
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploiting a silver–bismuth hybrid material as heterogeneous noble metal catalyst for decarboxylations and decarboxylative deuterations of carboxylic acids under batch and continuous flow conditions†‡
Rebeka
Mészáros
a,
András
Márton
b,
Márton
Szabados
 bc,
Gábor
Varga
*cd,
Zoltán
Kónya
ef,
Ákos
Kukovecz
e,
Ferenc
Fülöp
*ag,
István
Pálinkó§
bc and
Sándor B.
Ötvös
*gh
bc,
Gábor
Varga
*cd,
Zoltán
Kónya
ef,
Ákos
Kukovecz
e,
Ferenc
Fülöp
*ag,
István
Pálinkó§
bc and
Sándor B.
Ötvös
*gh
aInstitute of Pharmaceutical Chemistry, University of Szeged, Eötvös u. 6, Szeged, H-6720 Hungary
bDepartment of Organic Chemistry, University of Szeged, Dóm tér 8, Szeged, H-6720 Hungary
cMaterial and Solution Structure Research Group and Interdisciplinary Excellence Centre, Institute of Chemistry, University of Szeged, Aradi Vértanúk tere 1, Szeged, H-6720 Hungary
dDepartment of Physical Chemistry and Materials Science, University of Szeged, Rerrich Béla tér 1, Szeged, H-6720 Hungary. E-mail: gabor.varga5@chem.u-szeged.hu
eDepartment of Applied and Environmental Chemistry, University of Szeged, Rerrich Béla tér 1, Szeged, H-6720 Hungary
fMTA-SZTE Reaction Kinetics and Surface Chemistry Research Group, Hungarian Academy of Sciences, Rerrich Béla tér 1, Szeged, H-6720 Hungary
gMTA-SZTE Stereochemistry Research Group, Hungarian Academy of Sciences, Eötvös u. 6, Szeged, H-6720 Hungary. E-mail: fulop@pharm.u-szeged.hu
hInstitute of Chemistry, University of Graz, NAWI Graz, Heinrichstrasse 28, Graz, A-8010 Austria. E-mail: sandor.oetvoes@uni-graz.at
First published on 19th May 2021
Abstract
Herein, we report novel catalytic methodologies for protodecarboxylations and decarboxylative deuterations of carboxylic acids utilizing a silver-containing hybrid material as a heterogeneous noble metal catalyst. After an initial batch method development, a chemically intensified continuous flow process was established in a simple packed-bed system which enabled gram-scale protodecarboxlyations without detectable structural degradation of the catalyst. The scope and applicability of the batch and flow processes were demonstrated through decarboxylations of a diverse set of aromatic carboxylic acids. Catalytic decarboxylative deuterations were achieved on the basis of the reaction conditions developed for the protodecarboxylations using D2O as a readily available deuterium source.
1. Introduction
Carboxylic acids are of outstanding importance as inexpensive and easily accessible intermediates for the synthesis of an array of value-added products.1 Among carboxylic acid transformations, protodecarboxylations and related decarboxylative couplings play a crucial role in the formation of C–C, C–X and C–H bonds, and hence they are appealing for the generation of molecular diversity.2,3 The most common catalysts for protodecarboxylations contain copper, silver, gold, palladium or rhodium metals, typically as homogeneous sources in combination with various bases or ligands.4–6 For example, in copper- and rhodium-catalyzed examples, well-defined complexes are predominant over reusable heterogeneous sources.7–10 Palladium-catalyzed protodecarboxylations generally require high catalyst loading which severely limits their practical applicability.11–13 In addition, various hexaaluminate catalysts proved useful for decarboxylation of biomass-derived carboxylic acids.14–17 Due to the high costs involved, only a few studies have been reported for gold-catalyzed protodecarboxylations.18–20 Silver-catalyzed reactions have also emerged in the field of protodecarboxylations and decarboxylative transformations, such as decarboxylative allylations and azidations, and exhibited a highly beneficial reactivity trend, comparable to that of the more costly gold-catalyzed protocols.21–24 However, with a few exceptions,25 such reactions are promoted by soluble silver salts as non-reusable catalytic sources,26,27 typically in the presence of various ligands, which can be regarded as a considerable drawback from an environmental point of view.28–30Due to economic and environmental reasons, there is a continuously growing need for heterogeneous noble metal catalysts.31 However, immobilization of metal catalysts on various prefabricated supports is often accompanied by reduced selectivity or loss of activity, and in the case of inadequate catalyst-support interactions, leaching of the metal component may lead to substantial environmental concerns.32,33 Nowadays, in organic synthesis silver catalysis is considered as a significant methodology, which is due to its wide applicability, environmentally-benign nature and its lower costs compared with other precious noble metals such as gold, platinum or palladium.34,35 Typical synthetic applications of silver catalysis rely on Ag(I) salts or complexes as homogeneous sources for the catalytically active metal.36–38 As concerns heterogeneous silver sources, supported nanoparticles (nanosilver) are the most widely applied.33,39,40 Such heterogeneous materials are easily obtained via immobilization on various surfaces, however their main limitation is weak catalyst-support interactions which give rise to unsatisfactory stability and limits their practical synthetic utilities, especially under demanding reaction conditions, such as high-temperature continuous flow conditions or in the presence of coordinating ligands. On the basis of a naturally occurring mineral, called beyerite, we recently developed a heterogeneous silver–bismuth hybrid material (AgBi-HM) with structurally-bound silver catalytic centers.41 The material exhibited a layered structure and contained Ag(I) and Bi(III) cationic and carbonate anionic components with silver ion as the minor cationic component. As compared with traditionally immobilized catalysts, structurally-bound catalytic centres imply increased thermodynamic stability and robustness, and exhibit an increased tolerance against challenging reaction conditions and improved compatibility with various reactants and solvents.42
Continuous flow reaction technology in combination with heterogeneous catalysis have attracted significant attention in recent years,43–48 and now comprise a powerful methodology for the synthesis of an array of useful products.49–56 Heterogeneous catalysts can easily be handled, recycled and reused in packed-bed reactors, moreover, unlike in traditional batch processes, separation from the reaction products is really straightforward.57 Due to the enhanced control over the most important reaction conditions (e.g. residence time and temperature),58–60 reaction selectivity can easily be improved while less waste is generated.61,62 Moreover, in loaded catalyst columns, the continuous stream of reactants interacts with a superstoichiometric amount of catalyst species, which improves reaction rates significantly.63–65 On the downside, with increasing reactor dimensions scale-up may involve difficulties, such as insufficient intraparticle heat transfer rates, intraparticle diffusion limitations as well as susceptibility to liquid maldistribution.66 However, if catalyst deactivation and leaching can be eliminated, the scale of production becomes a direct function of the process time without modifying the reactor geometry (i.e. scale-out).67–69 In spite of these obvious benefits, there are very few precedents for heterogeneous silver-catalysts being utilized in continuous flow processes,70,71 which may be explained by the fact that stable and robust heterogeneous silver catalyst are at scarce.41,42,72,73
To the best of our knowledge, protodecarboxylations promoted by heterogeneous noble metal catalysts have not yet been achieved under efficient continuous flow conditions. We speculated that our silver-containing hybrid material may act as a ligand-free heterogeneous silver catalyst for protodecarboxylations, and because of its stability and robustness, not only under batch but also under more demanding flow conditions. We intended to investigate the flow reactions in a high-temperature packed-bed reactor system to exploit extended parameter spaces, and to study the possibility of chemical intensification as compared with the batch process. Considering the outstanding significance of deuterated compounds in chemistry, biochemistry, environmental sciences and also in pharmacological research,74,75 we were intrigued to explore not only protodecarboxylations but also decarboxylative deuterations as facile and site-specific access to valuable deuterium-labelled compounds.76,77 Our results are presented herein.
2. Experimental
2.1. General information
All chemicals used were analytical grade and were applied without further purification. Reaction products were characterized by NMR spectroscopy and mass spectrometry. 1H NMR and 13C NMR spectra were recorded on a Bruker Avance NEO 500 spectrometer, in CDCl3 as solvent, with tetramethylsilane as internal standard at 500.1 and 125 MHz, respectively. GC-MS analyses were performed on a Thermo Scientific Trace 1310 Gas Chromatograph coupled with a Thermo Scientific ISQ QD Single Quadrupole Mass Spectrometer using a Thermo Scientific TG-SQC column (15 m × 0.25 mm ID × 0.25 μm film). Measurement parameters were as follows. Column oven temperature: from 50 to 300 °C at 15 °C min−1; injection temperature: 240 °C; ion source temperature: 200 °C; electrospray ionization: 70 eV; carrier gas: He at 1.5 mL min−1 injection volume: 2 μL; split ratio: 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :33.3; and mass range: 25–500 m/z.
:33.3; and mass range: 25–500 m/z.
2.2. Synthesis and characterization of the AgBi-HM
AgBi-HM was synthesized by using the urea hydrolysis method according to a modified version of our procedure reported previously.42 AgNO3 (3.73 g) and Bi(NO3)3·5H2O (5.36 g) were dissolved in 50–50 mL 5 wt% nitric acid and the solutions were combined. Urea (7.05 g) dissolved in 100 mL of deionized water was next added to the mixture which was then placed into an oven for 24 h at 105 °C. The obtained material was next filtrated, washed with aqueous thiosulfate solution, water and ethanol four times, and dried at 60 °C to obtain the final product.The as-prepared material was fully characterized by means of diverse instrumental techniques as detailed earlier.40,41 The X-ray diffraction (XRD) patterns were recorded on a Rigaku XRD-MiniFlex II instrument applying CuKα radiation (λ = 0.15418 nm), 40 kV accelerating voltage at 30 mA. The morphology of the as-prepared and treated samples were studied by scanning electron microscopy (SEM). The SEM images were registered on an S-4700 scanning electron microscope (Hitachi, Japan) with accelerating voltage of 10–18 kV. The actual Ag/Bi metal ratios in the samples were determined with energy dispersive X-ray analysis (EDX) measurements (Röntec QX2 spectrometer equipped with Be window coupled to the microscope). More detailed images, both of the as-prepared and the used samples, were taken by transmission electron microscopy (TEM). For these measurements, an FEI Tecnai™ G2 20 X-Twin type instrument was applied, operating at an acceleration voltage of 200 kV. The thermal behaviour of the catalyst samples were investigated by thermogravimetry (TG) and differential thermogravimetry (DTG) using a Setaram Labsys derivatograph operating in air at 5 °C min−1 heating rate. For the measurements, 20–30 mg of the samples were applied. The amount of metal ions was measured by ICP–AES on a Thermo Jarell Ash ICAP 61E instrument. Before measurements, a few milligrams of the samples measured with analytical accuracy were digested in 1 mL cc. nitric acid; then, they were diluted with distilled water to 50 mL and filtered.
2.3. General procedure for the batch reactions
A typical procedure for the decarboxylation and decarboxylative deuteration reactions is as follows. N,N-Dimethylformamide (DMF, 3 mL), the appropriate carboxylic acid (0.45 mmol, 0.15 M, 1 equiv.), KOH (6 mg, 15 mol%) and AgBi-HM as catalyst (60 mg, corresponding to 5 mol% Ag loading) were combined in an oven-dried Schlenk tube equipped with a magnetic stir bar. In case of decarboxylative deuteration, 10 equiv. of D2O (90 μL) was also added to the reaction mixture. After stirring for 24 h at 110 °C, the reaction mixture was cooled to room temperature, and the catalyst was filtered off. The crude products were diluted with diethyl ether and were washed with aqueous NaHCO3 and brine. The combined organic layers were dried over Na2SO4, and concentrated under reduced pressure. The crude products were checked by NMR spectroscopy to determine conversion and selectivity. The products of the batch reactions were characterized by NMR and GC-MS techniques. In case deuterondecarboxylations, deuterium contents were determined from the relative intensities of the 1H NMR indicator signals. Characterization data can be found in the ESI.‡2.4. Investigation of the catalyst reusability under batch conditions
For investigation of catalyst reusability, the decarboxylation of 2-nitrobenzoic acid was carried out multiple times utilizing a single portion of catalyst. DMF (3 mL), 2-nitrobenzoic acid (0.45 mmol, 0.15 M, 1 equiv.), KOH (6 mg, 15 mol%) and AgBi-HM as catalyst (60 mg, corresponding to 5 mol% Ag loading) were combined in an oven-dried Schlenk tube equipped with a magnetic stir bar. The reaction mixture was stirred for 24 h at 110 °C. The mixture was next cooled to room temperature, and the solid material was removed by centrifugation. The liquid phase was extracted, dried and evaporated as detailed in section 2.3. The removed catalyst was washed with DMF (four times) and was dried in nitrogen flow before the next reaction cycle. Conversion and selectivity were determined after each cycle by using 1H NMR.2.5. General procedure for the flow reactions
To carry out the decarboxylation and decarboxylative deuteration reactions under flow conditions, a simple continuous flow set-up was assembled as shown in Fig. 1. The system consisted of an HPLC pump (JASCO PU-2085), a stainless steel HPLC column with internal dimensions of 4.6 × 100 mm as catalyst bed and a 5-bar backpressure regulator (BPR) from IDEX to prevent solvent boil over. The column encompassed 2 g of AgBi-HM as catalyst. For each reaction, the corresponding carboxylic acid (c = 0.1 M) and 15 mol% KOH were dissolved in acetonitrile (MeCN) or DMF. In order to achieve a clear solution, 20 equiv. of H2O was also added to the reaction mixture. In case of deuterodecarboxylation reactions, 20 equiv. D2O was added to the reaction mixture as deuterium source. In each run, 4 mL of product solution was collected under steady-state conditions. Between two experiments, the system was washed for 20 min by pumping the appropriate solvent at a flow rate of 0.5 mL min−1. When DMF was used as solvent, the crude product was worked-up similarly as detailed in section 2.3. In case of MeCN as solvent, the reaction mixture was simply evaporated. Samples were checked by NMR spectroscopy to determine conversion and selectivity. For scale-out, 2-nitrobenzoic acid (c = 0.1 M) and 15 mol% KOH was dissolved in MeCN together with 20 equiv. of H2O to achieve a clear solution. The reaction mixture was pumped continuously at 100 μL min−1 through the heated catalyst bed at 170 °C. The product solution was collected for 20 h under steady state conditions, and samples were taken in every hour to determine conversion and selectivity. The products of the flow reactions were characterized by NMR and GC-MS techniques. In case deuterondecarboxylations, deuterium contents were determined from the relative intensities of the 1H NMR indicator signals. Characterization data can be found in the ESI.‡ The residence time on the catalyst bed was determined experimentally by pumping a dye solution. The elapsed time between the first contact of the dye with the column and the moment when the coloured solution appeared at column the outlet was measured.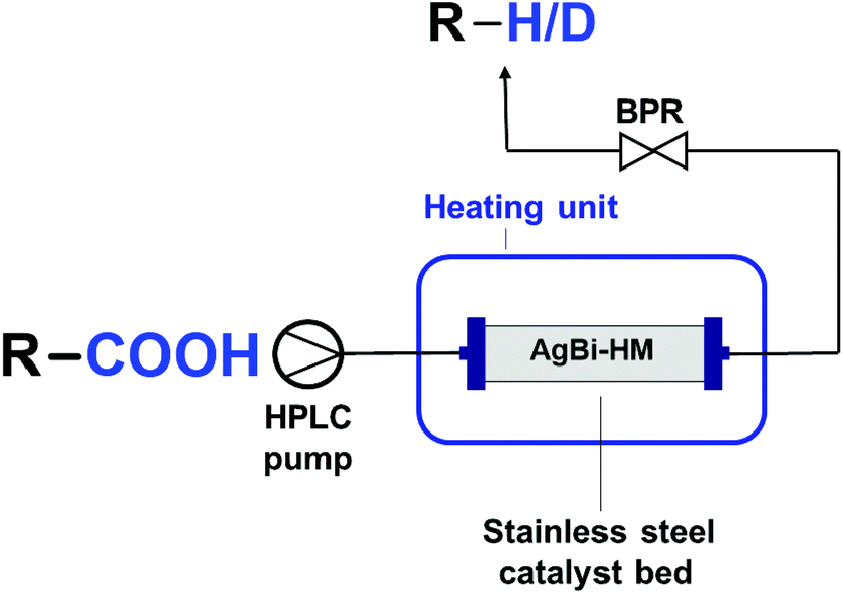 | ||
| Fig. 1 Experimental setup for the continuous flow experiments. | ||
3. Results and discussion
3.1. Decarboxylation of carboxylic acids under batch conditions
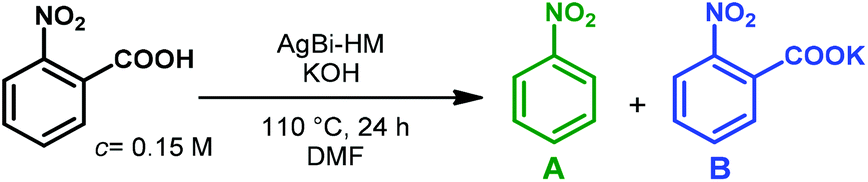
In order to achieve an initial picture on the catalytic activity of the silver-containing hybrid material in decarboxylation of carboxylic acids, batch reactions were explored first. The decarboxylation of 2-nitrobenzoic acid was chosen as model reaction to demonstrate the performance of the AgBi-HM in comparison with various commercially available silver and copper salts as the most typical homogeneous catalytic sources for this reaction type (Fig. 2). Based on literature data,25,78 DMF was selected as solvent, and the reaction mixture containing the substrate (0.15 M) together with 5 mol% of the appropriate catalyst and 15 mol% of KOH as base was stirred for 24 h at 110 °C. | ||
| Fig. 2 Investigation of various catalysts in the decarboxylation of 2-nitrobenzoic acid. | ||
It was corroborated, that product formation was not occurring without any catalyst present. Gratifyingly, the application of the hybrid material as catalyst resulted quantitative and selective decarboxylation to nitrobenzene. AgOAc, Ag2O, Ag2CO3 and AgNO3 as catalyst gave slightly lower conversions (95–97%) and 100% selectivity in each cases. In contrast to silver catalysts, copper salts performed poorer. In the presence of CuOAc and Cu(NO3)2, conversion was 68% and 70%, respectively, whereas CuBr2 was proven even less effective with a conversion of merely 39%. In all the copper-catalyzed reactions, potassium 2-nitrobenzoate appeared in the reaction mixture. Considering that the reaction is initiated by deprotonation of the carboxylic acid, the presence of the corresponding potassium salt as side product therefore indicates the incompleteness of the reaction.25 As corroborated by a test reaction carried out in the presence of 5 mol% of Bi(NO3)3·5H2O, the Bi(III) component of the hybrid material is inactive in decarboxylation of 2-nitrobenzoic acid.
After achieving promising preliminary results, the effects of the major reaction conditions were next explored. Upon investigation of solvent effects (Table 1), the best results were achieved by using DMF (entry 1). MeCN and N,N-dimethylacetamide (DMA) also gave acceptable conversions (85% and 62%, respectively) and high selectivities (100% and 85%, respectively; entries 2 and 3). In EtOAc and toluene only trace amounts of nitrobenzene formation was detected (entries 4 and 5), whereas in N-methyl-2-pyrrolidone (NMP) and dimethyl sulfoxide (DMSO), no decarboxylation occurred (entries 6 and 7).

As concerns reaction time, 24 h was required for completion, lower reaction times gave incomplete transformations (Fig. S1‡). As was expected, decarboxylation was not taking place at temperatures ≤50 °C, however conversion started to increase at 80 °C and reached completion at 110 °C (Fig. S1‡). The reaction gave the best results with substrate concentrations of 0.1 or 0.15 M (Table S1‡) The optimum value of the catalyst loading was 5 mol% as lower amounts resulted in decrease of the conversion (Table 2, entries 1–4). Upon investigation of the effects of the amount of the extraneous KOH (Table 2, entries 5–9), it was observed that without base the reaction gives only traces of the decarboxylated product; however only catalytic amounts are required for completion (e.g. 100% conversion was achieved with 15 mol% KOH). This is in accordance with the mechanistic proposal of Jaenicke and co-workers suggesting a negatively charged aryl–silver intermediate upon decarboxylation which is responsible for deprotonation after the base-promoted initiation of the reaction.25 In our study, KOH was selected as base as it involved no precipitation and ensured a pumpable clear solution when being combined with the substrate which is crucial when considering the upcoming continuous flow experiments.
Having established an optimal set of conditions for the decarboxylation of the model compound (5 mol% catalyst loading, 15 mol% KOH as base, DMF as solvent, 0.15 M substrate concentration, 110 °C temperature and 24 h reaction time), we set out to investigate the scope and applicability of the batch process (Table 3). Besides 2-nitrobenzoic acid (entry 1), its 5-methoxy-substituted derivative as well as 3,5-dinitrobenzoic acid underwent quantitative and selective protodecarboxylations (entries 2 and 3). The reaction tolerated well the replacement of the ortho-nitro substituent with bromine or methoxy groups, and gave good conversions (80% and 74%, respectively) and 100% selectivities in reactions of the corresponding benzoic acid derivatives (entries 4 and 5). Despite the higher steric hindrance, decarboxylation of 2,6-dimethoxybenzoic acid was also successful, although conversion was somewhat lower (65%) than in the case of the mono-substituted derivative (entry 6 vs. entry 5). Interestingly, decarboxylation of 2-chlorobenzoic acid and 2-hydroxybenzoic acid (salicylic acid) were not successful (entries 7 and 8), however 2,4-dichlorobenzoic acid proved as an excellent substrate and gave the corresponding dichlorobenzene with 92% conversion and 100% selectivity (entry 9). Selective decarboxylation of 1-naphtolic acid to naphthalene was also possible, however only with a moderate conversion of 49% (entry 10). To our delight, selective decarboxylation of heteroaromatic carboxylic acids, such as thiophene-2-carboxylic acid and nicotinic acid, proceeded with excellent conversions (100% and 86%, respectively; entries 11 and 12). Similarly high conversions (97–100%) and selectivities were achieved in decarboxylations of fused heteroaromatic substrates, such as indole-3-carboxylic acid, coumarin-3-carboxylic acid and chromone-3-carboxylic (entries 13–15). Decarboxylations of metha- and para-monosubstituted benzoic acid derivates, such as 3- and 4-nitrobenzoic acid, were also attempted, however in these cases no reaction occurred. These results are in accordance with earlier literature findings suggesting the formation of a metal-centered carboxylate intermediate which is stabilized by the electronic effects of the substituent(s) on the aromatic rings.79,80 Moreover, decarboxylation of aliphatic carboxylic acids, such as hexanoic acid and levulinic acid, was proven unsuccessful using this methodology. Note that isolated yield was determined in some representative instances.
|
|
|||
|---|---|---|---|
| Entry | Substrate | Conversiona,b (%) | Selectivitya (%) |
| a Determined by 1H NMR analysis of the crude product. b For representative examples, isolated yields are shown in parentheses. | |||
| 1 |

|
100 (98) | 100 |
| 2 |

|
100 | 100 |
| 3 |

|
100 (97) | 100 |
| 4 |

|
80 | 100 |
| 5 |
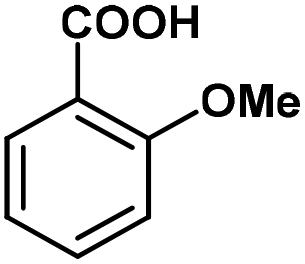
|
74 | 100 |
| 6 |

|
65 | 100 |
| 7 |

|
Traces | — |
| 8 |

|
Traces | — |
| 9 |

|
92 | 100 |
| 10 |

|
49 | 100 |
| 11 |

|
100 | 100 |
| 12 |

|
86 | 100 |
| 13 |
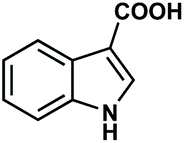
|
100 (97) | 100 |
| 14 |

|
100 | 100 |
| 15 |
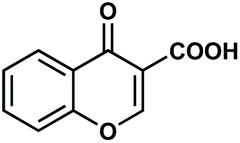
|
97 | 100 |

One of the main benefits of heterogeneous catalysis is the ability to reuse and recycle the catalytic material. In order to evaluate this sustainable feature of the AgBi-HM, protodecarbxylation of 2-nitrobenzoic acid was performed repeatedly under optimized conditions utilizing the same portion of catalyst for each reactions. The used hybrid material was removed between each cycle by centrifugation and after washing and drying, it was simply reused. Gratifyingly, no decrease in catalytic activity or selectivity occurred during the first 7 consecutive catalytic cycles, and conversion was around 90% even after the 10th reaction which implies the significant stability and robustness of the catalytic material (Fig. 3).
 | ||
| Fig. 3 Testing the reusability of the AgBi-HM catalyst in the decarboxylation of 2-nitrobenzoic acid. Selectivity was 100% in all reactions. (Reaction conditions: 0.15 M substrate concentration, 5 mol% catalyst, 15 mol% of KOH as base, DMF as solvent, 110 °C, 24 h reaction time.). | ||
3.2. Decarboxylation of carboxylic acids under continuous flow conditions
After achieving convincing batch results, we next turned our attention to continuous flow operation with the aim to improve the efficacy and sustainability of the catalytic process. As detailed in the Experimental, AgBi-HM was employed in a simple packed-bed reactor setup (see also Fig. 1). Similarly as in the batch study, the effects of the reaction conditions were investigated again using the decarboxylation of 2-nitrobenzoic acid as a model reaction. The effects of solvents which gave good results in the batch reactions were explored again under flow conditions. For this, the catalyst bed was heated to 170 °C, and the solution of the substrate together with 15mol% KOH was pumped continuously at 50 μL min−1 flow rate. Gratifyingly, under these conditions, MeCN performed superior compared to DMF and DMA, and resulted selective decarboxylation with conversions of 100% and 96% at 0.1 M and 0.15 M substrate concentrations, respectively (Table 4). This is a remarkable improvement considering that MeCN is much more acceptable from environmental aspects than DMF which performed best under batch conditions (Table 1, entry 1).81
Upon investigation of the effects of the residence time and reaction temperature, it was verified that a significant chemical intensification is possible under flow conditions. Due to the backpressure applied, it was possible to easily overheat the reaction mixture and to study the effects of temperatures far above the boiling point of MeCN. As shown in Fig. 4, quantitative and selective decarboxylation could be achieved at 170 °C while the reaction mixture (containing the substrate in 0.1 M concentration together with 15 mol% KOH) was streamed at 50 μL min−1 flow rate. Notably, this corresponded to a residence time of only 10.5 min which is a significant improvement compared to the batch reaction time of 24 h. When residence time was decreased to approximately 3.5 min (150 μL min−1 flow rate), the conversion of the decarboxylation was still 75% at 170 °C. When residence time was kept constant at 10.5 min, a rapid decrease of conversion was observed with the temperature; for example at 100 °C conversion was only 13%.
 | ||
| Fig. 4 Investigation of the effects of the reaction temperature (a) and residence time (b) in the AgBi-HM-catalyzed decarboxylation of 2-nitrobenzoic acid in a continuous flow reactor. (Reaction conditions: 0.1 M substrate concentration, 15 mol% of KOH as base, MeCN as solvent.). | ||
A range of aromatic carboxylic acids exhibiting diverse substitution patterns were next submitted to the optimized flow conditions (Table 5). Similarly as in the batch reactions, quantitative and selective decarboxylation was achieved in cases of 2-nitrobenzoic acid, its 5-methoxy-substituted derivative as well as 3,5-dinitrobenzoic acid (entries 1–3). To our delight, the flow protocol proved more effective in numerous reactions than the batch method. For example, 2-bromo-, 2-methoxy- as well as 2,6-dimethoxybenzoic acid furnished quantitative conversions (entries 4–6), whereas in batch, conversions were much lower. Notably, selective decarboxylations of 2-chloro- and 2-hydroxybenzoic acid were achieved successfully under flow conditions (conversions were 100% and 23%, respectively; entries 7 and 8), whereas these substrates remained inert under batch conditions. 2,4-Dichlorobenzoic acid and 1-naphtolic acid were also successfully decarboxylated and gave similar conversions than in the corresponding batch reactions (entries 9 and 10). Fused heteroaromatic substrates showed excellent reactivity, and gave quantitative conversion and 100% selectivity, similarly as under batch conditions (entries 11–13). Unfortunately, flow reactions of thiophene-2-carboxylic acid and nicotinic acid could not be evaluated due to possible deposition of the substrates and/or the products within the catalyst column. (Isolated yield was also determined for some representative examples.)
|
|
|||
|---|---|---|---|
| Entry | Substrate | Conversiona,b (%) | Selectivitya (%) |
| a Determined by 1H NMR analysis of the crude product. b For representative examples, isolated yields are shown in parentheses. c DMF was used as solvent due to solubility issues. | |||
| 1 |

|
100 (99) | 100 |
| 2 |

|
100 | 100 |
| 3 |
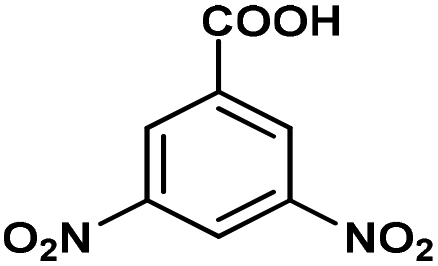
|
100 | 100 |
| 4 |
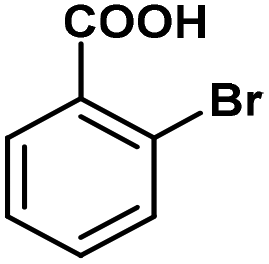
|
100 (98) | 100 |
| 5c |

|
100 | 100 |
| 6 |

|
100 | 100 |
| 7 |

|
100 | 100 |
| 8 |

|
23 | 100 |
| 9c |
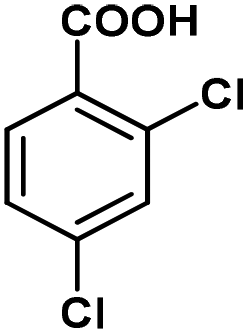
|
87 | 100 |
| 10 |

|
48 | 100 |
| 11c |
|
100 | 100 |
| 12c |

|
100 | 100 |
| 13c |
|
100 (97) | 100 |

In order to investigate the preparative capabilities of the AgBi-HM catalyzed protodecarboxylation under flow conditions, the reaction of 2-nitrobenzoic acid was scaled-out (Fig. 5). With the aim to maximize the productivity of the synthesis, the flow rate was increased to 100 μL min−1 (approx. 5 min residence time), all further reaction parameters were kept at the previously optimized values (0.1 M substrate concentration, 15 mol% of KOH as base, MeCN as solvent, 170 °C temperature). A 20 h reaction window was explored, with conversion and selectivity being determined in every hour to obtain a clear view of the actual catalyst activity. Gratifyingly, the packed-bed system proved highly stable. No decrease in activity or selectivity occurred in the first 18 h of the experiment: conversion remained steady around 80–85% and no side product formation occurred. In the last two hours, a slight loss of catalytic activity was detected, however after 20 h, at the end of the experiment, a satisfying conversion of 71% could still be achieved. Finally, as the result of the scale-out, 1.207 g of nitrobenzene was isolated which corresponded to an overall yield of 82%.
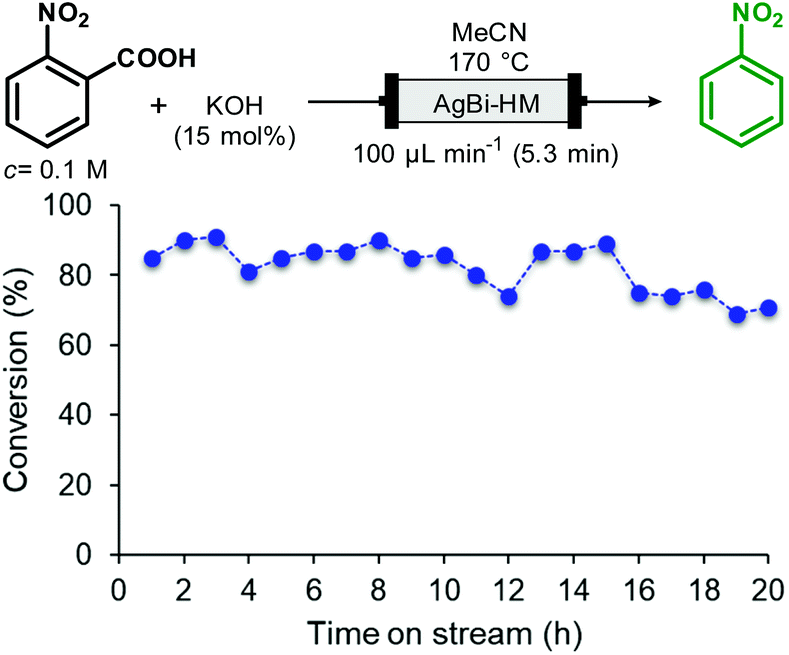 | ||
| Fig. 5 Scaling-out of the AgBi-HM-catalyzed decarboxylation of 2-nitrobenzoic acid in a continuous flow reactor. (Selectivity of the reaction was 100% in all points investigated.). | ||
3.3. Characterization of used AgBi-HM samples
With the aim to evaluate catalyst stability and robustness, AgBi-HM samples previously used in batch recycling experiments as well as in flow scale-out were examined extensively by various instrumental techniques. The materials were characterized by TG, SEM (SEM-EDX), TEM and XRD measurements, and the structure of the used catalyst samples was compared to that of the as-prepared material (Fig. 6).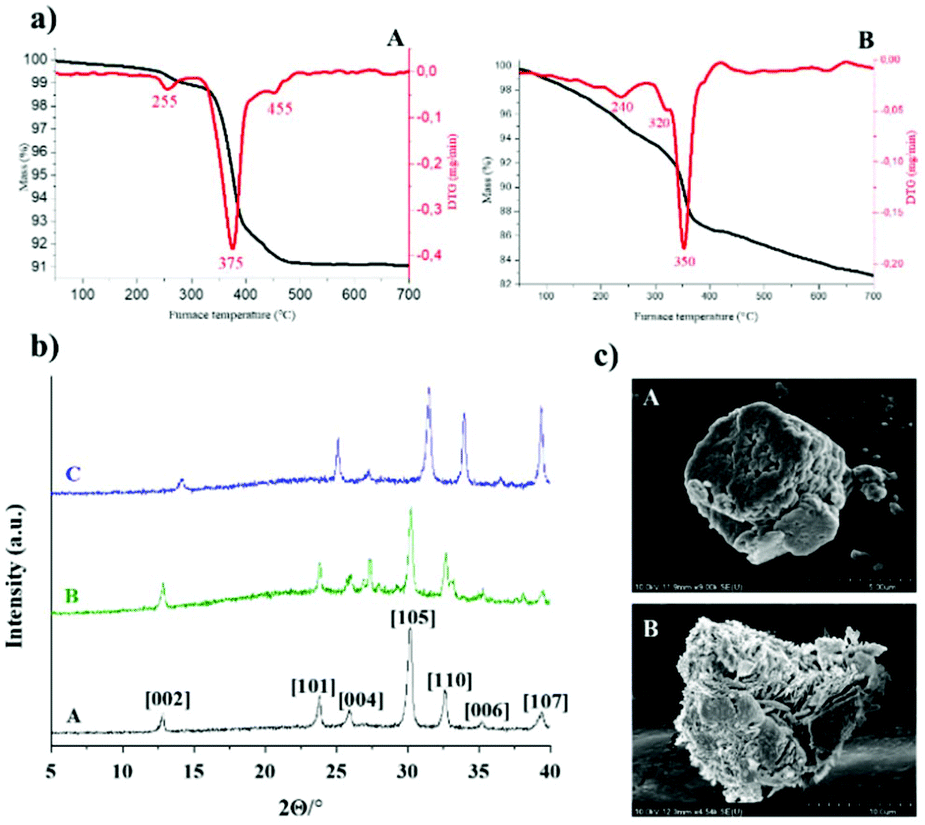 | ||
| Fig. 6 (a) Thermal behaviour of used AgBi-HM samples: sample used in flow scale-out (A), sample used in batch recycling experiment (B). (b) Comparison of the X-ray patterns of various AgBi-HM samples: as-prepared sample (A), sample used in batch recycling experiments (B), sample used in flow scale-out (C). (c) SEM images: as-prepared AgBi-HM sample – micrograph taken from ref. 41 (A), AgBi-HM sample used in flow scale-out (B). | ||
Thermal analysis revealed that the original structure was kept up to 380 °C, and weight losses occurred in three endothermic steps which was also observed in both used catalyst samples (Fig. 6a). In the case of the AgBi-HM sample used in the batch recycling experiment, slightly greater weight loss could be observed at lower temperatures which may be explained by trace amounts of organic deposition on the surface. The X-ray patterns of both used samples seemed to be the same as was experienced in case of the as-prepared sample (Fig. 6b), there was no evidence of structural degradation visible. Identification of the X-ray patterns were accomplished on the basis of our previous work.42 These results provided some further information about primer crystallite size of the composite calculated by using the well-known Scherrer equation. This resulted in an average primer crystallite size of 20.98 nm, not only for the as-prepared catalyst but also for the used ones. As shown earlier,41 the SEM image of the freshly-made catalyst displayed a lamellar (plate-like) morphology which was also observed in the used material (Fig. 6c). Additionally, this observation was also strengthened by TEM images, in which well-aggregated plates with a secondary particle size of around 100 nm could be seen for the as-prepared as well as for the used catalyst sample (Fig. S2‡). The SEM images also confirmed that organic contaminants in the form of larger aggregates (up to 10 μm) remained on the surface which makes more difficult to identify the original morphology. The SEM-EDX elemental maps demonstrated that the silver and bismuth ions are located evenly in the used sample as well (Fig. S3‡). ICP-AES measurements confirmed that the quantity of silver and bismuth ions are in arrangement with the as-prepared sample considering errors of measurements.41
Taking into account all the characterization data, it can be ascertained that the AgBi-HM is a highly robust heterogeneous catalyst which proved to be invariable in a structural point of view after extensive and demanding use under batch or flow conditions.
3.4. Decarboxylative deuterations
Due to its relatively good availability and also because of the considerably large isotope effect, deuterium is of outstanding importance among stable isotopes used for labelling studies.82,83 Synthetic protocols that incorporate deuterium into various organic substances have therefore many applications in medicinal, analytical or pharmaceutical chemistry.74 Deuterium-labelled compounds are typically applied as analytical standards, for the evaluation of the metabolic pathways or in tracer studies to investigate pharmacokinetics, catalytic cycles and reaction pathways.84–86 As exemplified by Austedo®, the first deuterated drug marketed, pharmaceutical ingredients may also be potentiated by deuterium exchange.87,88 In contrast to deuterations of C–C or C–X (X = hetero atom) multiple bonds,89–93 synthetic processes for the site-specific incorporation of a single deuterium into an aromatic ring are more challenging.74,76,77,94,95 In most cases, these methods involve halogen/D exchange and are commonly mediated by strong bases which severely limits the functional group tolerance.96 Furthermore, catalytic and acid- or base-mediated H/D exchange reactions are also available, however, unlike halogen/D exchange reactions, these often involve selectivity issues.97–99Inspired by these limitations, we were intrigued to explore decarboxylative deuterations of benzoic acid derivatives in the presence of the silver-containing hybrid material as catalyst. Initially, reactions were investigated in batch (Table 6), under conditions optimized for the protodecarboxylations earlier (0.15 M substrate concentration, 5 mol% AgBi-HM as catalyst, 15 mol% of KOH as base, DMF as solvent, 110 °C temperature and 24 h reaction time). As deuterium source, 10 equiv. of D2O was added to the reaction mixture. We were satisfied to find that with this simple protocol, deuterodecarboxylations of various nitrobenzoic acids as well as 2-bromo-, 2,6-dimethoxy- and 2,4-dichlorobenzoic acid went smoothly. Excellent conversions (79–100%) and 100% chemoselectivity were achieved in all cases. In all reactions, deuteration was highly favoured over incidental hydrogen incorporation as indicated by deuterium contents of 76–100%.






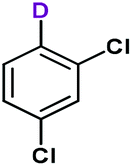
Continuous flow deuterodecarboxylations were next attempted in a packed bed reactor charged with AgBi-HM. Reaction conditions were simply taken from the protodecarboxylation experiments (0.1 M substrate concentration, 15 mol% of KOH as base, MeCN as solvent, 170 °C temperature, 50 μL min−1 flow rate, 10.5 min residence time). In these cases, 20 equiv. of D2O was used as deuterium source to achieve high deuterium contents. Gratifyingly, in all reactions investigated (Table 7), quantitative conversion and 100% chemoselectivity was achieved, and deuterium incorporation was also perfect in most cases.
|
|
||||
|---|---|---|---|---|
| Entry | Product | Conv.a (%) | Selectivitya (%) | D , (%) |
| a Determined by 1H NMR analysis of the crude product. b Deuterium content (represent deuterium incorporation rate over incidental hydrogen incorporation). c 10 equiv. of D2O was used as deuterium source. | ||||
| 1c |

|
100 | 100 | 31 |
| 2 | 100 | 100 | 100 | |
| 3 |

|
100 | 100 | 71 |
| 4 |

|
100 | 100 | 100 |
| 5 |

|
100 | 100 | 100 |
| 6 |

|
100 | 100 | 100 |

4. Conclusion
A silver-containing hybrid material with structurally-bound catalytic centers has been exploited as heterogenous noble metal catalyst for decarboxylations of carboxylic acids under batch and continuous flow conditions. It proved to be a robust, efficiently recyclable and highly active ligand-free catalyst which outperformed the most typical homogeneous catalytic sources in the decarboxylation of 2-nitrobenzoic acid as model reaction. Although, under batch conditions the catalyst performed best in DMF as solvent, the application of a simple packed-bed flow system enabled a solvent switch to the environmentally more acceptable MeCN. After the optimization of the most important reaction conditions, the selective decarboxylation of diversely substituted aromatic carboxylic acids were achieved with high conversions either in batch or in continuous flow mode. Importantly, the application of continuous flow conditions offered a marked chemical intensification as compared with the batch reactions (10 min residence time vs. 24 h reaction time) and ensured time-efficient syntheses. The preparative utility of the flow process was verified by a 20 h scale-out run in which the multigram-scale decarboxylation of 2-nitrobenzoic acid was achieved without notable decrease in the activity and without detectable degradation of the structure of catalyst. On the basis of the reaction conditions established for the protodecarboxylations, heterogeneous catalytic batch as well as flow methodologies were developed for decarboxylative deuterations in the presence of D2O as a readily available deuterium source.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Hungarian Ministry of National Economy, National Research Development and Innovation Office (GINOP2.3.2-15-2016-00034) and by TKP-2020. We are grateful to the Hungarian Research Foundation (OTKA No. K115731). R. M. was supported by the ÚNKP-19-3 New National Excellence Program of the Ministry for Innovation and Technology (Hungary). S. B. Ö. acknowledges the Premium Post Doctorate Research Program of the Hungarian Academy of Sciences. G. V. thanks for the postdoctoral fellowship under the grant number PD 128189.References
- L. J. Gooßen, N. Rodríguez and K. Gooßen, Angew. Chem., Int. Ed., 2008, 47, 3100–3120 CrossRef.
- T. Patra and D. Maiti, Chem. – Eur. J., 2017, 23, 7382–7401 CrossRef CAS.
- C. Shen, P. Zhang, Q. Sun, S. Bai, T. S. A. Hor and X. Liu, Chem. Soc. Rev., 2015, 44, 291–314 RSC.
- S. Cadot, N. Rameau, S. Mangematin, C. Pinel and L. Djakovitch, Green Chem., 2014, 16, 3089–3097 RSC.
- J. S. Dickstein, J. M. Curto, O. Gutierrez, C. A. Mulrooney and M. C. Kozlowski, J. Org. Chem., 2013, 78, 4744–4761 CrossRef CAS PubMed.
- S. Seo, J. B. Taylor and M. F. Greaney, Chem. Commun., 2012, 48, 8270–8272 RSC.
- Z. Li, Z. Fu, H. Zhang, J. Long, Y. Song and H. Cai, New J. Chem., 2016, 40, 3014–3018 RSC.
- M. T. Keßler, C. Gedig, S. Sahler, P. Wand, S. Robke and M. H. G. Prechtl, Catal. Sci. Technol., 2013, 3, 992–1001 RSC.
- L. J. Gooßen, W. R. Thiel, N. Rodríguez, C. Linder and B. Melzer, Adv. Synth. Catal., 2007, 349, 2241–2246 CrossRef.
- N. Chatani, H. Tatamidani, Y. Ie, F. Kakiuchi and S. Murai, J. Am. Chem. Soc., 2001, 123, 4849–4850 CrossRef CAS PubMed.
- S. Pan, B. Zhou, Y. Zhang, C. Shao and G. Shi, Synlett, 2016, 27, 277–281 CAS.
- M. H. Al-Huniti, M. A. Perez, M. K. Garr and M. P. Croatt, Org. Lett., 2018, 20, 7375–7379 CrossRef CAS PubMed.
- R. A. Daley and J. J. Topczewski, Synthesis, 2020, 365–377 CAS.
- G. J. S. Dawes, E. L. Scott, J. Le Nôtre, J. P. M. Sanders and J. H. Bitter, Green Chem., 2015, 17, 3231–3250 RSC.
- A. Bohre, U. Novak, M. Grilc and B. Likozar, Mol. Catal., 2019, 476, 110520 CrossRef CAS.
- A. Bohre, B. Hočevar, M. Grilc and B. Likozar, Appl. Catal., B, 2019, 256, 117889 CrossRef CAS.
- A. Bohre, K. Avasthi, U. Novak and B. Likozar, ACS Sustainable Chem. Eng., 2021, 9, 2902–2911 CrossRef CAS.
- H. Hikawa, F. Kotaki, S. Kikkawa and I. Azumaya, J. Org. Chem., 2019, 84, 1972–1979 CrossRef CAS PubMed.
- S. Dupuy, L. Crawford, M. Bühl and S. P. Nolan, Chem. – Eur. J., 2015, 21, 3399–3408 CrossRef CAS.
- S. Dupuy and S. P. Nolan, Chem. – Eur. J., 2013, 19, 14034–14038 CrossRef CAS PubMed.
- R. A. Crovak and J. M. Hoover, J. Am. Chem. Soc., 2018, 140, 2434–2437 CrossRef CAS PubMed.
- R. Grainger, J. Cornella, D. C. Blakemore, I. Larrosa and J. M. Campanera, Chem. – Eur. J., 2014, 20, 16680–16687 CrossRef CAS PubMed.
- L. Cui, H. Chen, C. Liu and C. Li, Org. Lett., 2016, 18, 2188–2191 CrossRef CAS PubMed.
- Y. Zhu, X. Li, X. Wang, X. Huang, T. Shen, Y. Zhang, X. Sun, M. Zou, S. Song and N. Jiao, Org. Lett., 2015, 17, 4702–4705 CrossRef CAS PubMed.
- X. Y. Toy, I. I. B. Roslan, G. K. Chuah and S. Jaenicke, Catal. Sci. Technol., 2014, 4, 516–523 RSC.
- L. J. Gooßen, C. Linder, N. Rodríguez, P. P. Lange and A. Fromm, Chem. Commun., 2009, 7173–7175 RSC.
- P. Lu, C. Sanchez, J. Cornella and I. Larrosa, Org. Lett., 2009, 11, 5710–5713 CrossRef CAS PubMed.
- R. A. Sheldon, Green Chem., 2017, 19, 18–43 RSC.
- M. C. Bryan, P. J. Dunn, D. Entwistle, F. Gallou, S. G. Koenig, J. D. Hayler, M. R. Hickey, S. Hughes, M. E. Kopach, G. Moine, P. Richardson, F. Roschangar, A. Steven and F. J. Weiberth, Green Chem., 2018, 20, 5082–5103 RSC.
- H. C. Erythropel, J. B. Zimmerman, T. M. de Winter, L. Petitjean, F. Melnikov, C. H. Lam, A. W. Lounsbury, K. E. Mellor, N. Z. Janković, Q. Tu, L. N. Pincus, M. M. Falinski, W. Shi, P. Coish, D. L. Plata and P. T. Anastas, Green Chem., 2018, 20, 1929–1961 RSC.
- C. Wen, A. Yin and W.-L. Dai, Appl. Catal., B, 2014, 160–161, 730–741 CrossRef CAS.
- A. K. Clarke, M. J. James, P. O'Brien, R. J. K. Taylor and W. P. Unsworth, Angew. Chem., Int. Ed., 2016, 55, 13798–13802 CrossRef CAS PubMed.
- X.-Y. Dong, Z.-W. Gao, K.-F. Yang, W.-Q. Zhang and L.-W. Xu, Catal. Sci. Technol., 2015, 5, 2554–2574 RSC.
- C.-J. Li and X. Bi, Silver catalysis in organic synthesis, Wiley-VCH, Weinheim, 2019 Search PubMed.
- Q.-Z. Zheng and N. Jiao, Chem. Soc. Rev., 2016, 45, 4590–4627 RSC.
- L. Maestre, R. Dorel, Ó. Pablo, I. Escofet, W. M. C. Sameera, E. Álvarez, F. Maseras, M. M. Díaz-Requejo, A. M. Echavarren and P. J. Pérez, J. Am. Chem. Soc., 2017, 139, 2216–2223 CrossRef CAS PubMed.
- J. Ozawa and M. Kanai, Org. Lett., 2017, 19, 1430–1433 CrossRef CAS PubMed.
- S. Guo, F. Cong, R. Guo, L. Wang and P. Tang, Nat. Chem., 2017, 9, 546–551 CrossRef CAS PubMed.
- N. Salam, A. Sinha, A. S. Roy, P. Mondal, N. R. Jana and S. M. Islam, RSC Adv., 2014, 4, 10001–10012 RSC.
- J. D. Kim, T. Palani, M. R. Kumar, S. Lee and H. C. Choi, J. Mater. Chem., 2012, 22, 20665–20670 RSC.
- S. B. Ötvös, R. Mészáros, G. Varga, M. Kocsis, Z. Kónya, Á. Kukovecz, P. Pusztai, P. Sipos, I. Pálinkó and F. Fülöp, Green Chem., 2018, 20, 1007–1019 RSC.
- R. Mészáros, S. B. Ötvös, G. Varga, É. Böszörményi, M. Kocsis, K. Karádi, Z. Kónya, Á. Kukovecz, I. Pálinkó and F. Fülöp, Mol. Catal., 2020, 498, 111263 CrossRef.
- A. Tanimu, S. Jaenicke and K. Alhooshani, Chem. Eng. J., 2017, 327, 792–821 CrossRef CAS.
- R. Ciriminna, M. Pagliaro and R. Luque, Green Energy Environ., 2021 DOI:10.1016/j.gee.2020.09.013 , (In Press).
- X. Liu, B. Unal and K. F. Jensen, Catal. Sci. Technol., 2012, 2, 2134–2138 RSC.
- C. G. Frost and L. Mutton, Green Chem., 2010, 12, 1687–1703 RSC.
- R. Munirathinam, J. Huskens and W. Verboom, Adv. Synth. Catal., 2015, 357, 1093–1123 CrossRef CAS.
- W.-J. Yoo, H. Ishitani, Y. Saito, B. Laroche and S. Kobayashi, J. Org. Chem., 2020, 85, 5132–5145 CrossRef CAS.
- R. Gérardy, N. Emmanuel, T. Toupy, V.-E. Kassin, N. N. Tshibalonza, M. Schmitz and J.-C. M. Monbaliu, Eur. J. Org. Chem., 2018, 2301–2351 CrossRef.
- R. Porta, M. Benaglia and A. Puglisi, Org. Process Res. Dev., 2016, 20, 2–25 CrossRef CAS.
- B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688–6728 CrossRef CAS PubMed.
- M. Baumann and I. R. Baxendale, Beilstein J. Org. Chem., 2015, 11, 1194–1219 CrossRef CAS PubMed.
- S. B. Ötvös and F. Fülöp, Catal. Sci. Technol., 2015, 5, 4926–4941 RSC.
- A. Hommes, H. J. Heeres and J. Yue, ChemCatChem, 2019, 11, 4671–4708 CrossRef CAS.
- R. Gérardy, D. P. Debecker, J. Estager, P. Luis and J.-C. M. Monbaliu, Chem. Rev., 2020, 120, 7219–7347 CrossRef PubMed.
- D. L. Riley and N. C. Neyt, Synthesis, 2018, 2707–2720 CrossRef CAS.
- K. Masuda, T. Ichitsuka, N. Koumura, K. Sato and S. Kobayashi, Tetrahedron, 2018, 74, 1705–1730 CrossRef CAS.
- Á. Georgiádes, S. B. Ötvös and F. Fülöp, Adv. Synth. Catal., 2018, 360, 1841–1849 CrossRef.
- I. M. Mándity, S. B. Ötvös and F. Fülöp, ChemistryOpen, 2015, 4, 212–223 CrossRef.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS.
- F. M. Akwi and P. Watts, Chem. Commun., 2018, 54, 13894–13928 RSC.
- L. Rogers and K. F. Jensen, Green Chem., 2019, 21, 3481–3498 RSC.
- A. Puglisi, M. Benaglia and V. Chiroli, Green Chem., 2013, 15, 1790–1813 RSC.
- C. Rodríguez-Escrich and M. A. Pericàs, Eur. J. Org. Chem., 2015, 1173–1188 CrossRef.
- T. Tsubogo, T. Ishiwata and S. Kobayashi, Angew. Chem., Int. Ed., 2013, 52, 6590–6604 CrossRef CAS PubMed.
- V. V. Ranade, R. Chaudhari and P. R. Gunjal, Trickle bed reactors: Reactor engineering and applications, Elsevier, Amsterdam, 2011 Search PubMed.
- S. B. Ötvös, M. A. Pericàs and C. O. Kappe, Chem. Sci., 2019, 10, 11141–11146 RSC.
- S. B. Ötvös, P. Llanes, M. A. Pericàs and C. O. Kappe, Org. Lett., 2020, 22, 8122–8126 CrossRef.
- C. De Risi, O. Bortolini, A. Brandolese, G. Di Carmine, D. Ragno and A. Massi, React. Chem. Eng., 2020, 5, 1017–1052 RSC.
- R. L. Papurello, J. L. Fernández, E. E. Miró and J. M. Zamaro, Chem. Eng. J., 2017, 313, 1468–1476 CrossRef CAS.
- L. Zhang, Z. Liu, Y. Wang, R. Xie, X.-J. Ju, W. Wang, L.-G. Lin and L.-Y. Chu, Chem. Eng. J., 2017, 309, 691–699 CrossRef CAS.
- J. Cao, G. Xu, P. Li, M. Tao and W. Zhang, ACS Sustainable Chem. Eng., 2017, 5, 3438–3447 CrossRef CAS.
- M. Jeganathan, A. Dhakshinamoorthy and K. Pitchumani, ACS Sustainable Chem. Eng., 2014, 2, 781–787 CrossRef CAS.
- J. Atzrodt, V. Derdau, W. J. Kerr and M. Reid, Angew. Chem., Int. Ed., 2018, 57, 1758–1784 CrossRef CAS PubMed.
- S. L. Harbeson and R. D. Tung, Med. Chem. News, 2014, 24, 8–22 Search PubMed.
- R. Grainger, A. Nikmal, J. Cornella and I. Larrosa, Org. Biomol. Chem., 2012, 10, 3172–3174 RSC.
- M. Rudzki, A. Alcalde-Aragonés, W. I. Dzik, N. Rodríguez and L. J. Gooßen, Synthesis, 2012, 184–193 CAS.
- S. Bhadra, W. I. Dzik and L. J. Gooßen, Angew. Chem., Int. Ed., 2013, 52, 2959–2962 CrossRef CAS PubMed.
- L. J. Gooßen, N. Rodríguez, C. Linder, P. P. Lange and A. Fromm, ChemCatChem, 2010, 2, 430–442 CrossRef.
- J. Cornella, C. Sanchez, D. Banawa and I. Larrosa, Chem. Commun., 2009, 7176–7178 RSC.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- A. J. Percy, M. Rey, K. M. Burns and D. C. Schriemer, Anal. Chim. Acta, 2012, 721, 7–21 CrossRef CAS PubMed.
- K. Gevaert, F. Impens, B. Ghesquière, P. Van Damme, A. Lambrechts and J. Vandekerckhove, Proteomics, 2008, 8, 4873–4885 CrossRef CAS PubMed.
- H. Wang, A. A. Hussain, J. S. Pyrek, J. Goodman and P. J. Wedlund, J. Pharm. Biomed. Anal., 2004, 34, 1063–1070 CrossRef CAS PubMed.
- K. Sanderson, Nature, 2009, 458, 269 CAS.
- D. M. Marcus, K. A. McLachlan, M. A. Wildman, J. O. Ehresmann, P. W. Kletnieks and J. F. Haw, Angew. Chem., Int. Ed., 2006, 45, 3133–3136 CrossRef CAS PubMed.
- P. Chen, S. Ren, H. Song, C. Chen, F. Chen, Q. Xu, Y. Kong and H. Sun, Bioorg. Med. Chem., 2019, 27, 116–124 CrossRef CAS.
- C. Schmidt, Nat. Biotechnol., 2017, 35, 493–494 CrossRef CAS.
- B. Dong, X. Cong and N. Hao, RSC Adv., 2020, 10, 25475–25479 RSC.
- M. Espinal-Viguri, S. E. Neale, N. T. Coles, S. A. Macgregor and R. L. Webster, J. Am. Chem. Soc., 2019, 141, 572–582 CrossRef CAS PubMed.
- R. Mészáros, B.-J. Peng, S. B. Ötvös, S.-C. Yang and F. Fülöp, ChemPlusChem, 2019, 84, 1508–1511 CrossRef PubMed.
- S. B. Ötvös, C.-T. Hsieh, Y.-C. Wu, J.-H. Li, F.-R. Chang and F. Fülöp, Molecules, 2016, 21(318), 1–11 Search PubMed.
- C.-T. Hsieh, S. B. Ötvös, Y.-C. Wu, I. M. Mándity, F.-R. Chang and F. Fülöp, ChemPlusChem, 2015, 80, 859–864 CrossRef CAS PubMed.
- A. Tlahuext-Aca and J. F. Hartwig, ACS Catal., 2021, 11, 1119–1127 CrossRef CAS.
- G. Orsy, F. Fülöp and I. M. Mándity, Green Chem., 2019, 21, 956–961 RSC.
- J. Atzrodt, V. Derdau, T. Fey and J. Zimmermann, Angew. Chem., Int. Ed., 2007, 46, 7744–7765 CrossRef CAS PubMed.
- G. Erdogan and D. B. Grotjahn, J. Am. Chem. Soc., 2009, 131, 10354–10355 CrossRef CAS PubMed.
- G. S. Coumbarides, M. Dingjan, J. Eames, A. Flinn and J. Northen, J. Labelled Compd. Radiopharm., 2006, 49, 903–914 CrossRef CAS.
- P. S. Kiuru and K. Wähälä, Steroids, 2006, 71, 54–60 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to the memory of our friend and colleague Prof. István Pálinkó. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc00924a |
| § Deceased. |
| This journal is © The Royal Society of Chemistry 2021 |