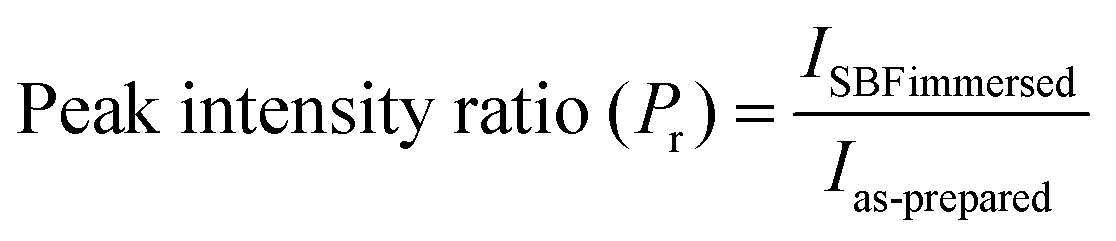DOI:
10.1039/D0MA00628A
(Paper)
Mater. Adv., 2021,
2, 413-425
Variation of the properties of sol–gel synthesized bioactive glass 45S5 in organic and inorganic acid catalysts
Received
21st August 2020
, Accepted 20th November 2020
First published on 21st November 2020
Abstract
In this study, bioactive glass (BG) 45S5 powder with a quaternary composition of 45 wt% SiO2, 24.5 wt% CaO, 24.5 wt% Na2O, and 6 wt% P2O5 was synthesized via a sol–gel route using 1 M nitric acid (HNO3), 1 M formic acid (HCOOH), and 1 M acetic acid (CH3COOH) as catalytic media for facilitating hydrolysis and polycondensation reactions. The prepared sols turned into gels within 2–6 days; they were subsequently dried at 70 °C for 120 min and then heated at 900 °C for 5 h for BG formation to occur. The gels were then crushed manually using a mortar and pestle followed by pulverization to obtain the final powder structure. The resulting powders were then characterized by thermogravimetric analysis (TGA), surface area measurements (BET), Raman spectroscopy, X-ray diffraction (XRD), and scanning electron microscopy in coalescence with energy dispersive spectroscopy (SEM-EDS). The in vitro bioactivity of the prepared BG powder was studied by immersion in simulated body fluid (SBF) to check for the formation of a hydroxyapatite layer. SEM-EDS and Raman spectroscopy were performed both before and after the immersion of BG in SBF to validate the formation of the surface hydroxyapatite layer. The crystallinity, porosity and in vitro bioactivity of the BG samples were then compared to check the effects of variation of the catalyst on the properties of the synthesized BG through the sol–gel route. Particle sizes varied between 1 and 10 μm, with a non-uniform morphology. The EDS results suggest that BG prepared with AA as a catalyst exhibits the highest Ca/P ratio of 1.76 when immersed in the SBF, indicating the formation of the apatite layer. TGA studies revealed that mass loss occurs at a higher temperature for the BG prepared with AA and FA as a catalyst. The BET results suggest that the porosities of all the prepared BG samples fall within the mesoporous range of 2–50 nm, with samples prepared with the AA catalyst displaying the highest specific surface areas (SSA).
1. Introduction
Since the conception of BG in 1969 by Hench, it has been an area of great interest and intensive research in the gamut of bone-tissue engineering applications.1,2 BG provides an alternative for integrating bone tissues, implant coatings, and adhesion between soft and hard tissues within the body compared to previous classes of orthopedic biomaterials.2,3 Bioactive glasses have been at the forefront of bio-ceramics used for orthopedic and bone-tissue engineering applications because of their superior bioactivity in terms of both in vivo and in vitro responses. The first BG discovered by Hench et al. was BG 45S5, a quaternary system with a composition of 45 wt% SiO2, 24.5 wt% CaO, 24.5 wt% Na2O and 6 wt% P2O5; this system exhibited class A bioactivity.4 Since then, numerous other compositions have emerged which display similar levels of bioactivity, such as 50S BG (50 wt% SiO2, 24.5 wt% Na2O, 25 wt% CaO),5 60S BG (60 wt% SiO2, 36 wt% CaO, 4 wt% P2O5),6 and 76S BG (76 wt% SiO2, 23 wt% CaO, 1 wt% P2O5).7 The bioactivity of these silica-based BGs is directly related to their dissolution rate; upon implantation within the body, the BG reacts with body fluid and gradually dissolves by an ion release mechanism, subsequently forming a carbonated hydroxyapatite (HCA) layer on the surface through a series of reactions.1,2,8 The dissolution steps have been proposed by Hench et al. and further explained by Hill et al.9 and De Aza et al.10 At first, leaching takes place, in which Na+ ions are exchanged for H+ ions and silanol (Si-OH) groups are formed. In this cation exchange procedure, the concentration of H+ ions increases at the BG/solution interface; subsequently, the pH rises to 10.5. This pH rise contributes to the breaking of Si–O–Si bonds in the BG structure, and additional silanol groups are formed. Further, soluble silica is lost in the form of Si(OH)4, which passes into the solution. In the next stage, repolymerization is carried out through the condensation of neighboring Si–OH groups, and a silica-rich layer is again formed. Ca2+ and PO43− ions migrate towards the surface, and an amorphous CaO–P2O5 film is formed on top of the silica-rich layer. Hydroxyl and carbonate ions from the solution aid the crystallization of the amorphous CaO–P2O5 film, and the HCA layer is formed. After the HCA layer is formed, it undergoes further bonding with biological moieties and orients to incorporate collagen fibrils.9,10 In the quaternary composition of Na2O–CaO–P2O5–SiO2-based BG, the molar percentage of SiO2 is critical. It has been established that up to ∼53% silica-enriched BG can bond to both soft and hard tissues. The formation of a HCA layer under exposure to biological fluid implies the reactivity of the BG particles and lessens the need for any surface nano-texturing of the particles. The higher specific surface area of the BG promotes the apatite-forming kinetics.11 In this regard, the sol–gel synthesis route is favored to achieve higher porosity, which is reflected in the higher specific surface area over the conventional melt-derived approach.11,12 As mentioned, the dissolution rate of BG in physiological fluid depends on the specific surface area of the BG powder and its inherent porosity.1,4 BG exhibits anti-bacterial and anti-inflammatory responses. The ions from the dissolution of BG aid the regeneration of soft tissues. The dissolute also aids the angiogenesis process. BG can also activate gene expression.13 Bahniuk et al.14 established that the porous structure obtained through the sol–gel route contributes to the protein adsorption of BG particles. Regardless of crystallinity, the sol–gel-derived powder adsorbed three to five times more protein than its melt-derived counterpart.14 Based on the empirical relationship described by Anderson et al. for predicting the response of a glass (RN) in a rabbit tibia model, it was proposed that BG with an RN value greater than 5 is biocompatible and can tightly bond with the implants. BG 45S5 exhibits an RN value of 6.10,15 Similarly, the concept of network connectivity is correlated with surface reactivity, and all reactive bioactive glasses have network connectivity values below 2. BG 45S5 reflects a network connectivity of 1.90. The extrapolation of this result is that the silicate structural units have low molar mass, which can dissolve easily without breaking the Si–O–Si bonds; this explanation is in correlation with Hench's degradation mechanism for BG.9
The conventional method of synthesizing BG is through a melt process involving mixing and melting of metal oxides and carbonation at high temperatures in the range of 1000–1400 °C.2,16,17 Li et al. demonstrated in 1991 that the sol–gel method can provide a better and more feasible alternative for BG synthesis; it involves mixing of metal salts and metalorganic precursors in an acidic or basic catalytic medium, followed by gelation via hydrolysis and polycondensation reactions.16–18 The formed gel is then thermally treated in the temperature range of 600–1000 °C to obtain the final BG form.1–3 In the formation of BG 45S5, alkoxide precursors undergo hydrolysis and partial condensation under the catalytic media to form the sol; later, the sol undergoes polycondensation reactions for gelling to occur (formation of metal–oxo–metal bonds), and the network structure is formed. In BG 45S5, the xerogel is formed through Si–O (NBO) bonds. Na2O and P2O5 precursors take a longer time for the completion of the hydrolysis and polycondensation reactions. In the ageing process, further condensation proceeds, and in the drying stage, densification of the xerogels occurs with the elimination of solvent. Finally, calcination occurs at a higher temperature for phase stabilization.19,20 However, the sol–gel mechanism for silica-based glasses is not inferred entirely, as the hydrolysis and condensation process begins almost simultaneously.21 The sol–gel technique provides a more viable method for BG synthesis, as it involves lower processing temperatures,1–3 higher purity and greater scope for compositional variation,17 more homogeneous compositions of particles,22 higher bioactivity due to the greater specific surface area and porous structure, and greater degradability compared to melt-derived BG.16,22,23 The apatite formation rate is also faster for sol–gel synthesized BG than for melt-derived BG.24 In general, porous structures are preferred for tissue repair engineering applications, as they facilitate a greater amount of nutrient flow and proper cellular adhesion.22 Significant research has been carried out to study the effects of the porous structures and pore sizes of BG on its bioactivity.16,22,25,26
Tetraethyl orthosilicate (TEOS) hydrolyses and condenses to form colloidal nanoparticles that later, under acidic conditions, form an inorganic silica network. The BG formed in the sol–gel process consists of coalesced nanoparticles, and the interstices of the particles provide added porosity over the melt-derived BGs. Triethylphosphate (TEP) and diammonium hydrogen phosphate are commonly used as phosphorous precursor chemicals in the sol–gel route.1 Nitrate salts of sodium and calcium are widely used for the sol–gel route due to their high solubility, low cost, and thermal decomposition ability. Heat treatment above 600 °C eliminates the toxic byproducts of nitrates. Nitric acid is traditionally used as the catalytic medium for the hydrolysis reactions. Heat treatment above 800 °C promotes crystallization, and in this process, the bioactive Na2Ca2Si3O9 (combite) phase is formed.24 The effect of Na2O in BG 45S5 has been further discussed in several articles.17,27 However, in a recent study, it was reported that complete elimination of nitrate was only possible at 850 °C.20 Several reports utilized organic acids such as lactic acid, citric acid, and acetic acid instead of strong inorganic acids to further enhance the efficacy of the sol–gel methodology.4,24,28,29 Lei et al. reported that acetic acid-catalyzed BG has faster apatite forming rates and superior specific surface areas.30
Initially, the sol–gel synthesis route for BG did not gain much attention due to its conversion to glass-ceramics during the heat treatment procedure. Sodium salt, such as sodium nitrate, has a decomposition temperature above 600 °C, which overlaps with the crystallization temperature of the formed glass. Further, sodium reacts with the nitric acid and forms additional sodium nitrate. Sodium also causes the formation of a crystalline combite phase around 600 °C. Hence, Na2O is intended to act as a network modifier and facilitate the formation of the crystalline HCA layer; however, it causes devitrification of glass to glass-ceramic during the stabilization of glass through the heat treatment procedure. Pirayesh et al.31 hypothesized that the crystalline structure facilitates sintering and other high-temperature processing of BG particles. They proposed that the same concentration of BG powder synthesized in the sol–gel route and a conventional melt-derived route will show similar dissolution in in vitro tests. The effects of crystallinity will be negated by the high specific surface area and superior surface texture of the sol–gel synthesized route, which is a more important factor in governing the dissolution rate of BG.31 Recently, Spirandeli et al. proposed that the non-bridging oxygen (NBO) bonds after calcination contribute to the bioactivity of the material.20
This study focuses mainly on determining the effects of the catalytic medium used for the sol–gel method on the structure and porosity of the final BG 45S5 powder, which are directly related to the bioactivity of the BG powder. Three different catalytic media were used for the poly-condensation of the BG sol; namely, 50 ml of 1 M nitric acid (HNO3) solution was taken for the inorganic medium, and for the organic medium, 50 ml of 1 M formic acid (FA) (HCOOH) and 50 ml of 1 M acetic acid (AA) (CH3COOH) were respectively taken. After gel formation from the sols, the gels were subjected to TGA studies to determine the transformations effected by temperature. The remaining gels were aged at 70 °C for 120 min and then thermally treated at 900 °C for 5 h of dwell time to complete the transformation into BG. The formed powders were first manually crushed and then pulverized to obtain a final powder structure with a finer size and were thereafter characterized to determine the average particle size distribution, chemical composition, pore size distribution and specific surface area (SSA), the nature of the chemical bonding, the major crystalline phases developed and the in vitro bioactivity of the powders in simulated body fluid (SBF) solution. The optimal samples were selected based on the results of these characterizations, and a study of the effects of the catalytic media on the BG was carried out.
2. Materials and methods
2.1. Materials
Sol–gel synthesis of BG was carried out using tetraethyl orthosilicate (TEOS) (purchased from Merck, Germany, >99% assay), triethyl phosphate (TEP) (purchased from Spectrochem, India, >98% pure), NaNO3 (purchased from Merck, India, 99.0 assay) and Ca(NO3)2·4H2O (purchased from Merck, India, 99.0 assay). The catalytic acid mediums used for accelerating the polycondensation reaction of the sols to occur were 69% HNO3 (purchased from Merck, India), 99% glacial acetic acid (AA) (M.W. ∼60.05 g mol−1, purchased from Merck, India) and formic acid (FA) (purchased from SRL Chemicals, India, 98% extra pure).
2.2. Sol–gel synthesis of BG powders
Sol–gel synthesis of the BG 45S5 powders was carried out in both inorganic and organic acid catalytic mediums. For the inorganic catalytic medium, 50 ml of 1 M HNO3 (0 pH) solution was taken, and for the organic medium, 50 ml of 1 M FA (1.88 pH) and 1 M AA (2.38 pH) were taken, respectively. The acidic mediums were placed inside separate glass beakers and maintained on a magnetic stirrer system. The precursors for the sol–gel synthesis of BG, namely tetraethyl orthosilicate (TEOS Si(OC2H5)4), triethyl phosphate (TEP (C2H5)3PO4), sodium nitrate (NaNO3), and calcium nitrate tetrahydrate (Ca(NO3)2·4H2O), were added at regular intervals of 30 min to the solution system under continuous stirring. The molar ratio of the (acid + water) system to the precursor chemicals used was maintained at 10 for all three systems. After mixing all the reactants, the solutions were maintained in a vacuum desiccator for drying at ambient temperature and kept undisturbed until gel formation occurred naturally. For the 1 M HNO3 media sol, the gel formation occurred within 3 days of maintenance in the desiccator. In the 1 M FA and 1 M AA sols, the gel formation time was longer and took 6 days. A similar result was observed by Arenas et al., in which they established that acetic acid reduces the gelation rates of BG sols.32 This can be attributed to the fact that organic acids are less potent than inorganic acids and thus take a long time to facilitate the hydrolysis and polycondensation reactions. An ageing time is necessary for the formation of a stable gel-like network. Further, the ageing time reduces water content in the gel phase and reduces the kinetic barrier for crystallization to occur. Zheng et al. further correlated the relationship between ageing time and bioactivity.33 Hence, a moderate ageing time was selected to balance between crystallization and bioactivity of the samples.
After complete gel formation, the gels were placed in a silica crucible and dried at 70 °C for 120 min, then heat-treated at 900 °C for 5 h (heating rate: 20 °C min−1) for BG formation to occur; they were then maintained for 5 h in a furnace (cooling rate: 20 °C min−1) to cool the BG, then subsequently removed and cooled at ambient temperature for 1 h. Although a range of temperatures was used for heat treatment of sol–gel synthesized BG between 600–1000 °C,1–3 the reasoning behind selecting the heat treatment temperature of 900 °C and a longer holding time of 5 h is that this ensures the complete elimination of cytotoxic nitrate salt residues from the synthesized BG along with complete crystallization with the formation of phases of pseudo-wollastonite and wollastonite, as shown by Catauro et al. in their work.34 The decomposition of sodium nitrate starts above 650 °C.33 Other advantages of high temperature sintering (>800 °C) include the formation of highly interconnected porous microstructures due to the formation of gaseous by-products and lower dissolution rates in aqueous solutions, as demonstrated by Cacciotti et al.2 The formed BG samples were then crushed manually using a mortar and pestle for 30 min and then pulverized in a Fritsch P0 pulverisette for a cumulative time of 1 h for each sample in a stainless steel vial containing a single tungsten carbide ball to obtain finer and more uniformly sized powder particles (a detailed breakdown is provided in Table 1).
Table 1 Breakdown of the milling operation time
| Sample |
Milling time (min) |
Rest time (min) |
Milling time (min) |
Rest time (min) |
Milling time (min) |
Rest time (min) |
Milling time (min) |
Rest time (min) |
Milling time (min) |
| A |
15 |
30 |
15 |
30 |
10 |
15 |
10 |
15 |
10 |
| B |
15 |
30 |
15 |
30 |
10 |
15 |
10 |
15 |
10 |
| C |
15 |
30 |
15 |
30 |
10 |
15 |
10 |
15 |
10 |
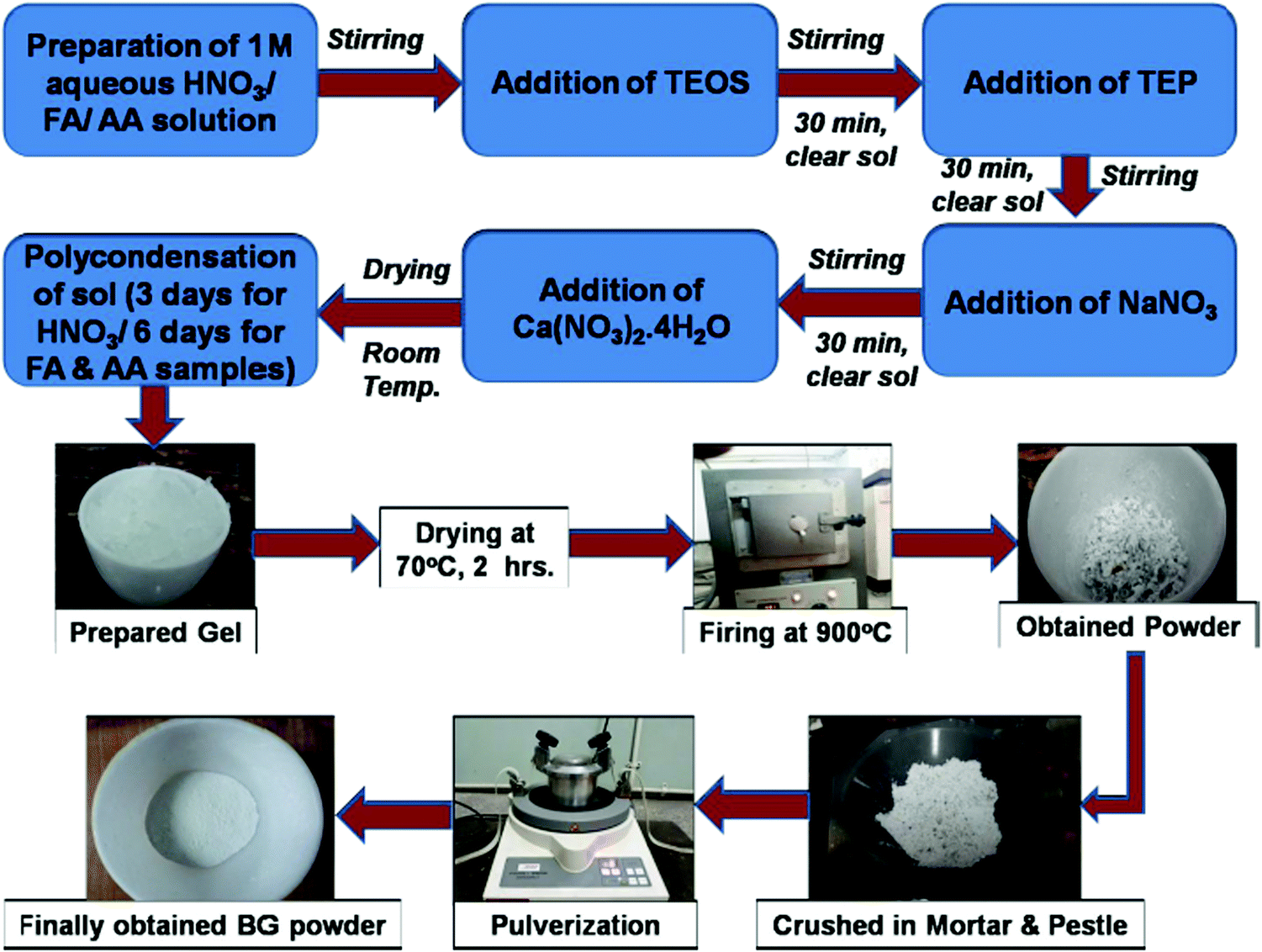
The BG powders prepared in 1 M HNO3, 1 M FA, and 1 M AA catalytic media were named samples A, B, and C, respectively. The step-by-step process of the synthesis of BG powder through the sol–gel route is shown in Fig. 1. These powder samples were then subjected to various characterizations, as described further in the study.
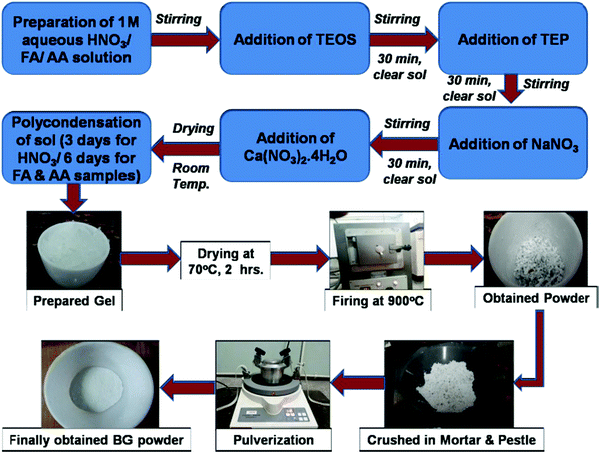 |
| | Fig. 1 Step-by-step process of the synthesis of BG powder through the sol–gel route. | |
2.3.
In vitro bioactivity tests
To carry out the in vitro bioactivity tests on the prepared BG powders, the SBF solution method described by Kokubu et al. was employed.35 The SBF was prepared by dissolving reagent grade NaCl, KCl, NaHCO3, MgCl2·6H2O, CaCl2, Na2SO4, and KH2PO4·3H2O in deionized water buffered at pH 7.4 with trishydroxymethyl aminomethane (Tris) and 1 N HCl at 37 °C. Table 2 shows the composition of this solution in comparison with that of human blood plasma. For the bioactivity test of the powders, 1 gm of powder samples were taken from each sample (A, B, and C, respectively) and were pelletized into 10 mm diameter pellets in an isostatic press under a pressure of 50 MPa for 5 min without the addition of any agglutinant.17 No further sintering was performed, as the powders seemed to have formed dense and mechanically stable pellet structures. The pellets were then immersed in the SBF solution and stored in polyethylene terephthalate (PET) sealed bottles at a constant temperature of 37 °C for 240 h (10 days). The ratio of the mass (m) of the pellets to the volume of SBF (Vs) was maintained as ∼0.01 g ml−1 according to the protocols used by Kokubo et al. in their work.4,35 This ratio was kept the same for all the pellets, as the same powder mass (m) was taken for all the samples. After SBF immersion, the pellets were found to be stable, with no substantial dissolution of the structure; they were washed for 10 s with acetone and dried in an open atmosphere for 15 min before characterization.
Table 2 Compositions of human blood plasma and SBF35
| Ion |
Blood plasma (mmol L−1) |
SBF (mmol L−1) |
| Na+ |
142.0 |
142.0 |
| K+ |
5.0 |
5.0 |
| Mg2+ |
1.5 |
1.5 |
| Ca2+ |
2.5 |
2.5 |
| Cl− |
148.8 |
103.0 |
| HCO3− |
4.2 |
27.0 |
| HPO42− |
1.0 |
1.0 |
| SO42− |
0.5 |
0.5 |
2.4. Characterization of the prepared BG powders
2.4.1. Thermal gravimetric analysis (TGA).
Mass loss during heating of the gel samples due to loss of water molecules and other aqueous compounds was quantified by employing TGA. The TGA was performed in a NETZSCH TG 209F1 Libra instrument on the BG gels, which were heated on alumina crucibles in N2 atmosphere with a heating rate of 10 °C per minute up to a maximum of 600 °C.
2.4.2. Scanning electron microscope and energy dispersive spectroscopy (SEM-EDS).
To analyze the morphology and chemical composition of the synthesized BG powders, a field emission gun scanning electron microscopy (FEG-SEM) (JSM 7800F, Japan) equipped with energy dispersive spectroscopy (EDS) was used. The samples were coated with gold before the examination. SEM was employed for microstructure analysis and particle size estimation of the synthesized powders, and EDS was used to evaluate their chemical compositions both before and after immersion in SBF.
2.4.3. BET analysis to determine the specific surface areas and pore size distributions.
The specific surface areas (SSAs) and pore size distributions of the prepared BG powders were analyzed using nitrogen adsorption–desorption isotherm data obtained at 77 K in a Micrometrics ASAP 2020 physisorption analyzer. In this method, the BG powder samples were first degassed under vacuum conditions (10−5 torr) at 523 K for 3 h to remove surface-adsorbed species and moisture from the powders. The SSA was determined by the adsorption isotherm of nitrogen in a relative pressure range of 0.05 < P/P0 < 0.03, where P is the partial pressure of nitrogen and P0 is the saturation pressure at the experimental temperature. The pore size distributions and pore volumes were determined by employing the Barrett–Joyner–Halenda (BJH) desorption method using the desorption isotherms.
2.4.4. X-ray diffraction (XRD).
The X-ray diffraction (XRD) patterns were recorded using a Rigaku Ultima IV X-ray diffractometer, Japan, using a powder diffraction method with a Cu source operating with Cu-Kα radiation (λ = 0.15418 nm) and a voltage of 40 kV. The diffraction patterns were obtained in the 2θ range of 20° to 60° in continuous mode at 2° min−1 using a Ni-Kβ filter.
2.4.5. Raman spectroscopy.
The Raman spectra were collected using a confocal Raman microscope (WITec Alpha 300R-confocal Raman imaging, Germany) with a charged coupled device (CCD) camera and equipped with a 600 lines per mm monochromator, utilizing a 532 nm laser source with 10 mW incident power on the sample surface. Images and spectra were transferred using a 50× Zeiss objective lens with a 0.65 numerical aperture, and the spectra were finally collected using a fiber-coupled ultra-high throughput spectrometer (UHTS). The Raman spectra were collected from the samples both before and after immersion in SBF.
3. Results and discussion
The results obtained from the different characterization experiments conducted are given below.
3.1. TGA studies
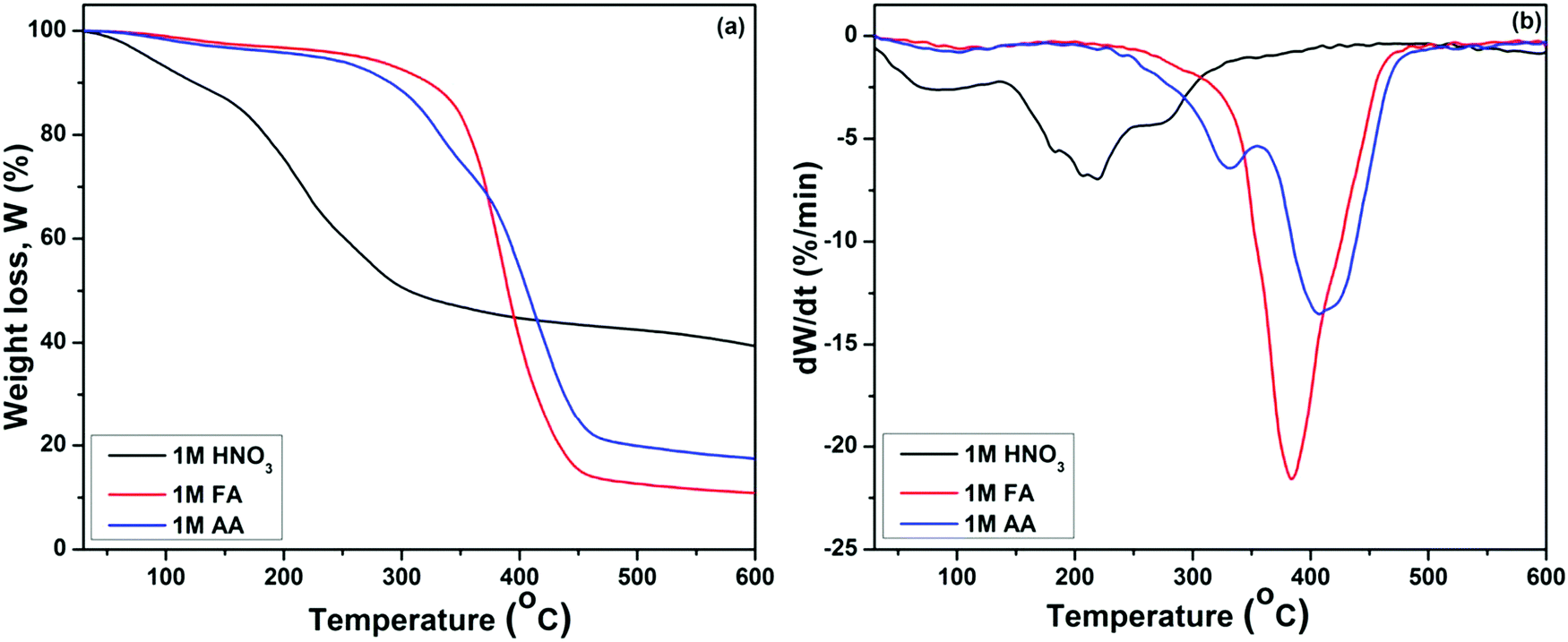
The TGA and DTGA graphs reflect three distinct stages of mass loss for all three samples, as shown in Fig. 2(a and b). The initial phase of mass loss in the regime up to 140 °C corresponds to the removal of physically adsorbed free water molecules and –OH groups. The second stage of mass loss is associated with removal of the chemically adsorbed water, and the third loss corresponds to the thermal decomposition of organic and nitrate precursors, condensation of silanols, and initiation of the crystallization in the samples.36–38 It was observed that the mass loss occurs at a higher temperature for BG samples prepared in FA and AA catalytic media than for the BG prepared in HNO3 catalytic media. The elimination of (CH3COO−) ion takes place around 420 °C, as observed in the BG samples prepared in AA as a catalyst,29 as shown in Fig. 2(a). Similarly, the maximum mass loss occurred in the BG sample prepared in the FA catalyst in that temperature range, corresponding to the removal of formate ions.
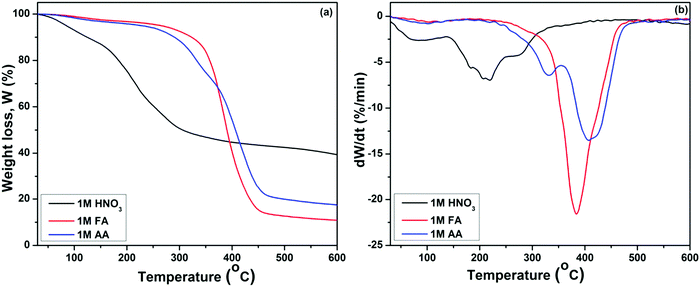 |
| | Fig. 2 (a) TGA and (b) DTGA graphs of BG gels prepared in different catalytic media. | |
3.2. Powder morphology and chemical composition
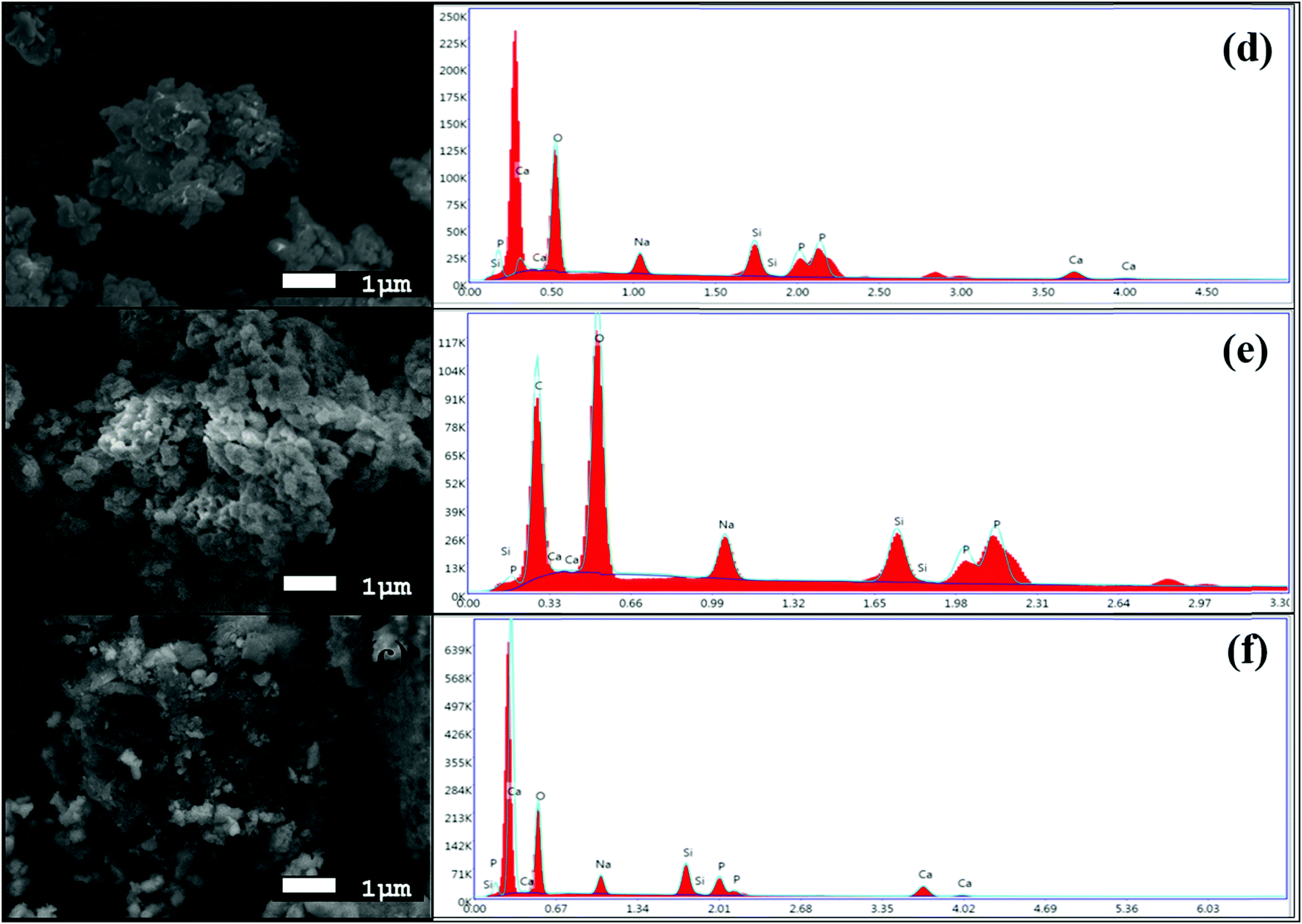
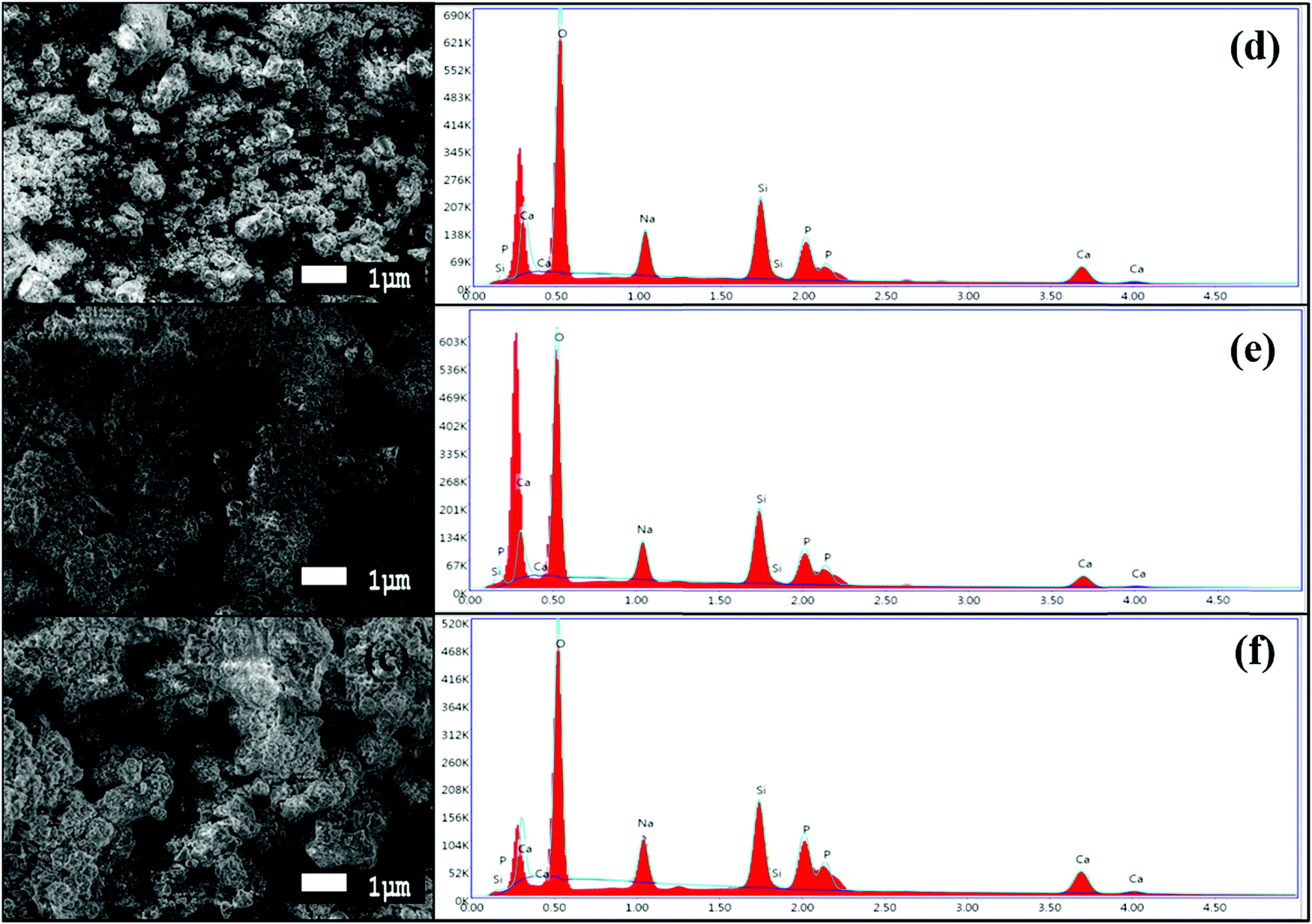
The morphologies of the synthesized BG powders are shown in Fig. 3(a–c), as obtained from SEM investigations at 5000× magnification. From the illustrations, we can observe that the particle size is non-uniform, has a rough texture, and has an irregular shape in each of the samples, namely A, B, and C. The BG powders also exhibit variations in size, mostly between 1 μm and 10 μm, although particles as large as 20 μm can be observed in sample C. This variation in size is due to the non-uniform pulverization of the powders. Also, from the overview of the particles, we can observe that there is significant agglomeration of the particles. This agglomeration of particles can be attributed to the finer sizes of the particles in conjunction with the effects of high energy milling under dry conditions.16,39 The chemical compositions of the BG samples were analyzed using EDS. Fig. 4(a–c) show the 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000× magnification images of focused granular particles of different BG samples and their respective chemical compositions obtained through EDS in Fig. 4(d–f). We can validate silicon, calcium, phosphorous, sodium, and oxygen within the samples from their corresponding peaks from the EDS spectra of the synthesized BG powder samples. This confirms the quaternary composition of the powders without the presence of any contamination. Thus, the purity of BG powders obtained from all three catalytic media can be confirmed through EDS analysis. Further, Lei et al. demonstrated weak acetic acid results in inhomogeneous hydrolysis of BG, which ultimately forms irregularly shaped structures.40 The elemental compositions of BG 45S5 powders are presented in Table 3. The theoretical values of the compositions were obtained from a study conducted by Faure et al.4 Variations of the elemental atomic percentages from the theoretical calculations were observed in the EDS analysis. This is probably because Bioglass 45S5 is a combination of both glass and glass-ceramic crystallites, and the bioactivity depends on both phases. As mentioned already, based on the literature, Na2O contributes to the formation of the Na2Ca2Si3O9 crystallite phase; thus, depletion of the Na2O phase occurs. Similarly, the formation of other crystalline phases may have created variability of the results from the calculated theoretical values. Literature suggests that full crystallization converts bioactive glasses to inert material, and this full conversion of the crystallite phase from the glass phase occurs during the extensive densification stage.26 Thus, in this study, we utilized green pellets (without sintering) for the SBF study to avoid additional crystallinity. 1 M AA best resembles the theoretical values.
000× magnification images of focused granular particles of different BG samples and their respective chemical compositions obtained through EDS in Fig. 4(d–f). We can validate silicon, calcium, phosphorous, sodium, and oxygen within the samples from their corresponding peaks from the EDS spectra of the synthesized BG powder samples. This confirms the quaternary composition of the powders without the presence of any contamination. Thus, the purity of BG powders obtained from all three catalytic media can be confirmed through EDS analysis. Further, Lei et al. demonstrated weak acetic acid results in inhomogeneous hydrolysis of BG, which ultimately forms irregularly shaped structures.40 The elemental compositions of BG 45S5 powders are presented in Table 3. The theoretical values of the compositions were obtained from a study conducted by Faure et al.4 Variations of the elemental atomic percentages from the theoretical calculations were observed in the EDS analysis. This is probably because Bioglass 45S5 is a combination of both glass and glass-ceramic crystallites, and the bioactivity depends on both phases. As mentioned already, based on the literature, Na2O contributes to the formation of the Na2Ca2Si3O9 crystallite phase; thus, depletion of the Na2O phase occurs. Similarly, the formation of other crystalline phases may have created variability of the results from the calculated theoretical values. Literature suggests that full crystallization converts bioactive glasses to inert material, and this full conversion of the crystallite phase from the glass phase occurs during the extensive densification stage.26 Thus, in this study, we utilized green pellets (without sintering) for the SBF study to avoid additional crystallinity. 1 M AA best resembles the theoretical values.
 |
| | Fig. 3 SEM images at 5000× magnification of BG prepared in catalytic media of (a) 1 M HNO3, (b) 1 M FA and (c) 1 M AA. | |
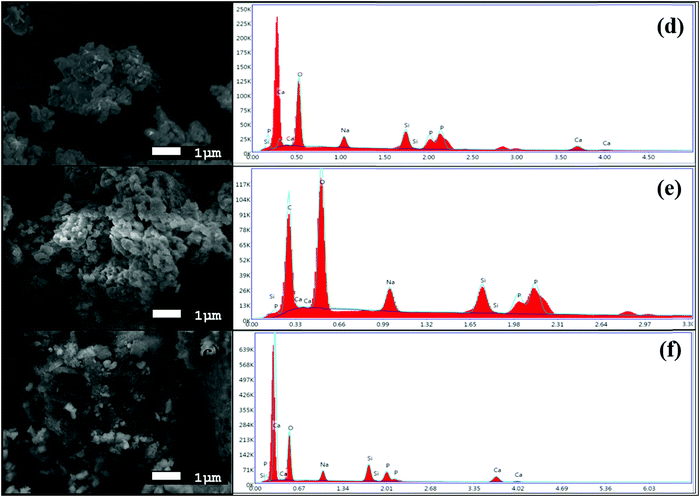 |
| | Fig. 4 High magnification SEM images at 5000× magnification of BG prepared in catalytic media of (a) 1 M HNO3, (b) 1 M FA, and (c) 1 M AA and EDS spectra of the BG powders prepared in catalytic media of (d) 1 M HNO3, (e) 1 M FA, and (f) 1 M AA. | |
Table 3 Elemental compositions of the BG 45S5 powders (average of 2 results). The theoretical values were obtained from ref. 4
| Sample |
O (at%) |
Na (at%) |
Si (at%) |
Ca (at%) |
P (at%) |
| Theoretical |
55.3
|
17.1
|
16.3
|
9.5
|
1.8
|
| A |
66.6 |
7.4 |
7.1 |
14.3 |
4.6 |
| B |
52.14 |
9.2 |
12.6 |
9.4 |
7.7 |
| C |
53.4
|
10.3
|
16.3
|
10.7
|
9.3
|
In contact with body fluid, bioactive glass first dissolves to form a silica gel layer that later transforms into amorphous calcium phosphate. The mechanism of this conversion remains the same for both transformations (glass and crystallite phase). However, the conversion of crystallite phase to amorphous calcium phosphate phase takes place slowly compared to conversion of glass phase to amorphous calcium phosphate phase under SBF immersion. In this context, a high surface area in the porous network is actually beneficial in expediting the kinetics required for transformation of crystal Na2Ca2Si3O9 phase to amorphous calcium phosphate phase at a much faster rate. The bioactivity of Na2Ca2Si3O9 phase has already been established in the literature.26
3.3. Porosity
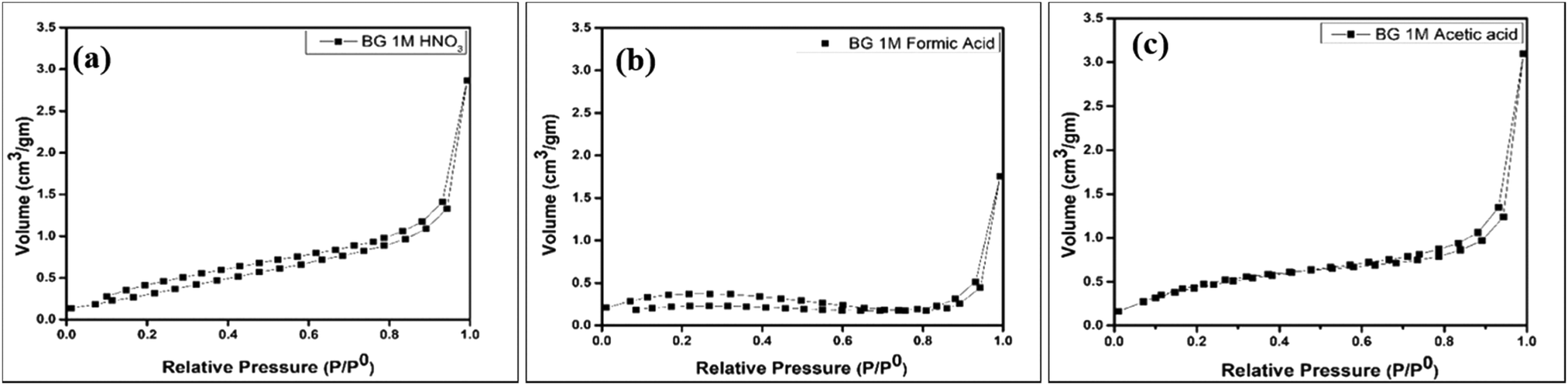
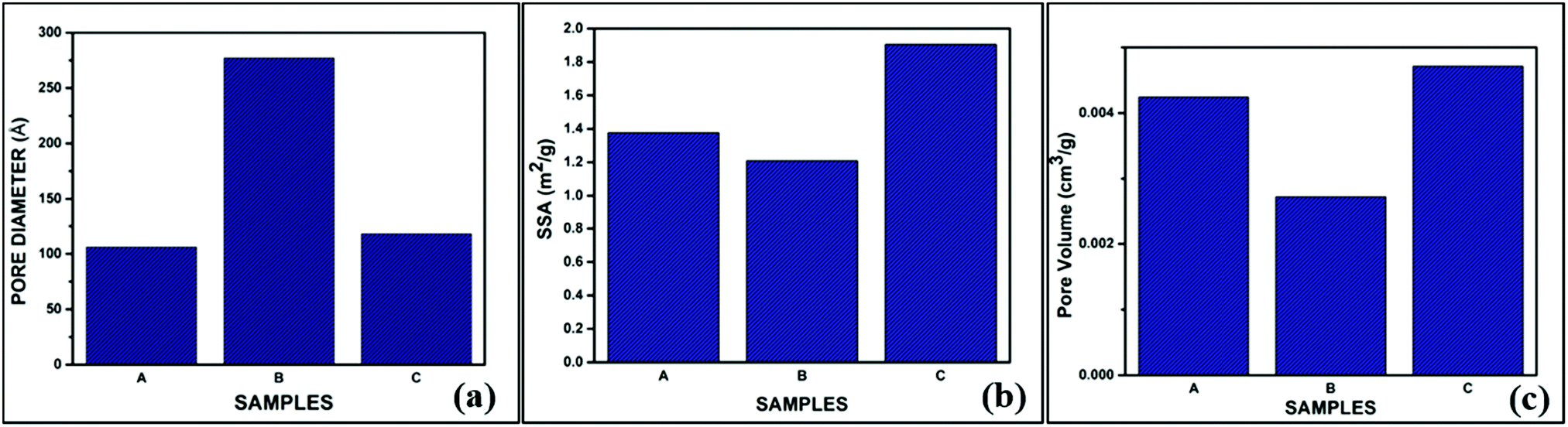
The BET isotherms of the powder samples A, B and C are shown in Fig. 5(a–c). The absorption isotherms were evaluated according to the absorption modes explained by Brunauer and IUPAC.41 From the adsorption isotherms, we can observe that all three sol–gel derived BG powders exhibited type II sigmoidal curves with low levels of adsorption and ultimately reached saturation levels, indicating monolayer coverage formation. This is indicative of a structure with minimal porosity.16,25 The pore diameters, specific surface areas (SSA), and pore volumes of the three BG samples obtained by the BET method are shown in Table 4.
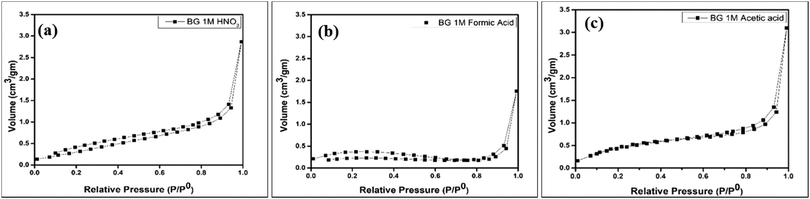 |
| | Fig. 5 Nitrogen adsorption–desorption isotherms for BG prepared in in (a) 1 M HNO3 catalytic media, (b) 1 M FA catalytic media and (c) 1 M AA catalytic media. | |
Table 4 Pore diameters, SSA and pore volumes obtained from the BET desorption curves for BG powder samples A, B and C
| Samples |
Pore diameter (Å) |
SSA (m2 gm−1) |
Pore volume (cm3 gm−1) |
| A |
106 |
1.3755 |
0.004244 |
| B |
277 |
1.2081 |
0.002723 |
| C |
118 |
1.9047 |
0.004711 |
Table 4 shows that the SSA obtained from the BET data is highest for BG sample C at 1.9047 m2 gm−1. Arenas et al. observed similar results, in which AA as the catalyst increased the surface area of BG particles synthesized in a sol–gel route.32 Amongst the samples under consideration, the pore diameter or size is highest for sample B, and the pore volume is highest for sample C. No apparent relation between pore diameter, pore volume, and SSA could be established with these data. It is a well-established fact that mild acids such as acetic acid can act as mesopore-forming agents in addition to their role as a catalyst in the hydrolysis reaction. Further, weak organic acids prevent the growth of particles. Hydrogen bonding interactions also take place among the inorganic precursors, which inhibits the cross condensation of silicates.40 The higher SSA for sample C (AA catalyst) is obviously the more desirable feature, as the bioactivity of the BG will depend on the higher measurement of the surface area due to the extensive porous structure.39 The pore sizes of all three samples fall within the mesoporous range, i.e., 2–50 nm.16,25,39 The pore size distribution, SSA, and pore volume variation of the BG samples A, B, and C are shown in graphical format in Fig. 6(a–c).
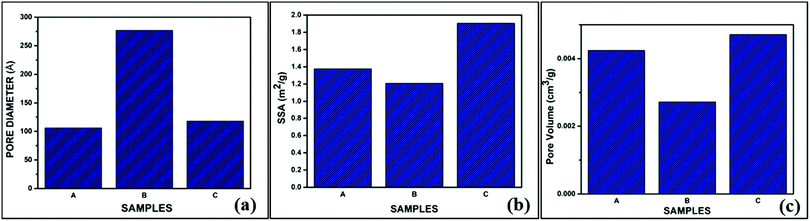 |
| | Fig. 6 Variations of the (a) pore size distributions, (b) pore volumes and (c) specific surface areas (SSAs) of the prepared BG samples A, B and C. | |
3.4. X-ray diffraction analysis
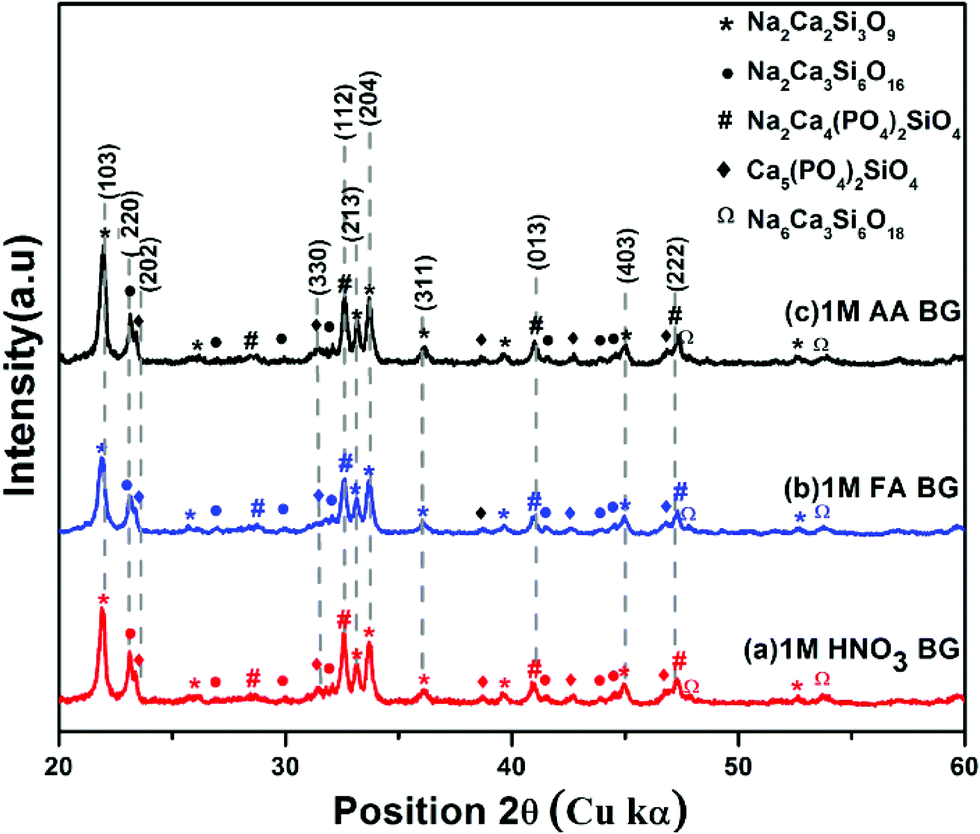
The XRD spectra were analyzed to identify the peaks obtained from X-Ray diffraction studies of samples A, B, and C of the sol–gel synthesized BG powders prepared in the aforementioned catalytic media. The XRD spectra of samples A, B, and C are shown in Fig. 7. All the samples exhibited distinct crystalline natures with minute traces of amorphous peaks. The dominant peaks identified in all the samples correspond to JCPDS ID 22-1455, which is that of Na2Ca2Si3O9 (combite), a form of highly bioactive sodium calcium silicate, usually found in BG powders at higher temperatures.2,4,17,18 Na2Ca2Si3O9 was identified as the major crystalline phase, as reported earlier by Cacciotti et al.2 Another sodium calcium silicate, namely Na2Ca3Si6O16 (devitrite), corresponding to JCPDS ID 23-0671 was also identified in the XRD spectra of the samples as a secondary crystalline phase.17,18,42 Along with these dominant peaks, other peaks of sodium calcium phosphate silicates were observed, such as JCPDS ID 32-1503 corresponding to β-Na2Ca4(PO4)2SiO4 and JCPDS ID 40-0393 corresponding to Ca5(PO4)2SiO4. Moreover, low intensity peaks of JCPDS ID 77-2189 were observed, which tally with the characteristic XRD peaks of Na2CaSi2O9, a variety of sodium calcium silicate which can be observed in a few varieties of BG powders.2 The different crystalline stoichiometric compounds identified through XRD support the elemental analysis through EDS. Additionally, the crystallite sizes (Dg) of the prepared powders were analyzed using the Scherrer formula,43–46Dg = Kλ/(βcosθ), where K is the dimensionless shape factor (∼0.94 for the Cu source), λ is the X-ray wavelength (for Cu kα 1, λ = 1.54060 Å) and β is the line broadening at half the maximum intensity (FWHM) in radians. The grain sizes in samples A and B vary within the range of 11–30 nm. In sample C, the pore sizes vary drastically, with grain sizes ranging from 10–45 nm. The larger size of the grains correlates with the BET data, which depicted higher pore volumes in sample C.
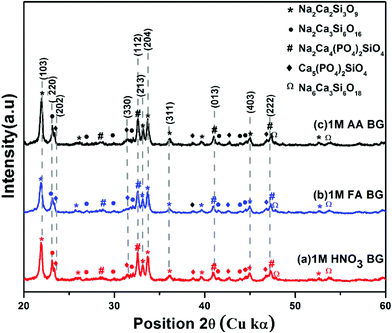 |
| | Fig. 7 X-Ray diffraction patterns of BG prepared in catalytic media of (a) 1 M HNO3, (b) 1 M FA, and (c) 1 M AA. | |
Further, to determine which of the samples can potentially have higher bioactivity based on the presence of bioactive Na2Ca2Si3O9 (combite) phase, the relative intensity of the phases was calculated by dividing the absolute intensity (y-axis count) of the most intense peak of Na2Ca2Si3O9 (combite phase–JCPDS. 21-1455) by the absolute peak intensity of Na2Ca3Si6O16 (devitrite phase–JCPDS. 23-0671) with the consideration that the absolute intensity of the most dominant peak of Na2Ca2Si3O9 (combite phase–JCPDS. 21-1455) is 100%. The intensities were calculated using Origin Pro 9.0 software and compared for each of the three BG samples (A, B, and C, respectively) (eqn (1)).
| | 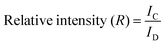 | (1) |
where
IC is the absolute peak intensity of the most intense peak of combite phase and
ID is the absolute peak intensity of the most intense peak of devitrite phase. A comparison of the relative intensities is provided in
Table 5 up to two decimal places, and it can be concluded that sample A and sample C both have the highest relative intensity of combite to devitrite and hence the highest potential to exhibit bioactive natures.
Table 5 Relative intensities of the Na2Ca2Si3O9 (combite–JCPDS 21-1455) and Na2Ca3Si6O16 (devitrite–JCPDS 23-0671) phases of BG powder samples A, B and C
| Samples |
Phase ratio of combite & devitrite |
| A |
0.75 |
| B |
0.69 |
| C |
0.75 |
3.5.
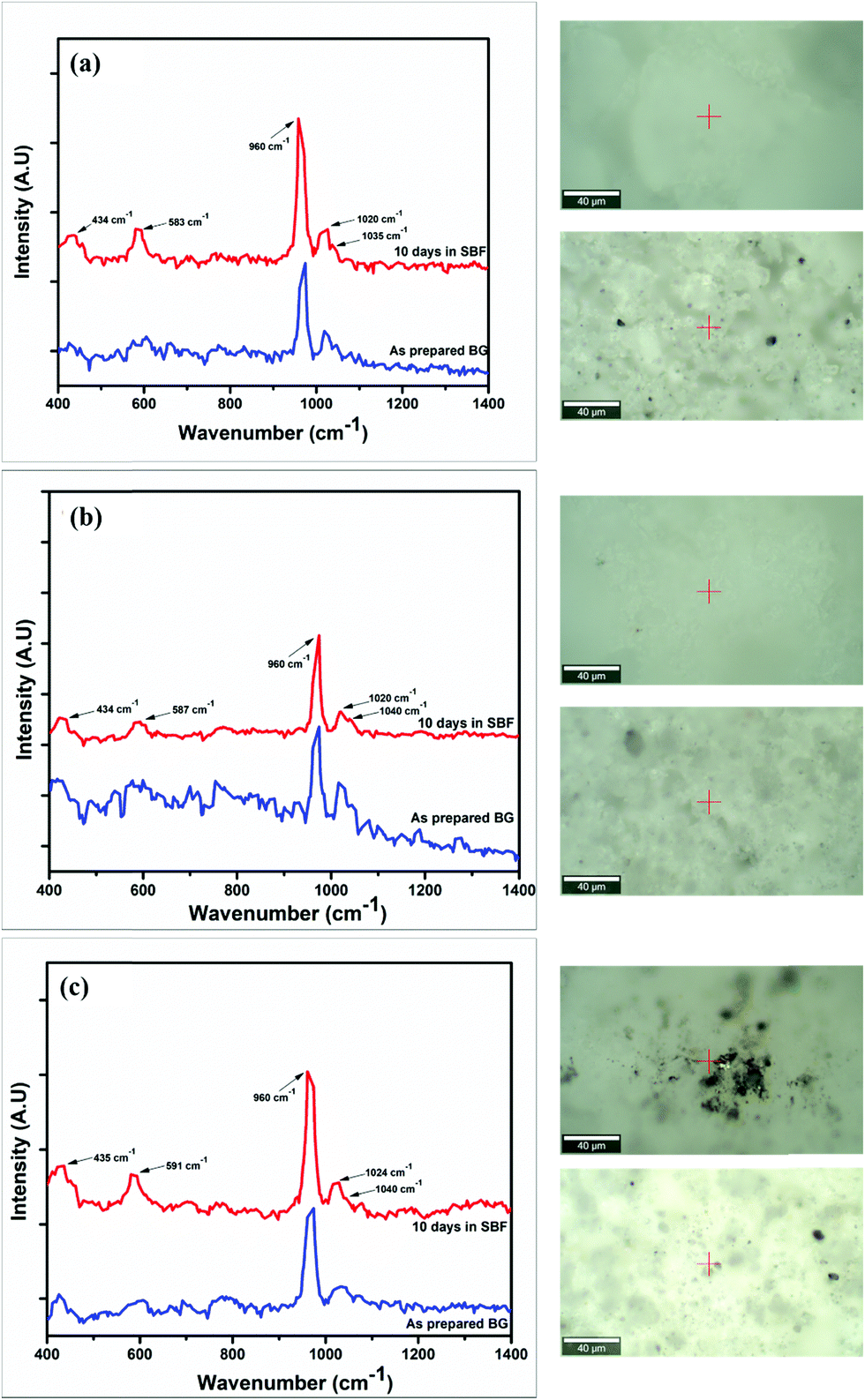
In vitro studies
The Raman spectra of the synthesized BG powders were collected both before and after immersion in SBF solution stored in polyethylene terephthalate (PET) sealed bottles at a constant temperature of 37 °C for 240 h (10 days). The collected spectra are illustrated in Fig. 8(a–c). Raman spectroscopy is a very powerful tool for monitoring and identification of Si–O–NBO bonds in BG.47,48 From the observed spectra, it was identified that a sharp and highly intense peak is present at 960 cm−1 in both the as-prepared and SBF-immersed variations of all of the BG powder samples synthesized in three different catalytic media. This characteristic peak at 960 cm−1 corresponds to the symmetric vibration of P–O bonds in the PO43− group.18,22,48,49 This peak is also present in the as-prepared samples of BG, but at a lesser intensity. The presence of this peak confirms the formation of HCA layer on the surface of the BG powders.22,49,50 The magnitude of intensity of the peak for HCA increases with immersion time in SBF, and a quantitative estimation for the formation of HCA layer can be given by comparing the absolute intensity (y-axis count) of the magnitudes of this particular peak in both the as-prepared and SBF-immersed samples (eqn (2)).| | 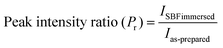 | (2) |
This comparison is depicted in Table 6 to provide an idea of the in vitro bioactivity of the prepared BG samples.
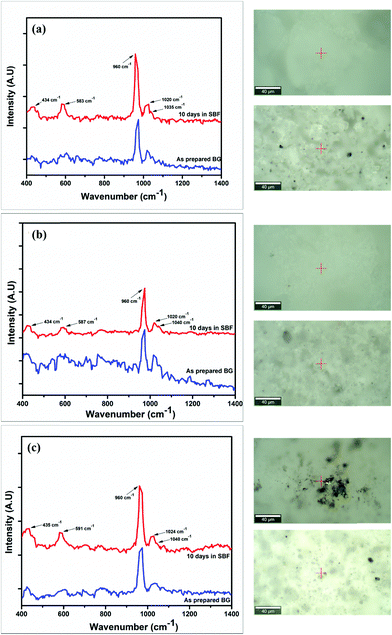 |
| | Fig. 8 Raman spectra of the as-prepared and SBF-immersed BG powders prepared in catalytic media of (a) 1 M HNO3, (b) 1 M FA, and (c) 1 M AA with the locations where the spectra were collected for the as-prepared (bottom inset) and SBF-immersed (top inset) samples. | |
Table 6 Comparison of the magnitudes of the 960 cm−1 peaks observed in the Raman spectra of the SBF-immersed and as-prepared BG powders
| Samples |
Ratio of 960 cm−1 peak in as-prepared BG to SBF immersed BG |
| A |
1.49 |
| B |
1.08 |
| C |
1.48 |
From the comparison of the magnitudes of the characteristic 960 cm−1 peak observed from the Raman spectra of the as-prepared and SBF-immersed BG powders, it can be inferred that the quantitative formation of HCA layer, which in turn is a qualitative indicator of the bioactivity of the BG powders, is the highest and nearly the same for samples A and C, which are the samples prepared in 1 M nitric acid and 1 M acetic acid catalytic media. In addition to this characteristic peak of HCA formation, other peaks were observed in the Raman spectra at around 435, 580–590 and 1020–1040 cm−1. The peak at 435 cm−1 indicates formation of the HCA layer and is present due to asymmetric bending of the P–O bonds of PO43− groups.22,49 The peaks between 500 and 600 cm−1 are due to the rocking vibrations of Si–O–Si bonds.48 The smaller peaks present at 1020 cm−1 and 1040 cm−1 are due to Si–O–Si asymmetric stretching of the silicate groups.18,49
The formation of an HCA layer over the surface of the BG powders can be seen in the SEM images, as shown in Fig. 9(a–c). After immersion in SBF for 240 h (10 days), the surface of the BG powders can be seen to be entirely covered by flaky apatite crystals with a cauliflower-like appearance.18,22,23 The change of the surface morphology is distinct when compared with the SEM images in Fig. 4(a–c). These results are coherent with the Raman spectroscopy results obtained previously, indicating the formation of the HCA layer over the BG powder samples. The adjacent EDS images in Fig. 9(d–f) prove that there have been significant increases in the calcium and phosphorous content in the BG powder surface, with slight decreases in the silicon content in each of the samples, namely samples A, B, and C, respectively. The EDS spectra also show that the silicon did not dissolve completely, as a substantial amount of silicon is still present in the powders. These results are coherent with previous results obtained by Zhou et al.,22 where they showed that the formation of apatite layer over BG surfaces in SBF occurs through a five-step process. The steps involve increasing the solution pH after SBF immersion, which motivates the formation of apatite nuclei over the silicon-rich glass surface followed by the dissolution of silicon from the surface through breaking of Si–O bonds and migration of Ca2+ and PO43− ions to the surface, replacing the silicon-rich layer. These then form the calcium and phosphorous-rich layers on the surface and incorporation of OH− and CO32− from the SBF solution to constitute the crystalline HCA layer over the surface. To get a better idea about the surface activity and bioactivity of the powder samples, the Ca/P ratios of the as-prepared and 10-day SBF-immersed samples were calculated from the EDS spectra and are given in Table 7.
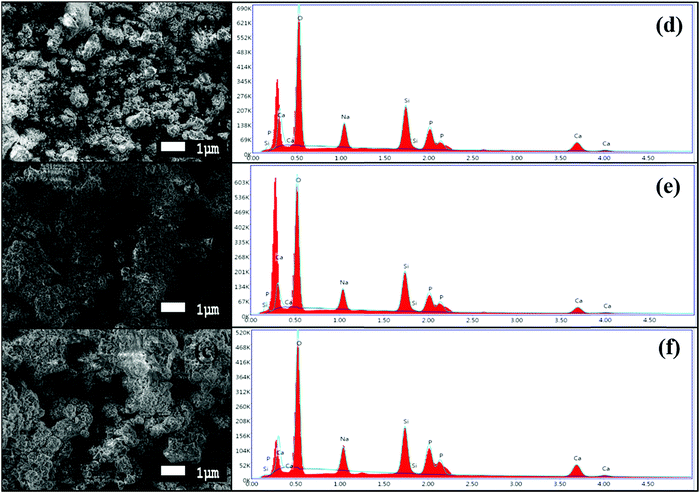 |
| | Fig. 9 High magnification SEM images at 5000× magnification of BG prepared in catalytic media of (a) 1 M HNO3, (b) 1 M FA, and (c) 1 M AA after 240 h of SBF immersion and EDS spectra of the corresponding BG powders (after 240 h of SBF immersion) prepared in catalytic media of (d) 1 M HNO3, (e) 1 M FA, and (f) 1 M AA. | |
Table 7 Ca/P ratios of the as-prepared and 10-day SBF-immersed BG samples
| Sample |
Ca/P ratio of As-prepared sample |
Ca/P ratio of 10 days SBF immersed sample |
| A |
1.23 |
1.67 |
| B |
1.34 |
1.38 |
| C |
1.62 |
1.76 |
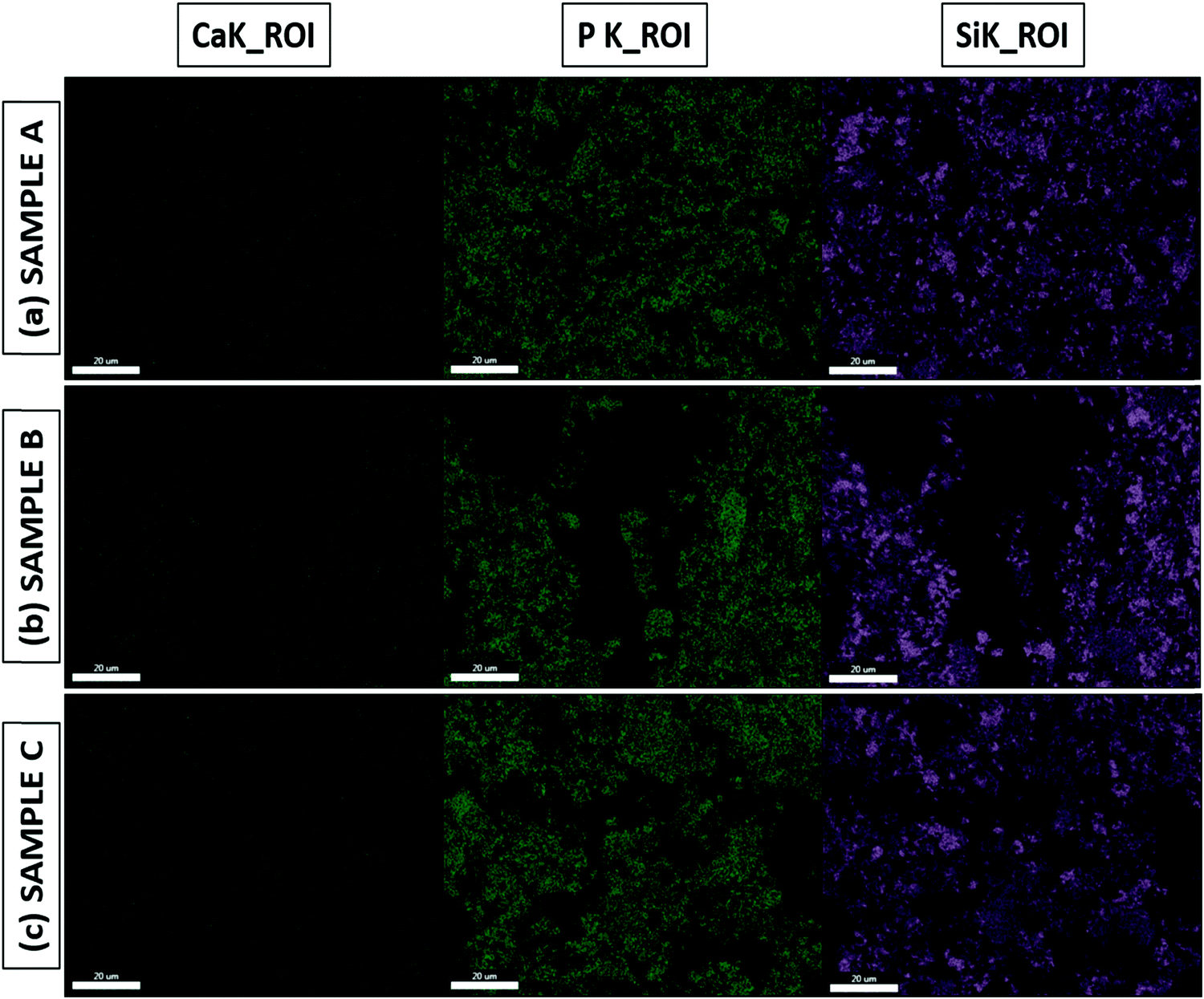
The Ca/P ratio is essential to the formation of the apatite layer, as it dictates bone formation and further bone cell growth. The Ca/P ratio of hydroxyapatite (HA), which has the composition Ca10(PO4)6OH2 and is present in human bones, is around 1.67.8,22,51,52 From the Ca/P ratios obtained from the EDS results for both the as-prepared and SBF-immersed samples, we can infer that SBF-immersed sample A has the exact same ratio of 1.67 as HA, which is quite remarkable, and forms a stable apatite layer. In contrast, SBF-immersed sample B shows a lower Ca/P ratio of 1.38, which indicates the formation of calcium-deficient apatite.53 SBF-immersed sample C exhibits the highest value of the Ca/P ratio of 1.76, which indicates the formation of a calcium-rich and much more stable apatite layer.22,54 EDS mapping images are shown in Fig. 10(a–c), mainly focusing on calcium, phosphorous, and silicon groups. We can validate both the presence and uniform distribution of the mentioned elements over the formed apatite layer from the mapping images.
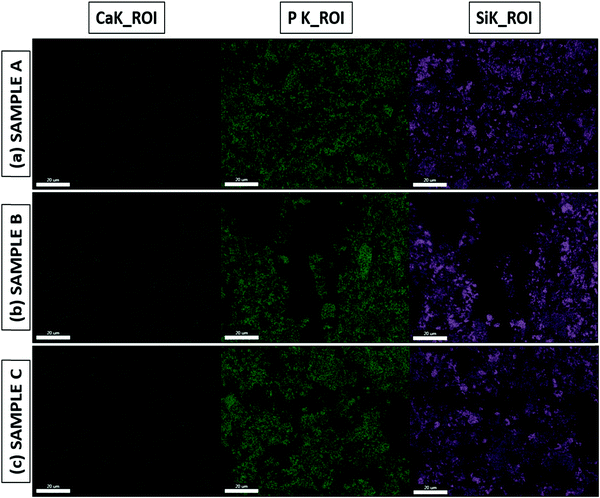 |
| | Fig. 10 EDS mapping of SBF-immersed BG prepared in catalytic media of (a) 1 M HNO3, (b) 1 M FA, and (c) 1 M AA. | |
The results of all the characterizations conducted are enlisted in Table 8 to reach a more precise inference about the optimal results obtained from the synthesized BG samples in different catalytic media, namely sample A, sample B, and sample C, respectively.
Table 8 Optimal results obtained from the characterization tests conducted on the BG samples A, B and C, respectively
| Characterization methods |
Optimal results |
Remarks |
| TGA and DTGA |
Mass loss occurs in all three samples in three stages due to loss of water molecules and thermal decomposition of organic and nitrate precursors, condensations of silanols, and initiation of the crystallization in the samples |
It was observed that the mass loss occurs at a higher temperature for BG samples prepared in FA and AA catalytic media as compared to the BG prepared in HNO3 catalytic media. The elimination of (CH3COO−) ion takes place around 420 °C in the BG samples prepared in AA as catalyst |
| SEM & EDS of as-prepared BG samples |
Average sizes ranging between 1–10 μm are observed in all BG samples with substantial particle agglomeration. Elemental analysis shows the presence of silicon, calcium, phosphorous, sodium and oxygen within all the samples without the presence of any contamination. |
Morphology observed in all BG samples is of highly irregular nature due to non-uniform pulverization. EDS results suggests that the highest Ca/P ratio is in as-prepared sample C |
| BET tests on as-prepared samples |
SSA obtained from the BET data is highest for BG sample C i.e. BG prepared in 1 M acetic acid catalytic medium. The pore size of all the three samples falls within the mesoporous range i.e. 2–50 nm |
The higher SSA for sample C is a desirable feature for enhanced bioactivity |
| X-ray diffraction studies on as-prepared samples |
Peaks of different sodium calcium phosphate silicates and sodium calcium silicates were observed through XRD studies. Na2Ca2Si3O9 (combite) & Na2Ca3Si6O16 (devitrite) were identified as primary and secondary phases respectively |
Sample A and sample C both have the highest combite to devitrite phase ratio and hence the highest potential to exhibit bioactive nature. Additionally, higher crystallite/grain size of sample C powders is resultant of its higher pore volume and hence higher bioactive nature |
| Raman spectroscopy of as-prepared and SBF immersed BG samples |
Sharp and high intense peak is present at 960 cm−1 in both the as-prepared and SBF immersed variations of all prepared samples confirming the presence and growth of carbonated apatite layer |
From the comparison of the magnitudes of the characteristic 960 cm−1 peaks, it can be inferred that the growth of HCA layer which in turn is an indicator for the bioactivity of the BG powders are highest and nearly same for samples A and C |
| SEM & EDS of SBF immersed BG samples |
SEM images confirmed the growth of flaky apatite layer over the surface of all BG powder samples after 10 days of SBF immersion. EDS spectra showed substantial increase in amount of calcium and phosphorous in the samples with meagre decrease in silicon content. EDS mapping confirms that the growth of apatite layer over the BG sample surfaces is of uniform nature |
SBF immersed sample C exhibits the highest value of Ca/P ratio of 1.76 and indicates the formation calcium-rich stable apatite layer |
From the inferences drawn from the observations of different characterization tests as listed in Table 8, we can safely deduce that the results support the bioactive nature of the powders and that the propensity of carbonated apatite layer formation inclines more towards the BG powders of sample C, i.e., the sample prepared in 1 M AA catalytic medium. Numerous studies have been conducted on the sol–gel synthesis of BG powders over the years,8,16–18,22,23,25,26 and the use of inorganic acid catalytic mediums such as HNO3 and HCL is mostly preferred for facilitating the hydrolysis and poly-condensation reactions that are essential for gel formation.2,8,17,22 Some studies have tested the use of organic acids as catalysts for the sol–gel synthesis of BG, such as citric acid catalytic medium,4 lactic acid catalytic medium,24,55 and AA catalytic medium.28,40,56 The results indicate that using organic acids as catalysts, namely FA and AA used in this work, successfully facilitates the formation of BG with a mesoporous and bioactive nature, which can successfully progress to apatite layer formation when immersed in SBF. The BG sample prepared in 1 M AA catalyst showed the highest SSA, the dominant presence of combite phase and the highest magnitude of the Ca/P ratio after SBF immersion. This indicates a higher level of in vitro bioactivity compared to the samples prepared in 1 M HNO3 and 1 M FA catalysts, which supports the results presented earlier by Lei et al.40
4. Conclusions
In this investigation, the inherent effects of different inorganic and organic acid catalysts on the structure, morphology, chemical composition, porous nature, bioactive phase forming ability, and apatite layer forming bioactivity of sol–gel synthesized 45S5 bioactive glasses have been studied. The BG 45S5 powder prepared in acetic acid showed superior porosity, more bioactive combite phase formation, and enhanced tendency towards carbonated apatite layer formation when immersed for 10 days in SBF. The results pointed towards the fact that the bioactive glass powders synthesized in acetic acid catalyst showed the best in vitro bioactivity by forming a calcium-rich, stable, and uniform apatite layer with a higher Ca/P ratio than that of the naturally occurring hydroxyapatite present in human bones. Further, the BG particles prepared with organic acid as a catalytic medium exhibited mass loss at a higher temperature than those prepared in inorganic catalytic medium. Mesoporous structures (porosity 2–50 nm) were obtained for all the particles in this sol–gel route. Na2Ca2Si3O9 (combite) and Na2Ca3Si6O16 (devitrite) were identified as the primary and secondary phases, respectively, in the XRD studies, indicating the influence of Na2O on the devitrification process in converting glass to glass-ceramic, which was in correlation with the literature. There was no substantial effect of the catalysts on the size and morphology of the powders fabricated in this study. Thus, organic acetic acid as a catalytic medium for sol–gel synthesis of bioactive glasses may be a more suitable candidate than the conventional inorganic acid catalysts in predominant use.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
Authors would like to acknowledge DST-SERB (ECR/2016/000083), Govt. of India for funding this research. PS would also like to acknowledge DST Inspire Faculty Scheme (DST/INSPIRE/04/2015/000742), Govt. of India. Author JA acknowledges the support of IIEST, Shibpur for providing Institute's PhD fellowship.
References
- C. Vichery and J.-M. Nedelec, Materials, 2016, 9, 288 CrossRef PubMed.
- I. Cacciotti, M. Lombardi, A. Bianco, A. Ravaglioli and L. Montanaro, J. Mater. Sci.: Mater. Med., 2012, 23, 1849–1866 CrossRef CAS PubMed.
- R. Li, A. Clark and L. Hench, J. Appl. Biomater., 1991, 2, 231–239 CrossRef CAS PubMed.
- J. Faure, R. Drevet, A. Lemelle, N. B. Jaber, A. Tara, H. El Btaouri and H. Benhayoune, Mater. Sci. Eng., C, 2015, 47, 407–412 CrossRef CAS PubMed.
- F. Naghizadeh, M. R. A. Kadir, A. Doostmohammadi, F. Roozbahani, N. Iqbal, M. M. Taheri, S. V. Naveen and T. Kamarul, J. Non-Cryst. Solids, 2015, 427, 54–61 CrossRef CAS.
- A. A. R. de Oliveira, B. B. de Carvalho, H. S. Mansur and M. de Magalhães Pereira, Mater. Lett., 2014, 133, 44–48 CrossRef CAS.
- A. Ramila, F. Balas and M. Vallet-Regi, Chem. Mater., 2002, 14, 542–548 CrossRef CAS.
- R. Ravarian, F. Moztarzadeh, M. S. Hashjin, S. Rabiee, P. Khoshakhlagh and M. Tahriri, Ceram. Int., 2010, 36, 291–297 CrossRef CAS.
- R. Hill, J. Mater. Sci. Lett., 1996, 15, 1122–1125 CrossRef CAS.
- P. N. De Aza, A. H. De Aza, P. Pena and S. De Aza, Bol. Soc. Esp. Ceram. Vidrio, 2007, 46, 45 CrossRef CAS.
- S. Kargozar, F. Kermani, S. Mollazadeh Beidokhti, S. Hamzehlou, E. Verné, S. Ferraris and F. J. M. Baino, Materials, 2019, 12, 3696 CrossRef CAS PubMed.
- I. Izquierdo-Barba and M. Vallet-Regí, Biomed. Glasses, 2015, 1, 140–150, DOI:10.1515/bglass-2015-0014.
- I. Cacciotti, J. Mater. Sci., 2017, 52, 8812–8831 CrossRef CAS.
- M. S. Bahniuk, H. Pirayesh, H. D. Singh, J. A. Nychka and L. D. Unsworth, Biointerphases, 2012, 7, 41 CrossRef CAS PubMed.
- Ö. H. Andersson, K. H. Karlsson and K. Kangasniemi, J. Non-Cryst. Solids, 1990, 119, 290–296 CrossRef.
- P. Sepulveda, J. R. Jones and L. L. Hench, J. Biomed. Mater. Res., 2001, 58, 734–740 CrossRef CAS PubMed.
- R. L. Siqueira, O. Peitl and E. D. Zanotto, Mater. Sci. Eng., C, 2011, 31, 983–991 CrossRef CAS.
- S. Chitra, P. Bargavi and S. Balakumar, J. Biomed. Mater. Res., Part B, 2020, 108, 143–155 CrossRef CAS PubMed.
- A. E. Danks, S. R. Hall and Z. Schnepp, Mater. Horiz., 2016, 3, 91–112 RSC.
- B. Spirandeli, T. Campos, R. Ribas, G. Thim and E. S. Trichês, Ceram. Int., 2020, 46, 20264–20271 CrossRef CAS.
- N. Shankhwar, G. Kothiyal and A. Srinivasan, RSC Adv., 2015, 5, 100762–100768 RSC.
- Z. Zhou, J. Ruan, J. Zou and Z. Zhou, J. Univ. Sci. Technol. Beijing, 2008, 15, 290–296 CrossRef CAS.
- A. Saboori, M. Rabiee, F. Moztarzadeh, M. Sheikhi, M. Tahriri and M. Karimi, Mater. Sci. Eng., C, 2009, 29, 335–340 CrossRef CAS.
- E. Rezabeigi, P. M. Wood-Adams and R. A. Drew, Mater. Sci. Eng., C, 2014, 40, 248–252 CrossRef CAS PubMed.
- K. Huang, S. Cai, G. Xu, X. Ye, Y. Dou, M. Ren and X. Wang, J. Alloys Compd., 2013, 580, 290–297 CrossRef CAS.
- F. Baino, E. Fiume, M. Miola and E. Verné, Int. J. Appl. Ceram. Technol., 2018, 15, 841–860 CrossRef CAS.
- Q.-Z. Chen, Y. Li, L.-Y. Jin, J. M. Quinn and P. A. Komesaroff, Acta Biomater., 2010, 6, 4143–4153 CrossRef CAS PubMed.
- B. Lei, X. Chen and Y.-H. Koh, J. Sol–Gel Sci. Technol., 2011, 58, 656–663 CrossRef CAS.
- R. L. Siqueira, L. C. Costa, M. A. Schiavon, D. T. de Castro, A. C. dos Reis, O. Peitl and E. D. Zanotto, J. Sol–Gel Sci. Technol., 2017, 83, 165–173 CrossRef CAS.
- B. Lei, X. Chen, Y. Wang, N. Zhao, C. Du and L. Fang, Biomed. Mater., 2010, 5, 054103 CrossRef PubMed.
- H. Pirayesh and J. A. Nychka, J. Am. Ceram. Soc., 2013, 96, 1643–1650 CrossRef CAS.
- L. T. Arenas, C. W. Simm, Y. Gushikem, S. L. Dias, C. C. Moro, T. M. Costa and E. V. Benvenutti, J. Braz. Chem. Soc., 2007, 18, 886–890 CrossRef CAS.
- K. Zheng, A. Solodovnyk, W. Li, O. M. Goudouri, C. Stähli, S. N. Nazhat and A. R. Boccaccini, J. Am. Ceram. Soc., 2015, 98, 30–38 CrossRef CAS.
- M. Catauro, A. Dell’Era and S. V. Ciprioti, Thermochim. Acta, 2016, 625, 20–27 CrossRef CAS.
- T. Kokubo and H. Takadama, Biomaterials, 2006, 27, 2907–2915 CrossRef CAS PubMed.
- G. S. Lázaro, S. C. Santos, C. X. Resende and E. A. dos Santos, J. Non-Cryst. Solids, 2014, 386, 19–28 CrossRef.
- E. Fiume, C. Migneco, E. Verné and F. Baino, Materials, 2020, 13, 540 CrossRef CAS PubMed.
- S. Chitra, P. Bargavi, D. Durgalakshmi, P. Rajashree and S. Balakumar, Process. Appl. Ceram., 2019, 13, 12–23 CrossRef CAS.
- B. A. Ben-Arfa, I. M. M. Salvado, J. M. Ferreira and R. C. Pullar, Mater. Sci. Eng., C, 2018, 91, 36–43 CrossRef CAS PubMed.
- B. Lei, X. Chen, Y. Wang, N. Zhao, C. Du and L. Zhang, J. Non-Cryst. Solids, 2009, 355, 2583–2587 CrossRef CAS.
- S. Brunauer, L. S. Deming, W. E. Deming and E. Teller, J. Am. Chem. Soc., 1940, 62, 1723–1732 CrossRef CAS.
- C. Liu, Z. Xia and J. Czernuszka, Chem. Eng. Res. Des., 2007, 85, 1051–1064 CrossRef CAS.
- A. Patterson, Phys. Rev., 1939, 56, 978 CrossRef CAS.
- Y. Orooji, M. Ghanbari, O. Amiri and M. Salavati-Niasari, J. Hazard. Mater., 2020, 389, 122079 CrossRef CAS PubMed.
- S. Gholamrezaei, M. Ghanbari, O. Amiri, M. Salavati-Niasari and L. K. Foong, Ultrason. Sonochem., 2020, 61, 104829 CrossRef CAS PubMed.
- S. Moshtaghi, S. Zinatloo-Ajabshir and M. Salavati-Niasari, J. Mater. Sci.: Mater. Electron., 2016, 27, 425–435 CrossRef CAS.
- A. Balamurugan, G. Sockalingum, J. Michel, J. Fauré, V. Banchet, L. Wortham, S. Bouthors, D. Laurent-Maquin and G. Balossier, Mater. Lett., 2006, 60, 3752–3757 CrossRef CAS.
- B. A. Ben-Arfa, H. R. Fernandes, I. M. Miranda Salvado, J. M. Ferreira and R. C. Pullar, J. Biomed. Mater. Res., Part A, 2018, 106, 510–520 CrossRef CAS PubMed.
- F. E. Ciraldo, L. Liverani, L. Gritsch, W. H. Goldmann and A. R. Boccaccini, Materials, 2018, 11, 692 CrossRef PubMed.
- T. J. Brunner, R. N. Grass and W. J. Stark, Chem. Commun., 2006, 1384–1386 RSC.
- G. Goller, H. Demirkıran, F. N. Oktar and E. Demirkesen, Ceram. Int., 2003, 29, 721–724 CrossRef CAS.
- B. A. Ben-Arfa, I. M. M. Salvado, R. C. Pullar and J. M. Ferreira, Ceram. Int., 2018, 44, 12754–12762 CrossRef CAS.
- H. Luo, D. Ji, W. Li, J. Xiao, C. Li, G. Xiong, Y. Zhu and Y. Wan, Mater. Chem. Phys., 2016, 176, 1–5 CrossRef CAS.
- L. A. Q. Sierra and D. M. E. Sierra, JOM, 2019, 71, 302–307 CrossRef.
- X. Chen, B. Lei, Y. Wang and N. Zhao, J. Non-Cryst. Solids, 2009, 355, 791–796 CrossRef CAS.
- A. Meiszterics and K. Sinkó, Colloids Surf., A, 2008, 319, 143–148 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Jaideep
Adhikari
ab,
Jaideep
Adhikari