
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxo-functionalised mesoionic NHC nickel complexes for selective electrocatalytic reduction of CO2 to formate†
Simone
Bertini
 a,
Motiar
Rahaman
a,
Abhijit
Dutta
a,
Philippe
Schollhammer
b,
Alexander V.
Rudnev
ac,
Fredric
Gloaguen
*b,
Peter
Broekmann
*a and
Martin
Albrecht
*a
a,
Motiar
Rahaman
a,
Abhijit
Dutta
a,
Philippe
Schollhammer
b,
Alexander V.
Rudnev
ac,
Fredric
Gloaguen
*b,
Peter
Broekmann
*a and
Martin
Albrecht
*a
aDepartment of Chemistry, Biochemistry &Pharmacy, Universität Bern, Freiestrasse 3, 3012 Bern, Switzerland. E-mail: martin.albrecht@dcb.unibe.ch; peter.broekmann@dcb.unibe.ch
bUMR 6521, CNRS, Université de Bretagne Occidentale, CS 93837, 29238 Brest, France. E-mail: frederic.gloaguen@univ-brest.fr
cA.N. Frumkin Institute of Physical Chemistry and Electrochemistry Russian Academy of Sciences, Leninskii pr. 31, 119071 Moscow, Russia
First published on 12th April 2021
Abstract
Strategies for the conversion of CO2 to valuable products are paramount for reducing the environmental risks associated with high levels of this greenhouse gas and offer unique opportunities for transforming waste into useful products. While catalysts based on nickel as an Earth-abundant metal for the sustainable reduction of CO2 are known, the vast majority produce predominantly CO as a product. Here, efficient and selective CO2 reduction to formate as a synthetically valuable product has been accomplished with novel nickel complexes containing a tailored C,O-bidentate chelating mesoionic carbene ligand. These nickel(II) complexes are easily accessible and show excellent catalytic activity for electrochemical H+ reduction to H2 (from HOAc in MeCN), and CO2 reduction (from CO2-saturated MeOH/MeCN solution) with high faradaic efficiency to yield formate exclusively as an industrially and synthetically valuable product from CO2. The most active catalyst precursor features the 4,6-di-tert-butyl substituted phenolate triazolylidene ligand, tolerates different proton donors including water, and reaches an unprecedented faradaic efficiency of 83% for formate production, constituting the most active and selective Ni-based system known to date for converting CO2 into formate as an important commodity chemical.
Introduction
The conversion of environmentally harmful CO2 into synthetically and industrially valuable products has become a pressing challenge for mitigating the threats associated with increased CO2 levels in the atmosphere.1–3 Among the various technologies developed for CO2 fixation,4–6 electrochemical reduction is particularly attractive7,8 since it allows for direct transformation of CO2 into synthetically or industrially valuable platform chemicals such as formate,9,10 alcohols,11–14 and unsaturated hydrocarbons (e.g., ethylene).15–17 In addition, electrochemical processes have the benefit to be fully sustainable, especially if they are powered by renewable energy sources (e.g., solar, wind, or hydro). The key critical parameter is then the nature of the catalyst, which is preferably derived from Earth-abundant metals in order to provide a truly sustainable process.18,19While several complexes based on Earth-abundant Mn, Fe, and Ni metals have been known to catalyze the reduction of CO2,20–25 the vast majority of these catalysts produce CO as a predominant product.26,27 Only two Ni systems have been reported to yield formate,28 namely Sauvage's Ni(cyclam) system from over 30 years ago,29–32 and Fontecave's Ni(III) catalyst,33 though selectivity is a major issue in both systems due to significant formation of CO. Formate formation is highly desirable as it constitutes a pathway to convert waste to a valuable product for synthesis, hydrogen storage, formic acid fuel cells, and for other industrial uses.
Inspired by the work of Kirchner and others on manganese pincer complexes,34–36 which demonstrated a key relevance of the metal-hydride intermediate to promote CO2 insertion rather than direct CO2 bonding, we became interested in exploiting the potential of triazolylidenes37,38 as a specific subclass of N-heterocyclic carbene (NHC) ligands39–41 for imparting such reactivity. In order to increase the robustness of the M–CNHC bond and hence the reliability of the carbene as a spectator ligand to the nickel active site,42 oxygen chelating groups were introduced.43 Here we show that this approach provides a set of new, tunable, and highly active catalysts for the electrochemical reduction of CO2 to formate. Catalyst screening in half-cell measurements revealed that the most active system accomplishes unrivalled faradaic efficiency and outstanding selectivity towards formate, largely outperforming currently known catalyst systems based on nickel.
Results and discussion
The new phenolate-substituted triazolium salts 2a–c were synthesized from 2-azidophenol 1 by [3 + 2] cycloaddition reaction through variation of the alkyne precursor (R = Ph, Bu, Mes; Scheme 1), followed by selective alkylation (>60% overall yield). Metalation was accomplished with NiCl2 as a simple and cheap nickel precursor in the presence of K2CO3. The new bis-carbene nickel(II) complexes 3a–c and 4 were obtained as yellow solids that are stable towards air and moisture for >2 months. | ||
| Scheme 1 Synthesis of NiII bis(carbene) complexes 3 and 4. | ||
The 1H NMR spectra of the complexes reveal the presence of only one isomer. While complexes 3a,b and 4 feature the phenolate proton resonances in the expected aromatic region (Fig. S20†), the spectrum of complex 3c is distinct with a markedly upfield shifted phenolate ortho proton (δH = 4.8; Fig. S20†). This shift of more than 2 ppm suggests a trans arrangement of the two C,O-bidentate ligands, with the phenolate Hortho influenced by ring current anisotropy of the mesityl group of the other ligand. No such effect has been observed in the NMR spectrum of complex 3b featuring a phenyl-substituted triazolylidene, which suggests a cis configuration. These ligand arrangements were unambiguously confirmed by single crystal X-ray diffraction (Fig. 1). In the trans configuration of 3c, the mesityl ring is essentially orthogonal to the carbene–Ni plane and close to Hortho (Hortho⋯arene 2.75 Å), which accounts for the ring anisotropy observed by NMR spectroscopy. The cis configuration of the other complexes places the carbene wingtip groups R in close proximity, which results in a substantial distortion from square-planar to tetrahedral (cf. τ4 ≥ 0.15).44 The distortion is larger for bulky wingtip groups (Bu, Ph, in 3a,b, respectively) than with methyl substituents (4), and negligible in the trans complex 3c (τ4 < 0.01; Table S7†).
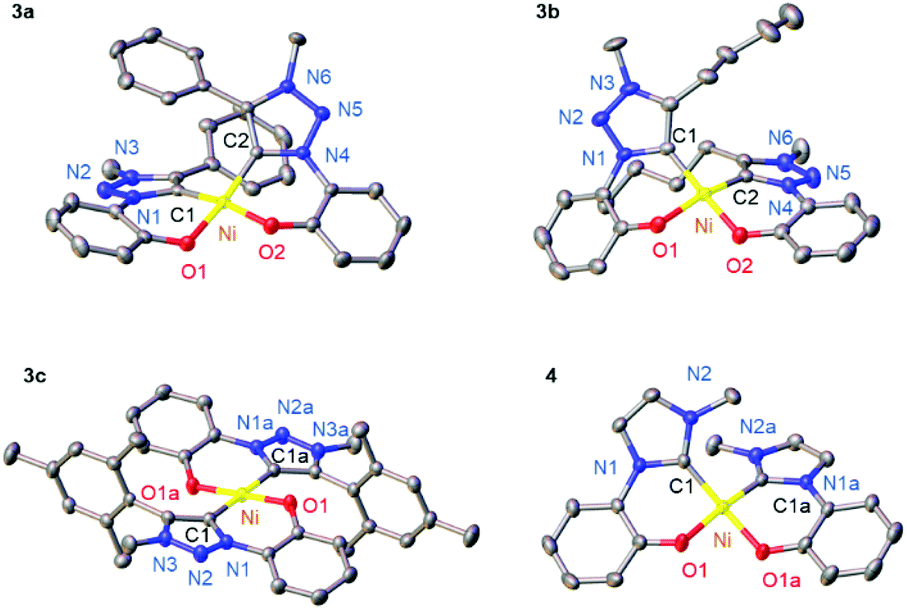 | ||
| Fig. 1 ORTEP diagram of Ni complexes 3a–c and 4; 50% probability level thermal ellipsoids; hydrogen atoms and co-crystallized solvent molecules (H2O for 3a, CHCl3 for 4) omitted for clarity; a denotes symmetry-related atoms. | ||
All four nickel complexes 3a–c, 4 display (quasi)reversible redox processes around +0.7 V and −2.0 V, tentatively attributed to NiII/NiIII and NiII/NiI transitions, respectively (Fig. 2, S21 and 22;† potentials vs. Ag/AgCl).33 Comparison of the redox potentials consistently indicates that triazolylidenes are stronger donor ligands than imidazolylidene,45 and that the wingtip group R directly affects the electron density on the nickel center with oxidation potentials increasing along the series R = Bu (E1/2 = 0.59) < R = Mes (E1/2 = 0.64) < R = Ph (E1/2 = 0.69; Table 1).
 | ||
| Fig. 2 Cyclic voltammograms of the Ni(II) complexes 3a (grey) and 4 (blue; both scans 1 mM in MeCN with 0.1 M (Bu4N)PF6 as supporting electrolyte, 250 mV s−1 scan rate, Fc+/Fc as internal standard with E1/2 = 0.36 V vs. Ag/AgCl). | ||
| Entry | Complex |
E
pc![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
E 1/2(NiII/III) b |
k
obs
[s−1] |
|---|---|---|---|---|
| a All values in V vs. Ag/AgCl; 1 mM MeCN solution of Ni complex with (Bu4N)PF6 as supporting electrolyte, 250 mV s−1 scan rate, Fc+/Fc as internal standard with E1/2 = 0.36 V vs. Ag/AgCl. b Cathodic peak potential Epc for NiII/NiI reduction and half-wave potential E1/2 (in parentheses ΔEp = Epa − Epc in mV for NiII/NiIII redox process) c Catalytic conditions; 1 mM complex in MeCN, AcOH (0.8 M), (Bu4N)PF6 as supporting electrolyte, kobs determined by foot of the wave analysis (see ESI for details†). | ||||
| 1 | 3a | –2.16 | 0.59 (130) | 440 |
| 2 | 3b | –2.09 | 0.69 (110) | 300 |
| 3 | 3c | –2.12 | 0.64 (120) | 10 |
| 4 | 4 | –1.92 (110) | 0.75 (120) | 200 |
The electrocatalytic performance of complexes 3a–c, 4 was first investigated in H+ reduction. A significant cathodic current was observed when acetic acid (AcOH) was present as proton donor in a MeCN solution of the Ni complex. Increasing the AcOH concentration from 5 to 400 eq. with respect to the Ni complex led to an enhanced current density, indicative of catalytic H+ reduction (Fig. 3). Extraction of kobs by foot-of-the-wave analysis (FOWA)46,47 reveals a direct influence of the steric and electronic properties of the ligand in promoting catalytic reduction (Table S8†). Specifically, the trans arrangement imposed by the very bulky mesityl wingtip groups is strongly deactivating, while all the cis-complexes are active. Moreover, the activity of the cis-complexes directly correlates with the ligand donor properties deduced from CV data: the alkyl-substituted triazolylidene induces more than a twice higher active than the analogous imidazolylidene (kobs = 440 s−1 for 3avs. 200 s−1 for 4).48
 | ||
| Fig. 3 Electrocatalytic reduction of H+ in MeCN as solvent (1 mM of complex 3a, (Bu4N)PF6 as supporting electrolyte), scan rate 250 mV s−1, HOAc as proton source Fc+/Fc used as internal standard (E1/2 = 0.36 V vs. Ag/AgCl); red dashed line: degassed solution of complex 3a (1 mM) under Ar; black dashed line: degassed solution of AcOH (0.1 M) in MeCN under Ar; olive to black solid lines: complex 3a (1 mM) in presence of increasing amounts of AcOH (5, 10, 40, 100, 200 and 400 mM, respectively) in MeCN solution). | ||
Prompted by the promising catalytic activities, complexes 3a–c, 4 were evaluated as catalyst precursors for electrochemical reduction of CO2. Initial experiments with a 1 mM MeCN solution of the nickel complex 3a under a CO2 atmosphere reveal a significant enhancement of the cathodic current upon complex reduction, indicative of CO2 transformation (Fig. 4). For complex 4 the reversibility of the NiII/NiI reduction was lost upon saturation with CO2 gas. All the complexes were active in the process, with only the trans isomer 3c induces low catalytic current, suggesting lower activity. The catalytic current enhances further when the reaction was performed in the presence of MeOH (40 eq. with respect to the Ni complex), suggesting a beneficial role of proton sources.49 Trifluoroethanol and phenol show similar effects, though the current increase is largest when using MeOH (Fig. 4). Blank measurements indicate no catalytic current with CO2-saturated solutions in the absence of the Ni complex, or when the complex is reduced in the presence of methanol yet without CO2 (Fig. S34†).
 | ||
| Fig. 4 Electrocatalytic reduction of CO2 in MeCN with complex 3a (0.1 M (Bu4N)PF6 as supporting electrolyte, 250 mV s−1 scan rate, glassy carbon working electrode); black line: CO2-saturated MeCN solution; blue line: degassed solution of complex 3a (1 mM) under Ar; red line: complex 3a (1 mM) in CO2-saturated MeCN solution; green line: complex 3a (1 mM) in CO2-saturated MeCN solution with 40 eq. MeOH. | ||
The robustness of the catalytically active species over time was investigated by chronoamperometry at −1.9 V vs. Ag/AgCl in MeOH/MeCN 1:50 v:v (Fig. S26–S29†). The observed catalytic current is constant over 2 h, suggesting no significant degradation during that time. More extended reaction reveals a gradual decrease of activity. Comparison of the different complexes reveals catalytic activity for CO2 reduction follows the same trends observed for H+ reduction, with highest rates for the nickel complex with the strongest donating triazolylidene ligand (3a, kobs = 280 s−1), which is essentially twice as fast as the corresponding imidazolylidene analogue 4 (kobs = 150 s−1; Table 2). Again, the cis ligand arrangement is essential for ensuing catalytic activity as the nickel complex 3c with the ligands in trans configuration is almost inactive (kobs = 10 s−1), and enhanced electron density at the nickel center increases catalytic activity (3a > 3b > 4).
| Entry | Complex | FEHCOO- (4 h) [%] | FEHCOO- (8 h) [%] | FEH2+CO (4 h) [%] |
k
obs
[s−1] |
|---|---|---|---|---|---|
|
a General conditions: 1 mM complex, at −1.9 V vs. Ag/AgCl, glassy carbon working electrode and Pt foil as counter electrode (see ESI for details†) in MeOH/MeCN 1:50 v:v with 0.1 M (Bu4N)PF6 as supporting electrolyte.
b Determined from foot of the wave data treatment (see ESI for details†).
|
|||||
| 1 | 3a | 54 | 68 | 3 | 280 |
| 2 | 3b | 43 | 47 | 4 | 220 |
| 3 | 3c | 10 | 10 | 2 | 10 |
| 4 | 4 | 25 | 25 | 3 | 150 |
Product identification focused first on gas-phase analysis of volatiles products as most hitherto known Ni catalysts for CO2 reduction produce CO.10,11 Remarkably, only traces of H2 were detected in the gas phase, and CO quantities were below the detection limit (less than 2% faradaic efficiency after 4 h, Table 2). Analysis of the solution phase by ion-exchange chromatography (IC), HPLC and NMR measurements identified formate as the principal product of CO2 reduction with complexes 3a–c, 4. Complex 3a does not display only the highest activity but also imparts the highest selectivity. Optimization of reaction times affords faradaic efficiencies up to 70% for formate formation, one of the highest known so far for homogeneous Ni-based electrocatalysts.10,11
Ligand tailoring has been used to further improve the catalytic activity of these nickel complexes, in particular through introduction of electron-donating substituents on the phenolate. Substitution of the 4,6-positions moreover avoids undesired radical reactions of the phenolate, which may infer from one-electron-reduction upon catalyst activation. To this end the 4,6-Me2-phenol-triazolium salt 5a was nickelated via the procedure established for the synthesis of 3a, while the tBu analogue 5b required nBuLi as a stronger base to accomplish nickel complexation (Fig. 5a). The new O,C-bidentate chelated trz Ni(II) complexes 6a and 6b showed NMR characteristic reminiscent of those of 3a,b, indicating the formation of the cis-isomers exclusively. This structural assignment was further confirmed by single crystal X-ray diffraction studies of complexes 6a,b (Fig. 5b). The distortion from square planar geometry in these complexes is slightly less than in the parent complex 3a with τ4 values of 0.15 and 0.18 for 6a and 6b, respectively (cf. 0.21 for 3a, Table S7†).
 | ||
| Fig. 5 (a) Synthesis of complexes 6a,b; (b) ORTEP diagrams of Ni complexes 6a,b (50% probability thermal ellipsoids, hydrogen atoms omitted for clarity, a denotes symmetry-related atoms). One tBu group in 6b is disordered about 2 conformations. Selected bond lengths (Å) for 6a: Ni–C1 = 1.862(2), Ni–O1 = 1.887(1). Selected bond lengths (Å) for 6b: Ni–C1 = 1.836(1), Ni–O1 = 1.899(1). | ||
The electrochemical properties of complexes 6a,b are comparable to those of complexes 3a–c, featuring a quasi-reversible oxidation at 0.49 and 0.45 V vs. Ag/AgCl, respectively, and an irreversible reduction below −2.0 V (Fig. S21 and 22†). The incorporation of donating groups on the phenolate lowers the redox potentials by about 90 (R = Me, 6a) and 150 mV (R = tBu, 6b relative to the parent phenolate complex 3a (Table 3). These potential shifts indicate enhanced electron density at the metal center and efficient electronic tailoring of the nickel center by ligand modifications.
| Entry | Complex |
E
pcb [V] |
FEHCOO- (8 h) [%] | FEH2+CO (4 h) [%] |
k
obs
[s−1] |
|---|---|---|---|---|---|
|
a General conditions: 1 mM complex, at −1.9 V vs. Ag/AgCl, glassy carbon working electrode and Pt foil as counter electrode (see ESI for details†) in MeOH/MeCN 1:50 v:v with 0.1 M (Bu4N)PF6 as supporting electrolyte.
b Cathodic peak potential Epc for NiII/NiI reduction in V vs. Ag/AgCl; 1 mM MeCN solution of Ni complex with (Bu4N)PF6 as supporting electrolyte, 250 mV s−1 scan rate, Fc+/Fc as internal standard with E1/2 = 0.36 V vs. Ag/AgCl.
c Determined from foot of the wave data treatment (see ESI for details†).
|
|||||
| 1 | 6a | –2.23 | 74 | 4 | 300 |
| 2 | 6b | –2.31 | 83 | 3 | 370 |
In these cis triazolylidene Ni complexes, lower reduction potentials are correlated with higher electrocatalytic CO2 reduction activity (cf.Table 2) Indeed, the stronger donor ligands in complexes 6a,b and the associated low reduction potential improved the catalytic performance. Foot of the wave data analysis revealed a higher kobs for CO2 reduction for 6a than for the parent complex 3a (300 vs. 280 s−1; Table 3), and more significantly for 6b with the lowest reduction potential (kobs = 370 s−1). Moreover, the robustness of the catalytically active species is demonstrated by a constant catalytic current over the first 2 h (Fig. 6 and S31†). Formate was identified as exclusive product of CO2 reduction also with complexes 6a,b with only traces of H2 and CO as side products. Complex 6b revealed the highest FE of 83% for formate formation and represents a new benchmark for Ni in this transformation. Moreover, these results demonstrate that ligand tailoring on a molecular level through incorporation of electron-donating substituents provides an efficient strategy for catalyst optimization for this CO2 reduction and leads in this case to a marked increase of the FE to unprecedented 83%.
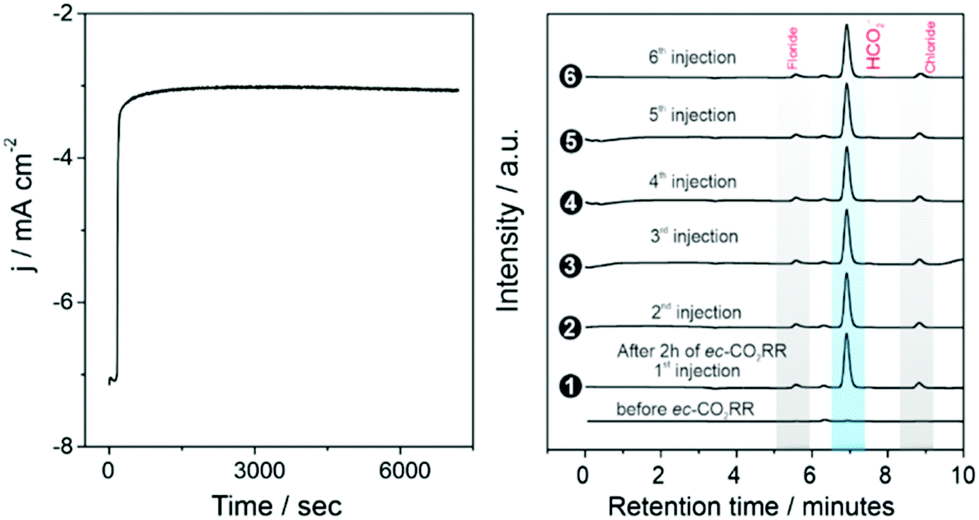 | ||
| Fig. 6 Electrolysis experiment performed with 1 mM complex 6b, at −1.9 V vs. Ag/AgCl, using glassy carbon working electrode, in a CO2 saturated solution of MeOH/MeCN 1:50 v:v with 0.1 M (Bu4N)PF6 as supporting electrolyte (left) and product analysis data on six different injections after 2 h by ion exchange chromatography (right). | ||
While this work is focused on catalyst optimization for the CO2 reduction half-reaction, obviously also the availability of protons is elementary as the efficiency of the CO2 to formate transformation is associated with a coupled electron/proton transfer. Variation of the proton source has been accomplished by varying the additive to the aprotic solvent (MeCN) from MeOH to different proton donors including PhOH, iPrOH, CF3CH2OH (TFE), and H2O (Fig. 7 and S32†). All these proton sources are mediating CO2 reduction, yet with variable efficiency. Notably, water as the cleanest proton source is tolerated and affords considerable yields of formic acid. Best performance was achieved with TFE, reaching FEs up to 82% within 2 h. These data indicate that formate is exclusively formed at the cathode and not anodically, e.g. by partial MeOH oxidation, and that MeOH is not required to achieve high formate yields. Moreover, the broad tolerance of a variety of proton sources provides ample opportunities for optimizing also the oxidation half-reaction for designing an efficient full cell electrolyzer.
 | ||
| Fig. 7 Comparison of the faradaic efficiencies (FE) for formate in a CO2-saturated solution containing 1 mM complex 6b with 0.5 M of different proton donors (in MeCN containing 0.1 M (Bu4N)PF6 as supporting electrolyte). Electrocatalytic CO2 reduction was performed at −1.9 V vs. Ag/AgCl for 2 h using glassy carbon working electrode; the formate yield was quantified by ion exchange chromatography (Fig. S32†). | ||
Total FE values below 100% suggest parasitic side reactions and catalyst deactivation, which might be triggered by several factors, including acidification of the reaction medium due to the generated formate, (cf. H+ reduction with high HOAc concentrations above). This limitation should be easily mitigated, for example, by using a flow reactor. Catalyst deactivation during CO2 electroreduction was evidenced by a combination of high-resolution scanning electron microscopy (HR-SEM), energy dispersive X-ray spectroscopy (EDX) and X-ray diffraction (XRD) analyses with an electrode containing complex 6b after 2 h of operation at −1.9 V vs. Ag/AgCl. Post-electrolytic HR-SEM and XRD analyses identified Ni oxide nanoparticles on the working electrode surface, while EDX suggests formation of films that are too thin for detection (Fig. S37†). Specifically, XRD revealed trace 2θ intensities that are characteristic of NiO and Ni2O3 on the electrode surface (Fig. S38†). Nickel oxide formation may be rationalized by partial reduction of the complexed nickel to Ni0 under the cathodic potential applied during electrolysis, which induces complex decomposition and formation of Ni nanoparticles. These electrochemically generated Ni nanoparticles were probably transformed to Ni oxides (NiO/Ni2O3) only when the electrode was exposed to air after the experiment. Such partial complex degradation thus provides a plausible rationale for the gradual loss of activity during extended reaction times and may account for the uncompensated charge from chronoamperometric experiments as some cathodic charge is involved in the in situ reduction of Nin+ to Ni0 during the CO2 reduction process. It must be noted, however, that Ni nanoparticles are unable to catalyze CO2 reduction and only induce H2 formation (HER), independent of size and shape of these nanoparticles.50 Therefore the highly selective reduction of CO2 to formate as observed here is confidently attributed to the distinct catalytic activity of the molecularly defined NHC Ni complexes.
The production of formate as valuable product from CO2 reduction is very rare for nickel-based catalysts, as most Ni systems produce CO.9 The best complex of the series, complex 6b shows high faradaic efficiency and exquisite formate vs. CO selectivity. The two other known Ni systems generating formate are less efficient in comparison and produce considerable amounts of CO as by-product (Table S11†).10,11 The triazolylidene system presented here offers unique potential, due to the intrinsically high selectivity towards formate production, and because the catalytic rate can be further optimized due to the correlation of rational ligand design and reduction potentials with catalytic activity. Moreover, a variety of proton sources are tolerated, including water. The largest drawback is probably the high overpotential required in comparison to the Kubiak-Sauvage electrocatalyst (ΔE = 0.5 V).11
The poor catalytic activity of the trans isomer points to a key role of the complex geometry. Either, the distortion from square-planar in the cis complexes may facilitate the formation of a penta-coordinate nickel(I) intermediate, or more likely, the cis-arrangement of the two oxygen donors produces a proton acceptor pocket, especially after one-electron reduction to either a formal nickel(I) complex or a phenoxide radical anion.51,52 Proton chelation by the two oxygen units is therefore suggested to form the reduced complex A (Scheme 2), presumably through a proton-coupled electron transfer (PCET). This intermediate facilitates the formation of a nickel(III) hydride intermediate B for CO2 insertion and generation of the formate complex C, which is then readily reduced to release the formate product. Formation of the nickel hydride intermediate is presumed to be key for the product selectivity,34–36,53–55 as most known nickel catalysts for CO2 reduction bind CO2 directly and therefore produce CO rather than formate.20,29–32,54–59 Alternatively, the proton scavenging may localize the negative charge on the oxygen, which facilitates the formation of the η1−OCO Ni adduct B′ as another potentially critical intermediate for formate formation.60 Attempts to characterize the putative nickel(I) intermediate by spectroelectrochemistry were not successful, as a solution of complex 3a or 4 did not produce any EPR signal after electrolysis at −2.0 V vs. Ag/AgCl, presumably because the nickel(I) species is not sufficiently stable in the absence of substrates. Nonetheless, this mechanistic proposal provides a rationale for the strong divergence of catalytic activity of the cis vs. trans complexes.
 | ||
| Scheme 2 Proposed mechanism for CO2 electroreduction with complexes 3 (and 6). | ||
Conclusions
In summary, we have synthesized and characterized a new class of bis-carbene nickel(II) complexes containing C,O-bidentate chelating phenolate-NHC ligands. These complexes are active in the selective electroreduction of CO2 to formate, reaching up to 83% faradaic efficiency, which is the highest value reported for a Ni-based electrocatalyst to date. Tailoring of the complexes substantially affects activity, with the cis isomers outperforming the trans analogue and a more nucleophilic nickel center achieving higher activities. The exquisite selectivity towards formate combined with the tolerance of a variety of proton sources hold great promises for optimizing also the anodic half-reaction and to develop a whole cell system for process operation. The unique aspects of this new class of complexes together with the viability of ligand modification, will provide new perspectives towards the design of novel electrocatalytic systems suitable for small molecules activation.Experimental section
General
2-Azidophenol 1, 1-(2-phenol)-imidazole 7 and 2-amino-4,6-di-tertbutylphenol were synthesized following procedures reported in literature.61–63 The synthesis of all triazolium salt ligand precursors (2a–c, 5a–b) is detailed in the ESI.† All other reagents were commercially available and used as received. Unless specified otherwise, NMR spectra were recorded at 25 °C with Bruker spectrometers operating at 300 or 400 MHz (1H NMR), and 100 MHz (13C NMR), respectively. Chemical shifts (δ in ppm, coupling constants J in Hz) were referenced to residual solvent signals (1H, 13C). Assignments are based on homo– and heteronuclear shift correlation spectroscopy. The purity of bulk samples of the complexes has been established by NMR spectroscopy, and by elemental analysis, which were performed at the University of Bern Microanalytic Laboratory by using a Thermo Scientific Flash 2000 CHNS–O elemental analyzer. Residual solvent was confirmed by NMR spectroscopy and also by X–ray structure determinations. High–resolution mass spectrometry was carried out with a Thermo Scientific LTQ Orbitrap XL (ESI–TOF).General procedure for synthesis of the nickel complexes 3a–c, 4 and 6a
The triazolium salt (0.5 mmol), K2CO3 (1 mmol) and NiCl2 (0.3 mmol) were suspended in dry MeCN under N2 atmosphere. The mixture was stirred at reflux temperature for 16 h and filtered through Celite (5 g). The filtrate was evaporated to dryness and the residue was extracted with CH2Cl2 and dried in vacuo. The residual powder was purified by column chromatography (Al2O3 basic; CH3CN/CH2Cl2 1:1). Single crystals suitable for X-ray diffraction analysis were obtained by slow diffusion of pentane into a CH2Cl2 solution of the complex.
Synthesis of cis-[Ni(trzBu^OPh)2] (3a)
According to the general procedure, reaction of 2a (200 mg, 0.52 mmol), NiCl2 (44 mg, 0.3 mmol), and K2CO3 (140 mg, 1 mmol) in MeCN (10 mL) afford 3a as a yellow crystalline powder (90 mg, 66%). 1H NMR (400 MHz, CD2Cl2): δ 7.68 (d, J = 8.2 Hz, 1H, HPhO), 7.14 (t, J = 8.2 Hz, 1H, HPhO), 7.01 (d, J = 8.2 Hz, 1H, HPhO), 6.56 (t, J = 8.2 Hz, 1H, HPhO), 3.98 (s, 3H, CH3–N), 2.40–2.22 (m, 1H, CtrzCH2), 2.03–1.83 (m, 2H, CtrzCH2CH2), 1.58–1.41 (m, 1H, CtrzCH2), 1.38–1.17 (m, 2H, CH2CH3), 0.73 (t, J = 6.5 Hz, 3H, CH3). 13C{1H} NMR (101 MHz, CD2Cl2): δ 159.92 (C–O), 146.40 (Ctrz–Bu), 141.66 (Ctrz–Ni), 129.73 (CPhO–N), 127.41 (CPhO–H), 122.35 (CPhO–H), 119.60 (CPhO–H), 113.16 (CPhO–H), 36.44 (CH3–N), 31.97 (CtrzCH2), 24.85 (CtrzCH2CH2), 22.78 (CH2CH3), 13.53 (CH3). HR-MS (ESI): calcd for C26H32O2N6Ni [M + Na]+m/z = 518.1935 (found 518.1916). Anal. calcd for C26H32O2N6Ni (519.28) × 0.5% CH2Cl2: C, 56.34; H, 5.89; N, 14.85. Found: C, 56.33; H, 5.67; N, 14.74.Synthesis of cis-[Ni(trzPh^OPh)2] (3b)
According to the general procedure starting from 2b (200 mg, 0.50 mmol), NiCl2 (42 mg, 0.3 mmol), and K2CO3 (140 mg, 1 mmol) in MeCN (10 mL) afforded complex 3b as a yellow crystalline powder (108 mg, 77%). 1H NMR (400 MHz, CD2Cl2): δ 7.70 (d, J = 8.5 Hz, 2H, HPh), 7.58 (d, J = 8.5 Hz, 1H, HPhO), 7.32–7.25 (m, 3H, HPh), 7.20 (d, J = 8.5 Hz, 1H, HPhO), 7.10 (t, J = 8.5 Hz, 1H, HPhO), 6.60 (t, J = 8.5 Hz, 1H, HPhO), 3.7 (s, 3H, N–CH3). 13C{1H} NMR (101 MHz, CD2Cl2): δ 159.37 (C–O), 145.07 (Ctrz–Ph), 144.41 (Ctrz–Ni), 130.15 (CPh–H), 129.67 (CPh–H), 129.40 (CPhO–N), 127.44 (CPh–H), 127.11 (CPhO–H), 126.88 (CPh–Trz), 122.70 (CPhO–H), 119.29 (CPhO–H), 113.55 (CPhO–H), 37.28 (N–CH3). HR-MS (ESI): calcd for C30H24N6NiO2 [M + Na]+m/z = 581.1206 (found 581.1205). Anal. calcd for C30H24N6NiO2 (559.26): C, 64.43; H, 4.33; N, 15.03. Found: C, 64.29; H, 4.08; N, 15.15.Synthesis of trans-[Ni(trzMes^OPh)2] (3c)
The reaction of triazolium salt 2c (200 mg, 0.45 mmol), NiCl2 (44 mg, 0.34 mmol) and K2CO3 (124 mg, 0.9 mmol) in MeCN (10 mL) according to the general procedure yielded complex 3c as a yellow crystalline powder (51 mg, 35%). 1H NMR (400 MHz, CD2Cl2): δ 7.80 (d, J = 8.5 Hz, 1H, HPhO), 7.02 (s, 2H, HMes), 6.64 (t, J = 8.5 Hz, 1H, HPhO), 6.35 (t, J = 8.5 Hz, 1H, HPhO), 4.97 (d, J = 8.5 Hz 1H, HPhO), 3.67 (s, 3H, CH3–N), 2.39 (s, 3H, CH3–Mes), 2.25 (s, 6H, CH3–Mes). 13C{1H} NMR (101 MHz, CD2Cl2): δ 157.79 (C–O), 148.98 (CPhO–H), 144.33 (Ctrz–Mes), 138.72 (Ctrz–Ni), 137.93 (CPhO-N), 128.40 (CMes–H), 127.59 (CPhO–H), 127.343 (CMes–CH3), 125.79 (CPhO–H), 122.91 (Ctrz–Mes), 118.23 (CPhO–H), 112.71 (CMes–CH3), 35.32 (CH3–N), 21.02 (CH3–Mes), 20.32 (CH3–Mes). HR-MS (ESI): calcd for C36H36N6NaNiO2 [M + Na]+m/z = 665.2130 (found 665.2145). Anal. calcd for C36H36N6NiO2 (643.42): C, 67.20; H, 5.64; N, 13.06. Found: C, 67.00; H, 5.98; N, 12.95.Synthesis of cis-[Ni(imiMe^OPh)2] (4)
According to the general procedure, reaction of 1-(2-phenol)-3-methyl-imidazolium iodide 9 (200 mg, 0.61 mmol), NiCl2 (42 mg, 0.3 mmol), and K2CO3 (200 mg, 1.4 mmol) in MeCN (10 mL) and purification by column chromatography (Al2O3; CH3CN/CH2Cl2 1:2) gave complex 4 as a yellow crystalline powder (90 mg, 70%). 1H NMR (400 MHz, CD2Cl2): δ 7.33–7.12 (m, 3H, 2HPhO + 1Himi), 7.03 (t, J = 8.5 Hz, 1H, HPhO), 6.92 (s, 1H, Himi), 6.61 (t, J = 8.5 Hz, 1H, HPhO), 3.19 (s, 3H, CH3–N). 13C{1H} NMR (101 MHz, CD2Cl2): δ 159.22 (C–O), 157.62 (Cimi–Ni), 127.77 (CPhO–N), 127.62 (CPhO–H), 124.06 (Cimi–H), 122.42 (CPhO–H), 118.23 (Cimi–H), 117.57 (CPhO–H), 113.64 (CPhO–H), 37.01 (CH3–N). HR-MS (ESI): calcd for C20H17N4NaNiO2 [M + Na]+m/z = 426.0597 (found 426.0603). Anal. calcd for C20H17N4NiO2 (404.08): C, 59.45; H, 4.24; N, 13.87. Found: C, 59.81; H, 3.95; N, 14.01.
Synthesis of cis-[Ni(trzBu^OPh(Me)2)2] (6a)
According to the general procedure, triazolium salt 5a (200 mg, 0.5 mmol), K2CO3 (140 mg, 1.0 mmol) and NiCl2 (42 mg, 0.3 mmol) were suspended in dry MeCN (10 mL). The residual powder was purified by column chromatography (Al2O3 basic; CH3CN/CH2Cl2 1:1) to afford complex 6a as a bright yellow crystalline solid (100 mg, 71%).1H NMR (400 MHz, CD2Cl2): δ 7.27 (s, 1H, HPh), 6.82 (s, 1H, HPh), 3.88 (s, 3H, N–CH3), 2.18 (s, 3H, CH3), 2.16 (s, 3H, CH3), 1.91 (s, b, 1H, CH–Pr), 1.81 (s, b, 1H, CH–Pr), 1.56 (s, b, 1H,CH–Et), 1.43 (s, b, 1H, CH–Et), 1.23–1.14 (m, 2H, CH2–CH3), 0.66 (t, J = 4 Hz, CH3). 13C{1H} NMR (101 MHz, CD2Cl2): δ 156.33 (C–O), 146.28 (Ctrz–Ph), 142.68 (Ctrz–Ni), 131.26 (CPh–H), 130.67 (CPh–H), 126.21 (CPhO–N), 121.10 (CPh–H), 117.13 (CPh–H), 36.47 (CH3–N), 32.25 (CH2–Pr), 24.99 (CH3), 22.98 (CH2–Et), 20.74 (CH3), 17.15 (CH2–CH3), 13.72 (CH3). HR-MS (ESI): calcd for C30H40N6NiO2 [M + H]+m/z = 575.2639 (found 575.2638). Anal. calcd for C30H24N6NiO2 (575.38): C, 62.62; H, 7.01; N, 14.61. Found: C, 62.33; H, 7.12; N, 14.54.
Synthesis of cis-[Ni(trzBu^OPh(tBu)2)2] (6b)
Triazolium salt 5b (300 mg, 0.61 mmol) was dissolved in 10 mL of THF in a Schlenk tube under inert atmosphere and the solution stirred at −78 °C for 5 min. After that time a 2.5 M solution of BuLi in hexane (0.56 mL, 1.40 mmol) was added, and the reaction mixture stirred for 30 min and then cannulated to another Schlenk tube containing NiCl2 (44 mg, 0.34 mmol) suspended in 5 ml of THF. The reaction mixture was stirred for 16 h at room temperature. After that time, the reaction was quenched, and the solvent removed under vacuum. DCM (30 mL) was added to the solid and the suspension was filtrated through a short Celite pad. The solvent was removed to leave a bright orange solid, which was then purified by column chromatography (basic Alox CH3CN/DCM 1:5) to obtain complex 6b as a dark yellow powder (280 mg, 62%). 1H NMR (400 MHz, CD2Cl2): δ 7.44 (d, J = 2.1 Hz, 1H, HPhO), 7.30 (d, J = 2.1 Hz, 1H, HPhO), 4.23 (s, 3H, CH3), 2.34 (t, J = 6.0 Hz, 2H, CH2–C3H7), 1.67 (s, b, 1H, CH2–C2H5), 1.47 (s, b, 1H, CH2–C2H5), 1.37–1.29 (m, 10H, (CH3)3–C + CH2–CH3), 1.28–1.24 (m, 10H, (CH3)3–C + CH2–CH3) 0.76 (t, 3H, J = 6.0 Hz, CH3). 13C{1H} NMR (100 MHz, CD2Cl2): δ 154.24 (C–O), 145.50 (CPh), 141.85 (CPh), 138.89 (CTrz–Ni), 128.53 (CPh–H), 126.22 (CPh–N), 122.42 (CTrz–Bu), 121.58 (CPh–H), 38.89 (C(CH)3), 36.19 (C(CH)3), 34.73 (CH3–N), 31.70 (CH3), 29.23 (CH3), 28.52 (CH2–C3H7), 23.49 (CH2–C2H5), 23.25 (CH2–CH3), 13.77 (–CH3). HR-MS (ESI): calcd for C42H64N6NiO2 [M + H]+m/z = 743.4517 (found 743.4495). Anal. calcd for C42H64N6NiO2 (743.70): C, 67.83; H, 8.67; N, 11.30. Found: C, 67.76; H, 8.88; N, 11.35.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank the Swiss National Science Foundation (200020_182663 and 200020_172507), the Marie Sklodowska Curie ITN NoNoMeCat (grant 675020-MSCA-ITN-2015-ETN), and the European Research Council (CoG 615653) for generous financial support of this work. We also thank the crystallography service unit of the University of Bern for analytical work (funded through SNSF R′equip 206021_128724). A.R. acknowledges support from the Ministry of Science and Higher Education of the Russian Federation.Notes and references
- M. Aresta, Carbon Dioxide as Chemical Feedstock, Wiley-VCH Verlag GmbH & Co. KGaA, 2010, Weinheim, Germany Search PubMed.
- C. Costentin, M. Robert and J.-M. Savéant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC.
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS PubMed.
- M. Mikkelsen, M. Jørgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 RSC.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114(3), 1709–1742 CrossRef CAS PubMed.
- W. Tu, Y. Zhou and Z. Zou, Adv. Mater., 2014, 26, 4607–4626 CrossRef CAS PubMed.
- J. P. Jones, G. K. S. Prakash and G. A. Olah, Isr. J. Chem., 2014, 54, 1451–1466 CrossRef CAS.
- Y. Matsubara, D. C. Grills and Y. Kuwahara, ACS Catal., 2015, 5, 6440–6452 CrossRef CAS.
- X. Min and M. W. Kanan, J. Am. Chem. Soc., 2015, 137, 4701–4708 CrossRef CAS PubMed.
- M. Rahaman, A. Dutta and P. Broekmann, ChemSusChem, 2017, 10, 1733–1741 CrossRef CAS PubMed.
- G. Reitz, J. Electrochem. Soc., 1985, 132, 8C CrossRef.
- K. P. Kuhl, T. Hatsukade, E. R. Cave, D. N. Abram, J. Kibsgaard and T. F. Jaramillo, J. Am. Chem. Soc., 2014, 136, 14107–14113 CrossRef CAS PubMed.
- D. Ren, Y. Deng, A. D. Handoko, C. S. Chen, S. Malkhandi and B. S. Yeo, ACS Catal., 2015, 5, 2814–2821 CrossRef CAS.
- M. Rahaman, A. Dutta, A. Zanetti and P. Broekmann, ACS Catal., 2017, 7, 7946–7956 CrossRef CAS.
- Y. Hori, K. Kikuchi and S. Suzuki, Chem. Lett., 1985, 14, 1695–1698 CrossRef.
- A. Dutta, C. E. Morstein, M. Rahaman, A. Cedeño López and P. Broekmann, ACS Catal., 2018, 8, 8357–8368 CrossRef CAS.
- A. S. Varela, W. Ju, A. Bagger, P. Franco, J. Rossmeisl and P. Strasser, ACS Catal., 2019, 9, 7270–7284 CrossRef CAS.
- B. Plietker, Iron Catalysis in Organic Chemistry: Reactions and Applications, 2008, John Wiley & Sons, NY Search PubMed.
- R. M. Bullock, Catalysis without precious metals, 2011, John Wiley & Sons, NY Search PubMed.
- N. Elgrishi, M. B. Chambers, X. Wang and M. Fontecave, Chem. Soc. Rev., 2017, 46, 761–796 RSC.
- (a) I. Bhugun, D. Lexa and J.-M. Savéant, J. Am. Chem. Soc., 1996, 118, 1769–1776 CrossRef CAS; (b) C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, Science, 2012, 338, 90–94 CrossRef CAS PubMed.
- M. Stanbury, J. D. Compain and S. Chardon-Noblat, Coord. Chem. Rev., 2018, 361, 120–137 CrossRef CAS.
- J. Gu, C. S. Hsu, L. Bai, H. M. Chen and X. Hu, Science, 2019, 364, 1091–1094 CrossRef CAS PubMed.
- Y. Sakaguchi, A. Call, M. Cibian, K. Yamauchi and K. Sakai, Chem. Commun., 2019, 55, 8552–8555 RSC.
- C. Steinlechner, A. F. Roesel, E. Oberem, A. Päpcke, N. Rockstroh, F. Gloaguen, S. Lochbrunner, R. Ludwig, A. Spannenberg, H. Junge, R. Francke and M. Beller, ACS Catal., 2019, 9, 2091–2100 CrossRef CAS.
- Notable exceptions: (a) H. Rao, L. C. Schmidt, J. Bonin and M. Robert, Nature, 2017, 548, 74–77 CrossRef CAS PubMed; (b) S. Roy, B. Sharma, J. Pécaut, P. Simon, M. Fontecave, P. D. Tran, E. Derat and V. Artero, J. Am. Chem. Soc., 2017, 139, 3685–3696 CrossRef CAS PubMed.
- For the use of CO as valuable product, see: T. Haas, R. Krause, R. Weber, M. Demler and G. Schmid, Nat. Catal., 2018, 1, 32–39 CrossRef CAS.
- For a recent overview, see: J.-W. Wang, W.-J. Liu, D.-C. Zhong and T.-B. Lu, Coord. Chem. Rev., 2019, 378, 237–261 CrossRef CAS.
- M. Beley, J. P. Collin, R. Ruppert and J.–P. Sauvage, J. Chem. Soc., Chem. Commun., 1984, 1315–1316 RSC.
- M. Beley, J. P. Collin, R. Ruppert and J.–P. Sauvage, J. Am. Chem. Soc., 1986, 108, 7461–7467 CrossRef CAS PubMed.
- J. P. Collin, A. Jouaiti and J.–P. Sauvage, Inorg. Chem., 1988, 27, 1986–1990 CrossRef CAS.
- T. Fogeron, T. K. Todorova, J.–P. Porcher, M. Gomez-Mingot, L.–M. Chamoreau, C. Mellot-Draznieks, Y. Li and M. Fontecave, ACS Catal., 2018, 8, 2030–2038 CrossRef CAS.
- J. D. Froehlich and C. P. Kubiak, Inorg. Chem., 2012, 51, 3932–3934 CrossRef CAS PubMed.
- C. A. Huff and M. S. Sanford, ACS Catal., 2013, 3, 2412–2416 CrossRef CAS.
- F. Bertini, M. Glatz, N. Gorgas, B. Stöger, M. Peruzzini, L. F. Veiros, K. Kirchner and L. Gonsalvi, Chem. Sci., 2017, 8, 5024–5029 RSC.
- F. Bertini, M. Glatz, B. Stöger, M. Peruzzini, L. F. Veiros, K. Kirchner and L. Gonsalvi, ACS Catal., 2019, 9, 632–639 CrossRef CAS.
- K. F. Donnelly, A. Petronilho and M. Albrecht, Chem. Commun., 2013, 49, 1145–1159 RSC.
- A. Vivancos, C. Segarra and M. Albrecht, Chem. Rev., 2018, 118, 9493–9586 CrossRef CAS PubMed.
- D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39–92 CrossRef CAS PubMed.
- W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290–1309 CrossRef CAS PubMed.
- F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3112–3172 Search PubMed.
- Y. Wei, A. Petronilho, H. Mueller-Bunz and M. Albrecht, Organometallics, 2014, 33, 5834–5844 CrossRef CAS.
- S. Hameury, P. de Frèmont and P. Braunstein, Chem. Soc. Rev., 2017, 46, 632–733 RSC.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 9, 955–964 RSC.
- C. J. Johnson and M. Albrecht, Organometallics, 2017, 36, 2902–2913 CrossRef CAS.
- J. M. Smieja, M. D. Sampson, K. A. Grice, E. E. Benson, J. D. Froehlich and C. P. Kubiak, Inorg. Chem., 2013, 52, 2484–2491 CrossRef CAS PubMed.
- C. Costentin, S. Drouet, M. Robert and J.–M. Savéant, J. Am. Chem. Soc., 2012, 134, 11235–11242 CrossRef CAS PubMed.
- Chronoamperometric measurements indicate a gradual decrease of the catalytic current when performing measurements over time periods larger than 10 min, which complicated the determination of catalytic efficiency, see ESI for details.†.
- A. V. Rudnev, U. E. Zhumaev, A. Kuzume, S. Vesztergom, J. Furrer, P. Broekmann and T. Wandlowski, Electrochim. Acta, 2016, 189, 38–44 CrossRef CAS.
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrochim. Acta, 1994, 39, 1833–1839 CrossRef CAS.
- P. Hemberger, G. da Silva, A. J. Trevitt, T. Gerbera and A. Bodi, Phys. Chem. Chem. Phys., 2015, 17, 30076–30083 RSC.
- E. R. Altwicker, Chem. Rev., 1967, 67, 475–531 CrossRef CAS.
- C. Gunanathan, Y. Ben-David and D. Milstein, Science, 2007, 317, 790–792 CrossRef CAS PubMed.
- S. Rösler, J. Obenauf and R. Kempe, J. Am. Chem. Soc., 2015, 137, 7998–8001 CrossRef PubMed.
- A. Mukherjee and D. Milstein, ACS Catal., 2018, 8, 11435–11469 CrossRef CAS.
- M. Fujihira, Y. Hirata and K. Suga, J. Electroanal. Chem., 1990, 292, 199–215 CrossRef CAS.
- J. A. Keith, K. A. Grice, C. P. Kubiak and E. A. Carter, J. Am. Chem. Soc., 2013, 135, 15823–15829 CrossRef CAS PubMed.
- N. J. Firet and W. A. Smith, ACS Catal., 2017, 7, 606–612 CrossRef CAS.
- L. Zhang, Z.–J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2017, 56, 11326–11353 CrossRef CAS PubMed.
- J. Song, E. L. Klein, F. Neese and Y. Shengfa, Inorg. Chem., 2014, 53, 7500–7507 CrossRef CAS PubMed.
- F. Dulong, O. Bathily, P. Thuery, M. Ephritikhine and T. Cantat, Dalton Trans., 2012, 41, 11980–11983 RSC.
- M. Liu, M. Nieger, E. G. Habner and A. Schmidt, Chem. – Eur. J., 2016, 22, 5416–5424 CrossRef CAS PubMed.
- Y. N. Kotovshchikov, G. V. Latyshev, M. A. Navasardyan, D. A. Erzunov, I. P. Beletskaya and N. V. Lukashev, Org. Lett., 2018, 20(15), 4467–4470 CrossRef CAS PubMed.
- Oxford Diffraction, CrysAlisPro (Version 1.171.38.41), Oxford Diffraction Ltd., Yarnton, Oxfordshire, UK, 2010 Search PubMed.
- P. Macchi, H. B. Bürgi, A. S. Chimpri, J. Hauser and Z. Gál, J. Appl. Crystallogr., 2011, 44, 763–771 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, A71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, C71, 3–8 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of all new triazolium salts, NMR spectra and cyclic voltammograms (CVs) of new compounds, NMR and CV experiments in acidic media, details on X-ray diffraction and on electrocatalytic set-up and product identification (PDF), crystallographic data for complexes 3a–c, 4, 6a–b (CIF). CCDC 2004181–2004184, 2050371 and 2050372. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1gc00388g |
| This journal is © The Royal Society of Chemistry 2021 |