
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of heparin and peptide conjugation on structure and functional properties of alginate in solutions and hydrogels†
Nir
Goldberg
a,
Yulia
Shmidov
a,
Olga
Kryukov
b,
Dina
Aranovich
a,
Smadar
Cohen
b and
Ronit
Bitton
 *ac
*ac
aDepartment of Chemical Engineering, Ben-Gurion University of the Negev, Beer-Sheva 84105, Israel. E-mail: rbitton@bgu.ac.il
bAvram and Stella Goldstein-Goren Department of Biotechnology Engineering and Regenerative Medicine and Stem Cell Center, Ben-Gurion University of the Negev, POB 653, Beer-Sheva 84105, Israel
cIlze Kats Institute for Nanoscale Science and Technology, Ben-Gurion University of the Negev, Beer-Sheva 84105, Israel
First published on 17th December 2020
Abstract
Alginate decoration with more than one type of biomolecule is becoming a prevalent process in the pursuit to turn alginate hydrogels into synthetic extracellular matrices (ECMs). Here we present a systematic study of the structural–physical properties–function relationship of alginate–G4RGDY–heparin aqueous solutions and hydrogels. Small-angle X-ray scattering (SAXS) and rheology were used to characterize the systems’ nanostructure, viscosity of the solutions, and storage modulus of the hydrogels. The bioactivity of these gels was explored by evaluating their ability to sustain the release of vascular endothelial growth factor (VEGF). We show that the mode and order of conjugating the heparin and the peptide to the alginate backbone greatly affect the modified alginate hydrogels’ structure–properties–function relations. Moreover, we show that a detailed structural analysis of the conjugated architecture in solution can be used as a tool to adapt the properties of alginate–heparin–peptide hybrid hydrogels.
Introduction
Alginate is a linear, hydrophilic, polysaccharide biopolymer consisting of β-D mannuronic and α-L guluronic acid blocks, which occur in the cell walls of brown seaweed.1 Alginate hydrogels have been proven to be promising scaffolds for tissue regeneration and repair2–4 for bone,5 skin,6 and nerve regeneration.7,8 An emerging trend in designing such scaffolds is recapitulating the natural extracellular matrix (ECM) traits by installing motifs capable of providing biochemical stimuli.9,10Functionalization of alginate hydrogels is often achieved by covalently attaching bioactive molecules (e.g., peptides, proteins, etc.) to the alginate backbone.11–13 The pioneering work in creating alginate synthetic ECMs focused on binding single peptides to the alginate backbone, the first being the G4RGDS peptide;13 nowadays, researchers focus on creating multifunctional hydrogels by functionalizing the alginate with more than one type of biological molecule.14
The amount and identity of the attached biomolecules are crucial for the scaffold's bioactivity; however, they are not the only factors determining its functionality. The scaffold's stiffness as well as the ligand's availability (e.g., density) have been shown to dictate the magnitude and arrangement of intracellular forces, hence affecting the materials’ cellular response to them.11,15,16 Therefore, understanding the structure–properties–function relationship of multicomponent alginate hydrogels is an important step toward bettering their rational design.
In previous studies, we have shown that covalently linking a peptide that contains arginine–glycine–aspartic acid sequence (RGD) to an alginate backbone affected the conformational state of the individual chain as well as chain assemblies of alginate in aqueous solutions.17,18 We have shown that the amount of bound peptide determines the behaviour of polysaccharide–peptide conjugates in solution, regardless of the specific nature of the polysaccharide.19 Furthermore, we have demonstrated that the sequence of the conjugated peptides is a significant factor in tuning the stiffness of the alginate/peptide hybrid hydrogels.19
In this follow up study, we aim to explore the structure–properties–function relationship of multifunctional alginate hydrogels, i.e., hydrogels composed of an alginate chain decorated with more than one type of biomolecule. In particularly, we are interested in studying the effect of mode and order of conjugation on the resulting properties. To this end, we chose to covalently bind to the alginate backbone both the G4RGDY peptide and heparin.
Heparin is a glycosaminoglycan (GAG), a stiff linear polysaccharide consisting of 1 to 4 linked disaccharide repeating units of uronic acid and glucosamine residues.20,21 Heparin has been shown to stabilize growth factors from denaturation while increasing the affinity of the complex to cell receptors, an important trait for tissue engineering applications.11–13 A variety of methods have been used for the covalent immobilization of heparin into hydrogels in the production of ECM-mimetic materials.22,23 Recently, conjugation of heparin to polysaccharides (i.e.,) alginate14 and chitosan24 has been suggested to improve their performance as synthetic ECMs.14,15,25 It has been suggested that when designing a hydrogel for promoting cell adhesion, growth, and function, synthesizing a surface that contains both RGD and heparin would provide a more desirable biomimetic material than hydrogels containing only one of the individual components.26,27 However, optimizing the function and physical properties of these multicomponent hydrogels could be a rigorous process; a better understanding of the interplay between the building block's structure and the properties of the resulting hydrogels may be used to simplify it.
Here, we present a systematic study of the structural–physical properties–function relationship of alginate–G4RGDY–heparin aqueous solutions and hydrogels. Using SAXS and rheology to characterize the systems’ nanostructure, the viscosity of the solutions, and the storage modulus of the hydrogels, we show that attaching heparin to an alginate–peptide molecule greatly affects its physical properties. Moreover, we show that a detailed structural analysis of the conjugated architecture in solution can be used as a tool to adapt the properties of alginate–heparin–peptide hybrid hydrogels.
Results and discussion
Synthesis of alginate–peptide–heparin
Conjugation of a peptide and/or heparin to the alginate backbone was performed using carbodiimide chemistry as described in the experimental section. Overall, four modifications upon alginate were performed: conjugation of alginate with G4RGDY was termed as Alg–RGD. Conjugation of alginate with heparin was termed as Alg–Hep. Conjugating of Alg–RGD with heparin was termed as Alg–RGD–Hep, and conjugating of Alg–Hep with G4RGDY was termed as Alg–Hep–RGD. The notation denotes the order of the conjugation and does not indicates the sequence (e.g. in Alg–RGD–Hep, heparin is not attached to RGD, but rather conjugated as the following step). In addition, a mixture of Alg–Hep and Alg–RGD (denoted as Alg–RGD + Alg–Hep) was prepared. The conjugation of the heparin and the peptide to the alginate was confirmed by FTIR spectroscopy (Fig. S1, ESI†) and XPS (Table S1, ESI†).Viscosity and nanostructure of modified alginate solutions
Differences in the features of polymer network in solution are manifested in its' rheological behavior. Thus, the steady shear viscosity as a function of the applied shear rate was monitored for the polysaccharide's aqueous solutions. Viscosity measurements of 2 wt% polysaccharide (natural and modified) in aqueous solution are displayed in Fig. 1. As can be seen, all tested polymers exhibit shear thinning behaviour typical of viscous polymers at the semi-dilute concentration regime,18,21,28,29 in which a Newtonian plateau (low shear rate) region is followed by a viscosity gradual decrease (higher shear rate). The lowest shear rate at which the decrease of viscosity becomes apparent is called the critical shear rate (![[small gamma, Greek, dot above]](https://www.rsc.org/images/entities/i_char_e0a2.gif) c). Shear thinning of fluids and weak gels can be described by
c). Shear thinning of fluids and weak gels can be described by| η = kn−1 | (1) |
is the shear rate (s−1), n is the power law exponent (for a Newtonian fluid n = 1), and k is a consistency index which is numerically equal to the viscosity at 1 s−1 (Table S2, ESI†).30
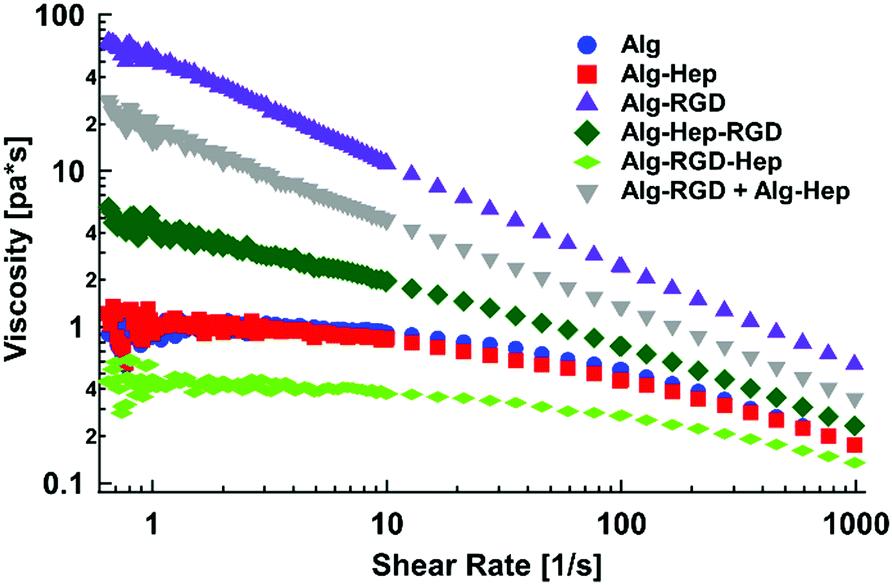 | ||
| Fig. 1 Steady shear viscosity as a function of the applied shear rate of modified alginate in water (2 wt%). | ||
As can be seen from Fig. 1, the behaviour of Alg–Hep is similar to that of alginate. This is expected due to the negative charge of the functional groups of both polymers, which prevents additional entanglements. In agreement with our previous work,17 Alg–RGD shows a much higher k and a smaller n, indicating a more pronounced shear thinning behaviour (i.e., more entanglements). The scan of Alg–Hep–RGD lies between those of Alg–Hep and Alg–RGD, indicating both molecules are affecting the polymers’ behaviour. Moreover, comparing the viscosity of Alg–RGD–Hep and Alg–Hep–RGD to the viscosity of a mixture of Alg–Hep and Alg–RGD (denoted as Alg–RGD + Alg–Hep) suggests a synergistic effect rather than an additive one, likely due to intermolecular interactions between Alg–RGD and Alg–Hep that are not substantial when the heparin and RGD are conjugated to the same alginate chain. The order of the molecules’ conjugation (i.e., peptide before heparin or heparin before peptide) also had a significant effect on the solution's viscosity; fitting the data to a power-law model yielded higher k and lower c (Table S2, ESI†) for the molecules in which the peptide was the last conjugated molecule, suggesting a more entangled polymer network is formed. Contrarily, the k of Alg–RGD–Hep is the lowest one.
Differences in the rheological behaviour of polymer solutions are usually caused by differences in the structural features of the polymer's network. Thus, SAXS measurements of the modified alginate in aqueous solution were conducted. The SAXS patterns of all of the investigated molecules are presented in Fig. 2. The visible differences between the scattering curves indicate that indeed, there are structural differences between the tested solutions.
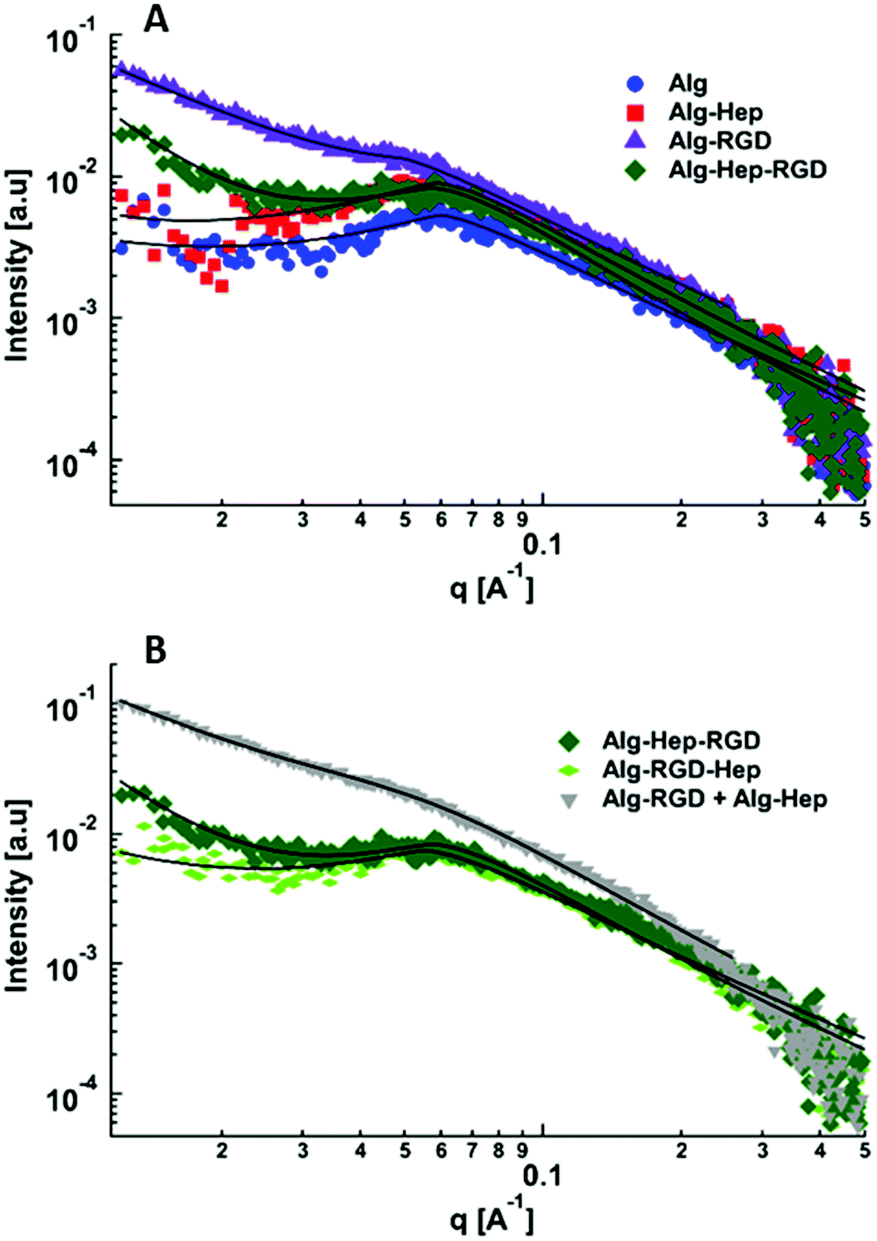 | ||
| Fig. 2 Small-angle X-ray scattering curves of: (A) Alg, Alg–Hep, Alg–RGD, Alg–Hep–RGD, (B) Alg–Hep–RGD, Alg–RGD–Hep and Alg–RGD + Alg–Hep in water (2 wt%). The curves were separated for better visualization. The black solid lines represent the best fits to eqn (2). | ||
Horkay and Hammouda described the scattering pattern of polyelectrolyte in salt-free solutions as the result of three contributions: spatial concentration fluctuations (clustering), which are apparent as an upturn in the low q range; thermal concentration fluctuations (high q); and a characteristic correlation peak (mid q) with a maximum, q0, corresponding to d0 = 2π/q0, which represents an average distance between the charged chains.31 This scattering pattern can be described by
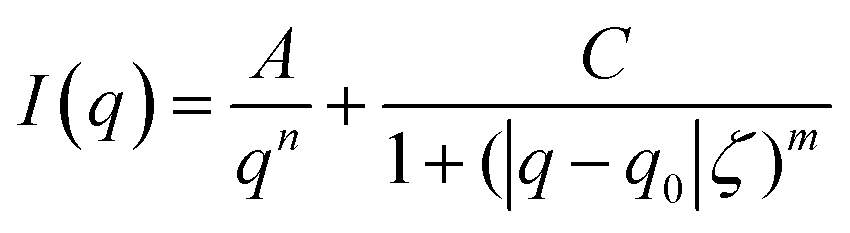 | (2) |
As can be seen in Fig. 2, the most pronounced differences between the scattering curves are in the mid- and low q-regimes. The characteristic correlation peak (mid-q) is clearly seen in the scattering pattern of alginate, Alg–Hep, Alg–Hep–RGD, and Alg–RGD–Hep; however, for Alg–RGD and Alg–RGD + Alg–Hep, while the correlation peak exists, it could not be well resolved. Fit of the data to eqn (2) shows that conjugating of both peptide and heparin to alginate chains did not significantly change the value of d0. The larger d0 value obtained for Alg–RGD + Alg–Hep is likely due to the fact that the correlation peak could not be well resolved and is not a true representation of the average distance between the charged domains (Table S3, ESI†).
As seen by SAXS, the structural feature that is most affected by the alginate modification is the clustering. Alg–Hep–RGD, Alg–RGD, and the Alg–RGD + Alg–Hep mixture all exhibit a clear upturn in the low q range. The clear distinction between the upturn and the correlation peak in the Alg–Hep–RGD curve is an indication that the clusters and the distance between the charged domains are of different length scales. The overlapping between the peak and the upturn in the scattering patterns of Alg–RGD and those of the Alg–RGD + Alg–Hep indicates the presence of clusters of various sizes, including some similar in size to d0.
Both SAXS and viscosity measurements show that conjugating a peptide, heparin, or both to an alginate backbone affects the alginate's spatial organization in aqueous solutions. Moreover, the results suggest that the order of the peptide conjugation plays a significant role in determining the network's structure.
By comparing the results of shear-dependent viscosity to the SAXS patterns, one can see that the solutions with the higher viscosities exhibit an upturn in the low q range of their scattering patterns. This observation implies that the higher viscosity of Alg–Hep–RGD, Alg–RGD + Alg–Hep, and Alg–RGD is due to the presence of large, dense clusters and not a smaller mesh size as would have been expected in the case of a homogeneous polymer network. Thus, we propose spatial organizations of heterogeneous networks, for the chain assemblies of modified alginates in aqueous solutions as depicted in Fig. 3.
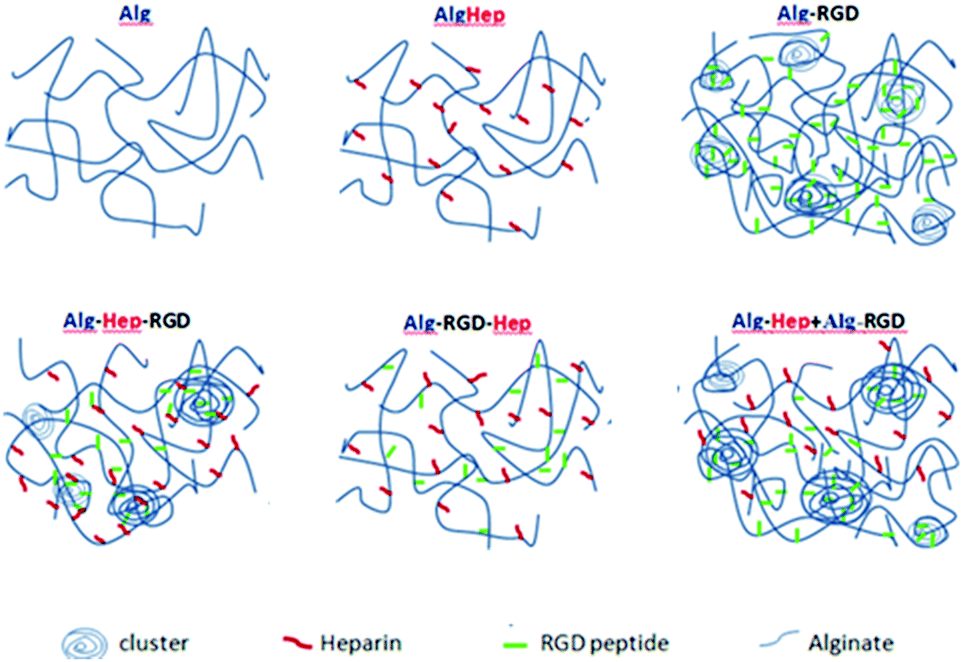 | ||
| Fig. 3 Illustration of the proposed spatial heterogeneous network organization of the modified alginate in aqueous solutions. The average distance between the charged domains is approximately the same for all the modifications. The structure of Alg–Hep is similar to that of Alg. The order of the peptide conjugation has a significant role on the tendency towards clustering; Alg–Hep–RGD, Alg–RGD + Alg–Hep, and Alg–RGD have large, dense clusters that are formed likely due to the attractive interactions either between two G4RGDY peptides or, more likely, between the peptide and the alginate backbone. The molecules are not drawn to scale. | ||
Formation of large clusters is typically caused due to attractive inter-particle interactions. Large clusters are observed only for Alg–RGD, Alg–Hep–RGD and Alg–RGD + Alg–Hep, indicating that the peptide is imperative for their formation and that the attractive interactions are either between two G4RGDY peptides or, more likely, between the peptide and the alginate backbone. Possible attractive interactions between these two molecules are hydrophobic interactions and hydrogen bonding, typical to polyphenol/polysaccharide interactions between the peptide's tyrosine and the alginate.17 However, for these interactions to take place, tyrosine needs to be accessible to the neighbouring molecules. In the case of Alg–RGD–Hep and Alg–Hep–RGD, one would expect the stronger electrostatic repulsion between the molecules, as a result of the presence of heparin, to prevent the formation of RGD-induced clusters.32 The fact that some clusters are formed in the latter (Alg–Hep–RGD) suggests that some of the peptides may have conjugated to the heparin instead of the alginate. Alg–Hep has a similar structure as unmodified alginate, since conjugation of heparin to the alginate backbone did not affect the electrostatic repulsion between the chains, as heparin is also a polyanion.
Mechanical properties and nanostructure of modified alginate hydrogels
Next, hydrogels of modified alginate were prepared (as described in experimental section). Briefly, CaCO3 and GDL (to allow homogeny gels) were added to the alginate solutions so that the final concentrations were 1.5% wt% alginate, 40 mM CaCO3, and 40 mM GDL. Rheological measurements were performed to quantify the mechanical properties (i.e., gel stiffness) of the different hydrogels. Frequency sweep scans of modified alginate hydrogels are presented in Fig. 4A. As expected from a gel's frequency sweep scan, both moduli G′ and G′′ are independent of the frequency of oscillation, and G′ > G′′ (Fig. 4A); that is, the elastic nature of the sample is stronger than its viscous nature, as typically, gel stiffness is represented by its storage modulus G′. A difference in G′ values of the different gels is clearly apparent (Fig. 4B), indicating that conjugation of heparin and G4RGDY affected the gels’ mechanical properties, with Alg–RGD and Alg–RGD + Alg–Hep being much stiffer than the rest of the gels.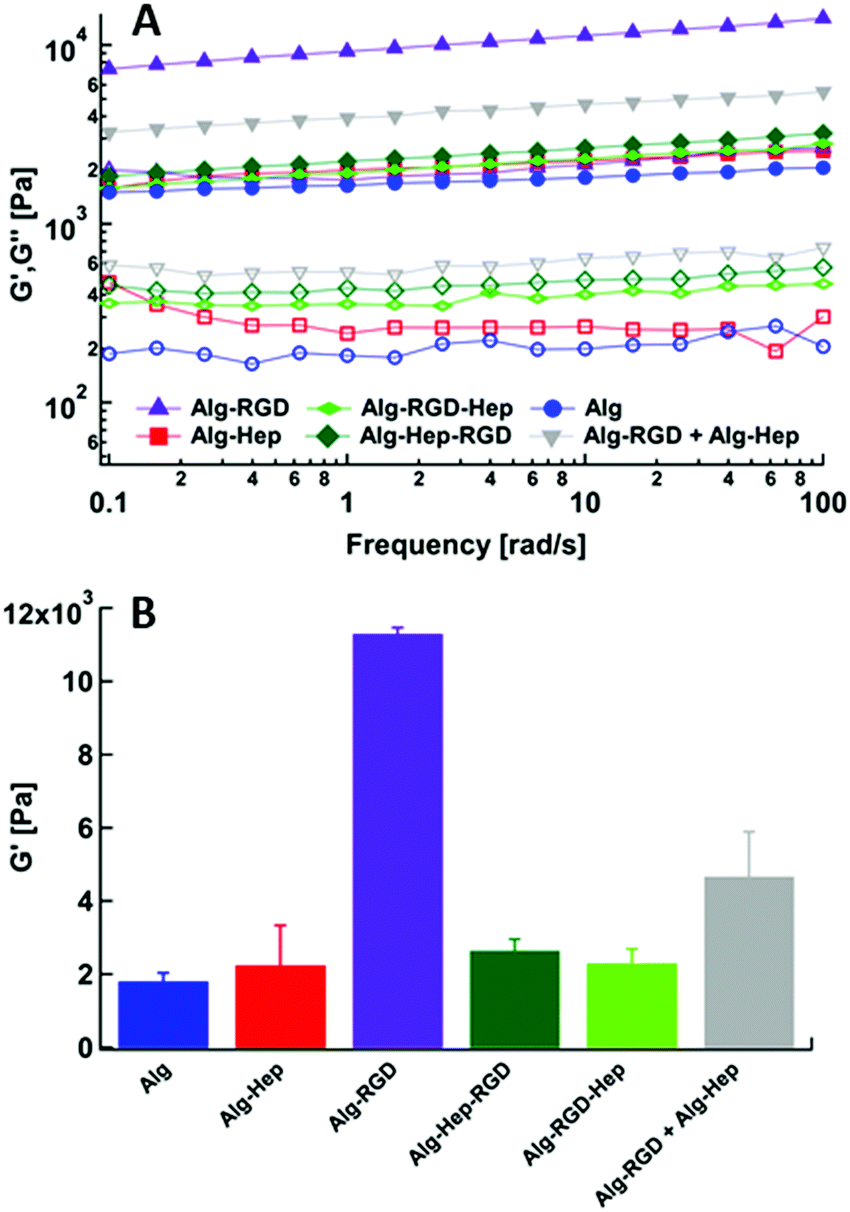 | ||
| Fig. 4 (A) Frequency sweep results of modified alginate gels. G′(filled), G′′(empty). (B) Storage modulus (G′) of modified alginate gels at frequency of 10 rad s−1. | ||
In previous studies of alginate–peptide gels, we attributed the increased stiffness of the gels to the presence of additional junction zones formed due to attractive interactions between the alginate and neighbouring peptides.17,33 The existence of these junction zones was revealed by SAXS, and more precisely by following the methodology of plotting the scattering curves of the alginate–peptide gels in a Kratky form,34 where a peak represents the presence of frozen inhomogeneities in the gel network. To identify whether the cause for the increased stiffness of the Alg–RGD and Alg–RGD + Alg–Hep also stems from the presence of additional/larger junction zones, we performed SAXS measurements and presented the scattering curves of all gels in a Kratky plot (Fig. 5).
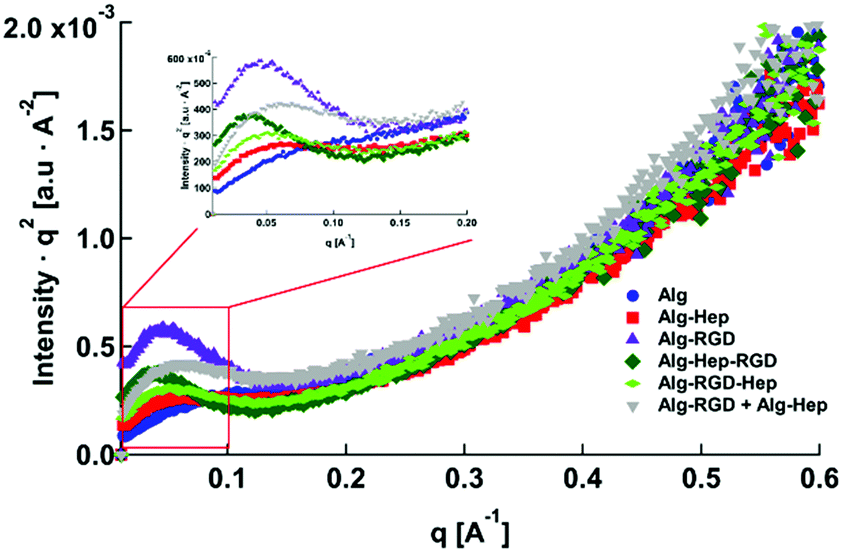 | ||
| Fig. 5 Kratky plot of modified alginate hydrogels. The inset is an elragment of the peaks. | ||
The pronounced peak that is evident in the Alg–RGD gel scattering curve, is evidence of the existence of additional (non-calcium) larger junction zones. The relatively less pronounced peaks of Alg–Hep–RGD, Alg–RGD–Hep and Alg–Hep indicate the existence of some non-calcium junction zones. Yet, the absence of a distinct peak implies that these junction zones are relatively small and evenly distributed.13,35,36 The scattering curve of the Alg–RGD + Alg–Hep gels lies between that of Alg–Hep and that of Alg–RGD. Indeed, it is comparable to a mathematical addition of the scattering curves of Alg–RGD gel and Alg–Hep gel (Fig. S2, ESI†), suggesting that the less pronounced peak of the Alg–RGD + Alg–Hep (compared to Alg–RGD) is not due to the presence of smaller non-calcium junction zones (compared to Alg–RGD), but rather due to an interpenetrating network structure in which the Alg–RGD and Alg–Hep gels coexist without affecting one another.
Not surprisingly, there is a correlation between the largest peak in the Kratky plot and the highest value of a gel's storage modulus. Though the conclusion of additional junction zones is similar to that of our previous study, it should be noted that the nature of these junction zones is different. G4RGDY does not form micelles in water; thus, the junction zones are not similar to those suggested for Alg–V6KRGDY or Alg–A6KRGDY. Moreover, G4RGDY is shorter and stiffer than the G6KRGDY; thus, it does not disrupt the continuity of the G blocks and hence does not prevent the formation of a large calcium junction zone. Here, the additional junction zones are most likely the clusters formed by the attractive interactions (described above) between the RGD peptide and the alginate backbone.
Taken together, our results thus far indicate that the structural features of the modified alginate chains as evident in solutions persist even after the gelation process, producing a correlation between the solution's and the gel's mechanical properties.
Kinetics of vascular endothelial growth factor (VEGF) release
It has been previously shown that structural features and mechanical properties of alginate hydrogels designed as ECM mimetics affect their functionality (i.e., bioactivity).37 The presence of heparin and/or the clusters within the hydrogels was expected to affect their ability to capture and release growth factors such as VEGF, due to its binding affinity to the heparin residues.26,38 Thus, in order to examine the structure–properties–function correlation in our system, we performed a preliminary study of the ability of the heparin containing gels to capture VEGF. VEGF was added to the modified alginate polymer solution and then gelation was induced (see Experimental section). VEGF molecules present in the medium during gelation, may or may not be captured in the gel, depending on the interactions between the VEGF molecules and the modified alginate polymer chains.39 In Fig. 6A it can be seen that Alg–Hep, Alg–RGD–Hep and Alg–RGD + Alg–Hep gels captured the highest amounts of VEGF. These results indicate that the accessibility of the heparin, i.e. having heparin at the “end” of the attached molecule, is essential for the ability of the gel to form interactions with VEGF and as a result, to capture it. The highest amount of VEGF captured by Alg–RGD + Alg–Hep suggests that the presence of the clusters within the gel plays a secondary role in determining the gels’ ability to capture VEGF, probably due to high local density of attractive interactions between the VEGF and alginate.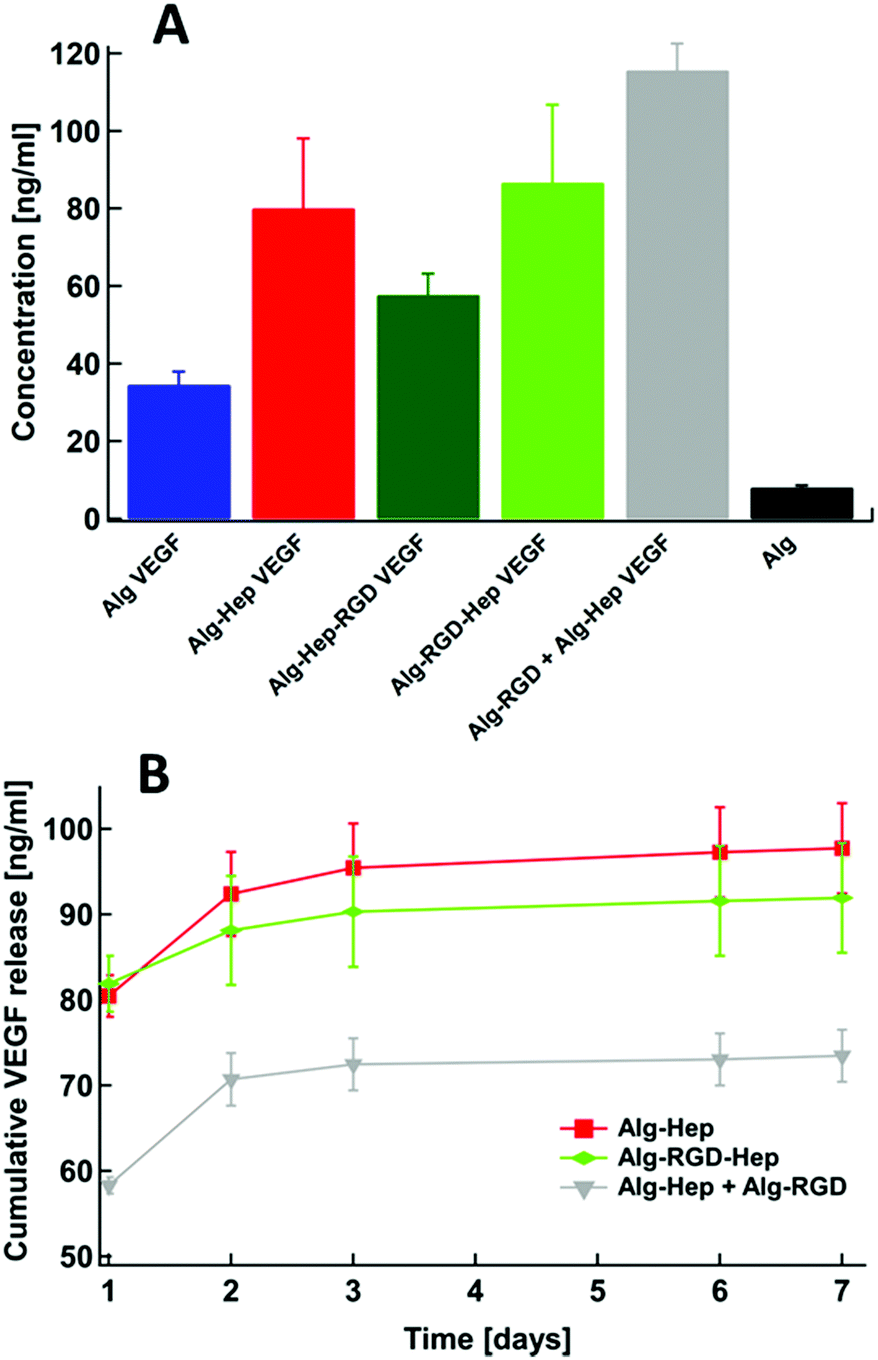 | ||
| Fig. 6 (A) The initial VEGF amount in the hydrogels (t = 0); (B) release profiles of VEGF from the Alg–Hep gel, Alg–RGD–Hep gel, and Alg–RGD + Alg–Hep gel. | ||
Next, we monitored VEGF release from the hydrogels during a period of 7 days (Fig. 6B). We were able to obtain VEGF release profiles only for Alg–Hep, Alg–RGD–Hep, and Alg–RGD + Alg–Hep gels (Fig. 6B) since the Alg and Alg–Hep–RGD hydrogels dissociated within 48 h. A dissociation of ionically crosslinked alginate hydrogels in tissue culture medium is a well-known phenomenon and typically is attributed to the presence of calcium chelators, monovalent ions, and competing non-cross-linking divalent ions present in the external medium.40,41 Thus, the degree of hydrogel dissociation was visually monitored (Table S4, ESI†). Alginate and Alg–Hep–RGD hydrogels, which had the lowest amount of captured VEGF, dissociated after 48 h. The fact that the hydrogels with attached heparin at the “end” of the bound molecule were the most stable implies that the accessibility of heparin to interact with the surrounding molecules is imperative for these hydrogels’ stability in medium.
Due to the dissociation of alginate and Alg–Hep–RGD hydrogels, VEGF release profiles were obtained only for Alg–Hep, Alg–RGD–Hep, and Alg–RGD + Alg–Hep gels (Fig. 6B). In all three gels, most of the encapsulated VEGF was released within 3 days. Comparing the release profiles of Alg–Hep and Alg–RGD–Hep shows that the release of VEGF from Alg–Hep is slightly faster than from Alg–RGD–Hep (Fig. 6B). The extent and rate of dissociation for Alg–Hep and Alg–RGD–Hep seems to be the same (Table S4, ESI†). Although Alg–RGD + Alg–Hep gel dissociates in a more pronounced manner comparing to the other two gels, it demonstrated lower cumulative VEGF release during 7 days. The high amount of VEGF captured in Alg–RGD + Alg–Hep as well as the lower release rate can be attributed to the presence of clusters in this gel. Diffusion of the growth factors through the clusters may be inhibited due to the high polymer density, explaining why even though the gel disintegrates, the release profile is very slow.
Experimental
Materials
Alginate (Protanal HF120RBS 55% guluronate, sodium salt,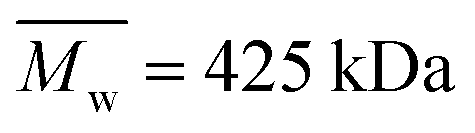 ) was a kind gift from FMC Biopolymers (Dramen, Norway). Heparin (183 USP units mg−1,
) was a kind gift from FMC Biopolymers (Dramen, Norway). Heparin (183 USP units mg−1, 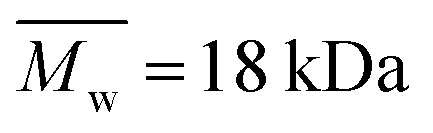 ), ethylenediamine (EDA, technical 75–80%), NaOH, CaCO3, 2-[N-morpholio] ethanesulfonic acid (MES buffer), N-hydroxysuccinimide sodium salt (NHS), (+)-gluconic acid δ-lactone (GDL) and N-(3-dimetylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) were purchased from Sigma-Aldrich. The peptide, G4RGDY (80–90% grade) was purchased from Bio-Sight/American Peptide Company (Vista, CA). Vascular endothelial growth factor (VEGF) and the ABTS Enzyme-Linked Immune-Sorbent Assay (ELISA) Buffer Kit for VEGF were purchased from Peprotech Ltd.
), ethylenediamine (EDA, technical 75–80%), NaOH, CaCO3, 2-[N-morpholio] ethanesulfonic acid (MES buffer), N-hydroxysuccinimide sodium salt (NHS), (+)-gluconic acid δ-lactone (GDL) and N-(3-dimetylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) were purchased from Sigma-Aldrich. The peptide, G4RGDY (80–90% grade) was purchased from Bio-Sight/American Peptide Company (Vista, CA). Vascular endothelial growth factor (VEGF) and the ABTS Enzyme-Linked Immune-Sorbent Assay (ELISA) Buffer Kit for VEGF were purchased from Peprotech Ltd.
Methods
Synthesis of Alg–Hep conjugation of heparin to alginate was performed following the literature42 with some modifications. To minimize hydrolysis of EDC, the reaction was carried out in a 0.1 M buffer of 2-morpholinoethane sulfonic acid (MES buffer). First, heparin sodium salt and sodium alginate (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :500 mg mg−1 heparin:polymer) were dissolved in an MES buffer at a concentration of 1 wt% alginate with overnight stirring at room temperature. Then, the carboxylic groups of alginate and heparin were activated using EDC and NHS (molEDC:molNHS = 0.6:1). After 5 min, 1 mL of ethylenediamine (3.1 × 10−2 M) was added to the solution under vigorous stirring at room temperature. After a reaction period of 4 h, the resulting polymer was dialyzed to remove excess unreacted EDC/NHS and aminated heparin molecules, using a dialysis tubing cellulose membrane (14000 MWCO), against 1 M NaCl for 24 h. The product was then frozen and lyophilized by freeze-drier for 72 h.
:500 mg mg−1 heparin:polymer) were dissolved in an MES buffer at a concentration of 1 wt% alginate with overnight stirring at room temperature. Then, the carboxylic groups of alginate and heparin were activated using EDC and NHS (molEDC:molNHS = 0.6:1). After 5 min, 1 mL of ethylenediamine (3.1 × 10−2 M) was added to the solution under vigorous stirring at room temperature. After a reaction period of 4 h, the resulting polymer was dialyzed to remove excess unreacted EDC/NHS and aminated heparin molecules, using a dialysis tubing cellulose membrane (14000 MWCO), against 1 M NaCl for 24 h. The product was then frozen and lyophilized by freeze-drier for 72 h.
Synthesis of Alg–RGD conjugation of a peptide to the alginate was performed using carbodiimide chemistry, according to Rowley et al.13,43 Alginate was dissolved in the stock MES buffer (0.1 M) to give a 1 wt% aqueous solution followed by stirring for 24 h. NHS was added in an amount that would give a ratio of NHS:EDC of 1:2. The peptide was added after 5 min (1:100 mg mg−1 peptide:polymer), and the solution was then stirred continuously for 24 h. Thereafter, the solution was dialyzed against triply distilled water in 3500 MWCO dialysis tubes for 96 h and then lyophilized for 72 h.
Synthesis of Alg–RGD–Hep was conducted by conjugating heparin to an Alg–RGD molecule rather than to pristine alginate.
Synthesis of Alg–Hep–RGD was conducted by conjugating G4RGDY to an Alg–Hep molecule instead of to pristine alginate.
For analysis, the requisite amount of the dried conjugated polymer was dissolved in triply distilled water (18.2 MΩ cm).
Using carbodiimide chemistry could result in additional undesirable outcomes besides binding of the heparin and the peptide to the alginate backbone. These include: binding of heparin directly to the RGD peptide on the modified alginate chain and vice versa; formation of other derivatives in addition to the desired one (e.g. Alg–RGD, Alg–Hep, Alg–Alg, Hep–Hep, Hep–RGD); crosslinking reactions during modification of alginate with Hep to create Alg–Hep–Alg; and derivatives with non-comparable amounts of each component. However, the amount of those compounds will be considerably lower than the desired product.
:GDL molar ratio was 1:1. The volume of the three solutions (alginate, CaCO3 and GDL) was designed to give a final volume of 0.5 mL. The hydrogels were prepared as discs in a 48 well plate (11 mm diameter). The gelling solutions were allowed to rest in room temperature to 12 hours in humid conditions before Rheology and SAXS measurements were performed, to allow for the completion of the gelation process.44
Measurements were carried out with a Reologica StressTech HR stress-controlled rheometer equipped with an extended temperature cell for temperature control and stainless steel parallel plate (d = 8 mm).
For the determination of initial amount of VEGF at each gel (t = 0), the gels were dissociated by adding 300 μL of Sodium citrate 6% in PBS and 600 μL Dulbecco's modified eagle medium (DMEM) with 0.1% bovine serum albumin (BSA, pH 7.2). The content of each well was transferred to low protein binding (LB) eppendorfs (1.5 mL) and stored at −80 °C until the ELISA was done.
For the release experiments, the modified alginate gels containing VEGF were placed in DMEM with 0.1 wt% BSA, pH 7.2 and incubated on an incubator at 37 °C. The supernatant was collected and replaced by fresh DMEM with 0.1% BSA at scheduled time points and kept frozen at −80 °C until all samples were collected and the amount of VEGF in releasing media was determined by an ELISA. The ELISA was performed following the manufacturer's instructions (Human VEGF165 Standard ABTS ELISA Development Kit, ABTS ELISA Buffer Kit, Peprotech Ltd).
Conclusions
We explored the conjugation manner of alginate modified with both heparin and the G4RGDY peptide. SAXS and rheology measurements showed that large clusters were formed only in the systems in which RGD was accessible to easily interact with a neighbouring alginate backbone (i.e., Alg–RGD and Alg–RGD + Alg–Hep). These large clusters also led to higher viscosity and a more pronounced shear thinning behavior of those solutions, indicating that the order of heparin and peptide conjugation to the alginate backbone plays a significant role in determining the structural–mechanical property relations of these modified alginate solutions.SAXS of the modified alginate gels shows that the large clusters formed in solutions remained intact during the calcium-induced gelation process, leading to gels with a higher storage modulus and slower release rates of the VEGF incorporated within them. Therefore, the mode and order of conjugating different molecules to the alginate backbone should be considered when designing such multicomponent alginate hydrogels. A detailed structural analysis of the conjugated architecture in solution can be used as a tool to adapt the properties of alginate-heparin-peptide hybrid hydrogels.
Conflicts of interest
There are no conflicts to declare.References
- T. Coviello, P. Matricardi, C. Marianecci and F. Alhaique, J. Controlled Release, 2007, 119, 5–24 CrossRef CAS PubMed.
- C. Borselli, C. A. Cezar, D. Shvartsman, H. H. Vandenburgh and D. J. Mooney, Biomaterials, 2011, 32, 8905–8914 CrossRef CAS PubMed.
- I. Freeman and S. Cohen, Biomaterials, 2009, 30, 2122–2131 CrossRef CAS PubMed.
- Y. M. Kolambkar, K. M. Dupont, J. D. Boerckel, N. Huebsch, D. J. Mooney, D. W. Hutmacher and R. E. Guldberg, Biomaterials, 2011, 32, 65–74 CrossRef CAS PubMed.
- R. Dittrich, G. Tomandl, F. Despang, A. Bernhardt, T. Hanke, W. Pompe and M. Gelinsky, J. Am. Ceram. Soc., 2007, 90, 1703–1708 CrossRef CAS.
- L. Shi, L. Xiong, Y. Hu, W. Li, Z. Chen, K. Liu and X. Zhang, Polym. Eng. Sci., 2018, 58, 1782–1790 CrossRef CAS.
- T. Hashimoto, Y. Suzuki, K. Suzuki, T. Nakashima, M. Tanihara and C. Ide, J. Mater. Sci.: Mater. Med., 2005, 16, 503–509 CrossRef CAS PubMed.
- D. Szarek, K. Marycz, P. Bednarz, P. Tabakow, W. Jarmundowicz and J. Laska, Biotechnol. Appl. Biochem., 2013, 60, 547–556 CrossRef CAS PubMed.
- Y. Sapir, O. Kryukov and S. Cohen, Biomaterials, 2011, 32, 1838–1847 CrossRef CAS PubMed.
- K. Uto, J. H. Tsui, C. A. DeForest and D.-H. Kim, Prog. Polym. Sci., 2017, 65, 53–82 CrossRef CAS PubMed.
- T. Andersen, P. Auk-Emblem and M. Dornish, Microarrays, 2015, 4, 133–161 CrossRef CAS PubMed.
- R. Censi, P. Di Martino, T. Vermonden and W. E. Hennink, J. Controlled Release, 2012, 161, 680–692 CrossRef CAS PubMed.
- J. A. Rowley, G. Madlambayan and D. J. Mooney, Biomaterials, 1999, 20, 45–53 CrossRef CAS PubMed.
- O. Jeon, C. Powell, L. D. Solorio, M. D. Krebs and E. Alsberg, J. Controlled Release, 2011, 154, 258–266 CrossRef CAS PubMed.
- O. Jeon, D. S. Alt, S. M. Ahmed and E. Alsberg, Biomaterials, 2012, 33, 3503–3514 CrossRef CAS PubMed.
- B. Trappmann and C. S. Chen, Curr. Opin. Biotechnol, 2013, 24, 948–953 CrossRef CAS PubMed.
- G. Ochbaum and R. Bitton, Polymer, 2017, 108, 87–96 CrossRef CAS.
- O. Bernstein-Levi, G. Ochbaum and R. Bitton, Colloids Surf., B, 2016, 137, 214–220 CrossRef CAS PubMed.
- G. Ochbaum, M. Davidovich-Pinhas and R. Bitton, Soft Matter, 2018, 14, 4364–4373 RSC.
- Y. Liang and K. L. Kiick, Acta Biomater., 2014, 10, 1588–1600 CrossRef CAS PubMed.
- D. L. Rabenstein, Nat. Prod. Rep., 2002, 19, 312–331 RSC.
- T. Nie, R. E. Akins Jr and K. L. Kiick, Acta Biomater., 2009, 5, 865–875 CrossRef CAS PubMed.
- T. Nie, A. Baldwin, N. Yamaguchi and K. L. Kiick, J. Controlled Release, 2007, 122, 287–296 CrossRef CAS PubMed.
- S. Boddohi, N. Moore, P. A. Johnson and M. J. Kipper, Biomacromolecules, 2009, 10, 1402–1409 CrossRef CAS PubMed.
- A. D. Baldwin and K. L. Kiick, J. Pept. Sci., 2010, 94, 128–140 CrossRef CAS PubMed.
- M. M. Adil, T. Vazin, B. Ananthanarayanan, G. M. Rodrigues, A. T. Rao, R. U. Kulkarni, E. W. Miller, S. Kumar and D. V. Schaffer, Biomaterials, 2017, 136, 1–11 CrossRef CAS PubMed.
- A. Tiwari, H. J. Salacinski, G. Punshon, G. Hamilton and A. M. Seifalian, FASEB J., 2002, 16, 791–796 CrossRef CAS PubMed.
- G. N. Liberman, G. Ochbaum, S. M. Arad and R. Bitton, Carbohydr. Polym., 2016, 152, 658–664 CrossRef PubMed.
- R. Tuinier, P. Zoon, M. C. Stuart, G. Fleer and C. De Kruif, Biopolymers, 1999, 50, 641–646 CrossRef CAS PubMed.
- J. F. Steffe, Rheological methods in food process engineering, Freeman press, 1996 Search PubMed.
- F. Horkay, P. J. Basser, A.-M. Hecht and E. Geissler, Phys. Rev. Lett., 2008, 101, 068301 CrossRef PubMed.
- C. Rodríguez-Rivero, L. Hilliou, E. M. M. del Valle and M. A. Galán, Rheol. Acta, 2014, 53, 559–570 CrossRef.
- G. Ochbaum, E. Chetrit, R. Berkovich and R. Bitton, Soft Matter, 2020, 16, 6155–6162 RSC.
- Y. Maki, K. Ito, N. Hosoya, C. Yoneyama, K. Furusawa, T. Yamamoto, T. Dobashi, Y. Sugimoto and K. Wakabayashi, Biomacromolecules, 2011, 12, 2145–2152 CrossRef CAS PubMed.
- E. M. Saffer, M. A. Lackey, D. M. Griffin, S. Kishore, G. N. Tew and S. R. Bhatia, Soft Matter, 2014, 10, 1905–1916 RSC.
- J. Zaragoza, N. Babhadiashar, V. O’Brien, A. Chang, M. Blanco, A. Zabalegui, H. Lee and P. Asuri, PLoS One, 2015, 10(8), e0136293 CrossRef PubMed.
- M. Ahearne, Interface Focus, 2014, 4, 20130038 CrossRef PubMed.
- U. Freudenberg, A. Hermann, P. B. Welzel, K. Stirl, S. C. Schwarz, M. Grimmer, A. Zieris, W. Panyanuwat, S. Zschoche and D. Meinhold, Biomaterials, 2009, 30, 5049–5060 CrossRef CAS PubMed.
- Y. Shmidov, M. Zhou, G. Yosefi, R. Bitton and J. B. Matson, Soft Matter, 2019, 15, 917–925 RSC.
- A. Martinsen, G. Skjåk-Bræk and O. Smidsrød, Biotechnol. Bioeng., 1989, 33, 79–89 CrossRef CAS PubMed.
- M. A. LeRoux, F. Guilak and L. A. Setton, J. Biomed. Mater. Res., 1999, 47, 46–53 CrossRef CAS PubMed.
- Q. Zuo, R. Guo, Q. Liu, A. Hong, Y. Shi, Q. Kong, Y. Huang, L. He and W. Xue, Biomed. Mater., 2015, 10, 035008 CrossRef PubMed.
- M. Daoud, J. Cotton, B. Farnoux, G. Jannink, G. Sarma, H. Benoit, C. Duplessix, C. Picot and P. De Gennes, Macromolecules, 1975, 8, 804–818 CrossRef CAS.
- B. Polyak, S. Geresh and R. S. Marks, Biomacromolecules, 2004, 5, 389–396 CrossRef CAS PubMed.
- S. R. Kline, J. Appl. Crystallogr., 2006, 39, 895–900 CrossRef CAS.
- B. T. Stokke, K. I. Drager, Y. Yuguchi, H. Urakawa and K. Kajiwara, Macromol. Symp., 1997, 120(1), 91–101 CrossRef CAS.
- B. T. Stokke, K. I. Draget, O. Smidsrød, Y. Yuguchi, H. Urakawa and K. Kajiwara, Macromolecules, 2000, 33, 1853–1863 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00669f |
| This journal is © The Royal Society of Chemistry 2021 |