8versus polysulfur catenates in the composite by comparing the integration of the S8 melt peak in the DSC analysis (Table 1) for pure sulfur and the composites. This analysis revealed that OSS90 and OSS95 are comprised by 82.7 ± 4.8% and 89 ± 4.8% S8, respectively, consistent with the results obtained from CS2 fractionation. The appearance of a cold crystallization peak in the DSC traces for both materials was also consistent with the presence of a network structure in which crystallization of sulfur is delayed due to limited mobility resulting from long polymeric chains.46,60 The more confined matrix of OSS90 is apparent from the broader cold crystallization peak as well as a later crystallization exotherm on cooling compared to OSS95 (Fig. 3 and Table 1). Thermogravimetric analysis (TGA) revealed only a slight decrease in decomposition temperature (<10 °C) for OSSx materials relative to sulfur and char yields consistent with the amount of organic (5 or 10%) present in the materials (ESI,† Fig. S5).
 | ||
| Fig. 3 Sample mass-normalized DSC traces for heating (left) and cooling (right) curves of OSS95 (red) and OSS90 (blue) for the third cycle. OSS90 exhibits a broader cold crystallization exotherm and a reduced sulfur melt peak on heating and a later crystallization exotherm on cooling compared to OSS95. | ||
Mechanical properties of biomass–sulfur composites
Some biopolymer–sulfur composites have shown flexural and compressive strengths that exceed those of familiar building materials such as Portland cement (Table 2). The mechanical properties of OSSx were measured to assess whether the starch composites could likewise serve as structural materials with lower degrees of olefin substitution than were required in previous biopolymer–sulfur composites. Flexural analysis in a dynamic mechanical analyser revealed that OSSx composites possessed strengths on par with other biopolymer–sulfur composites despite the lower crosslink densities (Table 2 and Fig. 4A). Compared to one another, OSS95 and OSS90 behaved almost identically within the regime of recoverable deformation. At higher stresses, however, OSS90 retained much of its rigidity whereas OSS95 deformed significantly, resulting in a near 40% greater toughness for OSS95 relative to OSS90 (Table 3).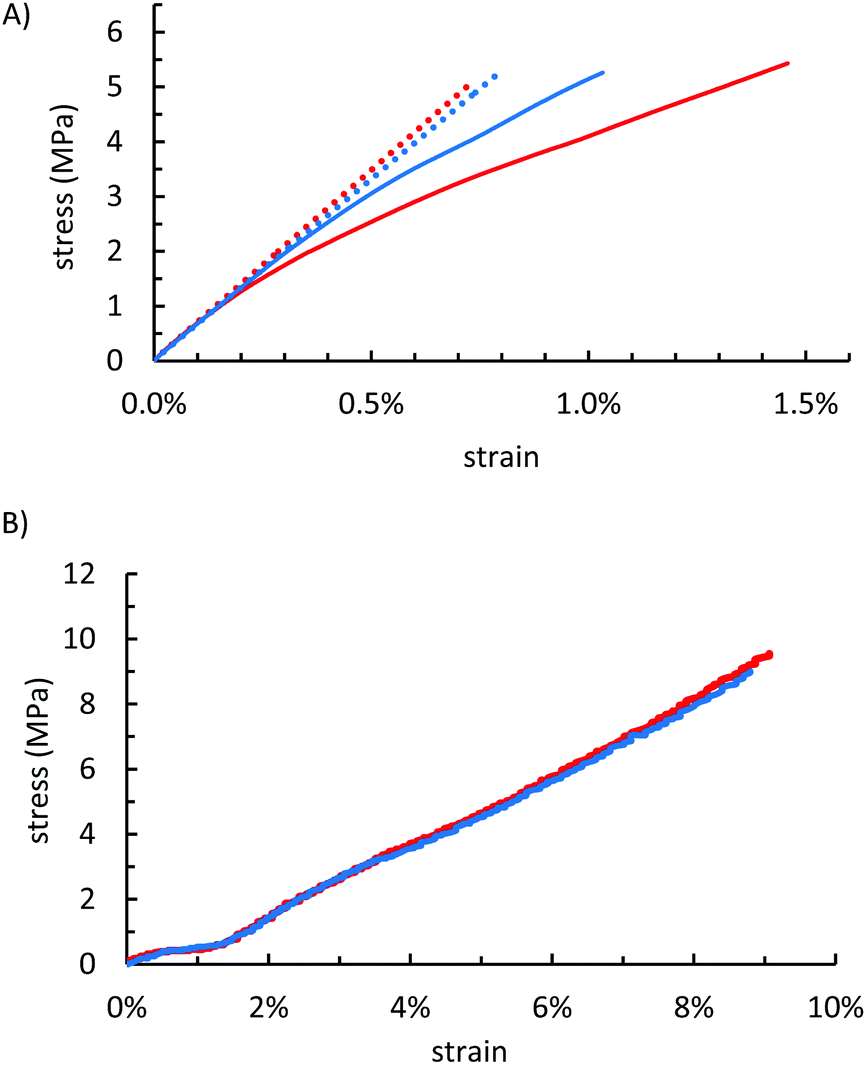 | ||
| Fig. 4 (A) Flexural stress–strain curves (solid lines) for OSS95 (red) and OSS90 (blue). Although the materials are similarly stiff under a recoverable deformation (linear region denoted by dotted lines), past the proportional limit OSS90 retains much of its rigidity due to a higher crosslink density whereas OSS95 deforms considerably resulting in toughness's of 42.9 and 30.2 kPa for OSS95 and OSS90, respectively although the materials exhibit similar flexural strengths. (B) Representative compressional stress–strain plots of OSS95 (red) and OSS90 (blue) showing nearly identical responses to compressive stresses. | ||
| OSS95 | OSS90 | |
|---|---|---|
| a The deformation at the linear limit as defined by a 0.05 MPa deviation from linearity. b The stress at the linear limit as defined by a 0.05 MPa deviation from linearity. c The integration at the linear limit. d The deformation at sample failure. e The integration at sample failure. | ||
| Linear straina (%) | 0.13 | 0.31 |
| Linear stressb (MPa) | 0.9 | 2.0 |
| Flexural strength (MPa) | 5.4 | 5.3 |
| Flexural modulus (MPa) | 690 | 660 |
| Modulus of resiliencec (kPa) | 0.6 | 3.2 |
| Max straind (%) | 1.46% | 1.03% |
| Toughnesse (kPa) | 42.9 | 30.2 |
Although the flexural strengths were quite impressive considering the limited crosslinkable sites, this phenomenon did not extend to the compressional strength characteristics. The compressive strengths of OSSx (10.9 and 11.9 MPa for OSS95 and OSS90, respectively) are reasonably strong considering that the compressive strength requirement of Portland cement is ≥17 MPa for residential building, but are less than half the values reported for lignocellulose–sulfur composites APSx that were prepared by the inverse vulcanization of allylated peanut shells. Interestingly, OSS90 and OSS95 had nearly identical strengths and behaviour under a compressional stress (Table 2 and Fig. 4B) when compared to one another.
Conclusions
Herein is delineated a mechanism for the valorisation of waste sulfur with corn starch. Starch was first modified to a low degree of substitution by reaction with OSA at room temperature in aqueous solution. Modified starch was then reacted via inverse vulcanization with sulfur to yield biomass–sulfur composites, OSSx. The limited concentration of olefin functionalities present in the modified starch were able to stabilize a surprisingly large amount of polymeric sulfur through crosslinking reactions, resulting in flexural strengths on par or better than previously-prepared biomass–sulfur composites that required more modification for their preparation. Both starch composites had similar strength and flexural behaviour under a recoverable deformation, but the addition of increased organic content resulted in a sustained stiffness with increasing applied force, ultimately reducing its toughness and potentially indicating a finite improvement in mechanical properties by increasing the ratio of organic content. Although the limited degree of crosslinking relative to previously modified biopolymers did not negatively impact flexural properties, it did have a more pronounced attenuation of compressional strength with both materials having nearly identical strengths and behaviour under a compressional deformation. Although this work confirms that starch–sulfur composite materials have potential to be used as structural materials even with their limited degrees of crosslinking, more affordable and sustainable derivatizing agents need to be explored before these types of materials become practically applicable components of sustainable building practices. The long-term stability of high sulfur-content materials is also an open question that must be addressed. Such studies are currently underway to further develop these and related materials for practical applications.Experimental
General considerations
Fourier transform infrared spectra were obtained using an IR instrument (Shimadzu IRAffinity-1S) with an ATR attachment. Scans were collected over the range 400–4000 cm−1 at ambient temperature with a resolution of 8. TGA was recorded (Mettler Toledo TGA 2 STARe System) over the range 20–800 °C with a heating rate of 10 °C min−1 under a flow of N2 (100 mL min−1). Each measurement was acquired in duplicate and presented results represent an average value. DSC was acquired (Mettler Toledo DSC 3 STARe System) over the range −60 to 150 °C with a heating rate of 5 °C min−1 under a flow of N2 (200 mL min−1). Each DSC measurement was carried out over five heat–cool cycles. Each measurement was acquired in triplicate to ensure consistent results were obtained.DMA was performed (Mettler Toledo DMA 1 STARe System) in single cantilever mode. DMA samples were cast from silicone resin moulds (Smooth-On Oomoo® 30 tin-cure). Samples were manually sanded to ensure uniform dimensions of approximately 15 × 8 × 1.5 mm but due to instrumental limitations (maximum force of 10 N), each sample differed slightly in thickness in order to obtain a stress at break. Sample dimensions were measured with a digital calliper with ±0.01 mm resolution. Clamping was done by hand due to the samples’ brittleness. The force was varied from 0 to 10 N with a ramp rate of 0.2 N min−1 measured isothermally at 25 °C.
Carbon disulfide extractions were performed by suspending 0.3 g of finely ground material (measured to 0.0001 g) in 20 mL of CS2, allowing the solid to settle for 30 minutes, pipetting off the supernatant into a separate vial, and adding another 20 mL of CS2. This process was repeated an additional 3 times so that a total of 5 washes was performed. The residual CS2 was evaporated under a flow of N2 and each vial was weighed to determine the fraction that was soluble (collected as supernatant) or insoluble (remained in the initial vial).
Compressional analysis was performed on a Mark-10 ES30 test stand equipped with a M3-200 force gauge (1 kN maximum force with ±1 N resolution) with an applied force rate of 3–4 N s−1. Compression cylinders were cast from silicone resin moulds (Smooth-On Oomoo® 30 tin-cure) with diameters of approximately 6 mm and heights of approximately 10 mm. Samples were manually sanded to ensure uniform dimensions and measured with a digital calliper with ±0.01 mm resolution. Compressional analysis was performed in triplicate and results were averaged.
Powder X-ray diffraction samples were placed on zero background sample holders and analysed using a Rigaku Ultima IV diffractometer with Cu Kα radiation (λ = 1.5406 Å). Data were collected from 5–65 degrees in 2-theta at a rate of 0.5 degree per minute with a sampling interval of 0.02 degrees. Crystallinity calculations were conducted by analysing the data from 5–30 degrees (crystalline peaks at ∼12, 15, 17, 18, 23, and 27 degrees and amorphous peaks at ∼19 and 27 degrees).61
Materials and methods
Corn starch (Sigma Aldrich) elemental sulfur (99.5%, Alfa Aesar), octenyl succinic anhydride (97%, mixture of cis and trans, Sigma Aldrich), sodium hydroxide (≥97% VWR Chemicals), hydrochloric acid (concentrated, VWR), potassium hydrogen phthalate (Aldrich Chemical Company), isopropyl alcohol (≥99.5%, Honeywell), and phenolphthalein (ACS grade, VWR) were all used as received unless otherwise specified.Details on the synthesis as well as characterization of OS can be found in the ESI† (pages, figures, etc.).
General composite synthesis
A quantity of OS (compensating for moisture as determined by duplicate TGA experiments) and sulfur totalling 10 g were weighed out and thoroughly mixed into a 20 mL scintillation vial equipped with a Teflon coated stir bar. The vials were sealed with rubber septa and flushed with N2 for ten minutes. The vials were placed in an oil bath set to 180 °C. Samples were heated for ∼9 hours, frequently stopping the reaction by cooling to room temperature under N2, manually scraping down the sides to help reincorporate the organic material, flushing for ten minutes with N2, and placing back in the oil bath until homogenized. OSS90 was prepared first and then the procedures were replicated for OSS95 to ensure identical heating procedures. Specific heating times can be found in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding for this project from the National Science Foundation (CHE-1708844) is gratefully acknowledged.Notes and references
- W. J. Chuang, J. J. Griebel, E. T. Kim, H. Yoon, A. G. Simmonds, H. J. Ji, P. T. Dirlam, R. S. Glass, J. J. Wie, N. A. Nguyen, B. W. Guralnick, J. Park, A. Somogyi, P. Theato, M. E. Mackay, Y.-E. Sung, K. Char and J. Pyun, Nat. Chem., 2013, 5, 518–524 CrossRef PubMed.
- W. J. Chung, J. J. Griebel, E. T. Kim, A. G. Shimmonds, H.-S. Kim, R. S. Glass, Y.-E. Sung, K. Char and J. Pyun, PMSE Preprints, 2012 Search PubMed.
- M. K. Lauer, T. A. Estrada-Mendoza, C. D. McMillen, G. Chumanov, A. G. Tennyson and R. C. Smith, Adv. Sustainable Syst., 2019, 3, 1900062 CrossRef CAS.
- C. V. Lopez, C. P. Maladeniya and R. C. Smith, Electrochem, 2020, 1, 226–259 CrossRef.
- J. H. Worthington Max, L. Kucera Renata, T. Gibson Christopher, A. Sibley, D. Slattery Ashley, A. Campbell Jonathan, F. K. Alboaiji Salah, J. Young, N. Adamson, R. Gascooke Jason, A. Lewis David, S. Quinton Jamie, V. Ellis Amanda, M. Chalker Justin, S. Albuquerque Ines, J. L. Bernardes Goncalo, A. Muller Katherine, A. Johs, D. Jampaiah, M. Sabri Ylias, K. Bhargava Suresh and J. Ippolito Samuel, Chemistry, 2017, 23, 16219–16230 CrossRef CAS PubMed.
- L. J. Esdaile and J. M. Chalker, Chemistry, 2018, 24, 6905–6916 CrossRef CAS PubMed.
- M. J. H. Worthington, C. J. Shearer, L. J. Esdaile, J. A. Campbell, C. T. Gibson, S. K. Legg, Y. Yin, N. A. Lundquist, J. R. Gascooke, I. S. Albuquerque, J. G. Shapter, G. G. Andersson, D. A. Lewis, G. J. L. Bernardes and J. M. Chalker, Adv. Sustainable Syst., 2018, 2, 1800024 CrossRef.
- Y. Zhang, T. S. Kleine, K. J. Carothers, D. D. Phan, R. S. Glass, M. E. MacKay, K. Char and J. Pyun, Polym. Chem., 2018, 9, 2290–2294 RSC.
- Y. Zhang, J. J. Griebel, P. T. Dirlam, N. A. Nguyen, R. S. Glass, M. E. MacKay, K. Char and J. Pyun, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 107–116 CrossRef CAS.
- P. T. Dirlam, R. S. Glass, K. Char and J. Pyun, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1635–1668 CrossRef CAS.
- T. S. Kleine, N. A. Nguyen, L. E. Anderson, S. Namnabat, E. A. LaVilla, S. A. Showghi, P. T. Dirlam, C. B. Arrington, M. S. Manchester, J. Schwiegerling, R. S. Glass, K. Char, R. A. Norwood, M. E. Mackay and J. Pyun, ACS Macro Lett., 2016, 5, 1152–1156 CrossRef CAS.
- P. T. Dirlam, A. G. Simmonds, T. S. Kleine, N. A. Nguyen, L. E. Anderson, A. O. Klever, A. Florian, P. J. Costanzo, P. Theato, M. E. Mackay, R. S. Glass, K. Char and J. Pyun, RSC Adv., 2015, 5, 24718–24722 RSC.
- J. J. Griebel, G. Li, R. S. Glass, K. Char and J. Pyun, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 173–177 CrossRef CAS.
- J. J. Griebel, N. A. Nguyen, S. Namnabat, L. E. Anderson, R. S. Glass, R. A. Norwood, M. E. MacKay, K. Char and J. Pyun, ACS Macro Lett., 2015, 4, 862–866 CrossRef CAS.
- V. P. Oleshko, J. Kim, J. L. Schaefer, S. D. Hudson, C. L. Soles, A. G. Simmonds, J. J. Griebel, R. S. Glass, K. Char and J. Pyun, MRS Commun., 2015, 5, 353–364 CrossRef CAS.
- J. J. Griebel, N. A. Nguyen, A. V. Astashkin, R. S. Glass, M. E. MacKay, K. Char and J. Pyun, ACS Macro Lett., 2014, 3, 1258–1261 CrossRef CAS.
- S. Namnabat, J. J. Gabriel, J. Pyun and R. A. Norwood, Proc. SPIE, 2014, 8983, 89830D CrossRef.
- A. G. Simmonds, J. J. Griebel, J. Park, K. R. Kim, W. J. Chung, V. P. Oleshko, J. Kim, E. T. Kim, R. S. Glass, C. L. Soles, Y.-E. Sung, K. Char and J. Pyun, ACS Macro Lett., 2014, 3, 229–232 CrossRef CAS.
- M. Mann, E. Kruger Jessica, F. Andari, J. McErlean, R. Gascooke Jason, A. Smith Jessica, J. H. Worthington Max, C. C. McKinley Cheylan, A. Campbell Jonathan, A. Lewis David, T. Hasell, V. Perkins Michael and M. Chalker Justin, Org. Biomol. Chem., 2019, 17, 1929–1936 RSC.
- T. Hasell, D. J. Parker, H. A. Jones, T. McAllister and S. M. Howdle, Chem. Commun., 2016, 52, 5383–5386 RSC.
- M. K. Lauer, A. G. Tennyson and R. C. Smith, ACS Appl. Polym. Mater., 2020, 2, 3761–3765 CrossRef CAS.
- M. S. Karunarathna, M. K. Lauer and R. C. Smith, J. Mater. Chem. A, 2020, 8, 20318–20322 RSC.
- T. Thiounn and R. C. Smith, J. Polym. Sci., 2020, 58, 1347–1364 CrossRef CAS.
- T. Thiounn, M. S. Karunarathna, L. M. Slann, M. K. Lauer and R. C. Smith, J. Polym. Sci., 2020, 58, 1347–1364 CrossRef CAS.
- S. J. Tonkin, C. T. Gibson, J. A. Campbell, D. A. Lewis, A. Karton, T. Hasell and J. M. Chalker, Chem. Sci., 2020, 11, 5537–5546 RSC.
- N. A. Lundquist, A. D. Tikoalu, M. J. H. Worthington, R. Shapter, S. J. Tonkin, F. Stojcevski, M. Mann, C. T. Gibson, J. R. Gascooke, A. Karton, L. C. Henderson, L. J. Esdaile and J. M. Chalker, Chem. – Eur. J., 2020, 26, 10035–10044 CrossRef CAS PubMed.
- T. Thiounn, M. K. Lauer, M. S. Bedford, R. C. Smith and A. G. Tennyson, RSC Adv., 2018, 8, 39074–39082 RSC.
- B. Zhang, H. Gao, P. Yan, S. Petcher and T. Hasell, Mater. Chem. Front., 2020, 4, 669–675 RSC.
- P. Yan, W. Zhao, B. Zhang, L. Jiang, S. Petcher, J. A. Smith, D. J. Parker, A. I. Cooper, J. Lei and T. Hasell, Angew. Chem., Int. Ed., 2020, 59, 13371–13378 CrossRef CAS PubMed.
- C. R. Westerman and C. L. Jenkins, Macromolecules, 2018, 51, 7233–7238 CrossRef CAS.
- C. R. Westerman, P. M. Walker and C. L. Jenkins, J. Visualized Exp., 2019, e59620 Search PubMed.
- T. Thiounn, M. K. Lauer, M. S. Karunarathna, A. G. Tennyson and R. C. Smith, Sustainable Chem., 2020, 1, 183–197 CrossRef.
- M. S. Karunarathna, M. K. Lauer, A. G. Tennyson and R. C. Smith, Polym. Chem., 2020, 11, 1621–1628 RSC.
- S. Park, D. Lee, H. Cho, J. Lim and K. Char, ACS Macro Lett., 2019, 8, 1670–1675 CrossRef CAS.
- M. S. Karunarathna, A. G. Tennyson and R. C. Smith, J. Mater. Chem. A, 2020, 8, 548–553 RSC.
- A. D. Smith, R. C. Smith and A. G. Tennyson, Sustainable Chem. Pharm., 2020, 16, 100249 CrossRef.
- A. D. Smith, C. D. McMillin, R. C. Smith and A. G. Tennyson, J. Polym. Sci., 2020, 58, 438–445 CrossRef CAS.
- A. D. Smith, T. Thiounn, E. W. Lyles, E. K. Kibler, R. C. Smith and A. G. Tennyson, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1704–1710 CrossRef CAS.
- A. D. Smith, R. C. Smith and A. G. Tennyson, Sustainable Chem., 2020, 1, 209–237 CrossRef.
- M. K. Lauer, M. S. Karunarathna, A. G. Tennyson and R. C. Smith, Mater. Adv., 2020, 1, 590–594 RSC.
- M. K. Lauer, M. S. Karunarathna, G. Tennyson Andrew and R. C. Smith, Mater. Adv., 2020, 1, 2271–2278 RSC.
- C. V. Lopez, M. S. Karunarathna, M. K. Lauer, C. P. Maladeniya, T. Thiounn, E. D. Ackley and R. C. Smith, J. Polym. Sci., 2020, 58, 2259–2266 CrossRef CAS.
- C. P. Maladeniya, M. S. Karunarathna, M. K. Lauer, C. V. Lopez, T. Thiounn and R. C. Smith, Mater. Adv., 2020, 1, 1665–1674 RSC.
- T. Thiounn, A. G. Tennyson and R. C. Smith, RSC Adv., 2019, 9, 31460–31465 RSC.
- M. S. Karunarathna and R. C. Smith, Sustainability, 2020, 12, 734–748 CrossRef CAS.
- M. S. Karunarathna, M. K. Lauer, T. Thiounn, R. C. Smith and A. G. Tennyson, J. Mater. Chem. A, 2019, 7, 15683–15690 RSC.
- M. K. Lauer and R. C. Smith, Compr. Rev. Food Sci. Food Saf., 2020, 1–53, DOI:10.1111/1541-4337.12627.
- L. Dai, J. Zhang and F. Cheng, Int. J. Biol. Macromol., 2019, 132, 897–905 CrossRef CAS PubMed.
- P. Paronen and M. Juslin, J. Pharm. Pharmacol., 1983, 35, 627–635 CrossRef CAS PubMed.
- N. A. S. Service, 2020 Corn Production: United States, https://www.nass.usda.gov/Charts_and_Maps/Field_Crops/cornprod.php.
- U. S. D. o. Agriculture, USDA Coexistence Fact Sheets: Corn, 2015.
- S. B. C. f. F. Agriculture, Back to Grass: The Market Potential for U.S. Grassfed Beef, 2017.
- J. B. Olayanju, Top Trends Driving Change in the Food Industry, 2019.
- B. Zhang, Q. Huang, F.-x. Luo, X. Fu, H. Jiang and J.-l. Jane, Carbohydr. Polym., 2011, 84, 1276–1281 CrossRef CAS.
- A. Naseri, H. Shekarchizadeh and M. Kadivar, J. Food Process. Preserv., 2019, 43, 1–10 CAS.
- F. Chang, X. He, X. Fu, Q. Huang and Y. Qiu, Food Chem., 2014, 152, 177–183 CrossRef CAS PubMed.
- Z.-Y. Yu, S.-W. Jiang, Z. Zheng, X.-M. Cao, Z.-G. Hou, J.-J. Xu, H.-L. Wang, S.-T. Jiang and L.-J. Pan, Int. J. Biol. Macromol., 2019, 137, 277–285 CrossRef CAS PubMed.
- M. K. Lauer, T. A. Estrada-Mendoza, C. D. McMillen, G. Chumanov, A. G. Tennyson and R. C. Smith, Adv. Sustainable Syst., 2019, 3, 1900062 CrossRef CAS.
- M. K. Lauer, A. G. Tennyson and R. C. Smith, ACS Appl. Polym. Mater., 2020, 2, 3761–3765 CrossRef CAS.
- A. Smith and W. B. Holmes, J. Am. Chem. Soc., 1905, 27, 979–1013 CrossRef CAS.
- K. Frost, D. Kaminski, G. Kirwan, E. Lascaris and R. Shanks, Carbohydr. Polym., 2009, 78, 543–548 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Proton NMR spectral data, FTIR spectra, TGA curves, analysis of char yield versus composition; DSC curves. See DOI: 10.1039/d0ma00948b |
| This journal is © The Royal Society of Chemistry 2021 |