
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
How interdiffusion affects the electrochemical performance of LiMn2O4 thin films on stainless steel†
Matthias
Rumpel
 a,
Michael
Machhaus
a,
Jordi
Sastre
b,
Simon
Ziegler
a,
Xubin
Chen
b,
Andreas
Flegler
a,
Yaroslav E.
Romanyuk
b and
Guinevere A.
Giffin
*a
a,
Michael
Machhaus
a,
Jordi
Sastre
b,
Simon
Ziegler
a,
Xubin
Chen
b,
Andreas
Flegler
a,
Yaroslav E.
Romanyuk
b and
Guinevere A.
Giffin
*a
aFraunhofer Institute for Silicate Research ISC, Neunerplatz 2, 97082 Wuerzburg, Germany. E-mail: guinevere.giffin@isc.fraunhofer.de
bLaboratory for Thin Films and Photovoltaics, Empa – Swiss Federal Laboratories for Materials Science and Technology, Überlandstrasse 129, 8600 Dübendorf, Switzerland
First published on 15th February 2021
Abstract
The sequential production process of thin-film solid-state batteries (TF-SSB) requires high temperatures up to 700 °C in order to achieve crystallized cathode films with high capacities and ceramic electrolytes with high ionic conductivities. This presents challenges for thermal interfacial stability between the electrode and electrolyte as well as electrode and substrate. Interdiffusion processes between the substrate and electrode at elevated temperatures are critical and may explain the low utilization of cathode active materials in TF-SSBs. This study examines the interdiffusion that occur between LiMn2O4 (LMO) thin films and the flexible stainless steel (StSt) substrate. Grazing incidence X-ray diffraction spectroscopy, time-of-flight secondary ion mass spectrometry and X-ray photoelectron spectroscopy reveal that during the crystallization at 600 °C and 700 °C, LMO undergoes lithium and manganese depletion whereas elements from the substrate (Fe, Cr, Al, Pt) diffuse into the LMO spinel structure. The combination of these effects, which are exacerbated by the higher crystallization temperature, results in a significant capacity loss during galvanostatic cycling. Various interlayers, including gold, platinum and indium tin oxide (ITO) were tested as conductive interdiffusion barriers. The use of ITO, unlike gold and platinum, as a barrier layer essentially prevents the interdiffusion, leading to an improved discharge capacity of 4.2 μA h cm−2 (600 °C) and 5.1 μA h cm−2 (700 °C). The results indicate that ITO is an effective interdiffusion barrier on flexible StSt current collector foils, which can enable higher performance TF-SSBs as well as cost effective roll-to-roll manufacturing.
Introduction
Thin-film batteries are of particular interest for various applications including microelectronics, such as flexible microdevices, wearables, medical implants, etc. Thin-film solid-state batteries (TF-SSB) promise better cell performance in terms of energy density, cycle stability, cycle life and safety than conventional batteries. One of the key elements of solid-state batteries is the ceramic or glassy electrolyte, which replaces the liquid electrolyte in conventional lithium-ion batteries. Without liquids, the risk of leakage and other safety related issues can be reduced or eliminated.1–7 Furthermore, Li-ion conductive ceramics, e.g. Li7La3Zr2O12 (LLZO)8–11 or Li1.3Al0.3Ti1.7(PO4)3 (LATP),12,13 and glassy lithium phosphorus oxynitrides (LIPON)14,15 form a stable solid electrolyte interface (SEI) or have a higher potential window up to 5 V vs. Li/Li+, so that less degradation of the electrolyte is expected and the implementation of high-voltage active materials might be feasible.16–18 Li et al. demonstrated, for instance, a high-voltage LiMn1.5Ni0.5O4 TF-SSB using a LIPON electrolyte with a capacity retention of 90.6% after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles and a coulombic efficiency of almost 100%. Although the performance was promising, the specific capacity of the cathode layer was about 83% of the theoretical value of 148 mA h g−1.15 This lack of utilization of the active material has been reported for many thin-film battery studies. It is reasonable to presume that the high temperature crystallization process of the deposited active material may influence the electrochemical performance due to interdiffusion processes of substrate and active material elements. Li et al. used platinum (Pt) on aluminum (Al) as substrate. Further studies with Pt,19,20 gold21,22 (Au), stainless-steel23 (StSt) or metal alloy24 substrates have been published and show similar behaviour.15 Komaba et al. reported a relationship between the temperature of the crystallization process and the cycling performance of LMO thin-layers on a StSt substrate.23 Diffusion of Fe and Cr atoms into the LMO layer was established via depth-profile X-ray photoelectron spectroscopy (XPS). Kim et al. slightly enhanced the utilization of LMNO on a Pt-coated StSt foil due to integration of an indium tin oxide (ITO) layer between Pt and StSt.20 A survey of these publications demonstrates the high impact of substrate material and the crystallization temperature on the battery performance.
000 cycles and a coulombic efficiency of almost 100%. Although the performance was promising, the specific capacity of the cathode layer was about 83% of the theoretical value of 148 mA h g−1.15 This lack of utilization of the active material has been reported for many thin-film battery studies. It is reasonable to presume that the high temperature crystallization process of the deposited active material may influence the electrochemical performance due to interdiffusion processes of substrate and active material elements. Li et al. used platinum (Pt) on aluminum (Al) as substrate. Further studies with Pt,19,20 gold21,22 (Au), stainless-steel23 (StSt) or metal alloy24 substrates have been published and show similar behaviour.15 Komaba et al. reported a relationship between the temperature of the crystallization process and the cycling performance of LMO thin-layers on a StSt substrate.23 Diffusion of Fe and Cr atoms into the LMO layer was established via depth-profile X-ray photoelectron spectroscopy (XPS). Kim et al. slightly enhanced the utilization of LMNO on a Pt-coated StSt foil due to integration of an indium tin oxide (ITO) layer between Pt and StSt.20 A survey of these publications demonstrates the high impact of substrate material and the crystallization temperature on the battery performance.
A cost effective production process for TF-SSBs is roll-to-roll manufacturing via sequential deposition and crystallization of the cathode and electrolyte on a flexible substrate. Each step requires a separate temperature treatment, which can cause interdiffusion. The highest temperature, ≥700 °C, is required after deposition of the ceramic electrolyte on top of the cathode layer to crystallize the ceramic electrolyte in order to achieve high ionic conductivities.11,25,26 These high temperatures enhance the interdiffusion processes at each interface and make their impact more significant.
This study takes a deeper look into the interdiffusion processes, their influence on the lattice structure of the active cathode material and on the subsequent electrochemistry. For this purpose, LMO cathode layers were deposited on flexible StSt foil via sol–gel dip-coating and crystallized at 600 °C and 700 °C. The three different conductive layers Pt, Au and ITO were tested as protection layers against interdiffusion between LMO and StSt. This systematic comparison elucidates the differences between the various substrates and reveals many phenomena like lithium depletion, interdiffusion, oxide layer formation and their impact on the electrochemistry. These results show that ITO is an excellent interdiffusion barrier between LMO and StSt and enables better electrochemical performance than using Au or Pt interlayers.
Experimental
Preparation of substrate and interlayers
The base substrate in this study was a flexible and heat-resistant ferritic chrome steel with 20 wt% Cr and 5 wt% Al (thickness = 30 μm, 1.4767, X8CrAl 20-5, H + S Präzesionsfolien GmbH). The StSt foil was cut into 5 × 5 cm2 samples. Au and Pt interlayers were deposited on top of the StSt substrate via DC sputtering (Bulzer Union 040) in an argon atmosphere with a pressure of 0.1 mbar. The sample–target distance was 7 cm and the sputter current was set to 40 mA. The deposition times were 10 min for Au and 15 min for Pt to achieve similar layer thicknesses of ca. 100 nm. ITO layers were coated onto StSt using a sol–gel technique.27 The solution was prepared by dissolving indium(III) acetate and diethanolamine in 1-butanol. This solution was refluxed for 10 min. Subsequently, ethoxyacetic acid and tin(II) 2-ethylhexanoate were added and refluxed for 1 h. The solvent was removed by a rotary evaporator and the sol adjusted to a defined solid content of 4.6 wt% by again adding 1-butanol. The coating of the ITO sol onto the StSt substrate was performed with a self-build dip-coater (Fraunhofer ISC) in a controlled air atmosphere with 25% relative humidity at 25 °C and with a withdraw velocity of 200 mm min−1. The ITO gel films were crystallized rapidly at 600 °C for 10 min (P330 muffle furnace, Nabertherm), which resulted in crystalline layers of ca. 100 nm with an electrical sheet resistivity of approx. 15 mΩ cm.Preparation of LMO layer
The LMO active material was coated on the various substrates using dip-coating. The LMO sol was synthesized by dissolving stoichiometric amounts of lithium acetylacetonate and manganese(II) acetate tetrahydrate in a solution of 2-(2-methoxyethoxy) acetic acid and ethanol. The mixture was stirred for 30 min under reflux at 120 °C. The solid content was 10 wt%. The dip-coating procedure was performed in a controlled air atmosphere with 25% relative humidity at 25 °C. The substrates were immersed for 10 s in the sol solution, withdrawn with a constant speed of 50 mm min−1 and immediately dried at 180 °C for 30 min. Subsequently, the LMO-coated substrates were rapidly annealed at 600 °C for 1 h or 700 °C for 1 h (P330 muffle furnace, Nabertherm) in an ambient air atmosphere. The samples were also rapidly cooled by opening the furnace at the crystallization temperature and removing the LMO-coated substrates.Cell assembly
Before cell assembly, the backside of the coated StSt was polished to remove the oxide layer formed during the annealing step. Cathodes with a diameter of 16 mm (2.01 cm2 area) were cut and dried in vacuum (10−2 mbar) at 110 °C for 10 h. Pouch cells were assembled in an argon-filled glovebox (H2O < 1 ppm and O2 < 1 ppm) with a Whatman GF/F glass microfiber separator and lithium metal as counter electrode. A mixture of ethylene carbonate and dimethyl carbonate (EC:DMC volume ratio of 1:1) with 1 M LiPF6 was used as electrolyte (LP30, BASF).
Electrochemical characterization
Galvanostatic cycling with limited potential (GCPL) and electrochemical impedance spectroscopy (EIS) were carried out with a galvanostat (VMP-300, Biologic) in a temperature-controlled climatic chamber (Memmert IPP260PLUS) at 25 °C. After a rest step of 12 h for acclimatization, galvanostatic charging and discharging were performed with a current density of 30 μA cm−2 between 4.4 V vs. Li/Li+ and 3.2 V vs. Li/Li+ for 24 cycles. After each constant current charge step, a constant voltage step at 4.4 V with the end criteria of either 5 min or a current density limit of 0.5 μA cm−2 was executed. After the 24 cycles, the cells were charged at 30 μA cm−2 to 4.4 V, followed by a constant voltage step at 4.4 V for either 3 h or until a current density limit of 0.75 μA cm−2 was reached to obtain quasi-stable conditions for the EIS measurements. The EIS measurements were carried out at 4.4 V in the frequency range between 1 MHz and 1 mHz with an amplitude of 5 mV. Two cells of each sample type were tested. The rate capability test was performed on a battery cycler (Maccor 4000) with the same cell set-up and test protocol as described above. The current densities were varied between 200 μA cm−2 (approx. 40C) and 10 μA cm−2 (approx. 2C). These measurements were carried out in triplicate.Structural characterization
Scanning electron microscopy (SEM) images have been collected on a ZEIS Supra 25 microscope with an accelerating voltage of 2.5 kV. The preparation of the cross sections were done by embedding the samples in an epoxy resin and polishing them with a JEOL SM-0910 cross-section polisher in argon atmosphere.Grazing incidence X-ray diffraction spectroscopy (GIXRD) was performed on a PANalytical Empyrean diffractometer with CuKα radiation between 15° and 60° 2θ with a step size of 0.02° and a scan duration per step of 5 s. The fixed incidence angle of the radiation was 0.5°.
Time-of-flight secondary ion mass spectrometry profiles (ToF-SIMS) were measured in a ToF.SIMS5 system from IONTOF. Depth profiling was performed by Cs+ ion sputtering with an acceleration voltage of 1 kV and a current of 50 nA on an area of 300 × 300 μm2. Bi+ primary ions, with an acceleration voltage of 25 kV and a current of 2 pA on a 100 × 100 μm2 area within the sputtering crater, were used for analysis.
X-ray photoelectron spectroscopy (XPS) measurements were performed with monochromatic Al Kα X-ray source (Surface Science Instruments, S-Probe). Single scans were done with a 32 eV pass energy, which provides high-resolution scans with the full-width half maximum of 1 eV. The scans were calibrated with the C1s peak at 284.60 eV.
Results and discussion
Morphology
The homogenous and dense ITO, Pt and Au interlayers with layer thickness of ca. 100 nm are shown in the SEM cross sections in Fig. 1. The LMO layer thickness does not depend on the substrate and is approx. 160 nm. When no interlayer material is used, an additional region of dark contrast can be seen between the LMO and StSt, which is indicative of the formation of oxide layers during annealing. This formation is confirmed in the ToF-SIMS depth profiles as will be discussed below. Such areas of dark contrast are also seen when ITO and Pt are used as the interlayer materials. In contrast to the other three samples, when Au is implemented as an interlayer, the oxide layer formation takes place at the LMO and Au interface. Cracks between the layers are not observed, therefore a good contact between the layers is ensured. The top view SEM images (Fig. S1, ESI†) show an increase in particle size with elevated temperature for all samples.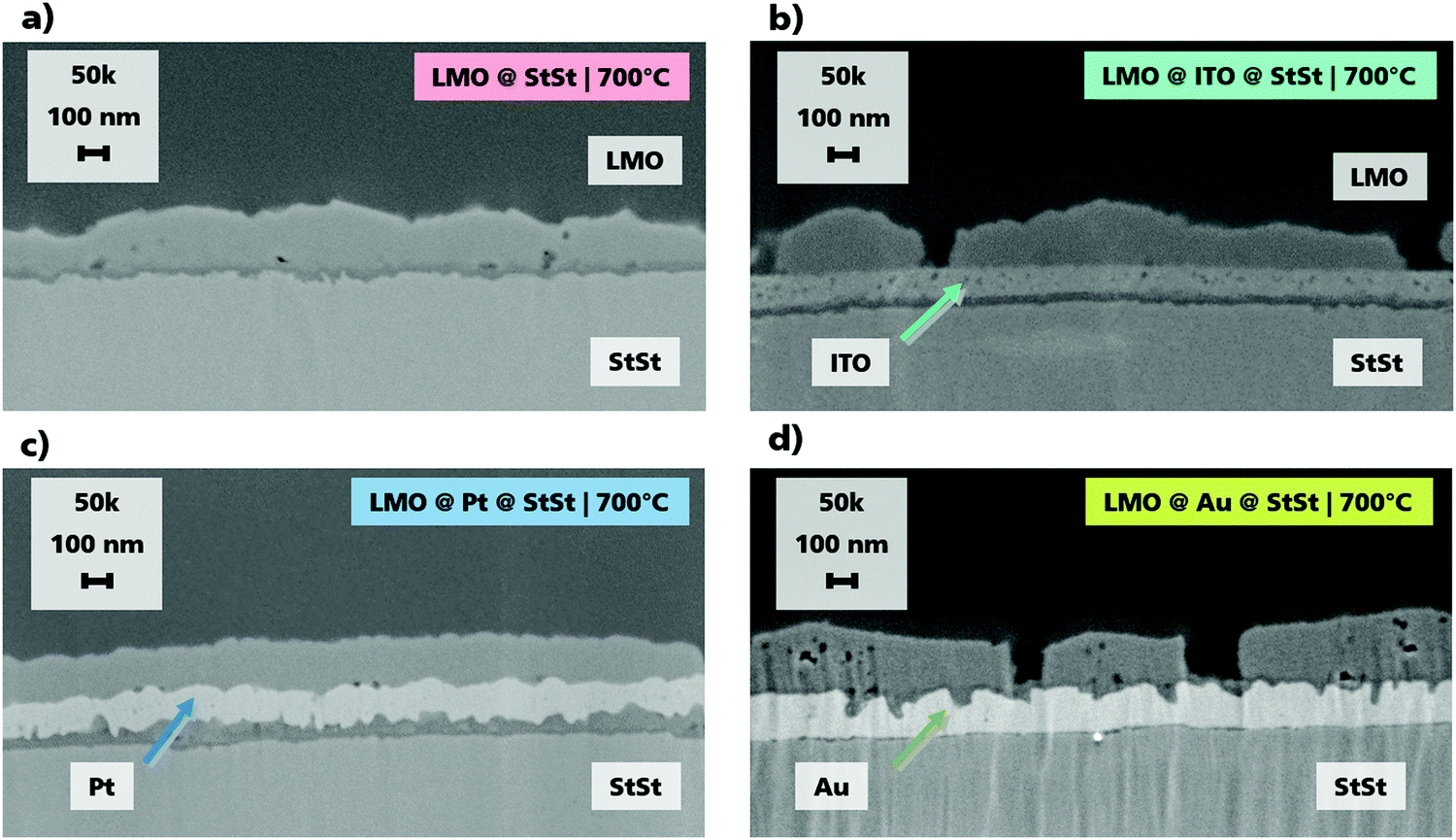 | ||
| Fig. 1 SEM cross sections of (a) LMO@StSt (LS700), (b) LMO@ITO (LIS700), (c) LMO@Pt (LPS700) and (d) LMO@Au (LAS700) annealed at 700 °C for 1 h. LMO layer thicknesses are approx. 160 nm. Layer thicknesses of Pt, Au and ITO are approx. 100 nm. | ||
Electrochemical performance
Galvanostatic cycling shows how the different interlayers and crystallization temperature influence the cell performance including the discharge capacity, coulombic efficiency and overvoltage (Fig. 2 and Table 1). With a crystallization temperature of 600 °C, LS600 has the lowest discharge capacity of 1.99 μA h cm−2 ± 0.03 μA h cm−2 (Table 1). Through the use of an interlayer, the LMO discharge capacities of the LIS600, LPS600 and LAS600 are improved (Fig. 2a and Table 1). Elevating the crystallization temperature to 700 °C causes the LMO capacity on StSt or Au (LS700 and LAS700) to be negligible and on Pt (LPS700) to be reduced by a factor of three. In contrast, the LMO discharge capacity on ITO increases to 5.06 μA h cm−2 ± 0.46 μA h cm−2 (LIS700). Furthermore, this combination had the highest coulombic efficiency (97.3% ± 0.3%) (Fig. 2e) and capacity retention (99.1% ± 2.3%) (Fig. 2b). Although LS600 and LAS600 have lower capacities than LIS700, the cycling behaviour is also stable. The capacity retention of LMO@Pt is substantially lower than all other samples (ca. 86% and ca. 46% for LPS600 and LPS700, respectively). This capacity retention, combined with a low coulombic efficiency of ca. 67%, indicates irreversible side reactions during charge. This behaviour is confirmed by the performance during the rate capability tests (Fig. 2f). The rate capability is evaluated by the change in the LMO discharge capacity between the 100 μA cm−2 (Cycle 19) and 30 μA cm−2 (Cycle 73) constant current steps. LPS600 and LPS700 show a decrease of the LMO discharge capacity of 9% and 16%, respectively. This capacity fade is unique for the Pt cells and cannot be explained by polarization processes. Consequently, it further suggests irreversible side reactions. All other samples demonstrate the expected increase of the discharge capacity with lower currents due to the current dependence of polarization processes of the LMO cathode and the ohmic resistance of the cell. The rate behaviour is reflected in the overvoltage from differential capacity plots, which is related to the concentration and charge transfer polarizations (Fig. 2c and d). The overvoltage is defined as difference between the peak positions of the first redox peak for charge and discharge. The overvoltage of LMO@ITO increases by a factor of 40 (14 mV ± 2 mV to 574 mV ± 143 mV) with elevated temperature, which results in an incomplete charge during the constant current step. LPS600, LPS700 and LAS600 show more moderate overvoltages in the range of ca. 66 mV to 79 mV.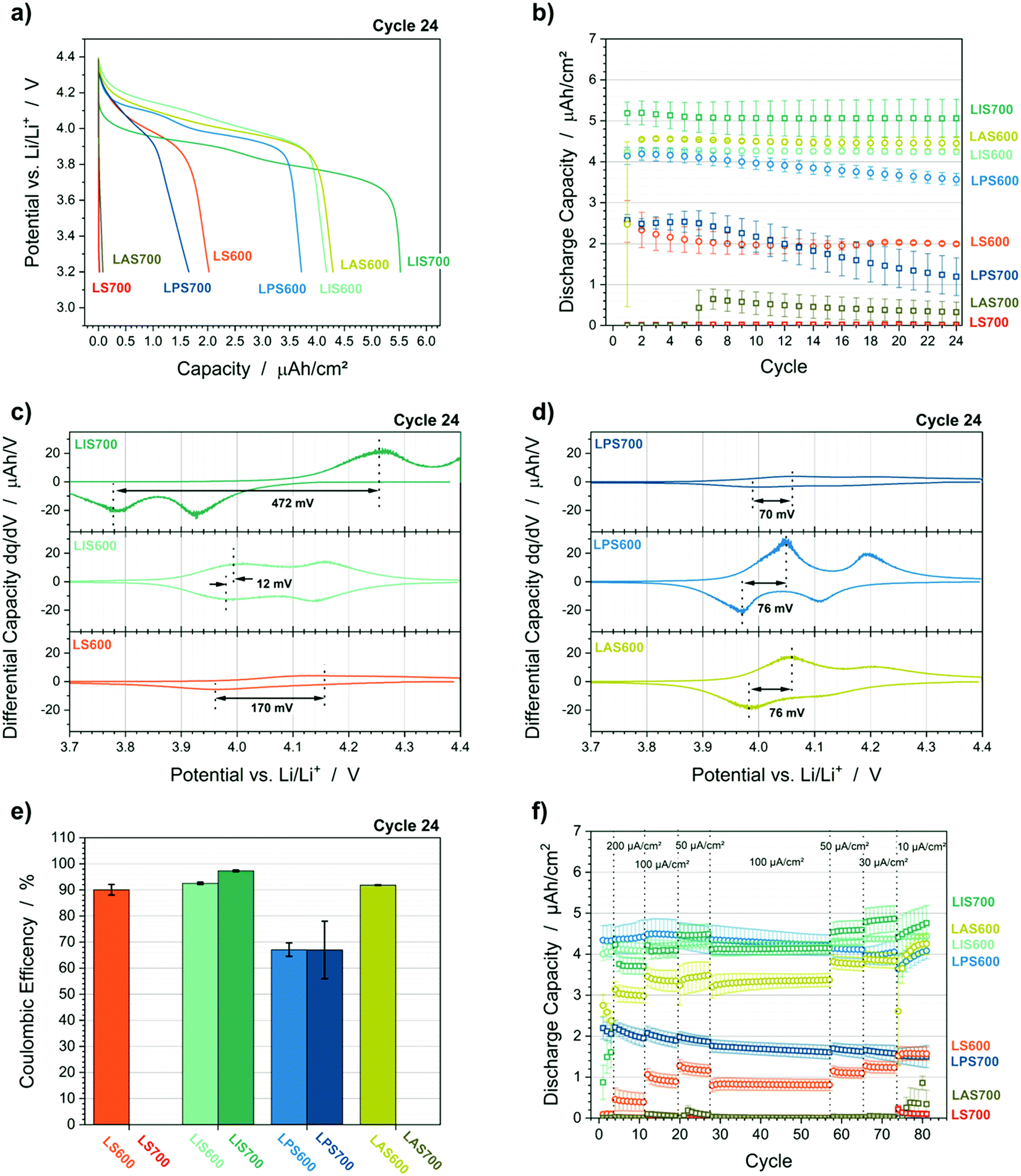 | ||
| Fig. 2 GCPL results of the LMO@StSt (red), LMO@ITO (cyan), LMO@Pt (blue) and LMO@Au (green). (a) Voltage profile of cycle 24 (representative cells), (b) discharge capacity as a function of cycles, (c and d) differential capacity plots of cycle 24 (representative cells), (e) Coulombic efficiencies of cycle 24 and (f) discharge capacities during C-rate test. | ||
| Sample | Discharge capacity cycle 24 (μA h cm−2) | Coulombic efficiency cycle 24 (%) | Overvoltage cycle 24 (mV) | Capacity retention after 24 cycles (%) | Rate capabilitya (%) |
|---|---|---|---|---|---|
| a Ratio of discharge capacity 30 μA cm−2: 100 μA cm−2. b n.a.: negative values due to side reaction, not reasonable to analyse. | |||||
| 600 °C crystallization temperature | |||||
| LS600 (LMO@StSt) | 1.99 ± 0.03 | 90.1 ± 2.0 | 139 ± 30 | 97.3 ± 16.1 | 38 |
| LIS600 (LMO@ITO) | 4.22 ± 0.07 | 92.5 ± 0.4 | 14 ± 2 | 99.3 ± 0.2 | 6 |
| LPS600 (LMO@Pt) | 3.57 ± 0.15 | 67.1 ± 2.5 | 66 ± 9 | 86.4 ± 0.1 | n.a.b |
| LAS600 (LMO@Au) | 4.45 ± 0.16 | 91.8 ± 0.2 | 79 ± 3 | 97.9 ± 4.0 | 15 |
| 700 °C crystallization temperature | |||||
| LS700 (LMO@StSt) | No capacity | ||||
| LIS700 (LMO@ITO) | 5.06 ± 0.46 | 97.3 ± 0.3 | 574 ± 143 | 99.1 ± 2.3 | 19 |
| LPS700 (LMO@Pt) | 1.19 ± 0.46 | 67.0 ± 11.0 | 76 ± 9 | 45.8 ± 13.6 | n.a.b |
| LAS700 (LMO@Au) | No capacity | ||||
A change in the ratio between the first and second redox peaks is also observable. This two-step redox process is related to the ordering of half of the Li-ions on the tetrahedral 8a sites in the spinel lattice. The electrochemical extraction takes place via diffusion of the Li atoms from one 8a site to another over the 16c sites. The diffusion activation energies depend on the atomic neighbours of the Li ion on the 8a-16c-8a diffusion pathway.28,29 Thus, a change in the ratio of the redox peaks might be explained by a change of the diffusion activation energies due to a change of the atomic neighbours in the crystal lattice, i.e., the substitution of Mn with elements of the interlayer or substrate. LIS600 shows a higher peak height for the second redox peak, which is reported in literature for LMO.29 With Au and Pt interlayers, the second redox peak has lower maximum and peak area than the first. Furthermore, the second redox peak vanishes in the plot of the LS600 sample (Fig. 1c). This might suggest interdiffusion, which causes in substitution of Mn in the spinel lattice with substrate or interlayer elements for the Au, Pt and StSt samples. This phenomenon will be discussed below with the help of ToF-SIMS, GIXRD and XPS.
Electrochemical impedance spectroscopy
The overvoltages can be correlated with the concentration polarization and charge transfer polarization, which can be further evaluated via electrochemical impedance spectroscopy. The resistance of a possible insulating layer at the interface of LMO and the electrolyte RIn and the charge transfer resistance RCT may be evaluated by fitting the impedance data with the equivalent circuit illustrated in Fig. 3b as demonstrated in the Nyquist plot in Fig. 3c and d. The resistances are summarized in Table 2. Further Nyquist plots including fit curves can be found in the supporting information (Fig. S3, ESI†).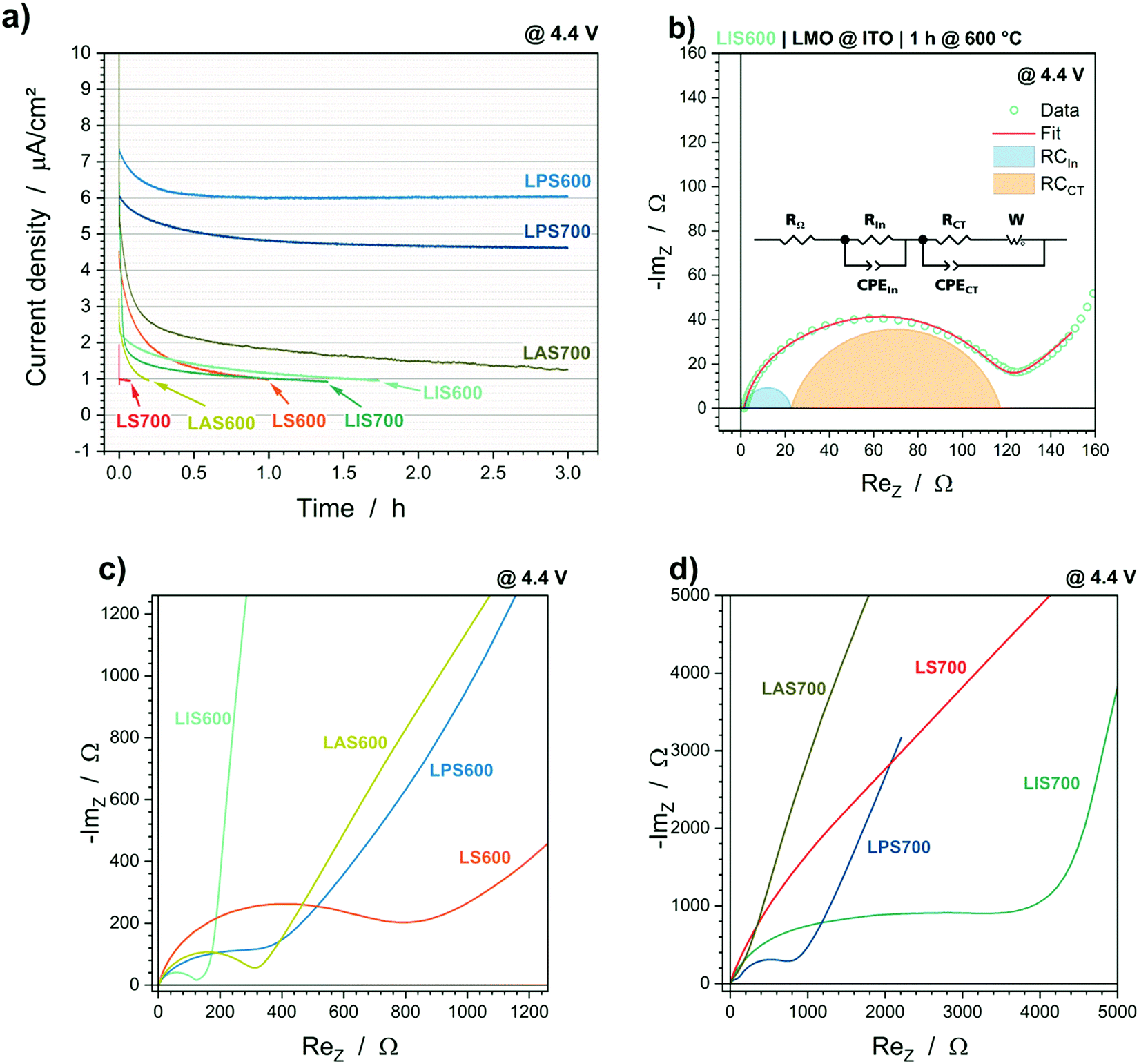 | ||
| Fig. 3 (a) Current decay during the CV step @ 4.4 V vs. Li/Li+ for 3 h and (b–d) Nyquist plots of the EIS data. (b) Fit of EIS data of LIS600 using the given equivalent circuit, (c) Nyquist plots of samples annealed at 600 °C and (d) Nyquist plots of samples annealed at 700 °C. LMO@StSt (red), LMO@ITO (cyan), LMO@Pt (blue) and LMO@Au (green). | ||
| Sample | R Ω (Ω) | R In (Ω) | R CT (Ω) |
|---|---|---|---|
| 600 °C crystallization temperature | |||
| LS600 (LMO@StSt) | 1.6 | 377.6 | 424.6 |
| LIS600 (LMO@ITO) | 1.4 | 21.3 | 94.7 |
| LPS600 (LMO@Pt) | 1.4 | 33.3 | 282.6 |
| LAS600 (LMO@Au) | 1.6 | 32.3 | 252.1 |
| 700 °C crystallization temperature | |||
| LS700 (LMO@StSt) | No capacity | ||
| LIS700 (LMO@ITO) | 1.4 | 1397.0 | 2129.2 |
| LPS700 (LMO@Pt) | 1.8 | 88.6 | 806.1 |
| LAS700 (LMO@Au) | No capacity | ||
The ohmic resistances RΩ are similar, so the oxide layer formation between substrate/interlayer and LMO has, comparatively, little impact on the electrochemical performance. Significant differences are observable in the values of RIn and RCT, which may be correlated with the overvoltages in the cycling experiments. When the samples are crystallized at 600 °C, LIS600 has the lowest RIn and RCT, followed by comparable values from LAS600 and LPS600; the highest resistances occur for LS600. The morphology of the LMO crystallized at 600 °C is comparable (top view SEM images in Fig. S1, ESI†), which suggests that differences in the electrochemical active surface area are not responsible for this result. In contrast, there is an increase in particle size of the LMO layer crystallized at 700 °C on ITO, which leads to a lower surface area and the increase in RIn and RCT. However, this increase cannot be completely explained by the change in morphology since RIn is a fifth of the total resistance for LIS600 and about a half for LIS700. Therefore, the formation of an insulating layer on the LMO surface with increasing temperature, which also depends on the interlayer material, is likely and will be discussed below.
The charge transfer resistance RCT is also highly dependent on the interlayer material. In addition to the high increase of RCT with higher particle size, LPS700 shows low RIn and RCT values, which suggest that the electrochemical reaction measured is not Li-ion extraction/insertion in the LMO cathode material, but a side reaction. In addition to the coulombic efficiency during cycling and impedance, this hypothesis is supported by the current decay during the CV step at the end of charge performed before the impedance measurement (Fig. 3a). The current density values of 6.0 μA cm−2 and 4.6 μA cm−2 throughout the entire CV step suggest that a faradaic current may be flowing for LPS600 and LPS700. In contrast, the cells containing the other interlayer materials have current density curves, which seem to be more capacitive in nature as they reach the current density limit of 0.75 μA cm−2 in less than 2 h. This unique behaviour of the LMO@Pt samples might be related to the electrochemical dissolution of metallic platinum. The top view SEM images (Fig. S1, ESI†) show that there are cracks in the LMO layers, so that a direct contact between the Pt interlayer and the liquid electrolyte exists. The redox reaction of Pt → Pt2+ + 2e− has a standard redox potential of 1.20 V vs. SHE (standard hydrogen electrode), which corresponds to 4.24 V vs. Li/Li+.30 Even when considering an overpotential due to the use of an organic electrolyte and the deviation from the standard conditions, the electrochemical dissolution of Pt at 4.4 V vs. Li/Li+ is likely. In the case of a solid-state electrolyte combined with a dense cathode layer up to 1 μm, the risk of Pt dissolution would likely be small, but still present.
Compositional depth profiling by ToF-SIMS
Changes in discharge capacity, overvoltage and resistance suggest changes in the LMO spinel lattice during the crystallization process that depend on the interlayer and substrate material. Therefore, dedicated ToF-SIMS measurements were performed to detect which elements diffused in- and out-of the LMO active layer. Throughout this section, the detected mass-to-charge ratios (m/z) will be described by Li− (m/z = 7 u), MnO− (m/z = 71 u), AlO− (m/z = 43 u), CrO− (m/z = 68 u), InO− (m/z = 131 u), Pt− (m/z = 195 u), PtO− (m/z = 211 u), Au− (m/z = 197 u) and AuO− (m/z = 213 u). FeO− cannot be reliably analysed because it has the same m/z value as MnOH− (m/z = 72 u).Fig. 4 shows the ToF-SIMS depth profiles of the samples crystallized at 700 °C. The results of the 600 °C crystallized samples can be found in the supporting information (Fig. S4, ESI†). The sputter time is normalized to one for the interface between LMO and interlayer because of the different sputter yields for Au, Pt, ITO and StSt. The LMO-interlayer interface is defined as the intercept of the MnO− signal and the interlayer element signal. Without an interlayer, i.e. the LMO@StSt sample, Fig. 4a demonstrates an overlap of the Li− and MnO− signals (from LMO) with the AlO− and CrO− signals (from StSt containing 20 wt% Cr and 5 wt% Al). This implies interdiffusion of the substrate elements into LMO, as well as Li and Mn into the substrate and results in Li depletion at the LMO surface. A Pt interlayer blocks the diffusion of Cr and Al into the LMO layer (Fig. 4c), but it cannot block Li diffusion. Furthermore, Pt is present throughout the LMO layer. Fig. 4d illustrates significant overlap of the ion signals derived from the LMO layer, Au interlayer and StSt. The Au interlayer does not block the diffusion of the substrate elements Cr and Al into the LMO layer nor the diffusion of Li and Mn into the StSt substrate, which is also demonstrated by the overlap of all element signals in the Au interlayer region between 1 and 2 normalized sputter time. In contrast, the ITO interlayer demonstrates excellent interdiffusion blocking characteristics. Fig. 4b shows a clear separation between the signals of the LMO and the StSt substrate. The ITO interlayer, evident from the InO− signal, seems to completely block Al and Cr diffusion and significantly reduces Li and Mn depletion. The SEM cross sections demonstrated the oxide layer formation (Fig. 1), which is confirmed here as peaks of CrO− and AlO− at the interface LMO/StSt, ITO/StSt or Pt/StSt. An Au interlayer, which cannot block Cr and Al diffusion, shows the oxide layer formation at the LMO/Au interface.
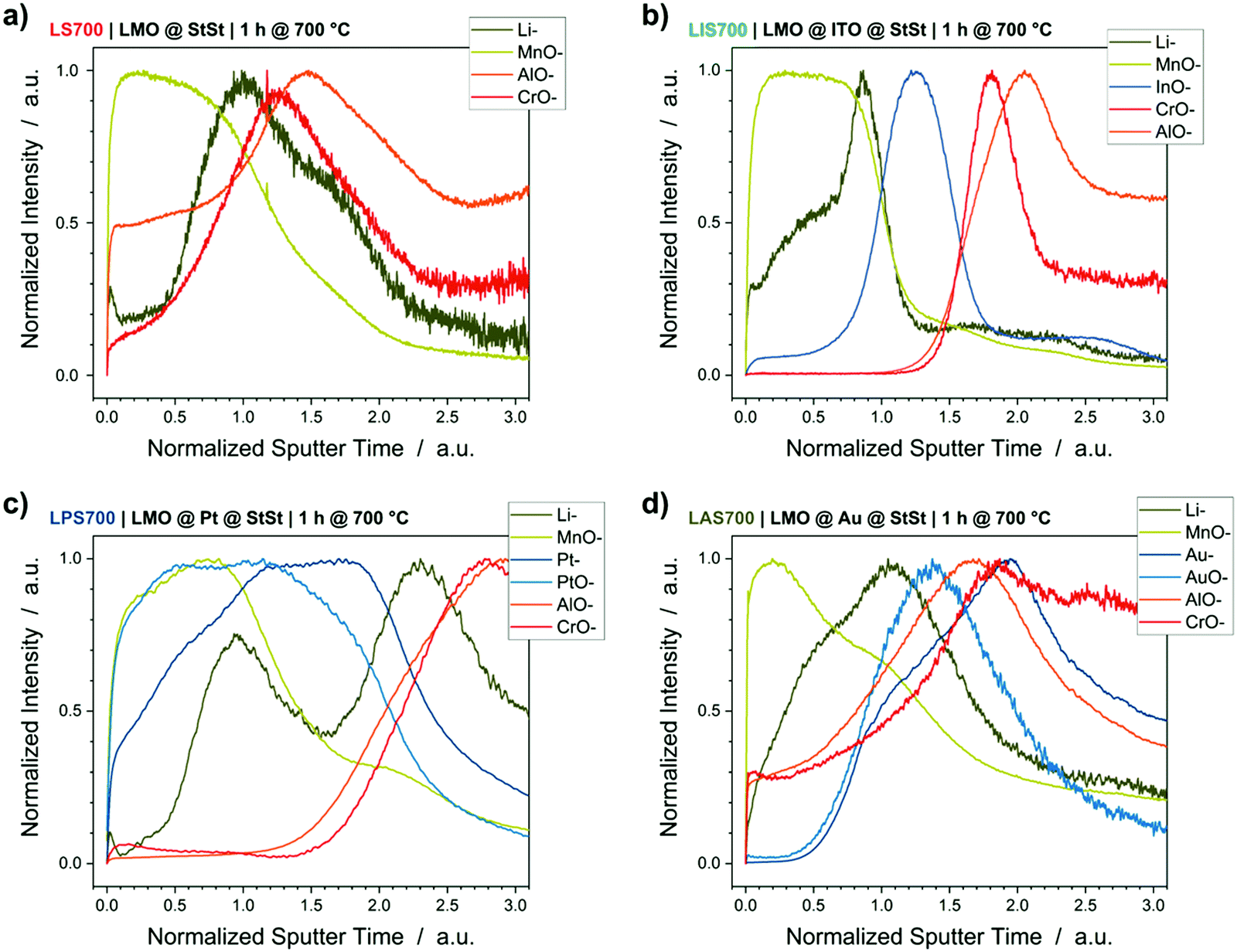 | ||
| Fig. 4 ToF-SIMS depth profiles of samples annealed at 700 °C: (a) LMO@StSt, (b) LMO@ITO, (c) LMO@Pt and (d) LMO@Au. Sputter time is normalized to 1 for the interface between LMO and interlayer. LMO elements (Li− and MnO−) in green, StSt elements (AlO− and CrO−) in red and interlayer elements (InO−, Pt−, PtO−, Au− and AuO−) in blue. Depth profiles of samples annealed at 600 °C are in the ESI† (Fig. S4). | ||
Crystalline phase
The effect of interdiffusion also affects the LMO crystallinity as can be deduced from GIXRD patterns (Fig. 5). LMO@StSt, LMO@Pt and LMO@ITO show crystalline LMO reflections and no mixed phases (Fig. 5a and b). LMO@Au exhibits crystalline LMO, but also the formation of AuLi (2θ = 28.9° and 41.0°, ICSD 58523), AuLi3 (2θ = 28.1°, ICSD 58524), Au2Mn (2θ = 28.1°, 37.8° and 41.3°, ICSD 657181) and AlAu2 (2θ = 20.0°, 38.1° and 43.3°, ICSD 606020) alloys due to interdiffusion processes (Fig. 5b). Additionally, MnO2 (ICSD 20228) and MnO (ICSD 18006) phases are detected. The LMO hkl 111 reflection at approx. 18.720° 2θ shows a shift to lower 2θ values with increasing interdiffusion. In Fig. 5c, LIS600 can be viewed as a benchmark. The 2θ position of 18.720° is comparable with the literature value of 18.714° (ICSD 94340). Shifts of 0.053° (LIS700) up to 0.1848° (LS700) to lower 2θ degrees are observable with increasing temperature and depending on interlayer. These shifts can be correlated to the formation of other spinel phases, such as Mn3O4, Fe3O4, FeMn2O4, MnFe2O4 or CrMn2O4. The 111 reflections are reported as 18.022° (ICSD 31094), 18.467° (ICSD 77592), 18.040° (ICSD 28666), 18.237° (ICSD 56121) and 18.097° (ICSD 74707), respectively. According to this data and ToF-SIMS depth profiles, interdiffusion of substrate elements and Li depletion in the LMO layer lead to the substitution of Li and Mn in the LMO spinel lattice.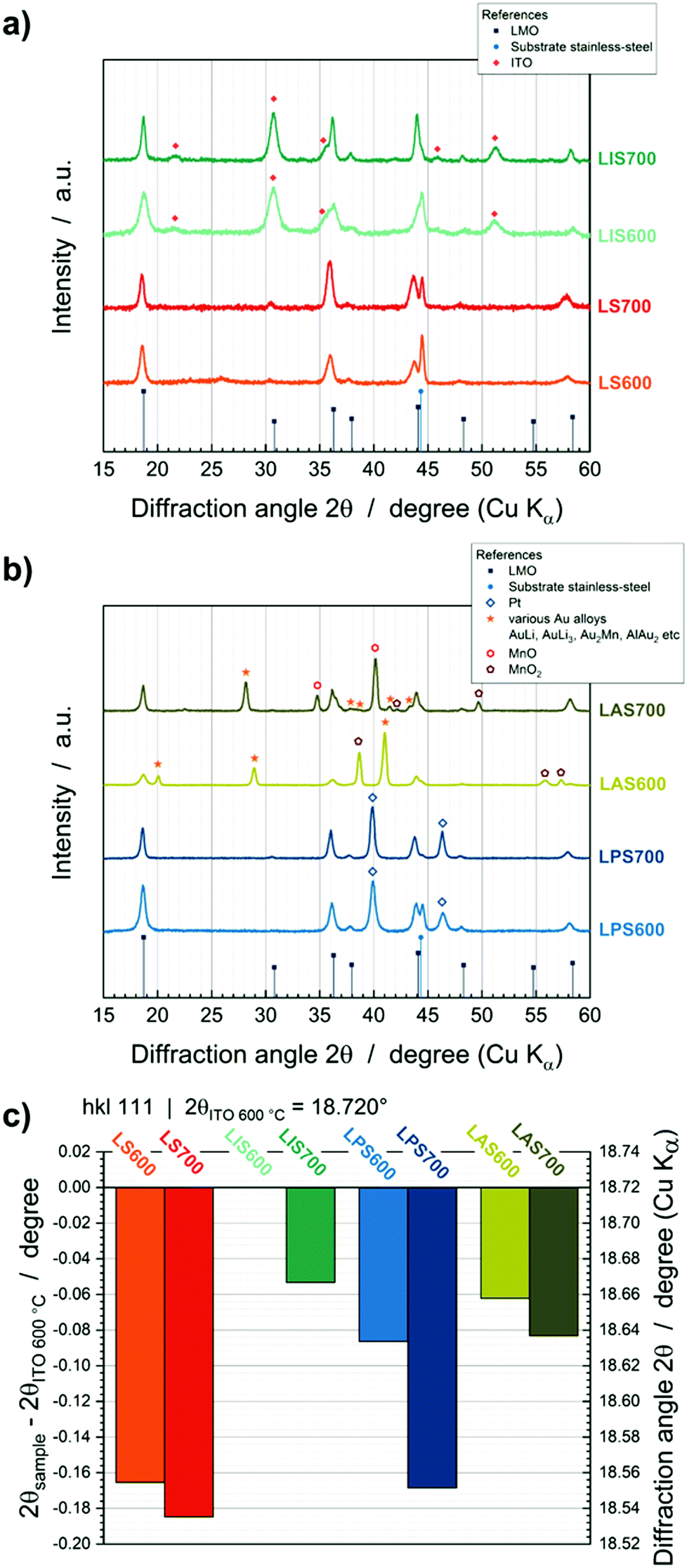 | ||
| Fig. 5 (a) GIXRD patterns of LMO StSt (red) and LMO@ITO (cyan) and (b) GIXRD patterns of LMO@Pt (blue) and LMO@Au (green). (c) Analysis of shift of the reflex positions of all samples: LMO@ITO is set as benchmark. | ||
Changes in oxidation states from XPS measurements
The substitution of Li and Mn in the LMO spinel lattice with substrate and interlayer elements would lead to changes in the Mn oxidation states. The Mn oxidation states in the LMO lattice are 3+ and 4+. In MnX2O4 spinels (X = Fe, Cr, Al, Pt, Mn), the oxidation state of Mn is 2+ and of X is 3+. Therefore, increased amounts of Mn2+ and decreased amounts of Mn3+ and Mn4+, as determined from the XPS Mn 2p3/2 peak could indicate the presence of MnX2O4 spinel phases. For the LMO@StSt samples, an increase in Mn2+ species at the surface from 18% to 24% with higher annealing temperature is detected (Fig. 6a). Lower amounts of Mn2+ are detected if an ITO interlayer is present, i.e. 11% and 17% for LIS600 and LIS700, respectively. The decrease of Mn4+ species also indicates that the formation of MnX2O4 (X = Fe, Cr, Al, Pt, Mn) spinels are favoured at the surface. Furthermore, Fe is detected at the surface of the LMO@StSt samples (Fig. 6b). In contrast, the results for the LMO@ITO samples shows a low Fe signal for LIS700 and no Fe signal from the surface of LIS600.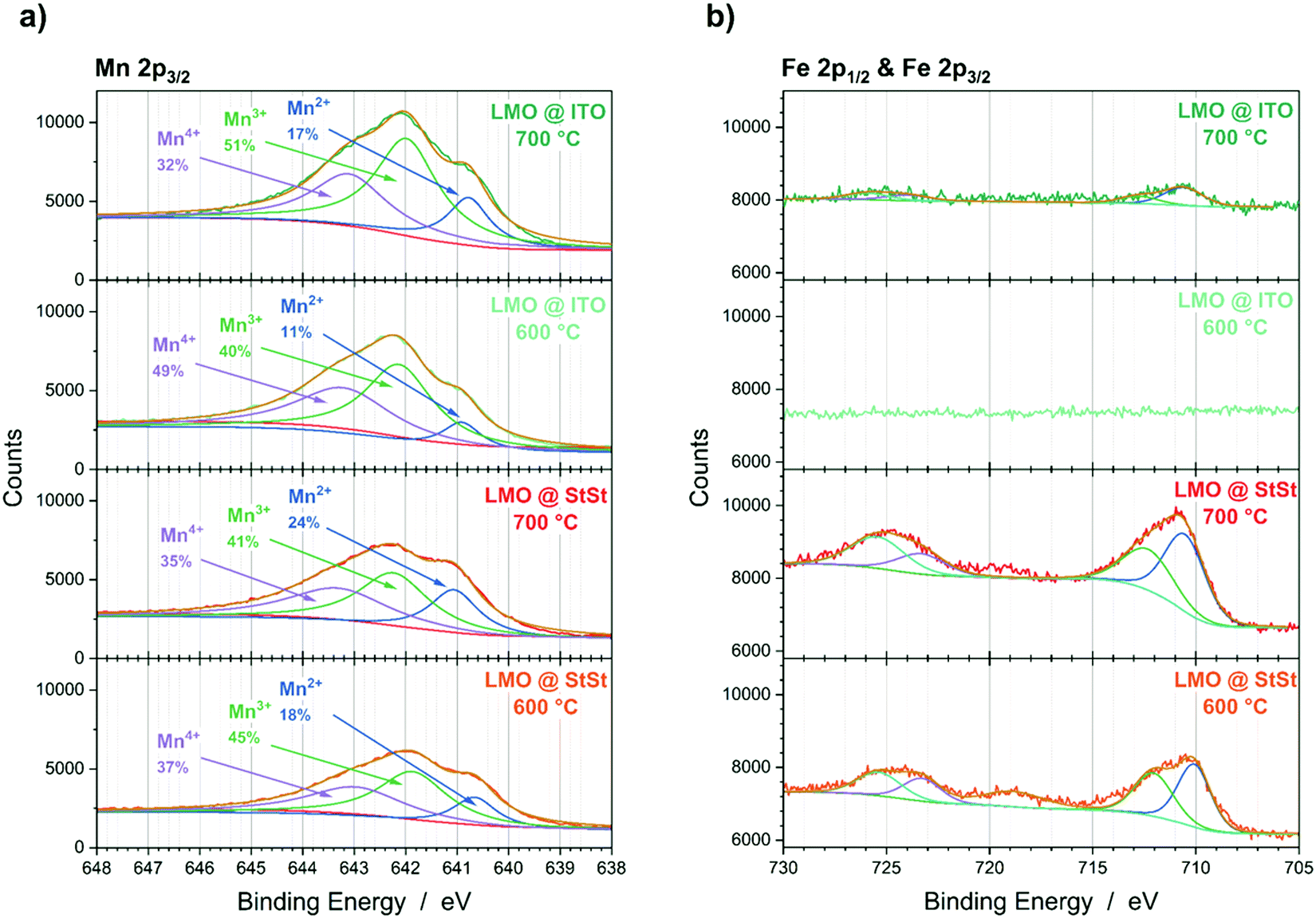 | ||
| Fig. 6 XPS data of LMO@StSt and LMO@ITO samples annealed at 600 °C and 700 °C. (a) Mn 2p3/2 region and (b) Fe 2p1/2 and Fe 2p3/2 region. | ||
Discussion
Interdiffusion processes between the LMO, the interlayer and the substrate cause changes in the LMO active material and therefore its electrochemical behaviour. Li and Mn diffuses into the interlayer and/or substrate, while the substrate elements (Fe, Cr, Al) and the interlayer element Pt diffuse into the LMO layer. This leads to substitution of Li and Mn in the LMO spinel lattice with Fe, Cr, Al and Pt. Li depletion leads to a change of the oxidation states of the Mn from 3+ and 4+ in the LMO spinel to 2+ and 3+ in MnX2O4 (X = Fe, Cr, Al, Pt, Mn) spinels.Ion radii between 0.55 Å and 0.80 Å are necessary for substitution of Li and Mn in the LMO crystal lattice.31,32 Various ions derived from the substrate, such as Fe2+ (0.78 Å), Fe3+ (0.65 Å), Fe4+ (0.59 Å), Al3+ (0.54 Å), Cr2+ (0.80 Å), Cr3+ (0.62 Å), Cr4+ (0.55 Å), and Pt4+ (0.60 Å), fill this criteria.33 As a result, any of these ions could diffuse into the LMO layer and were detected via ToF-SIMS. In contrast, Au3+ has an ion radius of 0.85 Å and consequently, Au diffusion into the LMO layer has not been detected. The general formula for a spinel structure is AB2O4, where A and B have the oxidations states of 2+ and 3+, respectively. Therefore, spinel compositions like FeMn2O4, CrMn2O4 or FeCr2O4etc. are also possible. The presence of such spinel phases and MnX2O4 (X = Fe, Cr, Al, Pt, Mn) spinels would result in the formation of an insulating layer at the surface of the LMO layer. The qualitative comparison of the ToF-SIMS depth profiles of the LIS600 (Fig. S4b, ESI†) and LIS700 (Fig. 4b) samples indicates a lower Li amount at the surface of the LIS700 samples, which confirms a higher amount of Mn2+-based spinel compositions. This insulating layer is then responsible for the increase in RIn and the overvoltage. The decrease in discharge capacities with higher interdiffusion degree is correlated with the thickness of the insulating layer. As the XPS measurements are only surface-sensitive, the thickness cannot be quantified.
The presence of spinel lattices with the composition LiMnXO4 (X = Fe4+, Cr4+, Pt4+) could explain the changes in the ratio of the first and second redox peaks in differential capacity plots. The substitution of the Mn4+ by other ions could change the diffusion activation energies for the Li ions, and thus the insertion/extraction process. Therefore, even the samples with interlayers not able to block interdiffusion have higher discharge capacities than the LS600 sample. At 700 °C, Au cannot block interdiffusion of StSt elements and Li depletion, while Pt tends to diffuse into the LMO layer. In both cases, the discharge capacities decrease. In contrast, ITO functions as interdiffusion barrier even at 700 °C and the increase in discharge capacity due to a higher crystallization degree of LMO is measurable. The higher overvoltage of LIS700 is induced by a number of factors including a decrease of the electrochemical active surface area due to an increase in particle size, the formation of insulating layer and a small amount of Fe in the LMO lattice. The larger particle size, and thus the longer diffusion pathways, affect the charge transfer and concentration polarization at 30 μA cm−2 (ca. 6C). In particular, the thickness and composition of the insulating layer has a significant impact on the overvoltage and electrochemical performance. This results in a higher overvoltage and a higher value for RIn.
Conclusion
The crystallization process of LMO on StSt leads to diffusion of Al, Cr and Fe into the LMO layer as well as the diffusion of Li and Mn into the substrate. Al, Cr and Fe replace Li and Mn in the spinel lattice and form new spinel compositions like MnX2O4 (X = Fe, Cr, Al, Pt, Mn) or LiMnXO4 (X = Fe4+, Cr4+, Pt4+). The loss of Li, the consequent change in the Mn oxidation states and the formation of an insulating layer at the LMO surface result in a reduction of the electrochemical performance including a decrease in discharge capacity and increases in RIn, RCT and the overvoltage.Three different conductive interlayers (Au, Pt and ITO) have been tested as potential temperature-resistant current collectors on StSt substrates. Au and Pt as an interlayer cannot satisfactory suppress the interdiffusion processes. LMO@Pt and LMO@Au crystallized at a temperature of 700 °C show a significant decrease in the discharge capacities compared to the 600 °C crystallized samples. In contrast, ITO exhibits the best diffusion barrier properties out of the three interlayers tested. Since ITO prevents Li and Mn depletion as well as interdiffusion of StSt elements (Fe, Cr, Al) into the LMO layer, the discharge capacity increases from 4.2 μA h cm−2 (600 °C) to 5.1 μA h cm−2 (700 °C).
The use of ITO as interlayer promises higher utilization of cathode active materials, which increases the performance and energy density of TF-SSBs. Its production via wet chemical processing, along with a flexible StSt foil as a substrate/current collector can enable cost-effective slot die roll-to-roll processes for TF-SSB manufacturing.
Conflicts of interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.Acknowledgements
The authors acknowledge the joint Empa-Fraunhofer ICON funding line (ICON: International Cooperation and Networking, IE4B project) and the funding of the German Federal Ministry of Education and Research (BMBF) for the project SOLID (grant number 03XP0129E). We thank the Laboratory for Surface Science and Coating Technologies at Empa for the access to ToF-SIMS equipment.References
- W. Hou, X. Guo, X. Shen, K. Amine, H. Yu and J. Lu, Nano Energy, 2018, 52, 279–291 CrossRef CAS.
- Y.-S. Hu, Nat. Energy, 2016, 1, 652 CrossRef.
- J. Janek and W. G. Zeier, Nat. Energy, 2016, 1, 1167 CrossRef.
- D. H. S. Tan, A. Banerjee, Z. Chen and Y. S. Meng, Nat. Nanotechnol., 2020, 15, 170–180 CrossRef CAS.
- R. C. Xu, X. H. Xia, S. Z. Zhang, D. Xie, X. L. Wang and J. P. Tu, Electrochim. Acta, 2018, 284, 177–187 CrossRef CAS.
- T. Zhang, W. He, W. Zhang, T. Wang, P. Li, Z. Sun and X. Yu, Chem. Sci., 2020, 334, 928 Search PubMed.
- F. Zheng, M. Kotobuki, S. Song, M. O. Lai and L. Lu, J. Power Sources, 2018, 389, 198–213 CrossRef CAS.
- H. Buschmann, J. Dölle, S. Berendts, A. Kuhn, P. Bottke, M. Wilkening, P. Heitjans, A. Senyshyn, H. Ehrenberg, A. Lotnyk, V. Duppel, L. Kienle and J. Janek, Phys. Chem. Chem. Phys., 2011, 13, 19378–19392 RSC.
- A. J. Samson, K. Hofstetter, S. Bag and V. Thangadurai, Energy Environ. Sci., 2019, 12, 2957–2975 RSC.
- J. Sastre, X. Chen, A. Aribia, A. N. Tiwari and Y. E. Romanyuk, ACS Appl. Mater. Interfaces, 2020, 36196–36207 CrossRef CAS.
- J. Sastre, A. Priebe, M. Döbeli, J. Michler, A. N. Tiwari and Y. E. Romanyuk, Adv. Mater. Interfaces, 2020, 2000425 CrossRef CAS.
- S. Breuer, D. Prutsch, Q. Ma, V. Epp, F. Preishuber-Pflügl, F. Tietz and M. Wilkening, J. Mater. Chem. A, 2015, 3, 21343–21350 RSC.
- R. DeWees and H. Wang, ChemSusChem, 2019, 12, 3713–3725 CrossRef CAS.
- N. J. Dudney, B. Neudecker, A. Ueda and C. D. Evans, Solid State Ionics, 2000, 135, 33–45 CrossRef.
- J. Li, C. Ma, M. Chi, C. Liang and N. J. Dudney, Adv. Energy Mater., 2015, 5, 1401408 CrossRef.
- Y. Zhu, X. He and Y. Mo, J. Mater. Chem. A, 2016, 4, 3253–3266 RSC.
- Y. Zhu, X. He and Y. Mo, ACS Appl. Mater. Interfaces, 2015, 7, 23685–23693 CrossRef CAS.
- F. Han, Y. Zhu, X. He, Y. Mo and C. Wang, Adv. Energy Mater., 2016, 6, 1501590 CrossRef.
- L. Baggetto, R. R. Unocic, N. J. Dudney and G. M. Veith, J. Power Sources, 2012, 211, 108–118 CrossRef CAS.
- J. H. Kim, J. Park, J. Y. Cheong, A. Song, K.-B. Chung, Y. C. Park, I.-D. Kim, Y. J. Kim, K. Park and H.-S. Kim, Ceram. Int., 2018, 44, 20093–20104 CrossRef CAS.
- M. Gellert, K. I. Gries, J. Sann, E. Pfeifer, K. Volz and B. Roling, Solid State Ionics, 2016, 287, 8–12 CrossRef CAS.
- M. Gellert, K. I. Gries, J. Zakel, A. Ott, S. Spannenberger, C. Yada, F. Rosciano, K. Volz and B. Roling, Electrochim. Acta, 2014, 133, 146–152 CrossRef CAS.
- S. Komaba, N. Kumagai, M. Baba, F. Miura, N. Fujita, H. Groult, D. Devilliers and B. Kaplan, J. Appl. Electrochem., 2000, 30, 1179–1182 CrossRef CAS.
- A. N. Filippin, T.-Y. Lin, M. Rawlence, T. Zünd, K. Kravchyk, J. Sastre-Pellicer, S. G. Haass, A. Wäckerlin, M. V. Kovalenko and S. Buecheler, RSC Adv., 2018, 8, 20304–20313 RSC.
- M. Rawlence, A. N. Filippin, A. Wäckerlin, T.-Y. Lin, E. Cuervo-Reyes, A. Remhof, C. Battaglia, J. L. M. Rupp and S. Buecheler, ACS Appl. Mater. Interfaces, 2018, 10, 13720–13728 CrossRef CAS.
- P. Hofmann, F. Walther, M. Rohnke, J. Sann, W. G. Zeier and J. Janek, Solid State Ionics, 2019, 342, 115054 CrossRef CAS.
- A. Prodi-Schwab, T. Lüthge, R. Jahn, B. Herbig and P. Löbmann, J. Sol-Gel Sci. Technol., 2008, 47, 68–73 CrossRef CAS.
- C. M. Julien, A. Mauger, K. Zaghib and H. Groult, Inorganics, 2014, 2, 132–154 CrossRef CAS.
- M. M. Thackeray, Prog. Solid State Chem., 1997, 25, 1–71 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical methods. Fundamentals and applications, John Wiley & Sons, New York, 1980 Search PubMed.
- K. E. Sickafus, J. M. Wills and N. W. Grimes, J. Am. Ceram. Soc., 1999, 82, 3279–3292 CrossRef CAS.
- M.-L.-P. Le, P. Strobel, C. V. Colin, T. Pagnier and F. Alloin, J. Phys. Chem. Solids, 2011, 72, 124–135 CrossRef CAS.
- U. Müller, Anorganische Strukturchemie, Vieweg + Teubner, Wiesbaden, 6th edn, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00893a |
| This journal is © The Royal Society of Chemistry 2021 |