
The facet-regulated oxidative dehydrogenation of lactic acid to pyruvic acid on α-Fe2O3†
Chunyu
Yin
a,
Xinli
Li
*a,
Yunsheng
Dai
b,
Zhi
Chen
a,
Dingfeng
Yang
a,
Ruixue
Liu
a,
Weixin
Zou
c,
Congming
Tang
 *a and
Lin
Dong
c
*a and
Lin
Dong
c
aSchool of Chemistry and Chemical Engineering, Chongqing University of Technology, Chongqing 400054, PR China. E-mail: lixinli@cqut.edu.cn; tcmtang2001@cqut.edu.cn
bCatalysis Sino-Platinum Metals Co., Ltd, Kunming 650106, PR China
cJiangsu Key Laboratory of Vehicle Emissions Control, Center of Modern Analysis, Nanjing University, Nanjing 210093, PR China
First published on 18th December 2020
Abstract
We propose a highly active α-Fe2O3 catalyst for the oxidative dehydrogenation of lactic acid to value-added pyruvic acid in air. The activity is determined by the utilized crystal face, due to the different adsorption energies of lactic acid molecules on different exposed planes. Furthermore, Fe sites show more preferential adsorption than crystal O sites.
Biomass and biomass derivatives, as renewable resources, are more desirable for the production of chemicals and fuels than fossil resources such as petroleum, coal and natural gas.1,2 Like ethene and acetylene, lactic acid (LA) can act as a platform molecule for producing various chemicals, such as pyruvic acid (PA),3,4 propionic acid,5 acetaldehyde,6 acrylic acid,7,8 2,3-pentanedione,9 and polylactic acid.10 Pyruvic acid (PA), a vital intermediate, is widely used in the chemical, pharmaceutical, food, agriculture, and environmental protection fields and in other areas.3 Compared with the traditional industrial production of PA from tartaric acid, which consumes a stoichiometric amount of potassium hydrogen sulfate and requires high energy, the direct catalytic synthesis of PA from LA via oxidative dehydrogenation is viewed as a potential environmentally friendly route.3,11
For the oxidative dehydrogenation of LA to PA, iron phosphates were used first, and M phases composed of Fe2+ and Fe3+ showed catalytic performance with LA conversion of 60% and PA selectivity of 62%.12,13 Later, metals and their oxides were investigated by several groups. Notably, a Pb–Pt bimetallic catalyst supported on carbon materials displayed better activity, achieving 70.7% LA conversion together with 80.1% PA selectivity.3 In addition, bimetallic oxides such as Nb–Ni–O11 and Mo–Ti–O4 have also been investigated for this reaction, with 30.5% LA conversion and 50.3% PA selectivity obtained with the former, and 60.6% LA conversion and 80.2% PA selectivity obtained with the latter. Recently, the activity for the oxidative dehydrogenation of LA to PA has been further enhanced using P-modified Fe–Mo bimetallic oxides, achieving 88.7% LA conversion and 75.3% PA selectivity.14 These investigations mainly focused on the relation between activity and chemical composition, and correlation between the crystal plane structure of the catalyst and the activity was not shown. Thus, continuing efforts are required to reveal the structure–activity relationship and gain a deep understanding of the mechanism of the oxidative dehydrogenation of LA to PA.
In recent years, nano-catalysts constructed with specific morphologies, particle sizes, and crystal structures have displayed promise whether applied in thermal catalysis15 or non-thermal catalysis, including in photocatalysis16–18 and electro-catalysis.19 Consequently, the basic principles relating to structure-selectivity correlation can be directly addressed. α-Fe2O316,17 is naturally abundant and environmentally benign, and it has bright prospects in the chemical industry. Herein, via catalyst characterization and first-principles calculations based on density functional theory, we discuss the selective oxidative dehydrogenation of LA on four regularly shaped α-Fe2O3 crystallites (nano-truncated hexagonal bipyramids (THB), nano-cubes (QC), nano-plates (HS), and nano-spheres (RC)) with well-defined facets and disclose the structure–activity relationship. The research findings will provide a very powerful method for the catalytic synthesis of oxy-organics from biomass and biomass derivatives.
In order to understand the true performances of the as-prepared catalysts, preliminary experiments were carried out to examine internal and external diffusion limits,20,21 and the results are shown in Fig. S1 and S2.† A catalyst particle size of 20–40 mesh along with a LA flow rate of 2 mL h−1 were selected to evaluate the catalytic performances of samples during the catalytic oxidative dehydrogenation of LA to PA, fully eliminating the influence of mass transfer. Initially, over α-Fe2O3 with different morphologies, the catalytic activity for the oxidative dehydrogenation of LA to PA was investigated, and the results are shown in Table 1 and Fig. S3a and b.† The conversion of LA drastically changed depending on the morphology of α-Fe2O3. LA was almost completely converted using RC and HS. However, the conversion of LA was less than 50% using THB and QC. Similar to the dependence of LA conversion on the morphology of α-Fe2O3, the PA selectivity decreased from 81.7% using RC to only 2.1% over THB. The formation of other products, such as acetaldehyde, acetic acid, and acrylic acid, was also influenced by the use of α-Fe2O3 with different morphologies (Table S1†). In terms of LA conversion and PA selectivity, RC offered more excellent performance than the other catalysts.
| Entry | Catalyst | Catalytic performance | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|---|
| LA conv. [%] | PA sel. [%] | k [h−1]a | k s [h−1 m−2]b | Stability [h] | SSA [m2 g−1]c | Dominant planed | |||
| a The apparent reaction rate constant (k) of the oxidative dehydrogenation of LA to PA on the α-Fe2O3 architectures is calculated based on a pseudo-first-order kinetics model. b k s represents the rate constant (k) normalized to the SSA. ks = k(catalyst mass × SSA)−1. c The specific surface area (SSA). d Dominant planes are confirmed using TEM and HRTEM. The experimental conditions in this work: catalyst, 0.30–0.40 g; reaction temperature, 230 °C; particle size, 20–40 mesh; carrier gas, air, 3 mL min−1; LA feedstock, 10 wt% in water; feed flow rate, 2 mL h−1; TOS, 4–5 h. | |||||||||
| 1 | 3Pb–1Pt/CB | 70.7 | 80.6 | N/A | N/A | N/A | 756 | N/A | 3 |
| 2 | Fe(PO4)3 | 60.2 | 62.3 | N/A | N/A | N/A | N/A | N/A | 12 and 13 |
| 3 | Mo/Fe(PO4)3 | 70.1 | 63.2 | N/A | N/A | N/A | N/A | N/A | 23 and 24 |
| 4 | Nb–Ni–O | 30.5 | 50.3 | N/A | N/A | 10 | 48 | N/A | 11 |
| 5 | Mo–Ti–O | 60.6 | 80.2 | N/A | N/A | N/A | N/A | N/A | 4 |
| 6 | FeMoO/P | 88.7 | 75.3 | N/A | N/A | 60 | 23.7 | N/A | 14 |
| 7 | THB | 36.7 | 2.1 | 1.27 | 0.23 | N/A | 12.82 | {113} | This work |
| 8 | QC | 43.1 | 4.2 | 1.28 | 0.49 | N/A | 7.97 | {012} | This work |
| 9 | HS | 94.0 | 55.3 | 7.49 | 0.95 | N/A | 21.26 | {001} | This work |
| 10 | RC | 94.6 | 81.7 | 13.13 | 2.02 | 100 | 16.91 | {001} | This work |
To evaluate the reactivities of the hematite architectures quantitatively, the apparent reaction rate constant (k)22 values of the conversion of LA to PA were calculated, and the results are also summarized in Table 1 and Fig. 1a. For the blank experiment without catalyst, the oxidative dehydrogenation of LA to PA was relatively slow at 230 °C with an apparent reaction rate constant k = 0.45 h−1. Under identical experimental conditions, the apparent reaction rates in the presence of catalysts were conspicuously elevated, and the ![[k with combining low line]](https://www.rsc.org/images/entities/i_char_006b_0332.gif) values were normalized to the specific surface area in order to explore the intrinsic catalytic activities, referred to ks.25,26 RC shows the highest catalytic activity, with ks = 2.02 h−1 m−2, while the ks values are 0.23, 0.49, and 0.95 h−1 m−2 for THB, QC, and HS, respectively. From the standpoint of industrial applications, stability testing is extremely significant during the study of heterogeneous catalysts.27 The stability testing of RC was carried out via controlling the initial LA conversion to less than 60% through reducing the amount of catalyst in order to realize the adequate saturation of catalyst active sites by reaction molecules and ensure authentic stability (Fig. 1b). We can clearly observe that LA conversion always remains at 50% and PA selectivity almost remains constant (∼81%) during 100 h on stream. It is generally difficult to obtain high conversion and high selectivity simultaneously. Thus, the stability at high levels of LA conversion was also investigated (Fig. S3c†). Encouragingly, at high LA conversion of 95%, the selectivity of PA still remained around 80% during 100 h on stream. With previously reported FeMoO/P,14 LA conversion was reduced by around 10% within 60 h and a longer reaction time was not studied. In comparison, the α-Fe2O3-RC catalyst displayed more excellent stability.
values were normalized to the specific surface area in order to explore the intrinsic catalytic activities, referred to ks.25,26 RC shows the highest catalytic activity, with ks = 2.02 h−1 m−2, while the ks values are 0.23, 0.49, and 0.95 h−1 m−2 for THB, QC, and HS, respectively. From the standpoint of industrial applications, stability testing is extremely significant during the study of heterogeneous catalysts.27 The stability testing of RC was carried out via controlling the initial LA conversion to less than 60% through reducing the amount of catalyst in order to realize the adequate saturation of catalyst active sites by reaction molecules and ensure authentic stability (Fig. 1b). We can clearly observe that LA conversion always remains at 50% and PA selectivity almost remains constant (∼81%) during 100 h on stream. It is generally difficult to obtain high conversion and high selectivity simultaneously. Thus, the stability at high levels of LA conversion was also investigated (Fig. S3c†). Encouragingly, at high LA conversion of 95%, the selectivity of PA still remained around 80% during 100 h on stream. With previously reported FeMoO/P,14 LA conversion was reduced by around 10% within 60 h and a longer reaction time was not studied. In comparison, the α-Fe2O3-RC catalyst displayed more excellent stability.
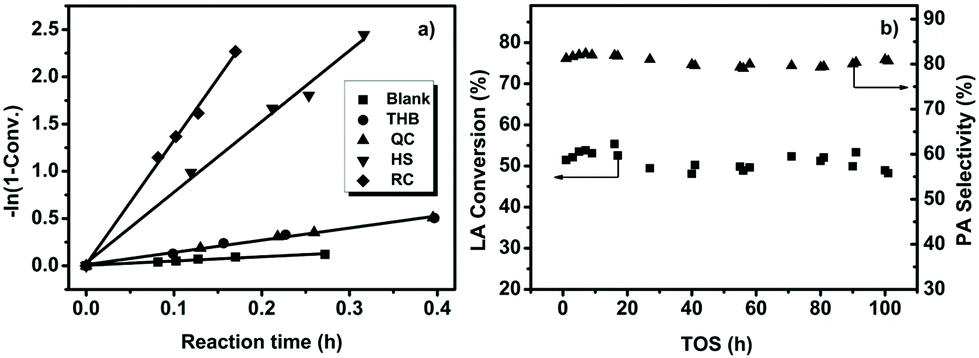 | ||
| Fig. 1 The excellent activity and stability of a-Fe2O3-RC. (a) A kinetics study of LA to PA conversion showing an activity order of RC > HS > QC > THB > blank. The error for each conversion test (after t h) was evaluated to be within ±2%. (b) The stability testing of a-Fe2O3-RC for LA to PA conversion; reaction conditions: reaction temperature, 230 °C; catalyst, 0.1 g; particle size, 20–40 mesh; carrier gas, air, 3 mL min−1; LA feedstock, 10 wt% in water; feed flow rate, 2 mL h−1. | ||
Why are the activities of α-Fe2O3 catalysts with different morphologies obviously different? We expected that α-Fe2O3 catalysts with different morphologies have different exposed crystal facets and that the Fe/O atomic ratios in these corresponding crystal facets are also different, resulting in different oxidation properties and reactivities. In order to further verify this assumption, the structures, morphologies, and oxidation states of the as-prepared α-Fe2O3 architectures were characterized. The different samples retained common characteristic diffraction peaks (Fig. 2a), suggesting phase purity in the form of α-Fe2O3 (JCPDS no. 33-0664). Furthermore, SEM images (Fig. 2b–e) show that the four samples present obviously different morphologies, namely, nano-truncated hexagonal bipyramids (THB), nano-cubes (QC), nano-plates (HS), and nano-spheres (RC).
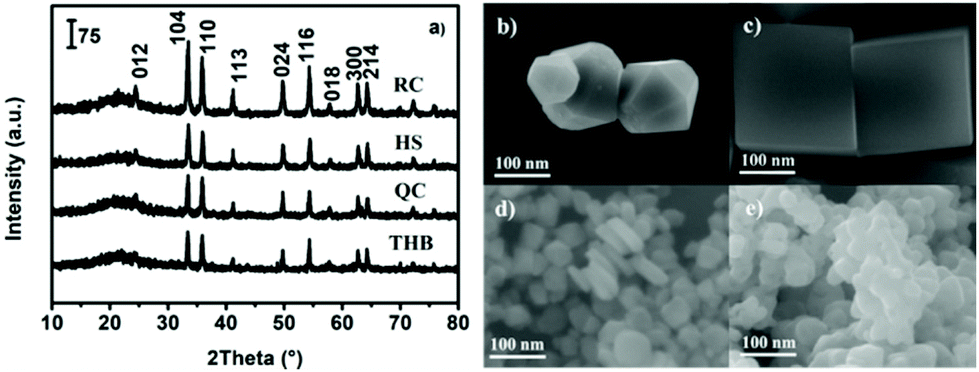 | ||
| Fig. 2 The XRD patterns of the α-Fe2O3 catalysts (a), and SEM images of (b) α-Fe2O3-THB, (c) α-Fe2O3-QC, (d) α-Fe2O3-HS, and (e) α-Fe2O3-RC. | ||
A TEM image of THB is shown in Fig. S4a† and an illustration of a structural model is given in Fig. S4b.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 28 All of the THB crystallites display a well-defined pseudo-hexagon shape. Based on SEM (Fig. 2b) and TEM analysis, the crystallites are seen to be composed of two truncated facets and twelve side planes. Classic HRTEM (Fig. S4b†) and fast-Fourier-transform (FFT; inset in Fig. S4b†) images show that four side facets were determined to be (−1−2−4), (124), (−1−24) and (12−4), belonging to {214}, the other eight side facets were confirmed to be (2−13), (−123), (−21−3), (−12−3), (1−2−3), (2−1−3), (−213), and (1−23), belonging to the {113}, and the two truncated facets belong to {104}. SEM (Fig. 2c) and TEM (Fig. S4c†) images indicate that the nano-cubes (QC) seem to be quasi-cubic in shape, and a diagram of a structural model is given in Fig. S4d.† HRTEM (Fig. S4d†) and fast-Fourier-transform (FFT; inset in Fig. S4d†) images show that QC consists of three pairs of parallel planes, and the lattice fringe are 0.25 nm and 0.22 nm. The dihedral angles between adjacent planes are 84°, 90°, and 96°. The above data show that QC had {012}, {104}, and {110} exposed.29 SEM (Fig. 2d) and TEM (Fig. S4e†) images reveal that the nano-plates (HS) seem to be hexagonally shaped, and an illustration of a structural model is given in Fig. S4f.† Based on SEM and TEM analysis of HS, the thickness and width are confirmed to be 460 ± 20.3 and 14.2 ± 2.1 nm, respectively. Representative HRTEM (Fig. S4f†) and fast-Fourier-transform (FFT; inset in Fig. S4f†) images show that HS involves (110), (−120), and (−210) planes. Also, the regular two hexagonal facets are confirmed to belong to {001}. The observed (Fig. S5†) vertically arranged plates are frequently wedge-shaped and the side surfaces are determined to belong to {110}.30Fig. 3 shows TEM and HRTEM (FFT; inset in Fig. 3b) images of RC, and the selected area electron diffraction (SAED) pattern is shown in the inset in Fig. 3a. RC displays well-defined roundness. Based on the SEM image (Fig. 2e), the crystallites comprise spheroidal particles. Furthermore, the TEM image (Fig. 3a) shows that the RC crystallites are composed of a large number of overlapping round particles. The microcrystals grow along the c-direction (the [001] axis),30 which is reflected in the SAED pattern (inset in Fig. 3a). The spherical surface is confirmed to belong to the {001} family of planes. HRTEM (Fig. 3b) and FFT (inset in Fig. 3b) images suggest that the three sets of lattice fringes (0.25 nm) match well with the (−120), (−210), and (110) planes. Therefore, the base plane of the α-Fe2O3 nano-spheres is {001}.16 The physical parameters of the prepared samples, including specific surface areas and main planes, are also summarized in Table 1.
28 All of the THB crystallites display a well-defined pseudo-hexagon shape. Based on SEM (Fig. 2b) and TEM analysis, the crystallites are seen to be composed of two truncated facets and twelve side planes. Classic HRTEM (Fig. S4b†) and fast-Fourier-transform (FFT; inset in Fig. S4b†) images show that four side facets were determined to be (−1−2−4), (124), (−1−24) and (12−4), belonging to {214}, the other eight side facets were confirmed to be (2−13), (−123), (−21−3), (−12−3), (1−2−3), (2−1−3), (−213), and (1−23), belonging to the {113}, and the two truncated facets belong to {104}. SEM (Fig. 2c) and TEM (Fig. S4c†) images indicate that the nano-cubes (QC) seem to be quasi-cubic in shape, and a diagram of a structural model is given in Fig. S4d.† HRTEM (Fig. S4d†) and fast-Fourier-transform (FFT; inset in Fig. S4d†) images show that QC consists of three pairs of parallel planes, and the lattice fringe are 0.25 nm and 0.22 nm. The dihedral angles between adjacent planes are 84°, 90°, and 96°. The above data show that QC had {012}, {104}, and {110} exposed.29 SEM (Fig. 2d) and TEM (Fig. S4e†) images reveal that the nano-plates (HS) seem to be hexagonally shaped, and an illustration of a structural model is given in Fig. S4f.† Based on SEM and TEM analysis of HS, the thickness and width are confirmed to be 460 ± 20.3 and 14.2 ± 2.1 nm, respectively. Representative HRTEM (Fig. S4f†) and fast-Fourier-transform (FFT; inset in Fig. S4f†) images show that HS involves (110), (−120), and (−210) planes. Also, the regular two hexagonal facets are confirmed to belong to {001}. The observed (Fig. S5†) vertically arranged plates are frequently wedge-shaped and the side surfaces are determined to belong to {110}.30Fig. 3 shows TEM and HRTEM (FFT; inset in Fig. 3b) images of RC, and the selected area electron diffraction (SAED) pattern is shown in the inset in Fig. 3a. RC displays well-defined roundness. Based on the SEM image (Fig. 2e), the crystallites comprise spheroidal particles. Furthermore, the TEM image (Fig. 3a) shows that the RC crystallites are composed of a large number of overlapping round particles. The microcrystals grow along the c-direction (the [001] axis),30 which is reflected in the SAED pattern (inset in Fig. 3a). The spherical surface is confirmed to belong to the {001} family of planes. HRTEM (Fig. 3b) and FFT (inset in Fig. 3b) images suggest that the three sets of lattice fringes (0.25 nm) match well with the (−120), (−210), and (110) planes. Therefore, the base plane of the α-Fe2O3 nano-spheres is {001}.16 The physical parameters of the prepared samples, including specific surface areas and main planes, are also summarized in Table 1.
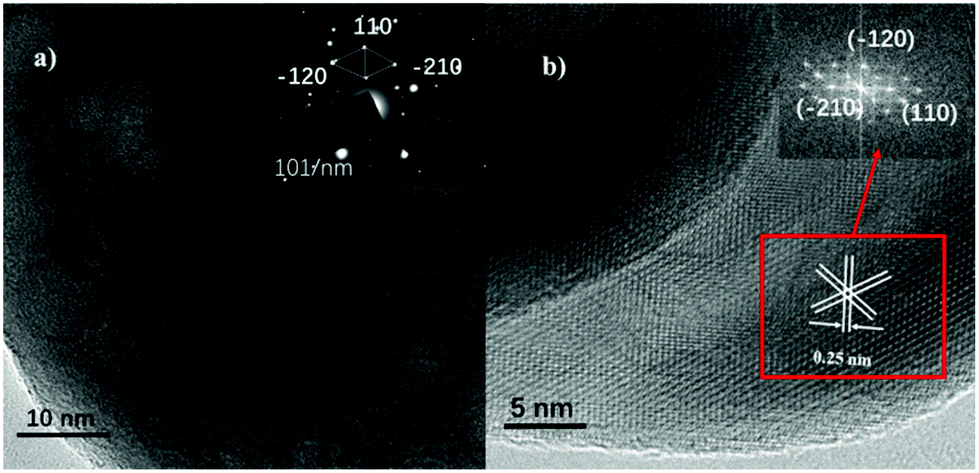 | ||
| Fig. 3 (a) A TEM image of α-Fe2O3-RC; inset: the SAED pattern. (b) A HRTEM image of α-Fe2O3-RC; inset: the FFT pattern. | ||
THB has the {113}, {214}, and {104} families of planes exposed, and they take up 4/7, 2/7, and 1/7 of the whole surface, respectively, judging from the area fraction of each crystal surface.28 QC has the {104}, {012}, and {110} families of planes exposed, and each occupies 1/3 of the whole surface.28 HS has the {001} and {110} families of planes exposed, and they take up 7/9 and 2/9 of the whole surface, respectively.28 RC has the {001} facets exposed, exclusively. Thus, the dominant exposed planes on the four α-Fe2O3 catalysts, THB, QC, HS and RC, are {113}, {012}, {001}, and {001}, respectively. The order of catalytic activities of the four α-Fe2O3 catalysts is THB < QC < HS < RC. It can be clearly concluded that the {001} facet is active for the oxidative dehydrogenation of LA to PA.
It is necessary for us to further study the relationship between activity and facets in order to understand why the {001} facet is active for the production of PA from LA. The adsorption energies of LA molecules on various α-Fe2O3 surfaces were evaluated via DFT calculations, and the results are shown in Table S2 and Fig. S6a.† The surface adsorption energy of LA on the {001} facet is −0.39 eV, which is clearly far lower than on the other {113} and {012} facets, which intensifies the adsorption. Lactic acid is adsorbed over α-Fe2O3, and the lower the energy required, the easier it is to adsorb it on the surface of the catalyst. Further calculations relating to the adsorption energy of LA molecules on different α-Fe2O3 {001} sites were performed, and the results are depicted in Tables S3, 4 and Fig. S6b–e.† The optimized adsorption structures of LA at different adsorption sites on α-Fe2O3 (001) facets show two stable adsorption configurations, A (Fe–O) and B (O–H); the adsorption energy of configuration A is lower than that of B, suggesting that type A shows preferential adsorption. It is noted that the ratios of Fe/O on the facets are 0.31, 0.46, and 0.60 for the {113}, {012}, and {001}, respectively, as shown in Table 2. This further suggests that the {001} facet is more beneficial for the adsorption of LA molecules than the other facets. The electronic structures of four fresh as-prepared samples were explored via diffuse-reflectance UV-vis spectra and derived Tauc plots. Fig. S7a† shows that all α-Fe2O3 samples possess comparable absorption edges at around 550–600 nm. The band gaps (Eg) are confirmed to be 2.13, 2.10, 2.14, and 2.15 eV for THB, QC, HS, and RC, respectively. A larger band gap indicates a smaller particle size in a sample, producing a larger specific area and favoring the transport of hot electrons. According to empirical formulae ECB = X − Ec − 1/2Eg (1) and EVB = ECB + Eg (2), where ECB is the conduction band (CB) energy, EVB is the valence band energy (VB), X is the absolute electronegativity of α-Fe2O3, and Ec is a constant relative to the NHE. EVB values of 2.455, 2.440, 2.460, and 2.465 eV are obtained for THB, QC, HS, and RC, respectively, when using Ec = 4.50 eV (as seen in Table S5†). A larger EVB value suggests stronger oxidizability. The binding energy of RC is the highest among the four catalysts from the Fe 2p XPS spectra shown in Fig. S7b,† and it has the strongest oxidation performance accordingly. At the same time, type A is the preferred adsorption configuration. As a result, an adsorbed LA molecule can be effortlessly oxidized by the Fe(III) ion located in this suitable position. Of the four catalysts, RC revealed the lowest-temperature reduction peak, centered at 385 °C, based on the H2-TPR results shown in Fig. S7c,† demonstrating that hydrogen easily reduces surface Fe–O of the catalyst. Similarly, a proton transferred from the −OH group of LA easily combines with surface Fe–O of the catalyst, leading to the formation of the pyruvic acid product via the removal of H2O, promoting the oxidative dehydrogenation of LA during the catalytic reaction.
| Atomic density [nm−2] | |||
|---|---|---|---|
| {113} | {012} | {001} | |
| Surface O | 8.4 | 10.8 | 7.7 |
| Surface Fe | 2.6 | 4.7 | 4.6 |
| Fe/O ratio | 0.31 | 0.46 | 0.60 |
On the basis of the above observations and DFT calculations relating to the adsorption of lactic acid molecules on the (001) facet, we propose a mechanism for the oxidative dehydrogenation of LA to PA (Scheme 1). The adsorption energies of iron and oxygen located at different positions on the (001) plane toward the hydroxyl and carboxyl functional groups of lactic acid molecules were calculated (detailed adsorption models and results are shown in Fig. S6a–e and Tables S2–4†). It is found that the top iron species (Fe3+) attacks the hydroxyl group of the lactic acid molecule with the lowest adsorption energy, indicating that lactic acid molecules are adsorbed and activated at the top iron site (1A); then, the O–H bond is broken and H+ is transferred to the adjacent oxygen to the iron site to form a new Fe-OH group. In this process, the enhancement of the iron oxidation state contributes significantly to activating the hydroxyl group. Among the four differently shaped α-Fe2O3 catalysts, the iron species in RC have a higher electron binding energy, which means stronger oxidizability. At the same time, UV-vis spectroscopy and valence band analysis show that RC has a higher valence band, indicating stronger oxidation, which is consistent with the XPS results. In addition, RC mainly exposed the (001) crystal plane, which has a higher Fe/O atomic ratio; this also is conducive to the Fe site adsorbing and activating the hydroxyl groups of lactic acid molecules. Then, the formed Fe-OH attacks C–H to break the C–H bond and form PA. At the same time, the dissociated H+ ion combines with Fe-OH to form water. Also, Fe3+ is reduced to Fe2+, which is supported by H2-TPR results, as RC has a lower-temperature reduction peak, and this is conducive to C–H activation and dissociation. The iron oxides with other morphologies, such as THB and QC, have higher-temperature reduction peaks, which are not conducive to the process. An increase in the reaction temperature is beneficial for enhancing the departure of oxygen from the iron oxide surface and promoting the process. In order to verify this assumption, we enhanced the reaction temperatures to observe the activity on THB and QC. As a result, the catalytic performances of THB and QC were improved (Table S6†), which demonstrated that this step has an extensive influence on the oxidative dehydrogenation of lactic acid. Fe2+ is oxidized to Fe3+ by O2 in air. Meanwhile the lost lattice oxygen is replenished. The catalyst returns to its initial state, completing the catalytic cycle. In the blank experiment, PA was barely detected under conditions involving lactic acid and air without catalyst, indicating that molecular oxygen could not directly oxidize LA to PA. In addition, using only lactic acid and catalyst, PA was observed, but the amount of PA decreased rapidly over time without air, indicating that the process of the oxidative dehydrogenation of PA consumed lattice oxygen from the catalyst. The departure of lattice oxygen directly affects the oxidation dehydrogenation of LA, and it is related to the exposed crystal surface of iron oxide. Therefore, the crystal surface of iron oxide can effectively regulate the activity during the oxidative dehydrogenation of lactic acid.
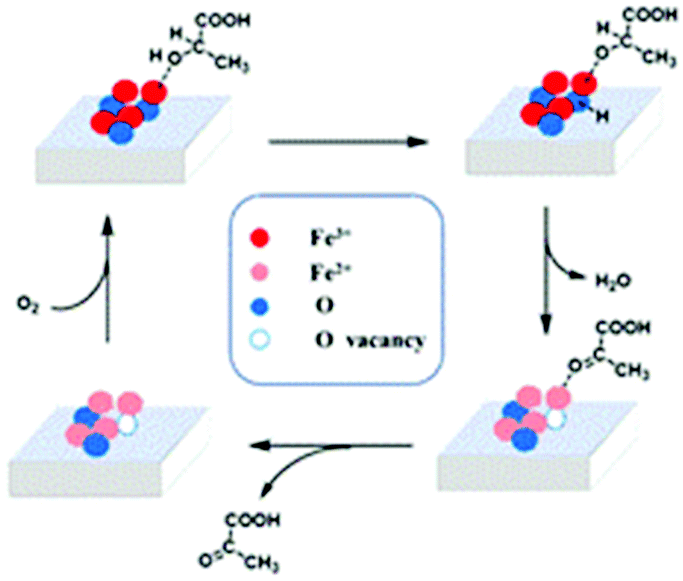 | ||
| Scheme 1 The reaction mechanism for the oxidative dehydrogenation of LA to PA over the RC catalyst. | ||
Comparing previous work focusing on amorphous and non-stoichiometric catalysts for the oxidative dehydrogenation of LA to PA,3,4,31 this work reveals that the structure of the thermo-catalyst on the nanoscale, with the exclusive construction of certain facets, is crucial for the design of high-performance nano-catalysts. The arrangement and coordination states of surface atoms have an important influence on the surface properties, including stability, hydrophilicity, adsorption capacity, etc., and these properties determine the catalytic activity.
Conclusions
In summary, the oxidative dehydrogenation of LA to PA with O2 at 230 °C over hematite is powerfully structurally dependent, and the reactivity trend can be reasonably interpreted as depending on the exposed facets in the sequence: {001} > {012} > {113 }. Hematite α-Fe2O3 (RC with dominantly exposed {001}) exhibited remarkable activity and stability for the oxidative dehydrogenation of LA to PA. This study demonstrates a prospective approach for the thermo-catalytic conversion of lactic acid. In addition, this study has broadened the applications of hematite α-Fe2O3 in areas relating to biomass and biomass derivatives.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22078033), Opening Foundation of Jiangsu Key Laboratory of Vehicle Emissions Control (OVEC 049), the fund of the State Key Laboratory of Advanced Technologies for Comprehensive Utilization of Platinum Metals (SKL-SPM-202010), Youth/Key Project of Science and Technology Research Program of Chongqing Education Commission of China (KJZD-K201901104, KJQN201801109), Natural Science Foundation of Chongqing (cstc2019jcyj-msxmX0198), and Special Project of Technology Innovation and Application Development of Chongqing (cstc2019jscx-msxmX0002).Notes and references
- L. J. Lei, Y. H. Wang, Z. X. Zhang, J. H. An and F. Wang, ACS Catal., 2020, 10, 8788–8814 CrossRef CAS.
- M. Sayed, N. Warlin, C. Hulteberg, I. Munslow, S. Lundmark, O. Pajalic, P. Tuna, B. Z. Zhang, S. H. Pyo and R. Hatti-Kaul, Green Chem., 2020, 22, 5402–5413 RSC.
- C. Zhang, T. Wang and Y. Ding, Appl. Catal., A, 2017, 533, 59–65 CrossRef CAS.
- K. Liu, X. Huang, E. A. Pidko and E. J. M. Hensen, Green Chem., 2017, 19, 3014–3022 RSC.
- X. L. Li, J. Pang, J. Zhang, C. Y. Yin, W. X. Zou, C. M. Tang and L. Dong, Ind. Eng. Chem. Res., 2019, 58, 101–109 CrossRef CAS.
- C. Tang, Z. Zhai, X. Li, L. Sun and B. Wei, J. Catal., 2015, 329, 206–217 CrossRef CAS.
- J. Peng, X. Li, C. Tang and W. Bai, Green Chem., 2014, 16, 108–111 RSC.
- B. Yan, L.-Z. Tao, Y. Liang and B.-Q. Xu, ACS Catal., 2014, 4, 1931–1943 CrossRef CAS.
- X. L. Li, L. W. Sun, W. X. Zou, P. Cao, Z. Chen, C. M. Tang and L. Dong, ChemCatChem., 2017, 9, 4621–4627 CrossRef CAS.
- X. H. Zhao, K. Li, Y. Wang, H. Tekinalp, G. Larsen, D. Rasmussen, R. S. Ginder, L. Wang, D. J. Gardner, M. Tajvidi, E. Webb and S. Ozcan, ACS Sustainable Chem. Eng., 2020, 8, 13236–13247 CrossRef CAS.
- S. Lomate, T. Bonnotte, S. Paul, F. Dumeignil and B. Katryniok, J. Mol. Catal. A: Chem., 2013, 377, 123–128 CrossRef CAS.
- M. Ai and K. Ohdan, Appl. Catal., A, 1997, 165, 461–465 CrossRef CAS.
- M. Ai and K. Ohdan, Appl. Catal., A, 1997, 150, 13–20 CrossRef CAS.
- C. Yin, X. Li, Z. Chen, W. Zou, Y. Peng, S. Wei, C. Tang and L. Dong, New J. Chem., 2020, 44, 5884–5894 RSC.
- G. Liu, A. W. Robertson, M. M. Li, W. C. H. Kuo, M. T. Darby, M. H. Muhieddine, Y. C. Lin, K. Suenaga, M. Stamatakis, J. H. Warner and S. C. E. Tsang, Nat. Chem., 2017, 9, 810–816 CrossRef CAS.
- X. Zhou, J. Lan, G. Liu, K. Deng, Y. Yang, G. Nie, J. Yu and L. Zhi, Angew. Chem., Int. Ed., 2012, 51, 178–182 CrossRef CAS.
- X. Zhou, Q. Xu, W. Lei, T. Zhang, X. Qi, G. Liu, K. Deng and J. Yu, Small, 2014, 10, 674–679 CrossRef CAS.
- K. Sivula, F. Le Formal and M. Gratzel, ChemSusChem., 2011, 4, 432–449 CrossRef CAS.
- Y. Z. Zhang, Q. Y. Chen, L. F. Liu, Y. Wang and M. K. H. Leung, Chem. Eng. J., 2020, 399, 11 Search PubMed.
- S. T. Oyama, X. M. Zhang, J. Q. Lu, Y. F. Gu and T. Fujitani, J. Catal., 2008, 257, 1–4 CrossRef CAS.
- A. Sachse, V. Hulea, A. Finiels, B. Coq, F. Fajula and A. Galarneau, J. Catal., 2012, 287, 62–67 CrossRef.
- Z. R. Sheng, Y. Watanabe, H. H. Kim, S. L. Yao and T. Nozaki, Chem. Eng. J., 2020, 399, 14 CrossRef.
- M. Ai, Appl. Catal., A, 2002, 234, 235–243 CrossRef CAS.
- K. Jerabek, L. Hankova, Z. Prokop and E. G. Lundquist, Appl. Catal., A, 2002, 232, 181–188 CrossRef CAS.
- J. J. Erbs, B. Gilbert and R. L. Penn, J. Phys. Chem. C, 2008, 112, 12127–12133 CrossRef CAS.
- A. J. Anschutz and R. L. Penn, Geochem. Trans., 2005, 6, 60–66 CrossRef CAS.
- P. Liu and E. J. Hensen, J. Am. Chem. Soc., 2013, 135, 14032–14035 CrossRef CAS.
- L. Gu, Q. Su, W. Jiang, Y. Yao, Y. Pang, W. Ji and C.-T. Au, Catal. Sci. Technol., 2018, 8, 5782–5793 RSC.
- G. S. Park, D. Shindo, Y. Waseda and T. Sugimoto, J. Colloid Interface Sci., 1996, 177, 198–207 CrossRef CAS.
- L. Chen, X. Yang, J. Chen, J. Liu, H. Wu, H. Zhan, C. Liang and M. Wu, Inorg. Chem., 2010, 49, 8411–8420 CrossRef CAS.
- C. Gao, J. Qiu, J. Li, C. Ma, H. Tang and P. Xu, Bioresour. Technol., 2009, 100, 1878–1880 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc03468a |
| This journal is © The Royal Society of Chemistry 2021 |