
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemisorption of CO2 on diaminated silica as bicarbonates and different types of carbamate ammonium ion pairs
Anthony E.
Szego
,
Aleksander
Jaworski
and
Niklas
Hedin
 *
*
Department of Materials and Environmental Chemistry, Stockholm University, Stockholm, SE-10691, Sweden. E-mail: niklas.hedin@mmk.su.se
First published on 7th December 2020
Abstract
The chemisorption of CO2 on aminated silica has a rich chemistry, and its details are important to research in relation to CO2 capture and catalytic chemistry. In this study, such chemisorption was investigated on aminated and diaminated silica with 1H and 13C solid state nuclear magnetic resonance (NMR) spectroscopy under dry and wet conditions. Fast magic-angle spinning (MAS) allowed us to obtain high resolution spectra. Porous silica was modified into a monoaminated version using (3-aminopropyl)triethoxysilane (APTS) and a diaminated one by using 3-(2-aminoethylamino)propyltriethoxysilane (AEAPTS). From the corresponding NMR spectra we could conclude that, under dry conditions, CO2 chemisorbed as carbamic acid and carbamate ammonium ion pairs and gave similar spectra for both directly-excited 13C and under cross-polarization (CP) {1H}13C MAS NMR. Under wet conditions, direct excitation and {1H}13C CPMAS NMR showed that carbamate ammonium ion pairs formed along with bicarbonates (HCO3−). For the wet conditions, the 13C and 1H NMR spectra were especially well resolved, and we could detect two different types of carbamate ammonium ion pairs forming on the diaminated silica. We conclude that carbamate ammonium ion pairs and HCO3− moieties can be detected by {1H}13C MAS NMR, at least qualitatively, in addition to the more time consuming direct excitation.
1. Introduction
Pilot plants for the capturing of carbon dioxide (CO2) are based on the use of established scrubbing technology with aminated solvents that allows the capturing and relatively easy release/regeneration of the captured CO2.1,2 There is a major issue with these practices though, which include the high energy demand for regeneration of the solvent, corrosion of equipment, and the evaporation and leaching of the amine or associated degeneration products. Immobilized amines on porous solid supports could possibly reduce the energy required for regeneration, limit the corrosion, and the evaporation of the amines.3–6Mesoporous silica, in particular, has been very popular as a solid to immobilize amine functions and reduce corrosion and leaching.7–10 One of three ways can be used to modify silica with amines: by physical immobilization of compounds on the surface, chemical tethering, or crosslinking of polyamines throughout the pores of the silica. The amine-modified solid adsorbents – similarly to liquid amines – have been shown to exhibit a highly selective uptake of CO2 and a high capacity for CO2 uptake at low partial pressures of the CO2.11–13
Despite the advantages these types of amine-modified sorbents have, full mechanistic understanding of the reaction of the amines with CO2 has yet to be achieved.14–20 It is known from previous works that in the reactions between CO2 and amines in the presence of water, three main species are involved: carbamic acid, carbamate ammonium ion pairs, and bicarbonates (HCO3−). Even though the formation of HCO3− has been shown to enhance the so-called amine efficiency,21–23 the formation of these species could potentially be considered a drawback in the context of CO2 capture due to the expected higher energy required for the regeneration of the material, and overall reduced adsorption and desorption rates, despite the increase in sorption capacity.24–26
The existence of HCO3− in CO2 chemisorption reactions on moist solid amines has been questioned in the past.14,15,23,27–29 In light of findings from the liquid state amine–CO2–H2O chemistries,30–32 it would seem obvious that HCO3− should form within pores of moist amine-modified sorbents. However, it has not been easy to detect HCO3− by solid-state NMR and, also, its IR signatures have been difficult to assign.11 Solid-state NMR is used extensively to characterize chemisorption products in general, because it can determine the structures of non-crystalline systems.33,34 This versatility also allows the method to be used in the identification and quantification of the analyzed products, in situ and ex situ.14,33,35 In NMR, in particular, the 13C chemical shift of HCO3− appears at a range of values, depending on the environment that contributes to the shielding experienced by the 13C nucleus.36 As a result, its NMR resonance may be masked by that of potentially more prominent carbamate or carbamic acid resonances. For this reason, the samples presented in this paper have been divided into “wet” and “dry” samples, as the absence of H2O is known to restrict the formation of HCO3−.37 Carbonates (CO32−) will not be observed in the present paper because conditions that favor its formation (pH > 10.3) are not present.
Despite the aforementioned debate regarding the involvement of HCO3− in these types of systems, previous work done recently by Chen et al.27 was able to find and assign a peak to this species. They could unambiguously show with the so called direct polarization 13C NMR method that HCO3− formed when the samples had been subjected to liquid H2O. The fast dynamics and, in our interpretation, the liquid-like character of the formed HCO3− dissolved in the pores largely filled with H2O, were the reasons why HCO3− was not easily detected at room temperature in that study and gave convincing insight as to why it had not earlier been detected by the more commonly used cross-polarization method, here called as {1H}13C NMR. The absence of the {1H}13C NMR signal for HCO3− in most studies has likely been related to that the details of the cross-polarization dynamics associated to its presumed liquid-state-type character are not fully taken into account, which in turn will hinder an effective magnetization transfer. H2O adsorbs in significant amounts on amine-modified adsorbents.15,22
The work presented here is meant to further contribute to this exploration of the presence of HCO3− in these systems. We show that {1H}13C NMR could be used at least qualitatively to identify HCO3− for samples being rich in co-adsorbed H2O and that two different types of carbamates formed in moist diaminated silica on the chemisorption of CO2.
2. Materials and synthesis
Chromatographic particles of porous silica, Davisil LC60 (Grace Davison), was used as a porous support. It had a particle size of 40–63 μm, and a surface area of 550 m2 g−1. The support was used as received after a drying treatment at a temperature of 110 °C for 16 h. Analytical- grade toluene ([CAS: 108-88-3], Sigma-Aldrich 99.8%) with 0.03 wt% H2O was used. The porous silica was modified with (3-aminopropyl)triethoxysilane (APTS) ([CAS: 919-30-2], Sigma-Aldrich >98%) and 3-(2-aminoethylamino)propyltriethoxysilane (AEAPTS) ([CAS: 5089-72-5], Sigma-Aldrich >96%). Labelled 13CO2 ([CAS: 1111-72-4], Sigma-Aldrich 99 atm% 13C) was used for the NMR experiments.2.1. Modifications with propylamine and a diamine groups
The porous silica was dried at a temperature of 110 °C before synthesis, and the synthesis procedure was adapted from methods previously reported in the group.38 In summary, for each synthesis 3 grams of porous silica and 180 cm3 of toluene were added to a three-necked flask equipped with a Dean–Stark reflux condenser. The solutions were heated to 50 °C under stirring for 30 min; a previously established amount of H2O was added and the mixtures were refluxed for a period of 1 h. After this period had elapsed, the required amount of silane monomer was added (APTS or AEAPTS) and left to reflux for 24 h. Finally, the solid was filtered off, washed with ethanol (50 cm3 × 3) and toluene (50 cm3 × 2) and dried overnight at a temperature of 100 °C. The sample modified with the monoamine monomer is called Silica_APTS, and the one with the diamine monomer Silica_AEAPTS.3. Characterization
3.1. Solid state nuclear magnetic resonance
In order to ensure that all chemisorbed species formed were detected, two protocols for the 13C NMR and one protocol for the 1H NMR were used for each sample. We studied 1H and 13C NMR responses using direct excitation, and {1H}13C NMR using cross-polarization (CP). 99% 13C-enriched CO2 was used for the experimentation. All NMR experiments were performed at the magnetic field strength of 14.1 T (Larmor frequencies of 600.1 and 150.9 MHz for 1H and 13C, respectively) with a Bruker Avance-III spectrometer.
1H MAS NMR experiments were carried out with a 1.3 mm probehead at the MAS rate of 60.00 kHz. Acquisitions involved rotor-synchronized, double-adiabatic spin-echo sequence with a 90° excitation pulse of 1.1 μs, followed by two 50.0 μs tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) h per tan high-power adiabatic pulses with 5 MHz frequency sweep.39,40 256 signal transients with 5 s relaxation delay were collected for each sample. Rotors were filled with powderous Silica_APTS and Silica_AEAPTS and pre-treated under high dynamic vacuum (turbo pump) on the degas port of a Micrometrics ASAP2020 volumetric adsorption analyzer at a temperature of 110 °C. After this degassing, the Silica_APTS and Silica_AEAPTS were divided into “dry” and “wet” samples. The dry samples were subjected to 1 bar of CO2 enriched in 13C, at 25 °C, for a period of 48 hours, and, finally, the rotors were capped in a glovebox under a continuous flow of N2 for these dry samples. For the wet samples, the degassed rotors were conditioned in an enclosed environment, for 16 h, where the air was saturated with H2O after which they were subjected to 1 bar of CO2 enriched in 13C in a fashion similar to that of the dry samples. The rotors were rapidly capped before each measurement within the glovebox.
h per tan high-power adiabatic pulses with 5 MHz frequency sweep.39,40 256 signal transients with 5 s relaxation delay were collected for each sample. Rotors were filled with powderous Silica_APTS and Silica_AEAPTS and pre-treated under high dynamic vacuum (turbo pump) on the degas port of a Micrometrics ASAP2020 volumetric adsorption analyzer at a temperature of 110 °C. After this degassing, the Silica_APTS and Silica_AEAPTS were divided into “dry” and “wet” samples. The dry samples were subjected to 1 bar of CO2 enriched in 13C, at 25 °C, for a period of 48 hours, and, finally, the rotors were capped in a glovebox under a continuous flow of N2 for these dry samples. For the wet samples, the degassed rotors were conditioned in an enclosed environment, for 16 h, where the air was saturated with H2O after which they were subjected to 1 bar of CO2 enriched in 13C in a fashion similar to that of the dry samples. The rotors were rapidly capped before each measurement within the glovebox.
Direct excitation 13C and {1H}13C CPMAS NMR spectra were recorded with a 4 mm probehead and 14.00 kHz MAS rate. 13C MAS NMR spectra involved a 4.0 μs 90° excitation pulse and Spinal64 1H decoupling sequence41 operated at proton nutation power of 65 kHz. 128 signal transients with 300 s relaxation delay were collected. For {1H}13C CPMAS acquisitions Hartmann–Hahn matched radiofrequency fields were applied for a contact interval of 0.5 ms and employed the same proton decoupling scheme. 16384 signal transients with 2 s relaxation delay were collected. Chemical shifts are referenced with respect to neat tetramethylsilane (TMS).
3.2. CO2 adsorption measurements
Adsorption and desorption isotherms of pure CO2 on Silica_APTS and Silica_AEAPTS were measured at a temperature of 0 °C using a Micrometrics ASAP2020 volumetric adsorption analyzer. These were compared to a sample of unmodified silica that had undergone the same toluene treatment without the inclusion of silane monomer. The volumetric uptake of CO2 was measured at CO2 pressures from 0.001 to 1 bar under equilibrium conditions. Thermal control was achieved by immersing the samples in a Dewar flask filled with ∼3 dm3 of H2O and ice, which equilibrated the temperature to 0 °C. For these measurements, the samples were pretreated under high dynamic vacuum conditions at 110 °C for 10 h before experimentation.3.3. Thermogravimetric analysis
Thermogravimetric analysis (TGA) was used to record the mass loss on heating Silica_APTS and Silica_AEAPTS using a TA Instruments Discovery (TA Instruments, Stockholm, Sweden) thermobalance in dry air, for which samples were heated from 50 to 950 °C at a rate of 10 °C min−1 in a platinum cup.4. Results and discussion
4.1. Solid-state NMR
In order to identify the species formed after the chemisorption of CO2 on Silica_APTS and Silica_AEAPTS under “wet” and “dry” conditions, isotopic labelling in 13C was used together with directly-excited 13C, {1H}13C, and 1H MAS NMR spectroscopy. Under dry conditions, carbamate ammonium ion pairs and carbamic acid groups were detected on Silica_APTS and Silica_AEAPTS, and the corresponding directly-excited 13C and {1H}13C MAS NMR spectra in Fig. 1 were similar. The broad resonance at a 13C chemical shift of about 165 ppm was assigned to the carbamate part of carbamate ammonium ion pairs,14,42–44 and the broad resonance at about 160 ppm to what seems to be carbamic acid.14,42,44,45 Under wet conditions, carbamate ammonium ion pairs and HCO3− species were detected in the directly excited 13C and {1H}13C NMR spectra in Fig. 1. The narrow 13C NMR signals at chemical shits of 161.4 and 161.7 ppm were assigned to HCO3− using the assignments of Chen et al.27 supported by assignments in related liquid state systems.46 The relative signal intensities for HCO3− in Fig. 1 were much smaller for the {1H}13C NMR spectra as compared to those observed in the directly excited 13C NMR spectra for both Silica_APTS and Silica_AEAPTS, which is in line with the general findings of the study by Chen et al.27 about a reduced or vanishing cross-polarization efficiency for these HCO3−. We ascribe that we, in contrast to Chen et al., were able to clearly detect the HCO3−, also at room temperature, either to the delicate details in the cross-polarization conditions or that we had equilibrated the samples with gaseous H2O (at 100% relative humidity). Note that the relative 13C NMR signal intensity for the HCO3− in the directly excited 13C NMR spectra was higher for Silica_AEAPTS than for Silica_APTS, which was related to a higher loading of organics in Silica_AEAPTS.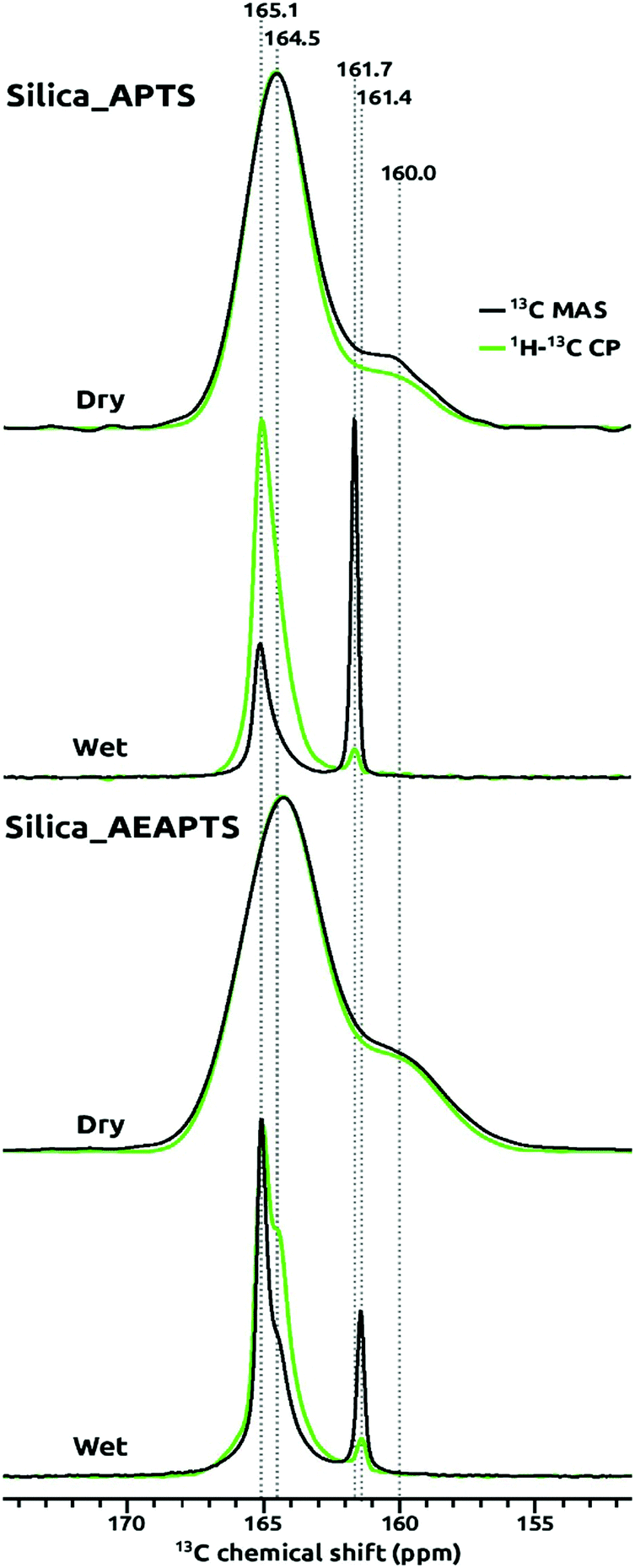 | ||
| Fig. 1 13C solid-state NMR spectra recorded in wet and dry conditions for samples modified with (3-aminopropyl)triethoxysilane (Silica_APTS) and 3-(2-aminoethylamino)propyltriethoxysilane (Silica_AEAPTS). Black lines correspond to direct excitation 13C NMR and the green ones to cross-polarization ({1H}13C) NMR. Labelled 13CO2 gas was used. | ||
Interestingly, signals in the 13C MAS NMR spectra for the chemisorbed moieties of CO2 under wet conditions were much narrower than for those recorded under dry conditions, as can be seen by comparing the line widths for the respective 13C NMR signals in Fig. 1. Our interpretation of the narrower distributions of 13C NMR chemical shifts for the wet samples relates to the fact that the mobility of the chemisorbed CO2 as both carbamates and HCO3− was much higher under the wet conditions as compared to dry conditions. The co-adsorbed H2O appears to solvate the tethered amines and their chemisorbed CO2. For the HCO3−, we even expect that a large fraction was dissolved in the H2O adsorbed on the samples. Amine modified silica adsorbs H2O well already at low relative humidity,15,22 and we had subjected Silica_APTS, and Silica_AEAPTS to 100% relative humidity before the chemisorption of 13CO2 for the wet samples. Under the dry conditions the spectroscopic dispersions of the carbamate moieties on Silica_APTS, and Silica_AEAPTS seem to have been larger and hence the corresponding 13C NMR resonances were broader. It is important to mention that the relatively sharp signals, especially for the HCO3− species, has been scarcely seen, if ever, for these type of materials.
As mentioned, carbamate ammonium ion pairs formed on chemisorption of CO2 under both dry and wet conditions for Silica_APTS and Silica_AEAPTS, in addition to carbamic acid and HCO3− that formed in parallel under dry and wet conditions, respectively. HCO3− moieties are not expected to be observed under dry conditions since H2O plays a role in the H-transfer that results in their formation, compare with Scheme 1(a).
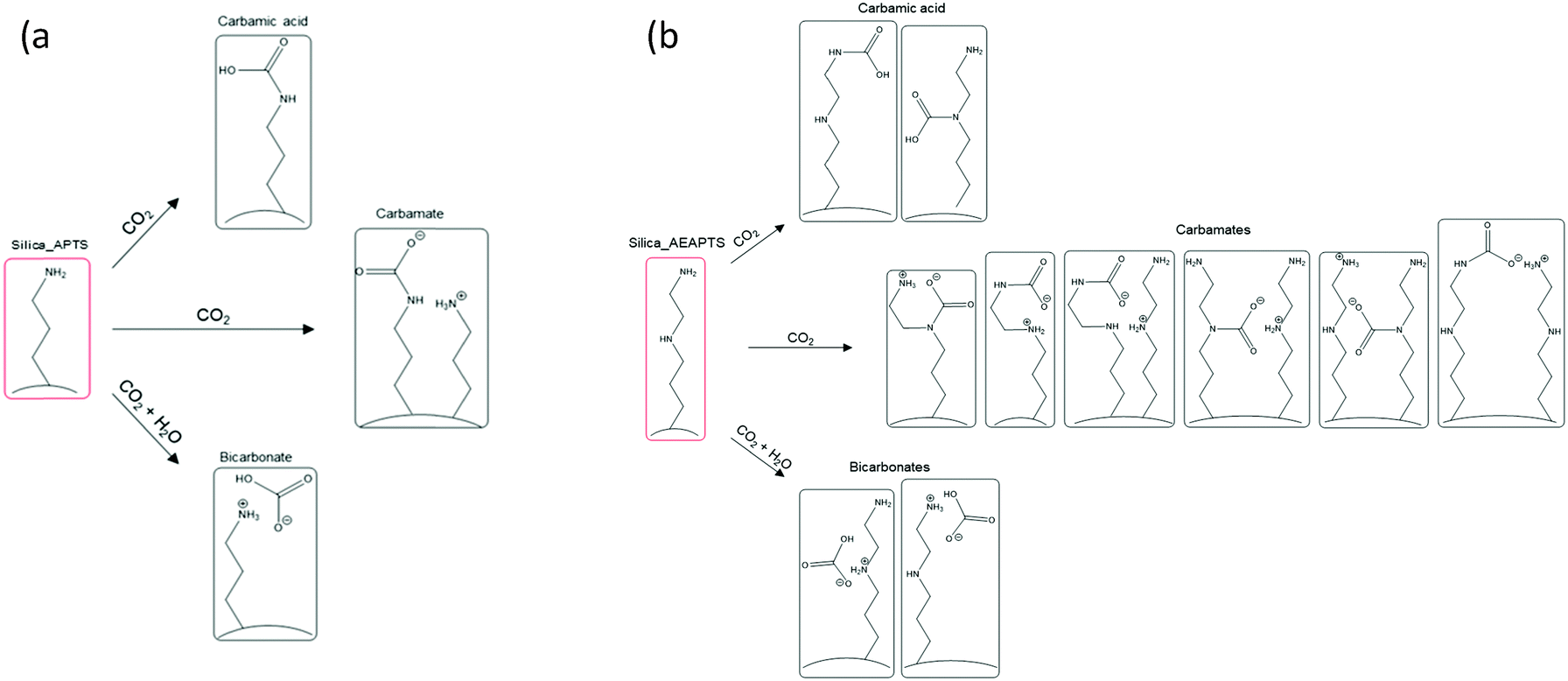 | ||
| Scheme 1 Scheme and structure of possible species formed during CO2 uptake of (a) the monoaminated Silica_APTS and (b) the diaminated Silica_AEAPTS. | ||
Notably, in the Silica_AEAPTS wet sample, the 13C NMR signal at ∼165 ppm ascribed to the carbamate species were split in two, cf.Fig. 1. This shoulder is thought to be due to the possibility of different moieties of carbamates being able to form in this case since the sample contains two amine groups, one being primary and the other being secondary14 (Scheme 1(b)).
Also in the 1H NMR spectra, the NMR resonances were narrower for the wet samples than for the dry ones, as can be observed from the 1H NMR signals in Fig. 2. This narrowing was ascribed to a more liquid-like character for the tethered amines and the chemisorbed CO2 in the wet samples. Co-adsorbed H2O has been shown to liberate the motion of the tethered and crosslinked molecules.47 It is also known that amine-modified silica materials have a higher hydrophilicity than bare silica leading to a situation where the conditions of the wet samples in this study were very close to those of liquid-filled pores, resulting in an enhancement of the mobility of the species.48
 | ||
| Fig. 2 1H NMR spectra in wet and dry conditions for samples modified with 3-aminopropyl)triethoxysilane (Silica_APTS) and 3-(2-aminoethylamino)propyltriethoxysilane (Silica_AEAPTS) after being subjected to CO2. The grey lines correspond to a zooming of the black ones and fast magic-angle spinning of 60.00 kHz was used. | ||
In the 1H NMR region of 0.6–4 ppm in Fig. 2, signals corresponding to the alkyl chains of the condensed moieties on the porous silica were observed. The 1H NMR spectra were similar for the Silica_APTS and Silica_AEAPTS samples due to the similar chemical nature of the condensed or tethered amines, and the resolution, albeit high under wet conditions, did not make it possible to distinguish every 1H NMR signal for each of the different CH2 groups in the Silica_AEAPTS sample. Nonetheless it was still possible to attribute the 1H NMR resonaces at around 0.6 ppm to the –CH2– species directly bonded to silicon atoms. At around 1.5 ppm, the.signals for the β-CH2– groups were observed, and at about 2.6–3.0 ppm the signals for the α-CH2– groups, next to the amine groups, were observed. For Silica_AEAPTS, the 1H NMR signal for the α-CH2– was split in the spectrum for the wet sample. It is noted that the diamine (AEAPTS) had three α-CH2– group, two of which were connected to the secondary amine and one to the primary amine. Hence, the signal at 3.0 ppm was tentatively assigned to the α-CH2– group of the primary amine. At 4.8–5.0 ppm, the 1H NMR resonace for adsorbed H2O was observed for the Silica_APTS and Silica_AEAPTS samples, and the signal intensities were particularly intense for the wet samples. The –NH– and –NH2 groups were, however, not detected directly in the 1H NMR spectra for Silica_APTS nor Silica_AEAPTS, likely due to chemical exchange with H2O or Si–OH groups in both the dry and wet samples.
4.2. CO2 adsorption measurements
Adsorption isotherms of CO2 for pure and amine modified silica are shown in Fig. 3, all of them following the treatment with toluene for comparison. At low pressures (0–0.02 atm) of CO2, the amine modified sorbents showed a higher uptake of CO2 than the unmodified silica did, with the diaminated sample (Silica_AEAPTS) showing the highest level of CO2 adsorption increase in this regime. For both amine modified silica samples, the CO2 isotherms had a much larger gradient at low pressures than for the unmodified silica as shown in Fig. 3. The difference in gradients support established findings49,50 of a higher heat of chemisorption of CO2 on amine modified silica than for physisorption of CO2 on unmodified silica surfaces. On pure silica, CO2 adsorbs by physisorption, which is mainly governed by electrostatic interaction between CO2 and the silica surface.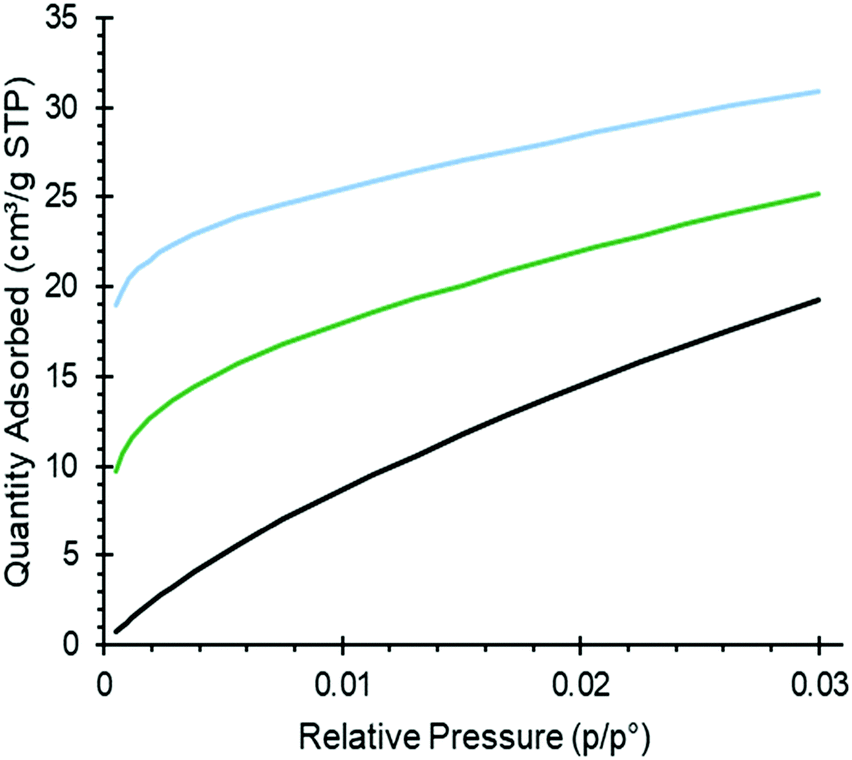 | ||
| Fig. 3 CO2 adsorption measurements at 0 °C. Black: pure silica; green: Silica_APTS; blue: Silica_AEAPTS. | ||
The interaction is caused by the significant quadrupole moment of CO2 molecules and the electrical field variation of the silica surface.51 On amine-modified silica, CO2 chemisorbs, as discussed in sections above, and in the literature,14,15,25,27,46 as carbamate-ion pairs, carbamic acid or alike, or HCO3− depending on the properties of the sorbent and conditions applied. A very high uptake of CO2 at a low pressure of CO2 for Silica_APTS shows a synergistic effect with carbamates forming across different propyl amine groups, which has been shown to occur above a critical amine density for monoamines.49,52,53 The very high CO2 uptake at a low pressure of CO2 for Silica_AEAPTS relates to intramolecular (and possibly intermolecular) carbamates being formed, which has been shown by e.g. Lashaki et al.54,55
4.3. Thermogravimetric analysis
The amount of organic groups in the Silica_APTS and Silica_AEAPTS were established with TGA by analyzing the mass loss recorded as the temperature increased within an appropriate temperature range. The temperature decomposition profiles can be seen in Fig. 4. From this, an estimation for the degree of modification of the amine-modified samples was extracted and presented in Table 1. It is also important to note that the diaminated Silica_AEAPTS presented an increased capacity to chemisorption of CO2 when compared to the monoaminated Silica_APTS, despite having a similar degree of modification molewise (Table 1), and simply ascribed to that each modification in the Silica_AEAPTS contained two reactive amine groups.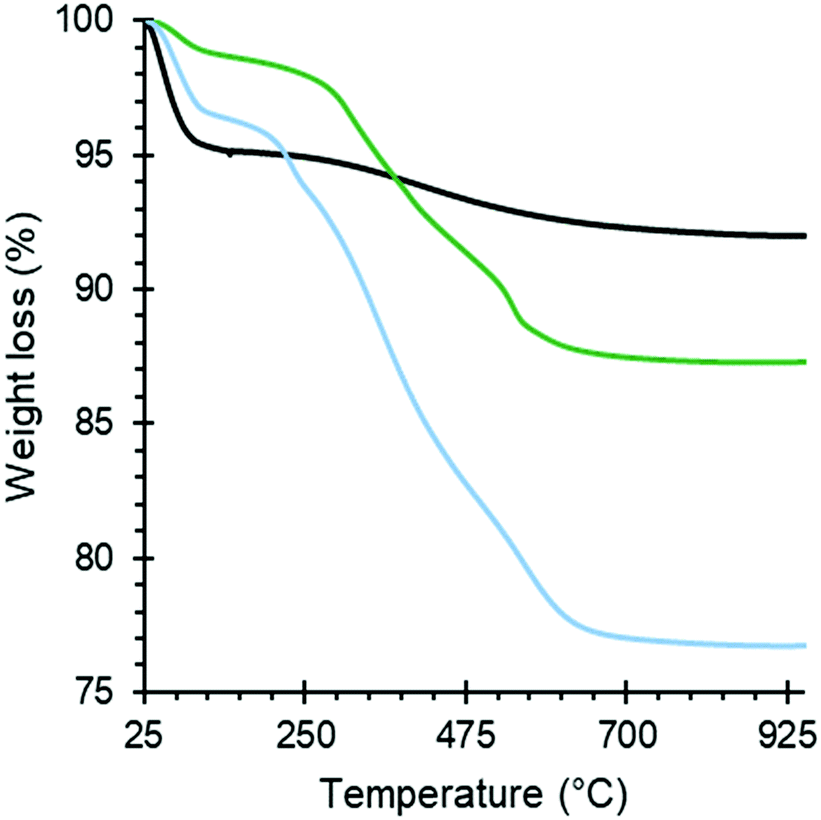 | ||
| Fig. 4 TGA curves in dry air. Black: pure silica; green: Silica_APTS; blue: Silica_AEAPTS. | ||
| Sample | Weight loss due to organics (%) | Monomer concentration (%) | Monomer/SiO2 |
|---|---|---|---|
| Silica_APTS | 11 | 13.1 | 7.5 |
| Silica_AEAPTS | 21 | 16 | 6.5 |
5. Conclusions
Here, mesoporous silica was modified with two different aminosilanes (the monoamine APTS and the diamine AEAPTS) and studied as CO2 sorbents. Systematic solid state NMR experiments were performed to study how CO2 chemisorbed under dry and moist conditions and the viability of using this technique under ultrahigh MAS conditions was tested. High quality spectra for both 13C and 1H NMR were detected especially in the case of the moist samples.For both modified samples, CO2 chemisorbed as carbamic acid and carbamate ammonium ion pairs under dry conditions, and the directly-excited 13C and {1H}13C MAS NMR spectra obtained were significantly similar. When the samples were allowed to equilibrate in a humid environment (wet samples), carbamate ammonium ion pairs were again detected together with significant amounts of HCO3− instead of carbamic acids, by using either direct excitation 13C or {1H}13C NMR spectroscopy. The cross-polarization efficiency to the HCO3− was reduced as compared to the directly detected 13C NMR. Still, with pre-knowledge of this tendency of reduction, {1H}13C NMR could be used to qualitatively establish the presence of HCO3− for these samples without having to cool them down. We predict that {1H}13C NMR can be used for qualitative assessment of HCO3− also without isotopic 13C enrichment for similar samples, which would be convenient.
The presence of H2O in the aminated samples lead to a sharpening of all signals (1H and 13C) under rapid MAS, and the sharpening was ascribed to an enhanced mobility of the species. This sharpening allowed for detection of two different types of carbamates formed on chemisorption of CO2 on the diaminated sample as well as resolving most of the signals from the alkyl chains in the 1H domain. These high resolutions of the 1H and 13C NMR spectra observed could open up for more understanding in the process of how amine-modified silicas capture CO2, can allow a better and more rational application of them, and sharpen their use in other important fields such as in catalysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by the EU-MSCA-ETN-GreenCarbon project 721991. The Swedish Research Council and the grant 2016-03568 is thanked for partial support of this study.References
- D. Aaron and C. Tsouris, Sep. Sci. Technol., 2005, 40, 321–348 CrossRef CAS.
- H. Yang, Z. Xu, M. Fan, R. Gupta, R. B. Slimane, A. E. Bland and I. Wright, J. Environ. Sci., 2008, 20, 14–27 CrossRef CAS.
- S. A. Didas, S. Choi, W. Chaikittisilp and C. W. Jones, Acc. Chem. Res., 2015, 48, 2680–2687 CrossRef CAS.
- E. Andreoli, E. P. Dillon, L. Cullum, L. B. Alemany and A. R. Barron, Sci. Rep., 2014, 4, 1–5 Search PubMed.
- J. F. Van Humbeck, T. M. McDonald, X. Jing, B. M. Wiers, G. Zhu and J. R. Long, J. Am. Chem. Soc., 2014, 136, 2432–2440 CrossRef CAS.
- X. Shi, H. Xiao, H. Azarabadi, J. Song, X. Wu, X. Chen and K. S. Lackner, Angew. Chem., Int. Ed., 2020, 59, 6984–7006 CrossRef CAS.
- G. Zhao, B. Aziz and N. Hedin, Appl. Energy, 2010, 87, 2907–2913 CrossRef CAS.
- A. Goeppert, S. Meth, G. K. S. Prakash and G. A. Olah, Energy Environ. Sci., 2010, 3, 1949–1960 RSC.
- R. W. Flaig, T. M. O. Popp, A. M. Fracaroli, E. A. Kapustin, M. J. Kalmutzki, R. M. Altamimi, F. Fathieh, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2017, 139, 39 CrossRef.
- F. Y. Chang, K. J. Chao, H. H. Cheng and C. S. Tan, Sep. Purif. Technol., 2009, 70, 87–95 CrossRef CAS.
- N. Hedin and Z. Bacsik, Curr. Opin. Green Sustainable Chem., 2019, 16, 13–19 CrossRef.
- S. Zhang, C. Chen and W. S. Ahn, Curr. Opin. Green Sustainable Chem., 2019, 16, 26–32 CrossRef.
- Y. Zhai, H. Jin and S. S. C. Chuang, Chemistry of Silica and Zeolite-Based Materials, Elsevier, 2019, pp. 121–142 Search PubMed.
- J. K. Moore, M. A. Sakwa-Novak, W. Chaikittisilp, A. K. Mehta, M. S. Conradi, C. W. Jones and S. E. Hayes, Environ. Sci. Technol., 2015, 49, 13684–13691 CrossRef CAS.
- Z. Bacsik, N. Ahlsten, A. Ziadi, G. Zhao, A. E. Garcia-Bennett, B. Martín-Matute and N. Hedin, Langmuir, 2011, 27, 11118–11128 CrossRef CAS.
- S.-J. Huang, C.-T. Hung, A. Zheng, J.-S. Lin, C.-F. Yang, Y.-C. Chang, F. Deng and S.-B. Liu, J. Phys. Chem. Lett., 2014, 5, 3 Search PubMed.
- G. S. Foo, J. J. Lee, C.-H. Chen, S. E. Hayes, C. Sievers and C. W. Jones, ChemSusChem, 2017, 10, 266–276 CrossRef CAS.
- A. Danon, P. C. Stair and E. Weitz, J. Phys. Chem. C, 2011, 115, 11540–11549 CrossRef CAS.
- C. Knöfel, C. Martin, V. Hornebecq and P. L. Llewellyn, J. Phys. Chem. C, 2009, 113, 21726–21734 CrossRef.
- N. Hiyoshi, K. Yogo and T. Yashima, Microporous Mesoporous Mater., 2005, 84, 357–365 CrossRef CAS.
- D. J. Fauth, M. L. Gray, H. W. Pennline, H. M. Krutka, S. Sjöström and A. M. Ault, Energy Fuels, 2012, 26, 2483–2496 CrossRef CAS.
- Y. Belmabkhout, R. Serna-Guerrero and A. Sayari, Chem. Eng. Sci., 2010, 65, 3695–3698 CrossRef CAS.
- S. A. Didas, M. A. Sakwa-Novak, G. S. Foo, C. Sievers and C. W. Jones, J. Phys. Chem. Lett., 2014, 5, 4194–4200 CrossRef CAS.
- V. Zelenak, D. Halamova, L. Gaberova, E. Bloch and P. Llewellyn, Microporous Mesoporous Mater., 2008, 116, 358–364 CrossRef CAS.
- A. C. C. Chang, S. S. C. Chuang, M. Gray and Y. Soong, Energy Fuels, 2003, 17, 468–473 CrossRef CAS.
- C.-H. Yu, C.-H. Huang and C.-S. Tan, Aerosol Air Qual. Res., 2012, 12, 745–769 CrossRef CAS.
- C. H. Chen, D. Shimon, J. J. Lee, F. Mentink-Vigier, I. Hung, C. Sievers, C. W. Jones and S. E. Hayes, J. Am. Chem. Soc., 2018, 140, 8648–8651 CrossRef CAS.
- M. W. Hahn, M. Steib, A. Jentys and J. A. Lercher, J. Phys. Chem. C, 2015, 119, 4126–4135 CrossRef CAS.
- X. Wang, V. Schwartz, J. C. Clark, X. Ma, S. H. Overbury, X. Xu and C. Song, J. Phys. Chem. C, 2009, 113, 7260–7268 CrossRef CAS.
- G. Richner and G. Puxty, Ind. Eng. Chem. Res., 2012, 51, 14317–14324 CrossRef CAS.
- R. Zhang, Z. Liang, H. Liu, W. Rongwong, X. Luo, R. Idem and Q. Yang, Ind. Eng. Chem. Res., 2016, 55, 3710–3717 CrossRef CAS.
- X. Yang, R. J. Rees, W. Conway, G. Puxty, Q. Yang and D. A. Winkler, Chem. Rev., 2017, 117, 9524–9593 CrossRef CAS.
- D. Bernin and N. Hedin, Curr. Opin. Colloid Interface Sci., 2018, 33, 53–62 CrossRef CAS.
- C.-H. Chen, D. Shimon, J. J. Lee, S. A. Didas, A. K. Mehta, C. Sievers, C. W. Jones and S. E. Hayes, Environ. Sci. Technol., 2017, 51, 26 Search PubMed.
- P. Rzepka, Z. Bacsik, A. J. Pell, N. Hedin and A. Jaworski, J. Phys. Chem. C, 2019, 123, 21497–21503 CrossRef CAS.
- F. Mani, M. Peruzzini and P. Stoppioni, Green Chem., 2006, 8, 995–1000 RSC.
- R. W. Flaig, T. M. Osborn Popp, A. M. Fracaroli, E. A. Kapustin, M. J. Kalmutzki, R. M. Altamimi, F. Fathieh, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2017, 139, 12125–12128 CrossRef CAS.
- B. Aziz, G. Zhao and N. Hedin, Langmuir, 2011, 27, 3822–3834 CrossRef CAS.
- T. L. Hwang, P. C. M. Van Zijl and M. Garwood, J. Magn. Reson., 1998, 133, 200–203 CrossRef CAS.
- G. Kervern, G. Pintacuda and L. Emsley, Chem. Phys. Lett., 2007, 435, 157–162 CrossRef CAS.
- B. M. Fung, A. K. Khitrin and K. Ermolaev, J. Magn. Reson., 2000, 142, 97–101 CrossRef CAS.
- T. Čendak, L. Sequeira, M. Sardo, A. Valente, M. L. Pinto and L. Mafra, Chem. – Eur. J., 2018, 24, 10136–10145 CrossRef.
- C. Te Hung, C. F. Yang, J. S. Lin, S. J. Huang, Y. C. Chang and S. Bin Liu, Microporous Mesoporous Mater., 2017, 238, 2–13 CrossRef.
- M. L. Pinto, L. Mafra, J. M. Guil, J. Pires and J. Rocha, Chem. Mater., 2011, 23, 1387–1395 CrossRef CAS.
- L. Mafra, T. Čendak, S. Schneider, P. V. Wiper, J. Pires, J. R. B. Gomes and M. L. Pinto, J. Am. Chem. Soc., 2017, 139, 389–408 CrossRef CAS.
- P. V. Kortunov, M. Siskin, L. S. Baugh and D. C. Calabro, Energy Fuels, 2015, 29, 5919–5939 CrossRef CAS.
- L. Phillips, F. Separovic, B. A. Cornell, J. A. Barden and C. G. dos Remedios, Eur. Biophys. J., 1991, 19, 147–155 CrossRef CAS.
- F. Rezaei, R. P. Lively, Y. Labreche, G. Chen, Y. Fan, W. J. Koros and C. W. Jones, ACS Appl. Mater. Interfaces, 2013, 5, 3921–3931 CrossRef CAS.
- Z. Bacsik, R. Atluri, A. E. Garcia-Bennett and N. Hedin, Langmuir, 2010, 26, 10013–10024 CrossRef CAS.
- M. W. Hahn, J. Jelic, E. Berger, K. Reuter, A. Jentys and J. A. Lercher, J. Phys. Chem. B, 2016, 120, 1988–1995 CrossRef CAS.
- R. Roque-Malherbe, R. Polanco-Estrella and F. Marquez-Linares, J. Phys. Chem. C, 2010, 114, 17773–17787 CrossRef CAS.
- P. D. Young and J. M. Notestein, ChemSusChem, 2011, 4, 1671–1678 CrossRef CAS.
- B. Aziz, N. Hedin and Z. Bacsik, Microporous Mesoporous Mater., 2012, 159, 42–49 CrossRef CAS.
- M. Jahandar Lashaki, S. Khiavi and A. Sayari, Chem. Soc. Rev., 2019, 48, 3320–3405 RSC.
- M. A. Alkhabbaz, P. Bollini, G. S. Foo, C. Sievers and C. W. Jones, J. Am. Chem. Soc., 2014, 136, 13170–13173 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |