DOI:
10.1039/D0MA00910E
(Paper)
Mater. Adv., 2021,
2, 1059-1071
Asymmetrical benzo[a]-fused N2O2-boron-chelated BODIPYs as red to near-infrared absorbing chromophores: synthesis, characteristics and device applications for opto-electronics†
Received
22nd November 2020
, Accepted 12th January 2021
First published on 13th January 2021
Abstract
Asymmetric benzo[a]-fused BODIPYs with benzo(thieno)[1,3,2]oxazaborinine units 1–4 were synthesized for the first time. The structural feature was elucidated by X-ray crystallography and NICS(0) calculation, indicating that the isoindole ring may possess azafulvene character. The photophysical properties of the dyes were investigated, demonstrating that they absorb far-red and NIR light with a λmax value of 663–709 nm in THF. Replacement of benzo[1,3,2]oxazaborinine with the thieno-counterpart caused a red-shift in the absorption band. Interestingly, the effect of thiophene insertion on the photophysical properties is dependent on the position of the thiophene in the chromophore, as determined from cyclic voltammetry (CV) measurement and theoretical calculations. Further, N2O2-ligated sp3-boron induces chirality into the chromophore, facilitating optical resolution of 2 by chiral high-pressure liquid chromatography; helical dyes P-2 and M-2 were successfully characterized. As a potential device application, a single component device (ITO/dye 2/Al) was fabricated to produce a photocurrent of 8.42 × 10−7 A cm−2 at a bias potential of 1 V under photoirradiation at 750 nm (128 μW cm−2). Compared to the dark current, the on/off current ratio at the same bias potential was determined to be 1.42 × 102. The result suggests that a sophisticated combination of asymmetrical BODIPY with a n-type acceptor would allow for NIR photodiodes with a p–n interface.
Introduction
Functional dyes with near-infrared (NIR) absorption bands have received much attention due to their tremendous utility in applications such as optoelectronics,1,2 photovoltaics,3,4 dye-sensitized solar cells,5,6 hydrogen production,7,8 chemosensing,9 bio-probes,3,10,11 photodynamic therapy,12,13 photoimmuno therapy,14etc. Although numerous NIR dyes have been prepared to date,15–17 they often suffer from low thermal and poor photo-stability. Accordingly, band-gap engineering is highly desired for the rational design of useful related materials. BODIPY (4,4-difluoro-4-bora-3a,4a-diaza-s-indacene)18–20 is among the most promising candidates due to its excellent photophysical properties and amenability to structural tuning. Structural modification at the periphery and at the boron center21 enables tuning of the photophysical properties.22 The traditional strategy is π-extension and fusion of aromatic rings such as benzene,23–25 thiophene,26–28 furan,29,30 naphthalene,31 phenanthrene,32 and others33 at the α-bond or β-bond of the pyrrole, which leads to a large red-shift in the absorption and fluorescence bands. Alternatively, π-extension by fusing and merging two and more BODIPY units have been studied.34–37 Furthermore, it was reported that oligomerization of BODIPY chromophores caused J-aggregation to give redshift in the absorption band.38 Given that electronic delocalization extended by intramolecular B,O-chelation leads to a dramatic red-shift in the absorption band,39–42 structural combination of benzene-annulation at the α-bond and B,O-chelation allowed for the production of NIR BODIPYs.43–45 Using that approach, we demonstrated that the related NIR dyes have great promise for use as NIR photosensitizers in optoelectronics devices.46–48 Given that the BODIPY skeleton is amenable to tuning, the synthesis and characterization of asymmetrical benzo[a]-fused BODIPYs are worthwhile pursuits.49–60 This is because the push–pull character of the asymmetrical π-conjugated structures would impact the photophysical properties.34,52 However, asymmetrical BODIPYs featuring NIR absorbing bands have rarely been reported. To the best of our knowledge, styryl-containing asymmetric aza-BODIPYs are the only known such species to date61 It is thus of interest to gain insight into the impact of cyclic fixation of asymmetric 3,5-diaryl (thienyl) BODIPY through B,O-chelation on the photophysical properties. In addition, the resultant tetrahedral geometry at boron can induce helicity62 in the chromophore to create enantiomeric BODIPYs with an asymmetrical structure.
From the standpoint of device applications, π-conjugated systems hold great promise for developing organic semiconductors.63,64 Notably, photodetectors that can convert incident NIR photons into an electrical signal have attracted considerable attention due to their many applications in image sensing, optical communication, night surveillance, etc.65–69 To this end, the development of sensitive photodetectors capable of capturing very weak NIR signals with fast response is highly desired. π-Conjugated polymers with a narrow-bandgap, which serve as organic semiconductors, make it possible to achieve high-performance photodetectors with spectral responses.70–75 On another front, given the advantage of small molecules, including their high purities and well-defined molecular structures, BODIPYs are among the most promising candidates.47,48,76 However, asymmetrical derivatives that can be used in optoelectronic materials with photocurrent generation properties have not yet been reported.
In this work, asymmetrical benzo(thieno)[1,3,2]oxazaborinine-containing BODIPYs 1–4 (Fig. 1) are synthesized and characterized. The related asymmetric BF2-dye 5 is also prepared as a control chromophore. X-ray crystallographic analysis of 2 reveals the packing structure, indicating that the enantiomer pair is present in a triclinic space group due to tetradentate N2O2-coordination to boron. The racemic mixture is successfully separated by using semi-preparative chiral HPLC. The photophysical properties of the dyes were investigated, demonstrating that they absorb far-red and NIR light with a λmax value of 663–709 nm in THF. Replacement of benzo[1,3,2]oxazaborinine with the thieno-counterpart caused a red-shift in the absorption band. Interestingly, the effect of thiophene insertion on the photophysical properties is dependent on the position of the thiophene in the chromophore, as determined from cyclic voltammetry (CV) measurement and theoretical calculations. Thiophene insertion led to longer-wavelength emission, with lower fluorescence quantum yields (ϕ). Nevertheless, dye 2 is a NIR fluorophore that emits at 721 nm with Φ value of 0.21 when excited at 610 nm in THF. Our continuous interest is to fabricate organic photodetectors with asymmetrical benzo[a]-fused BODIPY. Towards this end, a 2-containing film was prepared by vacuum deposition, where the film absorbed NIR light at 735 nm with a long spectral width and a λonset value of 787 nm. As a preliminary result, an indium tin oxide (ITO)/2/Al device allowed for photocurrent detection under light irradiation at 750 nm (128 μW cm−2). We demonstrate for the first time that asymmetrical BODIPY-based NIR dyes can serve as organic photodetectors for application in optoelectronics.
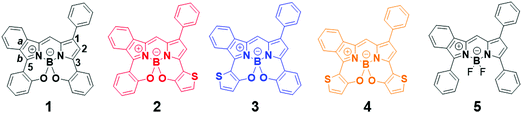 |
| | Fig. 1 Chemical structures of asymmetrical benzo[a]-fused N2O2-boron-chelated BODIPYs 1–4 and the control BF2 dye 5. | |
Experimental section
General
NMR spectra were measured on a Bruker Avance 500 spectrometer (1H: 500 MHz, 13C: 126 MHz, 11B: 160 MHz, 19F: 471 MHz) and JEOL JMN-ECS400 (1H; 400 MHz, 11B; 128 MHz). In 1H and 13C NMR measurements, chemical shifts (δ) are reported downfield from the internal standard Me4Si. BF3·OEt2 and C6F6 were used as external standard for 11B NMR and internal standard for 19F NMR, respectively. Mass spectrometry data were taken by using a JEOL JMS-700 fast atom bombardment (FAB) spectrometer where m-nitrobenzylalcohol was used as a matrix or a Bruker micrOTOF II-SDT1 spectrometer with atmospheric pressure chemical ionization (APCI) method. FT-IR spectra were recorded on a JASCO FT/IR-4600 equipped with a diamond ATR crystal (JASCO PRO 450S). Melting points for 1, 3, 4, 5, 12 and 13 were recorded on Seiko Instruments Inc EXSTAR6000. Differential scanning calorimetry (DSC) and thermal gravimetric analysis (TGA) for dye 2 were taken by using TGA/DSC 1 (Mettler Toledo). The absorption and fluorescence were measured using Shimadzu UV-3600 UV/vis/NIR spectroscopy and a JASCO FP-6500 spectrofluorometer, respectively. Fluorescence lifetimes were determined by Hamamatsu photonics Quantaurus-Tau C11367-21. Elemental analyses were performed on an Exeter Analytical, Inc. CE-440F Elemental Analyzer. Ionization potential data were measured by a Riken Keiki photoelectron spectroscopy instrument, AC-3. Atomic Force Microscope (AFM) was used AFM-5400 (Hitachi High-Technologies Corporations) in Dynamic Force mode using silicon probes with resonant frequency of 70 kHz and a force constant of 2 N m−1.
Materials
Reagents used for the synthesis were commercially available and used as supplied. Dry CH2Cl2 and dry toluene were prepared according to a standard procedure. 1-(3-Bromo-1H-isoindol-1-ylidene)-N,N-dimethylmethanamine 6,77 2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)anisole 7a,78 3-methoxythiophene-2-boronic acid pinacol ester 7b,79 1-(3-methoxythiophen-2-yl)-4-nitro-3-phenylbutan-1-one 10,48 3-phenyl-1-formylisoindole 8c,80 2-(2-methyloxyphenyl)-4-phenyl-1H-pyrrole 9a,81 and 2-phenyl-4-phenyl-1H-pyrrole 9c82 were prepared according to method previously reported.
Synthesis
(Z)-3-(2-Methoxyphenyl)-1-((5-(2-methoxyphenyl)-3-phenyl-1H-pyrrol-2-yl)methylene)-1H-isoindole (12a).
Under a N2 atmospheric icy condition, to a solution of 8a (2.52 g, 10.0 mmol) and 9a (2.50 g, 10.0 mmol) in dry CH2Cl2 (140 mL) was added dropwise POCl3 (1.0 mL, 11 mmol). The resultant solution was stirred for 2 h at room temperature. After quenching the reaction by adding water, the organic layer was extracted with CH2Cl2. Isolated product was washed with AcOEt to give 2.87 g of 12a as a red solid in 59% yield. M.p. 198 °C; 1H NMR (400 MHz, CDCl3): δ (ppm) 7.83 (1H, dd, J = 1.78 and 7.38 Hz), 7.77–7.74 (2H, m), 7.61 (2H, dd, J = 8.10 and 1.10 Hz), 7.57 (1H, d, J = 7.48 Hz), 7.52–7.46 (3H, m), 7.41–7.33 (2H, m), 7.36 (1H, s), 7.28–7.20 (2H, m), 7.11 (1H, td, J = 7.34 and 0.85 Hz), 7.09 (1H, d, J = 8.36 Hz), 7.00 (1H, td, J = 7.51 and 0.97 Hz), 6.93 (1H, dd, J = 8.24 and 0.68 Hz), 6.83 (d, J = 2.28 Hz, 1H); 13C NMR (126 MHz, CDCl3): δ (ppm) 164.4, 158.0, 156.1, 144.8, 141.0, 136.6, 136.1, 136.0, 133.6, 131.5, 130.8, 129.0, 128.7, 128.3, 128.0, 127.3, 127.2, 126.8, 125.7, 125.3, 123.0, 121.0, 120.6, 119.9, 119.0, 115.0, 111.5, 111.2, 109.1, 55.69, 55.46; ATR-FT-IR v (cm−1): 3400–3330 (N–H), 3052 (aromatic CH), 1603 (aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 1590 (aromatic CC), 1460–1386 (CN), 1220 (C–O); HRMS (APCI) calcd for C33H27N2O2 [M + H]+: 483.2067, found 483.2051.
C), 1590 (aromatic CC), 1460–1386 (CN), 1220 (C–O); HRMS (APCI) calcd for C33H27N2O2 [M + H]+: 483.2067, found 483.2051.
(Z)-3-(2-Methoxyphenyl)-1-((5-(3-methoxythiophen-2-yl)-3-phenyl-1H-pyrrol-2-yl)methylene)-1H-isoindole (12b).
Compound 12b was synthesized using 8a (1.75 g, 6.95 mmol) and 9b (1.78 g, 6.97 mmol) in a similar manner as the case of 12a. The product was purified by chromatography on silica gel using CH2Cl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane (1:1 v/v) as an eluent to give 2.47 g of 12b as a purplish red in 73%. M.p. 200 °C; 1H NMR (CDCl3, 400 MHz): δ = 7.89 (1H, dd, J = 7.40 and 1.76 Hz), 7.74 (1H, d, J = 7.64 Hz), 7.59–7.57 (2H, m), 7.51–7.45 (3H, m), 7.37 (1H, tt, J = 7.32 and 1.39 Hz), 7.33 (1H, d, J = 6.12 Hz), 7.32 (1H, s), 7.27–7.23 (1H, m), 7.12 (1H, td, J = 7.44 and 0.67 Hz), 7.09 (1H, d, J = 5.40 Hz), 7.09–7.07 (1H, m), 6.84 (1H, d, J = 5.52 Hz), 6.51 (1H, s), 3.87 (3H, s), 3.72 (3H, s); 13C NMR (126 MHz, CDCl3): δ = 163.8, 158.0, 154.2, 144.5, 140.9, 136.4, 135.7, 134.1, 132.4, 131.5, 130.7, 129.0, 128.7, 127.7, 127.1, 126.9, 125.5, 125.4, 123.2, 122.2, 120.6, 118.9, 116.3, 114.6, 112.3, 111.2, 107.7; ATR-FT-IR ṽ (cm−1): 3106 (aromatic CH), 1616 (aromatic CC), 1587 (aromatic CC), 1523–1375 (CN), 1209 (C–O); HRMS (APCI) calcd for C31H25N2O2S [M + H]+: 489.1631, found 489.1626.
:hexane (1:1 v/v) as an eluent to give 2.47 g of 12b as a purplish red in 73%. M.p. 200 °C; 1H NMR (CDCl3, 400 MHz): δ = 7.89 (1H, dd, J = 7.40 and 1.76 Hz), 7.74 (1H, d, J = 7.64 Hz), 7.59–7.57 (2H, m), 7.51–7.45 (3H, m), 7.37 (1H, tt, J = 7.32 and 1.39 Hz), 7.33 (1H, d, J = 6.12 Hz), 7.32 (1H, s), 7.27–7.23 (1H, m), 7.12 (1H, td, J = 7.44 and 0.67 Hz), 7.09 (1H, d, J = 5.40 Hz), 7.09–7.07 (1H, m), 6.84 (1H, d, J = 5.52 Hz), 6.51 (1H, s), 3.87 (3H, s), 3.72 (3H, s); 13C NMR (126 MHz, CDCl3): δ = 163.8, 158.0, 154.2, 144.5, 140.9, 136.4, 135.7, 134.1, 132.4, 131.5, 130.7, 129.0, 128.7, 127.7, 127.1, 126.9, 125.5, 125.4, 123.2, 122.2, 120.6, 118.9, 116.3, 114.6, 112.3, 111.2, 107.7; ATR-FT-IR ṽ (cm−1): 3106 (aromatic CH), 1616 (aromatic CC), 1587 (aromatic CC), 1523–1375 (CN), 1209 (C–O); HRMS (APCI) calcd for C31H25N2O2S [M + H]+: 489.1631, found 489.1626.
(Z)-1-((5-(2-Methoxyphenyl)-3-phenyl-1H-pyrrol-2-yl)methylene)-3-(3-methoxythiophen-2-yl)-1H-isoindole (12c).
Compound 12c was synthesized using 8b (0.565 g, 2.20 mmol) and 9a (0.548 g, 2.20 mmol) in a similar manner as the case of 12a. The product was purified by reprecipitation with CH2Cl2 and MeOH to give 0.741 g of 12c as a black solid in 69% yield. M.p. 177 °C; 1H NMR (500 MHz, CDCl3): δ (ppm) 12.63 (1H, s), 8.23 (1H, d, J = 7.80 Hz), 7.79 (1H, dd, J = 7.75 and 1.05 Hz), 7.74 (1H, d, J = 7.65 Hz), 7.61 (2H, d, J = 7.30 Hz), 7.50 (2H, t, J = 7.58 Hz), 7.42 (1H, d, J = 5.50 Hz), 7.39–7.26 (4H, m), 7.28 (1H, s), 7.06 (1H, t, J = 7.45 Hz), 7.03 (1H, d, J = 8.35 Hz), 7.01 (1H, dd, J = 5.65 and 0.50 Hz), 6.87 (1H, dd, J = 2.45 Hz), 4.02 (3H, s), 3.95 (3H, s); ATR-FT-IR ṽ (cm−1): 3095 (aromatic CH), 1608 (aromatic CC), 1592 (aromatic CC), 1528–1294 (CN), 1246 (C–O); HRMS (APCI) calcd for C31H25N2O2S [M + H]+: 489.1631, found 489.1629.
(Z)-3-(3-Methoxythiophen-2-yl)-1-((5-(3-methoxythiophen-2-yl)-3-phenyl-1H-pyrrol-2-yl)methylene)-1H-isoindole (12d).
Compound 12d was synthesized using 8b (1.33 g, 5.18 mmol) and 9b (1.30 g, 5.08 mmol) in a similar manner as the case of 12a. The product was chromatographed on silica gel (C-300) as toluene as an eluent to give 1.98 g of 12d as a gray solid in 60% yield. M.p. 184 °C; 1H NMR (500 MHz, CDCl3): δ (ppm) 12.23 (s, 1H), 8.26–8.24 (m, 1H), 7.72 (dt, J = 7.50 and 0.95 Hz, 1H), 7.58 (dt, J = 8.05 and 1.53 Hz, 2H), 7.49 (tt, J = 7.67 and 1.61 Hz, 2H), 7.42 (d, J = 5.45 Hz, 1H), 7.39–7.34 (m, 2H), 7.31 (td, J = 7.45 and 1.05 Hz, 1H), 7.23 (s, 1H), 7.15 (d, J = 5.45 Hz, 1H), 7.01 (d, J = 5.55 Hz, 1H), 6.93 (d, J = 5.50 Hz, 1H), 6.63 (s, 1H), 4.03 (s, 3H), 4.01 (s, 3H); 13C NMR (126 MHz, CDCl3): δ = 157.7, 157.0, 154.3, 144.4, 141. 7, 135.7, 135.1, 134.1, 132.1, 129.0, 128.7, 128.2, 127.8, 127.3, 126.9, 125.8, 123.8, 122.2, 118.9, 117.7, 117.0, 116.7, 113.0, 112.6, 108.8, 59.17, 58.81; ATR-FT-IR ṽ (cm−1): 3383–3208 (N–H), 3101 (aromatic CH), 1593 (aromatic CC), 1550 (aromatic CC), 1461–1299 (CN), 1246 (C–O) HRMS (APCI) calcd for C29H22N2O2S2 [M]+: 494.1117, found 494.1107.
(Z)-1-((3,5-Diphenyl-1H-pyrrol-2-yl)methylene)-3-phenyl-1H-isoindole (12e).
Compound 12e was synthesized using 8c (0.636 g, 2.88 mmol) and 9c (0.631 g, 2.88 mmol) in a similar manner as the case of 12a. The product was purified by reprecipitation with CH2Cl2 and hexane to give 1.07 g of 12e as a red solid in 88% yield. M.p. 206 °C; 1H NMR (500 MHz, CDCl3): δ (ppm) 12.84 (1H, s), 8.21 (2H, dd, J = 8.25 and 1.25 Hz), 7.98 (1H, d, J = 7.65 Hz), 7.82 (1H, d, J = 7.65 Hz), 7.72 (2H, dd, J = 8.28 and 1.13 Hz), 7.63–7.60 (2H, m), 7.56 (1H, tt, J = 7.28 and 1.72 Hz), 7.54–7.50 (2H, m), 7.48–7.44 (2H, m), 7.44–7.36 (m, 3H), 7.38 (1H, s), 7.32 (1H, tt, J = 7.40 and 1.26 Hz), 6.78 (s, 1H); 13C NMR (126 MHz, CDCl3): δ (ppm) 164.2, 144.8, 141.8, 138.3, 135.6, 135.3, 134.9, 134.8, 131.5, 130.0, 129.3, 129.1, 128.9, 128.8, 128.8, 128.6, 127.8, 127.6, 127.1, 126.4, 124.6, 122.5, 119.6, 114.8, 108.5; ATR-FT-IR ṽ (cm−1): 3320–3132 (N–H), 3041 (aromatic CH), 1615 (aromatic CC), 1610 (aromatic CC), 1495–1315 (CN); HRMS (APCI) calcd for C31H22N2 [M]+: 422.1778, found 422.1762.
Difluoroboron chelated (Z)-3-(2-methoxyphenyl)-1-((5-(2-methoxyphenyl)-3-phenyl-1H-pyrrol-2-yl)methylene)-1H-isoindole (13a).
To a solution of 12a (2.70 g, 5.60 mmol) and NEt3 (2.1 mL, 15.2 mmol) in dry toluene was added BF3·OEt2 (5.50 mL, 44.56 mmol) at 80 °C under a N2 atmosphere. The resultant solution was stirred at 100 °C for 2 h. After quenching the reaction by water, the solution was extracted with CH2Cl2. The organic phase was dried with Na2SO4 and evaporated. In this way, 2.59 g of 13a as crude product was obtained in 87% yield. A part of the sample was chromatographed on silica gel (Wakogel C-300) using CH2Cl2:hexane (2:1 v/v) as an eluent to give a red solid. M.p. 266 °C; 1H NMR (500 MHz, DMSO-d6): δ (ppm) 8.34 (d, J = 8.15 Hz, 1H), 8.11 (s, 1H), 7.69–7.67 (m, 2H), 7.64 (ddd, J = 6.31, 1.80 and 1.73 Hz, 1H), 7.56 (t, J = 7.63 Hz, 2H), 7.56–7.52 (m, 1H), 7.48–7.43 (m, 3H), 7.41–7.36 (m, 2H), 7.34 (ddd, J = 7.18, 1.28 and 1.28 Hz, 1H), 7.22 (d, J = 8.25 Hz, 1H), 7.08 (td, J = 7.49 and 0.78 Hz, 1H), 7.06 (dd, J = 8.55 and 0.75 Hz, 1H), 6.94 (td, J = 7.45 and 0.92 Hz, 1H), 6.62 (s, 1H), 3.74 (s, 3H), 3.70 (s, 3H); 13C NMR (126 MHz, CDCl3): δ (ppm) 157.6, 157.5, 157.0, 148.1, 139.8, 135.3, 134.8, 132.7, 132.0 (t, JC–F = 4.09 Hz), 131.8, 131.8, 131.6, 131.5, 130.4, 129.8, 129.0, 128.9, 127.7, 126.0, 124.4, 122.7, 120.5, 120.1, 119.8, 119.4, 119.3, 118.1, 111.2, 111.0, 55,81, 55.76; ATR-FT-IR ṽ (cm−1): 3049 (aromatic CH), 1603 (aromatic CC), 1588 (aromatic CC), 1464 (B–F), 1433–1291 (CN), 1308 (B–N), 1235 (C–O); HRMS (APCI) calcd for C33H25BF2N2O2 [M]+: 530.1977, found 530.1983.
Difluoroboron chelated (Z)-3-(2-methoxyphenyl)-1-((5-(3-methoxythiophen-2-yl)-3-phenyl-1H-pyrrol-2-yl)methylene)-1H-isoindole (13b).
Compound 13b was synthesized using 12b (3.00 g, 6.14 mmol) in a similar manner as the case of 13a. The product was purified by reprecipitation with CH2Cl2 and hexane to give 2.33 g of 13b in 70% yield as a bluish purple. M.p. 251 °C; 1H NMR (CDCl3, 400 MHz): δ (ppm) = 7.82 (1H, d, J = 8.08 Hz), 7.70 (1H, dt, J = 7.60 and 2.02 Hz), 7.58–7.56 (m, 2H), 7.53 (1H, s), 7.53–7.42 (4H, m), 7.40 (1H, d, J = 7.96 Hz), 7.28 (1H, d, J = 5.64 Hz), 7.28–7.24 (1H, m), 7.13 (1H, td, J = 7.50 and 0.79 Hz), 7.05 (1H, d, J = 8.52 Hz), 7.03 (1H, s), 6.83 (1H, d, J = 5.60 Hz), 3.90 (3H, s), 3.73 (3H, s); 13C NMR (CDCl3, 126 MHz): δ (ppm) = 157.8, 157.8, 155.8, 143.2, 140.4, 134.8, 134.7, 132.6, 132.0 (d, JC–F = 7.3 Hz), 131.6, 131.1, 130.9, 130.1, 129.0, 128.9, 127.8, 126.8, 125.9, 124.4, 120.5, 119.6, 119.3, 118.5, 118.3, 115.3, 111.8, 111.3; 11B NMR (CDCl3, 160 MHz): δ (ppm) = 1.54 (t, JB–F = 31.9 Hz); ATR-FT-IR ṽ (cm−1): 3043 (aromatic CH), 1617 (aromatic CC), 1587 (aromatic CC), 1519 (B–F), 1524–1317 (CN), 1313 (B–N), 1212 (C–O); HRMS (APCI) calcd for C31H24BF2N2O2S [M + H]+: 537.1620, found 537.1611.
Difluoroboron chelated (Z)-1-((5-(2-methoxyphenyl)-3-phenyl-1H-pyrrol-2-yl)methylene)-3-(3-methoxythiophen-2-yl)-1H-isoindole (13c).
Compound 13c was synthesized using 12c (0.723 g, 1.480 mmol) in a similar manner as the case of 13a. However, dry CH2Cl2 was used as reaction solvent for overnight stirring instead of dry toluene due to its solubility. The product was purified by reprecipitation with CH2Cl2 and MeOH to give 0.598 g of 13c as a purple solid in 75% yield. M.p. 224 °C; 1H NMR (500 MHz, CDCl3): δ (ppm) 7.82 (1H, d, J = 8.05 Hz), 7.78 (1H, dd, J = 7.63 and 0.98 Hz), 7.68 (1H, d, J = 8.15 Hz), 7.60 (2H, dd, J = 8.10 and 1.15 Hz), 7.57 (1H, s), 7.53–7.48 (4H, m), 7.42 (1H, tt, J = 7.41 and 1.44 Hz), 7.36–7.30 (2H, m), 7.04 (1H, td, J = 7.55 and 0.95 Hz), 6.96 (1H, d, J = 8.30 Hz), 6.92 (1H, d, J = 5.60 Hz), 6.64 (1H, s), 3.82 (3H, s), 3.81 (3H, s); 11B NMR (160 MHz, CDCl3): δ (ppm) 1.49 (t, JB–F = 31.79 Hz); ATR-FT-IR ṽ (cm−1): 3072 (aromatic CH), 1617 (aromatic CC), 1594 (aromatic CC), 1467 (B–F), 1509–1370 (CN), 1309 (B–N), 1219 (C–O); HRMS(APCI) calcd for C31H23BF2N2O2S [M]+: 535.1541, found 535.1543.
Difluoroboron chelated (Z)-3-(3-methoxythiophen-2-yl)-1-((5-(3-methoxythiophen-2-yl)-3-phenyl-1H-pyrrol-2-yl)methylene)-1H-isoindole (13d).
Compound 13d was synthesized using 12d (0.950 g, 1.92 mmol) in a similar manner as the case of 13a except for overnight stirring. The product was purified by reprecipitation with CH2Cl2 and MeOH to give 1.01 g of 13d as a gold solid in 91% yield. M.p. 232 °C; 1H NMR (500 MHz, CDCl3): δ (ppm) 7.80 (dd, J = 8.08 and 0.78 Hz, 1H), 7.69 (dd, J = 8.10 and 0.80 Hz, 1H), 7.60–7.56 (m, 3H), 7.53–7.50 (m, 2H), 7.48 (td, J = 7.53 and 1.28 Hz, 1H), 7.48 (s, 1H), 7.43 (tt, J = 7.36 and 1.42 Hz, 1H), 7.36 (d, J = 5.60 Hz, 1H), 7.31 (ddd, J = 7.11, 0.90 and 0.88 Hz, 1H), 7.11 (s, 1H), 6.98 (d, J = 5.60 Hz, 1H), 6.88 (d, J = 5.55 Hz, 1H), 3.93 (s, 3H), 3.83 (s, 3H); 13C NMR (126 MHz, CDCl3): δ (ppm) 158.9, 158.2, 148.3, 143.8, 140.5, 134.9, 134.6, 132.1, 131.5, 131.4, 130.1, 129.9, 129.0, 128.9, 127.9, 127.2, 125.9, 125.0, 119.2, 118. 5, 117.4, 115.8, 115.3, 111.8, 108.5, 59.05, 58.90; ATR-FT-IR ṽ (cm−1): 3087 (aromatic CH), 1614 (aromatic CC), 1588 (aromatic CC), 1523 (B–F), 1523–1349 (CN), 1311 (B–N), 1215 (C–O); HRMS(APCI) calcd for C29H21BF2N2O2S2 [M]+: 542.1105, found 542.1101.
Difluoroboron chelated (Z)-1-((3,5-diphenyl-1H-pyrrol-2-yl)methylene)-3-phenyl-1H-isoindole (5).
Compound 5 was synthesized using 12e (0.904 g, 2.14 mmol) in a similar manner as the case of 13a. The product was chromatographed on silica gel (Wakogel C-300) using CH2Cl2 as an eluent to give 0.849 g of 5 as a green solid in 84% yield. M.p. 250 °C; 1H NMR (500 MHz, CDCl3): δ (ppm) 7.89–7.83 (5H, m), 7.66–7.65 (1H, m), 7.65 (1H, s), 7.61–7.52 (1H, m), 7.45 (1H, tt, J = 7.38 and 1.51 Hz), 7.42–7.33 (4H, m), 6.62 (s, 1H); 13C NMR (126 MHz, CDCl3): δ (ppm) 158.7, 152.4, 140.8, 135.9, 134.5, 133.3, 132.2, 131.5, 131.3, 130.8, 130.5, 130.2 (t, JC–F = 2.95 Hz), 130.0, 129.1 (t, JC–F = 3.06 Hz), 129.0, 128.9, 128.6, 128.4, 128.1, 128.0, 126.7, 124.6, 120.0, 119.5, 116.8 (C14); 11B NMR (160 MHz, CDCl3): δ (ppm) 1.76; FAB-MS: m/z = 470 [M]+; ATR-FT-IR ṽ (cm−1): 3044 (aromatic CH), 1617 (aromatic CC), 1590 (aromatic CC), 1462 (B–F), 1515–1374 (CN), 1315 (B–N); UV/vis (THF) λmax = 589 nm (εmax = 7.25 × 104 M−1 cm−1); elemental analysis for C31H21BF2N2: C, 79.17; H, 4.50; N, 5.96. Found: C, 79.37; H, 4.54; N, 6.05.
Boron chelated (Z)-3-(2-hydroxyphenyl)-1-((5-(2-hydroxyphenyl)-3-phenyl-1H-pyrrol-2-yl)methylene)-1H-isoindole (1).
To a solution of 13a (2.01 g, 3.78 mmol) in dry CH2Cl2 (377 mL) under a N2 atmospheric icy condition was added 1 M BBr3 in CH2Cl2 (18 mL, 18 mmol). The resultant solution was stirred for 2 h and quenched by adding sat NaHCO3 aq. (150 mL) to partition it into organic and water phases. The organic phase was dried in Na2SO4 and evaporated to give 1.09 g of demethylated dipyrrin as a crude product (FAB-MS: m/z = 454[M]+). To a solution of the product (1.01 g, 2.20 mmol) and diisopropylethylamine (DIPEA) (4.1 mL, 24.1 mmol) in dry toluene (220 mL) under a N2 atmosphere was added BF3·OEt2 (4.0 mL, 32.4 mmol). The resultant solution was refluxed for 2 h. After the reaction was quenched by adding iced water and extracted with CH2Cl2. The organic phase was dried with Na2SO4 and evaporated. The residue was chromatographed on silica gel (Wakogel C-300) with toluene as an eluent and reprecipitated with CH2Cl2 and hexane to give 1 as purplish-red in 21% yield from 12a. M.p. 284 °C; 1H NMR (500 MHz, CDCl3): δ (ppm) 8.29 (2H, dd, J = 7.93 and 1.88 Hz), 7.96 (1H, d, J = 7.00 Hz), 7.82 (1H, dd, J = 7.75 and 1.60 Hz), 7.66–7.64 (m, 2H), 7.61 (1H, ddd, J = 8.01, 7.12 and 0.86 Hz), 7.54 (s, 1H), 7.54–7.42 (5H, m), 7.28 (1H, ddd, J = 8.53, 7.01 and 1.23 Hz), 7.19 (1H, td, J = 7.63 and 1.15 Hz), 7.09–7.05 (2H, m), 6.95 (1H, s), 6.91 (1H, dd, J = 8.23 and 0.93 Hz);13C NMR (126 MHz, CDCl3): δ (ppm) 156.0, 153.1, 148.3, 145.1, 140.9, 135.7, 134.3, 133.9, 133.9, 131.2, 130.8, 130.2, 130.0, 129.7, 129.1, 128.7, 128.0, 126.9, 125.1, 124.1, 120.7, 120.6, 120.4, 120.3, 120.2, 119.5, 119.4, 115.8, 110.5; 11B NMR (128 MHz, CDCl3): δ (ppm) −0.297; FAB-MS: m/z = 462[M]+; ATR-FT-IR ṽ (cm−1): 3049 (aromatic CH), 1600 (aromatic CC), 1563 (aromatic CC), 1481–1280 (CN), 1315 (B–O and B–N), 1237 (C–O); UV/vis (THF) λmax = 663 nm (εmax = 5.61 × 104 M−1 cm−1); Elemental analysis for C31H19BN2O2·0.25H2O: C, 79.76; H, 4.21; N, 6.00. Found: C, 79.46; H, 4.06; N, 5.97.
Boron chelated (Z)-3-(2-hydroxyphenyl)-1-((5-(3-hydroxythiophen-2-yl)-3-phenyl-1H-pyrrol-2-yl)methylene)-1H-isoindole (2).
Compound 2 was synthesized using 13b (1.60 g, 2.99 mmol) in a similar manner as 1. The purification was carried out by silica gel chromatography with benzene as an eluent. 0.331 g of 2 was obtained as a green solid in 24% from 13b. M.p. 286 °C; 1H NMR (CDCl3, 500 MHz): δ (ppm) = 8.28 (1H, d, J = 8.20 Hz), 8.25 (1H, dd, J = 7.90 and 1.55 Hz), 7.95 (1H, d, J = 8.05 Hz), 7.65–7.63 (2H, m), 7.57 (1H, ddd, J = 7.93, 7.08 and 0.88 Hz), 7.54–7.50 (2H, m), 7.50–7.43 (3H, m), 7.47 (1H, s), 7.31 (1H, d, J = 5.30 Hz), 7.18 (1H, td, J = 7.59 and 0.98 Hz), 7.12 (1H, dd, J = 8.33 and 0.98 Hz), 6.73 (1H, d, J = 5.10 Hz), 6.73 (s, 1H). 13C NMR (CDCl3, 126 MHz): δ (ppm) = 155.6, 155.4, 146.2, 144.0, 142.1, 135.1, 134.0, 133.1, 130.5, 130.1, 129.6, 129.5, 129.1, 128.8, 128.3, 126.8, 126.6, 123.7, 120.8, 120.3, 120.3, 120.2, 119.7, 114.6, 111.5, 109.6. 11B NMR (128 MHz, CDCl3): δ (ppm) 0.4467 vs. BF3·OEt2. FAB-MS: m/z = 468 [M]+; ATR-FT-IR ṽ (cm−1): 3094 (aromatic CH), 1610 (aromatic CC), 1561 (aromatic CC), 1485–1377 (CN), 1320 (B–O and B–N), 1188 (C–O); UV/vis (THF) λmax = 699 nm (εmax = 6.03 × 104 M−1 cm−1); elemental analysis for C29H17BN2O2S·0.2H2O: C, 73.80; H, 3.72; N, 5.94. Found: C, 73.70; H, 3.67; N, 5.96.
Boron chelated (Z)-1-((5-(2-hydroxyphenyl)-3-phenyl-1H-pyrrol-2-yl)methylene)-3-(3-hydroxythiophen-2-yl)-1H-isoindole (3).
Compound 3 was synthesized using 13c (0.551 g, 1.03 mmol) in a similar manner as the case of 1. The purification was carried out by silica gel chromatography with CH2Cl2 as an eluent and reprecipitated with CH2Cl2 and hexane to give 0.113 g of 3 as a black solid in 27% yield from 13c. M.p. 279 °C (determined using the sublimed sample); 1H NMR (500 MHz, CDCl3): δ (ppm) 8.08 (1H, d, J = 7.90 Hz), 7.92 (1H, d, J = 7.95 Hz), 7.79 (1H, dd, J = 7.67 and 1.63 Hz), 7.66 (1H, d, J = 5.30 Hz), 7.65–7.61 (3H, m), 7.53–7.49 (3H, m), 7.42–7.39 (1H, m), 7.41 (1H, s), 7.27–7.24 (1H, m), 7.06 (1H, td, J = 7.49 and 1.05 Hz), 6.98 (1H, dd, J = 8.10 and 0.95 Hz), 6.91 (1H, s), 6.87 (1H, d, J = 5.20 Hz); 13C NMR (126 MHz, CDCl3): δ (ppm) 162.1, 152.9, 148.3, 143.0, 138.5, 135.6, 134.8, 133.6, 132.1, 131.0, 129.9, 129.1, 129.0, 128.6, 127.6, 127.1, 124.7, 123.6, 120.7, 120.6, 120.4, 120.3, 119.5, 112.5, 110.5, 109.6. 11B NMR (128 MHz, CDCl3): δ (ppm) 0.5944 vs. BF3·OEt2; FAB-MS: m/z = 468[M]+; ATR-FT-IR ṽ (cm−1): 3070 (aromatic CH), 1605 (aromatic CC), 1558 (aromatic CC), 1507–1427(CN), 1313 (B–O and B–N), 1244 (C–O); UV/vis (THF) λmax = 669 nm (εmax = 4.82 × 104 M−1 cm−1); elemental analysis for C29H17BN2O2S·0.75H2O·0.75CH2Cl2: C, 65.50; H, 3.70; N, 5.14. Found: C, 65.71; H, 3.43; N, 5.17.
Boron chelated (Z)-3-(3-hydroxythiophen-2-yl)-1-((5-(3-hydroxythiophen-2-yl)-3-phenyl-1H-pyrrol-2-yl)methylene)-1H-isoindole (4).
To a solution of 13d (0.931 g, 1.72 mmol) in dry CH2Cl2 (175 mL) under a N2 atmospheric icy condition was added 1 M BBr3 in CH2Cl2 (8.7 mL, 8.8 mmol). The resultant solution was stirred for overnight and quenched by adding sat NaHCO3 aq. to partition it into organic and water phases. The organic phase was dried in Na2SO4 and evaporated to give 0.688 g of demethylated dipyrrin as a crude product (FAB-MS: m/z = 466 [M]+). To a solution of the product (0.614 g, 1.32 mmol) in dry THF (132 mL) under a N2 atmosphere was added B(OMe)3 (1.47 mL, 13.2 mmol). The resultant solution was refluxed overnight. After evaporation and adding hexane, obtained solid was reprecipitated with CH2Cl2. In this way, 0.480 of 4 was obtained as a green solid in 77% yield. The assignment data was collected employing 4 that was chromatographed on 6 wt% NH3-pretreated silica gel (C-300) using toluene as an eluent. M.p. 276 °C (determined using the sublimed sample); 1H NMR (500 MHz, CDCl3): δ (ppm) 8.06 (1H, d, J = 8.05 Hz), 7.91 (1H, d, J = 8.00 Hz), 7.63–7.58 (4H, m), 7.52–7.47 (3H, m), 7.41 (1H, tt, J = 7.41 and 1.47 Hz), 7.35 (1H, s), 7.24 (1H, d, J = 5.20 Hz), 6.90 (1H, d, J = 5.35 Hz), 6.78 (1H, d, J = 5.25 Hz), 6.67 (s, 1H); 13C NMR (126 MHz, CDCl3): δ (ppm) 160.9, 154.4, 146.9, 141.8, 139.6, 135.2, 134.5, 132.3, 131.6, 130.5, 129.3, 129.0, 128.7, 128.4, 127.8, 126.8, 125.1, 123.3, 120.4, 120.3, 112.0, 110.5, 108.4; 11B NMR (128 MHz, CDCl3): δ (ppm) 1.22; FAB-MS: m/z = 474 [M]+; ATR-FT-IR ṽ (cm−1): 3082 (aromatic CH), 1605 (aromatic CC), 1560 (aromatic CC), 1502–1430 (CN), 1310 (B–O and B–N), 1222 (C–O); UV/vis (THF) λmax = 709 nm (εmax = 4.10 × 104 M−1 cm−1); elemental analysis for C27H15BN2O2S2·0.67H2O: C, 66.69; H, 3.38; N, 5.76. Found: C, 66.78; H, 3.42; N, 5.79.
Determination of fluorescence quantum yield
The fluorescence quantum yield (Φexp) was calculated from the eqn (1).83| | 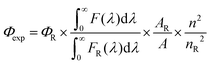 | (1) |
where F(λ) and FR(λ) describe the measured fluorescence intensities of the dye and the reference, respectively, and A and AR describe the corresponding absorbances at the excitation wavelength. The reference used was zinc phthalocyanine (ΦR = 0.23 in THF)84 for 1–4 and cresyl violet (ΦR = 0.54 in MeOH)85 for 5. The refractive indexes are n = 1.407 for THF and 1.329 for MeOH.
Theoretical calculation
All geometries of the dyes at the ground state were fully optimized by means of the CAM-B3LYP/6-31G(d,p) level method, Density functional theory (DFT) calculations at the CAM-B3LYP/6-31G(d,p) level were performed in the Gaussian 16. Gaussian 16 software.86 These molecular orbitals were visualized using Gauss view 6.0.16 program. NICS values were calculated using GIAO CAM-B3LYP/6-31G(d,p) level of theory at 0.0 Å [NICS(0)] from the center of in isoindole and pyrrole rings in the dyes.
Device fabrication
The dye 2 film with a thickness of 90 nm were formed on the surface of the indium tin oxide (ITO) glass substrate using a resistance heating type vacuum deposition method. Subsequently, the devices were completed by evaporating a 100 nm thick aluminum film as an electrode. Current–voltage (J–V) measurements of the dye-loaded devices were performed using a Keithley 4200-SCS source meter under irradiation at 750 nm (128 μW cm−2) by means of a PVL-3300 (Asahi Spectra Co., Ltd) for the device with 2.
Results and discussion
Synthesis and structural analysis
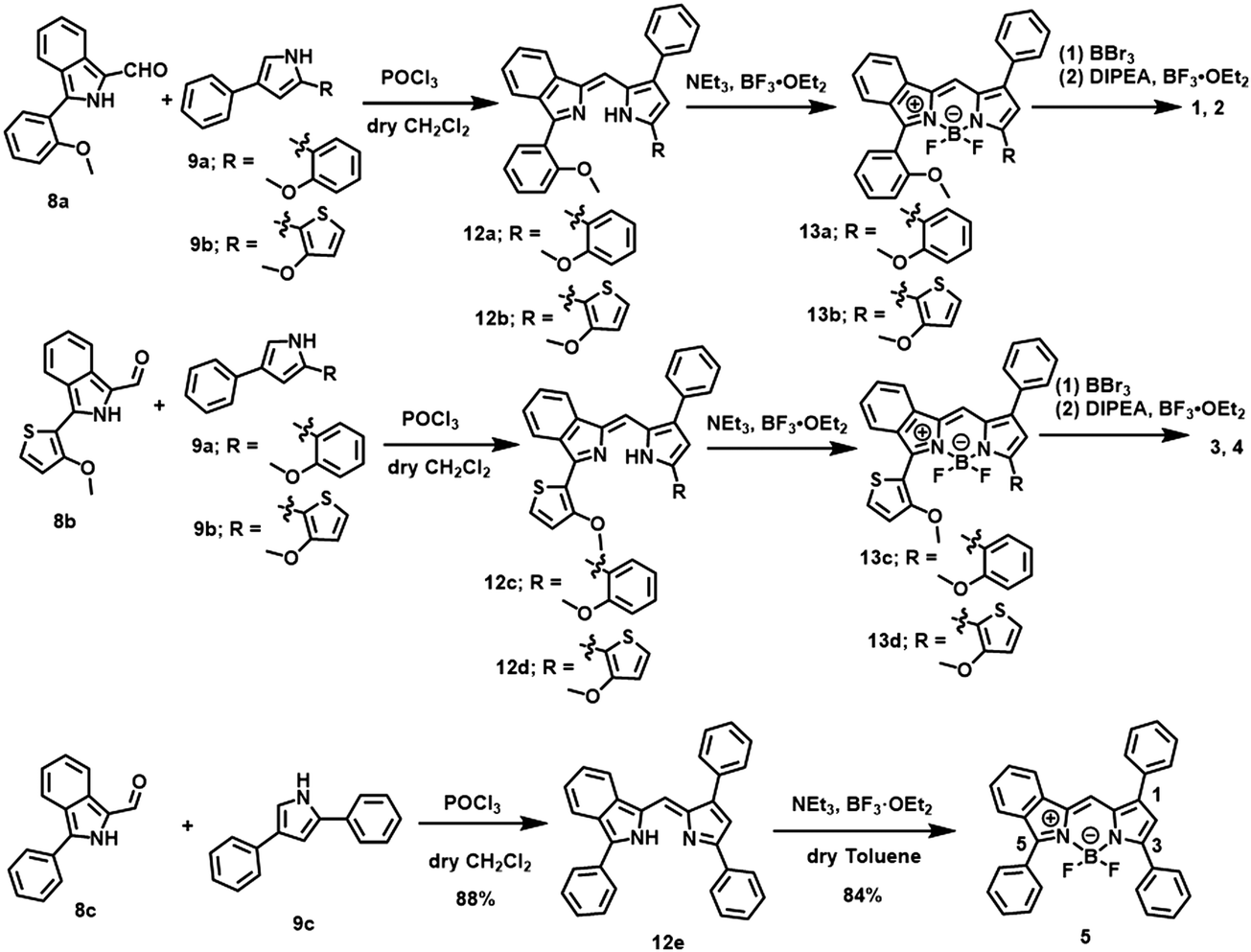
The path for synthesis of the targeted dyes is summarized in Scheme 1, where the preparation of the control dye 5 is also included. The key synthons, 1-formyl-3-(2-methoxyphenyl or 2-methoxythionyl)isoindoles 8a and 8b were prepared in 74% and 64% yield, respectively, by the Suzuki-cross coupling reaction of 677 with 7a and 7b (Scheme S1, ESI†). Another synthon, 2-(2-methoxythiony)-4-phenyl-1H-pyrrole 9b, was prepared from 10 through a two-step process48 (Scheme S1, ESI†). The subsequent nucleophilic addition reaction of 8 and 9 in dry CH2Cl2, followed by boron chelation, demethylation with BBr3, and N2O2 boron coordination in the presence of diisopropylethylamine (DIPEA) afforded targets 1–4. As a control, BF2 dye 5 was also prepared in a similar manner, except for demethylation and N2O2 boron coordination. The dyes were characterized by 1H and 13C NMR spectroscopy, mass spectrometry and elemental analysis.
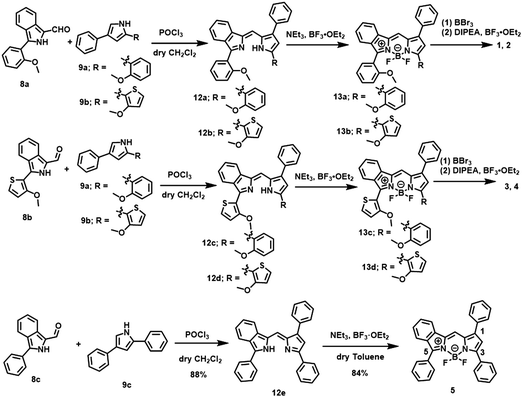 |
| | Scheme 1 Synthetic routes for of dyes 1–5. Reagent and reaction conditions; (i) POCl3, dry CH2Cl2, room temperature, 2 h. (ii) NEt3, BF3·OEt2, dry toluene, 100 °C, 2 h for 13a, 13b and 5. overnight for 13c and 13d (iii) BBr3, dry CH2Cl2, icy condition, 2 h and then DIPEA, BF3·OEt2, dry toluene, reflux, 2 h. (iv) BBr3, dry CH2Cl2, reflux, overnight and then B(OMe)3, dry THF, reflux, overnight. | |
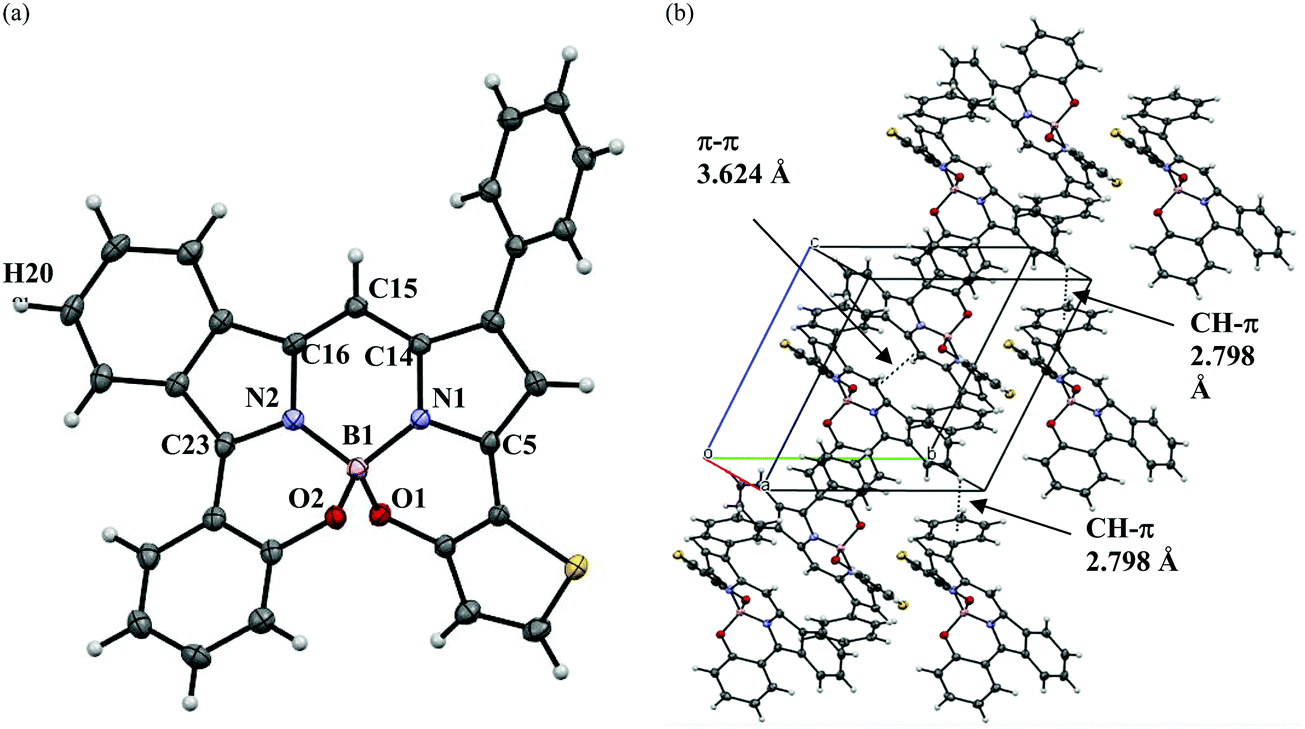
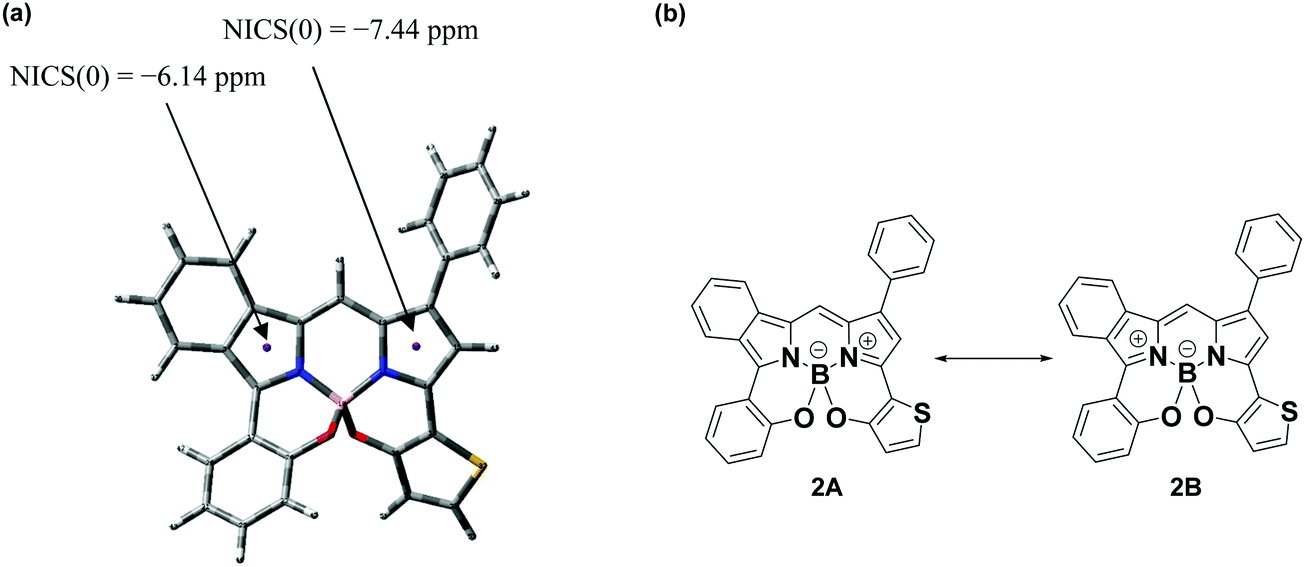
A suitable single crystal of 3-thieno-5-benzo-N2O2-type BODIPY 2 was obtained by the (solvent) vapor diffusion technique using CH2Cl2/hexane (1:1 v/v), where the crystals had triclinic crystal symmetry. The structure was analyzed by X-ray crystallography (CCDC no. 2024003†); the Oak Ridge thermal ellipsoid plot (ORTEP) diagram is presented in Fig. 2. Boron adopted a tetrahedral geometry in which it binds to the two nitrogen atoms of the isoindole and pyrrole rings, as well as two oxygens linked by thiophene and benzene rings. The N1–B1–O1 and N2–B1–O2 bite angles are 107.7(1)° and 106.4(1)°, respectively. Although the angles are somewhat smaller than that of prototype N2O2-boron chelated BODIPY reported by Burgess et al.,87 they are close to the ideal tetrahedral angle of 109.5°. The 3-thiophene ring is tilted by 29.6° relative to the pyrrole plane, where this angle is larger than the dihedral angle between the 5-benzene ring and the isoindole plane (Fig. S1, ESI†). Interestingly, the C15–C16 (1.385(2)) and C23–N2 (1.350(2)) bond distances (Å) are shorter than those of C15–C14 (1.405(3)) and C5–N1 (1.365(2)), respectively. NICS(0) calculation88 based on GIAO CAM B3LYP/6-31G(d,p) was conducted to understand the structural features. The NICS(0) value for the pyrrole unit of the isoindole ring was calculated to be −6.14, which is more positive than that of the pyrrole ring (−7.44), as shown in Fig. 3a. This suggests that the isoindole ring may possess azafulvene character, which means that dye 2 would primarily adopt resonance structure 2B in the resonance equilibration between 2A and 2B (Fig. 3b). Another interesting feature arises from the packing structure shown in Fig. 2b; the two dyes are present as a pair of enantiomers, where a more detailed representation is presented in Fig. S2 (ESI†). This is ascribable to the chiral peripherical environment at the sp3-boron center,89 where a dimer is formed through intermolecular π–π stacking interaction between the isoindole and pyrrole units, with a distance of 3.624 Å.90 Further, it was found that CH–π interactions were operative between isoindole-H (H20) and the phenyl ring at the 1 position of an adjacent dye with the same enantiomer (CH⋯benzene ring = 2.798 Å).
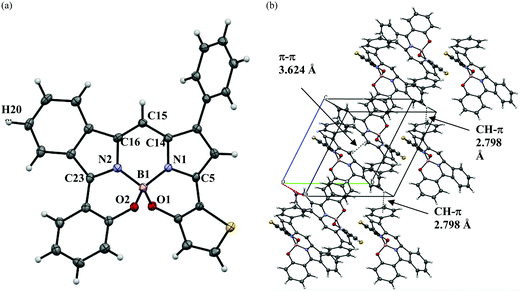 |
| | Fig. 2 ORTEP structure of 2, where thermal ellipsoids are drawn at the 50% probability levels. Front view (a) and packing structure (b). | |
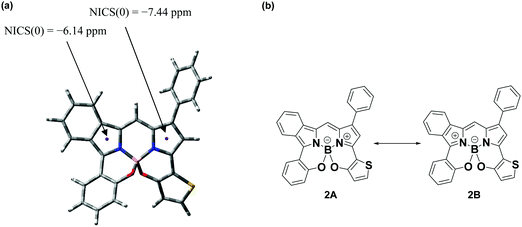 |
| | Fig. 3 NICS(0) value (a) and possible resonance structures of 2 (b). | |
Photophysical properties
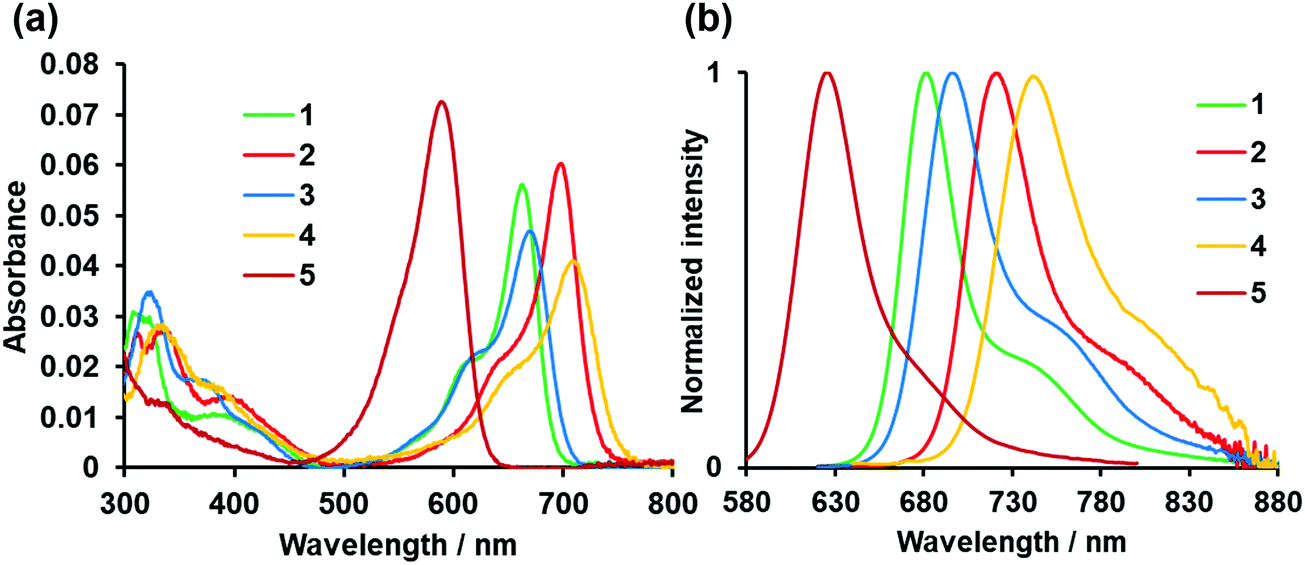
The photophysical properties of asymmetric N2O2-boron chelated dyes 1–4 and control dye 5 are shown in Fig. 4 and Table 1. Dye 1 absorbed visible light at 663 nm in THF, with a molar extinction coefficient (εmax) of 5.61 × 104 M−1 cm−1. A red-shift of 74 nm was observed relative to the absorption band of 5 as a result of intramolecular B,O chelation, and the εmax value declined somewhat. Replacement of the benzo[1,3,2]oxazaborinine unit with the corresponding thiophene counterpart further caused a red-shift in the absorption band, which depended on the position of thiophene in the chromophore; 2 with the thiophene ring at the 3 position absorbed far-red light with λmax at 699 nm (εmax = 6.03 × 104 M−1 cm−1), which is 30 nm longer than that of 5-thiophene derivative 3, for which the εmax value increased by 1.25 times. 3,5-Dithiophene-containing dye 4 showed the longest absorption band at 709 nm among the dyes tested herein, with longer wavelength absorption edges beyond 753 nm. This indicates that increasing the number of thiophene rings in the chromophore is beneficial for producing NIR dyes, although the εmax value was lower than that of dye 2. Fig. 4b displays the fluorescence spectra of the dyes when excited at 610 nm, showing a typical mirror-image relationship between the absorption and fluorescence spectra. Dye 1 emitted far-red light at 682 nm when excited at 610 nm in THF, with a fluorescence quantum yield (Φ) of 0.48. The lower Φ value compared to that of 5 is possibly due to increased nonradiative deactivation as the result of a smaller energy gap.16 It was also found that thiophene-based B,O chelation caused a red-shift in the emission spectra (Table 1). Consequently, 3,5-bis(thiophene)-incorporated dye 4 emitted NIR light at 741 nm, with a Φ value of 10% in THF. In this way, as λF was redshifted, Φ value was lowered, which is consistent with the trend of fluorescent lifetime (τ) (Table 1 and Fig. S3, ESI†). However, control dye 5 showed a relatively small value of τ (5.78 ns), which could be interrupted by the presence of free rotatable phenyl units at the 3,5 positions because of acceleration of non-radiative deactivation. Evaluation of the solvent dependency of the photophysical properties allowed us to find that the absorption and fluorescence spectra of the dyes were almost insensitive to the solvent polarity (Fig. S4, ESI†), indicating that these dyes undergo typical π–π* transition, as inferred from the small Stokes shift.
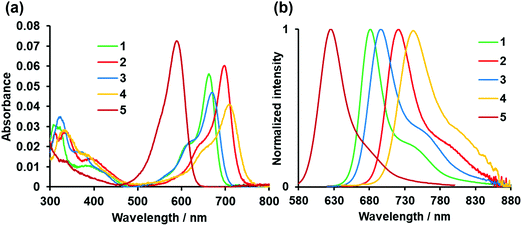 |
| | Fig. 4 Absorption (a) and fluorescence spectra (b) of dyes 1–5 (1 μM) in THF. λex = 610 nm for 1–4.λex = 546 nm for 5. | |
Table 1 Photophysical properties of dyes 1–5 (1 μM) in THF
| Dye |
λ
max/nm |
λ
onset/nm |
ε
max
/M−1 cm−1 |
FWHMb/nm |
λ
F
/nm |
Δsd/cm−1 |
Φ
F
/% |
τ
/ns |
|
Molar extinction coefficient.
Full-width at half maximum.
λ
ex = 610 nm for 1–4. λex = 546 nm for 5.
Δs = 1/λabs − 1/λem.
Fluorescence quantum yield.
Singlet state lifetime. λex = 630 nm.
λ
ex = 590 nm.
|
|
1
|
663 |
693 |
5.61 × 104 |
40 |
682 |
420 |
46 |
7.82 |
|
2
|
699 |
735 |
6.03 × 104 |
46 |
721 |
436 |
21 |
4.12 |
|
3
|
669 |
706 |
4.82 × 104 |
61 |
696 |
580 |
34 |
6.33 |
|
4
|
709 |
753 |
4.10 × 104 |
70 |
741 |
609 |
10 |
2.27 |
|
5
|
589 |
628 |
7.25 × 104 |
56 |
626 |
1003 |
91 |
5.78g |
Electrochemistry
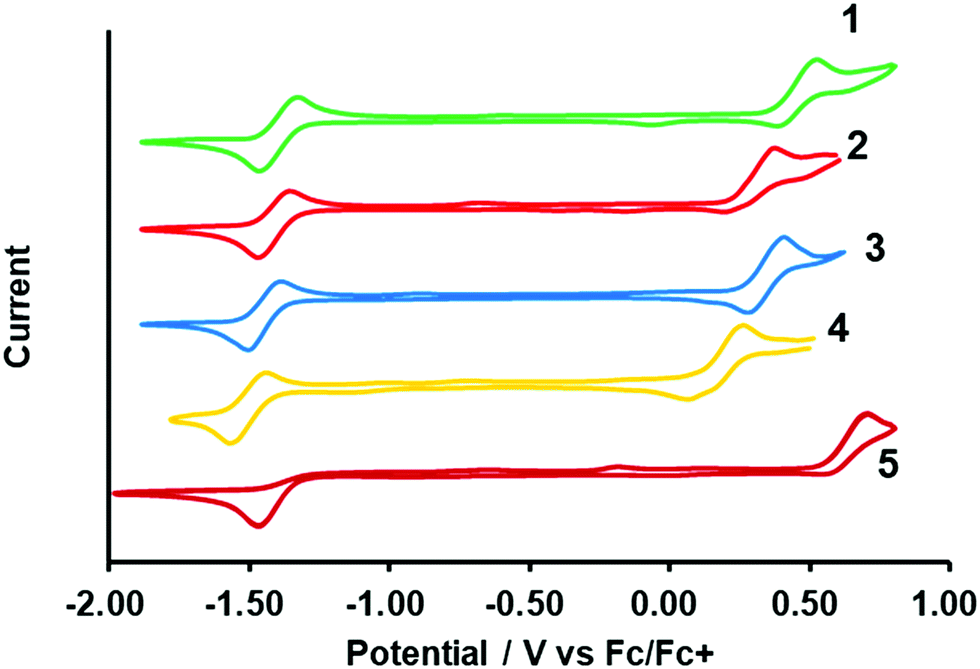
To better understand the photophysical character, the electrochemical properties of the dyes were investigated using cyclic voltammetry (CV), as shown in Fig. 5. The formal potential of Fc/Fc+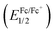 was 0.197 V versus Ag/Ag+. Reversible oxidation and reduction waves were observed for the N2O2-dyes (1–4) enabling us to determine the half-wave potentials ((Eox1/2) and (Ered1/2)) as summarized in Table 2. On another front, control 5 exhibited an irreversible reduction wave, from which the corresponding onset potentials were determined. The electrical gap of the dyes was inconsistent with the absorption bands observed in Fig. 4a, possibly due to some involvement of transitions other than the HOMO → LUMO transition in the absorption band. The HOMO and LUMO energy levels were estimated on the basis of those potential values (Table 2). When compared to BF2 dye 5, intramolecular B,O cyclization made the HOMO more negative than the LUMO, resulting in a narrower band gap and consequent red-shift in the absorption and fluorescence bands. It was also found that thiophene-insertion in the chromophore had a stronger impact on the HOMO level than on the LUMO. Notably, the HOMO level of 3-thiophene analogue 2 was higher-lying than that of 5-thiophene stereoisomer 3. Moreover, the HOMO level became more destabilized as the number of thiophene units in the chromophore increased. In this way, the HOMO levels were energetically higher in the following order: 4 > 2 > 3 > 1, resulting in a smaller HOMO–LUMO bandgap. The trend observed here was rationalized by theoretical calculation (vide infra).
was 0.197 V versus Ag/Ag+. Reversible oxidation and reduction waves were observed for the N2O2-dyes (1–4) enabling us to determine the half-wave potentials ((Eox1/2) and (Ered1/2)) as summarized in Table 2. On another front, control 5 exhibited an irreversible reduction wave, from which the corresponding onset potentials were determined. The electrical gap of the dyes was inconsistent with the absorption bands observed in Fig. 4a, possibly due to some involvement of transitions other than the HOMO → LUMO transition in the absorption band. The HOMO and LUMO energy levels were estimated on the basis of those potential values (Table 2). When compared to BF2 dye 5, intramolecular B,O cyclization made the HOMO more negative than the LUMO, resulting in a narrower band gap and consequent red-shift in the absorption and fluorescence bands. It was also found that thiophene-insertion in the chromophore had a stronger impact on the HOMO level than on the LUMO. Notably, the HOMO level of 3-thiophene analogue 2 was higher-lying than that of 5-thiophene stereoisomer 3. Moreover, the HOMO level became more destabilized as the number of thiophene units in the chromophore increased. In this way, the HOMO levels were energetically higher in the following order: 4 > 2 > 3 > 1, resulting in a smaller HOMO–LUMO bandgap. The trend observed here was rationalized by theoretical calculation (vide infra).
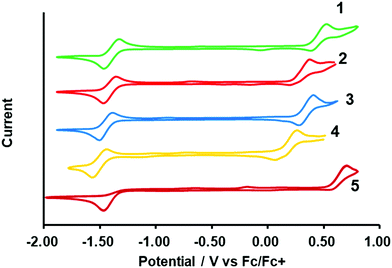 |
| | Fig. 5 Cyclic voltammograms of dyes (1 mM) in THF containing 0.1 M TBAPF6. Ferrocene was used as external standard. The scan rate is 0.05 V s−1. | |
Table 2 Electrochemical properties of dyes 1–5 in THF
| Dye |
E
red
1/2/V |
E
ox
1/2/V |
E
LUMO
/eV |
E
HOMO
/eV |
|
E
LUMO (eV) = −(4.8 + Ered1/2 [V vs. Fc/Fc+]).
E
HOMO (eV) = −(4.8 + Eox1/2 [V vs. Fc/Fc+]).
The value was calculated from each onset potential due to the irreversibility of the peaks in the voltammogram.
|
|
1
|
−1.40 |
0.446 |
−3.40 |
−5.25 |
|
2
|
−1.45 |
0.260 |
−3.35 |
−5.06 |
|
3
|
−1.44 |
0.341 |
−3.36 |
−5.14 |
|
4
|
−1.50 |
0.174 |
−3.30 |
−4.97 |
|
5
|
−1.33c |
0.629 |
−3.47 |
−5.43 |
Theoretical analysis
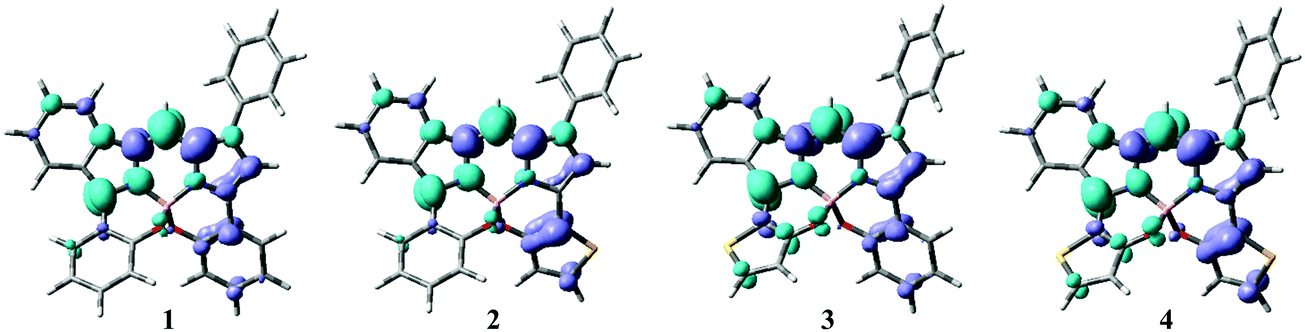
Time-dependent density functional theory (TD-DFT) and DFT analyses at the CAM-B3LYP/6-31G(d,p) level were conducted using Gaussian 16 software86 to understand the photophysical properties of the dyes. Tables S1–S5 (ESI†) summarize selected transitions from the theoretical data for the N2O2-type dyes (1–4) and 5. Based on the oscillator strength (f), the major absorption bands were from S0 → S1 transitions, corresponding to the HOMO → LUMO transition. Initially, the HOMO/LUMO energy gap of dye 1 was smaller than that of 5 by 0.20 eV. This observation supports the deduction that intramolecular B,O-chelation causes a red-shift in the absorption band. The electron-density distributions of the HOMO and LUMO levels are also presented in Tables S1–S5 (ESI†), which indicate that the π–π* transitions are primarily derived from both orbitals spread over the BODIPY backbone involving substituents at the 3 and/or 5 positions, as supported by the small Stokes shifts of the dyes (vide supra; Table 1). However, comparison of the surface plot of the HOMO with that of the LUMO suggests a decrease in the electron-density distributions of the 3-benzo(thieno)[1,3,2]oxazaborinine unit based on the energy of the HOMO → LUMO transition. In order to visualize the electron-density redistribution during the HOMO → LUMO transition, electron density difference plots (EDD) were employed (Fig. 6),91 indicative of partial charge-transfer character with a transition from the benzo(thieno)[1,3,2]oxazaborinine at the 3-position to the BODIPY core. Given the electron-donor character of thiophene, the electron density depletion of the 3-thiophene ring during the transition is larger than that of the 5-thiophene ring, indicating that introduction of the thiophene donor at the 3 position in dye 2 is more effective than that at the 5 position in 3. Accordingly, 3-thiophene dye 2 has a higher-lying HOMO than stereoisomer 3, which is consistent with the CV data. Such insight strongly supports the red-shift in the absorption and fluorescence bands of 2 compared to those of 3. Finally, the 3,5-dithiophene-containing dye 4 had the smallest calculated energy gap for the S0 → S1 transition, which reproduces the experimental result. Taken together, incorporation of thieno[1,3,2]oxazaborinine into the dye skeleton would not only contribute to expansion of the π–π* transition of the chromophore, but also tune the HOMO energy level. The broad absorption band ranging from 300 to 450 nm may include plural transitions of HOMO−1 → LUMO, HOMO → LUMO+1, and HOMO → LUMO+2.
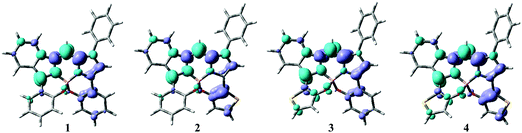 |
| | Fig. 6 EDD plots of HOMO → LUMO transition for the dyes. Turquoise and grey colors indicate electron density accumulation and depletion, respectively. | |
Optical resolution
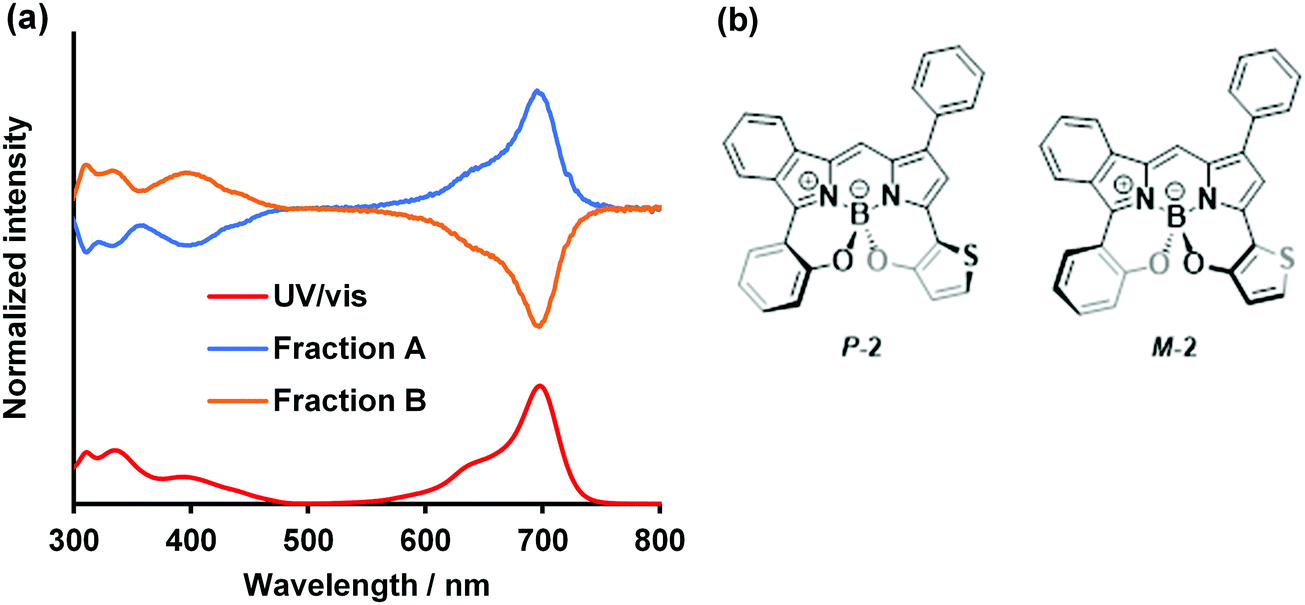
Another interesting feature of the N2O2-boron-chelated BODIPYs is the helicity with respect to the central boron atom (vide supra; Fig. 2).89 Optical resolution of dye 2 was conducted by preparative chiral HPLC (Daicel CHIRALPACK-IE) to successfully obtain the corresponding enantiomers (Fraction A and B; Fig. S11, ESI†). The normalized ECD spectra of each fraction, as well as the UV/vis absorption spectra in THF, are shown in Fig. 7. For Fraction A, a positive significant CD signal was observed in the far-red region with λmax of 699 nm, assignable to the S0 → S1 transition, whereas a relatively low-intensity negative CD signal was detectable in the shorter wavelength region (300–480 nm). In contrast, a mirror image CD spectrum was observed for Fraction B, indicating an enantiomeric counterpart of Fraction A. In order to determine the absolute configuration of each fraction, the CD spectrum of the P-isomer of 2 was calculated and compared with the experimental CD spectrum (Fig. S11, ESI†). Thus, Fraction A was assigned as the P-isomer. Accordingly, Fraction B is likely to be the M-isomer. Taken together, the CD intensity was derived from the BODIPY chromophore with a helical conformation of the peripherical boron center. The anisotropic factor g was determined to be 2.26 × 10−3 and 2.24 × 10−3 for the P-isomer and M-isomer, respectively. Chiral BODIPYs with a significant absorption band in the far-red region deserve further investigation in terms of chiroptical applications.92
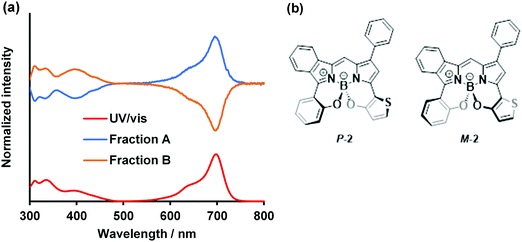 |
| | Fig. 7 (a) CD spectra and UV/vis spectrum (red) in THF. Blue and ocher lines indicate Fraction A and Fraction B, respectively. (b) Proposed structures of P-2 (blue line) and M-2 (orange line). [2] = 10 μM. | |
Fabrication of photodetector
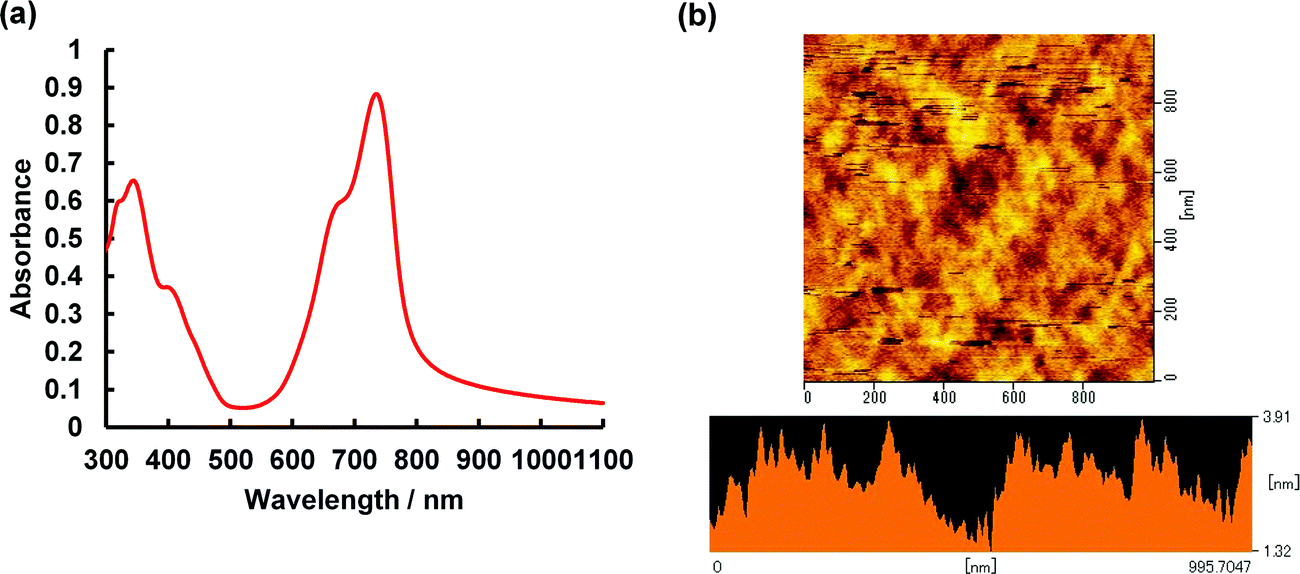
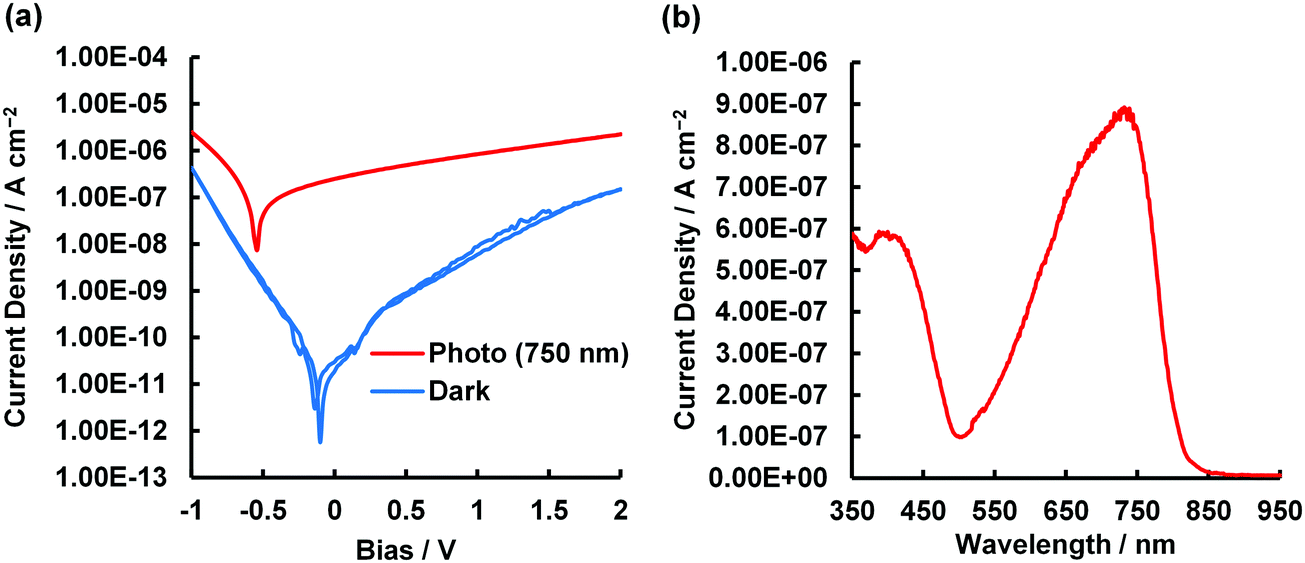
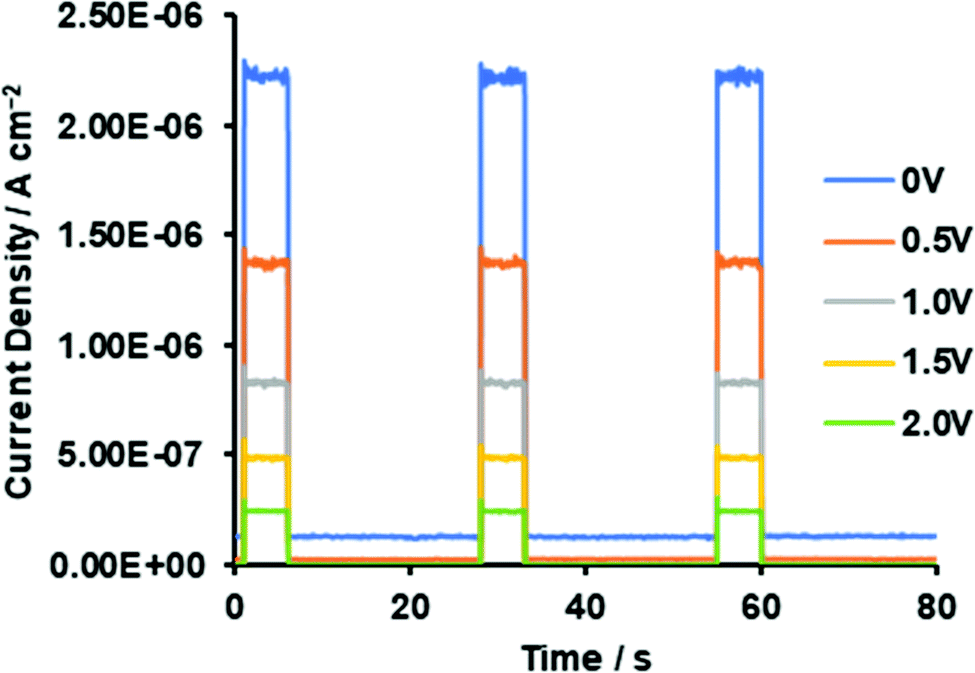
To determine if asymmetrical BODIPYs could serve as NIR photodetector materials, the 3-thiophene-5-benzene-containing dye 2 was selected as a plausible sensitizer for targeted device application. This is because 2 absorbs more strongly far-red and near infrared light, as shown in Fig. 4a (λmax = 699 nm, εmax = 6.03 × 104 M−1 cm−1). Prior to device fabrication, the thermal properties of 2 were investigated by differential scanning calorimetry (DSC) and thermal gravimetric analysis (TGA) (Fig. S12, ESI†); the observed melting point of 2 was 286 °C. Based on the thermal analysis, resistance-heating type vacuum deposition was employed to prepare a dye film on a quartz plate. The absorption spectrum of the film was measured, which showed a near-infrared absorption band at 735 nm and a λonset value of 787 nm (Fig. 8a). Compared to that in THF, a bathochromic shift of 36 nm was detected. An amorphous film was successfully formed on an ITO glass substrate in a similar method, and a flat film with a root mean square (RMS) thickness of 0.54 nm was obtained, as inferred from the atomic force microscopy (AFM) image of the 1 × 1 μm area (Fig. 8b). The ionization energy of 5.30 eV for the film was determined by photoelectron spectroscopy93 under ambient conditions (Fig. S13, ESI†), where the energy was transferred to the HOMO energy level as −5.30 eV. The value is lower than that of the ITO electrode (−5.0 eV). On another front, the LUMO energy level determined from the λonset value of the film was −3.72 eV, which is higher than the work function of Al (−4.3 eV). Taken together, photo-generated electron and hole injection would be energetically favorable enough to provide photodetector functionality. The current–voltage (J–V) measurements for the dye-loaded device under photoirradiation at 750 nm (128 μW cm−2) are shown in Fig. 9a. The 2-loaded device produced a photocurrent of 8.42 × 10−7 A cm−2 at a bias potential of 1 V. Compared to the dark current, the on/off current ratio at the same bias potential was determined to be 1.42 × 102. In this way, an increase in the forward injection of current under NIR irradiation was clearly detected. Fig. 9b shows the wavelength-dependent photocurrent densities of the 2-loaded device (ITO/2/Al); the spectral profile (with a maximum photocurrent at 735 nm) was almost the same as the absorption band of 2 in the film. Thus, near-infrared photons absorbed by 2 were converted to a photocurrent in the single-component device. Fig. 10 presents the light-associated cycle for photocurrent production and non-production, where the device showed a quick response to irradiation at 750 nm (128 μW cm−2). On the other hand, the photocurrent quickly fell to zero when the 750 nm irradiation was discontinued. This result demonstrates the potential of asymmetric benzo(thieno)[1,3,2]oxazaborinine-containing BODIPY in opto-electric applications such as NIR photodetectors. With this in mind, the specific detectivity (D*)71 was determined using the equation below:
| D* = R/(2qJd)1/2 = (Jph/Llight)/(2qJd)1/2 |
where R (A W−1) is the responsivity, which is defined as the ratio of photocurrent (Jph) to incident-light intensity (Llight), and q and Jd the absolute value of electron charge (1.6 × 10−19 Coulomb) and dark current, respectively. Given that Jph and Jd were measured to be 2.89 × 102 A cm−2 and 4.31 × 10−11 A cm−2 at 0.1 V bias potential, respectively. As a result, ITO/2/Al device showed a photodetectivity of 6.08 × 1011 Jones at 750 nm.
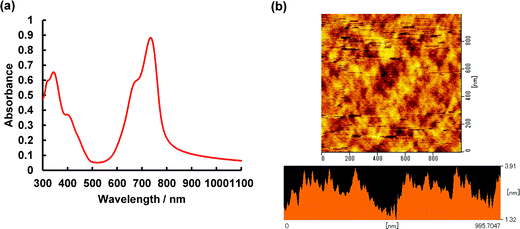 |
| | Fig. 8 (a) Absorption spectrum of film 2 on glass plate. (b) AFM image of film 2. | |
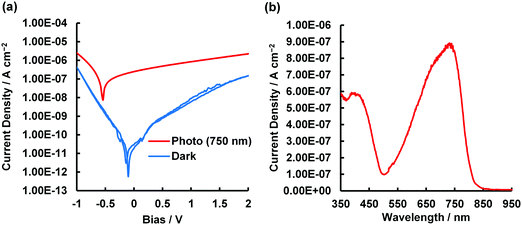 |
| | Fig. 9 (a) J–V curves of 2-loaded device (ITO/2/Al), measured in the dark and under illumination at 750 nm (128 μW cm−2). (b) Wavelength-dependent photocurrent densities of 2-loaded device (ITO/2/Al) at 1 V applied potential. | |
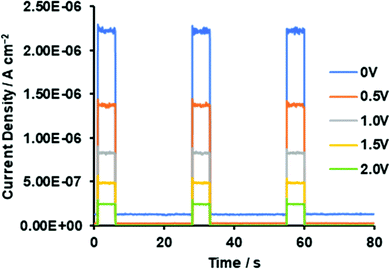 |
| | Fig. 10 Photocurrent–time trace for ITO/2/Al without and with light irradiation at 750 nm (128 μW cm−2) for ITO/2/Al at 750 nm. | |
Conclusion
In summary, this work for the first time presents the synthesis and characterization of a new family of B–O constrained asymmetric BODIPYs. The structural feature was elucidated by X-ray crystallography and NICS(0) calculation, indicating that the isoindole ring may possess azafulvene character. It was found that the structural rigidity imparted upon coordination to boron caused a significant red-shift in the absorbance and fluorescence properties. Based on theoretical calculations, the longest absorption band of the dyes is derived primarily from the π–π* transition within the BODIPY core, which is significantly influenced by thiophene insertion, accompanied by destabilization of the HOMO energy level. As a result, 3-benzo-5-thiophene-containing analogue 2 absorbed far-red and NIR light at λmax = 699 nm in THF. The electrochemical measurement and theoretical approach indicate that incorporation of thieno[1,3,2]oxazaborinine into the dye skeleton would not only contribute to the expansion of the π–π* transition of the chromophore, but also tune the HOMO energy level. In the latter case, the EDD plot suggests that the chromophore may possess partial charge-transfer character, with a transition from the constrained thiophene ring at the 3 position to the BODIPY core, which is ascribable to an increase in the HOMO level. This deduction is supported by the fact that the absorption band of 3-thiophene 2 is red-shifted relative to that of 5-thiophene 3. Further, as the number of thiophene units increased, the absorption band was more strongly red-shifted to the NIR region for 3,5-dithiophene 4 (λmax = 709 nm, λonset = 753 nm). Another interesting feature is that N2O2-type BODIPYs possess helicity with respect to the central boron atom. Optical resolution of 2 was conducted by preparative chiral HPLC to give both P and M-enantiomers, with an intense absorption band in the far-red region. Our ongoing program exploring NIR photodetectors in optoelectronics motivated us to fabricate a single-component device (ITO/2/Al). When illuminated by 750 nm NIR light (128 μW cm−2) the device produced a photocurrent of 8.42 × 10−7 A cm−2 at a bias potential of 1 V. Compared to the dark current, the on/off current ratio at the same bias potential was determined to be 1.42 × 102. The increase in the forward injection of current under NIR irradiation was ascribed to direct dissociation of the photoexcited electron–hole pairs. The results suggest that a sophisticated combination of dye 2 with an n-type acceptor would allow for NIR photodiodes with a p–n interface69 and deserve further investigation.
Conflicts of interest
There are no conflicts to declare.
Notes and references
- L. Dou, Y. Liu, Z. Hong, G. Li and Y. Yang, Chem. Rev., 2015, 115, 12633–12665 CrossRef CAS.
- R. R. Maar, R. Zhang, D. G. Stephens, Z. Ding and J. B. Gilroy, Angew. Chem., Int. Ed., 2019, 58, 1052–1056 CrossRef CAS.
- F.-C. Chen, Adv. Opt. Mater., 2019, 7, 1800662 CrossRef.
- K. Sakamoto, S. Yoshino, M. Takemoto and N. Furuya, J. Porphyrins Phthalocyanines, 2013, 17, 605–627 CrossRef CAS.
- S. Erten-Ela, Y. Ueno, T. Asaba and Y. Kubo, New J. Chem., 2017, 41, 10367–10375 RSC.
- H. Cheema, A. Baumann, E. K. Loya, P. Brogdon, L. E. McNamara, C. A. Carpenter, N. I. Hammer, S. Mathew, C. Risko and J. H. Delcamp, ACS Appl. Mater. Interfaces, 2019, 11, 16474–16489 CrossRef CAS.
- Y. Sun, Y. Sun, C. Dall’Agnese, X.-F. Wang, G. Chen, O. Kitao, H. Tamiaki, K. Sakai, T. Ikeuchi and S.-I. Sasaki, ACS Appl. Energy Mater., 2018, 1, 2813–2820 CrossRef CAS.
- O. Suryani, Y. Higashino, H. Sato and Y. Kubo, ACS Appl. Energy Mater., 2019, 2, 448–458 CrossRef CAS.
- T. Yudhistira, S. V. Mulay, Y. Kim, M. B. Halle and D. G. Churchill, Chem. – Asian J., 2019, 14, 3048–3084 CrossRef CAS.
- J. Mei, Y. Huang and H. Tian, ACS Appl. Mater. Interfaces, 2018, 10, 12217–12261 CrossRef CAS.
- S. Zhu, R. Tian, A. L. Antaris, X. Chen and H. Dai, Adv. Mater., 2019, 31, 1900321 CrossRef.
- S. He, J. Song, J. Qu and Z. Cheng, Chem. Soc. Rev., 2018, 47, 4258–4278 RSC.
- P. Chinna Ayya Swamy, G. Sivaraman, R. N. Priyanka, S. O. Raja, K. Ponnuvel, J. Shanmugpriya and A. Gulyani, Coord. Chem. Rev., 2020, 411, 213233 CrossRef CAS.
- M. Mitsunaga, M. Ogawa, N. Kosaka, L. T. Rosenblum, P. L. Choyke and H. Kobayashi, Nat. Med., 2011, 17, 1685–1691 CrossRef CAS.
- J. Fabian, H. Nakazumi and M. Matsuoka, Chem. Rev., 1992, 92, 1197–1226 CrossRef CAS.
- G. Qian and Z. Y. Wang, Chem. – Asian J., 2010, 5, 1006–1029 CrossRef CAS.
- Z. Guo, S. Park, J. Yoon and I. Shin, Chem. Soc. Rev., 2014, 43, 16–29 RSC.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS.
- G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184–1201 CrossRef CAS.
- N. Boens, B. Verbelen and W. Dehaen, Eur. J. Org. Chem., 2015, 6577–6595 CrossRef CAS.
- N. Boens, B. Verbelen, M. J. Ortiz, L. Jiao and W. Dehaen, Coord. Chem. Rev., 2019, 399, 213024 CrossRef CAS.
- H. Lu, J. Mack, Y. Yang and Z. Shen, Chem. Soc. Rev., 2014, 43, 4778–4823 RSC.
- G. Ulrich, S. Goeb, A. De Nicola, P. Retailleau and R. Ziessel, J. Org. Chem., 2011, 76, 4489–4505 CrossRef CAS.
- M. Wada, S. Ito, H. Uno, T. Murashima, N. Ono, T. Urano and Y. Urano, Tetrahedron Lett., 2001, 42, 6711–6713 CrossRef CAS.
- A. Nagai and Y. Chujo, Macromolecules, 2010, 43, 193–200 CrossRef CAS.
- S. G. Awuah, S. K. Das, F. D'Souza and Y. You, Chem. – Asian J., 2013, 8, 3123–3132 CrossRef CAS.
- S. G. Awuah, J. Polreis, V. Biradar and Y. You, Org. Lett., 2011, 13, 3884–3887 CrossRef CAS.
- Y. Yang, Q. Guo, H. Chen, Z. Zhou, Z. Guo and Z. Shen, Chem. Commun., 2013, 49, 3940–3942 RSC.
- K. Umezawa, A. Matsui, Y. Nakamura, D. Citterio and K. Suzuki, Chem. – Eur. J., 2009, 15, 1096–1106 CrossRef CAS.
- K. Umezawa, Y. Nakamura, H. Makino, D. Citterio and K. Suzuki, J. Am. Chem. Soc., 2008, 130, 1550–1551 CrossRef CAS.
- S. Yamazawa, M. Nakashima, Y. Suda, R. Nishiyabu and Y. Kubo, J. Org. Chem., 2016, 81, 1310–1315 CrossRef CAS.
- A. B. Descalzo, H.-J. Xu, Z.-L. Xue, K. Hoffmann, Z. Shen, M. G. Weller, X.-Z. You and K. Rurack, Org. Lett., 2008, 10, 1581–1584 CrossRef CAS.
- T. Sarma, P. K. Panda and J.-I. Setsune, Chem. Commun., 2013, 49, 9806–9808 RSC.
- A. Wakamiya, T. Murakami and S. Yamaguchi, Chem. Sci., 2013, 4, 1002–1007 RSC.
- J. Wang, Q. Wu, S. Wang, C. Yu, J. Li, E. Hao, Y. Wei, X. Mu and L. Jiao, Org. Lett., 2015, 17, 5360–5363 CrossRef CAS.
- L. J. Patalag, P. G. Jones and D. B. Werz, Angew. Chem., Int. Ed., 2016, 55, 13340–13344 CrossRef CAS.
- A. Patra, L. J. Patalag, P. G. Jones and D. B. Werz, Angew. Chem., Int. Ed., 2021, 60, 747–752 CrossRef CAS.
- L. J. Patalag, L. P. Ho, P. G. Jones and D. B. Werz, J. Am. Chem. Soc., 2017, 139, 15104–15113 CrossRef CAS.
- H. Kim, A. Burghart, M. B. Welch, J. Reibenspies and K. Burgess, Chem. Commun., 1999, 1889–1890 RSC.
- A. Loudet, R. Bandichhor, K. Burgess, A. Palma, S. O. McDonnell, M. J. Hall and D. F. O’Shea, Org. Lett., 2008, 10, 4771–4774 CrossRef CAS.
- E. Bodio and C. Goze, Dyes Pigm., 2019, 160, 700–710 CrossRef CAS.
- T. Nabeshima, M. Yamamura, G. J. Richards and T. Nakamura, J. Synth. Org. Chem., Jpn., 2015, 73, 1111–1119 CrossRef CAS.
- Y. Kubo, Y. Minowa, T. Shoda and K. Takeshita, Tetrahedron Lett., 2010, 51, 1600–1602 CrossRef CAS.
- Y. Tomimori, T. Okujima, T. Yano, S. Mori, N. Ono, H. Yamada and H. Uno, Tetrahedron, 2011, 67, 3187–3193 CrossRef CAS.
- T. Okujima, Y. Shida, K. Ohara, Y. Tomimori, M. Nishioka, S. Mori, T. Nakae and H. Uno, J. Porphyrins Phthalocyanines, 2014, 18, 752–761 CrossRef CAS.
- Y. Kubo, K. Watanabe, R. Nishiyabu, R. Hata, A. Murakami, T. Shoda and H. Ota, Org. Lett., 2011, 13, 4574–4577 CrossRef CAS.
- Y. Kubo, S. Tobinaga, Y. Ueno, T. Aotake, H. Yakushiji and T. Yamamoto, Chem. Lett., 2018, 47, 300–303 CrossRef CAS.
- Y. Kubo, T. Shimada, K. Maeda and Y. Hashimoto, New J. Chem., 2020, 44, 29–37 RSC.
- L. Jiao, C. Yu, M. Liu, Y. Wu, K. Cong, T. Meng, Y. Wang and E. Hao, J. Org. Chem., 2010, 75, 6035–6038 CrossRef CAS.
- C. Yu, Y. Xu, L. Jiao, J. Zhou, Z. Wang and E. Hao, Chem. – Eur. J., 2012, 18, 6437–6442 CrossRef CAS.
- C. Yu, Q. Wu, J. Wang, Y. Wei, E. Hao and L. Jiao, J. Org. Chem., 2016, 81, 3761–3770 CrossRef CAS.
- A. Bessette, T. Auvray, D. Désilets and G. S. Hanan, Dalton Trans., 2016, 45, 7589–7604 RSC.
- C. Maeda, T. Todaka, T. Ueda and T. Ema, Chem. – Eur. J., 2016, 22, 7508–7513 CrossRef CAS.
- Y. Wu, S. Wang, Z. Li, Z. Shen and H. Lu, J. Mater. Chem. C, 2016, 4, 4668–4674 RSC.
- S. Yuan, F. Wang, G. Yang, C. Lu, J. Nie, Z. Chen, J. Ren, Y. Qiu, Q. Sun, C. Zhao and W.-H. Zhu, Anal. Chem., 2018, 90, 3914–3919 CrossRef CAS.
- N. Chen, W. Zhang, S. Chen, Q. Wu, C. Yu, Y. Wei, Y. Xu, E. Hao and L. Jiao, Org. Lett., 2017, 19, 2026–2029 CrossRef CAS.
- X. Yang, L. Jiang, M. Yang, H. Zhang, J. Lan, F. Zhou, X. Chen, D. Wu and J. You, J. Org. Chem., 2018, 83, 9538–9546 CrossRef CAS.
- C. Zhao, Y. Zhou, Q. Lin, L. Zhu, P. Feng, Y. Zhang and J. Cao, J. Phys. Chem. B, 2011, 115, 642–647 CrossRef CAS.
- J. Thireau, J. Marteaux, P. Delagrange, F. Lefoulon, L. Dufourny, G. Guillaumet and F. Suzenet, ACS Med. Chem. Lett., 2014, 5, 158–161 CrossRef CAS.
- Y. Ni, W. Zeng, K.-W. Huang and J. Wu, Chem. Commun., 2013, 49, 1217–1219 RSC.
- X.-D. Jiang, D. Xi, J. Zhao, H. Yu, G.-T. Sun and L.-J. Xiao, RSC Adv., 2014, 4, 60970–60973 RSC.
- R. R. Maar and J. B. Gilroy, Chem. – Eur. J., 2018, 24, 12449–12457 CrossRef CAS.
- C. Wang, H. Dong, L. Jiang and W. Hu, Chem. Soc. Rev., 2018, 47, 422–500 RSC.
- S. Yoon, K. M. Sim and D. S. Chung, J. Mater. Chem. C, 2018, 6, 13084–13100 RSC.
- J. Chen, N. U. Rahman, Z. Mao, J. Zhao, Z. Yang, S. Liu, Y. Zhang and Z. Chi, J. Mater. Chem. C, 2019, 7, 8250–8254 RSC.
- R. D. Jansen-van Vuuren, A. Armin, A. K. Pandey, P. L. Burn and P. Meredith, Adv. Mater., 2016, 28, 4766–4802 CrossRef CAS.
- C. Wang, X. Zhang and W. Hu, Chem. Soc. Rev., 2020, 49, 653–670 RSC.
- L. Li, Y. Huang, J. Peng, Y. Cao and X. Peng, J. Mater. Chem. C, 2014, 2, 1372–1375 RSC.
- Q. Li, Y. Guo and Y. Liu, Chem. Mater., 2019, 31, 6359–6379 CrossRef CAS.
- Y. Yao, Y. Liang, V. Shrotriya, S. Xiao, L. Yu and Y. Yang, Adv. Mater., 2007, 19, 3979–3983 CrossRef CAS.
- X. Gong, M. Tong, Y. Xia, W. Cai, J. S. Moon, Y. Cao, G. Yu, C.-L. Shieh, B. Nilsson and A. J. Heeger, Science, 2009, 325, 1665 CrossRef CAS.
- K. H. Hendriks, W. Li, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2014, 136, 12130–12136 CrossRef CAS.
- X. Wang, L. Lv, L. Li, Y. Chen, K. Zhang, H. Chen, H. Dong, J. Huang, G. Shen, Z. Yang and H. Huang, Adv. Funct. Mater., 2016, 26, 6306–6315 CrossRef CAS.
- X. Zhou, D. Yang, D. Ma, A. Vadim, T. Ahamad and S. M. Alshehri, Adv. Funct. Mater., 2016, 26, 6619–6626 CrossRef CAS.
- S. Li, X. Deng, L. Feng, X. Miao, K. Tang, Q. Li and Z. Li, Polym. Chem., 2017, 8, 1039–1048 RSC.
- D. Ho, R. Ozdemir, H. Kim, T. Earmme, H. Usta and C. Kim, ChemPlusChem, 2019, 84, 18–37 CAS.
- P. Diana, A. Martorana, P. Barraja, A. Montalbano, A. Carbone and G. Cirrincione, Tetrahedron, 2011, 67, 2072–2080 CrossRef CAS.
- S. Morandi, E. Caselli, A. Forni, M. Bucciarelli, G. Torre and F. Prati, Tetrahedron: Asymmetry, 2005, 16, 2918–2926 CrossRef CAS.
-
T. Inouchi, T. Aotake, H. Yakushiji, M. Abe, S. Shinamura and Y. Sadamitsu, Preparation of dibenzopyrromethene boron chelate compounds as near infrared light absorbing materials for thin film and organic electronic device, WO2018079653 A1, 2017 Search PubMed.
- J. Wang, Q. Wu, Y. Xu, C. Yu, Y. Wei, X. Mu, E. Hao and L. Jiao, RSC Adv., 2016, 6, 52180–52188 RSC.
- A. Bessette, M. Cibian, F. Bélanger, D. Désilets and G. S. Hanan, Phys. Chem. Chem. Phys., 2014, 16, 22207–22221 RSC.
- M. J. Hall, S. O. McDonnell, J. Killoran and D. F. O'Shea, J. Org. Chem., 2005, 70, 5571–5578 CrossRef CAS.
- G. A. Crosby and J. N. Demas, J. Phys. Chem., 1971, 75, 991–1024 CrossRef CAS.
- A. Ogunsipe, D. Maree and T. Nyokong, J. Mol. Struct., 2003, 650, 131–140 CrossRef CAS.
- D. Magde, J. H. Brannon, T. L. Cremers and J. Olmsted, J. Phys. Chem., 1979, 83, 696–699 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revision A.03, Gaussian, Inc., 2016 Search PubMed.
- H. Kim, A. Burghart, M. B. Welch, J. Reibenspies and K. Burgess, Chem. Commun., 1999, 1889–1890 RSC.
- T. M. Krygowski and M. K. Cyrański, Chem. Rev., 2001, 101, 1385–1420 CrossRef CAS.
- R. B. Alnoman, S. Rihn, D. C. O'Connor, F. A. Black, B. Costello, P. G. Waddell, W. Clegg, R. D. Peacock, W. Herrebout, J. G. Knight and M. J. Hall, Chem. – Eur. J., 2016, 22, 93–96 CrossRef CAS.
- M. L. Główka, D. Martynowski and K. Kozłowska, J. Mol. Struct., 1999, 474, 81–89 CrossRef.
- M. Ponce-Vargas, C. Azarias, D. Jacquemin and B. Le Guennic, J. Phys. Chem. B, 2017, 121, 10850–10858 CrossRef CAS.
- H. Lu, J. Mack, T. Nyokong, N. Kobayashi and Z. Shen, Coord. Chem. Rev., 2016, 318, 1–15 CrossRef CAS.
- J. Sworakowski and K. Janus, Org. Electron., 2017, 48, 46–52 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2024003. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ma00910e |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Toshiki
Nozawa
a,
Kentaro
Maeda
b and
Yuta
Hashimoto
b
*a,
Toshiki
Nozawa
a,
Kentaro
Maeda
b and
Yuta
Hashimoto
b
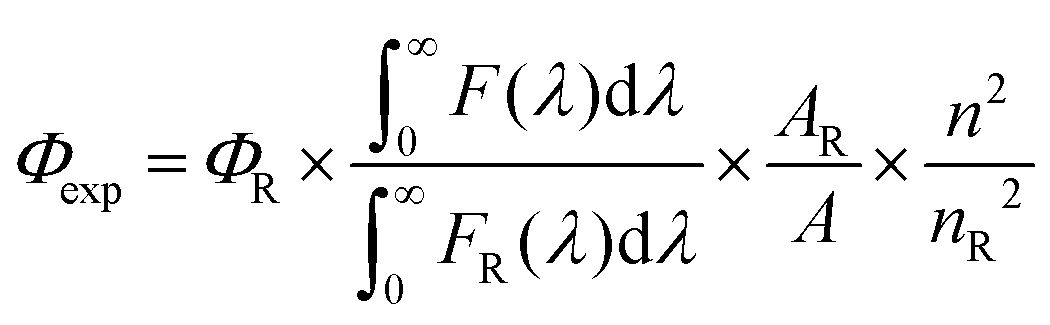
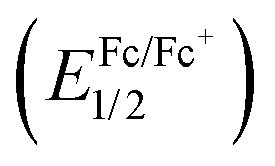 was 0.197 V versus Ag/Ag+. Reversible oxidation and reduction waves were observed for the N2O2-dyes (1–4) enabling us to determine the half-wave potentials ((Eox1/2) and (Ered1/2)) as summarized in Table 2. On another front, control 5 exhibited an irreversible reduction wave, from which the corresponding onset potentials were determined. The electrical gap of the dyes was inconsistent with the absorption bands observed in Fig. 4a, possibly due to some involvement of transitions other than the HOMO → LUMO transition in the absorption band. The HOMO and LUMO energy levels were estimated on the basis of those potential values (Table 2). When compared to BF2 dye 5, intramolecular B,O cyclization made the HOMO more negative than the LUMO, resulting in a narrower band gap and consequent red-shift in the absorption and fluorescence bands. It was also found that thiophene-insertion in the chromophore had a stronger impact on the HOMO level than on the LUMO. Notably, the HOMO level of 3-thiophene analogue 2 was higher-lying than that of 5-thiophene stereoisomer 3. Moreover, the HOMO level became more destabilized as the number of thiophene units in the chromophore increased. In this way, the HOMO levels were energetically higher in the following order: 4 > 2 > 3 > 1, resulting in a smaller HOMO–LUMO bandgap. The trend observed here was rationalized by theoretical calculation (vide infra).
was 0.197 V versus Ag/Ag+. Reversible oxidation and reduction waves were observed for the N2O2-dyes (1–4) enabling us to determine the half-wave potentials ((Eox1/2) and (Ered1/2)) as summarized in Table 2. On another front, control 5 exhibited an irreversible reduction wave, from which the corresponding onset potentials were determined. The electrical gap of the dyes was inconsistent with the absorption bands observed in Fig. 4a, possibly due to some involvement of transitions other than the HOMO → LUMO transition in the absorption band. The HOMO and LUMO energy levels were estimated on the basis of those potential values (Table 2). When compared to BF2 dye 5, intramolecular B,O cyclization made the HOMO more negative than the LUMO, resulting in a narrower band gap and consequent red-shift in the absorption and fluorescence bands. It was also found that thiophene-insertion in the chromophore had a stronger impact on the HOMO level than on the LUMO. Notably, the HOMO level of 3-thiophene analogue 2 was higher-lying than that of 5-thiophene stereoisomer 3. Moreover, the HOMO level became more destabilized as the number of thiophene units in the chromophore increased. In this way, the HOMO levels were energetically higher in the following order: 4 > 2 > 3 > 1, resulting in a smaller HOMO–LUMO bandgap. The trend observed here was rationalized by theoretical calculation (vide infra).