
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A facile spectroscopic method for measuring lignin content in lignocellulosic biomass†
Fachuang
Lu
 *ab,
Chen
Wang
a,
Mingjie
Chen
b,
Fengxia
Yue
a and
John
Ralph
b
*ab,
Chen
Wang
a,
Mingjie
Chen
b,
Fengxia
Yue
a and
John
Ralph
b
aState Key Laboratory of Pulp and Paper Engineering, South China University of Technology, Guangzhou, 510640, China
bDepartment of Biochemistry and The DOE Great Lakes Bioenergy Research Center, The Wisconsin Energy Institute, University of Wisconsin, Madison, WI 53726, USA. E-mail: fachuanglu@wisc.edu
First published on 9th June 2021
Abstract
Although measuring lignin contents is a routine operation for biomass compositional analysis in process development aiming at efficient utilization of woody biomass, it is still a challenging task requiring many steps, hazardous reagents, heating, and a significant time. A facile spectroscopic method, our CASA (Cysteine–Assisted Sulfuric Acid) method, was developed to quantify the lignin content of lignocellulosic biomass, based on an extraordinary system in which biomass samples are fully dissolved in 72% H2SO4 containing cysteine at 24 °C in 60 min. Using synthetic lignins, the lignin absorptivities were determined to be 17.25 g−1 L cm−1 for softwood lignin and 11.23 g−1 L cm−1 for hardwood lignin and monocot lignin. Seven softwoods, six hardwoods, and six monocots were tested using the CASA method. A high coefficient of determination (R2 = 0.95) was found between the CASA results and the acid-insoluble lignin contents, and an even better R2 (0.98) was obtained when the CASA data were correlated with the total lignin contents.
Introduction
Lignin is the most abundant aromatic polymer and the second most prominent renewable raw material after cellulose.1 In plant cells, lignin cross-links to hemicelluloses and cements cellulose microfibers, enhancing the mechanical strength of the plant stem, facilitating the transport of water and nutrients, and providing protection against biological attack.2–4 Due to the presence of lignin, it is difficult for microorganisms to fully utilize lignocellulosic feedstocks,5,6 although progress has been made in developing technologies aimed at achieving full and economical utilization of lignocellulosic biomass. These efforts include manipulating the lignification process by mis-regulating genes within the monolignol biosynthesis pathway in order to produce plants with desirable properties, including having low lignin contents and/or modified composition and structure, developing efficient biomass pretreatment processes, and proposing new strategies focused on converting and utilizing the lignin stream up-front.7–10 In all these activities, as well as in conventional pulping, irrespective of whether they are academic or industrial, lignin content measurement is one of the important routine practices for biomass compositional analysis in process development and optimization.11,12Many methods have been developed and modified over the past for quantitatively measuring the amount of lignin in certain kinds of plant tissues. The oldest and the most popular method for lignin quantitation, the Klason method, has been used for more than a century.13–15 During this period, other methods have been proposed, and the options for lignin quantitation have expanded from the traditional gravimetric ones to the rapid non-destructive methods using various instruments (UV, FTIR, NMR; NIR is a secondary method that will not be discussed here).16–18 Overall, methods for the measurement of lignin content can generally be grouped into two categories: direct and indirect quantitation.
The direct methods include the Klason lignin method and the recently proposed ALBTH method.19 These methods use acids to hydrolyze and solubilize carbohydrates in samples leaving the majority of the lignin as a solid residue to be determined by gravimetric measurement. The Klason procedure uses 72% H2SO4 followed by more dilute acid hydrolysis to dissolve away carbohydrates, leaving lignin as an insoluble residue.20 The small amount of lignin dissolved in the acidic solution, called acid-soluble lignin (ASL), is determined by UV spectrophotometry.21,22 The ALBTH method uses 60 wt% LiBr solution containing 40 mM HCl to dissolve polysaccharides leaving lignin as a solid residue, principally similar to the Klason method.19 The Klason procedure suffers from labor-intensive and tedious operation. Both the Klason method and the ALBTH method have some shortcomings including potentially insufficient hydrolysis of cellulose and the formation of humins from sugars, which often result in an overestimation of lignin.19,23,24 Additionally, a relatively large amount of sample is required to produce reliable results by gravimetric measurement and the acid-soluble lignin, which is significant in hardwood and grass lignins, needs to be measured by UV methods but from which the interference from furfurals is a concern.11 Indirect methods for lignin quantitation include invasive and non-invasive methods. Two invasive procedures (using thioglycolic acid or acetyl bromide) are based on the complete solubilization of whole cell wall material or lignin via sufficient derivatization, and the dissolved lignin in solution is measured by UV spectrophotometry.11 The thioglycolic acid lignin method has not been widely used perhaps because of the lengthy process, inconsistency with other methods, and the lack of suitable lignin standards required for calibration. The acetyl bromide method is the most popular indirect method for lignin quantitation, largely because of its relative simplicity and speed and the ability to use small sample weights.25 The method has been modified several times to determine the lignin content in non-woody plant samples.23,24,26 Briefly, a few milligrams of pre-extracted wood (CWR = cell wall residue after solvent extraction) are placed in a glass vial containing 5 mL of 25% v/v AcBr in acetic acid, sealed with Teflon-lined caps, and heated at 70 °C for 30 min. Lignin undergoes bromination of its α-hydroxy groups and acetylation of its γ-hydroxy groups and any free-phenolic groups and is consequently dissolved in acetic acid. The dissolved lignin is subsequently quantified by UV spectrophotometry at 280 nm. Dence27 cited the advantages of the procedure as being rapid and simple, appropriate for small sample sizes (5–25 mg), with no need to correct for acid-soluble lignin, providing precise absorbance values for determining total lignin content, and having less interference from non-lignin products. However, the furfural products derived from xylan interfere with the UV absorbance of lignin at 280 nm and AcBr lignin values often differ from those from Klason lignin analysis that is still considered to provide the best lignin measure.11
Noninvasive methods for lignin quantitation exploit the properties of lignin to absorb radiation in specific regions of the electromagnetic spectrum. Because lignin has stronger absorbance to UV light at a wavelength of 280 nm than carbohydrates, UV-microspectrophotometry was applied to measure lignin concentrations in cell walls of specific plant tissues.28,29 Infrared spectroscopy has been considered as a method for quantifying lignin in samples, particularly with the application of techniques such as diffuse reflectance Fourier transform spectrometry.30 Nuclear magnetic resonance spectroscopy (NMR) is a powerful tool frequently used to characterize structural features of lignin, particularly when the lignin-containing samples can be dissolved in, or swollen by, a suitable solvent for solution-state NMR.31,32 The improved spectral resolution of 13C-solid NMR, including cross-polarization/magic-angle-spinning (CP/MAS) NMR, allows this technique to be used to analyze lignin in lignocellulosic biomass samples;33 however, it has not been used routinely for quantification of lignin in whole plant samples.
Although many methods are available for directly and indirectly measuring lignin contents in lignocellulosic biomass, new alternative methods with advantages over the ones currently used are rather desperately required to streamline analyses. In this work, a new spectroscopic method, called the CASA lignin method, was established for measuring the lignin content of lignocellulosic biomass by UV spectrophotometry, based on the full dissolution of whole biomass in 72% sulfuric acid (SA) containing the amino acid cysteine.
Experimental
Materials
All chemicals and reagents are commercial products and used as supplied. L-Cysteine, microcrystalline cellulose and xylan were purchased from Sigma-Aldrich. Sulfuric acid (72%, 12 M, SA) was prepared by careful dilution of concentrated (95–98%) sulfuric acid purchased from Sigma-Aldrich or purchased from Fisher Scientific. The lignocellulosic biomass materials were ground with a Wiley mill and sieved to collect the fraction between 40 and 80 mesh for analysis.Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) for 4 h, followed by 95% ethanol extraction for 12 h; toluene could replace benzene for safety reasons, and simply using 80% ethanol extraction alone is quite effective especially for woody biomass.34 For monocot or grass biomass or non-woody dicots, extraction with water under sonication is recommended first to remove proteins and other water-soluble components. Each solvent-extracted biomass sample was kept in a sealed glass bottle after drying in an oven at 50 °C for 48 h and placed in a vacuum desiccator over P2O5.
:1) for 4 h, followed by 95% ethanol extraction for 12 h; toluene could replace benzene for safety reasons, and simply using 80% ethanol extraction alone is quite effective especially for woody biomass.34 For monocot or grass biomass or non-woody dicots, extraction with water under sonication is recommended first to remove proteins and other water-soluble components. Each solvent-extracted biomass sample was kept in a sealed glass bottle after drying in an oven at 50 °C for 48 h and placed in a vacuum desiccator over P2O5.
Preparation of model compounds and synthetic lignins (dehydrogenation polymers, DHPs) is described in the ESI.†
 | (1) |
(ES & H considerations: sulfuric acid is corrosive and should be handled with care.)
Results and discussion
Dissolution of lignocellulosic biomass
Lignocellulosic biomass is normally not soluble in any solvent. Although isolated or purified cellulose can be dissolved in many solvents and lignin can be partially extracted from lignocellulosic biomass under various conditions, complete dissolution of woody materials requires severe conditions such as intensive ball-milling and/or high temperature in ionic liquids. Acetyl bromide (25%) in acetic acid has been used for dissolving woody materials to measure lignin contents by UV spectrophotometry. However, acetyl bromide is a corrosive, volatile, and irritating compound such that the experiments are required to be performed within a fume hood.In an initial trial to dissolve whole wood meal, 10 mg of loblolly pine wood powder of less than 0.5 mm size was stirred in 1 mL of SA containing 0.1 g mL−1 of cysteine at 60 °C. A homogeneous, purple-colored solution was formed in 30 min, demonstrating that the woody biomass can be quickly and completely dissolved in SA in which cysteine had been added. When the solution was diluted to a volume of 10 mL, a clear colorless solution was obtained. In contrast, the SA alone produced a fine-solid suspension of a black residue, exactly as normally seen when performing the Klason lignin procedure (Fig. S1, ESI†). When the cysteine concentration in SA solution was reduced to 0.07 g mL−1, the wood meal could also be dissolved in 30 min at 60 °C although its diluted solution was pale-yellow colored. The dissolution of the same amount of wood meal was not complete in 30 min when the cysteine concentration in SA was reduced to 0.05 g mL−1.
The dissolving temperature was decreased to room temperature (24 °C in this study) in order to identify a milder condition, allowing convenient operation and minimizing interferences from carbohydrates. It was found that lignocellulosic biomass samples were completely dissolved in the cysteine–SA solution containing 0.1 g mL−1 cysteine at 24 °C after stirring for 60 min. When the solution was diluted with DI water to a volume of 100 mL to allow for appropriate UV absorbance values (0.1–0.9) at λ = 283, a clear colorless solution was formed (Fig. 1). Although the exact mechanism behind the dissolution of lignocellulosic biomass in 72% H2SO4 containing cysteine is not yet clear, discovering the extraordinary ability of the cysteine–SA combination to dissolve lignocellulosic materials under otherwise mild conditions (24 °C in 60 min) is striking, potentially allowing lignin contents to be measured in a particularly convenient way that requires no heating, only a few mg of sample, and minimal amounts of reagents/solvents.
 | ||
| Fig. 1 Illustration of the dissolution of wood meal in cysteine–SA for lignin quantification. | ||
The UV absorption spectrum of the dissolved loblolly pine wood had a local maximum absorption at λ = 283 nm, showing the aromatic characteristic of lignin. The milled wood lignin (MWL) isolated from the loblolly pine wood was also readily dissolved in the cysteine–SA solution under the same conditions, forming a grey-colored solution. The UV absorption spectrum of the dissolved MWL was similar to the one obtained from the dissolved loblolly pine wood or a synthetic guaiacyl lignin (G-DHP, dehydrogenation polymer) made from coniferyl alcohol (Fig. 2A), suggesting that the UV absorption of the dissolved wood was mainly contributed by lignin. When microcrystalline cellulose, glucose, or xylose were dissolved in 1 mL of the stock cysteine solution under the same conditions, their UV absorption spectra showed very low absorptivity at 283 nm (Fig. 2B). The UV-absorbance at 283 nm could therefore be used for the determination of lignin in lignocellulosic biomass by spectrophotometry following the full dissolution of the sample in cysteine–SA (followed by suitable dilution in water). It was therefore decided to use 0.1 g mL−1 of cysteine in SA as the stock solution to dissolve lignocellulosic biomass at 24 °C and 60 min as treatment time to produce a stable solution for measuring lignin content with a spectrophotometer.
 | ||
| Fig. 2 (A) UV spectra of dissolved G-DHP, loblolly pine MWL and loblolly pine wood. (B) UV spectra of loblolly pine MWL, xylan, glucose, xylose, and cellulose dissolved in cysteine–SA. | ||
Correlation of UV absorbance with the lignin content
Varying amounts (4–9 mg) of loblolly pine wood meals were stirred in the cysteine–SA (1 mL) solution at room temperature (24 °C) for 60 min and the resulting homogeneous solutions were diluted 100-fold to 100 mL. The absorbance of the diluted solution was measured at 283 nm (A283) in a 1 cm quartz cell using a UV spectrophotometer. A very good linear relationship was obtained (R2 = 0.9965) when a plot was made with the values of A283 against lignin concentrations calculated from the Klason lignins. When other lignocellulosic samples such as poplar and bamboo were applied to the cysteine–SA dissolving system, similar results were obtained (Fig. 3A–C). | ||
| Fig. 3 (A–C) Linear correlations between UV absorbance (at 283 nm) of the cysteine–SA-dissolved lignocellulosic biomass samples and their lignin concentrations calculated based on their Klason lignin contents. (D) Standard curve obtained from a G-DHP, showing its slope (absorptivity) of 17.25 g−1 L cm−1. | ||
The absorptivity of lignin dissolved in cysteine–SA
As a spectrophotometric method, the CASA method requires a reliable standard for developing standard (calibration) curves. However, finding an appropriate lignin preparation is challenging because lignin itself is not a pure compound, varying in composition and inter-unit linkage distribution. In the past, several materials have been tested as potential standards for the acetyl bromide method, including processed lignins,39,40 MWLs,41 native lignin,42 and lignins isolated by acetyl bromide or acidic dioxane.43,44 The lignin absorptivity can also be estimated by using wood samples of known lignin content, for instance by Klason lignin contents or total lignin contents.27 In this study, synthetic lignins, usually termed DHPs (dehydrogenation polymers) made from monolignols, were used as lignin standards to develop the standard curves. Using DHPs as standards has some advantages: (1) the synthesized DHPs are pure and can be readily prepared in adequate quantities; (2) DHPs with varying molar ratios of units can be assessed to study how G:S ratios affect the absorptivity of lignin. Thus, five DHP preparations including one G-DHP, three GS-DHPs with different G:S ratios, and one GSH-DHP (Table 1), were synthesized in a biomimetic system using peroxidase–H2O2 combination to produce phenolic radicals according to the conventional “Zutropf” protocol in which monomers are slowly dropped into the solution.45,46 The UV spectrum of the G-DHP treated or dissolved by the cysteine–SA solution was similar to that resulting from loblolly pine wood or its MWL (Fig. 2A). The standard curves produced from the G-DHP had a slope of 17.25, similar to that (17.61) obtained from loblolly pine based on its Klason lignin content (Fig. 3). Therefore, the absorption coefficient of 17.25 g−1 L cm−1 is recommended for the CASA method to determine lignin contents in softwood biomass.
| Molar ratios | ε, at 283 nm (g−1 L cm−1) | Recommended ε | |
|---|---|---|---|
| G-DHP | G | 17.25 ± 0.14 | 17.25 (G-lignin) |
| DHP-A | G:S = 3:7 |
11.21 ± 0.14 | 11.23 |
| DHP-B | G:S = 5:5 |
11.26 ± 0.23 | G:S ratios ≤1 |
| DHP-C | G:S = 7:3 |
12.20 ± 0.14 | 12.35 |
| DHP-D | G:S:H = 7.5:2:0.5 |
12.50 ± 0.08 | G:S ratios ≥2 |
The UV spectra of the SG-DHPs (DHP-A, DHP-B and DHP-C) dissolved in cysteine–SA had similar absorptivities at λ = 283 although their absorptivities varied over wavelengths ranging from 300 nm to 350 nm (Fig. 4). As G:S increases, the absorptivity of the DHP in this range decreases. Although the absorptivity values for DHP-A and DHP-B are almost the same, DHPs with higher G:S ratios generally had higher absorption coefficients. Considering that the G:S ratios for the majority of hardwood lignins (SG-lignin) or grass lignins (SGH-lignin) are equal to or less than 1, the absorption coefficient of 11.23 g−1 L cm−1 is recommended to be used for the CASA method quantification of lignin in hardwood or monocot lignocellulosic materials.
 | ||
| Fig. 4 UV spectra of DHP-A (7:3 S:G), DHP-B (5:5 S:G), DHP-C (3:7 S:G) and DHP-D (2:7.5:0.5 S:G:H). | ||
Absorptivity of lignin-related model compounds
To understand how much the various guaiacyl (G), syringyl (S), and p-hydroxyphenyl (H) units in lignin contribute to a lignin's UV absorptivity, eight lignin-related model compounds (Fig. 5) were treated with cysteine–SA following the same procedure as used for lignin quantification. The three G-type compounds (1, 3, and 6) had similar spectra, each with a local maximum absorption at around 280 nm and had the highest absorption coefficient at 283 nm among the three types of units (Table S2, ESI†); when dissolved in the cysteine–SA solution, syringaresinol 2 had a UV spectrum similar to those from hardwood lignins or GS-DHPs. The absorptivity at 283 nm for syringaresinol dissolved in cysteine–SA is 11.02 g−1 L cm−1, close to that obtained from DHP-A, although the S type monomers, S-glycerol 4 and ethyl sinapate 7, had the lowest absorption coefficients of 3.95 and 3.45 g−1 L cm−1 respectively. The local maxima at 280 nm were less pronounced for the S monomers tested. The spectra of H type monomeric compounds 5 and 8 treated with cysteine–SA showed local maximal absorption at around 276 nm in their UV spectra, although their absorption coefficients at 283 nm were relatively low compared to those of DHPs or lignins.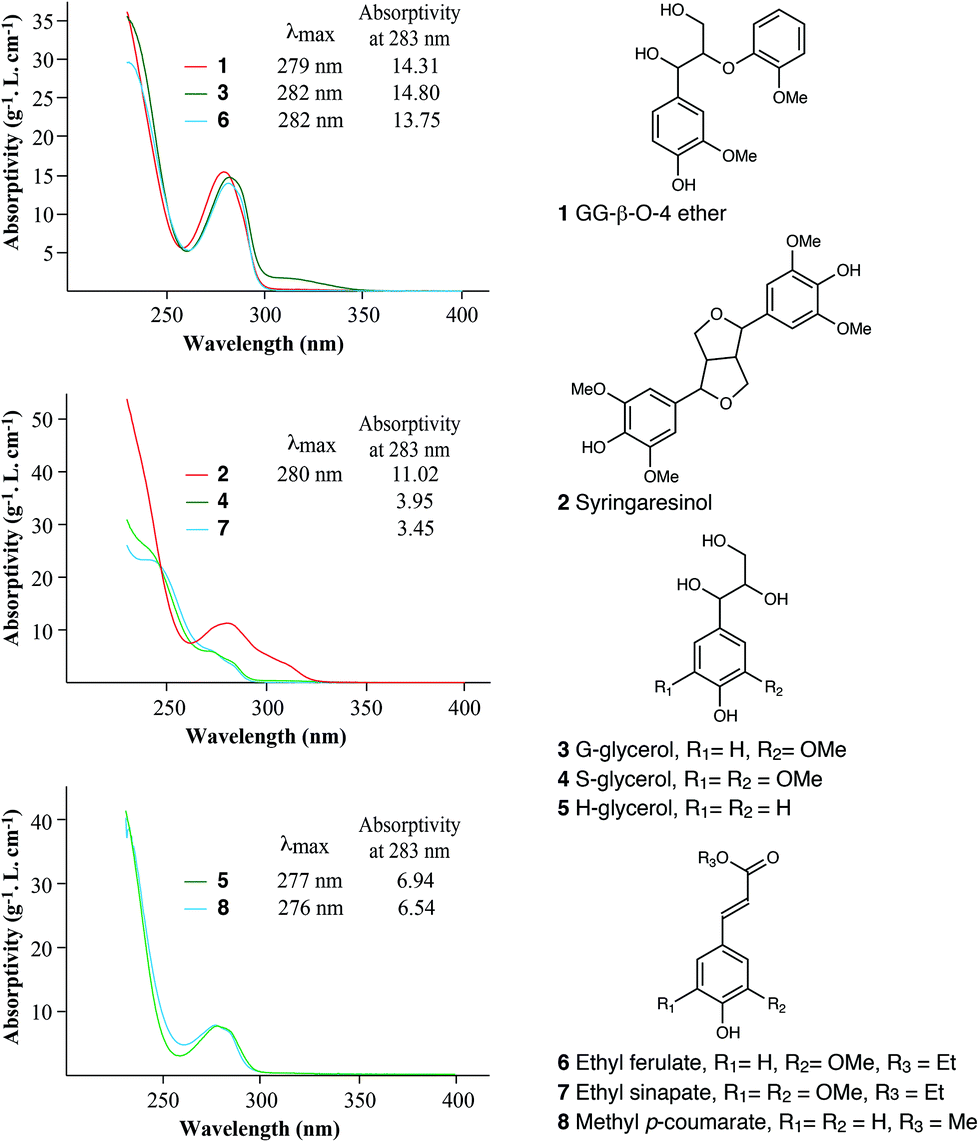 | ||
| Fig. 5 Chemical structures of lignin-related compounds and UV spectra of these compounds treated with cysteine–SA, showing the differences in their absorptivities at 283 nm. | ||
Our model study indicated that guaiacyl type compounds have higher UV absorptivities (at 283 nm) than those of syringyl or p-hydroxyphenyl compounds, which is consistent with the observation that softwood lignins have higher absorptivity than hardwood or monocot lignins.
Effects of cysteine–SA treatment on lignin spectral characteristics
To investigate the effect of cysteine–SA treatment on the spectral characteristics of lignins, three isolated lignin preparations (loblolly pine MWL, aspen MWL, and bamboo MWL) were dissolved in the cysteine–SA solution under the general conditions and their UV spectra were compared with those of the corresponding lignin samples dissolved in neutral aqueous dioxane solutions (Fig. 6). Generally, the cysteine–SA treatment changed lignin structures resulting in lower absorptivities, especially for bamboo lignin at λ from 275 nm to 350 nm. Two local maximal absorptions were observed for bamboo MWL in dioxane solution, one is at λ = 310 due to the conjugated double-bond of the hydroxycinnamates, mainly p-coumarate, present in the lignin; the other one at λ = 286 nm is caused by aromatic rings of lignin units and hydroxycinnamates. After being dissolved in the cysteine–SA solution, the local maximum at 310 nm disappeared and the local maximal absorption at 286 nm shifted to 282 nm, indicating that the conjugated double-bond was saturated in the strong acid environment with the strong nucleophile, cysteine. This result is consistent with those from the model compounds noted above. The UV spectrum of aspen MWL in dioxane had a local maximum at 277 nm, which became a shoulder with lower absorptivity after the CASA treatment. The UV spectrum of the loblolly pine CEL, a G-type lignin, was slightly changed by the cysteine–SA treatment, shifting the local maxima absorption from 281 to 283 nm but otherwise retaining its characteristics. | ||
| Fig. 6 UV spectra of three lignin preparations, showing the changes in their spectral characteristics caused by treatment with cysteine–SA. | ||
Verification of the proposed CASA lignin method
The accuracy or reliability of an indirect analytical method always needs to be verified by comparing or correlating to the established method. To verify the reliability of the CASA lignin method for lignin quantitation, the protocol was applied to various lignocellulosic biomass samples, including 7 softwoods, 6 hardwoods, and 6 grasses (monocots). The results were compared with the acid-insoluble lignin and total lignin contents obtained using the Klason method (using the NREL protocol).13,38 As summarized in Table S2,† the CASA-lignin contents are generally higher than those of Klason lignin and comparable to the total lignin contents. Fig. 7 shows the correlations between the lignin contents measured using the CASA method and those obtained using the classical Klason method from various lignocellulosic materials. All 19 samples were correlated with acid-insoluble lignin contents, and the results from 9 samples were compared with the total lignin contents (acid-soluble + acid-insoluble lignin). A high coefficient of determination (R2 = 0.945) was found between the CASA-lignin contents and acid-insoluble lignin contents, and an even better R2 (0.983) was obtained when the CASA-lignin contents were correlated with the total lignin contents. The latter point highlights another advantage of the CASA-lignin method – it does not require the two analytically different determinations to obtain the total aromatics content needed in the full Klason method. | ||
| Fig. 7 Correlations between the lignin contents measured using the CASA lignin method and the traditional Klason method. | ||
Conclusions
This study reports the discovery of a novel reagent combination (cysteine in SA) that completely dissolves lignocellulosic biomass under mild conditions, leading to the development of a new spectroscopic method, the CASA-lignin method, for the determination of lignin in lignocellulosic biomass. Compared with the currently used methods for lignin determination, the proposed CASA-lignin method has the following advantages: (1) the reagents used are easily accessible and environmentally friendly; (2) the procedure is short and is performed without heating; (3) it is sensitive; only a few milligrams of samples are needed; (4) the method provided results that are well correlated to the total lignin contents obtained using the Klason method; and (5) it is obvious that the method can be operated in a high-throughput mode.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support for this work by the National Natural Science Foundation of China (31770621 and 31870560) and the DOE Great Lakes Bioenergy Research Center (DOE BER Office of Science DE-SC0018409).Notes and references
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS.
- R. Vanholme, B. Demedts, K. Morreel, J. Ralph and W. Boerjan, Plant Physiol., 2010, 153, 895–905 CrossRef CAS.
- J. H. Grabber, Crop Sci., 2005, 45, 820–831 CrossRef CAS.
- L. Shuai, Q. Yang, J. Y. Zhu, F. Lu, P. J. Weimer, J. Ralph and X. J. Pan, Bioresour. Technol., 2010, 101, 3106–3114 CrossRef CAS.
- M. Kleinert and T. Barth, Energy Fuels, 2008, 22, 1371–1379 CrossRef CAS.
- A. M. Boudet, S. Kajita, J. Grima-Pettenati and D. Goffner, Trends Plant Sci., 2003, 8, 576–581 CrossRef CAS.
- B. M. Upton and A. M. Kasko, Chem. Rev., 2016, 116, 2275–2306 CrossRef CAS.
- C. O. Tuck, E. Perez, I. T. Horvath, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695–699 CrossRef CAS.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 1246843 CrossRef.
- J. Xu, C. Li, L. Dai, C. Xu, Y. Zhong, F. Yu and C. Si, ChemSusChem, 2020, 13, 4284–4295 CrossRef CAS PubMed.
- R. Hatfield and R. S. Fukushima, Crop Sci., 2005, 45, 832–839 CrossRef CAS.
- D. W. Templeton, C. J. Scarlata, J. B. Sluiter and E. J. Wolfrum, J. Agric. Food Chem., 2010, 58, 9054–9062 CrossRef CAS.
- J. B. Sluiter, R. O. Ruiz, C. J. Scarlata, A. D. Sluiter and D. W. Templeton, J. Agric. Food Chem., 2010, 58, 9043–9053 CrossRef CAS PubMed.
- K. Yoshihara, T. Kobayashi, T. Fujii and I. Akamatsu, Jpn. TAPPI J., 1984, 38, 86–95 Search PubMed.
- R. Katahira, J. B. Sluiter, D. J. Schell and M. F. Davis, J. Agric. Food Chem., 2013, 61, 3286–3692 CrossRef CAS PubMed.
- J. Rodrigues, O. Faix and H. Pereira, Holzforschung, 1998, 52, 46–50 CrossRef CAS.
- L. Fu, S. A. McCallum, J. J. Miao, C. Hart, G. J. Tudryn, F. M. Zhang and R. J. Linhardt, Fuel, 2015, 141, 39–45 CrossRef CAS PubMed.
- N. Jiang, Y. Q. Pu and A. J. Ragauskas, ChemSusChem, 2010, 3, 1285–1289 CrossRef CAS PubMed.
- N. Li, X. J. Pan and J. Alexander, Green Chem., 2016, 18, 5367–5376 RSC.
- B. L. Browning, Methods of Wood Chemistry, Wiley-Interscience, New York, 1967 Search PubMed.
- E. Maekawa, T. Ichizawa and T. Koshijima, J. Wood Chem. Technol., 1989, 9, 549–567 CrossRef CAS.
- W. E. Kaar and D. L. Brink, J. Wood Chem. Technol., 1991, 11, 465–477 CrossRef CAS.
- I. M. Morrison, J. Sci. Food Agric., 1972, 23, 455–463 CrossRef CAS PubMed.
- I. M. Morrison, J. Sci. Food Agric., 1972, 23, 1463–1469 CrossRef CAS.
- F. C. Moreira-Vilar, R. D. Siqueira-Soares, A. Finger-Teixeira, D. M. de Oliveira, A. P. Ferro, G. J. da Rocha, M. D. L. Ferrarese, W. D. dos Santos and O. Ferrarese, PLoS One, 2014, 9, e110000 CrossRef PubMed.
- M. Bagley, R. L. Cunningham and R. L. Maloney, Tappi, 1973, 56, 162–163 Search PubMed.
- C. W. Dence, in Methods in Lignin Chemistry, ed. S. Y. Lin and C. W. Dence, Springer-Verlag, Heidelberg, 1992, pp. 33–61 Search PubMed.
- J. Boutelje and U. Jonsson, Cellul. Chem. Technol., 1980, 14, 53–67 CAS.
- B. J. Fergus and D. A. I. Goring, Holzforschung, 1970, 24, 118–124 CrossRef CAS.
- T. Schultz, M. Templeteon and G. McGinnis, Anal. Chem., 1985, 57, 2867–2869 CrossRef CAS.
- F. Lu and J. Ralph, Plant J., 2003, 35, 535–544 CrossRef CAS PubMed.
- H. Kim and J. Ralph, Org. Biomol. Chem., 2010, 8, 576–591 RSC.
- J. F. Haw, G. E. Maciel and H. A. Schroeder, Anal. Chem., 1984, 56, 1323–1329 CrossRef CAS.
- O. Theander, Anim. Feed Sci. Technol., 1991, 32, 35–44 CrossRef CAS.
- S. Quideau and J. Ralph, J. Agric. Food Chem., 1992, 40, 1108–1110 CrossRef CAS.
- H. Kim and J. Ralph, J. Agric. Food Chem., 2005, 53, 3693–3695 CrossRef CAS PubMed.
- A. Björkman, Nature, 1954, 174, 1057–1058 CrossRef.
- A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter, D. Templeton and D. Crocker, Determination of structural carbohydrates and lignin in biomass, Report NREL/TP-510-42618, National Renewable Energy Laboratory, Golden, CO, USA, 2012 Search PubMed.
- A. Chesson, J. Sci. Food Agric., 1981, 32, 745–758 CrossRef CAS.
- J. M. Brillouet and D. Riochet, J. Sci. Food Agric., 1983, 34, 861–868 CrossRef CAS.
- K. Iiyama and A. F. A. Wallis, Wood Sci. Technol., 1988, 22, 271–280 CrossRef CAS.
- R. S. Fukushima, B. A. Dehority and S. C. Loerch, J. Anim. Sci., 1991, 69, 295–304 CrossRef CAS PubMed.
- R. S. Fukushima and B. A. Dehority, J. Anim. Sci., 2000, 78, 3135–3143 CrossRef CAS PubMed.
- R. S. Fukushima and R. D. Hatfield, J. Agric. Food Chem., 2001, 49, 3133–3139 CrossRef CAS PubMed.
- K. Freudenberg, Angew. Chem., 1956, 68, 508–512 CrossRef CAS.
- T. K. Kirk, W. J. Connors, W. D. Bleam, W. F. Hackett and J. G. Zeikus, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 2513–2519 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc01507a |
| This journal is © The Royal Society of Chemistry 2021 |