
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advancements in coinage metal nanostructures and bio-applications
Si Yin
Tee
and
Enyi
Ye
 *
*
Institute of Materials Research and Engineering, Agency for Science, Technology & Research 2 Fusionopolis Way, Singapore, 138634, Singapore. E-mail: yeey@imre.a-star.edu.sg
First published on 1st February 2021
Abstract
Coinage metals are best known for their monetary, adorning, and metallurgical values from a historical perspective, while their metal nanostructures and applications may be considered as products of modern science since the potential of nanotechnology has only been realized in recent years. Coinage metal nanostructures possess noteworthy properties such as size- and shape-dependent catalytic, optical, electrical, and chemical properties. Each of these attributes has inspired significant investigation toward the design and synthesis of coinage metal nanostructures as well as their potential application. For these reasons, this review summarizes the recent development of coinage metals such as Cu, Ag, Au, and their bimetallic nanostructures, particularly using solution-based approaches. It covers various synthetic methodologies including standard protocols as well as recent strategies that have been implemented to improve the functionality by effectively controlling the size, morphology, and composition of the coinage metal nanostructures. It also highlights the applications of these coinage metal nanostructures in bio-applications such as antibacterial activities, surface-enhanced Raman scattering (SERS)-based detection, and electrochemical sensing. Moving forward, this review provides a brief discussion on the challenges and future directions of these promising metal nanostructures, which will be essentially useful in designing new nanostructures for better functionalities in the future.
1. Introduction
Metal nanostructures have been gaining tremendous interests owing to their extremely small feature size for wide applications, which is more superior to their bulk counterparts. The distinct property of nanoscale materials that differentiates them from their bulk properties is the increase in the surface area to volume ratio. The benefit of having a large surface area to volume ratio is that the percentage of surface atoms becomes significant, which can lead to an increase in the reactivity of the material.1,2 Therefore, their unique size and shape-dependent properties are promising for widespread commercial applications in the field of electronics, information storage, biomedical devices, as well as environmental and energy technologies.3–8 Among various metallic nanostructures, group 11 coinage metals (i.e., Cu, Ag, and Au) have drawn a lot of interest and are being extensively studied by researchers within the large transition-metal family. The modern scientific evaluation of metallic nanoparticles did not begin until Michael Faraday pioneered the preparation of colloidal Au solution by reducing Au salt with phosphorus in water.9 Later, Enüstün and Turkevich prepared spherical nanoparticles of Au and Ag with tunable sizes by the reduction of Au or Ag salts using citrate in aqueous solutions in 1960s.10 The design and synthesis of these coinage metals are crucial for fundamental research and technology development; to date, there have been multiple synthetic methods for developing different types of transition metal nanostructures with well-controlled dimensions. This includes hydro/solvothermal methods, seed/template-mediated synthesis, polyol synthesis, and electrochemical synthesis; the ultimate goal of these fabrication methods is to introduce new nanostructures and to improve the existing ones for better functionalities. At present, manipulation of the size, shape, architecture, composition, and structure at the nanoscale are essentially key factors in enhancing the properties of metal nanostructures and thus their potential applications.In addition to monometallic nanostructures, bimetallic nanostructures are a new class of nanomaterials that may have better technological usefulness with distinct properties from those of individual atoms and molecules or bulk matter. Synergetic effects between two distinct elements make them superior to the monometallic counterpart, bringing superior or new physical/chemical properties and improved performances.11–15 The properties and the applicability of these bimetallic nanostructures not only depend on their size and shape but also on the combination of the component metals (composition) and their fine structure. Generally, bimetallic nanostructures are classified into three categories: alloyed structure, core–shell, and heterostructured, according to the mixing patterns of two different metals. The alloyed structure consists of the homogeneous distribution of the two types of metal atoms, which may be either ordered or randomly mixed, whereas the core–shell structure is made up of a shell of one type of metal ions surrounding a core of another metal. During the formation process, the metal ions that are reduced first form the inner core, while the other metal grows as a shell on the surface of the core, finally resulting in a core–shell structure. By controlling their core size and shell thickness, their properties can be well modulated. Nonetheless, under specific conditions, individual nucleation and growth of two types of metal atoms can independently occur to form the heterostructure. The mixed interface that they share during the growth process may play a significant role in controlling their behaviors.
The early scientific study of bimetallic nanoparticles was pioneered by Morriss and Collins, who synthesized the Au@Ag core–shell nanoparticles and studied their optical properties in the 1960s.16 The unique optical properties in these coinage nanostructures arise from the collective oscillation of conductive electrons (i.e., surface plasmon) as a result of the interaction of light waves and metallic surfaces. This fascinating finding has inspired further work on the development of bimetallic Au–Ag nanostructures, particularly on their preparation, characterization, properties, and application. In this review article, we summarize the recent progress in solution-based synthetic routes for the design and preparation of metallic and bimetallic nanostructures of Cu, Ag, and Au. In this aspect, more attention is focused on the tailoring of the particle size, shape, architecture, and composition of the nanostructured materials, which make them excellent candidates in antibacterial activities, SERS-based detection, and electrochemical sensing (Fig. 1). Lastly, we discuss the major challenges and future directions for these promising metal nanostructures.
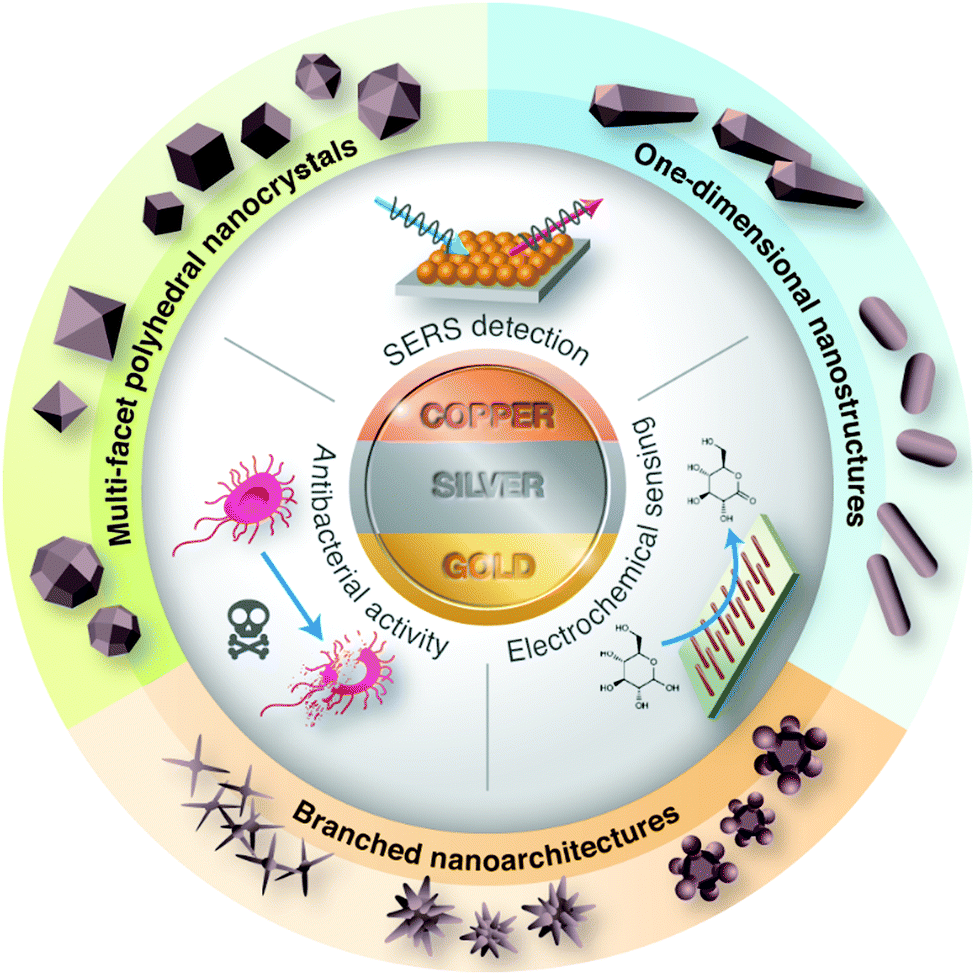 | ||
| Fig. 1 General presentation of coinage metal nanostructures and their applications. | ||
2. General synthetic methodology
The research on nanostructured materials has greatly intensified due to the advancements of the synthetic procedure, which enable good control over the size and shape. There are two general approaches for metal nanostructure synthesis—top-down approaches and bottom-up assembly. Most top-down approaches are physical methods (i.e., milling and lithography), which involve the use of expensive synthetic pathways that are not suitable for large-scale industrial production. Also, this approach limits the control over the size, size distribution, and particle shape. On the other hand, bottom-up approaches such as wet chemical synthesis are far more commonly used in the synthesis of nanostructured materials, which enable the preparation of metal nanostructures in high yield and structural purity with varying size, shape, structure, composition, and surface chemistry. Typically, colloidal nanostructures are synthesized in the liquid phase by the reduction of metal salts with chemical reducing agents.17 The size and shape of the metal nanoparticles are controlled by the concentration, reducing agents, and capping agents.6,18 Spherical metal nanoparticles are usually prepared by the direct reduction of a metal salt with a strong reducing agent in the presence of capping agents.18 On the contrary, anisometric metal nanostructures are usually prepared using a seed-mediated growth approach, in which the preformed metal seed particles are added to a solution containing the metal salt, a weak-reducing agent, and a shape-directing agent. This concept prevents the formation of new agglomerates and provides the growth in an ordered and desired manner.18,19The general path for the nucleation and growth of nanostructures from solution is summarized into five different stages, namely, (i) nucleation, (ii) growth, (iii) Ostwald ripening, (iv) surfactant capping, and (v) precipitation. The nucleation process represents the initial stage of any crystallization process, which refers to the production of monomers from the reaction of precursors. As the precursor decompose, the concentration of metal atoms increases gradually until it reaches a point of supersaturation, where the atoms start to aggregate into numerous small clusters and tend to quickly aggregate together to form larger, more thermodynamically stable particles.17,20 In most cases, Ostwald ripening happens at a later stage of crystal growth, which involves the dissolution of smaller particles and the consequent further growth of larger particles. During the formation process, a stabilizer or surfactant is added in order to prevent particle aggregation and mediate the growth process. Finally, the nanostructured products can be obtained by precipitation and further size-selective separation process can be performed to narrow the size distribution. With this colloidal synthesis method, nanostructures with various sizes, shapes, compositions, crystal structures, and surfactant coatings have been reported. For example, simple shapes such as spheres and cubes, elongated morphologies such as rods and wires, and more complicated faceted geometries such as plates and polyhedra have been fabricated.21–23 Hybrid nanostructures (i.e., heterostructures) as well as nanoscale alloys of metals with varying compositions have also been constructed.24,25
2.1. Copper nanostructures
Cu was one of the earliest metals to be exploited and is still considered as one of the important metals because of its excellent properties such as ductility, malleability, and conductivity. In comparison with another two coinage metals such as Au and Ag, the preparation of stable and monodisperse Cu nanostructures with controllable sizes, shapes, and surface properties has been relatively limited due to the difficulty of reducing Cu salts into metallic Cu and their inherent propensity for surface oxidation in ambient conditions.26,27 To address the oxidation limitation, various polymers or surfactants have been used as reducing agents and capping agents, which serve as a protective layer for the preparation of Cu nanostructures in aqueous solutions.Alternatively, glucose and ascorbic acid have been used as reducing agents to synthesize Cu nanostructures under elevated temperature for the reduction process to take place.32 Jin et al. carried out the synthesis of Cu nanostructures using aqueous-media reduction of Cu salts with glucose at 100 °C. In this approach, hexadecylamine (HDA) was employed as the capping agent to produce Cu nanostructures with controlled shapes and good uniformity.26 The study suggested that HDA was an effective capping agent for Cu and the concentration of HDA was one of the key factors in controlling the morphology of the final product. By changing the concentration of HDA, the distribution of the single-crystal versus multiply-twinned seeds could be manipulated at the nucleation stage. In addition to the concentration of HDA, the reduction kinetics induced by glucose also have an important impact on the morphology of the final product. Both factors contributed to the formation of four different kinds of Cu nanostructures encased by (100) facets (i.e., pentagonal nanowires, tadpole-like nanowires, pentagonal bipyramids, and nanocubes) via the selective adsorption of HDA on the (100) facets. In a recent study, this HDA-assisted glucose reduction was further modified by Fu et al. to synthesize Cu nanowires in high yield via in situ seed-mediated two-step approach.33 The process involved the nucleation of the nanoseeds, in which Cu ions were reduced to form nondecahedral Cu nanodots and further used to promote the formation of decahedral multiply-twinned particles for the subsequent growth of Cu nanowires. The seed-mediated two-step approach restricted the spontaneous ripening of undesired Cu nanoparticle by-products by segregating the nucleation step at a relatively lower temperature, thereby improving the Cu nanowire production yield.
In contrast to these approaches, our research group prepared the penta-twinned Cu nanowires through the controlled disproportionation of CuI facilitated by oleylamine and the subsequent disproportionation of the CuI complexes into metallic Cu0 and bivalent CuII (Fig. 2).36 In addition, these oleylamine-coated Cu nanowires could self-assemble by surface ligand exchange with trioctylphosphine. The trioctylphosphine-coated Cu nanowires also served to improve the oxidation resistance and allowed them to be well stored in air. The disproportionation reaction route was extended by Guo et al. to develop Cu nanospheres and Cu nanocubes using trioctylphosphine and trioctylphosphine oxide as the capping agents, respectively.37 The shape directing effect of the different capping agents was based on their coordination effects to Cu+ (Fig. 3). For instance, the stronger coordination effect of trioctylphosphine to Cu+ inhibited the metallic ions from being rapidly reduced to Cu0, resulting in nucleation at a higher temperature. This eventually led to the formation of isotropous crystals, which made the seeds grow equally in all directions into spherical-shaped crystals. On the other hand, the weaker coordination effect of trioctylphosphine oxide to Cu+ promoted the nucleation process to occur at a relatively lower temperature, leading to the (100) facets of the Cu crystal seeds selectively capped by the ligands. Consequently, the other facets grew at a faster rate than the (100) facets. The lower reaction temperature allowed a slow growth rate and hence the different reaction kinetics contributed to the formation of different Cu nanostructures. Huang et al. developed two distinctly-shaped Cu nanostructures (i.e., nanocubes and nanowires) by employing different shape-controlling additives.38 The use of NH4Cl and RuCl3 additives in the synthetic process has been recognized as an effective approach to control the growth of Cu nanostructures. Specifically, NH4Cl was employed in the oxidative etching process to selectively etch away the seeds containing twin defects and provide single-crystal seeds to form the nanocubes. Conversely, the role of RuCl3 was to consume O2 in the reaction and thus prevent the twinned nuclei from being oxidized and etched. As a result, this protection enabled the continuous growth of the multiply twinned seeds and thereby induced the formation of Cu nanowires.
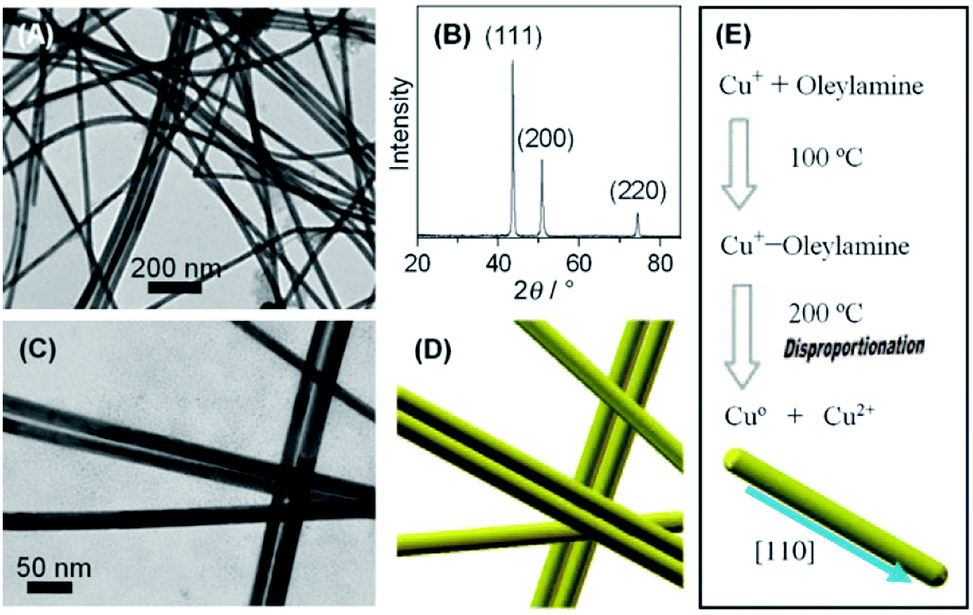 | ||
| Fig. 2 Fabrication of oleylamine-coated Cu nanowires by the disproportionation reaction. (A) TEM image and (B) XRD pattern of Cu nanowires. (C) Magnified TEM image and (D) schematic formation of Cu nanowires. (E) Schematic formation of Cu nanowires via the disproportionation reaction. Reproduced from ref. 36 with permission from Wiley-VCH. | ||
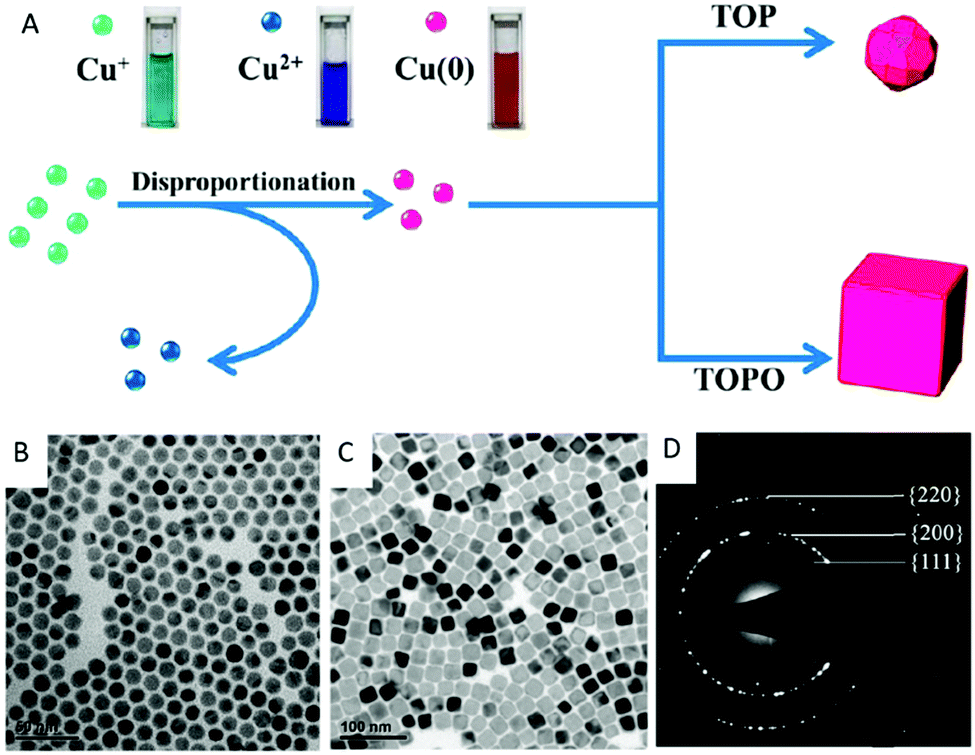 | ||
| Fig. 3 (A) Schematic formation of Cu nanospheres and nanocubes via the disproportionation reaction route. TEM image of (B) the Cu nanospheres with an average diameter of 12.6 nm, (C) Cu nanocubes with an average edge length of 24.1 nm, and (D) the corresponding SAED pattern. Reproduced from ref. 37 with permission from the American Chemical Society. | ||
2.2. Silver nanostructures
Historically, Ag metal has been widely used in jewellery and ornaments, high-value tableware and utensils, trade, and as the basis of many monetary systems. In the biomedical field, Ag-containing materials have been used in wound dressings and in topical treatments to heal burn wounds.39,40 However, the emergence of several antibiotics has resulted in the declined usage of Ag compounds. The recent advances in nanotechnology have opened up a whole new strategy to use Ag metal against a wide array of pathogens due to the capability of modulating metals into their nanosized counterparts, which significantly alter the chemical, physical, and optical properties of Ag. In this respect, numerous physical and chemical approaches have been used for synthesizing Ag nanostructures, including evaporation-condensation, laser ablation, and chemical reduction.17,41–44 In physical processes, metal nanoparticles generated are found to be inefficient in the materials and energy used.45The conventional borohydride-mediated or citrate-mediated reduction protocol (i.e., Lee–Meisel) have been developed for decades to reduce Ag ions to Ag nanoparticles but it can be a major challenge to obtain products with high monodispersity and stability. To address these limitations, numerous attempts have been made by many researchers to investigate the influence of reducing agents, capping agents, halides ions, and digestive ripening ligands in the synthesis of monodisperse Ag nanostructures of controlled size and shape. For instance, Agnihotri et al. adopted the co-reduction approach to produce Ag nanoparticles of different size ranges (5–100 nm), employing sodium borohydride as a primary reductant and trisodium citrate as the secondary reductant as well as stabilizing agent.59 The co-reduction approach provided a better balance between the nucleation and growth of Ag nanoparticles; therefore, the synthesis of size controlled Ag nanoparticles could be achieved. Li et al. developed a method to produce monodisperse, quasi-spherical Ag nanoparticles in the aqueous solution by reducing AgNO3 with both citrate and ascorbic acid in the presence of iodide ions.60 To obtain uniform quasi-spherical Ag nanoparticles, ascorbic acid was used to promote fast nucleation in order to inhibit secondary nucleation due to the slow reduction of Ag ions by citrate. On the other hand, iodide ions were employed as strong absorbates to tailor the growth of Ag into a quasi-spherical shape via its preferential adsorption on the (111) facets. Yang et al. synthesized uniform Ag nanoparticles of ∼10 nm using aniline as the reducing agent and dodecylbenzenesulfonic acid (DBSA) as the stabilizer.61 It has been demonstrated that the formation of Ag nanoparticles was accomplished in a three-stage model, starting with mesoscopic structures, in which Ag ions were confined in the dispersed phases of DBSA and aniline, subsequently generating Ag2O nanocrystallites in situ upon the addition of excess NaOH, followed by the reduction by aniline, leading to small uniform Ag nanoparticles.
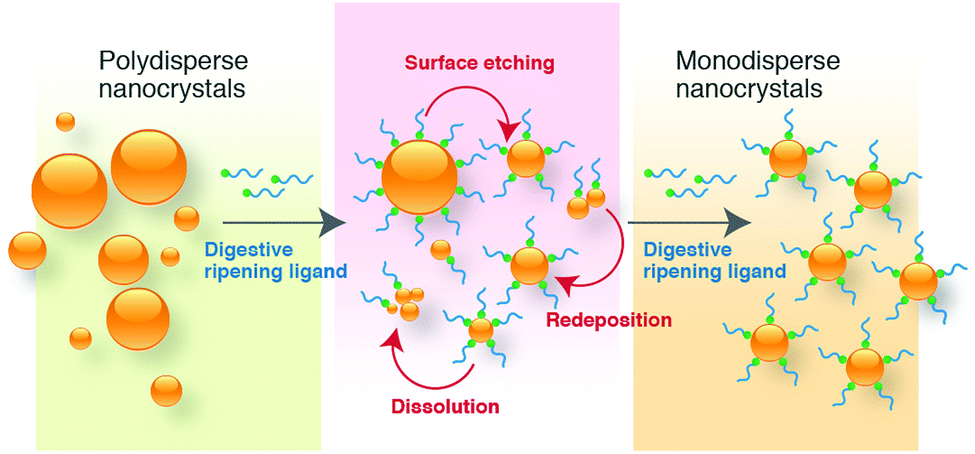 | ||
| Fig. 4 Schematic illustration of the formation mechanism of uniform monodisperse nanocrystals by digestive ripening. | ||
The seed-mediated growth method is also a common approach to prepare noble metal anisotropic nanostructures. Typically, a shaping agent is used in the solution to work as a suitable template that facilitates the growth process to yield shape-controlled nanostructures. For example, an early study was demonstrated by Jana et al. to prepare one-dimensional Ag nanostructures with a different aspect ratio via the seed-mediated growth approach.66 The growth of Ag nanorods and nanowires was achieved by the chemical reduction of Ag salt by ascorbic acid in the presence of the preformed Ag seed, cetyltrimethylammoniun bromide (CTAB), and NaOH. Notably, the seed and the base concentration relative to the Ag+ concentration were the key factors for producing large aspect ratio nanostructures. The seed-mediated growth approach was extended by Lee et al. to prepare Ag nanorods.67 It was reported that the preformed citrate-stabilized Ag seeds were added into the solution of Ag salt as well as ascorbic acid and CTAB. This work has demonstrated that the increase in the reaction temperature and pH led to a decrease in the aspect ratio of the Ag nanorods while improving the uniformity of sizes and shapes.
A recent work of Xia and co-workers demonstrated the preparation of Ag nanocubes in an aqueous system through the formation of the AgCl octahedra by mixing CF3COOAg with cetyltrimethylammonium chloride (CTAC), which subsequently served as a source of elemental Ag.69 The nucleation and growth of Ag nanocubes was facilitated by photolysis in the presence of ascorbic acid and FeCl3. The Fe3+/Fe2+ redox pair acted as an oxidative etchant to selectively dissolve the twinned seeds while chloride ions acted as a specific capping agent toward the (100) facets, leading to the formation of single-crystal Ag nanocubes with sharp corners and edges of an average edge length of 35–95 nm. Lately, Steiner et al. developed a method to synthesize Ag nanoparticles with rhombicuboctahedral shape via the combination of seed-mediated growth and the Cu-based etching process.68 The formation of thermodynamically stable Ag rhombicuboctahedra was assisted by CTAC and the process began with the synthesis of single-crystalline spherical Au nanoparticles, followed by the induced epitaxial overgrowth of Au spheres, and led to Ag nanocubes of the same crystallinity. The Ag nanocubes were then selectively etched on isotropic plasmonic Ag nanorhombicuboctahedra using Cu(NO3)2 as the etching agent in order to overcome the limitation of low-index facets and to achieve a final spherical morphology (Fig. 5).
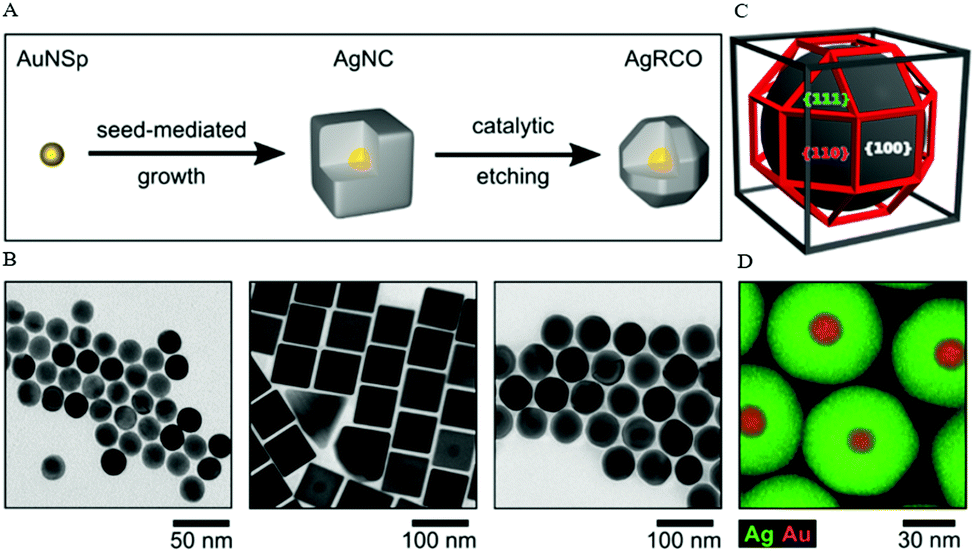 | ||
| Fig. 5 (A) Schematic formation of Ag nanorhombicuboctahedra via seed-mediated growth and Cu-based etching process. (B) TEM images of the intermediate particles: Au nanoparticles, Ag nanocubes, and Ag nanorhombicuboctahedra. (C) 3-D sketch of a rhombicuboctahedron geometry. (D) EDX elemental distribution of Ag and Au in the final rhombicuboctahedrically-shaped nanoparticles. Reproduced from ref. 68 with permission from the American Chemical Society. | ||
Following the early studies of Xia and co-workers, a comprehensively study has been made to develop Ag nanostructures with well-controlled morphologies (i.e., nanocubes, nanorods, nanowires, and nanospheres) by fine tuning the ratio of AgNO3 and PVP in the polyol process.46 In the PVP-mediated polyol process, the formation of a distinct morphology was determined by the crystallinity of the seeds through chemical interaction between the capping agent and various crystallographic planes of Ag and the extent of PVP coverage on the seed. By replacing the capping agent PVP with sodium citrate, the morphology of the Ag nanostructures was modified to thin triangular nanoplates and nanobelts due to the enlargement of the (111) rather than (100) planes. The polyol synthesis of controlling the specific seed structure to grow Ag nanostructures was later extended to selectively produce Ag pentagonal nanowires, cuboctahedra, nanocubes, nanobars, bipyramids, and nanobeams.74,75 These shape-defined Ag nanostructures were synthesized firstly through the reduction of Ag+ ions by EG. The reduced Ag atoms subsequently agglomerated to form nuclei of fluctuating structures, which are greatly dependent on their size and the thermal energy available. In many cases, the nuclei incorporated twin boundary defects because such defects enabled a lower surface energy. As the nuclei grew, the fluctuations stabilized and they were fixed as a multiply twinned, singly twinned, or single-crystal seed. Eventually, the crystallinity of the seeds governed the nanostructures of a specific shape.
Considering the long reaction time for the PVP-mediated polyol synthesis, the incorporation of a trace amount of sulfide anions (i.e., Na2S or NaHS) was reported to accelerate the production rate of Ag nanocubes due to an increase in the reduction rate of Ag ions.76 When compared to the conventional polyol process, the reaction time was significantly shortened from 16–26 h to 3–8 min. At this enhanced rate, the evolution of Ag nanocubes was dominated by the fast kinetic growth of single-crystal seeds. This sulfide-assisted polyol synthesis is useful for the preparation of Ag nanocubes to study the optical properties in the single isolated nanoparticles.77 Besides the conventional polyol process, a recent report of PVP-mediated polyol synthesis has also shown that the controlled growth of Ag nanoparticles could be performed in polyol solvent mixing with a co-solvent (i.e., propylene carbonate).78 This is to prevent heterogeneous nucleation when anionic additives or impurities are present in the reaction mixture; in particular, the formation of AgCl can occur when chloride is present and the surface of AgCl can serve as a nucleation site for Ag(0). Contrary to conventional polyol solvents such as EG and propylene glycol, the solubility of AgCl in the mixed solvent is sufficiently high such that heterogeneous nucleation on AgCl can be prevented.
Pastoriza-Santos et al. generated Ag nanoprisms in DMF with the assistance of PVP as a stabilizer. The size of the nanoprisms was tuned by the reaction time at reflux. However, this method produced a mixture of nanoprisms and nanospheroids during the synthetic process and the latter has to be removed through centrifugation.82 Gao et al. synthesized nanostructured Ag decahedrons via the PVP-mediated DMF reduction of AgNO3.83 This study revealed that Ag decahedrons capped by PVP contained five-twinning structures with distinct twinning boundaries, likely formed through the assembling of five tetrahedrons. An important suggestion in the present study is that the preferential facets of nanostructured Ag synthesized in DMF could be altered from the (100)-type to the (111)-type. The PVP-mediated DMF reduction process was extended by Tsuji et al. to prepare decahedral and icosahedral nanostructures as well as a series of their intermediate particles, which consist of a combination of two and more tetrahedra.84 It was proposed that the Ag polyhedra was synthesized via the stepwise growth of tetrahedral units on a specific (111) facet of intermediate species. In other words, the shape selective formation of decahedron and icosahedron was dependent on the concentration of PVP, which yielded a different extent of PVP coverage of each (111) facet. Later, the work was continued with PVP-assisted DMF reduction to produce nanoflag-type Ag nanostructures from nanorod seeds.79 These nanostructures were formed via a two-step process with EG and DMF as the reducing agent and solvent, starting with the synthesis of pentagonally twinned nanorods by the polyol reduction of AgNO3 in EG and PVP. In the second step, AgNO3 reduction took place in DMF with Ag nanorod seeds and PVP. The resulting products obtained were nanoflag-type structures having (111) facets. The shape evolution of these Ag nanostructures from trapezoid and triangular plates to tetrahedral structures was made possible by tuning the ratio of AgNO3 of the second step relative to the first step. The driving force of significant shape evolution was attributed to the changes in the preferential adsorption of PVP from (100) Ag facets in EG to (111) Ag facets in DMF.
2.3. Gold nanostructures
Traditionally, poorly reactive or chemically inert bulk Au is considered as a safe material for biomedical applications. To date, Au is one of the most studied materials owing to its unique chemical, electronic, and optical properties when the feature size decreases to the nanometer scale. A large number of studies are dedicated to the development of nanostructured Au with numerous morphologies to exploit the innovative nanoscale chemical effects for diverse applications such as biomedical, catalysis, energy, and environmental technologies.85–87The pioneering study of seed-mediated method was developed by Murphy and co-workers91 and later modified by El-Sayed and Nikoobakht.92 The work of Murphy demonstrated the preparation of Au nanorods using citrate-capped Au seeds synthesized from the reduction of HAuCl4 with borohydride. The ratio of the seed to the metal salt was then used to control the aspect ratio of the Au nanorods.91,93 The seed catalyzed the reduction of AuI to Au0 when added to HAuCl2 solution, which was previously prepared from HAuCl4 and ascorbic acid in the presence of CTAB and AgNO3.94 The role of Ag+ metal ions was significant for nanorod growth; in particular, the presence of metal ions increased the yield of Au nanorods from 20–40% to nearly 100%. On the other hand, the high yield of Au nanorods in the presence of Ag ions was also accompanied with a lower aspect ratio. The essential role of Ag+ was also demonstrated by Zhang et al. to synthesize concave nanocubes, enclosed by 24 high-index (720) facets via a modified seed-mediated growth method.95 Another key component in the synthesis was the chloride-containing surfactant that determined the product morphology. Specifically, this protocol has been modified from the usual bromide- to chloride-containing surfactant. Therefore, the use of the chloride counterion in the surfactant and combination with Ag+ resulted in the formation of concave cubes (i.e., CTAC) rather than a convex cubic structure (i.e., CTAB).
Likewise, the effect of halide ions during the seed-mediated growth has been demonstrated to prepare three different Au nanostructures (i.e., nanoprism, nanorod, and nanorice) by controlling the content of halide ions.96 It was reported that the addition of a small amount of iodide ions in the growth solution induced a change in the shape from Au nanorods to triangular nanoprisms. The presence of the iodide ions suppressed the overall crystal growth through selective binding on the Au(111) surface, resulting in the formation of Au(111)-faced triangular nanoprisms. On the contrary, a drastic change in the morphology from nanorod to nanorice was observed when the counterion of the surfactant was replaced with chloride ions (i.e., CTAB to CTAC). This study clearly demonstrated the influence of halide ion adsorption in the morphological transformation of Au nanostructures. Similarly, Chen et al. synthesized the triangular Au nanoplates through the iodide-assisted approach.97 The role of iodide facilitated the formation of Au nanoplates by selective binding onto the Au(111) facets and oxidative etching by tri-iodide ions to eliminate other shaped impurities, leaving behind dominant Au nuclei with planar-twined structures. Moreover, the edge length of triangular Au nanoplates were tunable (∼40 to ∼120 nm) by varying the concentration of the reagents such as ascorbic acid, KI, and NaOH, while keeping the concentrations of CTAC and Au3+ constant.
A comprehensive study on the effect of halide ions (chloride, bromide, and iodide) in the seed-mediated synthesis of Au anisotropic nanostructures was reported by Langille (Fig. 6).88 This comprised of two different sets of particle shapes through kinetic control, surface passivation, or a combination of both, either with or without Ag ions. Specifically, without Ag ions, the rate of reaction was the leading factor that governed the product growth. Slower rates of reaction yielded lower-energy surface facets and could be achieved by adding a lower amount of ascorbic acid or a larger halide (i.e., bromide or iodide). On the other hand, Ag ions could be used as an underpotential deposition agent to control the particle growth through surface passivation. Higher amounts of Ag on the particle surface yielded higher-index surfaces. Huang and co-workers reported a seed-mediated protocol for the preparation of Au nanostructures with systematic morphological evolution from truncated cubic to cubic, trisoctahedral, and rhombic dodecahedral structures.98 Shape transformation was achieved by adjusting the volume of ascorbic acid added into the growth solution to control the rate of Au atom addition on the crystal surface, leading to the formation of Au nanostructures. In this study, the use of CTAC surfactant and a tiny amount of NaBr into the growth solution was found to be critical to this morphological evolution. Later, the effect of bromide ions was replaced with iodide ions to induce the morphological evolution from rhombic dodecahedral to octahedral structures.99,100 Compared to chloride and bromide ions, iodide ions with a low reduction potential could act as a reducing agent in the presence of AuCl4 species to modulate the reduction rate along different crystallographic directions, resulting in the morphological evolution. Another work that explored the growth of Au nanocrystals with systematic shape evolution was studied by Kuo et al.101 Using seed-mediated method to grow Au nanocubes, rhombic dodecahedra, and octahedra as the starting points, shape transformation from rhombic dodecahedral to trisoctahedral and cubic structures could be achieved by adjusting the volume of HAuCl4 solution as well as incorporating NaBr in the process. On the other hand, the shape transformation from octahedral to corner-truncated octahedra, edge- and corner-truncated octahedra, and rhombic dodecahedra could be achieved in the presence of KI. The next examples describe the preparation of Au nanocrystals with unique shapes such as tetrahedral and trapezohedral through a seed-mediated growth process. Xia and co-workers reported the formation of Au tetrahedra with tunable sizes from 30 to 60 nm.89 The synthesis involved the use of single-crystal and spherical seeds in the presence of CTAB/CTAC as well as the manipulation of the reaction kinetics to promote an unsymmetrical growth pattern for the seeds, leading to the formation of tetrahedral products. On the hand, Niu et al. synthesized the trapezohedral Au nanocrystals by employing dimethyl sulfoxide (DMSO) as the solvent for shaping Au nanocrystals with (311) crystal facets.102 The success of this synthesis relied on the ambidentate nature of the DMSO solvent, where the lone-pair of electrons on the oxygen and sulphur atoms could coordinate to the Au surface, thus stabilizing the high-index (311) facets through a “two-center bonding” mode.
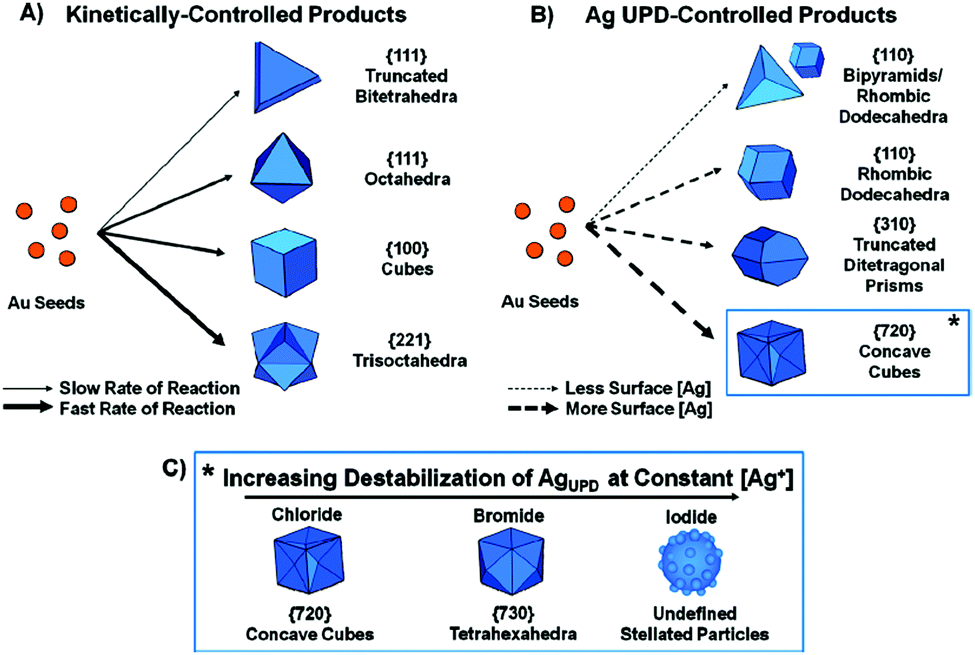 | ||
| Fig. 6 Schematic illustration of the seed-mediated synthesis of Au nancrystals through the use of halides and Ag ions. (A) Kinetically controlled products in the absence of Ag ions. (B) Ag underpotential deposition-controlled products. (C) High-index faceted products in the presence of halides and Ag ions. Reproduced from ref. 88 with permission from the American Chemical Society. | ||
In addition to seed-mediated synthesis, a purification protocol was developed by Scarabelli et al. to achieve shape monodispersity.103 This synthetic scheme for the preparation of monodisperse Au nanotriangles with the size of the edge length ranging from 50 to 150 nm was comprised of three consecutive steps: (1) generation of CTAC-coated Au seeds, (2) fast addition of the generated seeds to a final growth solution, and (3) purification of the products. Purification was carried out by flocculation and subsequent precipitation by CTAC micelles to remove the by-product particles. The high uniformity enabled the formation of the self-assembly and closely-packed Au nanotriangle monolayers. Apart from the Au nanostructures with well-controlled morphologies described above, it is worth mentioning that the seed-mediated method can also be extended to synthesize anisotropic Au nanocrystals with high chiroptical activity through the templating effect of chiral micelles formed in the presence of dissymmetric co-surfactants.104 This work was demonstrated by González-Rubio and co-workers to synthesize Au nanorods, employing 1,1′-bi(2-naphthol) as a cosurfactant with axial chirality to induce the assembly of surfactants into helical micelles, which tend to coil around the Au nanorods, leading to products with chiral features. Compared to the conventional nanorods, this surfactant-assisted seeded growth of Au nanorods possess chiral features and the chiral plasmonic nanostructures are promising candidates for real-life applications owing to their significantly larger optical chirality than that of natural chiral materials.105,106
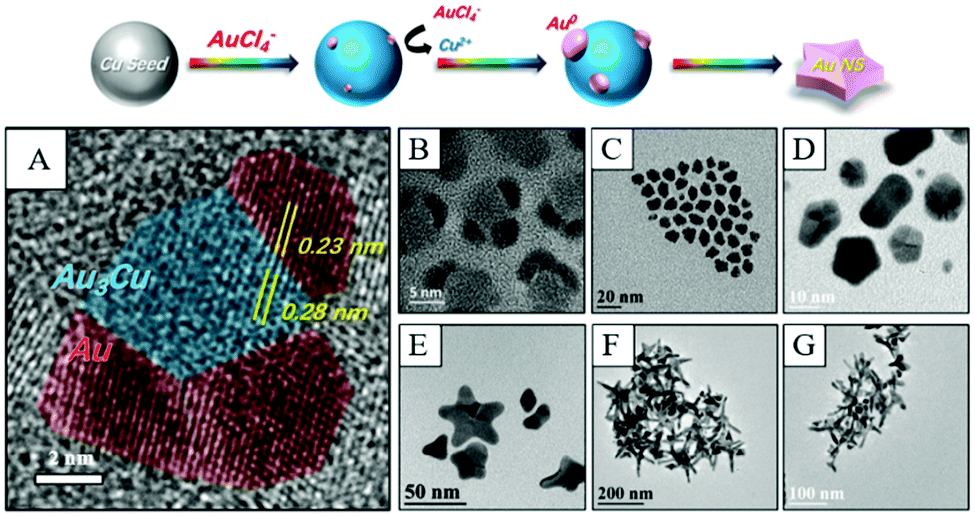 | ||
| Fig. 7 Schematic fabrication of Cu-induced formation of Au nanostars. (A) HRTEM image of nucleates from panel (B). (B)–(G) TEM images of nanocrystals obtained at different reaction times after the injection of the Au precursor. Reproduced from ref. 119 with permission from the Royal Society of Chemistry. | ||
2.4. Bimetallic metal nanostructures
Significant progress has been made on the controlled synthesis of single-metal nanostructures (as discussed in the previous section). On the contrary, the fabrication of bimetallic nanostructures consisting of Cu, Ag, and Au with controlled sizes/shapes has been less extensively studied and the strategies reported so far are relatively limited. The preparation methods using the solution-based approaches reported thus far are summarized in the following section.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 weight%), the co-reduction of Ag and Pd led to the formation of the Pd core–Ag shell clusters due to the higher redox potential of Pd.120 Conversely, co-precipitation in the ammonia solution containing the two metal salts and hydrazine led to the formation of the reverse Ag core–Pd shell cluster because of the stronger binding of NH3 to Pd than to Ag. In an example of Au–Ag core–shell nanocrystal synthesis, rhombic dodecahedral Au nanocrystals were used as the cores to produce the core–shell nanocrystals with cubic, truncated cubic, cuboctahedral, truncated octahedral, and octahedral structures.124 In this core–shell growth strategy, Au nanocrystals were added to an aqueous mixture of the CTAC surfactant, AgNO3, ascorbic acid, and NaOH to obtain the products. By adjusting the reaction temperature, the octahedra were obtained at a higher growth rate, while slower growth promoted cube formation.
:30 weight%), the co-reduction of Ag and Pd led to the formation of the Pd core–Ag shell clusters due to the higher redox potential of Pd.120 Conversely, co-precipitation in the ammonia solution containing the two metal salts and hydrazine led to the formation of the reverse Ag core–Pd shell cluster because of the stronger binding of NH3 to Pd than to Ag. In an example of Au–Ag core–shell nanocrystal synthesis, rhombic dodecahedral Au nanocrystals were used as the cores to produce the core–shell nanocrystals with cubic, truncated cubic, cuboctahedral, truncated octahedral, and octahedral structures.124 In this core–shell growth strategy, Au nanocrystals were added to an aqueous mixture of the CTAC surfactant, AgNO3, ascorbic acid, and NaOH to obtain the products. By adjusting the reaction temperature, the octahedra were obtained at a higher growth rate, while slower growth promoted cube formation.
In recent years, the green synthesis of nanoparticles has emerged as an attractive alternative route to traditional synthetic methods due to their potential in minimizing the toxicity of the nanoparticles. Specifically, there are three key aspects to consider with regard to green synthesis: solvent medium, non-toxic reducing agents, and non-hazardous stabilizing agents. The commonly used green sources include bacteria, fungi, actinomycetes, yeasts, viruses, polysaccharides, and plant extracts (leaves, flower, stem bark, fruit peels, seed, etc.), which have been reported to synthesize a series of metal nanoparticles.125–127 Among these, plant-mediated synthesis of metal nanoparticles appears to be a more favorable way because plant-modified materials are easy and inexpensive to be cultured as compared to those of microorganisms. Moreover, the eco-friendly synthetic approach is relatively simple, rapid, and inexpensive, which can be easily scaled up for larger production due to uncomplicated reaction procedures, which is typically done in an aqueous solution under ambient conditions.
Plant extracts contain a number of secondary metabolites and biomolecules in the form of coenzymes, which are vitamin-based intermediates that enable the reduction of metal ions to nanoparticles in a single step reduction process.128 In addition, these metabolites can have a dual role as reducing and stabilizing agents to prevent the aggregation and agglomeration of the nanoparticles during synthesis. The formation of metallic nanoparticles through biosynthesis is influenced by the type of plant extract, concentration (plant extract and metal salt), as well as the temperature and pH, which control the shape and size of the synthesized nanoparticles.129–131 Our group reported the flower-shaped Au nanostructures through aggregation-induced mechanism using star fruit (Averrhoa carambola) juice extract. The fruit extract is rich in in vitamin C and polyphenolic antioxidants, which can act as both reducing and stabilizing agents to synthesize branched gold nanoflowers, without additional shape-directing agents.132,133 Likewise, Nguyen et al. reported the biosynthesis of Ag nanostructures using three kinds of leaf extracts (Phyllanthus urinaria, Pouzolzia zeylanica, and Scoparia dulcis). When using Pouzolzia zeylanica leaf extracts as the reducing agents and stabilizers, diversified morphology was obtained, while the others showed a spherical morphology. To prepare Au–Ag bimetallic nanoparticles, various plant extracts have been reported as effective reducing agents and stabilizers, including Anacardium occidentale,134Gloriosa superba,135Solidago canadensis,136Commelina nudiflora,137Pulicaria undulata,138 and Muntingia calabura.139 In one example, Sheny et al. revealed that the biomolecules responsible for the reduction and stabilization of Au and Ag nanoparticles are found to be different. The proteins and polyols present in cashew leaves (Anacardium occidentale) are mostly responsible for the reduction.134 Both the formation of Au–Ag core–shell nanoparticles (6.5 nm) and Au–Ag alloy nanoparticles (8 nm) are in agreement with the UV-vis spectrum, featuring two plasmon bands and a single SPR band, respectively. Sharma et al. demonstrated the biosynthesis of Au–Ag bimetallic nanoparticles using clove bud extract.140 During the biosynthesis, the metal salt solutions containing Au(III) and Ag(I) are co-reduced competitively to form the Au–Ag alloy nanoparticles in the presence of the clove bud extract. The formation of hexagonal and polygonal shaped Au–Ag alloy nanoparticles with an average particle size of 16 nm was achieved through flavonoids and eugenol present in the extract. Alti et al. synthesized three kinds of Au–Ag bimetallic nanoparticles (10–12 nm) using fenugreek, coriander, and soybean leaf extracts.131 The biosynthesized Au–Ag bimetallic nanoparticles are in the form of an alloy and not a mixture of individual metal particles, as evident from the UV-vis spectrum.
Besides, the high-temperature reduction method can be used to prepare monodisperse Au–Ag alloy nanoparticles at 150 °C in organic solvent, with controlled diameter from 6 to 13 nm.143 In this method, the key is to control the surface plasmon resonance (SPR) frequency and the size of the Au–Ag alloy nanoparticles not only by varying the precursor ratio but also by varying the surfactant composition (i.e., oleylamine, dodecanethiol, and 1-hexadecylamine). The high-temperature reduction method was also used to successfully synthesize Cu–Ag bimetallic nanoparticles in the presence of trioctylphosphine and oleylamine.15 In a modification of the co-reduction method, Chen et al. reported a seed-based diffusion protocol driven by the difference in the redox potentials between Cu and Au metals for generating intermetallic Cu–Au nanoparticles.144 In the presence of oleic acid and tri-n-octylamine, Cu2+ ions were reduced to Cu atoms or clusters and subsequent collided with the active sites of preformed Au seeds, leading to the diffusion of active Cu atoms into the Au crystal lattice to form the Cu–Au alloy. In this approach, the size control of the Cu–Au nanoparticles could be fine-tuned by the selection of Au seeds while the selective synthesis of intermetallic Cu–Au and Cu3Au nanoparticles could be achieved by changing the molar ratio of the precursors.
 | ||
| Fig. 8 Preparation of Au–Ag double-walled nanoboxes from Ag nanocubes. (A) Optical images and schematic illustration of the morphological evolution of Au–Ag double-walled nanoboxes through simultaneous galvanic replacement and Kirkendall effect. (B) Corresponding high-angle annular dark field STEM images and EDX mapping. (C and D) TEM images of Au–Ag double-walled nanoboxes. Reproduced from ref. 151 With permission from the American Association for the Advancement of Science. | ||
Likewise, different Ag nanostructures have been adopted as templates to tune the morphology and shape of Au–Ag nanostructures by means of galvanic replacement. For example, twined Ag nanoparticles and single-crystalline Ag nanowires were used as the template to produce Au–Ag alloy rings and Ag@AuAg core–shell wires.152 Ag nanospheres were employed as the template for the synthesis of Au–Ag nanorings in the presence of AuCl4− and PVP aqueous solution.153 In another example of the Au–Ag nanoalloy, Camargo and co-workers synthesized Au–Ag nanodendrites through a combination of galvanic replacement and precursor reduction, employing Ag nanoparticles as seeds, hydroquinone as the reducing agent, and PVP as the stabilizer.154 The size and surface morphology of Au–Ag nanodendrites were tunable in the range of 45–148 nm by varying the concentration of the Ag nanoparticle seeds in the reaction mixture. A higher concentration of Ag nanoparticle seeds yielded smaller Au–Ag nanodendrites with respect to their outer diameters and size of the branches on the nanodendrite surfaces. Besides, a series of Cu–Au nanostructures (i.e. nanowires, nanotubes, and nanotriangles) were developed by our research group via a two-step approach.155 Firstly, the disproportionation reaction of the Cu+–oleylamine complex for the synthesis of Cu nanowires and the subsequent reaction with the Au3+ precursor at different temperatures to form the bimetallic Cu–Au nanostructures were carried out. The controlled addition of the Au3+ precursor to the Cu nanowires dispersion resulted in galvanic replacement due to the electrochemical potential difference between Cu and Au. In this process, the control of the reaction temperature was critical to yield the Cu–Au nanostructures of a desired morphology. Notably, the appropriate reaction temperature of 140 °C yields Cu–Au nanotubes through the surface passivation of the Cu nanowires when Cu0 is oxidized to a higher oxidation state of Cu2+ while Au3+ is reduced to Au0 (Fig. 9).
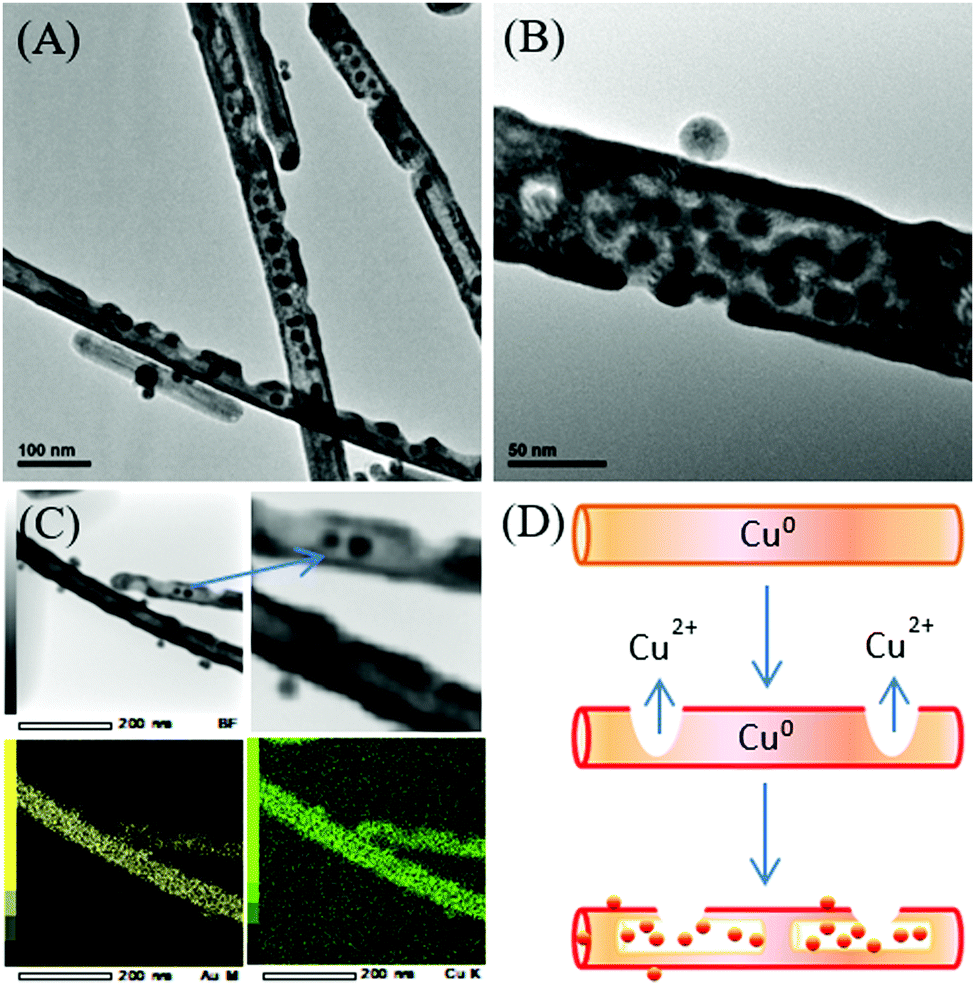 | ||
| Fig. 9 (A and B) TEM images of bimetallic Cu–Au nanotubes. (C) TEM EDX mapping. (D) Schematic formation of Cu–Au nanotubes with Au-incorporated nanoparticles. Reproduced from ref. 155 with permission from the Royal Society of Chemistry. | ||
3. Biomedical applications
The development of metal and bimetallic functional nanostructures has progressed tremendously over the past decades due to their unique structural features and outstanding properties, particularly in the field of viable biomedical materials and research in nanotechnology. The following discussions focus on a diverse range of available coinage metal nanostructures as a promising platform for a wide variety of bio-applications such as antibacterial activity, SERS-based detection, and electrochemical sensing.3.1. Antibacterial activities
Bacteria are among the most diverse groups of living organisms that differ in size, shape, habitat, and metabolism. These living organisms are capable of adapting to a great variety of ecological environments. On top of this, the serious problem is the bacterial adhesion to material surfaces and the subsequent formation of biofilms, which cause a major challenge in healthcare and industrial applications. As such, the prevention of bacterial attachment and biofilm growth is crucial, which has stimulated much efforts in the development of antibacterial metals. In general, antibacterial metals such as Cu, Ag, and Au are potentially used as a type of antibacterial agent.156–160 Significant progress in coinage metals relating to their antibacterial properties has been made through the design of metallic nanostructures as well as the understanding of their biological activity, structural, and functional changes induced in bacterial cells.158,160–165From these reports, Ag has been shown to display strong toxicity to an extensive range of microorganisms. Generally, the bactericidal activity is attributed to the Ag nanoparticles, which may attach and anchor to the surface of the bacterial cell wall and cause disruption of the cell function such as permeability and respiration, even cell death.167 Some studies have also shown that Ag nanoparticles are capable of penetrating inside the bacteria and causing further damage as well as the release of Ag ions from the nanoparticles, which promote the bactericidal activity.59,168 In addition to the size-dependent bactericidal properties of the nanoparticles, different shapes of Ag nanostructures may exhibit diverse antibacterial activities. A report has shown that truncated triangular Ag nanoplates exhibited a higher antibacterial activity against E. coli, relative to spherical, rod shaped nanoparticles, and also Ag ions.161 Similarly, Ag nanodendrites displayed stronger antimicrobial activities against E. coli, Candida albicans, and S. aureus, which were 10 times higher than that of Ag nanoparticles. These studies suggest that the antimicrobial efficiency of Ag is size- and shape-dependent; in particular, Ag nanostructures with highly active surfaces potentially function as multiple antibiotics.169
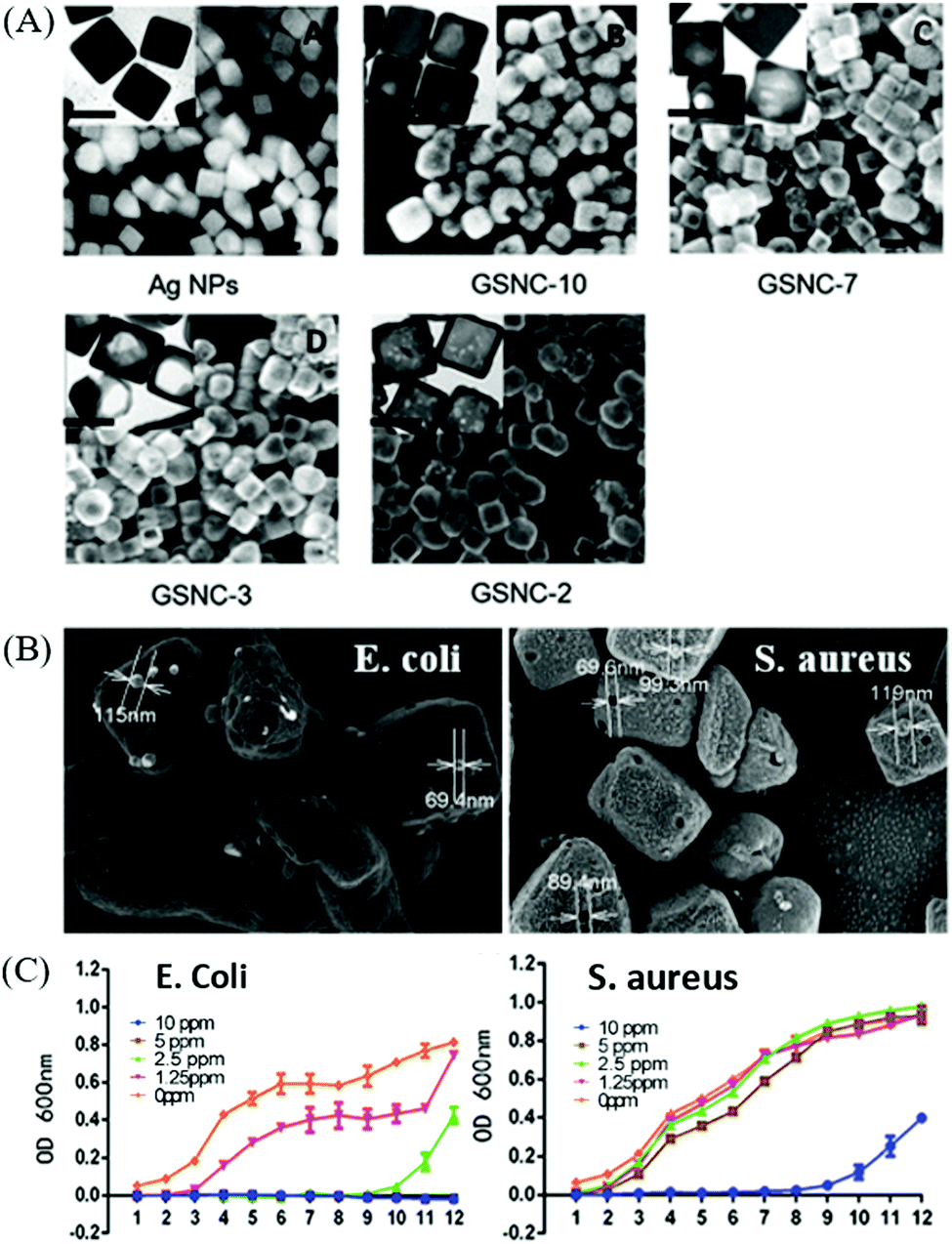 | ||
| Fig. 10 (A) SEM and TEM (inset) images of Au–Ag nanocages (scale bar is 50 nm) produced by reacting Ag nanocubes with varying amount of HAuCl4 to tailor different molar ratios of Ag/Au. (B) SEM images of E. coli and S. aureus to indicate cavities on the membrane, which matches with the Au–Ag nanocages, and (C) the corresponding E. coli and S. aureus growth curve with different concentrations of Au–Ag nanocages. Reproduced from ref. 172 with permission from the Royal Society of Chemistry. | ||
3.2. SERS sensing
SERS is a surface-sensitive technique that offers several orders of magnitude increase in the Raman intensity for detecting and identifying chemical and biological compounds at low concentrations. Since the discovery of the SERS effect in 1974 by Fleischman and colleagues,180 great efforts have been made toward understanding of the enhancement process. It is widely accepted that SERS enhancement is attributed to two primary mechanisms—electromagnetic field enhancement and chemical enhancement. The electromagnetic mechanism provides the main role for the SERS effect, which primarily results from the amplification of the local electromagnetic field due to localized surface plasmon resonance while the chemical enhancement is related to charge transfer between the chemisorbed molecule and the Raman-active platform.181 The signal enhancement enables detection with extraordinary sensitivity even at the single-molecule level.182To date, various nanostructures with different morphologies including nanorods,186–188 nanotriangles,189,190 nanocubes,191–193 nanostars,112,194 nanocages,195,196 and some other branched morphologies197–200 have been suggested for the fabrication of potential SERS-active platforms. For example, the correlation of the shape to SPR and SERS was studied with Au nanorods of different aspect ratios, which were prepared from the seed-mediated approach.201 The Au nanorods are particularly interesting because their anisotropy in dimensions lead their SPR to split into a transverse mode and a longitudinal mode. The longitudinal plasmonic bands can be finely tuned across the visible to the near-infrared region by increasing the aspect ratio of the nanorods. This spectral range (650–900 nm) is useful for developing near-infrared SERS nanoprobes in bioanalytical applications such as biomedical imaging and sensing. The shape-dependent SERS of the Au nanostructures has also been investigated on Au nanostars and spherical nanoparticles.202 This study illustrated that the spherical nanoparticles displayed a surface plasmon band centered at 527 nm, while star-shaped colloids of similar size exhibited a strongly red-shifted surface plasmon resonance with the maximum absorbance at 730 nm in the near-infrared region. The red-shifted band was associated to the plasmon mode localized at the nanostar tips. The SERS enhancement by the nanostars was enhanced several-fold when tested on analytes, thus demonstrating the high electromagnetic fields achieved by using nanostars as the optical enhancers.
A series of Au nanocrystals have been prepared by the seed-mediated approach and the facet-controlled products with four different shapes were assembled into an ordered array for the examination of facet-dependent SERS performance.203 The products obtained were cubic, concave cubic, octahedral, and rhombic dodecahedral Au nanocrystals, encased by (100), (200), (111), and (110), respectively. Among the Au nanocrystals, rhombic dodecahedra plasmonic monolayers exhibited the strongest SERS activity, with an analytical enhancement factor up to 1.2 × 106. In another SERS application of Au nanostructures, Au nanocages have been applied to serve as carriers for targeted and triggered drug delivery, while SERS was employed to monitor the release of Raman-active molecules such as dyes and drugs from the plasmonic nanocages.195 In this study, the Au nanocages obtained from the galvanic replacement of Ag nanocubes were loaded with the phase change material (1-tetradecanol) and Nile red or chemotherapeutic drug molecules (e.g., doxorubicin), followed by the release of encapsulated drugs along with melted 1-tetradecanol by thermal means. The release of the cargo from the Au nanocages was monitored by SERS, of which the intensity of the SERS spectra displayed a monotonic decrease in the intensity with longer exposure to elevated temperature, indicating the progressive release of the cargo from the Au nanocages. On the contrary, the control sample showed no change in the SERS intensity at room temperature (Fig. 11). In the SERS-based sensing applications, significant investigation have been conducted towards the detection of biomolecules and proteins.204–208 The SERS substrate composed of concave Au nanocubes was supported on a polydimethylsiloxane film and fabricated as a SERS active platform for protein detection (i.e., insulin and cytochrome c).209 The close assembly of the concave nanocubes promoted the formation of nanoholes through face-face and face-corner interactions between adjacent particles, which played the role of traps for effective protein detection. Once the proteins were trapped into the concave nanocubes arrays, the molecule experienced an electric field enhancement, which was expected to amplify the vibrational modes of a large part of the protein.
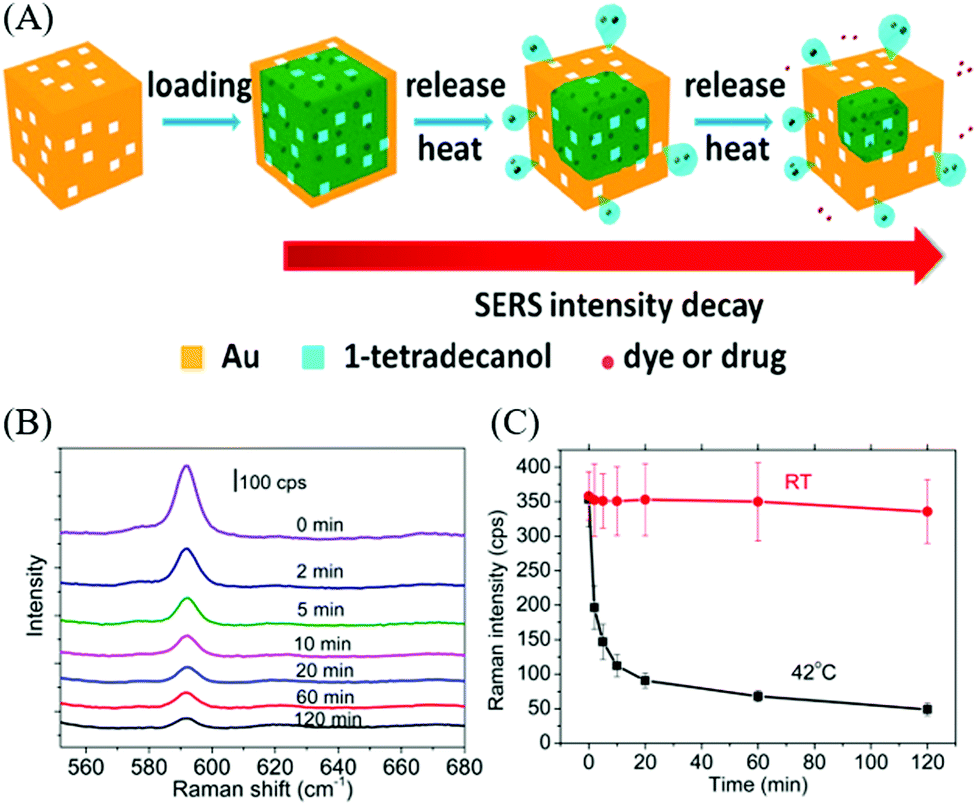 | ||
| Fig. 11 (A) Scheme illustrating drug delivery using Au nanocage. (B) Raman spectra of Au nanocages at different time intervals. (C) Raman intensity from Au nanocages exposed to water at 42 °C and water at room temperature during different time intervals. Reproduced from ref. 195 with permission from the American Chemical Society. | ||
3.3. Electrochemical sensing
Electrochemical sensing is commonly used for metabolic disorder detection in human body.218 A metabolic disorder occurs when abnormal chemical reactions in the body interfere with the normal metabolic process and cause either accumulation of large amounts of one metabolite or a deficiency of one or more metabolites in the body to stay healthy. For example, high level of uric acid, oxalic acid, and citric acid in the urine is associated with an increased risk of forming kidney stones, while an excess level of creatinine in the bloodstream and urine can be an indicator of impaired kidney function or kidney disease. Elevated levels of H2O2 are generated from many fundamental biological oxidative reactions as a by-product or an intermediate product, signifying the existence of an illness. Among these metabolic disorders, diabetes is the most common metabolic disease that is caused by the high level of blood glucose in the body over a prolonged period. Therefore, the study on glucose sensing is of practical significance and the development of an ideal glucose sensor is the most important issue in the biosensor field.The catalytic effects of metal-based nanostructured materials are highly dependent on the generation of electrocatalytically active sites and the enlargement of the surface area, which are closely related to the composition, structure, and morphology of the metal nanomaterials. For example, the electrochemical glucose sensor comprised of self-assembled Au nanoparticles on a 3-D silicate network demonstrated efficient glucose oxidation at a low potential (0.16 V vs. Ag/AgCl) in phosphate buffer solution at pH 9.2, showing a detection limit of 50 nM. There are also other Au nanostructures such as Au nanotube arrays222 and Au nanowire arrays,223 which have been studied as electrochemical glucose sensors due to their unique nanostructures. The high surface area of Au electrodes facilitated the large electrochemical response of glucose, which enabled glucose detection sensitively and selectively in the presence of electrochemical interference species. Despite the advantages of sensitivity toward glucose oxidation, the high-cost noble metal makes them less competitive in the practical application for the growing demand.
In search of new materials, Cu and their bimetallic nanostructures such as Cu–Au are promising materials for the mass production of glucose sensors in terms of the cost effectiveness, good sensitivity, and interference-free nature. To date, a wide range of Cu nanostructures such as Cu nanoparticles, Cu nanowires, Cu nanobelts, and Cu polyhedron pattern have been synthesized and studied as glucose sensors.220,224 Typically, during the oxidation of glucose to glucolactone on the Cu electrode, it is proposed that the catalytic component is Cu(III) oxyhydroxide species, the oxidized partner to the Cu(OH)2/CuOOH redox couple; hence, the catalytic reaction is highly dependent on the Cu(II)/Cu(III) redox couple. Our research group has developed a non-enzymatic glucose sensor based on various Cu–Au nanostructures from Cu nanowires template (Fig. 12). The bimetallic Cu–Au nanostructures exhibited superior sensing performance compared to the monometallic Cu nanowires, which clearly demonstrated their synergistic effect for electrocatalytic glucose oxidation. In particular, the sensing capability was certainly evident from the significantly improved electrocatalytic activity after adding a small quantity of Au of about 3 mmol% to the Cu nanowires. Furthermore, the high aspect ratio Cu–Au nanotubes with a large surface area provided abundant electroactive sites and facilitated mass transport, thereby improving the sensor response among other Cu–Au nanostructures.
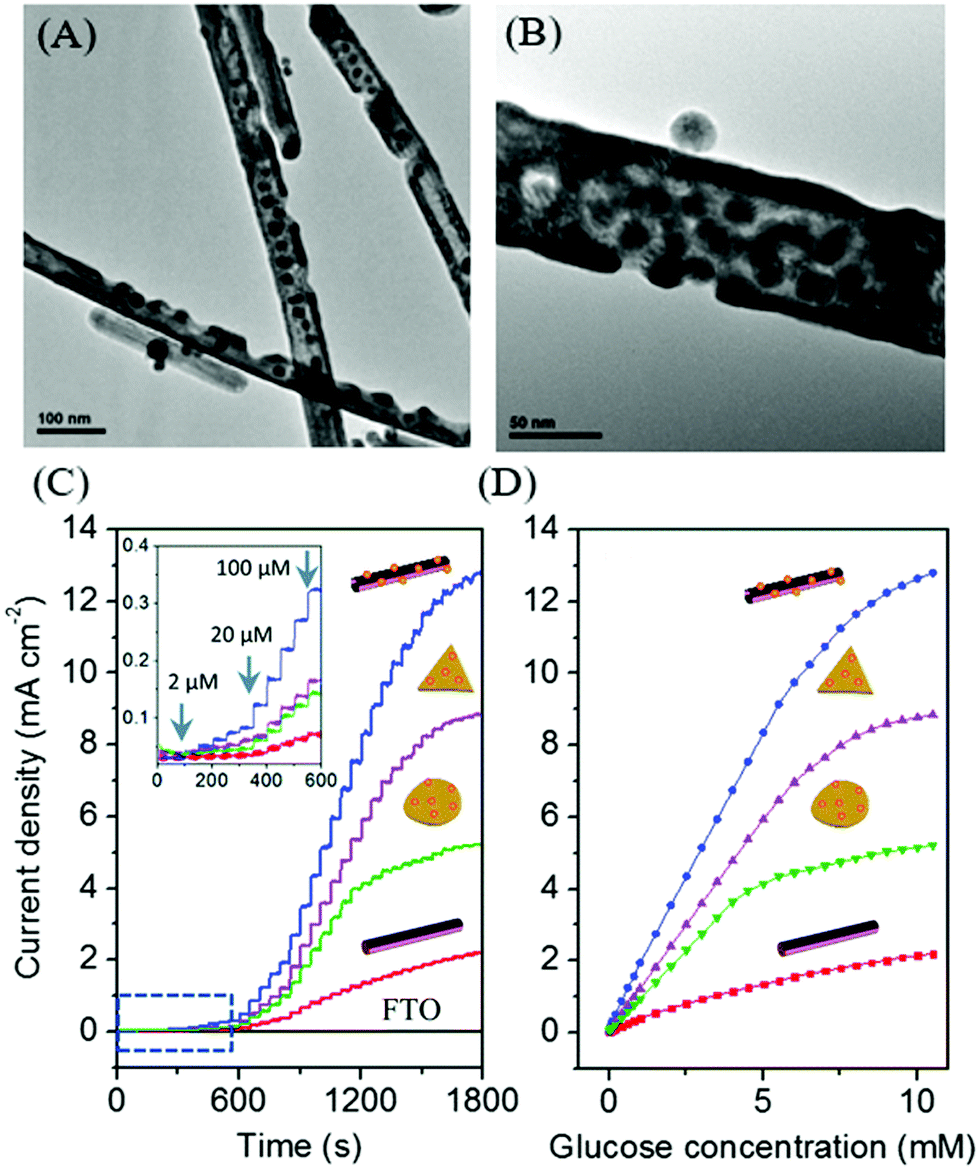 | ||
| Fig. 12 (A and B) TEM images of bimetallic Cu–Au nanotubes. (C and D) Electrocatalytic activities of Cu nanowires and Cu–Au nanostructures electrodes for glucose oxidation. Reproduced from ref. 155 with permission from the Royal Society of Chemistry. | ||
4. Conclusions and future prospects
The emergence of nanoscience and nanotechnology research have brought a large number of synthetic methods for the development of coinage metal nanostructures. As discussed in the review, these synthetic strategies such as the choice of a reducing agent, capping agent, the use of ionic impurity, secondary shape control agent (i.e., halide ions), and seed-mediated approach provide a means to control the size, shape, composition, and consequently the properties of the metal to be tuned for the application. The functional properties of coinage metal nanostructures are correlated to their surface area, whose reducing agents and capping agents are important to control the size and prevent particle aggregation. Examples of reducing agents include (but are not limited to) sodium borohydride, sodium citrate, ascorbic acid, saccharides, and polyols, which play a major role in the conversion of metal salts into metal nanoparticles. Nevertheless, Cu nanostructures are often synthesized using a variety of organic-phase-based methods due to the highly oxidizing nature of the metal. On the other hand, citrate anions have been regarded as one of the most widely accepted reductants for the size-controlled synthesis of Ag nanoparticles owing to the slow crystal growth as a result of the interaction between Ag and citrate ions. Capping agents such as PVP have shown promising results in preventing particle aggregation, which provides good stability and size/shape uniformity through good adsorption of the carbonyl group and nitrogen atom of the pyrrolidone ring with the metal surface. In addition, the presence of the halide ions affects the overall crystal growth, resulting in the formation of different shapes of Au nanoparticles through selective binding on the crystal facets. As for bimetallic nanostructures, the galvanic replacement reaction represents a simple and versatile approach for engineering the architectural nanostructures with hollow interiors and porous walls.The incorporation of coinage metal nanostructures in bio-applications has been extensively studied by the scientific community, of which the highlighted one in this review are the antibacterial activities, SERS-based detection, and electrochemical sensing. For instance, the size and shape of the metallic nanostructures have been finely tuned for the biocidal activity; in particular, the smaller particles with highly active surfaces potentially lead to higher antimicrobial efficiency. As for SERS sensing, the amplification of the SERS signal depends on a combination of the particle size, shape, as well as the increased sharpness and roughness in the metal nanostructures. In the context of electrochemical sensing, superior sensing performance has been demonstrated from the bimetallic nanostructures compared to the monometallic, which clearly exhibited the synergistic effect for electrocatalytic activity. Moreover, the porous nanostructures obtained from the dealloying process of the galvanic replacement reaction offer a larger surface area with abundant electroactive sites for detecting the analyte, thereby improving the sensor response. The properties of these coinage metal nanostructures are discussed here only to a limited extent and there are many promising applications in the chemical, energy, electronics, medicine, and healthcare fields. With regard to healthcare, the threat of the lethal Covid-19 viral infection has stressed healthcare systems around the world; coinage metal nanostructures may contribute significantly in the containment, diagnosis, and treatment of COVID-19. For example, Cu-based nanomaterials are potential candidates to be deployed for antiviral and antimicrobial nanocoatings, while Au and Ag nanomaterials are promising in drug delivery and therapeutics; in particular, Ag nanoparticles have significant virus-inhibitory properties.
To date, a large variety of protocols and approaches are available for the synthesis, assembly, fabrication, and the potential applications of coinage metal nanostructures. In the future, the challenge is to use such techniques reliably for large-scale production. Nevertheless, more effort is needed toward the scalable routes of synthesis before promising demonstrations can be transitioned to commercialization. This includes the precise control of the coinage metal nanostructures properties (i.e., optical, mechanical, electrical, and chemical) for each technological application, which are determined by a set of physical parameters such as the size, shape, surface functionality, and composition. Moreover, these metal nanostructures may have a tendency to aggregate during the growth or post-processing, which may affect the specific functionality of the nanostructures. Among the nanostructures of coinage metals, the low producibility and long-term stability of the Cu nanostructures remains a grand challenge for their large-scale production and a comprehensive solution is still lacking even though some shortcomings have been addressed.
Despite the promising future of coinage metal nanostructures, the uncontrolled and unregulated use of these nanostructures in a diverse range of applications will likely lead to adverse impacts on the human health and environment. To enable the safer use of these metal nanostructures, the toxicological investigation of nanoparticles and surface coating as well as the in-depth understanding of their interactions with various biological compounds, cells, tissues, or bioenvironment are essential. Notably, these studies will provide important insight for the further investigation of the nanostructures in in vivo applications such as medical diagnostics and therapeutics. Moving forward, there is still room for the intensive investigation of new metallic/bimetallic systems because of the importance to develop multifunctional nanoparticles with numerous properties that enable greater ability than conventional nanoparticles in order to accomplish several objectives synergistically (Table 1).
| Metal | Synthesis method | Reducing agent | Capping agent | Shape and size | Application | Ref. |
|---|---|---|---|---|---|---|
| Cu | Chemical reduction (aqueous) | Glucose | Hexadecylamine | Wire and cube (50–200 nm) | — | 26 |
| Cu | Chemical reduction (organic) | Oleylamine | Trioctylphosphine | Wire | — | 36 |
| Cu | Chemical reduction (organic) | Oleylamine | Trioctylphosphine oxide | Cube (24.1–76.4 nm) | — | 37 |
| Cu | Seed-mediated growth | Glucose | Hexadecylamine | Wire | — | 33 |
| Cu | Chemical reduction (aqueous) | Ascorbic acid | Trisodium citrate | Spherical (200–500 nm) | Antifungal property against plant pathogens | 229 |
| Cu | Green synthesis | Green and black tea leaves extracts | — | Spherical (26–40 nm) | Antibacterial/antifungal property against S. aureus and Aspergillus species. | 230 |
| Cu | Green synthesis | Leaf extracts (Celastrus paniculatus) | — | Spherical (2–10 nm) | Antifungal property against plant pathogenic fungi (Fusarium oxysporum) | 231 |
| Ag | Chemical reduction (aqueous) | NaBH4, trisodium citrate | Trisodium citrate | Spherical (5–100 nm) | Bactericidal activity against E. coli, B. subtilis and S. aureus | 59 |
| Ag | Seed-mediated growth | Ascorbic acid | CTAC | Cubes (35–95 nm) | SERS-based detection | 69 |
| Ag | Seed-mediated growth + catalytic etching | NaBH4, ascorbic acid | CTAB, CTAC | Rhombi-cuboctahedral (60 nm) | — | 68 |
| Ag | Polyol process | Ascorbic acid | PVP | Spherical (10–90 nm) | — | 78 |
| Ag | Green synthesis | Leaf extracts (Ocimum Sanctum) | — | Spherical (10–20 nm) | Antibacterial property against E. coli | 232 |
| Ag | Green synthesis | Leaf extracts (Phyllanthus urinaria, Pouzolzia zeylanica, and Scoparia dulcis) | — | Spherical (4–52 nm) | Antifungal effect against Aspergillus niger, Aspergillus flavus, and Fusarium oxysporum | 233 |
| Au | Seedless growth process | Ascorbic acid | CTAC | Triangular plates (40–120 nm) | SERS-based detection | 97 |
| Au | Seed-mediated growth | NaBH4, ascorbic acid | CTAC | Concave cube (130 nm) | SERS-based detection | 209 |
| Au | Seed-mediated growth | NaBH4, ascorbic acid | CTAC | Polyhedral (69–96 nm) | — | 101 |
| Au | Template-directed approach | Oleylamine | Oleylamine | Cross (100–350 nm) | Photothermal therapy, photothermal ablation of multidrug-resistant bacteria | 114, 115, 118 |
| Au | Galvanic replacement reaction | Oleylamine | Oleylamine | Star (50–200 nm) | Photothermal cancer therapy | 119 |
| Au | Galvanic replacement reaction | EG | PVP | Cage (73 nm) | Drug delivery + SERS | 195 |
| Au | Polyol process | 1,5-Pentanediol | Poly(diallyldi-methylammonium) chloride | Octahedra (30–110 nm) | — | 109 |
| Au | Green synthesis | Star fruit juice (Averrhoa carambola) | — | Flower (100 nm) | Photothermal therapy | 132 |
| Au–Ag | Galvanic replacement reaction | EG | PVP | Cages (92–107 nm) | Antibacterial properties against E. coli and S. aureus | 172 |
| Au–Ag | Galvanic replacement reaction | NaBH4, ascorbic acid | CTAB | Stars (55 nm) | SERS-based detection | 211 |
| Au–Ag | Green synthesis | Plant extract (Commelina nudiflora) | — | Spherical (20–80 nm) | Bactericidal activity against tested oral pathogenic bacteria | 137 |
| Au–Ag | Green synthesis | Herbal leaf extracts (fenugreek, coriander, and soybean) | — | Spherical (10–12 nm) | Antileishmanial effects against promastigotes | 131 |
| Cu–Au | Galvanic replacement reaction | Oleylamine | Oleylamine | Tube | Glucose sensing | 155 |
| Cu–Au | Galvanic replacement reaction | Oleylamine | Oleylamine | Tube | SERS substrate | 217 |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Institute of Materials Research and Engineering (IMRE), Agency for Science, Technology and Research (A*STAR).References
- Z. Y. Zhou, N. Tian, J. T. Li, I. Broadwell and S. G. Sun, Chem. Soc. Rev., 2011, 40, 4167–4185 RSC.
- K. Zhou and Y. Li, Angew. Chem., Int. Ed., 2012, 51, 602–613 CrossRef CAS.
- P. K. Jain, X. Huang, I. H. El-Sayed and M. A. El-Sayed, Acc. Chem. Res., 2008, 41, 1578–1586 CrossRef CAS.
- H. M. Chen and R.-S. Liu, J. Phys. Chem. C, 2011, 115, 3513–3527 CrossRef CAS.
- E. C. Dreaden, A. M. Alkilany, X. Huang, C. J. Murphy and M. A. El-Sayed, Chem. Soc. Rev., 2012, 41, 2740–2779 RSC.
- S. E. Lohse and C. J. Murphy, J. Am. Chem. Soc., 2012, 134, 15607–15620 CrossRef CAS.
- C. N. Rao, H. S. Ramakrishna Matte, R. Voggu and A. Govindaraj, Dalton Trans., 2012, 41, 5089–5120 RSC.
- I. Khan, K. Saeed and I. Khan, Arabian J. Chem., 2019, 12, 908–931 CrossRef CAS.
- M. Faraday, Philos. Trans. R. Soc. London, 1857, 147, 145–181 CrossRef.
- B. V. Enüstün and J. Turkevich, J. Am. Chem. Soc., 1963, 85, 3317–3328 CrossRef.
- L. Au, Y. Chen, F. Zhou, P. H. Camargo, B. Lim, Z. Y. Li, D. S. Ginger and Y. Xia, Nano Res., 2008, 1, 441–449 CrossRef CAS.
- C. L. Bracey, P. R. Ellis and G. J. Hutchings, Chem. Soc. Rev., 2009, 38, 2231–2243 RSC.
- J. Yang, X. Chen, X. Yang and J. Y. Ying, Energy Environ. Sci., 2012, 5, 8976 RSC.
- G. Guisbiers, S. Mejia-Rosales, S. Khanal, F. Ruiz-Zepeda, R. L. Whetten and M. Jose-Yacaman, Nano Lett., 2014, 14, 6718–6726 CrossRef CAS.
- G. Darabdhara, B. Sharma, M. R. Das, R. Boukherroub and S. Szunerits, Sens. Actuators, B, 2017, 238, 842–851 CrossRef CAS.
- R. H. Morriss and L. F. Collins, J. Chem. Phys., 1964, 41, 3357–3363 CrossRef CAS.
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS.
- J. A. Dahl, B. L. Maddux and J. E. Hutchison, Chem. Rev., 2007, 107, 2228–2269 CrossRef CAS.
- Y. Xia, K. D. Gilroy, H. C. Peng and X. Xia, Angew. Chem., Int. Ed., 2017, 56, 60–95 CrossRef CAS.
- S.-Y. Zhang, M. D. Regulacio, K. W. Shah, T. Sreethawong, Y. Zheng and M.-Y. Han, J. Mol. Eng. Mater., 2014, 02, 1430001 CrossRef.
- A. R. Tao, S. Habas and P. Yang, Small, 2008, 4, 310–325 CrossRef CAS.
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS.
- K. Zhou and Y. Li, Angew. Chem., Int. Ed., 2012, 51, 602–613 CrossRef CAS.
- P. D. Cozzoli, T. Pellegrino and L. Manna, Chem. Soc. Rev., 2006, 35, 1195–1208 RSC.
- R. Costi, A. E. Saunders and U. Banin, Angew. Chem., Int. Ed., 2010, 49, 4878–4897 CrossRef CAS.
- M. Jin, G. He, H. Zhang, J. Zeng, Z. Xie and Y. Xia, Angew. Chem., Int. Ed., 2011, 50, 10560–10564 CrossRef CAS.
- D. Zhang, R. Wang, M. Wen, D. Weng, X. Cui, J. Sun, H. Li and Y. Lu, J. Am. Chem. Soc., 2012, 134, 14283–14286 CrossRef CAS.
- I. Lisiecki, F. Billoudet and M. P. Pileni, J. Phys. Chem., 1996, 100, 4160–4166 CrossRef CAS.
- B. K. Park, S. Jeong, D. Kim, J. Moon, S. Lim and J. S. Kim, J. Colloid Interface Sci., 2007, 311, 417–424 CrossRef CAS.
- Y. Chang, M. L. Lye and H. C. Zeng, Langmuir, 2005, 21, 3746–3748 CrossRef CAS.
- A. R. Rathmell, S. M. Bergin, Y. L. Hua, Z. Y. Li and B. J. Wiley, Adv. Mater., 2010, 22, 3558–3563 CrossRef CAS.
- S. Bhanushali, P. Ghosh, A. Ganesh and W. Cheng, Small, 2015, 11, 1232–1252 CrossRef CAS.
- Q. Q. Fu, Y. D. Li, H. H. Li, L. Xu, Z. H. Wang and S. H. Yu, Langmuir, 2019, 35, 4364–4369 CrossRef CAS.
- D. Mott, J. Galkowski, L. Wang, J. Luo and C. J. Zhong, Langmuir, 2007, 23, 5740–5745 CrossRef CAS.
- F. Cui, Y. Yu, L. Dou, J. Sun, Q. Yang, C. Schildknecht, K. Schierle-Arndt and P. Yang, Nano Lett., 2015, 15, 7610–7615 CrossRef CAS.
- E. Ye, S. Y. Zhang, S. Liu and M. Y. Han, Chem. – Eur. J., 2011, 17, 3074–3077 CrossRef CAS.
- H. Guo, Y. Chen, M. B. Cortie, X. Liu, Q. Xie, X. Wang and D.-L. Peng, J. Phys. Chem. C, 2014, 118, 9801–9808 CrossRef CAS.
- X. Huang, Y. Chen, C. Y. Chiu, H. Zhang, Y. Xu, X. Duan and Y. Huang, Nanoscale, 2013, 5, 6284–6290 RSC.
- N. George, J. Faoagali and M. Muller, Burns, 1997, 23, 493–495 CrossRef CAS.
- B. S. Atiyeh, M. Costagliola, S. N. Hayek and S. A. Dibo, Burns, 2007, 33, 139–148 CrossRef.
- C. Chen, L. Wang, H. Yu, J. Wang, J. Zhou, Q. Tan and L. Deng, Nanotechnology, 2007, 18, 115612 CrossRef.
- J. Zhang, M. R. Langille and C. A. Mirkin, J. Am. Chem. Soc., 2010, 132, 12502–12510 CrossRef CAS.
- K. M. M. Abou El-Nour, A. a. Eftaiha, A. Al-Warthan and R. A. A. Ammar, Arabian J. Chem., 2010, 3, 135–140 CrossRef CAS.
- Z. Huang, X. Jiang, D. Guo and N. Gu, J. Nanosci. Nanotechnol., 2011, 11, 9395–9408 CrossRef CAS.
- Y.-H. Chen and C.-S. Yeh, Colloids Surf., A, 2002, 197, 133–139 CrossRef CAS.
- B. Wiley, Y. Sun, B. Mayers and Y. Xia, Chem. – Eur. J., 2005, 11, 454–463 CrossRef CAS.
- J. A. Scholl, A. L. Koh and J. A. Dionne, Nature, 2012, 483, 421–427 CrossRef CAS.
- R. Jin, Nanoscale, 2010, 2, 343–362 RSC.
- J. Xiao and L. Qi, Nanoscale, 2011, 3, 1383–1396 RSC.
- L. Qu, G. Shi, X. Wu and B. Fan, Adv. Mater., 2004, 16, 1200–1203 CrossRef CAS.
- J. Q. Hu, Q. Chen, Z. X. Xie, G. B. Han, R. H. Wang, B. Ren, Y. Zhang, Z. L. Yang and Z. Q. Tian, Adv. Funct. Mater., 2004, 14, 183–189 CrossRef CAS.
- S. H. Zhang, Z. X. Xie, Z. Y. Jiang, X. Xu, J. Xiang, R. B. Huang and L. S. Zheng, Chem. Commun., 2004, 9, 1106–1107 RSC.
- D. Yu and V. W. Yam, J. Am. Chem. Soc., 2004, 126, 13200–13201 CrossRef CAS.
- B. Pietrobon and V. Kitaev, Chem. Mater., 2008, 20, 5186–5190 CrossRef CAS.
- B.-H. Lee, M.-S. Hsu, Y.-C. Hsu, C.-W. Lo and C.-L. Huang, J. Phys. Chem. C, 2010, 114, 6222–6227 CrossRef CAS.
- X. He, X. Zhao, Y. Li and X. Sui, J. Mater. Res., 2011, 24, 2200–2209 CrossRef.
- K.-J. Wu and L. Torrente-Murciano, React. Chem. Eng., 2018, 3, 267–276 RSC.
- S. Zhang, L. Zhang, K. Liu, M. Liu, Y. Yin and C. Gao, Mater. Chem. Front., 2018, 2, 1328–1333 RSC.
- S. Agnihotri, S. Mukherji and S. Mukherji, RSC Adv., 2014, 4, 3974–3983 RSC.
- H. Li, H. Xia, D. Wang and X. Tao, Langmuir, 2013, 29, 5074–5079 CrossRef CAS.
- J. Yang, H. Yin, J. Jia and Y. Wei, Langmuir, 2011, 27, 5047–5053 CrossRef CAS.
- J. R. Shimpi, D. S. Sidhaye and B. L. V. Prasad, Langmuir, 2017, 33, 9491–9507 CrossRef CAS.
- N. Razgoniaeva, M. Yang, P. Garrett, N. Kholmicheva, P. Moroz, H. Eckard, L. Royo Romero, D. Porotnikov, D. Khon and M. Zamkov, Chem. Mater., 2018, 30, 1391–1398 CrossRef CAS.
- L. Yu, H. You, Q. Zhang, L. Zhang and J. Fang, CrystEngComm, 2019, 21, 1529–1533 RSC.
- Y. Wan, Z. Guo, X. Jiang, K. Fang, X. Lu, Y. Zhang and N. Gu, J. Colloid Interface Sci., 2013, 394, 263–268 CrossRef CAS.
- N. R. Jana, L. Gearheart and C. J. Murphy, Chem. Commun., 2001, 7, 617–618 RSC.
- G. Lee, Mater. Chem. Phys., 2004, 84, 197–204 CrossRef CAS.
- A. M. Steiner, M. Mayer, D. Schletz, D. Wolf, P. Formanek, R. Hübner, M. Dulle, S. Förster, T. A. F. König and A. Fery, Chem. Mater., 2019, 31, 2822–2827 CrossRef CAS.
- S. Zhou, J. Li, K. D. Gilroy, J. Tao, C. Zhu, X. Yang, X. Sun and Y. Xia, ACS Nano, 2016, 10, 9861–9870 CrossRef CAS.
- I. Washio, Y. Xiong, Y. Yin and Y. Xia, Adv. Mater., 2006, 18, 1745–1749 CrossRef CAS.
- Y. Xiong, I. Washio, J. Chen, H. Cai, Z. Y. Li and Y. Xia, Langmuir, 2006, 22, 8563–8570 CrossRef CAS.
- T. Zhao, R. Sun, S. Yu, Z. Zhang, L. Zhou, H. Huang and R. Du, Colloids Surf., A, 2010, 366, 197–202 CrossRef CAS.
- J.-Y. Lin, Y.-L. Hsueh, J.-J. Huang and J.-R. Wu, Thin Solid Films, 2015, 584, 243–247 CrossRef CAS.
- B. J. Wiley, Z. Wang, J. Wei, Y. Yin, D. H. Cobden and Y. Xia, Nano Lett., 2006, 6, 2273–2278 CrossRef CAS.
- B. Wiley, Y. Sun and Y. Xia, Acc. Chem. Res., 2007, 40, 1067–1076 CrossRef CAS.
- A. R. Siekkinen, J. M. McLellan, J. Chen and Y. Xia, Chem. Phys. Lett., 2006, 432, 491–496 CrossRef CAS.
- N. Grillet, D. Manchon, F. Bertorelle, C. Bonnet, M. Broyer, E. Cottancin, J. Lerme, M. Hillenkamp and M. Pellarin, ACS Nano, 2011, 5, 9450–9462 CrossRef CAS.
- S. Chen and R. L. Penn, Cryst. Growth Des., 2019, 19, 4332–4339 CrossRef CAS.
- M. Tsuji, X. Tang, M. Matsunaga, Y. Maeda and M. Watanabe, Cryst. Growth Des., 2010, 10, 5238–5243 CrossRef CAS.
- Y. Sun, Chem. Soc. Rev., 2013, 42, 2497–2511 RSC.
- K. Eid, H. Wang and L. Wang, Supra-Materials Nanoarchitectonics, 2017, 6, 135–171 Search PubMed.
- I. Pastoriza-Santos and L. M. Liz-Marzán, Nano Lett., 2002, 2, 903–905 CrossRef CAS.
- Y. Gao, P. Jiang, L. Song, J. X. Wang, L. F. Liu, D. F. Liu, Y. J. Xiang, Z. X. Zhang, X. W. Zhao, X. Y. Dou, S. D. Luo, W. Y. Zhou and S. S. Xie, J. Cryst. Growth, 2006, 289, 376–380 CrossRef CAS.
- M. Tsuji, M. Ogino, R. Matsuo, H. Kumagae, S. Hikino, T. Kim and S.-H. Yoon, Cryst. Growth Des., 2010, 10, 296–301 CrossRef CAS.
- A. S. Thakor, J. Jokerst, C. Zavaleta, T. F. Massoud and S. S. Gambhir, Nano Lett., 2011, 11, 4029–4036 CrossRef CAS.
- N. Li, P. Zhao and D. Astruc, Angew. Chem., Int. Ed., 2014, 53, 1756–1789 CrossRef CAS.
- L. Y. Chen, C. W. Wang, Z. Yuan and H. T. Chang, Anal. Chem., 2015, 87, 216–229 CrossRef CAS.
- M. R. Langille, M. L. Personick, J. Zhang and C. A. Mirkin, J. Am. Chem. Soc., 2012, 134, 14542–14554 CrossRef CAS.
- Y. Zheng, W. Liu, T. Lv, M. Luo, H. Hu, P. Lu, S. I. Choi, C. Zhang, J. Tao, Y. Zhu, Z. Y. Li and Y. Xia, Chem. – Asian J., 2014, 9, 2635–2640 CrossRef CAS.
- Y. Zheng, X. Zhong, Z. Li and Y. Xia, Part. Part. Syst. Charact., 2014, 31, 266–273 CrossRef CAS.
- N. R. Jana, L. Gearheart and C. J. Murphy, Adv. Mater., 2001, 13, 1389–1393 CrossRef CAS.
- B. Nikoobakht and M. A. El-Sayed, Chem. Mater., 2003, 15, 1957–1962 CrossRef CAS.
- N. R. Jana, L. Gearheart and C. J. Murphy, J. Phys. Chem. B, 2001, 105, 4065–4067 CrossRef CAS.
- C. J. Murphy, T. K. Sau, A. M. Gole, C. J. Orendorff, J. Gao, L. Gou, S. E. Hunyadi and T. Li, J. Phys. Chem. B, 2005, 109, 13857–13870 CrossRef CAS.
- J. Zhang, M. R. Langille, M. L. Personick, K. Zhang, S. Li and C. A. Mirkin, J. Am. Chem. Soc., 2010, 132, 14012–14014 CrossRef CAS.
- T. H. Ha, H.-J. Koo and B. H. Chung, J. Phys. Chem. C, 2007, 111, 1123–1130 CrossRef CAS.
- L. Chen, F. Ji, Y. Xu, L. He, Y. Mi, F. Bao, B. Sun, X. Zhang and Q. Zhang, Nano Lett., 2014, 14, 7201–7206 CrossRef CAS.
- H. L. Wu, C. H. Kuo and M. H. Huang, Langmuir, 2010, 26, 12307–12313 CrossRef CAS.
- P. J. Chung, L. M. Lyu and M. H. Huang, Chem. – Eur. J., 2011, 17, 9746–9752 CrossRef CAS.
- C.-Y. Chiu, P.-J. Chung, K.-U. Lao, C.-W. Liao and M. H. Huang, J. Phys. Chem. C, 2012, 116, 23757–23763 CrossRef CAS.
- B.-H. Kuo, C.-F. Hsia, T.-N. Chen and M. H. Huang, J. Phys. Chem. C, 2018, 122, 25118–25126 CrossRef CAS.
- W. Niu, Y. Duan, Z. Qing, H. Huang and X. Lu, J. Am. Chem. Soc., 2017, 139, 5817–5826 CrossRef CAS.
- L. Scarabelli, M. Coronado-Puchau, J. J. Giner-Casares, J. Langer and L. M. Liz-Marzan, ACS Nano, 2014, 8, 5833–5842 CrossRef CAS.
- G. Gonzalez-Rubio, J. Mosquera, V. Kumar, A. Pedrazo-Tardajos, P. Llombart, D. M. Solis, I. Lobato, E. G. Noya, A. Guerrero-Martinez, J. M. Taboada, F. Obelleiro, L. G. MacDowell, S. Bals and L. M. Liz-Marzan, Science, 2020, 368, 1472–1477 CrossRef CAS.
- M. Hentschel, M. Schaferling, X. Duan, H. Giessen and N. Liu, Sci. Adv., 2017, 3, e1602735 CrossRef.
- L. Nguyen, M. Dass, M. F. Ober, L. V. Besteiro, Z. M. Wang, B. Nickel, A. O. Govorov, T. Liedl and A. Heuer-Jungemann, ACS Nano, 2020, 14, 7454–7461 CrossRef CAS.
- D. Seo, J. C. Park and H. Song, J. Am. Chem. Soc., 2006, 128, 14863–14870 CrossRef CAS.
- C. Li, K. L. Shuford, M. Chen, E. J. Lee and S. O. Cho, ACS Nano, 2008, 2, 1760–1769 CrossRef CAS.
- Y. Lu, H. Zhang, F. Wu, H. Liu and J. Fang, RSC Adv., 2017, 7, 18601–18608 RSC.
- H. Zhang, Y. Lu, C. Guan, N. Song, Y. Zhang, H. Liu and J. Fang, J. Mater. Chem. C, 2017, 5, 645–653 RSC.
- O. M. Bakr, B. H. Wunsch and F. Stellacci, Chem. Mater., 2006, 18, 3297–3301 CrossRef CAS.
- C. G. Khoury and T. Vo-Dinh, J. Phys. Chem. C, 2008, 112, 18849–18859 CrossRef CAS.
- H.-L. Wu, C.-H. Chen and M. H. Huang, Chem. Mater., 2009, 21, 110–114 CrossRef CAS.
- E. Ye, K. Y. Win, H. R. Tan, M. Lin, C. P. Teng, A. Mlayah and M. Y. Han, J. Am. Chem. Soc., 2011, 133, 8506–8509 CrossRef CAS.
- E. Ye, M. D. Regulacio, M. S. Bharathi, H. Pan, M. Lin, M. Bosman, K. Y. Win, H. Ramanarayan, S. Y. Zhang, X. J. Loh, Y. W. Zhang and M. Y. Han, Nanoscale, 2016, 8, 543–552 RSC.
- Z. Li, W. Li, P. H. Camargo and Y. Xia, Angew. Chem., Int. Ed., 2008, 47, 9653–9656 CrossRef CAS.
- D. Y. Kim, T. Yu, E. C. Cho, Y. Ma, O. O. Park and Y. Xia, Angew. Chem., Int. Ed., 2011, 50, 6328–6331 CrossRef CAS.
- C. P. Teng, T. Zhou, E. Ye, S. Liu, L. D. Koh, M. Low, X. J. Loh, K. Y. Win, L. Zhang and M. Y. Han, Adv. Healthcare Mater., 2016, 5, 2122–2130 CrossRef CAS.
- D. Zhu, Y. Liu, M. Liu, X. Liu, P. N. Prasad and M. T. Swihart, J. Mater. Chem. B, 2020, 8, 5491–5499 RSC.
- D. V. Goia and E. Matijević, New J. Chem., 1998, 22, 1203–1215 RSC.
- H. Bönnemann and R. M. Richards, Eur. J. Inorg. Chem., 2001, 2455–2480 CrossRef.
- C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025–1102 CrossRef CAS.
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef CAS.
- Y. C. Tsao, S. Rej, C. Y. Chiu and M. H. Huang, J. Am. Chem. Soc., 2014, 136, 396–404 CrossRef CAS.
- M. S. Akhtar, J. Panwar and Y.-S. Yun, ACS Sustainable Chem. Eng., 2013, 1, 591–602 CrossRef CAS.
- S. Ahmed, M. Ahmad, B. L. Swami and S. Ikram, J. Adv. Res., 2016, 7, 17–28 CrossRef CAS.
- P. Khandel, R. K. Yadaw, D. K. Soni, L. Kanwar and S. K. Shahi, J. Nanostruct. Chem., 2018, 8, 217–254 CrossRef CAS.
- P. Malik, R. Shankar, V. Malik, N. Sharma and T. K. Mukherjee, J. Nanopart., 2014, 2014, 1–14 CrossRef.
- R. Sankar, P. Manikandan, V. Malarvizhi, T. Fathima, K. S. Shivashangari and V. Ravikumar, Spectrochim. Acta, Part A, 2014, 121, 746–750 CrossRef CAS.
- H. R. El-Seedi, R. M. El-Shabasy, S. A. M. Khalifa, A. Saeed, A. Shah, R. Shah, F. J. Iftikhar, M. M. Abdel-Daim, A. Omri, N. H. Hajrahand, J. S. M. Sabir, X. Zou, M. F. Halabi, W. Sarhan and W. Guo, RSC Adv., 2019, 9, 24539–24559 RSC.
- D. Alti, M. Veeramohan Rao, D. N. Rao, R. Maurya and S. K. Kalangi, ACS Omega, 2020, 5, 16238–16245 CrossRef CAS.
- D. P. Yang, X. Liu, C. P. Teng, C. Owh, K. Y. Win, M. Lin, X. J. Loh, Y. L. Wu, Z. Li and E. Ye, Nanoscale, 2017, 9, 15753–15759 RSC.
- M. D. Regulacio, D.-P. Yang and E. Ye, CrystEngComm, 2020, 22, 399–411 RSC.
- D. S. Sheny, J. Mathew and D. Philip, Spectrochim. Acta, Part A, 2011, 79, 254–262 CrossRef CAS.
- K. Gopinath, S. Kumaraguru, K. Bhakyaraj, S. Mohan, K. S. Venkatesh, M. Esakkirajan, P. Kaleeswarran, N. S. Alharbi, S. Kadaikunnan, M. Govindarajan, G. Benelli and A. Arumugam, Microb. Pathog., 2016, 101, 1–11 CrossRef CAS.
- T. L. Botha, E. E. Elemike, S. Horn, D. C. Onwudiwe, J. P. Giesy and V. Wepener, Sci. Rep., 2019, 9, 4169 CrossRef.
- P. Kuppusamy, S. Ilavenil, S. Srigopalram, D. H. Kim, N. Govindan, G. P. Maniam, M. M. Yusoff and K. C. Choi, J. Inorg. Organomet. Polym. Mater., 2017, 27, 562–568 CrossRef CAS.
- M. Khan, K. Al-Hamoud, Z. Liaqat, M. R. Shaik, S. F. Adil, M. Kuniyil, H. Z. Alkhathlan, A. Al-Warthan, M. R. H. Siddiqui, M. Mondeshki, W. Tremel, M. Khan and M. N. Tahir, Nanomaterials, 2020, 10, 1885 CrossRef CAS.
- N. Patra, A. C. Taviti, A. Sahoo, A. Pal, T. K. Beuria, A. Behera and S. Patra, RSC Adv., 2017, 7, 35111–35118 RSC.
- C. Sharma, S. Ansari, M. S. Ansari, S. P. Satsangee and M. M. Srivastava, Mater. Sci. Eng., C, 2020, 116, 111153 CrossRef CAS.
- P. Hou, H. Liu, J. Li and J. Yang, CrystEngComm, 2015, 17, 1826–1832 RSC.
- D. Kim, J. Resasco, Y. Yu, A. M. Asiri and P. Yang, Nat. Commun., 2014, 5, 4948 CrossRef CAS.
- S. Liu, G. Chen, P. N. Prasad and M. T. Swihart, Chem. Mater., 2011, 23, 4098–4101 CrossRef CAS.
- W. Chen, R. Yu, L. Li, A. Wang, Q. Peng and Y. Li, Angew. Chem., Int. Ed., 2010, 49, 2917–2921 CrossRef CAS.
- S. E. Skrabalak, J. Chen, Y. Sun, X. Lu, L. Au, C. M. Cobley and Y. Xia, Acc. Chem. Res., 2008, 41, 1587–1595 CrossRef CAS.
- B. Goris, L. Polavarapu, S. Bals, G. Van Tendeloo and L. M. Liz-Marzan, Nano Lett., 2014, 14, 3220–3226 CrossRef CAS.
- S. W. Chee, S. F. Tan, Z. Baraissov, M. Bosman and U. Mirsaidov, Nat. Commun., 2017, 8, 1–8 CrossRef CAS.
- Y. Sun and Y. Xia, J. Am. Chem. Soc., 2004, 126, 3892–3901 CrossRef CAS.
- X. Lu, L. Au, J. McLellan, Z. Y. Li, M. Marquez and Y. Xia, Nano Lett., 2007, 7, 1764–1769 CrossRef CAS.
- Y. Ma, W. Li, E. C. Cho, Z. Li, T. Yu, J. Zeng, Z. Xie and Y. Xia, ACS Nano, 2010, 4, 6725–6734 CrossRef CAS.
- E. Gonzalez, J. Arbiol and V. F. Puntes, Science, 2011, 334, 1377–1380 CrossRef CAS.
- C. H. Tsai, S. Y. Chen, J. M. Song, M. Haruta and H. Kurata, Nanoscale Res. Lett., 2015, 10, 1–9 CrossRef.
- T. S. Rodrigues, A. G. M. da Silva, A. B. L. de Moura, I. G. Freitas and P. H. C. Camargo, RSC Adv., 2016, 6, 62286–62290 RSC.
- A. G. da Silva, T. S. Rodrigues, T. J. Slater, E. A. Lewis, R. S. Alves, H. V. Fajardo, R. Balzer, A. H. da Silva, I. C. de Freitas, D. C. Oliveira, J. M. Assaf, L. F. Probst, S. J. Haigh and P. H. Camargo, ACS Appl. Mater. Interfaces, 2015, 7, 25624–25632 CrossRef CAS.
- S. Y. Tee, E. Ye, P. H. Pan, C. J. Lee, H. K. Hui, S. Y. Zhang, L. D. Koh, Z. Dong and M. Y. Han, Nanoscale, 2015, 7, 11190–11198 RSC.
- A. Panacek, L. Kvitek, R. Prucek, M. Kolar, R. Vecerova, N. Pizurova, V. K. Sharma, T. Nevecna and R. Zboril, J. Phys. Chem. B, 2006, 110, 16248–16253 CrossRef CAS.
- Y. Zhou, Y. Kong, S. Kundu, J. D. Cirillo and H. Liang, J. Nanobiotechnol., 2012, 10, 19 CrossRef CAS.
- N. M. Zain, A. G. Stapley and G. Shama, Carbohydr. Polym., 2014, 112, 195–202 CrossRef CAS.
- F. Paladini, M. Pollini, A. Sannino and L. Ambrosio, Biomacromolecules, 2015, 16, 1873–1885 CrossRef CAS.
- Y. N. Slavin, J. Asnis, U. O. Hafeli and H. Bach, J. Nanobiotechnol., 2017, 15, 65 CrossRef.
- S. Pal, Y. K. Tak and J. M. Song, Appl. Environ. Microbiol., 2007, 73, 1712–1720 CrossRef CAS.
- Z. M. Xiu, Q. B. Zhang, H. L. Puppala, V. L. Colvin and P. J. Alvarez, Nano Lett., 2012, 12, 4271–4275 CrossRef CAS.
- X. Yan, B. He, L. Liu, G. Qu, J. Shi, L. Hu and G. Jiang, Metallomics: Integrated Biometal. Sci., 2018, 10, 557–564 RSC.
- M. Ramezani, A. Kosak, A. Lobnik and A. Hadela, Text. Res. J., 2019, 89, 5130–5143 CrossRef CAS.
- S. Tang and J. Zheng, Adv. Healthcare Mater., 2018, 7, e1701503 CrossRef.
- A. Ivask, A. Elbadawy, C. Kaweeteerawat, D. Boren, H. Fischer, Z. Ji, C. H. Chang, R. Liu, T. Tolaymat, D. Telesca, J. I. Zink, Y. Cohen, P. A. Holden and H. A. Godwin, ACS Nano, 2014, 8, 374–386 CrossRef CAS.
- G. Franci, A. Falanga, S. Galdiero, L. Palomba, M. Rai, G. Morelli and M. Galdiero, Molecules, 2015, 20, 8856–8874 CrossRef CAS.
- J. R. Morones, J. L. Elechiguerra, A. Camacho, K. Holt, J. B. Kouri, J. T. Ramirez and M. J. Yacaman, Nanotechnology, 2005, 16, 2346–2353 CrossRef CAS.
- H. B. Li, P. Liu, Y. Liang, J. Xiao and G. W. Yang, Nanoscale, 2012, 4, 5082–5091 RSC.
- C. M. Goodman, C. D. McCusker, T. Yilmaz and V. M. Rotello, Bioconjugate Chem., 2004, 15, 897–900 CrossRef CAS.
- P. Nalawade, P. Mukherjee and S. Kapoor, J. Nanostruct. Chem., 2014, 4, 1–8 Search PubMed.
- Y. Wang, J. Wan, R. J. Miron, Y. Zhao and Y. Zhang, Nanoscale, 2016, 8, 11143–11152 RSC.
- M. Banerjee, S. Sharma, A. Chattopadhyay and S. S. Ghosh, Nanoscale, 2011, 3, 5120–5125 RSC.
- N. Cioffi, L. Torsi, N. Ditaranto, G. Tantillo, L. Ghibelli, L. Sabbatini, T. Bleve-Zacheo, M. D'Alessio, P. G. Zambonin and E. Traversa, Chem. Mater., 2005, 17, 5255–5262 CrossRef CAS.
- L. A. Tamayo, P. A. Zapata, N. D. Vejar, M. I. Azocar, M. A. Gulppi, X. Zhou, G. E. Thompson, F. M. Rabagliati and M. A. Paez, Mater. Sci. Eng., C, 2014, 40, 24–31 CrossRef CAS.
- M. Raffi, S. Mehrwan, T. M. Bhatti, J. I. Akhter, A. Hameed, W. Yawar and M. M. Ul Hasan, Ann. Microbiol., 2010, 60, 75–80 CrossRef CAS.
- P. S. Harikumar, Int. J. Sci., 2016, 2, 83–90 Search PubMed.
- R. Lakshminarayanan, E. Ye, D. J. Young, Z. Li and X. J. Loh, Adv. Healthcare Mater., 2018, 7, e1701400 CrossRef.
- S. Shankar and J.-W. Rhim, Mater. Lett., 2014, 132, 307–311 CrossRef CAS.
- M. Fleischmann, P. J. Hendra and A. J. McQuillan, Chem. Phys. Lett., 1974, 26, 163–166 CrossRef CAS.
- G. Demirel, H. Usta, M. Yilmaz, M. Celik, H. A. Alidagi and F. Buyukserin, J. Mater. Chem. C, 2018, 6, 5314–5335 RSC.
- I. Bruzas, W. Lum, Z. Gorunmez and L. Sagle, The Analyst, 2018, 143, 3990–4008 RSC.
- S.-Y. Ding, J. Yi, J.-F. Li, B. Ren, D.-Y. Wu, R. Panneerselvam and Z.-Q. Tian, Nat. Rev. Mater., 2016, 1, 1–16 Search PubMed.
- A. I. Pérez-Jiménez, D. Lyu, Z. Lu, G. Liu and B. Ren, Chem. Sci., 2020, 11, 4563–4577 RSC.
- S. Laing, L. E. Jamieson, K. Faulds and D. Graham, Nat. Rev. Chem., 2017, 1, 1–19 CrossRef.
- B. Saute, R. Premasiri, L. Ziegler and R. Narayanan, The Analyst, 2012, 137, 5082–5087 RSC.
- K.-Q. Lin, J. Yi, S. Hu, B.-J. Liu, J.-Y. Liu, X. Wang and B. Ren, J. Phys. Chem. C, 2016, 120, 20806–20813 CrossRef CAS.
- M. J. Seo, G. W. Kim, P. V. Tsalu, S. W. Moon and J. W. Ha, Nanoscale Horiz., 2020, 5, 345–349 RSC.
- C. Wang, B. Liu and X. Dou, Sens. Actuators, B, 2016, 231, 357–364 CrossRef CAS.
- C. Kuttner, M. Mayer, M. Dulle, A. Moscoso, J. M. Lopez-Romero, S. Forster, A. Fery, J. Perez-Juste and R. Contreras-Caceres, ACS Appl. Mater. Interfaces, 2018, 10, 11152–11163 CrossRef CAS.
- R. Kodiyath, S. T. Malak, Z. A. Combs, T. Koenig, M. A. Mahmoud, M. A. El-Sayed and V. V. Tsukruk, J. Mater. Chem. A, 2013, 1, 2777 RSC.
- T. J. Dill, M. J. Rozin, S. Palani and A. R. Tao, ACS Nano, 2016, 10, 7523–7531 CrossRef CAS.
- J. E. Park, Y. Lee and J. M. Nam, Nano Lett., 2018, 18, 6475–6482 CrossRef CAS.
- S. He, J. Chua, E. K. M. Tan and J. C. Y. Kah, RSC Adv., 2017, 7, 16264–16272 RSC.
- L. Tian, N. Gandra and S. Singamaneni, ACS Nano, 2013, 7, 4252–4260 CrossRef CAS.
- C. Liu, L. Tan, L. Li, J. Dong and W. Qian, CrystEngComm, 2017, 19, 3233–3236 RSC.
- J. Li, J. Wu, X. Zhang, Y. Liu, D. Zhou, H. Sun, H. Zhang and B. Yang, J. Phys. Chem. C, 2011, 115, 3630–3637 CrossRef CAS.
- P. C. Angelomé, H. Heidari Mezerji, B. Goris, I. Pastoriza-Santos, J. Pérez-Juste, S. Bals and L. M. Liz-Marzán, Chem. Mater., 2012, 24, 1393–1399 CrossRef.
- Y. Zhang, B. Wang, S. Yang, L. Li and L. Guo, New J. Chem., 2015, 39, 2551–2556 RSC.
- M. Bosman, E. Ye, S. F. Tan, C. A. Nijhuis, J. K. Yang, R. Marty, A. Mlayah, A. Arbouet, C. Girard and M. Y. Han, Sci. Rep., 2013, 3, 1312 CrossRef.
- H. Guo, F. Ruan, L. Lu, J. Hu, J. Pan, Z. Yang and B. Ren, J. Phys. Chem. C, 2009, 113, 10459–10464 CrossRef CAS.
- L. Rodríguez-Lorenzo, R. N. A. Álvarez-Puebla, F. J. G. A. de Abajo and L. M. Liz-Marzán, J. Phys. Chem. C, 2010, 114, 7336–7340 CrossRef.
- S. Lin, X. Lin, Y. Shang, S. Han, W. Hasi and L. Wang, J. Phys. Chem. C, 2019, 123, 24714–24722 CrossRef CAS.
- Y. C. Cao, R. Jin and C. A. Mirkin, Science, 2002, 297, 1536–1540 CrossRef CAS.
- D. K. Lim, K. S. Jeon, J. H. Hwang, H. Kim, S. Kwon, Y. D. Suh and J. M. Nam, Nat. Nanotechnol., 2011, 6, 452–460 CrossRef CAS.
- H. Xu, E. J. Bjerneld, M. Käll and L. Börjesson, Phys. Rev. Lett., 1999, 83, 4357–4360 CrossRef CAS.
- X. X. Han, B. Zhao and Y. Ozaki, Anal. Bioanal. Chem., 2009, 394, 1719–1727 CrossRef CAS.
- B. Fazio, C. D'Andrea, A. Foti, E. Messina, A. Irrera, M. G. Donato, V. Villari, N. Micali, O. M. Marago and P. G. Gucciardi, Sci. Rep., 2016, 6, 26952 CrossRef CAS.
- P. Matteini, M. de Angelis, L. Ulivi, S. Centi and R. Pini, Nanoscale, 2015, 7, 3474–3480 RSC.
- F. Zhang, N. Wu, J. Zhu, J. Zhao, G.-J. Weng, J.-J. Li and J.-W. Zhao, Sens. Actuators, B, 2018, 271, 174–182 CrossRef CAS.
- Y. Su, S. Xu, J. Zhang, X. Chen, L. P. Jiang, T. Zheng and J. J. Zhu, Anal. Chem., 2019, 91, 864–872 CrossRef CAS.
- S. Tian, W. You, Y. Shen, X. Gu, M. Ge, S. Ahmadi, S. Ahmad and H.-B. Kraatz, New J. Chem., 2019, 43, 14772–14780 RSC.
- X. Zhao, M. Deng, G. Rao, Y. Yan, C. Wu, Y. Jiao, A. Deng, C. Yan, J. Huang, S. Wu, W. Chen, T. Lei, P. Xu, W. He and J. Xiong, Small, 2018, 14, e1802477 CrossRef.
- A. V. Markin, N. E. Markina, J. Popp and D. Cialla-May, TrAC, Trends Anal. Chem., 2018, 108, 247–259 CrossRef CAS.
- N. E. Markina, S. N. Ustinov, A. M. Zakharevich and A. V. Markin, Anal. Chim. Acta, 2020, 1138, 9–17 CrossRef CAS.
- N. E. Markina, E. K. Volkova, A. M. Zakharevich, I. Y. Goryacheva and A. V. Markin, Mikrochim. Acta, 2018, 185, 481 CrossRef.
- Z. Jiang, Q. Zhang, C. Zong, B.-J. Liu, B. Ren, Z. Xie and L. Zheng, J. Mater. Chem., 2012, 22, 18192 RSC.
- C. Zhu, G. Yang, H. Li, D. Du and Y. Lin, Anal. Chem., 2015, 87, 230–249 CrossRef CAS.
- M. M. Rahman, A. J. Ahammad, J. H. Jin, S. J. Ahn and J. J. Lee, Sensors, 2010, 10, 4855–4886 CrossRef CAS.
- S. Y. Tee, C. P. Teng and E. Ye, Mater. Sci. Eng., C, 2017, 70, 1018–1030 CrossRef CAS.
- D. W. Hwang, S. Lee, M. Seo and T. D. Chung, Anal. Chim. Acta, 2018, 1033, 1–34 CrossRef CAS.
- Y.-G. Zhou, S. Yang, Q.-Y. Qian and X.-H. Xia, Electrochem. Commun., 2009, 11, 216–219 CrossRef CAS.
- S. Cherevko and C.-H. Chung, Sens. Actuators, B, 2009, 142, 216–223 CrossRef CAS.
- P. Si, Y. Huang, T. Wang and J. Ma, RSC Adv., 2013, 3, 3487–3502 RSC.
- X. Gao, L. Jin, Q. Wu, Z. Chen and X. Lin, Electroanalysis, 2012, 24, 1771–1777 CAS.
- G. A. El-Nagar, R. M. Sarhan, A. Abouserie, N. Maticiuc, M. Bargheer, I. Lauermann and C. Roth, Sci. Rep., 2017, 7, 12181 CrossRef.
- P. Miao, B. Wang, J. Yin, X. Chen and Y. Tang, Electrochem. Commun., 2015, 53, 37–40 CrossRef CAS.
- N. Wang, Y. Han, Y. Xu, C. Gao and X. Cao, Anal. Chem., 2015, 87, 457–463 CrossRef CAS.
- N. Pariona, A. I. Mtz-Enriquez, D. Sánchez-Rangel, G. Carrión, F. Paraguay-Delgado and G. Rosas-Saito, RSC Adv., 2019, 9, 18835–18843 RSC.
- M. A. Asghar, E. Zahir, S. M. Shahid, M. N. Khan, M. A. Asghar, J. Iqbal and G. Walker, Lwt, 2018, 90, 98–107 CrossRef CAS.
- S. C. Mali, A. Dhaka, C. K. Githala and R. Trivedi, Biotechnol. Rep., 2020, 27, e00518 CrossRef.
- S. Jain and M. S. Mehata, Sci. Rep., 2017, 7, 15867 CrossRef.
- D. H. Nguyen, J. S. Lee, K. D. Park, Y. C. Ching, X. T. Nguyen, V. Phan and T. T. Hoang Thi, Nanomaterials, 2020, 10, 542 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |