
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHuman neuromuscular junction on micro-structured microfluidic devices implemented with a custom micro electrode array (MEA)†
Pauline
Duc
 *a,
Michel
Vignes
b,
Gérald
Hugon
c,
Audrey
Sebban
d,
Gilles
Carnac
c,
Eugene
Malyshev
d,
Benoît
Charlot
d and
Florence
Rage
*a
*a,
Michel
Vignes
b,
Gérald
Hugon
c,
Audrey
Sebban
d,
Gilles
Carnac
c,
Eugene
Malyshev
d,
Benoît
Charlot
d and
Florence
Rage
*a
aIGMM, University of Montpellier, CNRS, Montpellier, France. E-mail: pauline.duc@igmm.cnrs.fr; florence.rage@igmm.cnrs.fr
bIBMM UMR5247, University of Montpellier, CNRS, Montpellier, France
cPhyMedExp, INSERM U1046, CNRS UMR9214, Université de Montpellier, Montpellier, France
dIES, CNRS University of Montpellier, Montpellier 34095, France
First published on 24th September 2021
Abstract
In the neuromuscular system, signal transmission from motor neurons (MNs) to innervated muscle fibers is crucial for their synaptic function, viability, and maintenance. In order to better understand human neuromuscular junction (hNMJ) functionality, it is important to develop on-a-chip devices with human cells. To investigate this cell network, microfluidic platforms are useful to grow different cell types in isolated compartments. Such devices have already been developed to study in vitro neuronal circuitry. Here, we combined microfluidics with two techniques: soft lithography and custom microelectrodes array (MEA). Our goal was to create hNMJs on a specific pattern of electrodes to stimulate pre-synaptic axons and record post-synaptic muscle activity. Micromachining was used to create structurations to guide muscle growth above electrodes, without impairing axon propagation, therefore optimizing the effectiveness of activity recording. Electrodes were also arranged to be aligned with the microfluidic chambers in order to specifically stimulate axons that were growing between the two compartments. Isolation of the two cell types allows for the selective treatment of neurons or muscle fibers to assess NMJ functionality hallmarks. Altogether, this microfluidic/microstructured/MEA platform allowed mature and functional in vitro hNMJ modelling. We demonstrate that electrical activation of MNs can trigger recordable extracellular muscle action potentials. This study provides evidence for a physiologically relevant model to mimic a hNMJ that will in the future be a powerful tool, more sensitive than calcium imaging, to better understand and characterize NMJs and their disruption in neurodegenerative diseases.
Introduction
For decades, approaches to understand the function of the nervous system and human neurodegenerative diseases were restricted to animal models or animal-derived tissues.1 Despite our vast knowledge of molecular mechanisms and the composition of neuronal transmission, there remains a gap to develop personalized humanized synapses. The emergence of human-induced pluripotent stem cells (hiPSCs), and the considerable progress made to differentiate them in multiple cell types, provide a wide variety of human genetic models, and a tool to recapitulate specific diseases, such as neurodegenerative ones.2–4 In particular, neuromuscular disorders can arise from defects in the motor neurons (MNs), as in amyotrophic lateral sclerosis (ALS) and spinal muscular atrophy (SMA), from muscle defects as in Duchenne muscular dystrophy (DMD), or from Schwann cell disorders, as in Charcot–Marie–Tooth disease and Guillain–Barré syndrome. Among these various disorders, neuromuscular junction (NMJ) represents one of the impacted components.5–7 To date, morphological and molecular data on neuromuscular junction (NMJ) from experimental animal models or animal-derived cells are well documented8 but there is a paucity of informations concerning human NMJ. A nice recent study provides evidence that, while no morphological differences between rats and mouse NMJs were distinguishable, human NMJ was unique by its size, with only half the area of mouse endplate.9In addition, proteomic analysis suggests that hNMJ displays distinct molecular composition compared to other species. More recently, the same group used a systematic morphometric platform analysis to compare NMJ morphology, muscle fiber diameter, or body size. Surprisingly, data provide evidence that human NMJ is closer to the ovine one rather than to lower mammalian such as rodent.10 This study reveals how it became clear that an in vitro model of human NMJ needed to be developed. In vertebrates, NMJ shares the structural characteristics and features of many central synapses. Indeed, action potentials associated with calcium influx propagate from MNs to muscles and chemical signals are provided at the synapse by the release of acetylcholine (ACh) from synaptic vesicles. Once released in the synaptic cleft, ACh binds the acetylcholine receptors (AChRs) that are organized in clusters on the mature muscle membrane. However, the achievement of mature synapse relies on a set of well-coordinated events including a fine-tuned cross-talk between neurons and muscles during development that can be called the “Agrin-Lrp4-MuSK signaling pathway”.11,12 It has been postulated that during development, agrin released by motor neurons enables the aggregation of AchRs.13,14 Concomitantly, proteins expressed in muscle fibers, such as muscle-specific kinase (MuSK), and low-density lipoprotein receptor-related protein 4 (Lrp4), participate in the formation and the organization of the NMJ.15 Agrin binding on the Lrp4 protein triggers formation of the agrin-Lrp4-MuSK-Dok7 complex, which is essential for the formation of NMJ.16 Altogether, combined synchronized events lead to functional in vivo adult NMJ.
Discriminating neuronal compartments during NMJ in vitro modelling and recreating the physiological spatial organization are of special interest since NMJs are composed of two cell types growing in different microenvironments. The implementation of microfluidic devices has appeared to be a powerful and useful tool for their ability to precisely control, monitor and manipulate cellular microenvironments.17–19 Such approaches can bring some valuable insights to investigate and decipher molecular mechanisms occurring during NMJ formation in normal and pathological conditions. In order to mimic the physiological NMJ development, in vitro attempt to grow mouse NMJ on microfluidic devices was first achieved using mouse MN primary culture and skeletal muscle, or mouse embryonic stem cell (mESC)-derived MNs and c2c12 myotubes.20–22 The relevance of compartmentalized cell cultures was highlighted by the work of Zahavi,23 showing the spatial differentiating effect of GDNF, that could promote axonal growth and muscle innervation only when applied to distal axon and not to the soma. To date, a few studies have completed these pioneering works to investigate different pathways involved in the NMJ establishment.24,25 They were further emphasized in 2018 when Santhanam et al. reported the first entire human NMJ microfluidic model.26 In this study, the platform was designed to provide electrical stimulation of motor neurons without affecting the muscle compartment. Indeed, the system allowed selective electrical activation of motor neurons by submerging silver electrodes in the MN compartment, whereas muscle contraction activity was imaged. By combining electrical stimulation and pharmacology, the authors demonstrated dose-dependent responses to pre-and post-synaptic inhibitors. In the last three years, there has been a considerable increase in the number of papers presenting several models of hNMJs. For example, a functional NMJ platform has been validated by combining microfluidic chips and pseudotyped ΔG-rabies virus for retrograde monosynaptic tracing27 or chips coupled with MEA for spontaneous activity recording.28
Most of the time, muscle activity is measured by evaluating contraction after electrical stimulation using patch-clamp29 or optogenetic neuronal stimulation.30 Recently, planar micro electrode arrays, or MEAs31,32 were used as an in vitro systems that records the extracellular electrical activity of excitable cells seeded on their surface. They can, for instance, be used to assess neuron, muscle or cardiomyocyte activities. Unlike single electrode recordings, as patch-clamp, MEAs are very useful to record electrical activities propagation in networks of living cells both in cultures and tissue sections. MEAs measure the extracellular potential changes generated in the vicinity of the cells. The measured signal, although weaker than intracellular (a few hundred μV), still allows the quantification of electrophysiological activity such as single action potential generation or bursting activities within cell network. These systems have been extensively used in neurobiology to study neuronal network development activity in healthy and disease models such as Alzheimer's disease, amyotrophic lateral sclerosis (ALS) or Huntington's disease.33–35 Moreover, MEAs have been employed for drug application to identify their effects on electrical activity.36,37
Another important function of MEAs is their ability to evoke action potentials or depolarizations by electrically stimulating cells through current or voltage steps applied to individual electrodes. Most MEAs encompass regular arrays of flat metallic microelectrodes, typically 64 to 256 microelectrodes of a few tens of microns in diameter. A passivation layer, usually made of silicon nitride, is used to locate the measurement site and also to isolate the routing tracks. To improve the positioning of the electrodes as close as possible to the active cells, particularly in brain slices, they are conical38,39 or vertical40 which allows penetration into the tissue. Finally, in parallel to those so-called passive technologies, high density micro electrode arrays (HDMEA)41 have been developed and are based on the use of complementary metal oxide semi-conductor (CMOS) substrates integrating transistors to allow line and column switching, as well as pre-amplification as close as possible to the measurement sites. These systems allow the integration of several thousand microelectrodes but at the expense of substrate transparency, which prevents the combination with fluorescence.
The goal of our study was to create mature, functional and reliable human NMJs using microfluidic devices combined with custom MEA, in order to record, for the first time, muscle action potential triggered by evoked neuronal electrical stimulation. By combining MEAs with microfluidic circuits,42 we took advantage of the spatial organization provided by the compartmentalized microfluidic circuit. A strategic design of microelectrodes underneath the microchannels and each cell compartment, enabled the direct stimulation of distinct axons and the recording of induced muscle activity. Moreover, we designed an electrode layout that would fit with microstructurations added by lithography in order to align muscle fibers on top of electrodes, and we used the fastest growth factor-induced differentiation protocol of iPSCs into MNs. Thus, we developed a humanized platform providing a complete mature and functional NMJ, which represents a proof of concept to study NMJ electrical activity.
Results and discussion
Design of microstructurations to guide muscle orientation while being compatible with co-culture
Spatial organization of myotubes and motor neurons, either derived from murine primary cultures or stem cells, has been successfully achieved in the past using microfluidic chambers, focusing on the ability of MNs to connect skeletal muscles. In vivo studies suggested that muscle cues, delivered to MNs, induced guiding of neural innervation. Reciprocal crosstalk occurs during synaptogenesis to form NMJ. Enhancement of muscle fibers alignment was shown to increase their maturation and ability to organize acetylcholine receptors (AChRs) to act as a pattern for innervating neurons.43 In order to guide muscle growth, we created microstructurations using lithography on coverslips. We first designed three patterns: continuous grooves of varied spacing, ranging from 10 μm (noted 10c), 20 μm (20c) and 30 μm (30c), and PDMS microfluidics were aligned on these substrates. After cell mask labelling (Fig. 1a), the orientation of the fibers was first evaluated using the directionality plugin of the Fiji software. The rate of myotubes, distributed between 0° and 180°, was counted (Fig. 1b) and no significant differences of muscles angular orientation (±90°) were observed regardless of the spacing of the grooves, whereas control muscles were randomly oriented with a mean angular orientation close to 60°. The control group showed a high scatter value and a low goodness of fit (R2 = 0.69), reflecting a random orientation, while a single sharp peak showed a low dispersion value when myotubes were seeded on grooved substrates (10c, 20c and 30c) with a high goodness of fit (R2 > 0.9) (Fig. 1c). This confirmed that grooves lead to muscle alignment. In addition, α-actinin staining labels mature fibers and enables to calculate fusion index and proportion of striations, reflecting the level of fiber maturation. There were no significant differences between the four types of grooves concerning the number of nuclei per fiber (fusion index) (Fig. 1d), but we observed that the number of total nuclei as well as the myotube area were maximal for the 20c condition and similar to the control (Fig. 1e and f). Since the number of myoblasts seeded in the microfluidic devices was similar for each condition, and we observed better viability (less cells were detaching and dying during culture) for the 20c condition, we assumed that it was the best set up to continue the development of our custom platform. This observation is consistent with the percentage of the striated fibers, reflecting muscle maturation, which is better for 20c compared to 10c and 30c conditions (Fig. 1g). | ||
| Fig. 1 Assessment of muscle growth on microsctructured substrates. (a) Alignment of myoblasts labelled with cell mask on continuous microstructurations interspaced of 10, 20 or 30 μm (noted 10c, 20c and 30c) vs. no structuration (ctrl). (b) Statistical analysis of the fiber orientation according to the type of grooves, n = 18 fields from 6 chips. (c) Dispersion of fiber orientation depending on the goodness of fit (R2) in each condition, n = 18 fields from 6 chips. (d) Scatter plots of index fusion, (e) number of nuclei, (f) myotube area (n = 18 fields from 6 chips) and (g) percentage of striated fibers accounting for muscle maturation (n = 90 fields from 6 chips. (***p < 0.001); (**p < 0.01); (*p < 0.05). Scale bars: 100 μm. | ||
However, the implementation of fiber alignment using continuous lines appeared to produce a barrier to the growth of axons in the muscle compartment (perpendicularly to muscle fibers), which could have impaired hNMJ formation. The addition of 20c continuous lines has been replaced by the introduction of broken lines (noted 20b4) with a 4 μm wide opening every 100 μm, keeping a shift between each hole from one line to the other (Fig. 2a inset). The rationale behind the implementation of a shift was to avoid muscles to align perpendicularly to the other ones. Difference of axon invasion of the muscle compartment can be observed between 20c and 20b4 conditions (Fig. 2a). The length of axons has been measured as the distance between the exit point of axons from the microchannels to the farthest extension in the muscle compartment. Results indicated that the axons seeded in the 20b4 broken lines substrates, reached the double of length in the muscle compartment compared with continuous lines (Fig. 2b). No significant differences in muscle fiber angular orientation have been observed between 20c and 20b4 substrates (Fig. 2c). These results emphasized that it is possible to combine the use of microstructurations to align muscles without impairing the invasion of motor neurons into the muscle chamber, and the possibility of synapse formation between both components of the NMJ.
 | ||
| Fig. 2 Microstructuration optimization for MN invasion. (a) Staining of axons using anti-Smi32 antibody to visualize their growth in the muscle compartment containing either continuous lines (20c) or broken lines (20b4); inset depicts the lithography of 20b4 broken lines used in this study, n = 650 measures in 18 fields from 6 chips. (b) Statistical analysis of the axonal invasion of these two different muscle compartments. (c) Comparison of fiber directionality between 20c and 20b4 grooves, n = 18 fields from 6 chips. (****p < 0.0001). Scale bars: 100 μm. | ||
Endplate formation and NMJ maturation
We next used the 20b4 microfluidic device to grow human MNs derived from iPSCs and myotubes derived from human myoblasts. To evaluate the stage of maturation of the endplate, we labeled the presynaptic nerve with choline-acetyl-transferase (ChAT). As for the postsynaptic plate, it was labeled with alpha-bungarotoxin (BTX) to visualize the AChR clusters, and muscle fibers were observed by immunostaining of troponin. Fig. 3a shows ChAT-positive axons connected to troponin-positive multinuclear myotubes. Recruitment of proteins such as bassoon (Bsn) colocalizing with AChRs is consistent with the assumption that we obtained human active synapses, since Bsn mediates the synaptic vesicle fusion to the pre-synaptic membrane and the release of ACh in the synaptic cleft44 (Fig. 3b).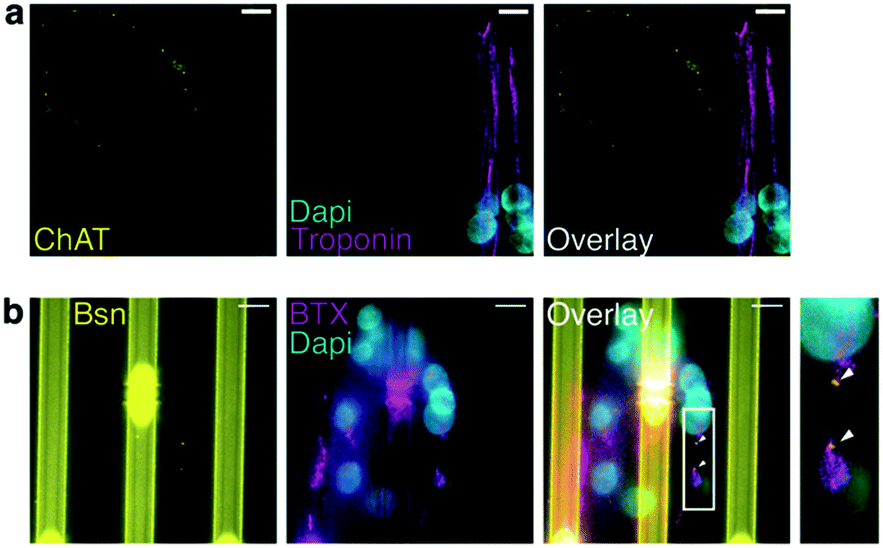 | ||
| Fig. 3 NMJ visualization by colocalization of pre- and post-synaptic markers. (a) Confocal image on MN axon stained with ChAT antibody (yellow) contacting a muscle fiber stained with troponin antibody (magenta). (b) Image showing clusters of AChRs (BTX: magenta) at the active zone of the synapse (Bsn: yellow). Arrows indicate sites of the pre- and post-synaptic markers colocalization. Note that the vertical Bsn-positive staining is the autofluorescence of 20b4 miscrostructurations, the 4 μm gaps are even brighter, as visible in the center of this image. Scale bars: 100 μm. | ||
Functionality of the NMJ revealed by calcium imaging
We then evaluated the functionality of the NMJ by GCaMP6f calcium live imaging under epifluorescence microscopy. GCaMP6f is a genetically encoded calcium indicator (GECI), expressed by the cells after lentiviral infection, consisting of the fusion of a calmodulin domain, a GFP domain (that produces minimal fluorescence in the absence of Ca2+), and a M13 domain. When Ca2+ binds to the calmodulin domain, it triggers a change of conformation producing detectable fluorescence.45 Co-culture was differentiated in 20b4 microfluidic devices as described above and equilibrated in BrainPhys medium without phenol red (StemCell Technologies) prior recording. After time-lapse imaging, regions of interests (ROIs) were created for each time point in the myofibers compartment, in order to depict the pattern of signal under different conditions, either in the basal condition or after drug treatment. Results of individual experiments indicated that in basal condition, cells display spontaneous activity evidenced by GCaMP6f fluorescence changes. This activity was abolished by D-tubocurarine (curare) treatment (25 nM), which is a competitive antagonist of acetylcholine (ACh) blocking nicotinic acetylcholine receptors (nAChRs). As shown in Fig. 4a, the effect of the drug on muscles was rapid since calcium activity was extinguished in less than 30s after curare was added. Statistical analysis showed that absolute values of fluorescence amplitude (|ΔF/F|) in the basal condition (0.065 ± 0.003 SEM) were significantly abolished after curare treatment (0.036 ± 0.002 SEM). Curare effect was reversible to the extent that the calcium activity recovered after washes and |ΔF/F| returned to basal rates (0.056 ± 0.002 SEM) (Fig. 4b). In another set of experiments, tetrodotoxin (TTX) (1 μM) was applied in the pre-synaptic compartment to block MN action potentials. By comparing the fluorescence activity during 1 minute prior and 1 minute after treatment, we observed that GCaMP6f detection was abolished in some myotubes, while others did not show notable changes. Among the number of muscles analyzed in 5 microfluidic devices, a mean value of 54.2% (ranging from 29% to 100%) responded to the inhibition of neuronal action potential by TTX. Response was assessed by measuring |ΔF/F| of each muscle and pooling them in “responsive” or “unresponsive” groups, which statistically confirmed the effect of TTX (Fig. 4c). These results confirmed that muscle activity is dependent on MN activity and validated the presence of functional hNMJs in our devices. Note that TTX treatment also inhibited MN-dependent muscle contractions, thus supporting the calcium imaging validation of functional hNMJs (Fig. S1†). | ||
| Fig. 4 Effect of curare and TTX on intracellular calcium activity to assess NMJ functionality. (a) Normalized fluorescence of muscle calcium waves when treated with curare (0 represent the start of treatment) and statistical analysis (n = 4 chips). (b) Normalized fluorescence of muscle calcium waves after curare recovery (0 represent the moment of wash) and statistical analysis (n = 4 chips). (c) TTX responsive muscles from all analysed muscles expressed in number and in percentage, n = 48 muscles in 5 chips (left), which highlights two distinct groups of muscles responsiveness after TTX treatment, represented by 2 scatter plots (middle and right). (****p < 0.0001). | ||
Microelectrode array design fitting with microstructured 20b4 microfluidic devices
>We then designed a custom MEA to align the functional hNMJ-on-chip on a unique platform, with a specific configuration developed to match with neuronal and muscle positions. Indeed, until now MEA platform was designed to study organization of neuronal network.42 The layout of the MEA is shown in Fig. 5a. It is composed of a set of 59 recording/stimulating electrodes (30 pre and 29 postsynaptic electrodes) plus 1 line of grounded reference. More specifically, it consists of three blocks of electrodes: a set of 50 μm-diameter circular electrodes (so-called ‘MN cell body electrodes’) that are located in the motor neuron chamber. A set of linear electrodes located at the entrance of microchannels, along a ground line, were designed to stimulates axons entering microchannels (so-called ‘axon electrodes’). In the second chamber, facing the axon electrodes, a set of 24 rectangular electrodes (200 × 60 μm) was used for recording of myofiber activity (so-called ‘muscle electrodes’). All these electrodes were aligned with the 20b4 broken line microgrooves, which ensured the presence of muscles specifically above their dedicated electrodes, and the insulation of the routing connection of these electrodes (Fig. 5b). The axon electrodes were also designed to be placed under the microchannels that connect the chambers. These electrodes were used to stimulate motor neuron axons passing through the microchannels. One of these microelectrodes could stimulate axons crossing about a dozen of microchannels (Fig. 5b zoom). Therefore, once the MEAs were aligned with the microstructuration and the microfluidic PDMS, we obtained a platform pictured in Fig. 5c. The specific microfabrication process has been developed and standardized in order to build series of microelectrode arrays that fitted both the MEA2100-60 amplifier (MultiChannel System, MCS, Germany) and the compartmentalized microfluidic circuit (see Materials and methods). Therefore, each electrode was connected to a pad that came in contact with the amplifier to further assess hNMJ electrical activity.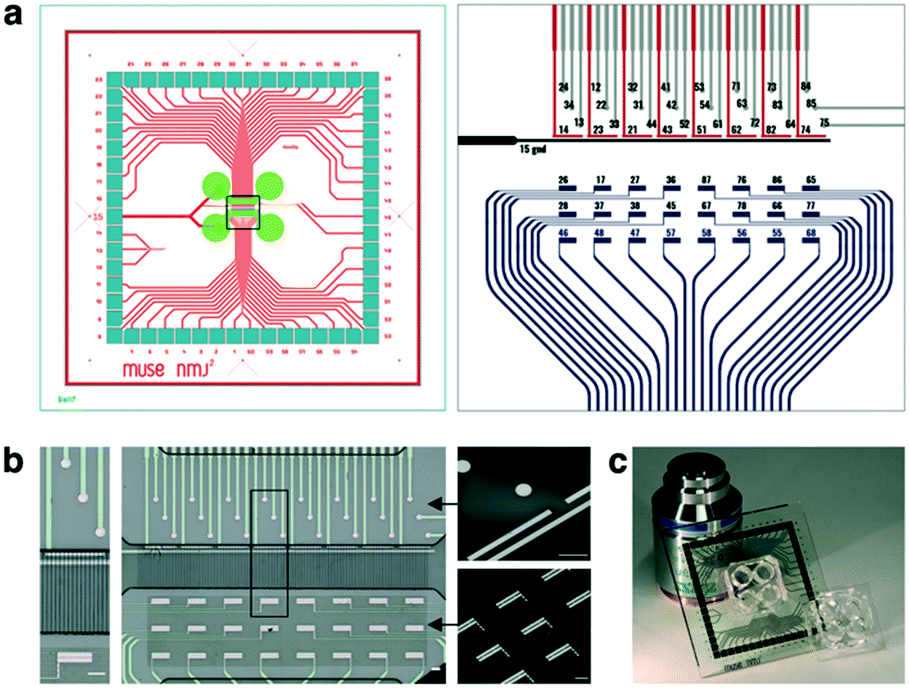 | ||
| Fig. 5 Microelectrode array design (a) global MEA layout for 49 × 49 mm glass substrate, with the localisation of the microfluidic chamber in green (left), and a zoom-in of the MEA central area (right), with the neuronal electrodes (cell bodies in grey, and axon in red) and the muscle electrodes (in blue). On the MCS-experimenter software, “MN cell body electrodes” were numbered 24, 34, 13, 12, 22, 33, 32, 31, 44, 41, 42, 53, 64, 54, 61, 71, 63, 72, 73, 83, 64, 84, 85, 76), “axon electrodes” were numbered 14, 23, 21, 43, 51, 62, 82, 74) and “muscle electrodes” were numbered 26, 28, 46, 17, 37, 48, 27, 38, 47, 36, 45, 57, 87, 67, 58, 76, 78, 56, 86, 66, 55, 65, 77, 68. (b) Alignment of the electrodes with the microstructured chip: neuron compartment is at the top of the images, and muscle compartment at the bottom, connected by microchannels for axon growth (inset). (c) Assembly of MEA and PDMS, compared to classic microfluidic chips. Scale bars: 100 μm. | ||
Electrical functionality using MEA recording
Since recording intracellular Ca2+ changes may integrate a number of action potentials, we evaluated whether the custom MEA; described above, could detect faster individual action potentials.46 The goal was to assess if it was possible to trigger action potentials (APs or spikes) extracellularly in distinctive axons, and record electrical activity at the muscle level, thanks to their alignment by microgrooves on the electrodes. Spontaneous neuronal APs were consistently recorded by the electrodes localized underneath the axon compartment i.e., ‘axon electrodes’ according to the layout of the MEA. Axon electrodes exhibiting more than 10 spikes per minute were considered as ‘active’ and further used to apply stimuli. Spontaneous APs were recorded in the muscle compartment when optimal culture conditions were reached, i.e., cell survival, density and connectivity. Fig. 6a shows typical spontaneous APs recorded from axons and muscles, the latter showing negative–positive or positive–negative phases. This difference in signal shapes could be due to the distance between the recording electrode and the cells emitting APs,47 to the direction of AP propagation (i.e. toward or away from the recording electrode), or to specific local regulation of extracellular ion concentration in the vicinity of the cells.48 Regardless of the shape, we were able to record spontaneous muscle APs, so we went ahead to record muscle APs triggered by MN stimulation. For that purpose, fifteen individual MEAs, obtained from 3 independent co-cultures, have been used to test and optimize the stimulus pattern, described in detail below, that would allow us to further analyze the connectivity between motor neurons and muscle cells. Firstly, the effectiveness of the stimulation was visually confirmed by monitoring intracellular Ca2+ changes in motor neurons expressing GCaMP6f. Single stimulus elicited transient intracellular Ca2+ increase in the axons underneath the stimulated electrode, evidencing neuronal depolarization and AP generation and propagation (Fig. 6b). Interestingly, to obtain Ca2+ increase, single stimulation of at least 200 μA was required, while we could detect AP activity on the MCS-recorder from a single 100 μA stimulation. Secondly, we defined the optimal number of pulses required to increase muscle activity. Indeed, single electrical stimulus elicited muscular spike frequency changes rather inconsistently. Therefore, the number of stimuli were increased from 1 to 6 at 1 Hz. As illustrated in Fig. 6c, incrementing the number of pulses generally resulted in an increase in AP occurrence recorded by muscle electrodes. Four pulses at 1 Hz elicited consistent increase in muscle activity. It is noteworthy that APs recorded by axon electrodes were rather unsensitive to stimuli increments but, when 4 pulses were applied, the number of spikes was in the same range as the one recorded by the muscle electrodes (Fig. 6d). Then, this protocol (4 pulses at 1 Hz) was applied repetitively in order to assess if it elicited consistent changes in muscle AP occurrence over time with different intervals between the cumulative stimulations. We observed that shorter intervals (30 seconds) were not sufficient to observe a recovery of spike frequency increase, elicited by the stimulation, whereas 60 seconds was sufficient for a complete recovery (Fig. 6e). Thus, the final protocol (4 pulses at 1 Hz and 60 second intervals) was used to test if NMJ fatigue could eventually be observed in subsequent experiments. Fatigue is defined by the state of a muscle that cannot perform any more contractions due to repeated stimulations.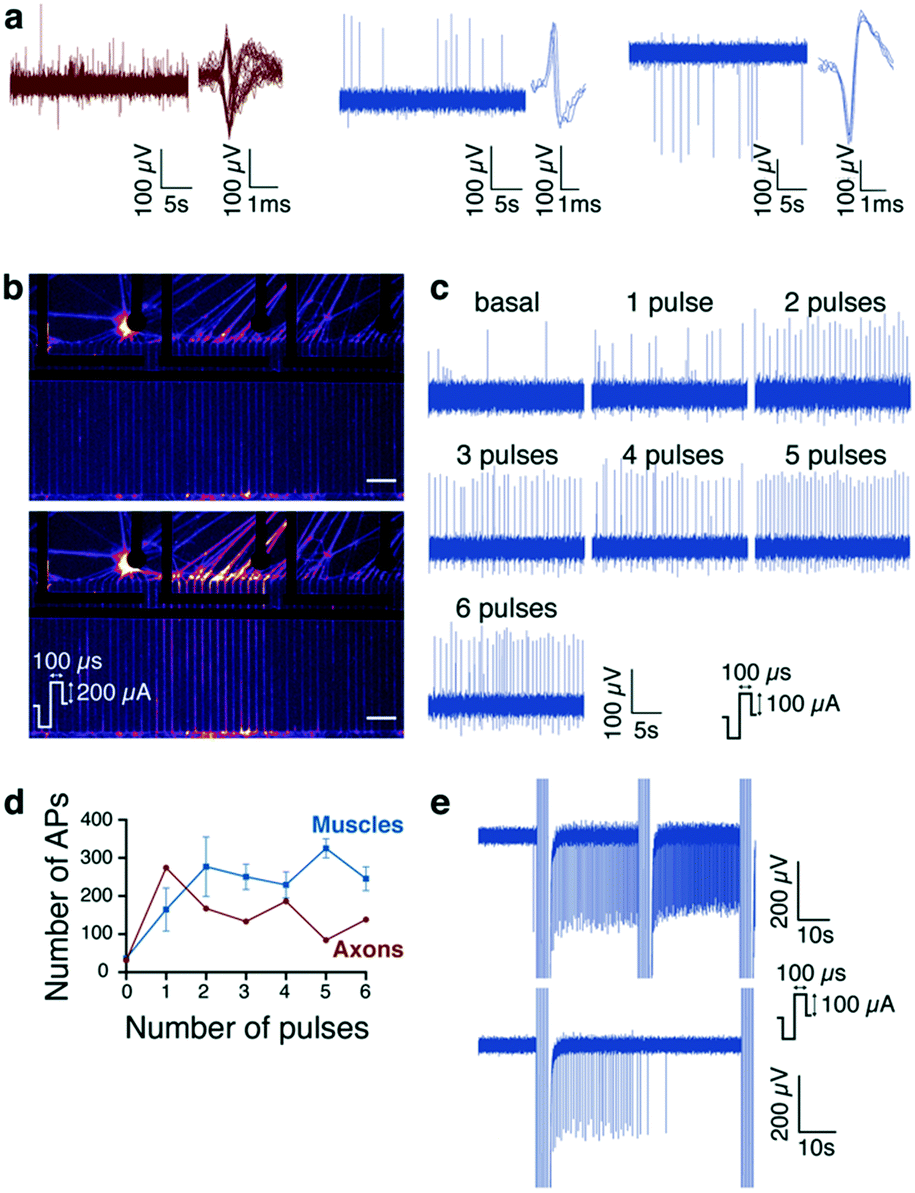 | ||
| Fig. 6 Custom MEA stimulation and recording set up (a) illustrative examples depicting spiking activity and superimposed traces of spikes recorded in axon (left) and muscle electrodes (middle and right). Note that both positive and negative ongoing signals were recorded by axon electrodes since one electrode records a dozen of microchannel-containing axons. (b) Intracellular Ca2+ increases in axons of neurons expressing GCaMp6f and is elicited by one single stimulus (200 μA) applied to one axon electrode (fire layout). (c) Representative spiking activity recorded in one muscle electrode in response to increments of the stimuli (from 1 to 6, 1 Hz) applied to one axon electrode. (d) Pooled data of spiking activity changes recorded in muscles and axons in response to incremented stimuli (from 1 to 6, at 1 Hz). (e) Comparison of the spiking activity elicited by axon electrodes stimulations repeated either every 30 or 60 seconds (respectively upper panel and down panel). An interval of one minute between two stimuli allowed a complete recovery from the spiking activity increase. Scale bars: 100 μm. | ||
Using the stimulation protocol determined above, 3 MEAs exhibiting optimal functional NMJs were further studied. One of them, representative of all co-cultures, is pictured in Fig. 7a. Taking into account these 3 MEAs, 14 active muscles were considered active since they displayed more than 10 spikes per minute. It appeared that delivering 15 times the 4-pulse stimulation resulted in rather different patterns in AP muscle frequency, and that some muscle electrodes even displayed an “on–off” pattern (Fig. 7b), which was expected considering that the myoblast culture was a mix of different fiber populations (Fig. S2†). When looking at the number of spikes induced by the cumulated stimulations, muscle induced activity were gathered in Fig. 7c based on 3 responses: 10 out of 14 muscle electrodes displayed the “on–off” pattern previously mentioned, with the synchronization of the alternate activity. This type of synchronization could occur because a single axon AP is able to activate several muscle fibers at the same time.49 It is noteworthy that spike frequency increased until the 4th stimulation to reach a maximum, then every other time, the muscle was silent, suggesting that the stimulation occurred during a refractory period due either to the muscle itself or to a complete depletion of ACh vesicles at the active zone that must be refilled in order to trigger a new AP. In 2 out of 14 muscles, no changes in the number of spikes were observed. However, we noticed a decrease for the 2 remaining activated muscle electrodes depicting stimulation-induced “fatigue”. For each active electrode of every group, a threshold was set between basal frequencies and stimulation-induced frequencies and the number of spikes under or above this threshold were counted to assess the effect of stimulations for recorded low and high frequencies (Fig. 7d for example). This analysis supported the previous observation done with global number of APs, since “on–off” pattern is prominent for recorded high frequency APs of 10 muscles. Stable activity is visible for recorded low frequency APs of 2 muscles and decreased activity impacted both low and high frequency AP of the 2 remaining muscles (Fig. 7e). Moreover, the response of muscle over time seemed unrelated to a potential axonal fatigue, since global number of APs, as well as numbers of low and high frequency APs were rather stable during the course of the experiment (Fig. 7f). Taken together, our observations indicate that the outcome of the electrical stimulation of axons on muscle cell spiking activity was variable and unrelated to the chip co-culture quality, as the 3 different types of responses could be observed on the same chip. On the contrary, the different muscle activity response indicated that the presence of different muscle fibers could be detected. Here, the culture was mainly composed of slow fibers (type I), since it is known that they are less sensitive to fatigue, compared to fast fibers (type II) that exhibits a constant decreased response after repeated stimulations.50 Interestingly, it has been described that these different types of fibers depend on the motor neuron innervation during development, that can modulate the initial fibers properties, giving rise to distinct motor units, respectively named slow and fast motor units.51 However in human, fast fibers are separated in fast-fatigue resistant fibers IIa and fast-fatigue sensitive fibers IIx,52 giving the possibility that the model of co-culture established in this study might be relevant to electrically assess a phenotype change when muscles are co-cultured with MNs from patients.
 | ||
| Fig. 7 hNMJ-on-chip electrical assessment using custom MEA (a) Example of a co-culture grown on the MEA platform before stimulation and recording. (b) ‘On–off’ spiking activity changes elicited by axon stimulation. (c) Effect of repeated stimulation on global number of APs, means from 10 distinct muscle electrodes displaying an “on/off” pattern (left), from 2 distinct muscle electrodes displaying a constant pattern (center), and from 2 distinct muscle electrodes displaying “muscle fatigue” (right). (d) Representative traces of spiking activity recorded from one muscle electrode (left). The red line represents the threshold set to compare basal activity from stimulated activity by counting the number of low frequencies (under the threshold) and high frequency (above the threshold) to obtain the graph on the right. (e) Effect of repeated stimulations on the number of low and high frequencies for “on/off” muscles (left, n = 10) constant muscles (center, n = 2) and fatigued muscles (right, n = 2) based on analyses depicted in Fig. 6d. (f) Effect of repeated stimulations on neuronal spiking activity recorded by axon electrodes (n = 13 electrodes from 3 MEAs). Scale bars: 100 μm. | ||
Conclusions
Here we report the implementation of a platform that allows the formation of mature and functional human NMJs using MNs derived from hiPSCs and myoblasts from biopsies. This study uncovered an important role of muscular orientation, which allows single muscle activity recording after MN stimulation, with different muscle activities depending on muscle fibers. So far, most of the studies reported the formation of murine NMJs or used a mix of human and murine cells co-culture. Only a few reports rely on human NMJ, but none of them combine both microfluidics, microstructurations and MEA. Here, we have addressed these technical issues and give new insights for an in vitro model that recapitulates reliable human NMJs. To our knowledge, this platform represents a unique tool to record electric activity of post-synaptic endplate, close to the physiological system, where the fast and slow muscles fibers can be discriminated after evoked axonal stimulation. It is noteworthy, that the biological results described above were obtained using a motor neurons and myoblasts, each differentiated from a unique cell source, and the co-culture was optimized to obtain mature hNMJs. If the device developed here were to be used with other cell source, the timing of cell seeding might need to the optimized to obtain mature and functional hNMJ in order to further assess their AP activity. However, this limitation does not question the microfabrication and application of the custom device that was developed here. Indeed, once optimized with any cell source, studying hNMJ at the level of the action potential might be of great interest since it is the first event occurring after nicotinic receptor stimulation, activating voltage-dependent Na+ and Ca2+ channels, triggering a more general intracellular mobilization of calcium that in turn leads to contraction. This process is known as the excitation–contraction coupling.53Besides healthy hNMJ, such a platform could be extremely useful to better understand neurodegenerative diseases affecting motor neurons and achieve drug screening approaches. To study diseases mentioned in the introduction (SMA, ALS, DMD, Charcot–Marie–Tooth disease and Guillain–Barré syndrome), it might be of great interest to measure what occurs at the level of voltage-dependent channels. When considering neurodegenerative diseases, the model we established, could be used to detect electrical defects both at the presynaptic and postsynaptic zone. This custom MEA could also be used to study muscle diseases to highlight the role of ion channels or protein regulating them, considering that such channels or proteins are disturbed in channelopathies, such as Myasthenia gravis54 (one of the most common autoimmune NMJ channelopathy), Myotonia congenita55 (a congenital neuromuscular channelopathy), and the newly described channelopathy called TRPV4-pathy that includes congenital distal spinal muscular atrophy (dSMA), scapuloperoneal SMA, and hereditary motor and sensory neuropathy type IIC.56 For diseases where neuromuscular disorders arise from muscle defects, such as DMD, our device could detect the level of muscle disturbance impairing its calcium activity or its contraction: MN defaults, NMJ defaults, signal transmission defaults, or defaults proper to the muscle.
Materials and methods
Microstructured substrates
24 × 24 mm #1.5 thick glass slides were covered with a 2.7 μm thick layer silicon nitride (Si3N4) using a plasma enhanced chemical vapor deposition (PECVD, Corial D250, CORIAL S.A, France). Deposition was made at 280 °C for 20 min. The patterns of the micro grooves were then transferred to the glass slides by photolithography. For this purpose, a positive photoresist layer (AZ 1518, Microchemicals GmbH, Ulm, Germany) was spincoated and exposed through a photomask. After development, the patterns were transferred into the silicon nitride by plasma etching. The samples were placed under reactive ion etching (Corial 200 IL, CORIAL S.A, France) using a mixture of CHF3, Ar and oxygen gases during 7 min and the etching was controlled by laser interference. After photoresist stripping, the patterned glass blades were cleaned with a piranha solution (H2SO4 and hydrogen peroxide 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to remove any organic materials.
:1) to remove any organic materials.
Primary muscle cell culture and analysis on microstructured substrates
Quadriceps muscle biopsy was performed on one healthy adult at the Centre Hospitalier Universitaire Lapeyronie (Montpellier, France). The donor signed an informed written consent after the description of the protocol (authorization N° DC-2008-594). Myoblasts (muscle progenitor cells) were purified from the muscle biopsy and were cultured on collagen-coated dishes in DMEM/F12 medium with 10% fetal bovine serum (FBS), 0.1% Ultroser G and 1 ng ml−1 of human basic fibroblast growth factor (proliferation medium), as previously described. Myoblasts can differentiate morphologically by fusing with each other into long multinucleated cells and biochemically, these myotubes expressing specific skeletal muscle proteins. For cell differentiation, confluent cells were cultured in DMEM with 5% FBS (differentiation medium).57 For analysis of cellular orientation myoblasts were differentiated during 5 days in microfluidics devices without motor neurons, then, washed with PBS, fixed in 4% PFA for 20 minutes and washed again with PBS for further studies. Cells were stained with cell mask (CellMask™ Green Actin Tracking Stain, Thermo Fisher Scientific, cat. no. H32714) with a 10 mg ml−1 stock solution freshly diluted 1:1000 in PBS for 5 minutes and washed with PBS before adding Vectashield containing DAPI. Epifluorescence Nikon Ti2 inverted microscope was used to image 3 fields by chips, 6 chips were used for each condition with a 10× Plan Apochromat 0.45NA air objective. Using the directionality plugin of Fiji software, muscle orientation and goodness of fit R2 were obtained as described in Ko et al.43 For analysis of muscle fusion and maturation, cells were stained with anti-actinin (see Immunostaining section). Images were acquired with the same microscope and the same objectives to obtain the same number of replicates. Fiji software was used to select large region of interests (ROIs) to exclude muscle growing outside of the microstructuration. Images were then cropped and α-actinin and DAPI channels were split. DAPI images were analyzed using the Fiji software to obtain the number of nuclei per image, normalized with the total image area to compare all conditions. For this automatic counting, manual threshold was set to tell the nuclei apart from the background, and the number of nuclei was provided in the “Analyze > Analyze particles” command. α-Actinin images were analyzed using Fiji software to obtain the total mature myotube area by image (i.e., unorganized α-actinin positive cells), by setting a threshold to discriminate the stained cells from the background and measuring it thanks to the “Analyze > Measure” command, also normalized. The fusion index was calculated as the ratio of the nuclei number in unorganized α-actinin positive cells with three or more nuclei versus the total number of nuclei. This ratio was calculated by setting a threshold selection, inverting the selection, and pasting it on the DAPI images so that only the internal nuclei of myotubes appear, which were then counted with the same technique used to determine the total number of nuclei. For the percentage of striated fibers (organized α-actinin positive cells), the same chips were acquired with the inverted confocal Zeiss LSM780 microscope using the 40× Plan Apo oil 1.4NA objective. The maximum intensity projection was performed on Z series stacks, striated areas were measured using ROIs and compared to muscle total area as mentioned above (i.e., organized/unorganized α-actinin ratio).
Analysis of hIPSC-derived MN invasion in the muscle chamber
Motor neurons were cultured as described in the “hIPSC-derived MN and Myoblast co-culture” section and stained with Smi32 antibody as described in the Immunostaining section. Using the inverted confocal Zeiss LSM780 microscope with a Plan Apo 0.8NA, 4 to 5 fields were acquired for each chip, and 6 chips were used for each condition. Images were rotated with Fiji software so that the edge of the muscle chamber, where axons come in, were vertical. Distance of axon invasion was measured by tracing the longest horizontal lines starting at the exit of MN from each microchannels and ending at the point where axons were the farthest in the muscle chambers.hIPSC-derived MN and myoblast co-culture
Motor neurons differentiation was achieved following Maury's protocol58 with slight modifications. Briefly, iPSCs purchased from Coriell Institute (#GM24474) were grown in mTseR plus medium (StemCell Technologies) on vitronectin-coated dishes. After 5 days of iPSC expansion, embryonic bodies (EBs) were produced by iPSC dissociation and suspension in flasks containing mTesR plus medium. After one day, they were filtered on 37 μm reversible strainer (StemCell Technologies) and grown in suspension in BrainPhys medium, containing 0.02% NeuroCult SM1 without vitamin A and 0.01% N2 supplement-A (N2 medium), supplemented with LDN-193189 (0.1 μM), CHIR (3 μM), Ascorbic acid (10 μM) and SB-431542 (20 μM). On day 2, medium was replaced by fresh N2 medium supplemented with retinoic acid (RA, 100 nM) on top of the molecule cocktail previously used. On day 4, CHIR was replaced by SAG (500 nM). On day 7, N2 medium was only supplemented with AA, RA and SAG, and on day 9 with AA, DAPT (10 μM), RA and SAG. Also on day 9, microfluidic devices were precoated with poly-L-ornithine (pLO) O/N followed by laminin coating O/N. Depending on the experiment, coating concentration was optimized: for classical microfluidic devices, 20% of pLO stock (Sigma-Aldrich) and 10 μg ml−1 of laminin; for MEA devices, 26% of pLO stock and 5 μg ml−1 of laminin were used. On day 11, EBs were enzymatically dissociated by accutase treatment followed by mechanical dissociation, using tips, in N2 medium containing AA, BDNF (10 ng ml−1), DAPT, GDNF (10 ng ml−1), RA, RI (Y-27632 at 10 μM) and SAG. Isolated cells were then counted and plated in the microfluidic devices, at the density of 200000 cells per chip. For the MN compartment, medium was then changed 3 times a week with N2 medium supplemented with AA, BDNF, DAPT, GDNF and RA until maturation (from day 13 to day 24). Meanwhile, after two days of MN culture, myoblasts (10000 cells) from human biopsies (as described above) were seeded in the opposite compartment for two days in proliferation medium, then differentiated for nine additional days. After 24 days from the stage EB0, NMJ was subjected to further experiments.
Immunostaining
Cells on coverslips and chips integrated with PDMS chambers were fixed in 4% paraformaldehyde in 1× PBS for 30 min and washed twice with PBS at room temperature. For immunofluorescence studies, cells were washed twice in B1 buffer (0.1 M Tris-HCl at pH 7.5, 0.15 M NaCl), Cy3-conjugated α-bungarotoxin (Life Technologies; diluted 1 in 100) was added before permeabilization for 30 min. After PBS washing, permeabilization was performed with B1 buffer containing 0.1% Triton X-100 for 10 min, and then blocked with B1, 0.1% Triton X-100 containing either 2% donkey serum, 2% goat serum or 2% bovine serum albumin for 30 min at room temperature. Primary antibodies were diluted in the same buffer, mouse anti-α-actinin (Sigma-Aldrich, cat. no. A7811 1:100), mouse-anti-troponinT (Sigma-Aldrich, cat. no. T6277, 1:200), mouse-Smi-32 (Biolegend, 1:1000 or 1:500), mouse-anti-Bassoon (Antibodies Inc. 1:1000), goat-anti ChAT (Sigma-Aldrich, 1:250), mouse anti-myosin slow (Sigma-Aldrich, cat. no. M8421, 1:200) and mouse-anti-myosin fast (Sigma-Aldrich, cat. no. M4276) antibodies, O/N at 4 °C. Cells were washed twice in B1 buffer and secondary antibodies labeled with Alexa 488, Cy3 or Cy5 were used at 1:500 or 1:1000 dilution. After two PBS washing, Vectashield containing DAPI was added in the chambers or used for coverslip mounting on microscope slides for further imaging.
Neurofluidic circuits
The compartmentalized microfluidic circuit is composed of two main culture chambers, namely motor neuron and muscle chambers, of 4 mm long, 1 mm wide and 100 μm thick and open at each end by inlets and outlets made with a punched cylinder of 6 mm diameter. These two chambers are connected together by a set of 500 μm long microchannels of 5 × 5μm section. A master mold was made with SU-8 photoresist on a silicon wafer using a dual thickness photolithography process. A first layer of SU-8 2005 (5 μm high) was spin-coated on a 3 inches silicon wafer, then baked, exposed and developed for defining the microchannels. A second layer of SU-8 2100 (50 μm high) was subsequently spin-coated and aligned over the first level, and was developed to build seeding chambers and the synaptic compartment. Note that a short silicon over-etching was performed with reactive ion etching between the two steps so that the microchannel pattern was visible for alignment of the second mask. PDMS microfluidic chips were fabricated with standard procedures. A 10:1 mix of PDMS and curing agents were prepared and mixed together (Dow chemical, Sylgard 184). The mixture was then poured on the samples, degassed for 30 min under vacuum and cured at 80 °C for two hours. After dissection and extraction, PDMS circuits were punched to create 6 mm diameter seeding wells, and intensely washed with DI water. Finally, chips were treated by oxygen plasma (Plasmasystem Nano, Diener electronic GmbH, Ebhausen, Germany) allowing a permanent bonding microstructured glass slides. Alignment was made manually under binocular lense. The microfluidic chip on 24 × 24mm glass slides were then placed in 6 well plastic chambers and sterilized using UVs.
Lentivirus production
pAAV-Syn-GCaMP6f-WPRE-SV40 (plasmid #100837 Addgene) was subcloned into a lentivirus backbone plasmid under p-synapsin or p-ubiquitin promoters. These plasmids were delivered into cells by retroviral infection. The HEK293T cells were transiently transfected with a set of plasmids encoding retroviral proteins and producing genomic retroviral p-synapsin-GCaMP6f or p-ubiquitin-GCaMP6f particules. Lentiviral particles were collected 24–72 hours after transfection, passed through a 0.45 μm filter and precipitated using Lenti-X™ Concentrator (Clontech) before infection of recipient MNs or myocytes, respectively.Calcium imaging and fluorescence analyses
Genetically encoded sensors like GCaMP6f producing fluorescent calcium signal are widely used for monitoring the neuronal activity. It is among the best calcium indicators with high sensitivity, high fluorescence yield and relatively fast response time.45 Here, fluorescent sensors were delivered through lentiviral transduction in both differentiated MNs and myocytes on day 15 of culture. On day 24, cells stably expressed the calcium sensor, and microfluidic co-culture were imaged using Epifluorescence Nikon Ti2 inverted microscope and 20× Plan Apochromat 0.75NA air objective. For curare treatment (Tocris cat. no. 2820), calcium activity of muscles was recorded for 3 minutes before treatment, for 3 minutes after treatment, and for 3 minutes after two washes. Because curare is completely effective after 30 seconds, we only compared 2.5 minutes of recording for each condition. To assess global calcium activity in 4 chips, regions of interest (ROIs) were manually drawn on Fiji software to contain several muscles, and the raw fluorescence signals were extracted as the max fluorescence from individual ROIs at each time point of the recording, as well as the area of the ROIs. Maximum fluorescence signals were normalized to the area so that we could compare the replicates. For each, ΔF was measured for every time point and divided by the fluorescence baseline. The spikes of calcium were either positive or negative, so we calculated the absolute values |ΔF/F|. As the range of this values varied greatly between chips, we normalized it by setting the highest |ΔF/F| of each chip at 1. We then grouped all |ΔF/F|values of all chips for basal condition, curare condition and recovery condition. For TTX (Tocris cat. no. 1069) treatment, muscle calcium activity was recorded for 1 minute before treatment and 1 minute after. Since the TTX effect is not global, ROIs of same area were traced on 54 muscle fibers from 5 chips. The analysis was the same, except for the surface normalization that was not needed in this case.Contraction imaging and analyses
Microfluidic co-culture was imaged using Epifluorescence Nikon Ti2 inverted microscope and 20× Plan Apochromat 0.75NA air objective. Muscle contraction was recorded for 30 seconds before TTX (Tocris cat. no. 1069) treatment and 30 seconds after as described in Ionescu et al.24 region of interest (ROI) and normalized ROI intensity was performed as described in the same article.RNA extraction and RT-PCR for myosin isoform detection
Total RNA was extracted from the muscle compartment of 6 chips, using Direct-zol RNA MicroPrep with Zymo-Spin IC Columns (Zymo Research, Ozyme cat. no. ZR2060S) according to manufacturer's recommendations. First-strand cDNAs was synthesized from 400 ng of total RNA using 200 U M-MLV reverse transcriptase and 2.5 nM of N6 random primers. cDNA (1/5 of RT reaction) was used as a template for PCR reaction, and amplified with the GoTaq Polymerase (Promega), 20 pmol of primer pairs, 200 μM dNTPs and 1.5 mM MgCl2 in 1× GoTaq buffer, sequentially cycled 35 times. Primer sequences that were used are: Fw 5′-CTGCAAGCAAAGGTGAAATCCTAC-3′ and Rev 5′-CATTTTGTGCATTTCTTTGGTCACC-3′ (human MYH1 to detect type I fibers); Fw 5′-GGTCTCCATTTACAAGCTCACG-3′ and Rev 5′-GGACACCTGTTCTACAGTCTGG-3′ (human MYH2 to detect type IIa); Fw 5′-GGAGGTTCACACAAAAGTCATAAG-3′ and Rev 5′-CTTGATATACAGGACAGTGACAAAG-3′ (human MYH4 to detect type IIb); and Fw 5′-TACCAGACGGAGGAGGACAG-3′ and Rev 5′-CAAGATGTGGCAAAGCTACTCC-3′ (human MYH7 to detect type IIx). PCR products then were separated on 2.5% agarose gel containing ethidium bromide and visualized using Amersham Imager 680.Microelectrode array custom fabrication for NMJ
Square 49 × 49 mm, 500 μm-thick borofloat glass substrates (Paul Marienfeld GmbH & Co. KG, Germany) were first cleaned with acetone and isopropyl alcohol followed by N2 gun drying and plasma cleaning (nano low-pressure plasma system, Diener electronic GmbH, Germany) using a mixture of 75% oxygen and 25% nitrogen at 0,6 mbar for 6 min. The first lithography step consisted in a lift-off of metal electrodes. A 2 μm-thick layer of AZ nLOF 2020 negative photoresist (Microchemicals Gmbh, Germany) was spin-coated on glass samples at 3000 rpm and baked at 110 °C for 90 s. The first photomask was aligned with glass edges and exposed under UV light (EVG 680, EV Group, Austria) at a dose of 10 mJ cm−2. Samples were then submitted to an inversion bake at 110 °C for 90 s and developed using an AZ 726MIF developer for 50 s. After a short oxygen plasma ashing, samples were placed in a vacuum for metal deposition of a 10 nm Titanium adhesion layer and 150 nm Platinum using a Univex 350 electron gun evaporator (Leybold GmbH, Germany). Microchips were then processed for lift-off in a bath of Nano Remover PG (Microchemicals Gmbh, Germany) at 70 °C for 10 min followed by another bath in a clean solution of Remover PG and finally thoroughly washed using deionized water (DI) water and isopropyl alcohol (IPA). Surfaces were careful examined; another bath of remover was eventually made in order to completely strip off the PR and metal lines. A thin layer of 500 nm Silicon nitride (Si3N4) was deposited using plasma enhanced chemical vapor deposition (PECVD, Corial D250, CORIAL S.A, France) at 280 °C for 5 min under a mixture of SiH4, N2 and NH3 gazes to create the passivation layer. The second lithography step consisted in opening the passivation layer to create recording electrode sites, microgrooves and connection pads. An AZ nLOF 2020 negative photoresist was used with higher exposure dose to avoid undercut for lift-off. Substrate alignments were made using an EVG mask aligner. After development the samples were placed under reactive ion etching (Corial 200 IL, CORIAL S.A, France) using a mixture of CHF3, Ar and oxygen gazes. The etching was monitored by laser interference that allows etching end point detection. MEA chips were then cleaned using oxygen plasma etching (Corial 200 IL, CORIAL S.A, France) for 4 min. The chips were finally cleaned with IPA and DI water and final piranha wet etch and were carefully examined to detect eventual defects. The microfabrication process global yield reached 80%. The compartmentalized microfluidic circuit is finally manually aligned and permanently bonded on top of the MEA using oxygen plasma.Electrophysiological recordings and analysis
Experiments were carried out at 37 °C by placing the MEAs into a temperature-controlled incubator under humidified CO2 (5%) atmosphere. Spontaneous action potentials were recorded with the amplifier MEA2100-Mini-60-System (Multichannelsystems, Harvard Bioscience). Stimuli were applied to a selected microelectrode thanks to an integrated stimulator. They consisted of biphasic pulses of 100 or 200 μA amplitude and 100 μseconds duration.59 They were delivered at 1 Hz frequency. Data storage and analyses were performed with the MCS-Experimenter and MCS-Analyzer softwares, respectively (Multichannelsystems, Harvard Bioscience). Data were high pass filtered at 200 Hz (Butterworth filter). Action potential count and frequency calculation were obtained by setting detection threshold at 50 μV for both positive and negative ongoing signals with 5 millisecond deadtime. Electrical activity of every electrode was recorded for 16 minutes (1 minute in basal condition, and 15 after stimulations). For each minute and each MEA, data were exported from the MCS-Analyzer software in two ASCII files containing numerical results and instantaneous parameters. Numerical results contained the number of spikes for each electrode. These sheets were pasted in a homemade Excel template to automatically sort MN cell body electrodes, from axon ones, and from muscle ones. The template also discriminated active and inactive electrodes with a threshold of 10 spikes per minute. Instantaneous parameters contained all the frequencies registered for each electrode at each time point. These sheets were also pasted in a homemade Excel template. By writing the number of one active electrode, it automatically extracted and ordered the frequencies of the mentioned electrode for the basal condition and the 15 stimulations from all the data. The analysis of spontaneous activity revealed low and high spiking activity, so the same template was used to set a threshold between the low values observed in the basal condition and higher values obtained after stimulation and to count the number of frequencies present in both groups. Spiking frequencies were thus separated into two groups of high or low frequencies.Statistical analysis
Statistical analyses of data sets were performed using GraphPad Prism version 9.0.2 for macOS, GraphPad Software, La Jolla, CA, USA. Unless indicated, data are presented as the mean ± SEM. One-way ANOVA or unpaired t-test was used for muscle directionality depending on the number of samples compared; Kruskal–Wallis test for the fusion index, number of nuclei, myotube area and striated fiber percentage; Mann–Whitney test was used for MN invasion and curare effect; Welch's test was used for TTX effect; and 2 way ANOVA for MEA analysis.Microscopy
Fluorescence acquisition was performed either with the inverted Epifluorescence Nikon Ti2 microscope controlled by the NIS Element software or with the inverted confocal Zeiss LSM780 microscope controlled the Zeiss Zen Software from the CRBM-MRI facility. Images were analyzed by Fiji software. Brightfield acquisition was performed with Evos FL microscope using a 10× objective and images were reconstituted as one mosaic using the Stitching plugin on Fiji software.60Author contributions
Conceptualization, PD, BC and FR. Formal analysis, PD supported by MV and GH. Funding acquisition, BC and FR. Investigation, PD. Methodology, PD, MV and FR. Project Administration, PD and FR. Resources GH, AS, GC, EM. Supervision, PD and FR. Validation, PD and FR. Visualization PD. Writing – original draft, PD, MV, BC and FR. Writing – review & editing, all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a grant from i-Site MUSE (ANR-16-IDEX-0006). PD was supported by a PhD fellowship from the same i-Site MUSE grant. The authors thank the staff of the imaging facility MRI, Biocampus. The authors acknowledge Frederic Pichot and Jean Marie Peiris from Centrale de Technologie en Micro et nanoélectronique (CTM) for their support in the clean room engineering. The authors would also like to acknowledge Dr Frank Hoffmann (Multichannelsystems) for his invaluable help for setting up electrophysiological recordings with MEAs. The authors are grateful to Drs Johann Soret and Eran Perslon for their critical review of the manuscript, and are specifically grateful for the useful support and discussion provided by Dr Johann Soret during the early stages of this project.Notes and references
- R. G. Webster, Int. J. Mol. Sci., 2018, 19, 1326 CrossRef PubMed.
- A. D. Ebert, J. Yu, F. F. Rose Jr, V. B. Mattis, C. L. Lorson, J. A. Thomson and C. N. Svendsen, Nature, 2009, 457, 277–280 CrossRef CAS PubMed.
- M. Yoshida, S. Kitaoka, N. Egawa, M. Yamane, R. Ikeda, K. Tsukita, N. Amano, A. Watanabe, M. Morimoto, J. Takahashi, H. Hosoi, T. Nakahata, H. Inoue and M. K. Saito, Stem Cell Rep., 2015, 4, 561–568 CrossRef CAS PubMed.
- X. Guo, M. Gonzalez, M. Stancescu, H. Vandenburgh and J. Hickman, Biomaterials, 2011, 32, 9602–9611 CrossRef CAS PubMed.
- L. M. Murray, L. H. Comley, D. Thomson, N. Parkinson, K. Talbot and T. H. Gillingwater, Hum. Mol. Genet., 2008, 17, 949–962 CrossRef CAS PubMed.
- E. B. Moloney, F. de Winter and J. Verhaagen, Front. Neurosci., 2014, 8, 252 Search PubMed.
- R. M. Lovering, S. R. Iyer, B. Edwards and K. E. Davies, Neurosci. Lett., 2020, 737, 135304 CrossRef CAS PubMed.
- C. R. Slater, Prog. Neurobiol., 2015, 134, 55–103 CrossRef PubMed.
- R. A. Jones, C. Harrison, S. L. Eaton, M. Llavero Hurtado, L. C. Graham, L. Alkhammash, O. A. Oladiran, A. Gale, D. J. Lamont, H. Simpson, M. W. Simmen, C. Soeller, T. M. Wishart and T. H. Gillingwater, Cell Rep., 2017, 21, 2348–2356 CrossRef CAS PubMed.
- I. Boehm, A. Alhindi, A. S. Leite, C. Logie, A. Gibbs, O. Murray, R. Farrukh, R. Pirie, C. Proudfoot, R. Clutton, T. M. Wishart, R. A. Jones and T. H. Gillingwater, J. Anat., 2020, 237, 827–836 CrossRef PubMed.
- B. Zhang, W. C. Xiong and L. Mei, Dev. Cell, 2009, 16, 325–327 CrossRef CAS PubMed.
- A. Barik, B. Zhang, G. S. Sohal, W.-C. Xiong and L. Mei, Dev. Neurobiol., 2014, 74, 828–838 CrossRef CAS PubMed.
- R. M. Nitkin, M. A. Smith, C. Magill, J. R. Fallon, Y. M. Yao, B. G. Wallace and U. J. McMahan, J. Cell Biol., 1987, 105, 2471–2478 CrossRef CAS PubMed.
- U. J. McMahan, Cold Spring Harbor Symp. Quant. Biol., 1990, 55, 407–418 CrossRef CAS PubMed.
- N. Kim, A. L. Stiegler, T. O. Cameron, P. T. Hallock, A. M. Gomez, J. H. Huang, S. R. Hubbard, M. L. Dustin and S. J. Burden, Cell, 2008, 135, 334–342 CrossRef CAS PubMed.
- Y. Zong and R. Jin, Cell. Mol. Life Sci., 2013, 70, 3077–3088 CrossRef CAS PubMed.
- S. Torino, B. Corrado, M. Iodice and G. Coppola, Inventions, 2018, 3, 65 CrossRef.
- A. M. Taylor, S. W. Rhee and N. L. Jeon, Methods Mol. Biol., 2006, 321, 167–177 CAS.
- A. M. Taylor, M. Blurton-Jones, S. W. Rhee, D. H. Cribbs, C. W. Cotman and N. L. Jeon, Nat. Methods, 2005, 2, 599–605 CrossRef CAS PubMed.
- K. A. Southam, A. E. King, C. A. Blizzard, G. H. McCormack and T. C. Dickson, J. Neurosci. Methods, 2013, 218, 164–169 CrossRef PubMed.
- H. S. Park, S. Liu, J. McDonald, N. Thakor and I. H. Yang, in 2013 35th Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC), 2013, pp. 2833–2835 Search PubMed.
- T. Osaki, S. G. M. Uzel and R. D. Kamm, Nat. Protoc., 2020, 15, 421–449 CrossRef CAS PubMed.
- E. E. Zahavi, A. Ionescu, S. Gluska, T. Gradus, K. Ben-Yaakov and E. Perlson, J. Cell Sci., 2015, 128, 1241–1252 CAS.
- A. Ionescu, E. E. Zahavi, T. Gradus, K. Ben-Yaakov and E. Perlson, Eur. J. Cell Biol., 2016, 95, 69–88 CrossRef CAS PubMed.
- R. Mills, H. Taylor-Weiner, J. C. Correia, L. Z. Agudelo, I. Allodi, C. Kolonelou, V. Martinez-Redondo, D. M. S. Ferreira, S. Nichterwitz, L. H. Comley, V. Lundin, E. Hedlund, J. L. Ruas and A. I. Teixeira, Mol. Metab., 2018, 7, 12–22 CrossRef CAS PubMed.
- N. Santhanam, L. Kumanchik, X. Guo, F. Sommerhage, Y. Cai, M. Jackson, C. Martin, G. Saad, C. W. McAleer, Y. Wang, A. Lavado, C. J. Long and J. J. Hickman, Biomaterials, 2018, 166, 64–78 CrossRef CAS PubMed.
- U. S. Bauer, R. van de Wijdeven, R. N. Raveendran, V. Fiskum, C. Kentros, I. Sandvig and A. Sandvig, bioRxiv, 2019, DOI:10.1101/745513.
- J. Bellmann, R. Y. Goswami, S. Girardo, N. Rein, Z. Hosseinzadeh, M. R. Hicks, V. Busskamp, A. D. Pyle, C. Werner and J. Sterneckert, Biomaterials, 2019, 225, 119537 CrossRef CAS PubMed.
- X. Guo, V. Smith, M. Jackson, M. Tran, M. Thomas, A. Patel, E. Lorusso, S. Nimbalkar, Y. Cai, C. W. McAleer, Y. Wang, C. J. Long and J. J. Hickman, Adv. Ther., 2020, 3 DOI:10.1002/adtp.202000133.
- C.-Y. Lin, M. Yoshida, L.-T. Li, A. Ikenaka, S. Oshima, K. Nakagawa, H. Sakurai, E. Matsui, T. Nakahata and M. K. Saito, JCI Insight, 2019, 4 DOI:10.1172/jci.insight.124299.
- C. M. Didier, A. Kundu, D. DeRoo and S. Rajaraman, J. Micromech. Microeng., 2020, 30, 103001 CrossRef.
- Y. Nam and B. C. Wheeler, Crit. Rev. Biomed. Eng., 2011, 39, 45–61 CrossRef PubMed.
- S.-A. Chong, I. Benilova, H. Shaban, B. De Strooper, H. Devijver, D. Moechars, W. Eberle, C. Bartic, F. Van Leuven and G. Callewaert, Neurobiol. Dis., 2011, 44, 284–291 CrossRef CAS PubMed.
- B. J. Wainger, E. Kiskinis, C. Mellin, O. Wiskow, S. S. W. Han, J. Sandoe, N. P. Perez, L. A. Williams, S. Lee, G. Boulting, J. D. Berry, R. H. Brown, M. E. Cudkowicz, B. P. Bean, K. Eggan and C. J. Woolf, Cell Rep., 2014, 7, 1–11 CrossRef CAS PubMed.
- A. Virlogeux, E. Moutaux, W. Christaller, A. Genoux, J. Bruyere, E. Fino, B. Charlot, M. Cazorla and F. Saudou, Cell Rep., 2018, 22, 110–122 CrossRef CAS PubMed.
- D. Jans, G. Callewaert, O. Krylychkina, L. Hoffman, F. Gullo, D. Prodanov and D. Braeken, J. Pharmacol. Toxicol. Methods, 2017, 87, 48–52 CrossRef CAS PubMed.
- G. H. Kim, K. Kim, E. Lee, T. An, W. Choi, G. Lim and J. H. Shin, Materials, 2018, 11, 1995 CrossRef PubMed.
- M. O. Heuschkel, M. Fejtl, M. Raggenbass, D. Bertrand and P. Renaud, J. Neurosci. Methods, 2002, 114, 135–148 CrossRef PubMed.
- M. O. Heuschkel, C. Wirth, E.-M. Steidl and B. Buisson, in Advances in Network Electrophysiology: Using Multi-Electrode Arrays, ed. M. Taketani and M. Baudry, Springer US, Boston, MA, 2006, pp. 69–111 Search PubMed.
- D. A. Soscia, D. Lam, A. C. Tooker, H. A. Enright, M. Triplett, P. Karande, S. K. G. Peters, A. P. Sales, E. K. Wheeler and N. O. Fischer, Lab Chip, 2020, 20, 901–911 RSC.
- M. E. J. Obien, A. Hierlemann and U. Frey, Sci. Rep., 2020, 10, 4847 CrossRef CAS PubMed.
- E. Moutaux, B. Charlot, A. Genoux, F. Saudou and M. Cazorla, Lab Chip, 2018, 18, 3425–3435 RSC.
- E. Ko, S. J. Yu, G. J. Pagan-Diaz, Z. Mahmassani, M. D. Boppart, S. G. Im, R. Bashir and H. Kong, Adv. Sci., 2019, 6, 1801521 CrossRef PubMed.
- E. D. Gundelfinger, C. Reissner and C. C. Garner, Front. Synaptic Neurosci., 2016, 7, 19 Search PubMed.
- T.-W. Chen, T. J. Wardill, Y. Sun, S. R. Pulver, S. L. Renninger, A. Baohan, E. R. Schreiter, R. A. Kerr, M. B. Orger, V. Jayaraman, L. L. Looger, K. Svoboda and D. S. Kim, Nature, 2013, 499, 295–300 CrossRef CAS PubMed.
- L. Huang, P. Ledochowitsch, U. Knoblich, J. Lecoq, G. J. Murphy, R. C. Reid, S. E. de Vries, C. Koch, H. Zeng, M. A. Buice, J. Waters and L. Li, eLife, 2021, 10, 1–19 Search PubMed.
- M. E. J. Obien, K. Deligkaris, T. Bullmann, D. J. Bakkum and U. Frey, Front. Neurosci., 2015, 8 DOI:10.3389/fnins.2014.00423.
- I. V. Kubasov and M. Dobretsov, J. Physiol., 2012, 590, 2937–2944 CrossRef CAS PubMed.
- H. Nishimune and K. Shigemoto, Neurol. Clin., 2018, 36, 231–240 CrossRef PubMed.
- R. E. Burke, D. N. Levine and F. E. Zajac, Science, 1971, 174, 709–712 CrossRef CAS PubMed.
- S. Schiaffino, M. Sandri and M. Murgia, Physiology, 2007, 22, 269–278 CrossRef CAS PubMed.
- V. Smerdu, I. Karsch-Mizrachi, M. Campione, L. Leinwand and S. Schiaffino, Am. J. Physiol., 1994, 267, C1723–1728 CrossRef CAS PubMed.
- E. O. Hernández-Ochoa and M. F. Schneider, Skeletal Muscle, 2018, 8, 22 CrossRef PubMed.
- M. Bardhan, H. Dogra and D. Samanta, in StatPearls, StatPearls Publishing, Treasure Island (FL), 2021 Search PubMed.
- E. S. Bryan and M. Alsaleem, in StatPearls, StatPearls Publishing, Treasure Island (FL), 2021 Search PubMed.
- J. Dai, T.-J. Cho, S. Unger, E. Lausch, G. Nishimura, O.-H. Kim, A. Superti-Furga and S. Ikegawa, J. Hum. Genet., 2010, 55, 400–402 CrossRef PubMed.
- M. Kitzmann, A. Bonnieu, C. Duret, B. Vernus, M. Barro, D. Laoudj-Chenivesse, J. M. Verdi and G. Carnac, J. Cell. Physiol., 2006, 208, 538–548 CrossRef CAS PubMed.
- Y. Maury, J. Côme, R. A. Piskorowski, N. Salah-Mohellibi, V. Chevaleyre, M. Peschanski, C. Martinat and S. Nedelec, Nat. Biotechnol., 2015, 33, 89–96 CrossRef CAS PubMed.
- F. Lanté, M.-C. de J. Ferreira, J. Guiramand, M. Récasens and M. Vignes, Hippocampus, 2006, 16, 345–360 CrossRef PubMed.
- S. Preibisch, S. Saalfeld and P. Tomancak, Bioinformatics, 2009, 25, 1463–1465 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1lc00497b |
| This journal is © The Royal Society of Chemistry 2021 |