
An integrated microfluidic system for early detection of sepsis-inducing bacteria†
Yen-Ling
Fang
a,
Chih-Hung
Wang
a,
Yi-Sin
Chen
a,
Chun-Chih
Chien
b,
Feng-Chih
Kuo
 c,
Huey-Ling
You
b,
Mel S.
Lee
*c and
Gwo-Bin
Lee
*ade
c,
Huey-Ling
You
b,
Mel S.
Lee
*c and
Gwo-Bin
Lee
*ade
aDepartment of Power Mechanical Engineering, National Tsing Hua University, Hsinchu 30013, Taiwan. E-mail: gwobin@pme.nthu.edu.tw; Tel: +886 3 5715131 Ext. 33765
bLaboratory Medicine, Kaohsiung Chang Gung Memorial Hospital, Taiwan
cDepartment of Orthopaedic Surgery, Kaohsiung Chang Gung Memorial Hospital, Chang Gung University, Kaohsiung 83301, Taiwan. E-mail: mellee@cgmh.org.tw; Tel: +886 7 7317123 Ext. 3286
dInstitute of Biomedical Engineering, National Tsing Hua University, Taiwan
eInstitute of Nano-Engineering and Microsystems, National Tsing Hua University, Taiwan
First published on 16th November 2020
Abstract
Since early diagnosis of sepsis may assist clinicians in initiating timely, effective, and prognosis-improving antibiotic therapy, we developed an integrated microfluidic chip (IMC) for rapid isolation of both Gram-positive and Gram-negative bacteria from blood. The device comprised a membrane-based filtration module (90 min operating time), a bacteria-capturing module using a micro-mixer containing magnetic beads coated with “flexible neck” regions of mannose-binding lectin proteins for bacteria capture (20 min), and a miniature polymerase chain reaction (PCR) module for bacteria identification (90 min via TaqMan® probe technology). The filter separated all white blood cells and 99.5% of red blood cells from bacteria, which were captured at rates approaching 85%. The PCR assay's limit of detection was 5 colony-forming units (CFU) per reaction, and the entire process was completed in only 4 h. Since this is far less than that for culture-based approaches, this IMC may serve as a promising device for detection of sepsis.
Introduction
Sepsis is a life-threatening disease caused by the immune system's response to blood pathogens, including bacteria, fungi, and viruses.1,2 It can lead to insufficient blood flow, organ failure, and even death.3 Unfortunately, sepsis is often diagnosed too late because its symptoms are also associated with other pathologies, leading to misdiagnosis of this “hidden killer”. Therefore, fast and precise identification of sepsis-inducing bacteria is key to proper administration of therapy. Blood-borne pathogens are normally at low concentrations relative to white and red blood cells (white blood cells (WBCs) and red blood cells (RBCs), respectively) and must be cultured for 24–72 h prior to detection and use in antibiotic susceptibility tests (ASTs).4,5 By the time a positive result is obtained, the patient may already be suffering from severe sepsis, septic shock, or even organ failure. Additionally, some bacteria multiply slowly and generate low microbial activity signals and even false-negative results. If the patient was administered with antibiotics prior to blood collection, AST results become even more difficult to interpret.6 The acquisition of a sufficient blood volume for culture is also a challenge for some patients, especially neonates.7 Although the lengthy, labor-intensive, culture-based method is the current gold standard, it is clear that a superior approach is needed.Recently, microfluidic chips have been demonstrated to rapidly isolate bacteria from blood. For instance, a simple, low-cost microfluidic chip with a curved microchannel was capable of separating airborne microorganisms from large particles/cells based on inertial differences.8 Large particles/cells migrated into the outer outlet while small particles remained in the streamline due to inertial forces, and 70% of 3 μm particles were separated at the first outlet while 70% of Staphylococcus epidermidis and Adenoviridae cells were transported into the second and third outlets, respectively. Alternatively, an elasto-inertial microfluidic chip was reported to separate bacteria from whole blood.9 In this device, viscoelastic flow enabled size-based migration of blood cells into a non-Newtonian solution, while smaller bacteria remained in the blood. With this approach, 76% of the Escherichia coli were recovered from the side outlet while 92% of the WBCs were separated into the middle outlet. However, owing to the fact that a synergistic effect of the elastic and inertial forces occurs at modest flow rates, relatively low volumetric flow rates must be used with the elasto-inertial microfluidic devices. For instance, it would take about 17 h to process 1 mL of blood, too lengthy a time for diagnosis of sepsis.
Affinity probes can also isolate microorganisms from solutions, including blood.10 Target microorganisms were first captured by probes immobilized on magnetic beads or in microchannels and were thereafter extracted. For instance, antibody-functionalized super-paramagnetic beads captured 71% of Salmonella typhimurium from raw milk.11 This approach utilized specifically-functionalized surfaces to improve capture efficiencies at low working pressures (and consequently low clogging rates). Furthermore, membrane filtration approaches have been used to separate particles/cells of different sizes, such as extracellular vesicles (EVs) and leukocytes, from blood.12–16
Porous polymer monoliths have also been used to remove cells and contaminating debris from blood while allowing small, EV-sized particles to pass through for downstream analysis.17 Although this method does not require lengthy centrifugation steps or probes, clogging is common and may limit the throughput to the nL level.17 Therefore, the processing time needed to ensure that clogging is limited diminishes recovery rates and throughput, nor can these monoliths be readily integrated with other microfluidic devices. A vortex-type micro-mixer utilizing pneumatically driven membranes that induced tangential velocity and swirling flow for fluid mixing somewhat alleviated this clogging issue; when supplied with compressed air, the oppositely positioned polydimethylsiloxane (PDMS) membranes were deflected in tandem to generate a vortex flow field, and the mixing efficiency was ∼95% within 0.6 s.18 In another work, an integrated microfluidic chip (IMC) with a membrane-based filtration module and a micro-mixer was capable of continuously agitating cells to prevent filter clogging; filtration (with a pore size of 0.2 μm) was then performed to capture circulating EVs from blood via antibody-coated magnetic beads for consequent quantification with an on-chip sandwich-like assay.19
PCR has also been used to detect microbial genes in just a few hours, and miniaturized PCR devices have attracted particular interest in recent years due to their compactness, speed, efficiency, and ease of integration with other modules.20–22 For instance, a miniaturized PCR system could detect influenza A virus genes in only 45 min.23 Herein, we reported an IMC consisting of three modules: 1) a membrane-based filtration module (improved upon a prior design), 2) a bacteria-capturing module featuring a micro-mixer containing magnetic beads surface-coated with a novel affinity probe, and 3) a PCR module for bacteria identification. We hypothesized that, with this device, we could detect low concentrations of sepsis-inducing bacteria from human blood in a significantly shorter amount of time than culture-based approaches while avoiding the membrane filter clogging issues associated with prior technologies.19
Materials and methods
Experimental procedure and setup
The experimental procedures (Fig. 1) included sample treatment with the stirring-enhanced filtration module, mixing of magnetic beads and bacteria by a micro-mixer, bead collection, on-chip PCR, and bacterial identification by fluorescence detection (TaqMan® probes, Applied Biosystems, USA). Whole blood was first collected using 21-gauge needles (BD Vacutainer® Precision Glide™, catalog no. 360213, USA) from healthy donors that gave informed consent and stored in citrate-based anticoagulant tubes (BD Vacutainer, acid citrate dextrose solution A, catalog no. 364606) at 4 °C to prevent clotting. Within 3 h, the blood was spiked with bacteria and injected into the opening of the stirring-enhanced filtration module. In other experiments, sepsis-causing bacteria were instead used as positive controls. Clinically-isolated bacterial samples of Escherichia coli, Klebsiella pneumoniae, Pseudomonas aeruginosa, Staphylococcus epidermidis, and Staphylococcus saprophyticus were provided by the Department of Laboratory Medicine at Kaohsiung Chang Gung Memorial Hospital under the approval of the Institutional Bio-safety Committee. Note that these are the most common bacteria for causing sepsis.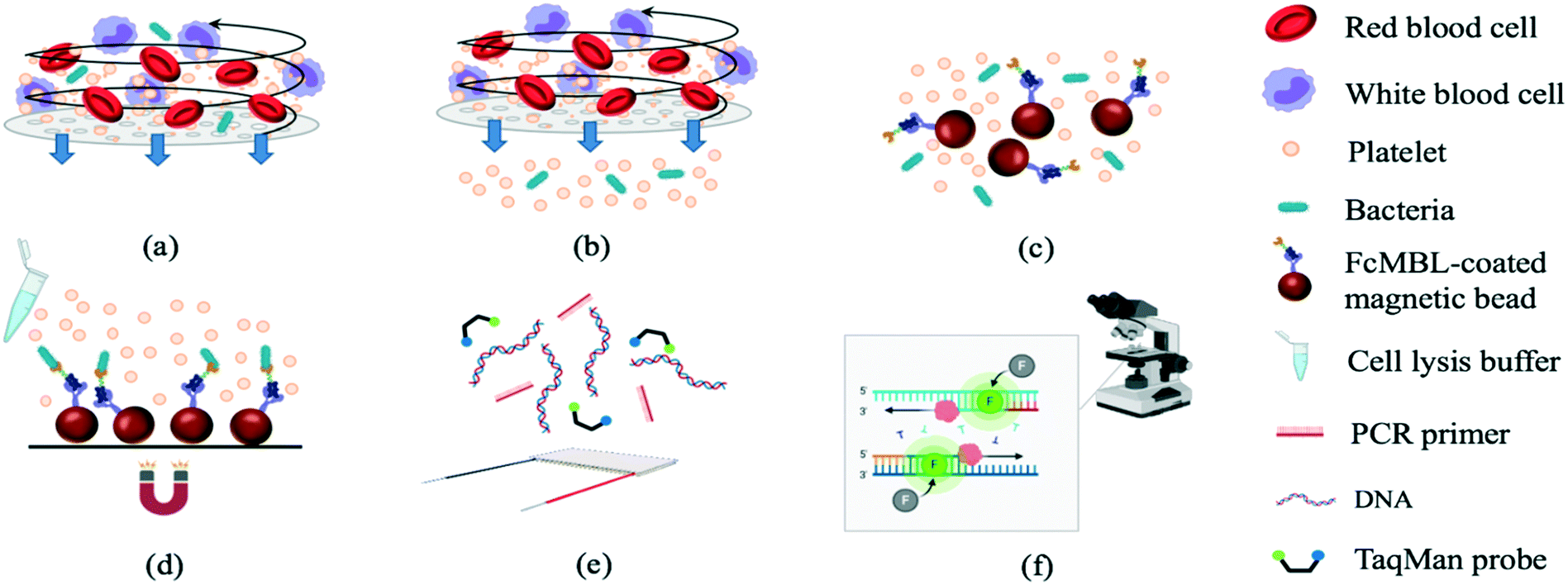 | ||
| Fig. 1 Isolation and identification of bacteria from human blood. (a) Human blood with bacteria loaded into the chip. (b) Plasma enriched with bacteria separated from off-target cell types. (c) Isolated bacteria transferred into a micro-mixer chamber and mixed gently with FcMBL-coated magnetic beads. (d) Bead–bacteria complexes collected using an external magnet. (e) Target bacterial DNA amplified by PCR. (f) Fluorescence signals detected using a PMT (bacteria identification). | ||
The stirring-enhanced filtration module was activated using compressed air (15 kPa) and vacuum (−20 kPa) pressures controlled by electro-magnetic valves (EMVs; SMC, S070M-5BG-32, Japan) at a certain driving frequency. Blood cells and bacteria were agitated by the vortex-type micro-stirrer, and only cells <1 μm (i.e. bacteria) passed through a nucleopore track-etched polycarbonate membrane (WHA110610 [1 μm], Whatman, UK). The vortex flow generated by the deflected PDMS membranes kept the membrane from being clogged by blood cells such that gentle and continuous separation of bacteria from the blood could be achieved. All experiments were performed in accordance with the guidelines from Kaohsiung Chang Gung Memorial Hospital (KCCMH), and approved by the ethics committee at KCCMH (IRB no. 201800535B0). The study participants were fully informed regarding the purposes of the study and consent was obtained.
The WBC/RBC-free plasma was drawn by a vacuum and deposited into a collection chamber at the bottom of the chip prior to transport into the micro-mixer, which was loaded with magnetic beads coated with a probe targeting the “flexible neck” regions of mannose-binding lectin (FcMBL; Fig. 2) through a micro-pump. Since FcMBL can be biotinylated at the/ N-termini to permit attachment onto magnetic beads, they tend to have high bacterial capture rates.24 FcMBL proteins (40 μg in 266 μL, Sino Biological, China) were incubated with 50 μL of 20 mg mL−1 protein A-coated magnetic beads (diameter = 500 nm, So-Fe Biomedicine, China) for 1 h on a wheeling rotator at 20 revolutions per minute (RPM) at room temperature.
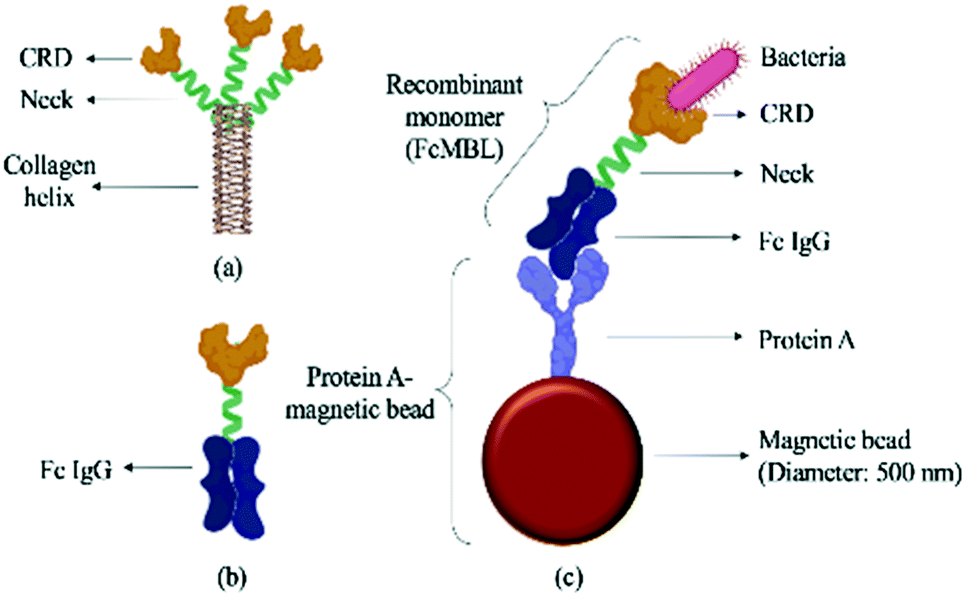 | ||
| Fig. 2 Schematic illustration of a FcMBL-coated magnetic bead. (a) The molecular structure of mannose-binding lectin, which features a collagen-like domain, a neck region, and a carbohydrate recognition domain (CRD).36 Single molecules of MBL associate to form a functional trimeric subunit that can further associate to form a hexamer of trimers. (b) FcMBL is an engineered version of MBL created by fusing the CRD to the flexible neck of the Fc portion of IgG. (c) FcMBL was biotinylated at its N-terminus to permit oriented attachment to protein A-coated magnetic beads. | ||
Afterwards, unbound FcMBL was washed out with 500 μL of 0.02% Tween-20 (Sigma, USA), and the solution was resuspended in 1000 μL of 1× phosphate buffered saline (PBS). After gently mixing bacteria with these beads for 20 min, the bead–bacteria complexes were collected using a magnet. The unbound material was washed away, and distilled water was added to the micro-mixer to resuspend the bead–bacteria complexes. This solution was then distributed equally to four chambers containing PCR reagents designed to detect genes from target bacteria: 0.5 μL of 10 mM dNTPs (Promega, USA), 3 μL of 10× SuperMix buffer (GeneDireX, Taiwan), 1 μL of bacterial gene specific primers (10 μM, 0.5 μL of each of the forward and reverse primers; Table 1), 0.5 μL of Taq DNA polymerase (5 U μL−1, Premix Ex Taq, Takara, Japan), 0.5 μL of 10 μM TaqMan probe (Takara, Japan; Table 1), and 4.5 μL of double-distilled water (ddH2O); either bacterial DNA (in optimization experiments) or the bead–bacteria complex solution comprised the remaining 20 μL.25 Note that captured bacteria were thermally lysed to release genomic DNA before performing on-chip PCR. Both 6-carboxyfluorescein (FAM) and Fluor-Red 610 (DLO) probes were designed so that two bacteria types could be detected within the same chamber. Thermocycling was as follows: 95 °C for 5 min followed by 30 cycles at 95 °C for 20 s, 53, 56, or 59 °C for 15 s (for optimization tests), and 72 °C for 20 s. Slab-gel electrophoresis was further used to confirm the PCRs. Briefly, two grams of Low EEO agarose (FocusBio, Taiwan) were dissolved in 100 mL tris/borate/ethylenediaminetetraacetic acid (TBE) buffer (Amresco, USA) which resulted in a 2% agarose gel. Furthermore, a PCR primer set targeting a conserved region of the bacterial 16S ribosome ribonucleic acid (16S rRNA) gene was used as a control to detect bacteria.2
| Bacteria | Amplified gene | Primer sequence |
|---|---|---|
| Escherichia coli | uidA | Forward: 5′-TGGTAATTACCGACGAAAACGGC-3′ |
| Reverse: 5′-ACGCGTGGTTACAGTCTTGCG-3′ | ||
| TaqMan: 5′-FAM-ACACCACGCCGAACACCTGG-BHQ-1-3′ | ||
| Klebsiella pneumoniae | Rob | Forward: 5′-CGACGGTGTGGTTACTGACG-3′ |
| Reverse: 5′-TCTACGAAGTGGCCGTTTTC-3′ | ||
| TaqMan: 5′- DLO-CCTGTCTGCTATCGAAGAAGGC-BHQ-2-3′ | ||
| Pseudomonas aeruginosa | DUF484 family protein | Forward: 5′-ACGAACTGGCGTTCCTCTT-3′ |
| Reverse: 5′-GTGCCGAGGGAACTCTTGTA-3′ | ||
| TaqMan: 5′- FAM-CACGGCGTGCTCGCCATCGG-BHQ-1-3′ | ||
| Staphylococcus epidermidis | Sep | Forward: 5′-GGCAAATTTGTGGGTCAAGA-3′ |
| Reverse: 5′-TGGCTAATGGTTTGTCACCA-3′ | ||
| TaqMan: 5′-FAM-CCGTATCCTGGTAATAGTGATTTAGCA-BHQ-1-3′ | ||
| Staphylococcus saprophyticus | HrcA | Forward: 5′-GACCTTTCCTCTACATTGAG-3′ |
| Reverse: 5′-CCTGATGTAAACACAACCAC-3′ | ||
| TaqMan: 5′-DLO-TTGATTAGAGCAAATGCTTATTTGGTT-BHQ-2-3′ |
A thermoelectric (TE) cooler (TECI 241.10, Tande Energy and Temperature Associates, Taiwan) and a thermocouple (TP-01-1M, Centenary Materials, Taiwan) for temperature feedback control (both placed underneath the PCR chambers) controlled PCR thermocycling. Note that captured bacteria were lysed thermally at 95 °C for 5 min by using this TE cooler. An Arduino micro-controller (UNO, Italy) was used to control the EMVs and the TE cooler. The resulting fluorescence signals emitted by the TaqMan probes permitted bacteria identification upon switching the optical filters, and fluorescence signals corresponding to successful bacterial identification were acquired with a photomultiplier tube (PMT; C3830, Hamamatsu Photonics, Japan) attached to a microscope (BX43, Olympus, Japan). Then, optical signals were captured using a DS-Qi1Mc camera (Nikon, Japan) and converted into electrical signals. As a benchtop comparison to the IMC, 50 μL of magnetic beads (20 mg mL−1) were added to 200 μL of bacteria (102 CFU per mL) and incubated via a wheeling rotator (DRM-36, Double Eagle Enterprise, Taiwan) at 20 RPM (C2 mode) for 1 h. Afterwards, the supernatant was removed, and the magnetic beads were collected via a DynaMag™-2 magnet (Thermo Fisher Scientific, USA).
Fabrication and design of the microfluidic chip
The 85 × 71 × 23 mm (L × W × H) IMC (Fig. 3a–c) was fabricated using a standard PDMS soft lithography process with polymethyl methacrylate (PMMA) master molds (Da Teh Hong, Taiwan).27 The geometrical patterns of the stirring-enhanced filtration, bacteria-capturing, and micro-PCR modules were designed using AutoCAD 2018 (Autodesk, USA). Regarding the latter, four detection chambers (three for target samples & one for negative controls) were designed to be symmetrically oriented around the inlet of the micro-pump. Another two chambers were designed for positive (100 ng bacterial genomic DNA) and negative (ddH2O) PCR controls. The master molds were carved on the PMMA plates with a computer-numerical-control machine (EGX-400, Roland DGA, USA) featuring a 0.5 mm drill bit at 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 RPM. The engraved molds were cleaned and polished. A PDMS (Sylgard, Dow Corning, USA) liquid prepolymer mixture of 10:1 (w:w) 184A:184B was poured into the PMMA molds. After degassing under vacuum for 10 min to remove air bubbles, the PDMS replica was cured for 90 min at 80 °C, peeled mechanically from the master mold, and bonded via O2 plasma treatment (90 W for 90 s, CUTE-MPR, Femto Science, Korea) to the other PDMS layers as well as the glass substrate (0.7 mm thick, Ruilong Photoelectric, Taiwan). Double-sided tape (68552, Tesa SE, Germany) was used to bind the stirring-enhanced filtration module to the PDMS layers. Since PDMS is hydrophobic, 90 W O2 plasma treatment (90 s) was used to clean the surface and increase the adhesivity of the double-sided tape to the PDMS. Afterwards, the PDMS–tape layers were placed on a hot plate (80 °C) to increase stability. Pluronic F-68 solution (10% poloxamer 188 solution, Sigma-Aldrich) was injected into the IMC channels and chambers to further increase hydrophilicity (thereby preventing protein adsorption) after the O2 plasma treatment at 90 W for 90 s.28 After baking at 60 °C overnight, the chip was cleaned with 75% ethanol, deionized water, and PBS. Additionally, via-holes were drilled into the glass under the filtered plasma collection chamber and the chamber for storing the filtrate (Fig. 3a). Note that the bacteria-containing solution was transported into the microchambers for PCR in two stages, one for the upper and lower ones and another for the two in the middle (Fig. 3b), to achieve uniform liquid transport. Fig. 3(c) and (d) show photographs of the IMC and an assembled IMC with packaging devices.
000 RPM. The engraved molds were cleaned and polished. A PDMS (Sylgard, Dow Corning, USA) liquid prepolymer mixture of 10:1 (w:w) 184A:184B was poured into the PMMA molds. After degassing under vacuum for 10 min to remove air bubbles, the PDMS replica was cured for 90 min at 80 °C, peeled mechanically from the master mold, and bonded via O2 plasma treatment (90 W for 90 s, CUTE-MPR, Femto Science, Korea) to the other PDMS layers as well as the glass substrate (0.7 mm thick, Ruilong Photoelectric, Taiwan). Double-sided tape (68552, Tesa SE, Germany) was used to bind the stirring-enhanced filtration module to the PDMS layers. Since PDMS is hydrophobic, 90 W O2 plasma treatment (90 s) was used to clean the surface and increase the adhesivity of the double-sided tape to the PDMS. Afterwards, the PDMS–tape layers were placed on a hot plate (80 °C) to increase stability. Pluronic F-68 solution (10% poloxamer 188 solution, Sigma-Aldrich) was injected into the IMC channels and chambers to further increase hydrophilicity (thereby preventing protein adsorption) after the O2 plasma treatment at 90 W for 90 s.28 After baking at 60 °C overnight, the chip was cleaned with 75% ethanol, deionized water, and PBS. Additionally, via-holes were drilled into the glass under the filtered plasma collection chamber and the chamber for storing the filtrate (Fig. 3a). Note that the bacteria-containing solution was transported into the microchambers for PCR in two stages, one for the upper and lower ones and another for the two in the middle (Fig. 3b), to achieve uniform liquid transport. Fig. 3(c) and (d) show photographs of the IMC and an assembled IMC with packaging devices.
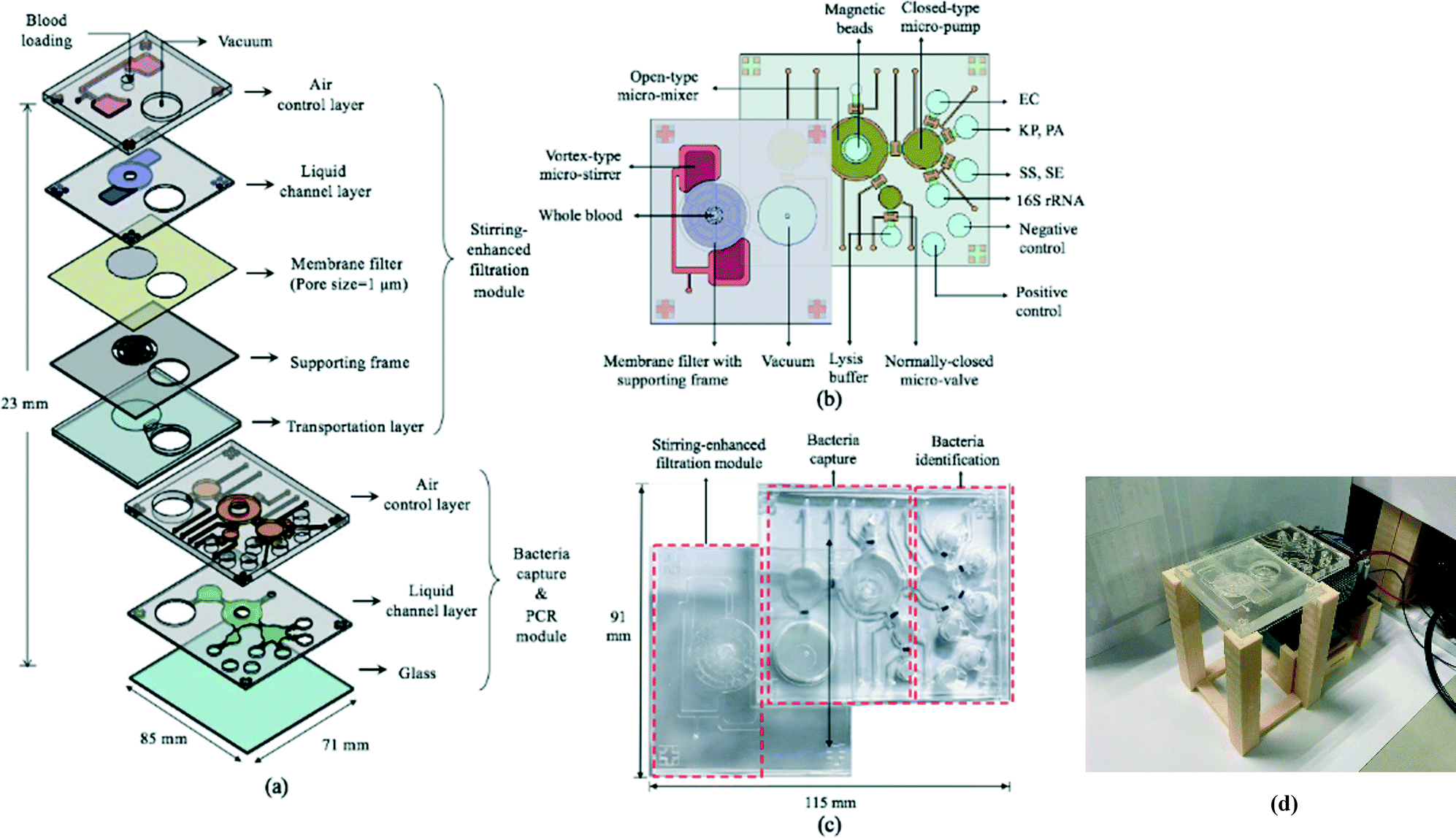 | ||
| Fig. 3 (a) An exploded view of the integrated microfluidic chip (IMC) featuring a stirring-enhanced filtration module, a bacteria-capturing module, and a micro-PCR module. (b) The chip was equipped with microfluidic components (e.g. micro-stirrers, micro-pumps, micro-mixers, micro-valves, & microchambers). A magnet was placed underneath the micro-mixer for bead collection (not shown), and a TE cooler was placed under the four microchambers for PCR thermocycling (not shown). Microchambers for positive and negative PCR controls were also incorporated. EC, KP, PA, SS and SE stand for E. coli, K. pneumoniae, P. aeruginosa, S. saprophyticus, and S. epidermidis respectively. (c) A photograph of the IMC. (d) A photograph of an assembled IMC with packaging devices. | ||
Design and working principles of the microfluidic devices
The 69 mm × 61 mm × 6.5 mm (L × W × H) vortex-type micro-stirrer used for filtration (Fig. 3b and 4) was composed of two PDMS layers and a glass substrate.14 Two air chambers with connecting air channels were located in the top PDMS layer while a mixing chamber with two fluidic chambers comprised the bottom layer. The diameters of the circular mixing chamber and blood injection port were 22 and 5 mm, respectively. Note that the smaller the opening for blood injection, the higher the resulting mixing and agitation efficiency in the fluid layer; this is due to the larger contact area between the PDMS membrane and the sample. When compressed air was applied to the two air chambers via the connecting channels, the 200 μm thick PDMS membranes were deflected, and a swirling flow was generated inside the mixing chamber.29 When a vacuum was instead applied, the PDMS membranes were deflected away from the chamber, thereby providing a larger space for liquid agitation. The filtered, bacteria-containing plasma was collected by applying a vacuum to the chamber beneath the membrane at 0.5 Hz (1 s vacuum durations); this periodic cessation of the vacuum prevented larger particles (i.e. WBCs and RBCs) from clogging the membrane. | ||
| Fig. 4 (a) Schematic illustration of the vortex-type micro-mixer for filtration. (b) Cross-sectional view of bacteria-containing blood added to the mixing chamber (see Fig. 1 legend for identity of mixture constituents). (c) PDMS membranes were deflected upon injecting compressed air, thereby inducing swirling flow. (d) A vacuum was applied to raise the membrane. (e) Suction was applied to the chamber under the membrane filter such that particles with smaller diameters passed through. (f) The suction was intermittent (frequency = 0.5 Hz). (g) During stoppage, the swirling flow agitated the particles, thus preventing larger ones from clogging the filter. | ||
A pneumatically-driven micro-mixer mixed the filtered plasma with the FcMBL-coated magnetic beads so that bacteria could be captured while an external magnetic field was applied (Fig. 3b). When compressed air was applied to deflect the PDMS membrane, gentle mixing was generated; when the compressed air flow was terminated, the resulting suction drew the fluid into the chambers beneath the membrane.30 In other words, the pumping volume and rate were correlated with the deflection of the membrane. By repeatedly deflecting the PDMS membrane bidirectionally, the magnetic beads and bacteria-containing plasma were mixed efficiently by the vortex flow generated in the chamber. Upon bead collection, cells were lysed during the initial denaturation step of the PCR (described above), and PCR was carried out in the micro-PCR module, which consisted of micro-pumps, micro-valves, and microchambers (Fig. 3b).
Results and discussion
Stirring-enhanced filtration
Three filters with different pore sizes (1, 2, and 3 μm for WHA110610, WHA110611, and WHA110612, respectively) were tested to optimize particle removal efficiency (Table 2). Note that the porosity was calculated from the pore densities and the actual pore sizes provided by the manufacturer. Experimental results showed that the throughput for the 1 μm filter was the highest due to the higher porosity, as expected. After activating the IMC, cells in the filtered plasma were quantified using a TC20 automated cell counter (Bio-Rad, USA), and no WBCs were observed for any filter size. However, most RBCs passed through the 2- and 3 μm filters (removal rates of only 3 and 4%, respectively). For this reason, we used the 1 μm filters for all subsequent experiments, in which case, 99.5% of RBCs were removed at an optimal filtering frequency of 3 Hz (Fig. 5) and optimal positive and negative gauge pressures of 5 and −10 kPa, respectively (with a filtration rate of 20.74 μL min−1). Higher suction levels forced RBCs through the membrane.31| Pore size | Porosity | Conditions | Throughput | Removal rate | Bacteria passing rate |
|---|---|---|---|---|---|
| P p = positive gauge pressure, Pn = negative gauge pressure. | |||||
| 1 μm | 13.10% | P p: 5 kPa, 3 HzPn: −10 kPa, 0.5 Hz | 20.74 μL min−1 | 99.47% |
Escherichia coli: 76%
Pseudomonas aeruginosa: 69%
Klebsiella pneumoniae: 72% Staphylococcus saprophyticus: 67% Staphylococcus epidermidis: 68% |
| 2 μm | 5.64% | P p: 5 kPa, 3 HzPn: −6 kPa, 0.5 Hz | 36.42 μL min−1 | 3.06% | NC |
| 3 μm | 10.19% | P p: 5 kPa, 3 HzPn: −1 kPa, 0.5 Hz | 78.23 μL min−1 | 4.25% | NC |
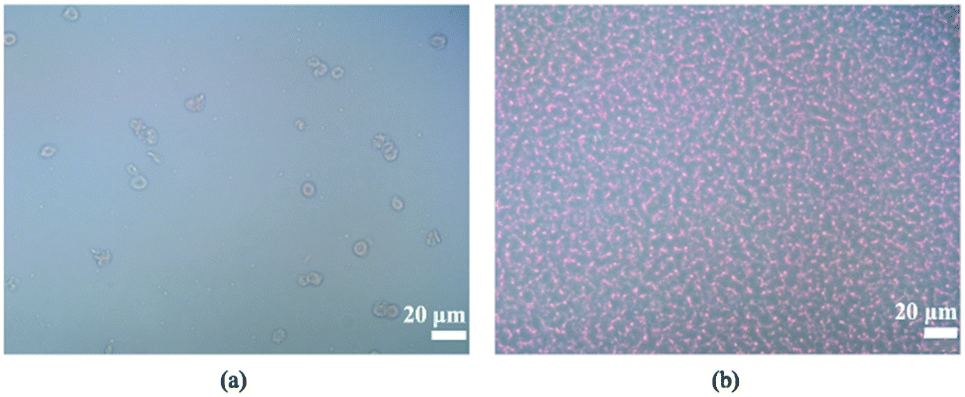 | ||
| Fig. 5 2000-fold-diluted (a) filtered plasma (1 μm) and (b) blood under an optical microscope. | ||
Under these conditions, 76, 69, 72, 67, and 68% of E. coli, P. aeruginosa, K. pneumoniae, S. saprophyticus, and S. epidermidis cells passed through the filters. The 30% left on the membrane could be due to variability in cell shapes or simply because, as the filter volume increases, so does the number of larger blood cells above the filter; this could potentially thwart bacterial cell passage.32 Regardless, the intermittent vacuum method decreased membrane clogging.
A challenge in sepsis diagnostics is the need to detect as few as 10–100 CFU of bacteria among approximately 109 RBCs, 107 WBCs, and 108 platelets per mL of blood. A continuous blood filtration device utilizing inertial lift forces captured 80% of bacteria while removing 90% of RBCs; at 200 μm min−1, it took 17 h to process 1mL of undiluted blood.33 An acoustophoretic isolation-based microfluidic device depleted 99.8% of RBCs yet only captured 10% of the target bacterial cells.34 Our IMC captured more bacteria in a shorter period of time than either of these devices, though its throughput (20.74 μL min−1) was lower than that for acoustophoretic isolation. Nevertheless, bacteria could be detected within 90 min at concentrations as low as 10–100 CFU per mL.
Additionally, the spiked samples with bacteria concentrations as low as 10–100 CFU per mL were used to demonstrate the performance of the developed devices (will be discussed later). Experimental results showed that the developed membrane-based filtration module has great potential in developing new and faster methods for sepsis diagnostics.
Capture rate of the FcMBL-coated magnetic beads
The capture rates of the FcMBL-coated magnetic beads were tested on-bench (60 min) and on-chip (20 min), and rates of 69, 77, 56, 58, and 64% were achieved for E. coli, P. aeruginosa, K. pneumoniae, S. saprophyticus, and S. epidermidis, respectively; these were similar to those of the on-bench bacteria approach: 71, 85, 13, 52, and 67%, respectively (Fig. 6). As a comparison, vancomycin-coated beads do not efficiently bind to Gram-negative species since binding occurs via certain specific amino acid sequences on the cell walls.35 The FcMBL-coated magnetic beads are consequently better universal bacterial probes. The on-chip and on-bench capture rates were found to be consistent except that there was a significant difference for K. pneumoniae which was reduced from 56 to 13%. K. pneumoniae exhibited a pronounced polysaccharide capsule covering the entire bacterial surface, resulting in a mucoid phenotype when grown on agar plates. It is speculated that this may lead to adhesion of the bacteria–bead complexes to the Eppendorfs and the pipette tips, which caused the loss of the beads. As a result, the capture efficiency for on-bench experiments was much lower than the on-chip data owing to some loss during bead collection.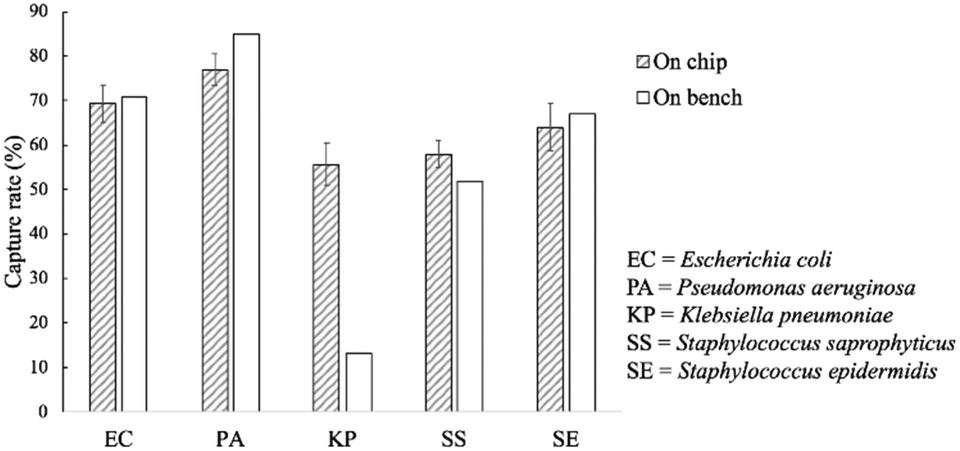 | ||
| Fig. 6 On-chip and on-bench capture rates of the FcMBL-coated beads. All experiments were repeated thrice, and error bars represent standard variations. | ||
Optimization of PCR conditions and PCR-based limits of detection (LODs)
Since the PCRs were successful at all three annealing temperatures (Fig. 7), the highest, most stringent temperature of 59 °C was used for all 90 min assays, and each primer only amplified the target gene (Fig. 8). It is worth mentioning that bacterial 16S rRNA was also targeted to prevent false-negative results because of its presence in all prokaryotes, and it was successfully amplified in all targets with high specificity (Fig. 7 and 8).26 Primers could be designed for other pathogenic bacteria in the future if necessary.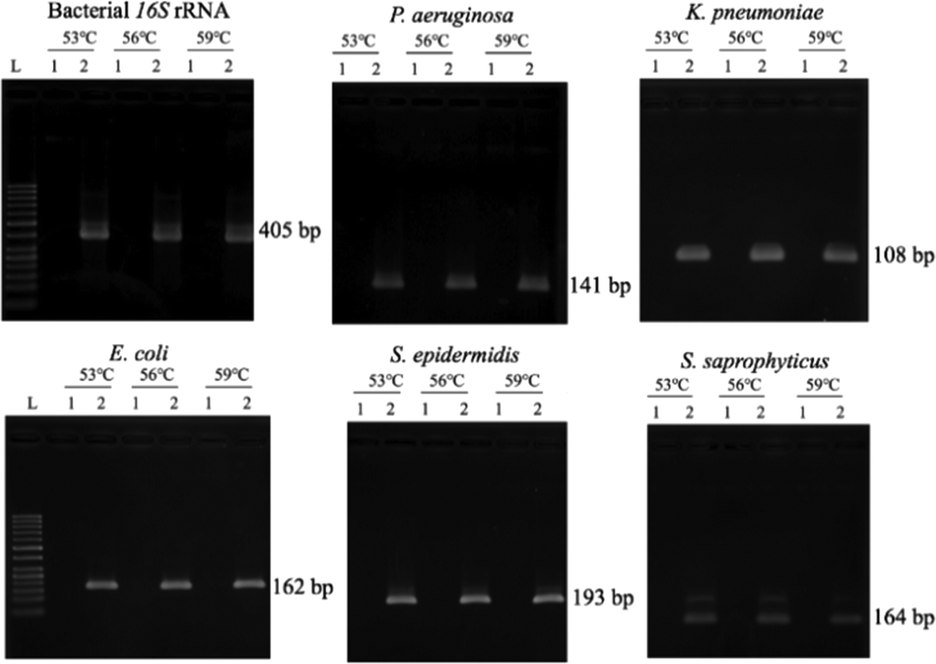 | ||
| Fig. 7 Optimization of the annealing temperature. In all panels, L = 50 base pair (bp) DNA marker ladder, lane 1 = negative control (double-distilled water), and lane 2 = positive control (106 copies of bacterial DNA). | ||
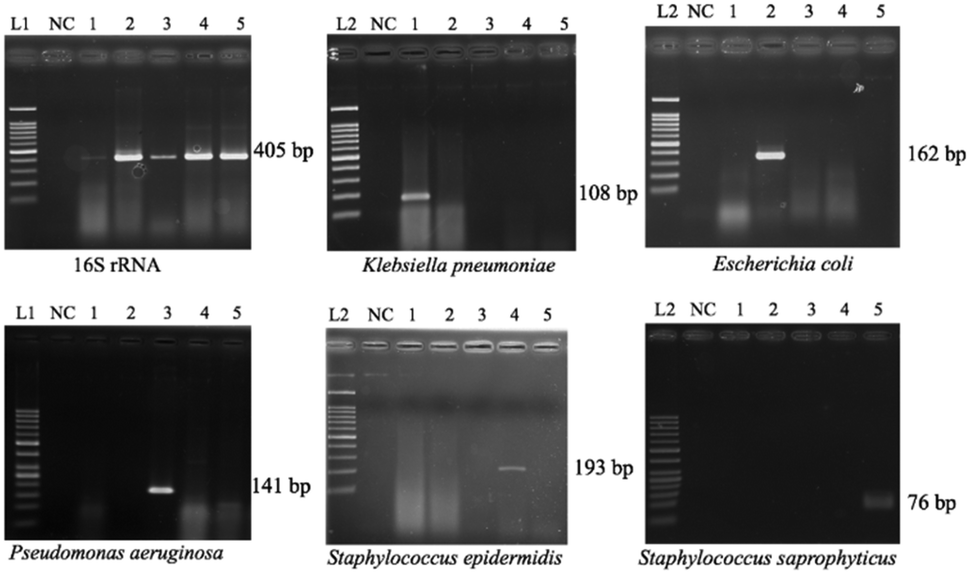 | ||
| Fig. 8 Specificity tests of the primers for five target bacterial genes. In all panels, lanes L1, L2, and NC correspond to 100 bp DNA marker ladders, 50 bp DNA ladders, and negative controls (distilled water), respectively. Lanes 1–5 = gDNA (equivalent to 106 gene copies) of Klebsiella pneumoniae, Escherichia coli, Pseudomonas aeruginosa, Staphylococcus epidermidis, and Staphylococcus saprophyticus, respectively. The results were similar when including the TaqMan probe in the reaction mixture. | ||
When using these PCR conditions, the limits of detection (LODs) for E. coli, K. pneumoniae, P. aeruginosa, S. epidermidis, and S. saprophyticus were experimentally found to be 5, 5, 5, 1 and 1 CFU per reaction on-bench, respectively (Fig. 9). The E. coli LOD in a prior study was higher, i.e. 10 to 100 CFU per mL; the superiority of our device could be due to the near-complete removal of interfering substances prior to the PCR.36 With the minimum bacteria passing rate (67% for S. saprophyticus), minimum on-chip bacteria capturing rate (56% for K. pneumoniae), mean bacteria concentration in a septic patient (i.e. 10 CFU per mL), and the maximum LOD (5 CFU per reaction for E. coli, K. pneumoniae and P. aeruginosa), a minimum volume of 5.4 mL of human blood would be needed to make an accurate sepsis diagnosis. This is much lower than that for the conventional culture-based method (∼20 mL).
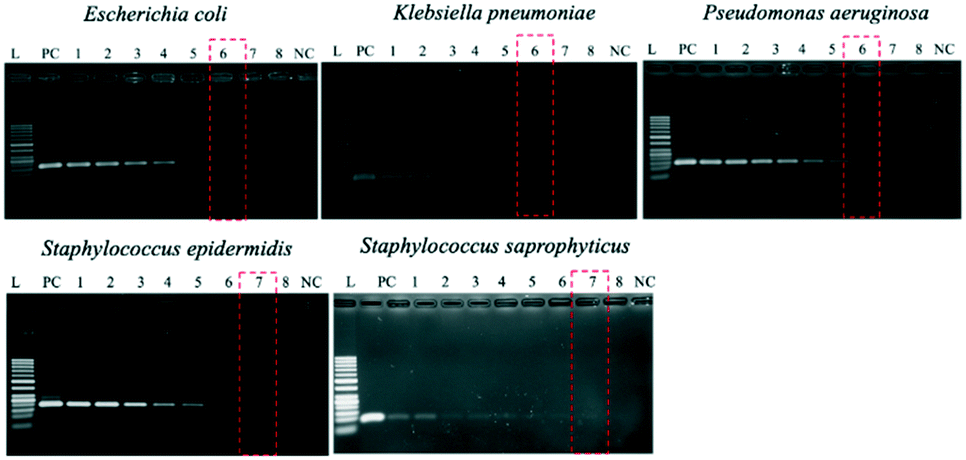 | ||
| Fig. 9 Limits of detection (LODs) for the five types of sepsis-inducing bacteria. In all panels, lanes L and NC correspond to 50 bp DNA marker ladders and negative controls (distilled water), respectively. Lane PC is the positive control (106 copies of genomic DNA). Lanes 1–8 = 1000, 500, 100, 50, 10, 5, 1, and 0.5 CFU per reaction. Lane 8 was used as an internal control to ensure that the dilution was accurate. | ||
Micro PCR-based limits of detection (LODs)
The on-chip LODs were further explored. Three of the targeted bacteria (Escherichia coli, Staphylococcus epidermidis, and Staphylococcus saprophyticus) with concentrations ranging from 104 to 101 CFU per mL were tested to verify the capability of the bacteria identification module and the results of fluorescence detection are shown in Fig. 10. The fluorescence optical signals were detected under a microscope equipped with a PMT, captured using a DS-Qi1Mc camera and then converted into electric signals. By switching the microscopy filters, light of different colors can be generated for the detection of the corresponding bacteria. The TaqMan probes of Escherichia coli and Staphylococcus epidermidis were labeled with FAM fluorophores and therefore they could be identified once excited by blue light. On the other hand, the TaqMan probe of Staphylococcus saprophyticus was labeled with the DLO fluorophore so that red light could be emitted once it was excited by green light. The fluorescence signals were found to decrease once the concentration of the bacteria was lowered. As shown in Fig. 10, the on-chip LODs for Escherichia coli, Staphylococcus epidermidis, and Staphylococcus saprophyticus were found to be 10, 10, and 10 CFU per mL, respectively. This was attributed to the fact that the temperature control and the fluorescence detection modules for on-chip PCR could be successfully performed, revealing that bacteria could be identified on this developed microfluidic chip.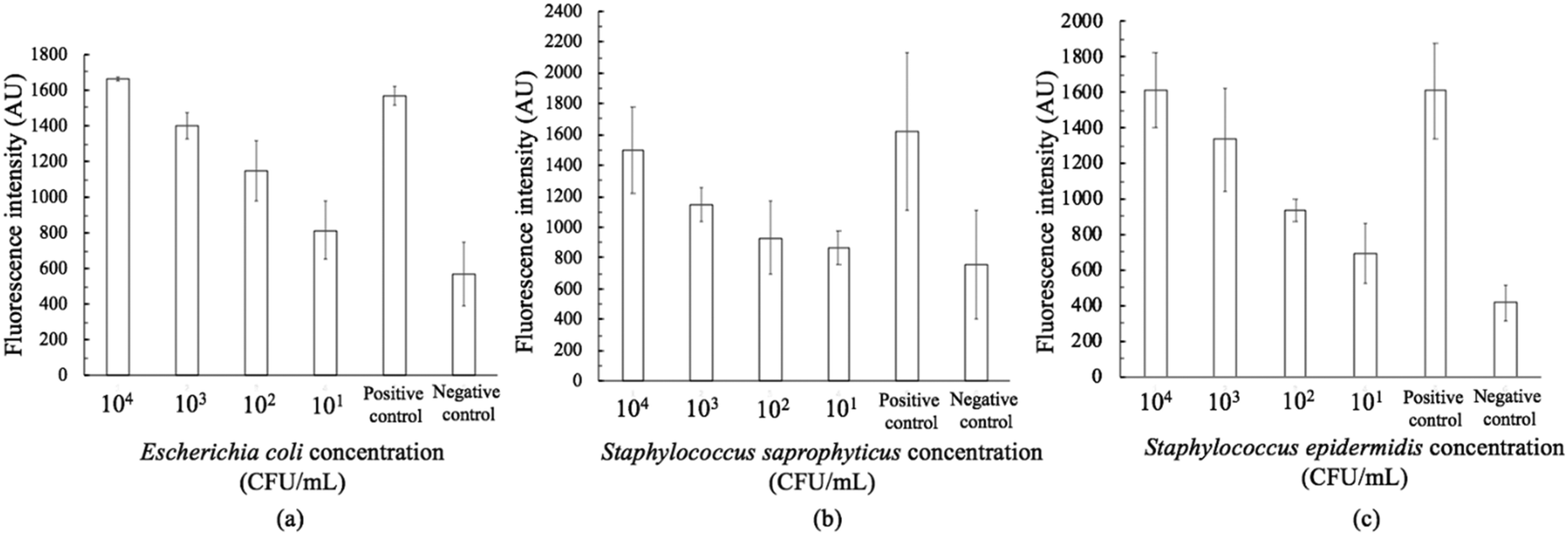 | ||
| Fig. 10 On-chip limits of detection (LODs) for (a) Escherichia coli, (b) Staphylococcus epidermidis, and (c) Staphylococcus saprophyticus. 106 copies of genomic DNA were used for positive control and distilled water was used for negative control. | ||
Spiked samples were further tested for the integrated microfluidic system. Whole blood containing 103 CFU per mL Escherichia coli was spiked to verify the performance of the developed device. All processes including filtration, bacteria capture and bacteria identification could be automatically conducted on this microfluidic system. The result is shown in Fig. 11, indicating a positive result of bacteria identification on this integrated microfluidic device.
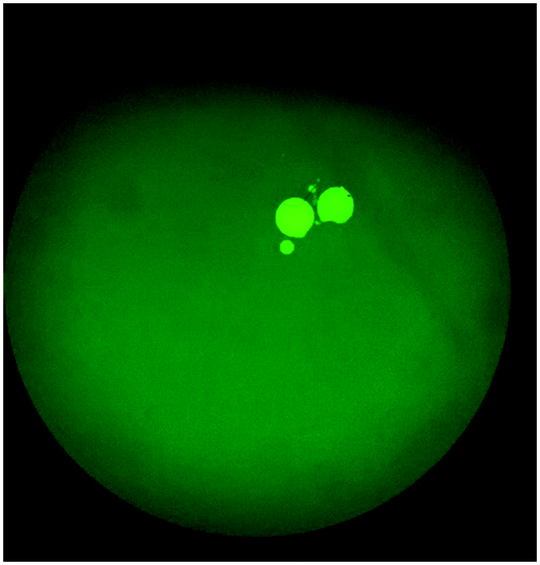 | ||
| Fig. 11 Fluorescence image of Escherichia coli spiked whole blood which had been detected on the IMC. The result indicated that this developed device was capable of performing sample pretreatment, bacteria isolation and bacteria identification automatically. | ||
Conclusions
We have presented an IMC capable of removing 100% of WBCs and 99.5% of RBCs via a filtration-enhanced micro-stirrer equipped with a 1 μm filter. Protein A–FcMBL-coated magnetic beads captured five types of target bacteria at rates ranging from 56 to 85% in only 20 min, and 5 CFU per mL of bacteria could be detected within 4 h, far less than the 72 h needed for culture-based methods. The capture rate could be improved by optimizing the filter rate and/or the capture rate of the beads. For instance, the filtering time could be extended. Ultimately, this IMC should be used with clinical samples to ensure that it may serve as a promising tool for future diagnosis of sepsis.Author contributions
Y. L. Fang designed the microfluidic chips, implemented the experiments, and prepared the manuscript. Y. S. Chen designed the experimental setup. C. H. Wang prepared the samples and designed the PCR primers. C. C. Chien, H. L. You, F. C. Kuo and M. S. Lee provided useful comments and suggestions regarding clinical conditions and clinical samples. G. B. Lee proposed the design of the chips, supervised the experiments, and proofread the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by Taiwan's Ministry of Science and Technology (MOST107-2221-E-007-013-MY3 & 106-2221-E-007-029-MY3 to GBL). Partial financial support from Kaohsiung Chang Gung Hospital, Taiwan under grant number CMRPG8K0501 is highly appreciated.References
- R. C. Bone, R. A. Balk, F. B. Cerra, R. P. Dellinger, A. M. Fein, W. A. Knaus, R. M. H. Schein and W. J. Sibbald, Chest, 1992, 101, 1644–1655 CrossRef CAS.
- J. Delaloye and T. Calandra, Virulence, 2014, 5, 161–169 CrossRef.
- R. P. Dellinger, et al. Surviving Sepsis Campaign, Crit. Care Med., 2013, 41, 580–637 CrossRef.
- F. Yu, Y. Li, M. Y. Li, L. H. Tang and J. J. He, Biosen. Bioelectron., 2017, 89, 880–885 CrossRef CAS.
- A. W. Bauer, W. M. M. Kirby, J. C. Sherris and M. Turck, Am. J. Clin. Pathol., 1966, 45, 493–496 CrossRef CAS.
- M. T. Johnson, R. Reichley, J. Hoppe-Bauer, W. M. Dunne, S. Micek and M. Kollef, Crit. Care Med., 2011, 39, 1859–1865 CrossRef CAS.
- A. Zea-Vera and T. J. Ochoa, J. Trop. Pediatr., 2015, 61, 1–13 CrossRef.
- S. C. Hong, J. S. Kang, J. E. Lee, S. S. Kim and J. H. Jung, Lab Chip, 2015, 15, 1889–1897 RSC.
- M. A. Faridi, H. Ramachandraiah, I. Banerjee, S. Ardabili, S. Zelenin and A. Russom, J. Nanobiotechnol., 2017, 15, 3 CrossRef.
- J. Wu, Q. Chen and J. M. Lin, Analyst, 2017, 142, 421–441 RSC.
- I. Pereiro, A. Bendali, S. Tabnaoui, L. Alexandre, J. Srbova, Z. Bilkova, S. Deegan, L. Joshi, J. L. Viovy, L. Malaquin, B. Dupuy and S. Descroix, Chem. Sci., 2017, 8, 1329–1336 RSC.
- J. Y. Han, J. P. Fu and R. B. Schoch, Lab Chip, 2008, 8, 23–33 RSC.
- J. Kim, M. Johnson, P. Hill and B. K. Gale, Integr. Biol., 2009, 1, 574–586 CrossRef CAS.
- R. T. Davies, J. Kim, S. C. Jang, E. J. Choi, Y. S. Gho and J. Park, Lab Chip, 2012, 12, 5202–5210 RSC.
- R. A. Bowden, S. J. Slichter, M. H. Sayers, M. Mori, M. J. Cays and J. D. Meyers, Blood, 1991, 78, 246–250 CrossRef CAS.
- G. Sirchia, A. Parravicini, P. Rebulla, N. Greppi, M. Scalamogna and F. Morelati, Vox Sang., 1982, 42, 190–197 CrossRef CAS.
- X. Chen, D. F. Cui, C. C. Liu and H. Li, Sens. Actuators, B, 2008, 130, 216–221 CrossRef CAS.
- S. Y. Yang, J. L. Lin and G. B. Lee, J. Micromech. Microeng., 2009, 19, 035020 CrossRef.
- Y. S. Chen, Y. D. Ma, C. C. Chen, S. C. Shiesh and G. B. Lee, Lab Chip, 2019, 19, 3305–3315 RSC.
- M. A. A. Valones, R. L. Guimaraes, L. A. C. Brandao, P. R. E. deSouza, A. D. T. Carvalho and S. Crovela, Braz. J. Microbiol., 2009, 40, 1–11 CrossRef CAS.
- M. U. Kopp, A. J. deMello and A. Manz, Science, 1998, 280, 1046–1048 CrossRef CAS.
- C. S. Zhang, J. L. Xu, J. Q. Wang and H. P. Wang, Chin. J. Anal. Chem., 2006, 34, 1197–1202 CAS.
- K. A. Hagan, C. R. Reedy, M. L. Uchimoto, D. Basu, D. A. Engel and J. P. Landers, Lab Chip, 2011, 11, 957–961 RSC.
- N. Nath, B. Godat, H. Benink and M. Urh, J. Immunol. Methods, 2015, 426, 95–103 CrossRef CAS.
- R. K. Saiki, S. Scharf, F. Faloona, K. B. Mullis, G. T. Horn, H. A. Erlich and N. Arnheim, Science, 1985, 230, 1350–1354 CrossRef CAS.
- J. M. Janda and S. L. Abbott, J. Clin. Microbiol., 2007, 45, 2761–2764 CrossRef CAS.
- C. Matellan and A. E. D. Hernandez, Sci. Rep., 2018, 8, 6971 CrossRef.
- S. Pinto, P. Alves, C. M. Matos, A. C. Santos, L. R. Rodrigues, J. A. Teixeira and M. H. Gil, Colloids Surf., B, 2010, 81, 20–26 CrossRef CAS.
- M. A. Eddings and B. K. Gale, J. Micromech. Microeng., 2006, 16, 2396–2402 CrossRef.
- Y. C. Chung, Y. L. Hsu, C. P. Jen, M. C. Lu and Y. C. Lin, Lab Chip, 2004, 4, 70–77 RSC.
- D. Kuzman, S. Svetina, R. E. Waugh and B. Zeks, Eur. Biophys. J., 2004, 33, 1–15 CrossRef CAS.
- Y. Y. Wang, F. Hammes, M. Duggelin and T. Egli, Environ. Sci. Technol., 2008, 42, 6749–6754 CrossRef CAS.
- A. J. Mach and D. Di Carlo, Biotechnol. Bioeng., 2010, 107, 302–311 CrossRef CAS.
- B. Ngamsom, M. J. Lopez-Martinez, J. C. Raymond, P. Broyer, P. Patel and N. Pamme, J. Microbiol. Methods, 2016, 123, 79–86 CrossRef.
- Y. Cetinkaya, P. Falk and C. G. Mayhall, Clin. Microbiol. Rev., 2000, 13, 686–707 CrossRef CAS.
- C. Auriti, G. Prencipe, M. Moriondo, I. Bersani, C. Bertaina, V. Mondi and R. Inglese, J. Immunol. Res., 2017, 7045630 Search PubMed.
Footnote |
| † Partial preliminary results had been presented at the IEEE MEMS 2020. |
| This journal is © The Royal Society of Chemistry 2021 |