
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of a multi-method analytical approach based on the combination of synchrotron radiation X-ray micro-analytical techniques and vibrational micro-spectroscopy methods to unveil the causes and mechanism of darkening of “fake-gilded” decorations in a Cimabue painting†
Letizia
Monico
 *abc,
Silvia
Prati
*d,
Giorgia
Sciutto
d,
Emilio
Catelli
d,
Aldo
Romani
ab,
Diego Quintero
Balbas
d,
Zelan
Li
d,
Steven
De Meyer
c,
Gert
Nuyts
c,
Koen
Janssens
ch,
Marine
Cotte
ef,
Jan
Garrevoet
g,
Gerald
Falkenberg
g,
Vanessa Isabel
Tardillo Suarez
e,
Remi
Tucoulou
e and
Rocco
Mazzeo
d
*abc,
Silvia
Prati
*d,
Giorgia
Sciutto
d,
Emilio
Catelli
d,
Aldo
Romani
ab,
Diego Quintero
Balbas
d,
Zelan
Li
d,
Steven
De Meyer
c,
Gert
Nuyts
c,
Koen
Janssens
ch,
Marine
Cotte
ef,
Jan
Garrevoet
g,
Gerald
Falkenberg
g,
Vanessa Isabel
Tardillo Suarez
e,
Remi
Tucoulou
e and
Rocco
Mazzeo
d
aCNR-SCITEC, Via Elce di Sotto 8, I-06123 Perugia, Italy. E-mail: letizia.monico@cnr.it
bSMAArt Centre, Department of Chemistry, Biology, and Biotechnology, University of Perugia, Via Elce di Sotto 8, I-06123 Perugia, Italy
cAXIS Research Group, NANOlab Centre of Excellence, University of Antwerp, Groenenborgerlaan 171, B-2020 Antwerp, Belgium
dMicrochemistry and Microscopy Art Diagnostic Laboratory (M2ADL), Department of Chemistry “G. Ciamician”, University of Bologna – Ravenna Campus, Via G. Guaccimanni 42, I-48121 Ravenna, Italy. E-mail: s.prati@unibo.it
eESRF, Avenue des Martyrs 71, F-38000 Grenoble, France
fLAMS, CNRS UMR 8220, Sorbonne Université, UPMC Univ. Paris 06, Place Jussieu 4, F-75005 Paris, France
gDeutsches Elektronen-Synchrotron DESY, Notkestraße 85, D-22603 Hamburg, Germany
hRijksmuseum, Conservation & Restoration – Scientific Research, Hobbemastraat 22, 1071 ZC Amsterdam, Netherlands
First published on 18th November 2021
Abstract
Redox processes activated by environmental factors have been identified as the main cause of the chromatic alterations of a number of artists' pigments, including the yellow pigment orpiment (As2S3). Although a general comprehension of the mechanisms has been provided through characterization of degradation compounds of As2S3, experimental evidences to prove how other paint components and how different environmental agents influence the formation pathways of specific secondary compounds are still lacking. Thus, it becomes fundamental to develop a methodological strategy which enable achieving a discrimination among the causes affecting the chemical stability of more heterogenous As2S3-based paints and defining the mechanism through which the alteration establishes and evolves, with the ultimate goal of optimizing the preventive conservation measures of unique masterpieces. In this paper, we propose a comprehensive multi-material and multi-method approach based on the combination of synchrotron radiation X-ray micro-analytical techniques (i.e., X-ray diffraction, X-ray fluorescence and X-ray absorption near edge structure spectroscopy at S K-/Ag L3-/As K-edges) and vibrational micro-spectroscopy methods to unveil the causes and mechanism of darkening of “fake-gilded” decorations in tempera paintings, originally consisting of an unusual mixture of As2S3 and metallic silver (Ag0). Such degradation process is a not yet understood phenomenon threatening a series of Old Master paintings, including those by the Italian painters Cimabue and Pietro Lorenzetti. The high specificity, sensitivity and lateral resolution of the employed analytical methods allowed providing first-time evidence for the presence of black acanthite (α-Ag2S), mimetite [Pb5(AsO4)3Cl] and syngenite [K2Ca(SO4)2·H2O] as degradation products of the “fake-gilded” decorations in the Maestà by Cimabue (Church of Santa Maria dei Servi, Bologna, Italy). Furthermore, the study of the painting combined with that of tempera paint mock-ups permitted to explore and define the environmental agents and internal factors causing the darkening, by proving that: (i) Ag0 and moisture are key-factors for triggering the transformation of As2S3 to α-Ag2S and As-oxides; (ii) S2--ions arising from the degradation of As2S3 are the main responsible for the formation of α-Ag2S; (iii) light exposure strengthens the tendency of the paint components towards alteration. Based on our findings, we finally propose a degradation mechanism of As2S3/Ag0-based tempera paints.
1. Introduction
In chemical sciences the study of the causes triggering the color change of paintings is a grand challenge. Over the past decades, investigations of micro-samples taken from historical paintings and artificially aged paint mock-ups have established that redox processes driven by environmental agents (light, humidity, temperature and/or atmospheric pollutants) are the main responsible for irreversible chromatic alterations of a number of inorganic pigments, such as cadmium yellows (Cd1−xZnxS, with 0 ≤ x≤ 0.3),1–6 chrome yellows (PbCr1−xSxO4, with 0 ≤ x≤ 0.8),7–13 vermilion red (HgS),14,15 Prussian blue (MFeIII[FeII(CN)6] × H2O, with M = K+, NH4+ or Na+)16,17 and zinc yellow (K2O·4ZnCrO4·3H2O).18Similar to the above-mentioned compounds, also the yellow pigment orpiment (As2S3) is characterized by a low chemical stability. In aerobic environments, the photo-degradation of the pigment occurs via a structural rearrangement of As2S3, leading to the production of white arsenolite (As2O3) and S-species.19–21 The latter may trigger the formation of organosulfur derivatives and/or H2S, by initiating a series of reactions that may explain the formation of black sulfides in Cu- and Pb-containing paintings under the influence of adjacent As2S3 paints.22 Investigations of 17th century Flemish and Dutch artworks led to the hypothesis that moisture and some intrinsic chemical properties of the paint (e.g., local pH and matrix composition) may also promote the fading of As2S3 even in the absence of light, thus giving rise to As2O3 and/or other arsenite (AsIII)-species. In a following step, AsIII-species can be further oxidized to arsenate (Asv)-compounds.23–27
From a methodological point of view, synchrotron radiation (SR)-based X-ray methods employing micro-probes, including micro X-ray fluorescence (μ-XRF), micro X-ray diffraction (μ-XRD) and micro X-ray absorption near edge structure (μ-XANES) spectroscopy (in mapping and point analysis mode), were used for their capabilities to provide spatially resolved elemental speciation and structural information down to the (sub)micrometer scale length.1,2,4–18,21–26 The specificity of vibrational spectroscopy techniques (i.e., IR and Raman) to render complementary molecular information with respect to SR-based X-ray methods were also sometime exploited to study the chemical nature of secondary compounds associated with the degradation of different pigments.3,5,6,12,20–25 In addition, electrochemical techniques were proposed as alternative methods for a fast monitoring of the photo-degradation mechanism of a selection of semiconductor pigment powders.21,22,28,29
In the context of orpiment-based paints, despite a large quantity of data on the general characterization and distribution of degraded compounds has been recently acquired,19–27 very little is still known about the interactions that take place among As2S3 and other paint components (e.g., other pigments, additives, binder) and their reactivity with the surrounding environment. Furthermore, such data, being in most of the cases limited to the study of historical paintings, do not prove directly how and why such alterations activate. Therefore, it becomes highly relevant to establish a methodological strategy which permit to obtain a discrimination among the causes that affect paint stability and to define the mechanism through which the alteration establishes and evolves, with the ultimate goal to optimize and develop preventive conservation measures and actions aimed at safeguarding unique works of art.
In this paper, we propose the use of a comprehensive multi-material and multi-method approach, based on the combination of SR X-ray micro-analytical techniques and vibrational micro-spectroscopy methods, to study the darkening issue affecting As2S3-based areas of the painting Maestà of Santa Maria dei Servi attributed to Cimabue (1240–1302) (Fig. 1a) and of tempera paint mock-ups aged with either light or moisture. Earlier macroscale investigations30,31 have revealed that the darkened decorations of the Cimabue painting, assumed to have originally had a gilded appearance, are composed of an unusual mixture of As2S3 and silver powder. It has been reasonably hypothesized that, as consequence of the visible darkening, the “fake-gilded” areas were later covered, probably by Cimabue himself, with another oily brownish layer containing Pb-based pigments, on top of which traces of a gold leaf are present as well.31 The combined use of orpiment and silver (in the form of either powder or leaf) was found in a fair number of artworks, this being a quite common medieval practice for imitating gildings.32–35 Similar to the Maestà by Cimabue, also some of the “fake-gilded” areas of a series of paintings by Pietro Lorenzetti (ca. 1280–1348) show a visible darkening nowadays.36
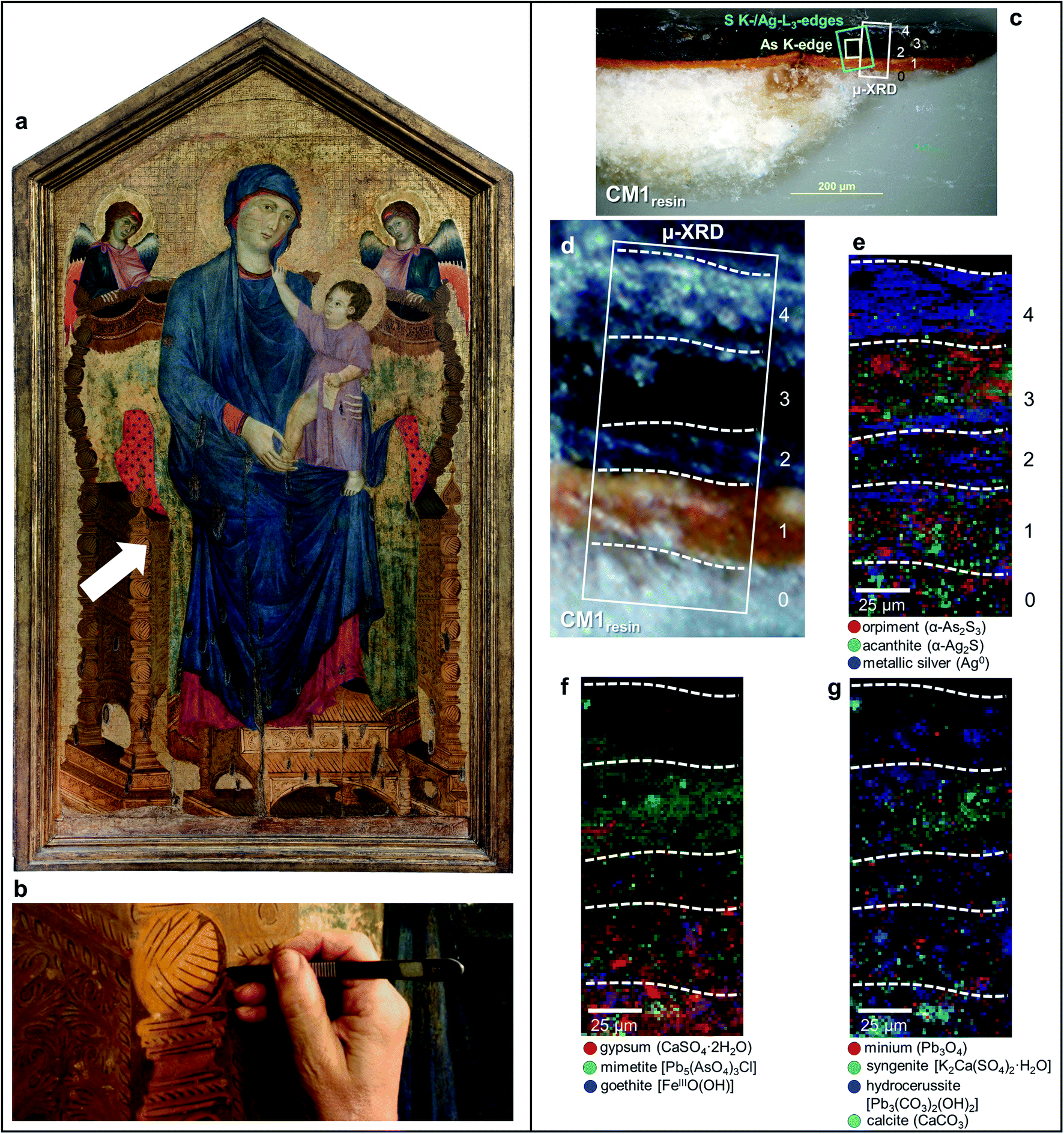 | ||
| Fig. 1 (a) Photograph of Maestà of Santa Maria dei Servi (tempera and gold on panel painting, ca. 1280–1285; Bologna, Italy) attributed to Cimabue, before the restoration undertaken in 2015; the white arrow indicates the sampling area. (b) Detail image of the sampling spot of sample CM1. (c) Micrograph of cross-sections CM1resin taken with visible light (magnification: 200×) and (d) corresponding detail image where SR μ-XRD mapping was performed. (e–g) Composite SR μ-XRD images of the identified crystalline phases [map size (v × h): 140 × 75 μm2; step size (v × h): 1.5 × 1.5 μm2; exp. time: 1 s per pixel; energy: 21 keV] (see Fig. S1† for a selection of the XRD patterns). In (c) the yellow and cyan rectangles show the areas where SR μ-XRF/μ-XANES investigations were performed (see Fig. 2 for the results), while numbers indicate the paint stratigraphy. | ||
All such observations suggest that a synergy between As2S3 and metallic silver (Ag0) may have prompted chemical transformations, through which both chemicals have changed their nature. In this regard, three reasonable starting hypotheses can be formulated on the causes of darkening of “fake-gilded” paints:
(i) Based on earlier studies,37,38 under aerobic conditions, the Ag+ ions, formed superficially by oxidation of Ag0 particles, interact with either organosulfur derivatives or H2S [arising from the (photo)degradation of As2S3] forming the black compounds acanthite (α-Ag2S) and/or argentite (β-Ag2S).
(ii) Alternatively, it is possible to assume that, under oxidative circumstances, As2S3 is converted to other AsIII- and AsV-based compounds, while the parallel oxidation of Ag0 to Ag+ might occur locally, followed by the precipitation of a number of dark materials, such as AgS-compounds, Ag3AsO4 and Ag3AsO3.39
(iii) As a third pathway, we can hypothesize the photo-dissolution of Ag+ cations in the As2S3 matrix, with a first step of the reaction leading to the formation of Ag2S as a result of the direct photo-oxidation of Ag0 to Ag+, and the subsequent formation of various Ag–As–S ternary compounds as final products.40–42
In order to elucidate which of the above mechanisms actually may have taken place, on the one hand, SR-based μ-XRD and μ-XRF mapping along with Ag L3-/As K-edges μ-XANES and μ-Raman spectroscopy analyses at selected spots of two darkened paint micro-fragments taken from the Maestà by Cimabue and artificially aged tempera paint mock-ups permitted to gain complementary insights into the chemical nature, stratigraphic distribution and relative-abundances of Ag- and As-based compounds. On the other hand, single-point S K-edge μ-XANES analysis and attenuated total reflection (ATR) mode μ-FTIR mapping allowed characterizing and semi-quantifying S-based compounds and other secondary products formed as a consequence of the chemical alteration of pigments and the pigment–binder interactions.
2. Experimental
2.1 Paint cross-sections from Maestà
A sample taken from a darkened “fake-gilded” decoration area of the painting Maestà was divided in two different fragments [sizes (h × v) of about 600–700 × 200–300 μm2], that were analyzed as cross-sections (Fig. 1b, c and 3a). For SR-based X-ray measurements, one of the fragments was embedded only in Sody33 polyester resin (henceforth called CM1resin). After curing of the resin for 24 h at room temperature, the embedded sample was cut down to a thickness of ca. 50 mm and hand-polished with 120- to 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-grit sandpaper to expose the cross-section.
000-grit sandpaper to expose the cross-section.
To avoid the interference of resin during FTIR analysis, the second fragment was before embedded in a KBr pellet (hereafter referred to as CM1KBr) and then in a synthetic resin by following the experimental procedure described in previous studies.43,44
2.2 Preparation of paint mock-ups and artificial aging protocols
Three sets of paint mock-ups were prepared over polycarbonate supports. The first two sets were obtained by mixing either commercial orpiment (Kremer, 10700) or metallic silver powders (Sigma-Aldrich) with whole egg as binder (sample names: Orp and Ag, respectively). To mimic the unusual “fake-gilded” decoration found in the Cimabue painting,31 the third set was prepared by mixing orpiment and metallic silver powders in the weight ratio of 3:1 and then by laying out such mixture as a paint layer with whole egg as binder (sample name: Orp–Ag). All Orp-based paint mock-ups were prepared in triplicate to be subjected to two different types of aging treatments. Ag mock-ups were prepared in duplicate to be exposed to one type of aging.
UVA-visible photochemical aging was performed by allocating the touch-dry paints (i.e., after ∼1 month since their preparation; hereinafter called OrpUVA-vis, AgUVA-vis and Orp–AgUVA-vis) inside an in-house–made aging chamber, equipped with a UV-filtered 300 W Cermax xenon lamp (λ ≥ 300 nm; see ref. 9 for the emission spectrum of the source) at a relative humidity (RH) of ∼25% (average indoor humidity level value, measured daily by a thermohygrometer). The irradiance and temperature at the sample position were ∼1 × 103 W m−2 and 30 °C, respectively. Paints were irradiated for 123 h, thus reaching radiant exposure values of ∼1.2 × 105 W m−2 hour−1.
High humidity aging treatment was performed by placing the touch-dry Orp-based paints (henceforth denoted as Orp95%RH and Orp–Ag95%RH) in a vessel maintained in the dark at RH ≥ 95% (achieved using distilled water) and at 40 °C for an overall period of 288 h.
Cross-sectioned samples of each mock-up were prepared using KBr pre-embedding (see above) for μ-FTIR and μ-Raman analyses,43,44 while they were embedded in polyester resin for SR-based X-ray investigations.
2.3 Optical microscopy
Microphotographs of cross-sections CM1resin and CM1KBr (Fig. 1c and 3a) were taken by an Olympus BX51M microscope equipped with a digital camera Olympus DP70, while those of the paint mock-ups were obtained by employing a Canon Powershot S50 digital camera coupled to a Leica MZ6 stereomicroscope equipped with a tungsten lamp. Images were processed via Canon ZoomBrowser Ex 4.5 software.2.4 SR μ-XRD
μ-XRD measurements on CM1resin and paint mock-up cross-sections were carried out at the microprobe hutch of the Hard X-ray Micro/Nanoprobe beamline P06 of the PETRA III storage ring (DESY, Hamburg, Germany).45 The incident energy of the beam (i.e., 21 keV) was selected by means of a Si(111) double-crystal monochromator. A Kirkpatrick–Baez (KB) mirror system was employed to focus the beam down to 0.46 × 0.46 μm2 (h × v). Diffraction patterns were collected by a PILATUS 300 K area detector (Dectris Ltd., CH). Calibration of the diffraction setup was performed using a LaB6 standard. Crystalline phases were identified using the XRD patterns of reference compounds that are available in the American Mineralogist database.46 Bidimensional distribution maps of each crystalline phase were obtained by full pattern refinement using XRDUA.47 Notably, the relative positions and intensities of the diffraction peaks for every compound were kept fixed during the fitting procedure, while the angle-independent parameter W of the Caglioti width function was used to model the peak width. Refined values for the scaling, displacement, and width parameters were obtained for each compound. The intensity scaling parameters obtained in each pixel of the image were used to visualize the spatial distribution of the different compounds. A more detailed explanation of the modeling procedure is offered elsewhere.472.5 SR μ-XRF/μ-XANES at S K-, Ag L3-, As K-edges
S, Ag, and As speciation investigations of CM1resin, of mock-ups and of a set of reference powders were performed at the scanning X-ray microscope end station of beamline ID21 and at the hard X-ray nanoprobe beamline ID16b of the European Synchrotron Radiation Facility (ESRF, Grenoble, France).48,49At both beamlines, measurements were carried out by means of a Si(111) double-crystal monochromator. A KB mirror system was used to focus the beam down to ∼0.6 × 0.4 μm2 (h × v) and ∼0.1 × 0.08 μm2 (h × v) during the investigations at ID21 and ID16b, respectively. At these beamlines, XRF signals were collected by a single energy-dispersive silicon drift detector (Xflash 5100, Bruker) and a Si drift-diode array detector, respectively.
Single point μ-XANES spectra were acquired in XRF mode. At ID21, data were recorded by scanning the primary energy around the S K-edge (2.46–2.53 keV; step: 0.17 eV) and the Ag L3-edge (3.34–3.42 keV; step: 0.25 eV), while at ID16b spectral profiles were collected by scanning the primary energy around the As K-edge (11.78–12.16 keV; step: 1 eV). The energy calibration was performed using CaSO4·2H2O, an Ag foil and As2S3 as standards and by setting the position of the peak maximum of their first-order derivative spectrum at 2.4817 keV, 3.3547 keV and 11.8693 keV, respectively.
ATHENA50 was employed for the normalization and the linear combination fit (LCF) of the spectra. The LCF procedure, which was carried out using the spectra of a set of S, Ag, and As references, permitted to quantitatively determine the average relative amount of sulfide (S−II) and sulfate (SVI) compounds (expressed as %[S−II]/[Stotal] and %[SVI]/[Stotal]) and of different Ag compounds. The best description of each S K-edge μ-XANES spectrum was obtained by including in the LCF model three or four S-reference spectra [i.e., of α-As2S3, α-Ag2S and of one or two sulfates], while only two reference spectra [i.e., of α-Ag2S and either Ag2SO4 or CH3COOAg] were required for fitting the Ag L3-edge μ-XANES spectra.
At ID21, maps of the same region of interest (ROI) were collected with a 100 ms per pixel dwell time at the three following energies: (i) 2.4718 keV and (ii) 2.482 keV to favour the excitation of S−II- and SVI-species, respectively and (iii) 3.4 keV to obtain the XRF intensity of all S- and Ag-species (cf.Fig. 2d). At ID16b, maps of the same ROI were recorded by employing a 100 ms per pixel dwell time at the three following energies: (i) 11.870 keV and (ii) 11.8754 keV to favour the excitation of As2S3 and As2O3/AsV-species, respectively and (iii) 12.0 keV to record the XRF intensity of all As-species (cf.Fig. 2b). PyMca51 was used to fit the XRF spectra and to separate the contributions of different elements. The experimental procedure used for recording and producing the S and As speciation maps is described in previous studies.1,25
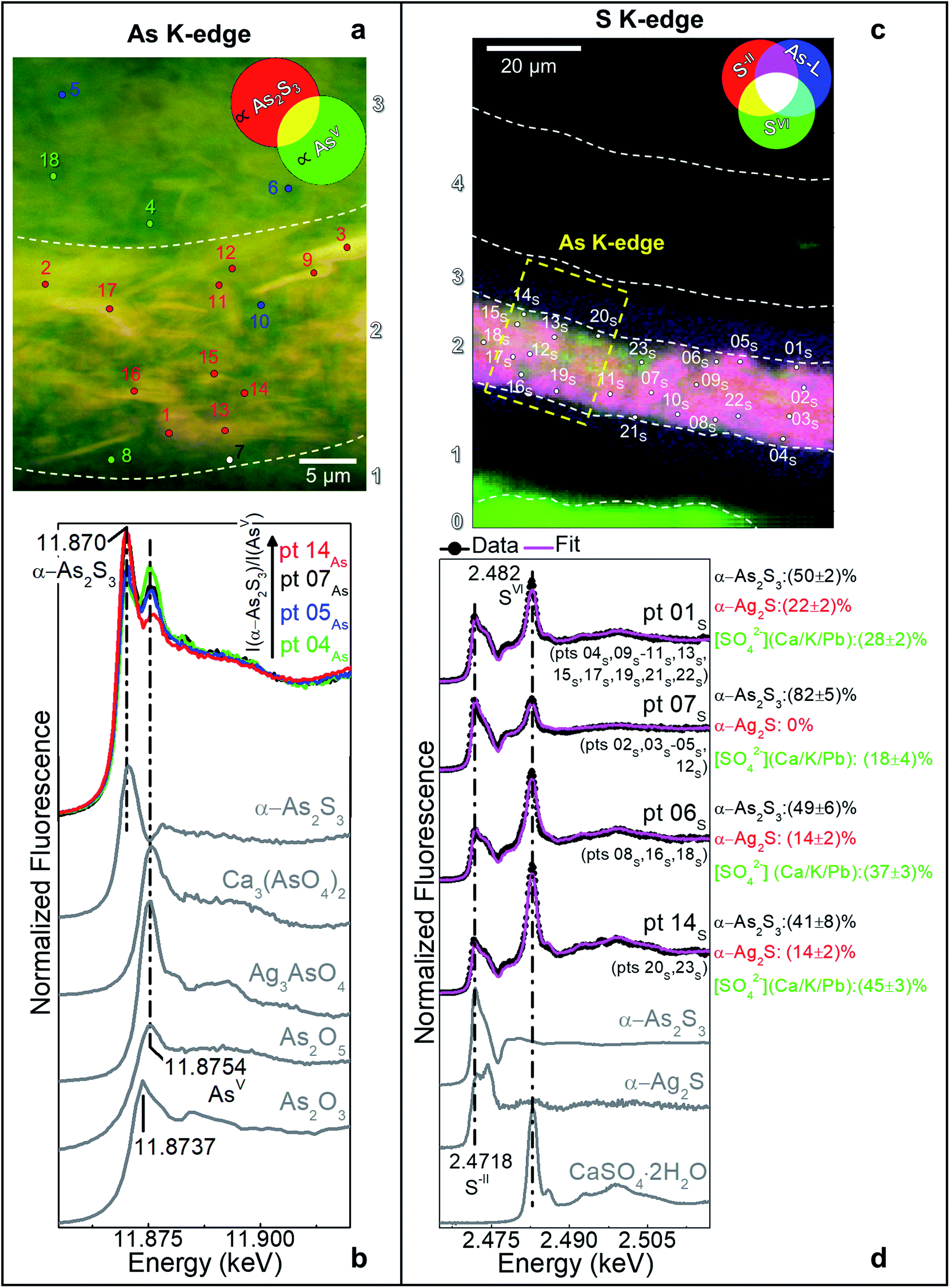 | ||
| Fig. 2 (a) Composite SR μ-XRF images of AsIII (∝α-As2S3) (red) and AsV (green) [map size (v × h): 30.7 × 38.1 μm2; step size (v × h): 0.1 × 0.1 μm2; exp. time: 100 ms per pixel; energy (AsIII) = 11.870 keV, energy (AsV) = 11.8754 keV] and (b) selection of the As K-edge μ-XANES spectra acquired from the spots shown in (a) compared to those of a set of As-reference compounds. Different colors refer to spectra characterized by similar features and showing a variable ratio between the intensity of the white lines of α-As2S3 and AsV-compounds. (c) RGB SR μ-XRF images of S−II/SVI/As-L [map size (v × h): 99.5 × 74.2 μm2; step size (v × h): 0.5 × 0.7 μm2; exp. time: 100 ms per pixel; energy (S−II) = 2.4718 keV, energy (SVI) = 2.482 keV] and (d) selection of the S K-edge μ-XANES spectra (black) recorded from the spots shown in (c) with LCF results (magenta) of different S-reference compounds. In grey, spectra of selected S-reference compounds. Numbers in brackets refer to the spectral profiles showing similar features to those reported. | ||
Considering the sensitivity of pigments/paints under the exposure to SR-based X-ray micro-beams,52–54 preliminary tests at increasing fluences were performed at a few spots of selected reference powders and mock-ups to ensure that the collected data were not affected by any artefacts due to X-ray beam exposure.
2.6 ATR mode μ-FTIR
μ-FTIR mapping investigations on CM1KBr from Maestà and paint mock-ups were performed in ATR mode by employing a Thermo Scientific Nicolet iN10MX spectrometer equipped with a Ge crystal and a Mercury cadmium telluride (MCT) detector.Maps from selected regions of CM1KBr and paint mock-up cross-sections were obtained with an optical aperture of either 40 × 40 μm2 or 48 × 48 μm2 (effective investigated areas: 10 × 10 μm2 or 12 × 12 μm2) and with a step size down to 8 μm. Data were recorded in the 4000–675 cm−1 range, with 4 cm−1 spectral resolution and 64 scans. Datasets were processed with the Omnic Picta and Omic32 software.
2.7 μ-Raman
μ-Raman analyses at selected spots of paint mock-ups were carried out by a Bruker Senterra Raman Microscope coupled to an Olympus BX 40 microscope and equipped with a CCD camera. Spectra were recorded using a 785 nm diode laser with excitation powers of either 1 mW or 10 mW and a slit of 25 × 1000 μm2. Data were acquired in the 2630–72 cm−1 and 1518–72 cm−1 ranges, with 3–5 cm−1 spectral resolution, 3 s exposure time and 15–20 scans. A minimum number of six spectra was recorded from each sample. Data were processed with the OPUS and/or Omnic32 software.2.8 Colorimetry
A Konica-Minolta CM700D portable colorimeter was used for the analysis of color changes of paint mock-ups. The instrument is equipped with a pulsed xenon lamp emitting in the visible spectral range, a silicon photodiode array detector and an integrating sphere with a 40 mm internal diameter. The software interfaced with the instrumentation permitted for the conversion of the spectra (recorded in the 360–740 nm range and with 10 nm spectral resolution) into CIE L*a*b* chromatic coordinates under D65 standard illuminant and 10° angle observer. Total color changes were calculated according to the CIE 1976 formula, ΔE*=(ΔL*2 + Δa*2 + Δb*2)1/2.3. Results and discussion
3.1 Evaluation of the alteration state of the “fake-gilded” decorations in the Maestà
The selective micro-sampling of the darkened decoration areas offered us the opportunity to study the alteration state of the orpiment-silver-based paint at this specific location. Two similar cross sections (CM1resin and CM1KBr) were studied in parallel by SR-based X-ray methods and μ-FTIR mapping. An overview of the obtained results is shown in Table 1.| Layer | SR μ-XRD | S speciation [SVI]/[Stotal] (%) (SR μ-XRF, μ-XANES)c | Ag speciation (SR μ-XRF, μ-XANES)c | As speciation (SR μ-XRF, μ-XANES)c | μ-FTIR (ATR mode) |
|---|---|---|---|---|---|
| a Only a few crystals are present (see Fig. 1f and g). b In this layer the abundance of Ag0, α-Ag2S, Pb5(AsO4)3Cl and α-As2S3 is much lower than that found in layer 3 (see Fig. 1e and f). c μ-XANES measurements performed only in layers 2 and 3 (see Fig. 2). d According to the LCF results, the presence of sulfates of Ca, Pb and/or K is more likely (see Fig. 2d). e The presence of minor abundances of other Ag-compounds, such as silver sulfate and/or acetate, is likely (see Fig. S2). | |||||
| 4 (Repaint) | Metallic silver (Ag0); hydrocerussite [Pb3(CO3)2(OH)2] | — | — | — | Lipids; lead carboxylates; Pb3(CO3)2(OH)2; CaCO3; silicates |
| 3 (“Fake-gilded”) | Acanthite (α-Ag2S); mimetite [Pb5(AsO4)3Cl]; α-As2S3; Pb3(CO3)2(OH)2; syngenite [K2Ca(SO4)2·H2O] | — | — | α-As2S3; AsV-species | Proteinaceous material; silicates; sulfates |
| 2 (“Fake-gilded”) | Ag0; α-Ag2S; α-As2S3; minium (Pb3O4); Pb3(CO3)2(OH)2; calcite (CaCO3)a | ∼20–45d | α-Ag2Se | Proteinaceous material; sulfates; silicates | |
| 1 (Paint) | Pb3O4; Pb3(CO3)2(OH)2;CaCO3; gypsum (CaSO4·2H2O); goethite [FeIIIO(OH)]a,b | — | — | — | Proteinaceous material; silicates; CaCO3 |
| 0 (Ground) | CaSO4·2H2O; CaCO3; FeIIIO(OH); Pb3O4; Pb3(CO3)2(OH)2b | Proteinaceous material; CaSO4·2H2O; CaCO3; silicates | |||
In the original “fake-gilded” layers (n. 2 and 3), SR μ-XRD mapping (Fig. 1d, e and S1†) in combination with As K- and Ag L3-edges μ-XANES spectroscopy (Fig. 2a, b and S2†) reveals the co-localized presence of α-As2S3 and acanthite (α-Ag2S). In line with previous studies,37,38 black α-Ag2S can be considered a secondary product arising from the corrosion of the original Ag0 powder and is the main responsible for the observed darkening. In the innermost darkened gilded layer (n. 2) and in the outermost α-As2S3-free repaint layer (n. 4), Ag0 is still present (Fig. 1e). The darkening of Ag-based decorations due to the formation of α-Ag2S was observed also in the absence of As2S3 as a possible result of the interaction between the paint and either S-gaseous pollutants, or S-species of the binder, or both.55,56 Nevertheless, the detection of α-Ag2S mainly in the uppermost original gilded layer (n. 3) and in the As2S3-containing layers of the Maestà (n. 2 and 3) strongly suggests that S2− anions arising from the degradation of orpiment might be the main source for the formation of achantite.
Interestingly, next to α-As2S3, AsV- and SVI-species were also found in layers 2–3 (Fig. 2). The relative abundances of AsV-compounds decrease when going from layer 3 to 2, as pointed out by the increasing of the intensity ratio between As K-edge XANES signals at 11.870 keV (assigned to As2S3) and 11.8754 keV (due to AsV-species) (Fig. 2b). In the uppermost layer 3, SR μ-XRD clearly reveals the presence of a yellowish lead-arsenate based rare mineral, mimetite [Pb5(AsO4)3Cl] (Fig. 1f). As documented by previous studies in wall and oil paintings,26,27,57 we can reasonably assume that this compound has formed as a result of the interaction among the oxidation products of orpiment, Cl−-ions and Pb2+-ions released by Pb-based pigments, such as minium (Pb3O4) and lead white [PbCO3 and Pb3(CO3)2(OH)2], all components that are widespread throughout layers 2–3 (Fig. 1g and S2†). Based on the fact that in layer 4 no As-compounds are present, while in previous studies arsenate species have been shown to exhibit a high mobility inside paint layer stacks,23,24,26 we hypothesize that the layer 4, rich in lead white and Ag0, is likely to have been applied on top of the other paint layers after that the oxidation of Ag0 and α-As2S3 took place in layer 3, possibly to counterbalance or completely cover the darkening of the “fake-gilded” surface.
According to the LCF results of each S K-edge μ-XANES spectrum (Fig. 2c and d), S-phases are present in layer 2 in the following variable abundances: α-As2S3 (∼40–80%), sulfates (likely of Ca, Pb and/or K) (∼20–45%) and α-Ag2S (up to ∼20%). The presence of a sulfate compound in such layer is further corroborated by μ-FTIR, through the sulfate asymmetric stretching mode [ν3(SO42−)] at 1122 cm−1 (Fig. 3b, c and S3†). Syngenite [K2Ca(SO4)2·H2O] (Fig. 1g) was identified by μ-XRD in layer 3. Based on previous studies,26,27 it is likely that the latter material was not originally present but has resulted from the reaction between sulfate ions that either formed in situ by oxidation of sulfidic ions of orpiment or by local solubilization of other constitutive materials (e.g., gypsum), and K+ and Ca2+ cations present within the paint.
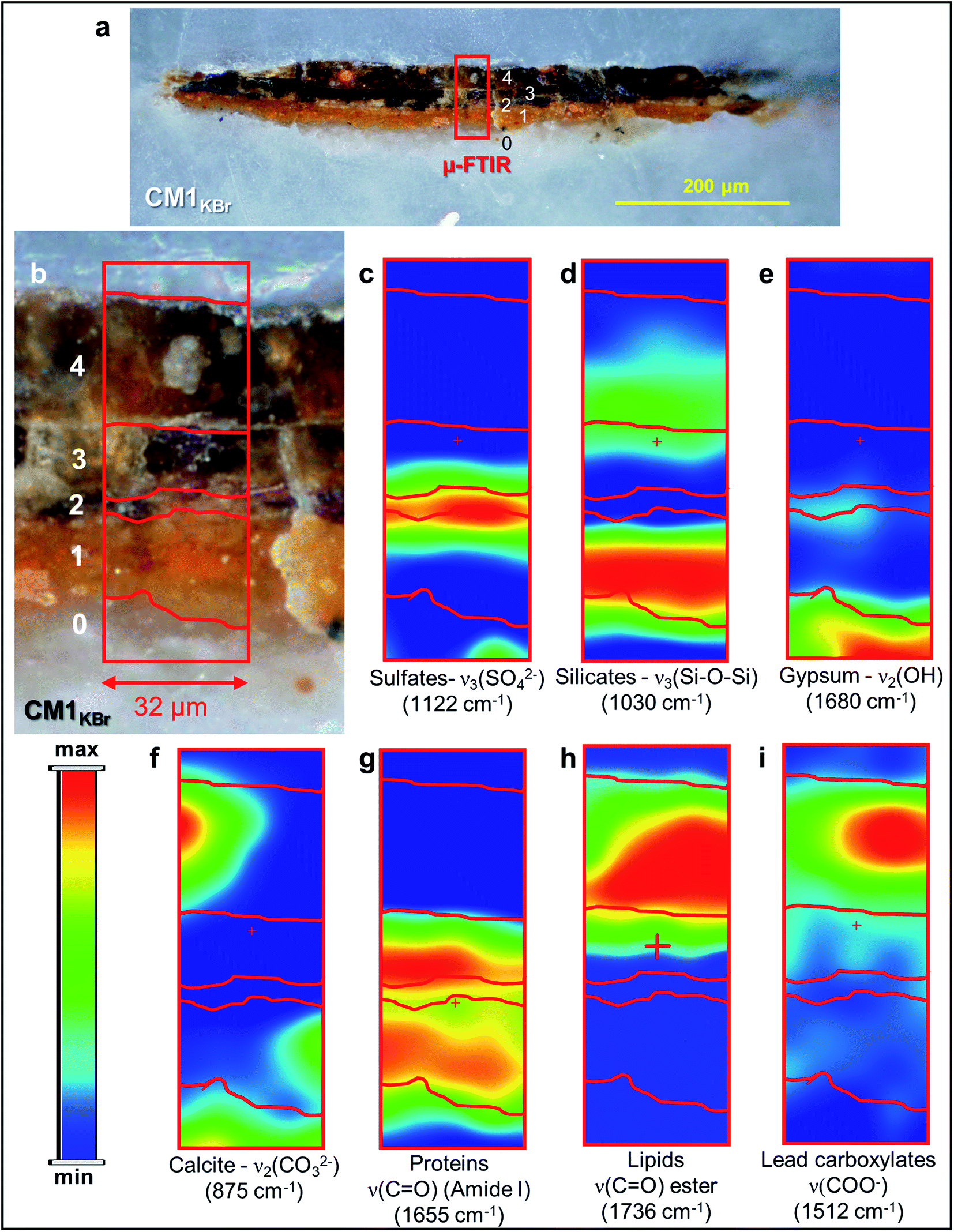 | ||
| Fig. 3 (a) Micrograph of cross-section CM1KBr taken with visible light (magnification: 200×) from the sampling spot shown in Fig. 1a and (b) corresponding detail image where ATR mode μ-FTIR mapping was performed. (c–i) μ-FTIR distribution maps of the identified compounds (see Fig. S3† for a selection of the corresponding spectra). | ||
In the ground (layer 0), gypsum is the dominant phase along with lower amounts of α-As2S3, α-Ag2S, Ag0 and silicates [ν3(Si–O–Si) at 1030 cm−1] (Fig. 1e,f and 3d,e). Crystals of goethite [FeIIIO(OH)], calcite (CaCO3), hydrocerussite [Pb3(CO3)2(OH)2] and Pb3O4 are visible too. The three latter components are also widespread throughout layer 1, where silicates are present too (Fig. 1f, g and 3f).
Regarding the binding media, the FTIR bands at 1655 and 1541 cm−1, visible in the spectra acquired from layers 0–3, are associated to proteinaceous materials (Fig. 3g). Such result provides evidence of the original use of a tempera technique and is consistent with painting technique of the 13th century. In the Ag0-rich repainted layer 4, assumed to be more recent than the layers below, the identification of the characteristic ester ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) band at 1736 cm−1 (Fig. 3h) denotes instead the use of a lipidic binder, whose interaction with Pb3(CO3)2(OH)2 (Fig. 1g) can be considered responsible for the occurrence of lead carboxylates [ν(COO−) signal at 1512 cm−1; Fig. 3i].
O) band at 1736 cm−1 (Fig. 3h) denotes instead the use of a lipidic binder, whose interaction with Pb3(CO3)2(OH)2 (Fig. 1g) can be considered responsible for the occurrence of lead carboxylates [ν(COO−) signal at 1512 cm−1; Fig. 3i].
Overall, the distributions of the identified Ag-phases and As-/S-compounds firmly suggest that interactions among the components of the paint and of the original painting surface materials with environmental agents (i.e., light and humidity) may have induced the corrosion of Ag0 to α-Ag2S and the oxidation of α-As2S3 to AsV- and SVI-compounds in the original “fake-gilded” layers of the studied cross-sections. The identified arsenate- and sulfate-species can be linked to the oxidation process of α-As2S3, in which the AsIII- species, that are originally present in the pigment, become AsV-species, while the sulfidic counter ions are either involved in the formation of black α-Ag2S or become oxidized to sulfates, as already previously described in detail.23–27
3.2. Effects of Ag0 and environmental agents on the chemical transformations of orpiment tempera paints
To assess the effect of Ag0 and of different environmental parameters on the degradation process of orpiment, thus to provide direct insights into which factors are responsible for the darkening process of the “fake-gilded” decoration areas of the painting Maestà, we studied a series of tempera paint mock-ups prepared using commercial powders of (i) orpiment, (ii) metallic silver and (ii) mixtures of orpiment and Ag0 (in Table 2 and Fig. S4–S6 and S9† referred to as Orp, Ag, and Orp–Ag). The mock-ups were subjected to accelerated aging under UVA-vis light at RH ∼25% and high humidity conditions (RH ≥ 95%) at T = 40° (in Table 2, Fig. 4–6, S7 and S9† denoted with the subscripts UVA-vis and 95%RH, respectively). In what follows, a selection of the most meaningful SR-based X-ray and vibrational spectroscopy data for the discussion is provided and a summary of the obtained results is shown in Table 2.| Sample (Fig. n.) | Colorimetric data | μ-FTIR (ATR mode) | μ-Raman | SR μ-XRD | S speciation SR μ-XRF, (μ-XANES) | Ag speciation (μ-XANES) | As speciation (SR μ-XRF, μ-XANES) | |||
|---|---|---|---|---|---|---|---|---|---|---|
| ΔL* | Δa* | Δb* | ΔE* | |||||||
| a Analysis performed directly at the paint surface (results not shown). b Present as aggregates of 5–40 μm diameter throughout the paint stratigraphy. c Present as a layer of about 15–20 μm thickness at the paint surface. d Widespread throughout the paint stratigraphy and detected in lower amount with respect to the equivalent UVA-vis aged paint. | ||||||||||
| Unaged | ||||||||||
 Orp (Fig. S4) Orp (Fig. S4) |
— | — | — | — | — | α-As2S3 | α-As2S3 | α-As2S3 | — | α-As2S3 |
 Orp–Ag (Fig. S5 and S6) Orp–Ag (Fig. S5 and S6) |
— | — | — | — | Traces of As2O3 | α-As2S3; possible Ag2S | α-As2S3;Ag0; α-Ag2Sb | α-As2S3; traces of sulfates | Ag0 | α-As2S3 |
 Ag (Fig. S9) Ag (Fig. S9) |
— | — | — | — | — | — | Ag0; traces of AgCl | — | — | — |
|
||||||||||
| Photochemical aging | ||||||||||
 OrpUVA-vis (Fig. 4) OrpUVA-vis (Fig. 4) |
3 ± 1 | 1 ± 1 | 3 ± 2 | 4 ± 1 | Traces of As2O3 | α-As2S3a | α-As2S3; As2O3(b) | α-As2S3; sulfatesb | — | α-As2S3; As2O3 |
 Orp–AgUVA-vis (Fig. 5, S7 and S8) Orp–AgUVA-vis (Fig. 5, S7 and S8) |
−17 ± 2 | −0.8 ± 0.8 | −27 ± 4 | 32 ± 2 | As2O3 | α-As2S3; possible Ag2S; As2O3 | α-As2S3; Ag0; α-Ag2S; c As2O3c | α-As2S3; sulfatesb | Ag0; α-Ag2Sc | α-As2S3; As2O3 |
 AgUVA-vis (Fig. S9) AgUVA-vis (Fig. S9) |
−5 ± 1 | 0.6 ± 0.2 | 0 ± 1 | 5 ± 1 | — | — | Ag0; traces of AgCl | — | — | — |
|
||||||||||
| High moisture aging | ||||||||||
 Orp95%RH (Fig. 6a and b) Orp95%RH (Fig. 6a and b) |
4 ± 1 | 0 ± 2 | 3 ± 2 | 5 ± 1 | Traces of As2O3a | α- As2S3; | α-As2S3; As2O3b | α-As2S3a | — | — |
 Orp–Ag95%RH (Fig. 6c and d) Orp–Ag95%RH (Fig. 6c and d) |
−3 ± 2 | 2.6 ± 0.9 | −4 ± 3 | 5 ± 2 | Traces of As2O3 | α-As2S3; possible Ag2S; As2O3 | α-As2S3; Ag0; α-Ag2S;d As2O3d | α-As2S3a | — | — |
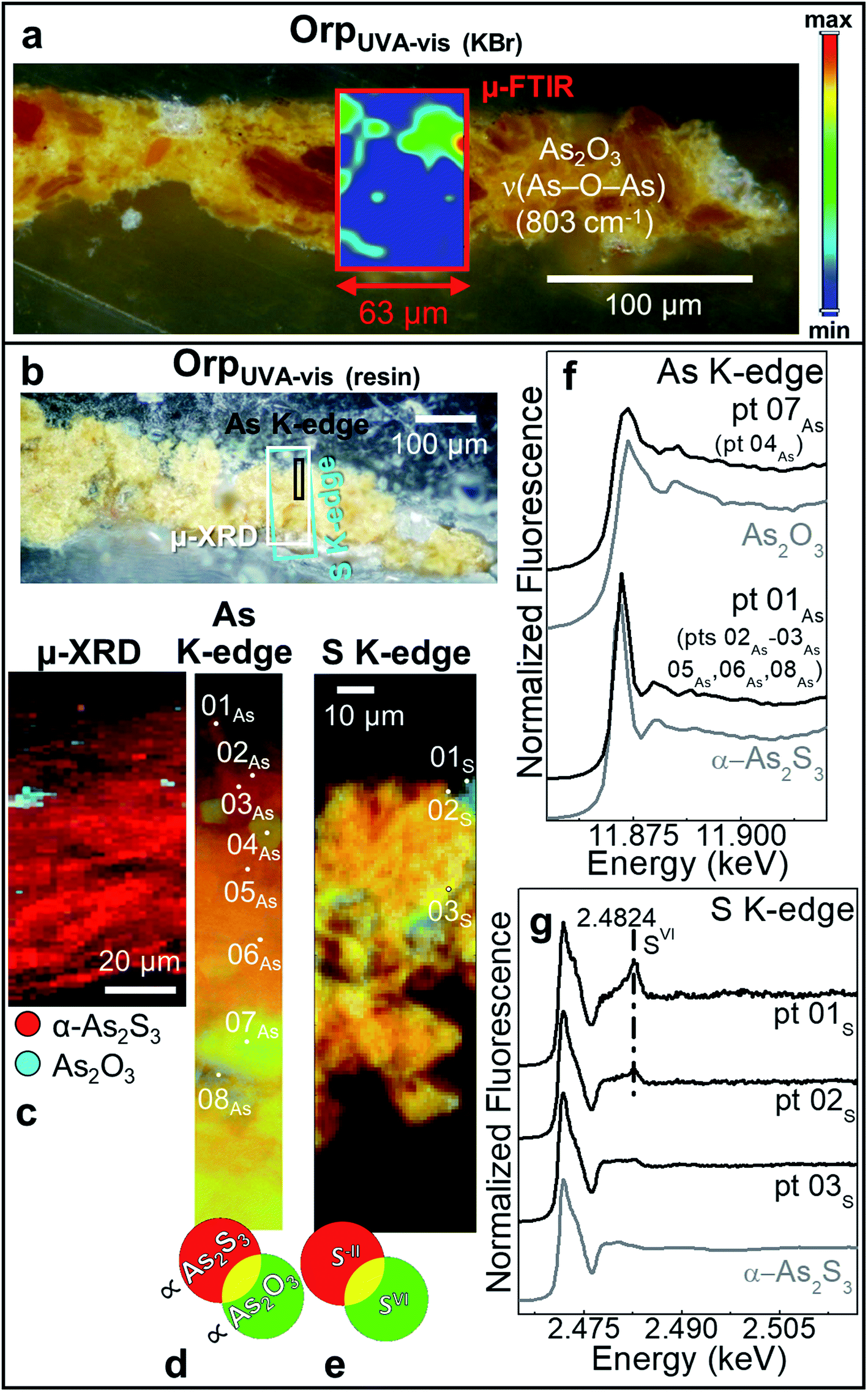 | ||
| Fig. 4 Photomicrographs of (a) OrpUVA-vis (KBr) and (b) OrpUVA-vis (resin) cross-sections. In (a) the μ-FTIR map of As2O3 is also shown. (c) Composite SR μ-XRD maps of α-As2S3 (red) and As2O3 (cyan) [map size (v × h): 145.5 × 50 μm2; step size (v × h): 1.5 × 2 μm2; exp. time: 1 s per pixel; energy: 21 keV]. Composite SR μ-XRF images of (d) AsIII (∝α-As2S3) (red) and AsIII (∝As2O3) (green) [map size (v × h): 63 × 9.9 μm2; step size (v × h): 0.1 × 0.15 μm2; exp. time: 100 ms per pixel; energy (AsIII, ∝α-As2S3) = 11.870 keV, energy (AsIII, ∝α-As2O3) = 11.8754 keV] and (e) S−II (red) and SVI (green) [map size (v × h): 159 × 46 μm2; step size (v × h): 1 × 1 μm2; exp. time: 100 ms per pixel; energy (S−II) = 2.4718 keV, energy (SVI) = 2.482 keV]. Selection of the μ-XANES spectra recorded at the (f) As K-edge and (g) S K-edge from the spots reported in (d and e), compared to those of a set of reference compounds (grey). In (b), rectangles show the areas where maps of (c–e) were recorded. In (f and g), numbers in brackets refer to the spectra showing similar features to those reported. | ||
 | ||
| Fig. 5 Photomicrographs of (a) Orp–AgUVA-vis (KBr) and (c) Orp–AgUVA-vis (resin) cross-sections. (b) ATR mode μ-FTIR map of As2O3. (d) Composite SR μ-XRD images of α-As2S3 (red), α-Ag2S (green), Ag0 (blue) and As2O3 (cyan) [map size (v × h): 150 × 60 μm2; step size (v × h): 1.5 × 2 μm2; exp. time: 1 s per pixel; energy: 21 keV] (see Fig. S8† for the corresponding XRD patterns). (e) RGB SR μ-XRF images of S−II/SVI/Ag-L [map size (v × h): 130 × 100 μm2; step size (v × h): 0.97 × 1.1 μm2; exp. time: 100 ms per pixel] and (f) selection of S K-edge μ-XANES spectra (black) acquired from the spots shown in (e). In (a and c), rectangles show the areas where maps of (b,d and e) were recorded. In (f), numbers in brackets refer to the spots showing similar spectral features to those reported. | ||
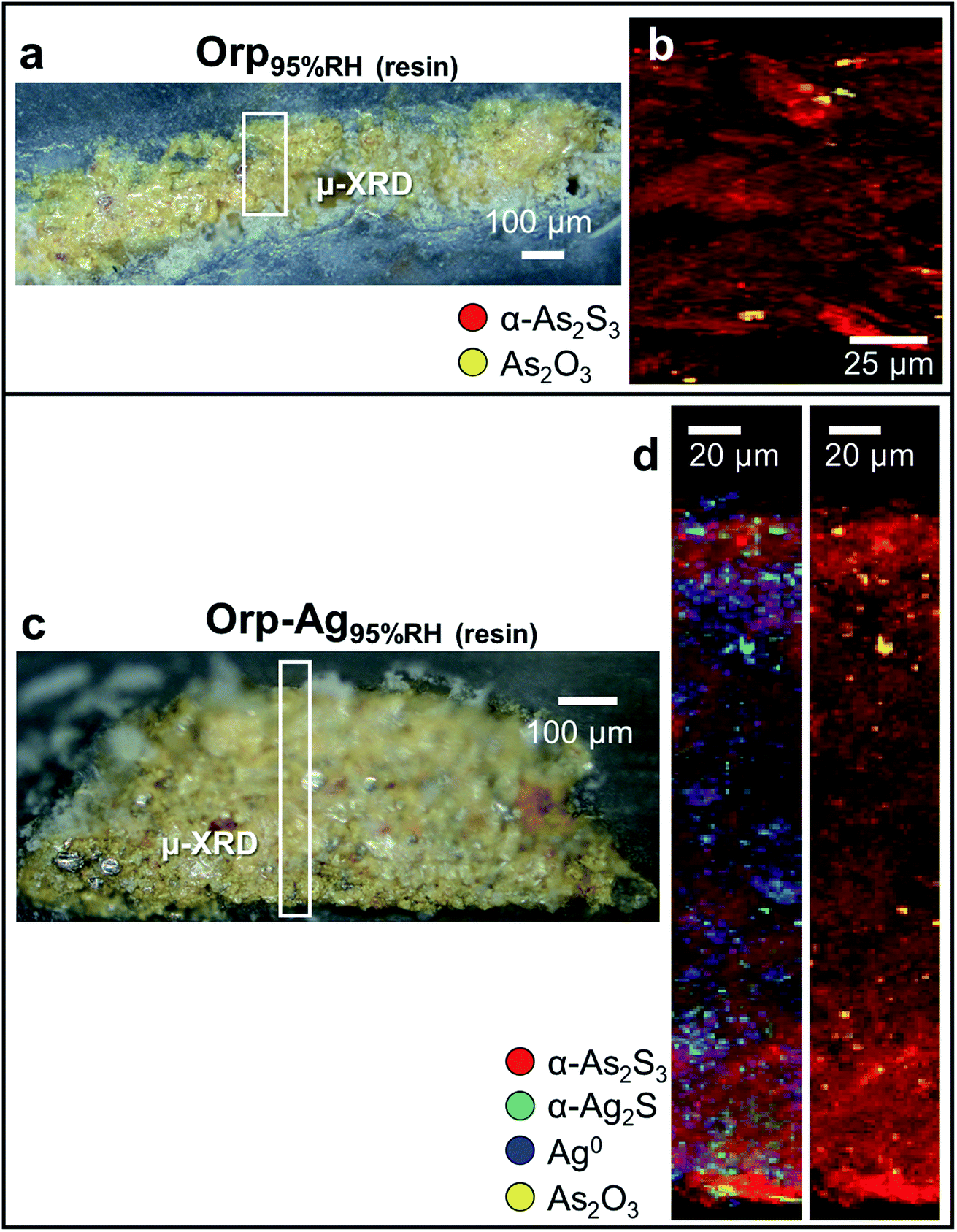 | ||
| Fig. 6 Photomicrographs of (a) Orp95%RH (resin) and (c) Orp–Ag95%RH (resin) cross-sections. (b and d) Composite SR μ-XRD images of α-As2S3 (red), α-Ag2S (green), Ag0 (blue) and As2O3 (yellow) recorded from (b) Orp95% RH [map size (v × h): 239 × 100 μm2; step size (v × h): 1 × 2 μm2; exp. time: 0.5 s per pixel; energy: 21 keV] and (d) Orp–Ag95% RH mock-ups [map size (v × h): 430.5 × 50 μm2; step size (v × h): 1.5 × 2 μm2; exp. time: 1 s per pixel; energy: 21 keV]. In (a and c), rectangles show the regions where maps of (b and d) were acquired. | ||
As a first step, we have evaluated the stability of the paint mock-ups after exposure to UVA-vis light at RH ∼ 25% (Fig. 4 and 5).
In sample OrpUVA-vis, in which only α-As2S3 has been identified before aging (Fig. S4†), photo-oxidation has induced a slight color change of the paint surface (Table 2: ΔE = 4 ± 1). In line with literature,20–22 such a phenomenon can be associated to the in situ conversion of α-As2S3 to As2O3 (Fig. 4a–d and f: pts 04As, 07As), that is present in the form of aggregates of about 5 to 30 μm diameter within the paint (Fig. 4c and d). In addition, the local formation of sulfates is visualized via S-oxidation state maps and the signal at 2.482 keV in the S K-edge μ-XANES spectra collected at selected spots (Fig. 4e,g: pts 01s,02s). Sulfates may result from the oxidative degradation of S-groups of amino acids of the proteinaceous binder58–60 and/or from that of α-As2S3 in contact with the organic matrix.21,26
The photoaging of Orp–AgUVA-vis led to a profound darkening of the paint surface (Table 2: ΔE = 32 ± 2), resulting in a significant phenomenon: the formation of a α-Ag2S- and As2O3-rich layer within the uppermost 15–20 μm of the α-As2S3/Ag0-based paint stratigraphy (Fig. 5a–d; see Fig. S5 and S6† for the data recorded before light exposure). The distribution of these phases was also corroborated via single point μ-Raman and Ag L3- and As K-edges μ-XANES investigations (Fig. S7†). In addition, newly formed sulfates aggregates were identified throughout the α-As2S3 paint matrix (Fig. 5e and f: pts 01S–03S). Such results clearly highlight that, under the synergistic effect of exposure to light and presence of Ag0, the α-As2S3 originally present was transformed to As2O3, while the sulfidic counter ions precipitated with the Ag+ ions derived from the oxidation of Ag0.
In a second step, we have investigated the effects of high humidity conditions (at RH ≥ 95%) in the absence of light exposure on the chemical reactivity of As2S3/Ag0 tempera paints. The exposure to moisture of the Orp95%RH mock-up (Fig. 6a and b) induced a colour change (Table 2: ΔE = 5 ± 1) that is comparable to those obtained via photochemical aging along with the formation of As2O3 aggregates of ∼5 μm diameter within the yellow α-As2S3 matrix. For sample Orp–Ag95%RH, high moisture conditions promoted a less clear darkening of the paint surface (Table 2: ΔE = 5 ± 2) than that observed in the equivalent light-exposed sample. Smaller abundances of both α-Ag2S and As2O3 are also visible in the corresponding μ-XRD maps (Fig. 6c and d). Such newly-formed compounds are often co-localized in the same regions, but, differently from the equivalent light-exposed mock-up, they are quite homogenously distributed throughout the entire paint stratigraphy. For both moisture-aged mock-ups, no evidence of the presence of sulfate-species was revealed by S K-edge XANES analysis (results not reported). It is relevant to note that under none of the employed aging conditions, the formation of AsV-species could be observed.
In summary, our results prove that Orp–Ag mock-ups present a higher reactivity than those containing only orpiment. Indeed, in the Orp–Ag cross-section As2O3 and α-Ag2S were identified in few spots even before the artificial aging (Fig. S5 and S6†). In addition, the observation that the photoaging of tempera paint mock-ups composed of only Ag0 has induced a slight color change of the paint surface (Table 2: ΔE = 5 ± 1) and no formation of α-Ag2S (Fig. S9†) is an extra prove of the key-role played by As2S3 for the formation of α-Ag2S.
3.3. Degradation mechanism of Ag0/orpiment tempera paints
By combining the micro-analytical results obtained from the study of the altered “fake-gilded” decorations of the Maestà by Cimabue with those arising from artificially paint mock-ups, we have demonstrated that, before the application of the more recent repainted layer (Fig. 1–3; layer 4), interactions among the component of the paint and of the painting surface with environmental agents (i.e., light and humidity) have induced the corrosion of Ag0 to α-Ag2S and the oxidation of α-As2S3 to AsV- and SVI-compounds (Fig. 1–3; layers 2 and 3). Intriguingly, Ag0 triggers the degradation of α-As2S3, while S2− ions resulting from the degradation of α-As2S3 are the main responsible for the formation of α-Ag2S in orpiment and Ag0 tempera paint mixtures (Fig. 4–6, S7 and S8†).Based on our experimental findings and earlier research,22,23,57,61–75 a mechanism for the darkening process of Ag0/orpiment-based tempera paints close to the mechanism (i) described in the Introduction,37,38 can thus be put forward.
As a first step of the transformation, in aerobic conditions, As2S3 gives rise to the formation of As2O3 (Fig. 4–6) and polysulfides (Sn2−, with 2 ≤ n ≤ 9) according to the following reaction [eqn (1)]:61
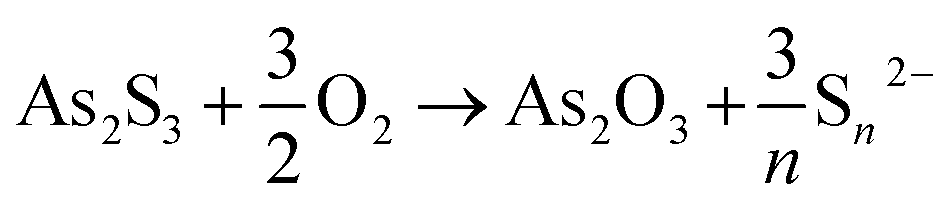 | (1) |
The speciation of polysulfides in aqueous/moist environments depends on different parameters, including the sulfur content, the pH and the temperature. At around neutral pH values, such are those of tempera paints,62 the formation of Sn2− with 3 ≤ n≤ 5 is more likely.61,63,64
So far, no direct evidence has been provided regarding the exact nature of the S-based compounds in eqn (1). Whether and how polysulfides or other S-species (e.g., radicals)22 form as one of the primary degradation products of As2S3, it remains to be addressed. Nevertheless, in the context of this work it is reasonable to assume their presence in view of the tendency of Sn2− to react (at 20 °C) with moisture (entrapped in the paint matrix and/or from the environment), giving rise to other Sn+12− compounds and to HS− [eqn (2)],61 the latter being one of the S-species responsible for the α-Ag2S formation:37,38
| xS2−n + H2O → HS− + OH− + (x − 1)Sn+12− | (2) |
Under neutral conditions the following HS−/H2S equilibrium reaction can be established (eqn (3)):65
| HS− + H+ ⇌ H2S | (3) |
The interaction between HS− and/or H2S and Ag0 results in the formation of α-Ag2S (Fig. 1, 5 and 6) according to the following reactions [eqn (4) and (5)]:38,66–68
| 2Ag0 + HS− (aq) + ½O2 → Ag2S + OH− | (4) |
| 2Ag0 + H2S (aq) + ½O2 → Ag2S + H2O | (5) |
The above-mentioned sequence of reactions [eqn (1)–(5)] explains the role of moisture in the darkening process of Ag0/orpiment tempera paints, thus also providing an explanation why the in situ formation of the alteration products can already be observed prior to the start of the aging treatments of the paint mock-ups (Fig. S5 and S6†).
Under exposure to light, the higher efficiency of the degradation process towards the formation of α-Ag2S and As2O3 (Fig. 4 and 5) can be explained by taking into account the fact that the photoaging may promote the formation of (organo)sulfur radicals (e.g., HS˙, 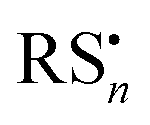 ) via two different pathways: (i) direct photolysis of S-compounds (e.g., H2S/HS−) arising either from the degradation of orpiment and/or S-containing amino acids of the binder;60,69,70 (ii) reactions between S-containing compounds and radicals (e.g., R˙, RO˙, ROO˙, OH˙), the latter arising from the photo-oxidative degradation of the binding medium.60,71–75
) via two different pathways: (i) direct photolysis of S-compounds (e.g., H2S/HS−) arising either from the degradation of orpiment and/or S-containing amino acids of the binder;60,69,70 (ii) reactions between S-containing compounds and radicals (e.g., R˙, RO˙, ROO˙, OH˙), the latter arising from the photo-oxidative degradation of the binding medium.60,71–75
In Cimabue's Maestà (Fig. 1f and g), mimetite and syngenite are not expected to have been present originally in the paint samples. Both compounds have been recently identified in other artworks and have been interpreted as secondary products of the degradation of orpiment,26,27 a process that probably in the Maestà has occurred slowly over time. Under aerobic and moist environment, As2O3, the primary alteration product of As2S3, may dissolve and oxidize to AsV-species. Pb2+ is one of dominating cations present in the paint (due to the presence of lead white and minium) and can act as a counter ion forming mimetite. The source of Cl−-ions required for the formation of such compound is not easily identified; however, a number of painters' materials, including the binding medium76 and the specific formulation of some pigments (e.g., lead white),77 generally contain an abundance of Cl-species.
Sulfates originate either from materials originally used from the painter (e.g., gypsum; Fig. 1f and 3e) or from the oxidation of sulfur groups present in the amino acids of the binder and/or sulfur-containing-radicals arising from the degradation of orpiment. Such species interact then with K+ and Ca2+ of the paint matrix and readily precipitate as syngenite. Only very rarely, syngenite has been mentioned as a primary material in the plaster of a Chinese wall painting, together with calcite, quartz and gypsum.78 Thus, the presence of such phase in the degraded “fake-gilded” layer of the Maestà (Fig. 1g) seems to indicate its formation as a secondary product.
In summary, we can assume that the darkening process of the “fake-gilded” decorations of the Maestà has developed via conversion of As2S3/Ag0 tempera paint to α-Ag2S and As2O3 as a result of the interaction of the original “fake-gilded” layer with moisture and light before the application of the oil-based current repaint layer. The conversion of AsIII- to AsV-species may have occurred even after the application of the repaint layer leading to the complete transformation of As2O3. The kinetic of this reaction would probably require a long time and this may explain why we didn't observe arsenates in the artificially aged paint mock-ups.
4. Conclusions
A comprehensive multi-material and multi-method approach based on SR X-ray micro-analytical techniques and vibrational micro-spectroscopy methods has been presented here and successfully exploited to establish the causes and mechanism of darkening of “fake-gilded” decorations, made up of a As2S3/Ag mixture, in the 13th century Maestà by Cimabue.The employed analytical methods permitted to discriminate and visualize down to the sub-micrometre scale length the distribution of the main components of the darkened “fake-gilded” tempera layers, namely: α-Ag2S, arsenates (mimetite) and sulfates (syngenite). These results, combined with the findings obtained from artificially aged paint mock-ups, led us to conclude that such compounds can be interpreted as alteration products of the paint and that the observed darkening is due to black α-Ag2S. Even more interestingly, we observed that Ag0 prompts the degradation of α-As2S3 to As2O3, whereas S2− ions arising from the degradation of α-As2S3 are the main responsible for the formation of α-Ag2S in orpiment and Ag0 tempera paint mixtures. In high moisture conditions (RH ≥95%), the conversion of the original As2S3/Ag0 tempera paint to α-Ag2S and As2O3 can take place also in the absence of light, while UVA-visible irradiation contributes to further increases the reactivity of As2S3/Ag0 tempera paint, possibly via the formation of (organo)sulfur radical intermediates, but with no changes in the nature of the final degradation products.
Based on our results, we can assume that the darkening process of the “fake-gilded” layers of the Maestà has developed via two main steps:
(i) Conversion of As2S3/Ag0 tempera paint to α-Ag2S and As2O3 as a result of the interaction of the original “fake-gilded” layer with moisture and light of the environment. It is most likely that the process took place prior to the application of the oil-based current repaint layer, containing both Ag0 and hydrocerussite;
(ii) Slow dissolution and oxidation of As2O3 to arsenates under aerobic and moist conditions. Such transformation may have progressed after the application of the repainting layer.
The above-mentioned findings are expected to have meaningful implications for the preventive conservation of the Maestà by Cimabue. In particular, the degradation of the As2S3/Ag0 paints might be mitigated by minimizing the exposure of the painting to excessively high moisture levels (i.e., RH <30%) and by keeping the lighting at values foreseen for light-sensitive painting materials.79
Overall, the multi-material and microscale multi-method analytical approach developed here paves the way for the study of darkening of “fake-gilded” decorations of several other old master paintings and for a targeted investigation into research on the degradation mechanism of As2S3/Ag0-based mixtures also in lipidic media. These studies are still ongoing and their results will be published in a follow-up paper. Furthermore, considering the crystalline nature of the alteration phases identified, this study lays the basis for the integration of micro-analysis on a limited number of micro-samples with non-invasive macro-scale XRD mapping investigations27 of a larger number of darkened “fake-gilded” paint areas of the Maestà and many other artworks realized with a similar technique.
Author contributions
Letizia Monico: investigation, formal analysis, validation, methodology, conceptualization, writing – original draft. Silvia Prati, Aldo Romani: conceptualization, supervision, writing – review & editing. Giorgia Sciutto, Emilio Catelli, Diego Quintero Balbas, Steven De Meyer, Gert Nuyt: investigation, formal analysis, validation, methodology, writing – review & editing. Koen Janssens, Rocco Mazzeo: supervision, writing – review & editing. Marine Cotte, Jan Garrevoet, Gerald Falkenberg, Vanessa Isabel Tardillo Suarez, Remi Tucoulou: setting up of synchrotron radiation-based experiments, investigation, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was financially supported by the EU projects IPERION-CH (H2020-INFRAIA-2014-2015, GA no. 654028) and IPERION-HS (H2020-INFRAIA-2019-1, GA no. 871034), the project AMIS (Dipartimenti di Eccellenza 2018–2022, funded by MUR and Perugia University) and the program “Ricerca di Base 2017” (funded by Perugia University). K. J. would like to thank the FWO Brussels for financial support through grants G056619N, G054719N and I001919N. For the beamtime grants received, we thank ESRF-ID21/ID16b (experiment nos. HG64, HG117 and in-house beamtimes). We acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities (experiment nos. I-20170101 EC, I-20190179 EC). Parts of this research was performed at the P06 beamline of Petra III. The research leading to this result has been supported by the project CALIPSOplus (GA no. 730872 from the EU Framework Programme for Research and Innovation HORIZON 2020). The Authors wish to thank Dr Franco Faranda for allowing the analytical investigation on the painting by Cimabue.References
- G. Van der Snickt, J. Dik, M. Cotte, K. Janssens, J. Jaroszewicz, W. De Nolf, J. Groenewegen and L. van der Loeff, Anal. Chem., 2009, 7, 2600 CrossRef PubMed.
- G. Van der Snickt, K. Janssens, J. Dik, W. De Nolf, F. Vanmeert, J. Jaroszewicz, M. Cotte, G. Falkenberg and L. van der Loeff, Anal. Chem., 2012, 84, 10221 CrossRef CAS PubMed.
- J. Mass, J. Sedlmair, C. S. Patterson, D. Carson, B. Buckley and C. Hirschmugl, Analyst, 2013, 138, 6032 RSC , and references therein.
- E. Pouyet, M. Cotte, B. Fayard, M. Salomé, F. Meirer, A. Mehta, E. S. Uffelman, A. Hull, F. Vanmeert, J. Kieffer, M. Burghammer, K. Janssens, F. Sette and J. Mass, Appl. Phys. A: Mater. Sci. Process., 2015, 121, 967 CrossRef CAS , and references therein.
- L. Monico, A. Chieli, S. De Meyer, M. Cotte, W. de Nolf, G. Falkenberg, K. Janssens, A. Romani and C. Miliani, Chem.–Eur. J., 2018, 24, 11584 CrossRef CAS PubMed.
- L. Monico, L. Cartechini, F. Rosi, A. Chieli, C. Grazia, S. De Meyer, G. Nuyts, F. Vanmeert, K. Janssens, M. Cotte, W. De Nolf, G. Falkenberg, I. Crina Anca Sandu, E. Storevik Tveit, J. Mass, R. Pereira de Freitas, A. Romani and C. Miliani, Sci. Adv., 2020, 6, eaay3514, DOI:10.1126/sciadv.aay3514.
- L. Monico, G. Van der Snickt, K. Janssens, W. De Nolf, C. Miliani, J. Verbeeck, H. Tian, H. Tan, J. Dik, M. Radepont and M. Cotte, Anal. Chem., 2011, 83, 1214 CrossRef CAS PubMed.
- L. Monico, K. Janssens, C. Miliani, G. Van der Snickt, B. G. Brunetti, M. Cestelli Guidi, M. Radepont and M. Cotte, Anal. Chem., 2013, 85, 860 CrossRef CAS PubMed.
- L. Monico, K. Janssens, M. Cotte, A. Romani, L. Sorace, C. Grazia, B. G. Brunetti and C. Miliani, J. Anal. At. Spectrom., 2015, 30, 1500 RSC.
- L. Monico, K. Janssens, E. Hendriks, F. Vanmeert, G. Van der Snickt, M. Cotte, G. Falkenberg, B. G. Brunetti and C. Miliani, Angew. Chem., Int. Ed., 2015, 54, 13923 CrossRef CAS PubMed.
- L. Monico, K. Janssens, M. Cotte, L. Sorace, F. Vanmeert, B. G. Brunetti and C. Miliani, Microchem. J., 2016, 124, 272 CrossRef CAS.
- V. Otero, M. Vilarigues, L. Carlyle, M. Cotte, W. De Nolf and M. J. Melo, Photochem. Photobiol. Sci., 2018, 17, 266 CrossRef CAS PubMed.
- L. Monico, L. Sorace, M. Cotte, W. de Nolf, K. Janssens, A. Romani and C. Miliani, ACS Omega, 2019, 4, 6607 CrossRef CAS , and references therein.
- M. Radepont, W. de Nolf, K. Janssens, G. Van der Snickt, Y. Coquinot, L. Klaassen and M. Cotte, J. Anal. At. Spectrom., 2011, 26, 959 RSC.
- M. Radepont, Y. Coquinot, K. Janssens, J. J. Ezrati, W. de Nolf and M. Cotte, J. Anal. At. Spectrom., 2015, 30, 599 RSC.
- L. Samain, F. Grandjean, G. J. Long, P. Martinetto, P. Bordet, J. Sanyova and D. Strivay, J. Synchrotron Radiat., 2013, 20, 460 CrossRef CAS PubMed.
- L. Samain, G. Silversmit, J. Sanyova, B. Vekemans, H. Salomon, B. Gilbert, F. Grandjean, G. J. Long, R. P. Hermann, L. Vincze and D. Strivay, J. Anal. At. Spectrom., 2011, 26, 930 RSC.
- L. Zanella, F. Casadio, K. A. Gray, R. Warta, Q. Ma and J. F. Gaillard, J. Anal. At. Spectrom., 2011, 26, 1090 RSC.
- L. Bindi and P. Bonazzi, Am. Mineral., 2007, 92, 617 CrossRef CAS.
- K. Trentelman and L. Stodulski, Anal. Chem., 1996, 68, 1755 CrossRef CAS PubMed.
- M. Vermeulen, K. Janssens, J. Sanyova, V. Rahemi, C. McGlinchey and K. De Wael, Microchem. J., 2018, 138, 82 CrossRef CAS.
- M. Vermeulen, J. Sanyova, K. Janssens, G. Nuyts, S. De Meyer and K. De Wael, J. Anal. At. Spectrom., 2017, 32, 1331 RSC.
- K. Keune, J. Mass, A. Mehta, J. Church and F. Meirer, Heritage Sci., 2016, 4, 10 CrossRef.
- K. Keune, J. Mass, F. Meirer, C. Pottasch, A. van Loon, A. Hull, J. Church, E. Pouyet, M. Cotte and A. Mehta, J. Anal. At. Spectrom., 2015, 30, 813 RSC.
- M. Vermeulen, G. Nuyts, J. Sanyova, A. Vila, D. Buti, J. P. Suuronen and K. Janssens, J. Anal. At. Spectrom., 2016, 31, 1913 RSC.
- J. Simoen, S. De Meyer, F. Vanmeert, N. de Keyser, E. Avranovich, G. Van der Snickt, A. van Loon, K. Keune and K. Janssens, Heritage Sci., 2019, 7, 83 CrossRef.
- F. Vanmeert, N. de Keyser, A. van Loon, L. Klaassen, P. Noble and K. Janssens, Anal. Chem., 2019, 91, 7153 CrossRef CAS PubMed.
- W. Anaf, S. Trashin, O. Schalm, D. van Dorp, K. Janssens and K. De Wael, Anal. Chem., 2014, 86, 9742 CrossRef CAS PubMed.
- W. Anaf, K. Janssens and K. De Wael, Angew. Chem., Int. Ed., 2013, 125, 12800 CrossRef.
- R. D'Amico, La Terra promessa. Conoscenza e Conservazione. Attività di catalogazione e di restauro nelle Chiese della città e della Diocesi di Bologna, Alfa, Bologna, 1981 Search PubMed.
- G. Sciutto, T. Frizzi, E. Catelli, N. Aresi, S. Prati, R. Alberti and R. Mazzeo, Microchem. J., 2018, 137, 277 CrossRef CAS.
- T. Dias, E. Murta, C. Barrocas Dias and V. Serrão, Conservar Patrimonio, 2015, 22, 29 CrossRef CAS.
- D. V. Thompson, The Materials and Techniques of Medieval Painting, Dover Publications, New York, USA, 2012 Search PubMed.
- A. Daveri, B. Doherty, P. Moretti, C. Grazia, A. Romani, E. Fiorin, B. G. Brunetti and M. Vagnini, Spectrochim. Acta, Part A, 2015, 135, 398 CrossRef CAS PubMed.
- C. Cennini, Il Libro dell'arte, Neri Pozza Editore, Milano, 5th edn, 2009 Search PubMed.
- H. Howard and J. Najorka, in ICOM Committee for Conservation 18th Triennial Meeting Copenhagen Denmark 4-8 September 2017, Pulido & Nunes and ICOM Committee for Conservation, Copenhagen, 2017, https://www.icom-cc-publications-online.org/1764/ Search PubMed.
- M. B. Mcneil and B. J. Little, J. Am. Inst. Conserv., 1992, 31, 355 CrossRef.
- T. E. Graedel, J. Electrochem. Soc., 1992, 139, 1963 CrossRef CAS.
- X. Zheng-miao and R. Naidu, in: Managing Arsenic in the Environment: from Soil to Human Health, ed. R. Naidu, E. Smith, G. Owens and P. Bhattacharya, CSIRO publishing, Collingwood (VIC), 2006, pp. 223–234 Search PubMed.
- D. Tsiulyanu and I. Stratan, J. Non-Cryst. Solids, 2010, 356, 147 CrossRef CAS.
- M. Mitkova and M. N. Kozicki, in Technologies in Optoelectronic Amorphous Materials and Devices, ed. G. Lucovsky and M. Popescu, INOE Publ. House, Bucharest, 2004, pp. 30–40 Search PubMed.
- M. Popescu, F. Sava, A. Lorinczi, A. Velea, M. Leonovici and S. Zamfira, J. Optoelectron. Adv. Mater., 2009, 11, 1586 CAS.
- S. Prati, F. Rosi, G. Sciutto, R. Mazzeo, D. Magrini, S. Sotiropoulou and M. Van Bos, Microchem. J., 2012, 103, 79 CrossRef CAS.
- S. Prati, G. Sciutto, E. Catelli, A. Ashashina and R. Mazzeo, Anal. Bioanal. Chem., 2013, 405, 895 CrossRef CAS PubMed.
- C. G. Schroer, P. Boye, J. M. Feldkamp, J. Patommel, D. Samberg, A. Schropp, A. Schwab, S. Stephan, G. Falkenberg, G. Wellenreuther and N. Reimers, Nucl. Instrum. Methods Phys. Res., Sect. A, 2010, 616, 93 CrossRef CAS.
- R. T. Downs and M. Hall-Wallace, Am. Mineral., 2003, 88, 247 CrossRef CAS.
- W. De Nolf, F. Vanmeert and K. Janssens, J. Appl. Crystallogr., 2014, 47, 1107 CrossRef CAS.
- M. Cotte, E. Pouyet, M. Salome, C. Rivard, W. De Nolf, H. Castillo-Michel, T. Fabris, L. Monico, K. Janssens, T. Wang, P. Sciau, L. Verger, L. Cormier, O. Dargaud, E. Brun, D. Bugnazet, B. Fayard, B. Hesse, A. P. del Real, G. Veronesi, J. Langlois, N. Balcar, Y. Vandenberghe, V. A. Sole, J. Kieffer, R. Barrett, C. Cohen, C. Cornu, R. Baker, E. Gagliardini, E. Papillon and J. Susini, J. Anal. At. Spectrom., 2017, 32, 477 RSC.
- G. Martínez-Criado, J. Villanova, R. Tucoulou, D. Salomon, J.-P. Suuronen, S. Labouré, C. Guilloud, V. Valls, R. Barrett, E. Gagliardini, Y. Dabin, R. Baker, S. Bohic, C. Cohen and J. Morse, J. Synchrotron Radiat., 2016, 23, 344 CrossRef PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537 CrossRef CAS PubMed.
- M. Cotte, T. Fabris, G. Agostini, D. M. Meira, L. De Viguerie and V. A. Solé, Anal. Chem., 2016, 88, 6154 CrossRef CAS PubMed.
- L. Bertrand, S. Schöeder, D. Anglos, M. B. Breese, K. Janssens, M. Moini and A. Simon, TrAC, Trends Anal. Chem., 2015, 66, 128–145 CrossRef CAS , and references therein.
- M. Ganio, E. S. Pouyet, S. M. Webb, C. M. S. Patterson and M. S. Walton, Pure Appl. Chem., 2018, 90, 463 CrossRef CAS.
- L. Monico, M. Cotte, F. Vanmeert, L. Amidani, K. Janssens, G. Nuyts, J. Garrevoet, G. Falkenberg, P. Glatzel, A. Romani and C. Miliani, Anal. Chem., 2020, 92, 14164 CrossRef CAS PubMed.
- N. Salvadó, S. Butí, A. Labrador, G. Cinque, H. Emerich and T. Pradell, Anal. Bioanal. Chem., 2011, 399, 3041 CrossRef PubMed.
- I. Crina Anca Sandu, M. H. de Sá and M. C. Pereira, Surf. Interface Anal., 2011, 43, 1134 CrossRef.
- D. Hradil, J. Hradilová, P. Bezdička, S. Švarcová, Z. Čermáková, V. Košařová and I. Němec, J. Raman Spectrosc., 2014, 45, 848 CrossRef CAS.
- C. Duce, L. Ghezzi, M. Onor, I. Bonaduce, M. P. Colombini, M. R. Tine and E. Bramanti, Anal. Bioanal. Chem., 2012, 402, 2183 CrossRef CAS PubMed.
- M. Odlyha, N. S. Cohen, G. M. Foster and R. H. West, Thermochim. Acta, 2000, 365, 53 CrossRef CAS.
- A. Karpowicz, Stud. Conserv., 1981, 26, 153–160 CAS.
- R. Steudel, in Elemental Sulfur und Sulfur-Rich Compounds II, ed. R. Steudel, Springer, Berlin, Heidelberg, 2003, pp. 127–152 Search PubMed.
- J. S. Pozo-Antonio, C. Cardell, D. Barral, A. Dionisio and T. Rivas, Minerals, 2020, 10, 424 CrossRef CAS.
- A. Kamyshny, I. Ekeltchik, J. Gun and O. Lev, Anal. Chem., 2006, 78, 2631 CrossRef CAS PubMed.
- R. Steudel and T. Chivers, Chem. Soc. Rev., 2019, 48, 3279 RSC.
- C. Yongsiri, J. Vollertsen and T. Hvitved-Jacobsen, J. Environ. Eng., 2004, 130, 104 CrossRef CAS.
- H. Kim and J. H. Payer, Corros. Sci. Technol., 2006, 5, 206 Search PubMed.
- Y. Huo, S. W. Fu, Y. L. Chen and C. C. Lee, J. Mater. Sci.: Mater. Electron., 2016, 27, 10382–10392 CrossRef CAS.
- J. Novakovic, P. Vassiliou and E. Georgiza, Int. J. Electrochem. Sci., 2013, 8, 7223 CAS.
- J. D. Sinclair, J. Electrochem. Soc., 1982, 129, 33–39 CrossRef CAS.
- J. Xu, C. Li, P. Liu, D. He, J. Wang and Q. Zhang, Chemosphere, 2014, 109, 202 CrossRef CAS PubMed.
- D. Benchoam, E. Cuevasanta, M. N. Möller and B. Alvarez, Antioxidants, 2019, 8, 48 CrossRef CAS PubMed.
- K. Xu and J. Wang, Microchem. J., 2019, 149, 103934 CrossRef CAS.
- B. D. Darwent, R. L. Wadlinger and M. J. Allard, J. Phys. Chem., 1967, 71, 2346 CrossRef CAS.
- G. Mills, K. H. Schmidt, M. S. Matheson and D. Meisel, J. Phys. Chem., 1987, 91, 1590 CrossRef CAS.
- F. Denes, M. Pichowicz, G. Povie and P. Renaud, Chem. Rev. (Washington, DC, U. S.), 2014, 114, 2587 CrossRef CAS PubMed.
- H. H. Sunwoo and N. Gujral, in Handbook of Food Chemistry, ed. P. C. K. Cheung and B. M. Mehta, Springer, Berlin, Heidelberg, 2015, pp. 331–363 Search PubMed.
- N. Eastaugh, V. Walsh, T. Chaplin and R. Siddall, Pigment Compendium: a Dictionary of Historical Pigments, Routledge, Taylor and Francis group, London, 2007 Search PubMed.
- Q. G. Zeng, G. X. Zhang, C. W. Leung and J. Zuo, Microchem. J., 2010, 96, 330 CrossRef CAS.
- C. C. I. de LEclairage, Control Of Damage To Museum Objects By Optical Radiation. Commision Internationale de l'Eclairage. Standards DS, Vienna, Austria, CIE Ed, 30, 2004 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ja00271f |
| This journal is © The Royal Society of Chemistry 2022 |