
Waste-minimized synthesis of C2 functionalized quinolines exploiting iron-catalysed C–H activation†
Francesco
Ferlin
 ,
Agnese
Zangarelli
,
Simone
Lilli
,
Stefano
Santoro
and
Luigi
Vaccaro
*
,
Agnese
Zangarelli
,
Simone
Lilli
,
Stefano
Santoro
and
Luigi
Vaccaro
*
Laboratory of Green S.O.C. – Dipartimento di Chimica, biologia e Biotecnologie, Università degli Studi di Perugia, Via Elce di Sotto 8, 06123 – Perugia, Italy. E-mail: luigi.vaccaro@unipg.it; Web: http://www.dcbb.unipg.it/greensoc
First published on 26th November 2020
Abstract
Herein we present an efficient and regioselective iron-catalyzed methodology for the external oxidant-free functionalization of quinoline-N-oxides. The protocol, based on the use of inexpensive and easily accessible FeSO4, showed broad applicability to a wide range of substrates. An additional green feature of this synthetic methodology is H2O being the only by-product. Experimental and computational investigations provide support to a mechanism based on a facile C–H activation event. The green efficiency of the process has also been carefully assessed using: (i) metrics related to the synthetic process (AE, Yield, 1/SF, MRP and RME); (ii) safety/hazard metrics (SHZI and SHI); and (iii) metrics related to the metal used as the catalyst (Abundance, OEL and ADP). In addition to the many advantages of this protocol related to the green iron catalyst used and the safety/hazard features of the process, an E-factor value of ca. 0.92 (84 to >99% reduction compared to known protocols) evidently confirms the sustainable efficiency of the procedure presented. Practical utility has also been demonstrated by performing the reaction efficiently on a multi-gram scale.
Introduction
The direct C–H functionalization of quinoline-N-oxides represents an interesting synthetic strategy,1 offering the possibility of a direct modification of an important heterocyclic moiety that can be found in different bioactive molecules (Scheme 1a) and natural products.2 Moreover, quinoline-N-oxides also offer the opportunity to realize “external oxidant-free” oxidative functionalizations, exploiting the inherent ability of N-oxides to reoxidize the catalyst, apart from serving as a weakly-coordinating directing group.3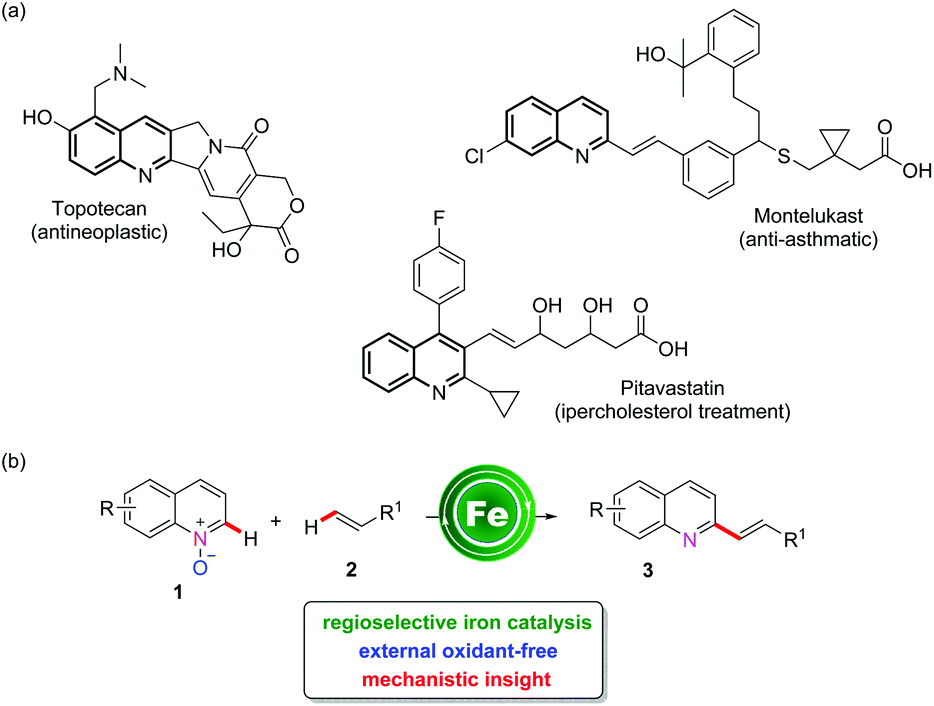 | ||
| Scheme 1 Examples of biologically active molecules with a quinoline core and features of iron-catalyzed C–H alkenylation. | ||
The direct C–H functionalization of quinoline-N-oxides has been mainly reported using different transition metal catalysts.4 Among these, palladium,3b,e rhodium,5 copper,6 and cobalt7 catalysts have been exploited to achieve either C2 or C8 olefination of quinoline-N-oxide, and in a unique case, C2 alkenylation has been performed using a heterogeneous catalytic system.3a In addition, alkenylation strategies also comprise metal-free or Brønsted acid catalyzed protocols.8 In recent years, there has been growing interest in the development of efficient catalytic processes based on 3d transition metals, in place of the more expensive 4d and 5d elements.9 From this perspective, iron represents an ideal choice, as one of the most abundant elements in the earth's crust and being associated with very low toxicity.10 Excellent recent contributions have reported the efficiency of iron to catalyze C–H functionalization processes, including allylation,11 alkylation,12 arylation,13 alkynylation14 and amination reactions.15 Despite these considerable advances, the iron-catalyzed C–H bond alkenylation reaction remains limited to a few examples,16 and specifically to the use of waste-producing organo-zinc or Grignard reagents, but with no possibility for direct C–H bond alkenylation. In our program aimed at the development of sustainable C–H activation reactions,17 we are also interested in the use of earth-abundant non-toxic transition metals, and in this contribution we report our result on the development of the first iron-catalyzed C2-selective alkenylation of quinoline-N-oxides (Scheme 1b). In terms of sustainability, this synthetic methodology features H2O as the sole by-product with excellent atom economy.
Results and discussion
We initially investigated various reaction conditions for the envisioned iron-catalyzed C–H alkenylation of quinoline-N-oxide 1a with methyl acrylate 2a (Table 1). After considerable preliminary experimentation (see also the ESI† for additional data), we observed that FeSO4 is the most viable catalyst for the desired C–H alkenylation of 1a. The optimal results were obtained using as little as 1 mol% of FeSO4 in a highly concentrated toluene solution [20 M] at 100 °C for 16 h (Table 1, entry 1). Very importantly, the reaction afforded selectively the C2-alkenylated quinoline (3a).18|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Cat. | C [%] | Selectivity (3a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4a) :4a) |
3a [%] |
| a Reaction conditions: 1a (1 mmol), 2a (2 mmol), catalyst (1 mol%), solvent (50 μL, 20 M), 16 h at 100° C. b GLC conversion has been determined using samples of pure compounds as reference standards. c Isolated yield. d Degradation products have been detected. e Iron(II) phthalocyanine. | |||||
| 1 | Toluene | FeSO4 | 98 | 100:0 |
86 |
| 2 | Solvent-free | FeSO4 | 83 | 95:5 |
77 |
| 3 | DMF | FeSO4 | 90 | 52:48 |
30 |
| 4 | Dioxane | FeSO4 | 68 | 45:55 |
18 |
| 5 | Toluene | — | 30 | 15:85 |
9 |
| 6 | Toluene | FeCl3 | 95 | 52:48 |
15 |
| 7 | Toluene | CuSO4 | 82 | 5:95 |
— |
| 8 | Toluene | Cu(OAc)2 | 84 | 17:83 |
13 |
| 9 | Toluene | Pd(OAc)2 | 56 | 60:40 |
30d |
| 10 | Toluene | CoSO4 | 91 | 50:50 |
23 |
| 11 | Toluene | BrMn(CO)5 | 98 | 25:75 |
18 |
| 12 | Toluene | FePce | 68 | 85:15 |
12 |
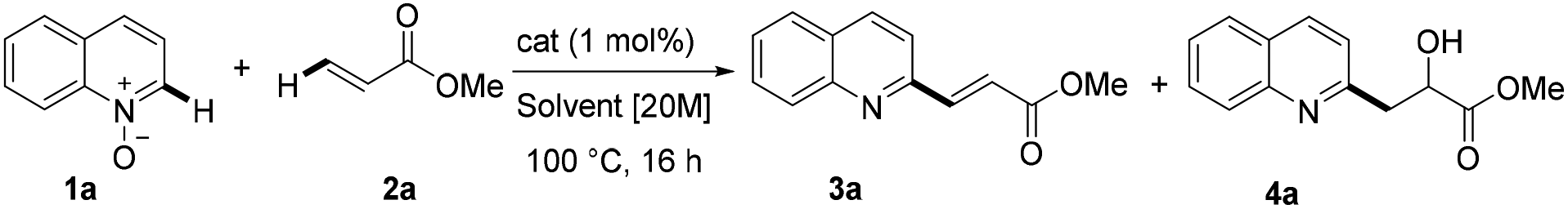
Performing the reaction under solvent-free conditions or by changing the medium to DMF or 1,4-dioxane resulted in lower yields and most importantly with lower selectivity. In fact, in the latter cases, the formation of the hydroxylated product was observed, probably derived from an alternative cycloaddition/ring opening sequence.8 Conducting the reaction without the catalyst led to very poor conversion. The use of different iron, copper, palladium, cobalt or manganese catalysts resulted in variable conversions and poor selectivities and isolated yields.
With the optimized reaction conditions in hand, we explored the reaction scope (Scheme 2) by reacting a variety of quinoline-N-oxides (1) with different acrylates (2).
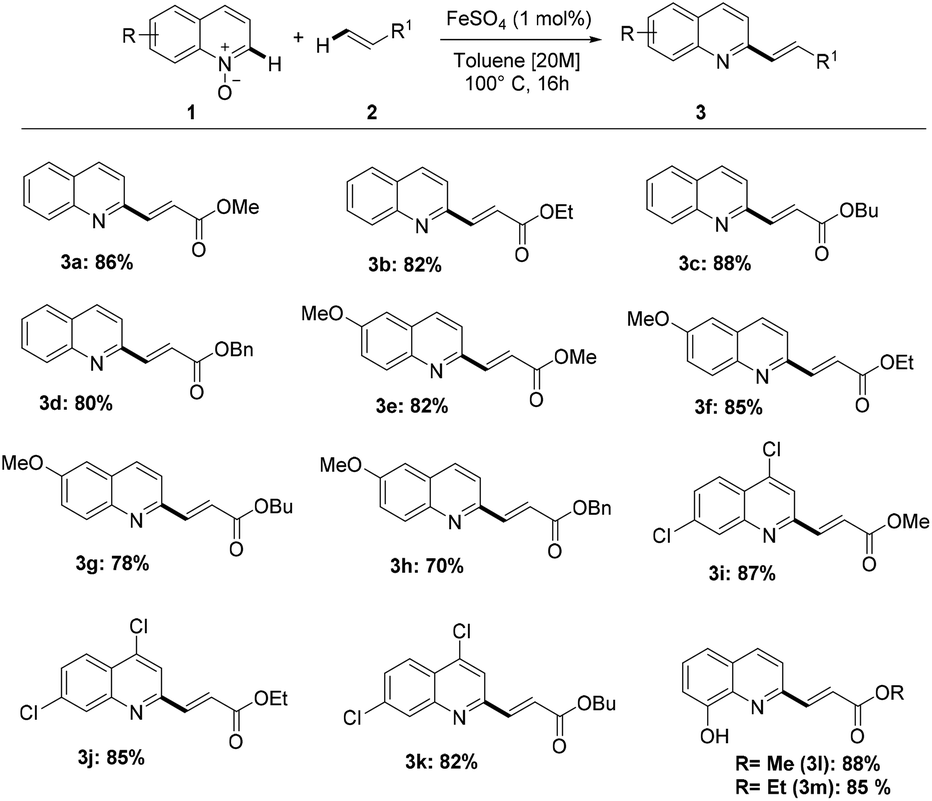 | ||
| Scheme 2 Scope of C–H alkenylation of quinoline-N-oxides 1 with acrylates 2. | ||
Gratifyingly, quinoline-N-oxides containing either electron-donating or electron-withdrawing groups were olefinated regioselectively in good yields. Substitution at the C6 and C8 positions by either methoxy or hydroxy groups and 4,7-dichlorosubstitution were tolerated, giving good results in terms of selectivity and yield. It is worth noting that methoxy, chloride, and hydroxy groups are versatile functionalities that allow further transformations. Also, changing the alkyl substituent on the acrylate only resulted in marginal differences in product yields.
Next, we investigated whether the methodology is compatible with different alkenes, other than acrylates. Indeed, the reaction is very efficient with substituted or unsubstituted styrene derivatives, showing also in this case excellent compatibility with different functionalities present on quinoline-N-oxide; in this protocol, acid labile substrates (6m and 6n) have also been obtained in excellent yields (Scheme 3).
 | ||
| Scheme 3 Scope of C–H alkenylation of quinoline-N-oxides 1 with styrene 5. | ||
Furthermore, the methodology also enabled the direct alkenylation of isoquinoline-N-oxide, with an excellent level of selectivity, allowing to obtain 1-styrylisoquinoline 6k. Notably, this iron-catalyzed process has been found to allow the direct alkenylation of quinoxaline-N-oxide 7, and the methodology herein reported offers, to the best of our knowledge, a very rarely explored and step economical access to C2-functionalized quinoxalines (Scheme 4).
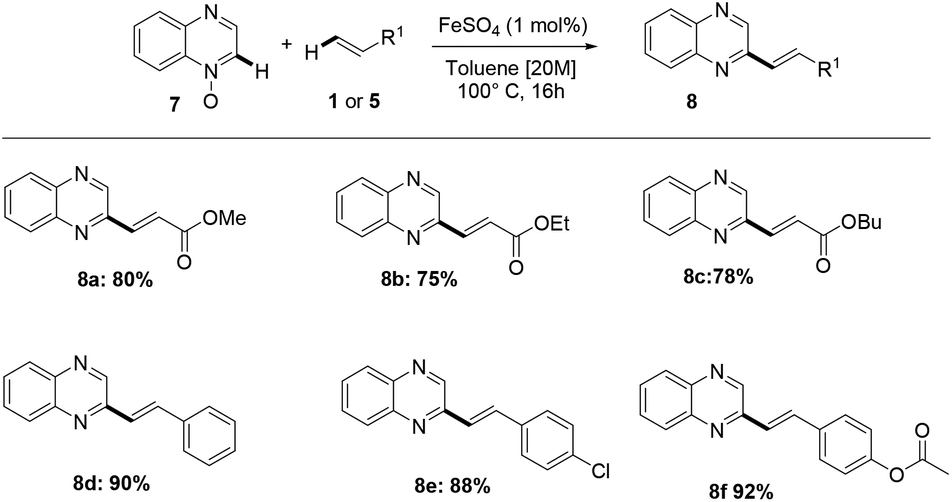 | ||
| Scheme 4 Scope of C–H alkenylation of quinoxalines 7 and acrylates 2 or styrene 5. | ||
The practical usefulness of the methodology is also demonstrated by its prompt scalability, which allowed us to perform multigram-scale reactions on two different substrates, further improving the associated yields (Scheme 5).
 | ||
| Scheme 5 Gram-scale reactions. | ||
Mechanistic studies of iron-catalyzed reactions are particularly challenging, for both experimental and computational methods. Intrigued by the efficiency of the proposed methodology, we decided to investigate its mechanism (Scheme 6). First, we heated substrate 1a in D2O in the presence of 1 mol% of the catalyst, observing an almost complete deuteration of C2 in 5 hours (Scheme 6a). This result suggests that the C–H activation event should be relatively fast. Next, we measured the deuterium kinetic isotope effect of the reaction by subjecting 1a and the completely deuterated analogue 1ad7, either in separate reactions or in one pot, to the optimized reaction conditions (Scheme 6b and c). In both cases, we obtained KIE ≈2.1. Overall, these results are compatible with a mechanism involving a relatively fast and reversible C–H activation step preceding an irreversible rate-determining step.19 Furthermore, we also conducted the reaction between 1a and 2a in the presence of 1 equivalent of TEMPO and under otherwise standard reaction conditions (Scheme 6d). The alkenylation product 3a was formed in very small amount (12%), with the major product being quinoline 1a′. This might indicate that the inhibiting effect of TEMPO is exerted through a cleavage of the directing group, rather than through an inhibition of a radical pathway.
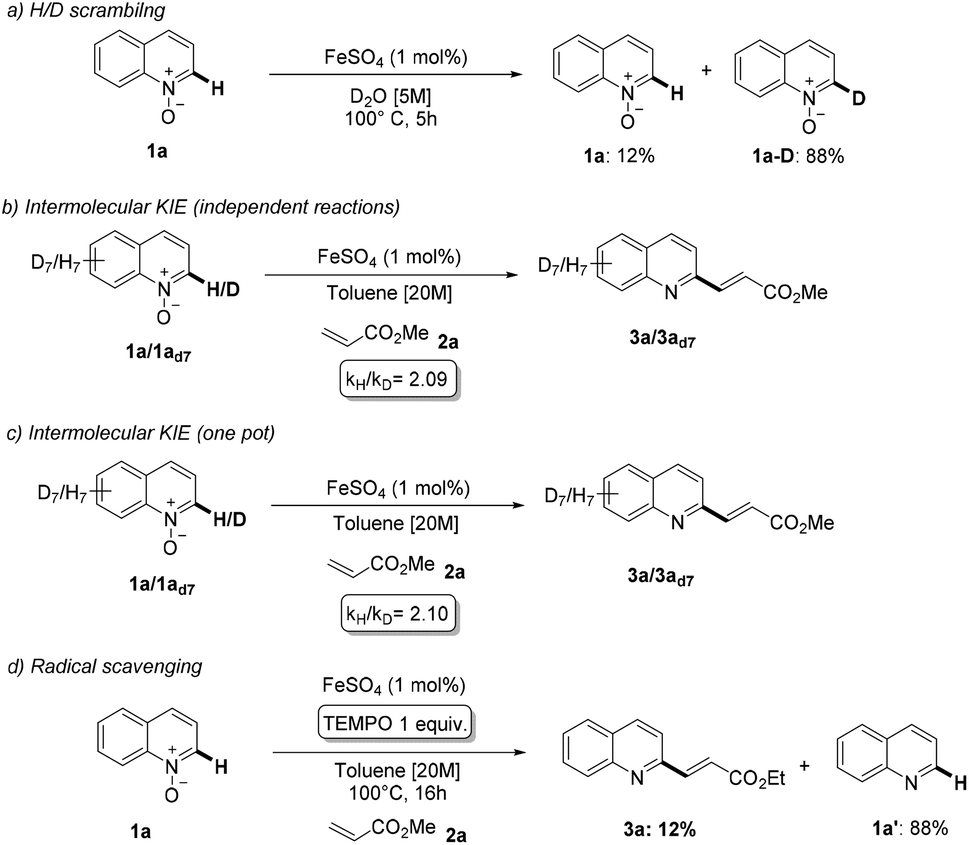 | ||
| Scheme 6 Mechanistic investigation on the iron-catalyzed C–H alkenylation. | ||
To gain more insight, we also studied the complete reaction mechanism by means of DFT calculations (see Scheme 7 and the ESI†). The most energetically favorable pathway involves a reversible ligand-promoted C2 deprotonation mediated by sulfate (TS1C2), followed by the oxidation of iron, a plausibly rate-determining insertion of the olefin into the C2–Fe bond (TS2C2) and a final H-abstraction to regenerate the double bond (TS3C2). Very importantly, this mechanistic proposal is also able to account for the experimentally observed regioselectivity, since the pathway for the reaction to occur on C8 is associated with significantly higher energy barriers. Furthermore, our DFT calculations suggest that alternative mechanistic scenarios involving an initial [3 + 2] cycloaddition8 are associated with significantly higher energy barriers, essentially ruling out this possibility for the present reaction (additional details are available in the ESI†).
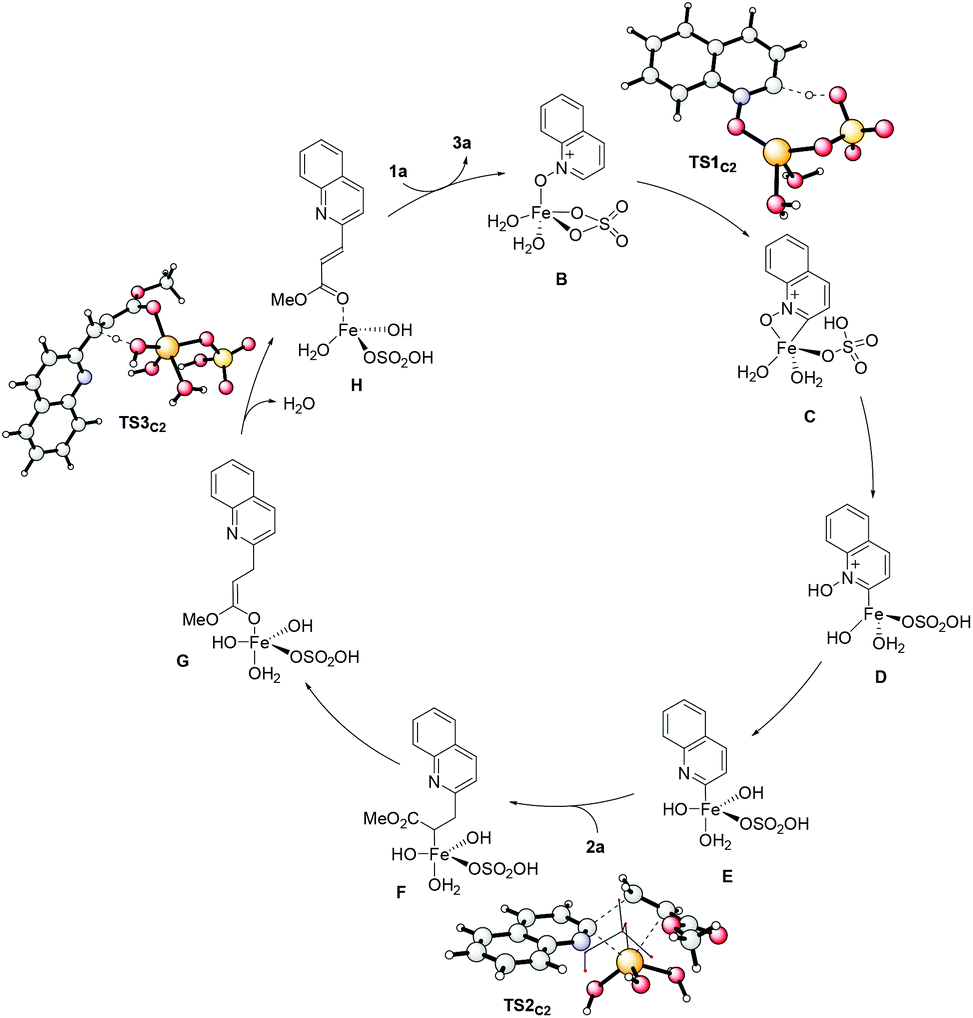 | ||
| Scheme 7 Proposed mechanism based on DFT calculations. | ||
Finally, we analyzed in depth the greenness of our protocol by using radial polygons (Fig. 1).
 | ||
| Fig. 1 Radial polygons for metrics evaluation. | ||
We carefully assessed three categories of metrics: (i) metrics related to the synthetic process (AE, Yield, 1/SF, MRP and RME); (ii) safety/hazard metrics (SHZI and SHI); and (iii) metrics related to the metal used as the catalyst (Abundance, OEL and ADP).
First of all, it is worth noting that our protocol has an associated E-factor value of 0.92 which is very low compared to other protocols available in the literature (an 84–>99% reduction considering values from 6 to 37 of catalyst-free protocols to 131–457 of palladium-catalyzed protocols, see the ESI†). This result is evidently due to the use of highly concentrated conditions and to the small amount of the solvent needed for the work-up procedure. As a consequence, the other mass-related metrics also support the efficiency and greenness of the process.
Furthermore, analyzing the safety/hazard metrics, we found that the safety hazard index (SHZI) related to the input and to the excess of starting materials, which finally results in waste, is, in the case of our protocol, the lowest resulting in an optimal Safety Hazard Impact (SHI) for the newly developed process.
These results and also the green features of iron, the metal used as the catalyst, reported in Table 2, strongly support the sustainability of the green iron-catalyzed process, indeed with iron being decisively more abundant in the earth's crust compared to palladium and its very low depletion (ASP).
| Catalyst | Metal abundance (%) | RPP | OEL (mmol m−3) | ADP |
|---|---|---|---|---|
| RPP: risk phrase potential; OEL: occupational exposure limit; ADP: abiotic resource depletion. | ||||
| FeSO4 | 5.6 | 1.56 | 0.17 | 0.00035 |
| PdOAc2 | 0.015 | 1.73 | 0.004 | 0.32 |
| PdCl2 | 0.015 | 1.85 | 0.004 | 0.32 |
In addition, by combining the toxicity data present in the literature (exposure limits, risk phrases and acute toxicity), the expected reduced hazard impact of iron compared to palladium can be quantified.
Conclusions
In conclusion, here we reported the first examples of external oxidant-free, C2-selective iron-catalyzed C–H alkenylation of quinolines. The methodology makes use of the inexpensive, earth-abundant FeSO4 catalyst and N-oxides as the directing group and internal oxidant. The power of this approach was demonstrated by the efficient, chemo- and regioselective coupling of variously substituted quinoline-N-oxides with acrylates and styrenes, even on a gram scale. Experiments and DFT calculations allowed us to propose a mechanism based on an initial ligand promoted C–H activation, followed by internal metal oxidation, insertion and H-abstraction. Further studies on the environmental and hazard impact showed that the iron-catalyzed process allowed an environmentally benign access to widely interesting C2 alkenylated quinolines and quinoxalines with consistent waste minimization. The E-factor value of ca. 0.92 evidently confirms the sustainable efficiency of the procedure presented.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research leading to these results has received funding from the NMBP-01-2016 Programme of the European Union's Horizon 2020 Framework Programme H2020/2014-2020/under grant agreement no. [720996]. The Università degli Studi di Perugia and MIUR are acknowledged for financial support to the project AMIS, through the program “Dipartimenti di Eccellenza – 2018–2022”. S. S. acknowledges CINECA for computational resources provided in the framework of ISCRA calls (project IsC64_AMUCHFR).Notes and references
- (a) V. V. Kouznetsov, L. Y. Vargas Méndez, C. E. M. Puerto Galvis and C. Ortiz Villamizar, New J. Chem., 2020, 44, 12–19 RSC; (b) M. H. Muhammad, X.-L. Chen, B. Yu, L.-B. Qu and Y.-F. Zhao, Pure Appl. Chem., 2019, 91, 33–41 CAS; (c) E. N. da Silva Júnior, G. A. M. Jardim, R. S. Gomes, Y. F. Liang and L. Ackermann, Chem. Commun., 2018, 54, 7398–7411 RSC; (d) T. Iwai and M. Sawamura, ACS Catal., 2015, 5, 5031–5040 CrossRef CAS.
- (a) M. G. Savelieff, G. Nam, J. Kang, H. J. Lee, M. Lee and M. H. Lim, Chem. Rev., 2019, 119, 1221–1322 CrossRef CAS; (b) O. Afzal, S. Kumar, M. R. Haider, M. R. Ali, R. Kumar, M. Jaggi and S. A. Bawa, Eur. J. Med. Chem., 2015, 97, 871–910 CrossRef CAS.
- (a) D. Sciosci, F. Valentini, F. Ferlin, S. Chen, Y. Gu, O. Piermatti and L. Vaccaro, Green Chem., 2020, 22, 6560–6566 RSC; (b) T. Guo, Y. Liu, Y.-H. Zhao, P.-K. Zhang, S.-L. Han and H.-M. Liu, Tetrahedron Lett., 2016, 57, 3920–3923 CrossRef CAS; (c) N. Barsu, M. Sen, R. Premkumar and B. Sundaraju, Chem. Commun., 2016, 52, 1338–1341 RSC; (d) X. Huang, J. Huang, C. Du, X. Zhang, F. Song and J. You, Angew. Chem., Int. Ed., 2013, 52, 12970–12974 CrossRef CAS; (e) J. Wu, X. Cui, L. Chen, G. Jiang and Y. Wu, J. Am. Chem. Soc., 2009, 131, 13888–13889 CrossRef CAS.
- For examples of palladium catalysis, see: (a) X. Chen, X. Cui and Y. Wu, Org. Lett., 2016, 18, 2411–2414 CrossRef CAS; (b) D. E. Stephens, J. Lakey-Beitia, G. Chavez, C. Ilie, H. D. Arman and O. V. Larionov, Chem. Commun., 2015, 51, 9507–9510 RSC; (c) D. E. Stephens, J. Lakey-Beitia, A. C. Atesin, T. A. Atesin, G. Chavez, C. Ilie, H. D. Arman and O. V. Larionov, ACS Catal., 2015, 5, 167–175 CrossRef CAS; (d) Z. Wu, C. Pi, X. Cui, J. Bai and Y. Wu, Adv. Synth. Catal., 2013, 355, 1971–1976 CrossRef CAS. For examples of rhodium catalysis, see: (e) C. You, C. Pi, Y. Wu and X. Cui, Adv. Synth. Catal., 2018, 360, 4068–4072 CrossRef CAS; (f) B. Wang, C. Li and H. Liu, Adv. Synth. Catal., 2017, 359, 3029–3034 CrossRef CAS. For examples of copper catalysis, see: (g) G.-H. Li, D.-Q. Dong, Y. Yang, X.-Y. Yu and Z.-L. Wang, Adv. Synth. Catal., 2019, 361, 832–835 CAS; (h) Z. Wu, H. Song, X. Cui, C. Pi, W. Du and Y. Wu, Org. Lett., 2013, 15, 1270–1273 CrossRef CAS; (i) G. Li, C. Jia and K. Sun, Org. Lett., 2013, 15, 5198–5201 CrossRef CAS; (j) X. Chen, C. Zhu, X. Cui and Y. Wu, Chem. Commun., 2013, 49, 6900–6902 RSC. For examples of cobalt catalysis, see: (k) D. Kalsi, R. A. Laskar, N. Barsu, J. R. Premkumar and B. Sundaraju, Org. Lett., 2016, 18, 4198–4201 CrossRef CAS.
- (a) R. Sharma, R. Kumar and U. Sharma, J. Org. Chem., 2019, 84, 2786–2797 CrossRef CAS; (b) T. Shibata and Y. Matsuo, Adv. Synth. Catal., 2014, 356, 1516–1520 CrossRef CAS; (c) J. Ryu, S. H. Cho and S. Chang, Angew. Chem., Int. Ed., 2012, 51, 3677–3681 CrossRef CAS.
- H. Hu, X. Hu and Y. Liu, Angew. Chem., 2020, 59, 18975–18979G CrossRef CAS.
- R. K. Shukla, A. M. Nair, S. Khan and C. M. R. Volla, Angew. Chem., 2020, 59, 17042–17048 CrossRef CAS.
- (a) Z. Zhang, C. Pi, H. Tong, X. Cui and Y. Wu, Org. Lett., 2017, 19, 440–443 CrossRef CAS; (b) R. Kumar, I. Kumar, R. Sharma and U. Sharma, Org. Biomol. Chem., 2016, 14, 2613–2617 RSC; (c) G. E. M. Crisenza, E. M. Dauncey and J. F. Bower, Org. Biomol. Chem., 2016, 14, 5820–5825 RSC.
- For selected recent reviews, see: (a) P. Gandeepan, T. Mueller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS; (b) Y. Hu, B. Zhou and C. Wang, Acc. Chem. Res., 2018, 51, 816–827 CrossRef CAS; (c) R. Shang, L. Ilies and E. Nakamura, Chem. Rev., 2017, 117, 9086–9139 CrossRef CAS; (d) M. Moselage and L. Ackermann, ACS Catal., 2016, 6, 498–525 CrossRef CAS.
- (a) T. E. Boddie, S. H. Carpenter, T. M. Baker, J. C. DeMuth, G. Cera, W. W. Brennessel, L. Ackermann and M. L. Neidig, J. Am. Chem. Soc., 2019, 141, 12338–12345 CrossRef CAS; (b) J. Mo, T. Muller, J. C. A. Oliveira and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 7719–7723 ( Angew. Chem. , 2018 , 130 , 7845–7849 ) CrossRef CAS; (c) M. C. White and J. Zhao, J. Am. Chem. Soc., 2018, 140, 13988–14009 CrossRef CAS; (d) A. Furstner, ACS Cent. Sci., 2016, 2, 778–789 CrossRef; (e) L. Ilies and E. Nakamura, Top. Organomet. Chem., 2015, 56, 1–18 CrossRef.
- (a) A. M. Messinis, L. H. Finger, L. Hu and L. Ackermann, J. Am. Chem. Soc., 2020, 142, 13102–13111 CrossRef CAS; (b) G. Cera, T. Haven and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 1484–1488 CrossRef CAS.
- (a) J. Loup, T. Parchomyk, S. Luelf, S. Demeshko, F. Meyer, K. Koszinowski and L. Ackermann, Dalton Trans., 2019, 48, 5135–5139 RSC; (b) E. R. Fruchey, B. M. Monks and S. P. Cook, J. Am. Chem. Soc., 2014, 136, 13130–13133 CrossRef CAS; (c) L. Ilies, T. Matsubara, S. Ichikawa, S. Asako and E. Nakamura, J. Am. Chem. Soc., 2014, 136, 13126–13129 CrossRef CAS; (d) J. Norinder, A. Matsumoto, N. Yoshikai and E. Nakamura, J. Am. Chem. Soc., 2008, 130, 5858–5859 CrossRef CAS.
- (a) B. Zhou, H. Sato, L. Ilies and E. Nakamura, ACS Catal., 2018, 8(1), 8–11 CrossRef CAS; (b) Z. Shen, G. Cera, T. Haven and L. Ackermann, Org. Lett., 2017, 19, 3795–3798 CrossRef CAS; (c) R. Shang, L. Ilies, A. Matsumoto and E. Nakamura, J. Am. Chem. Soc., 2013, 135, 6030–6032 CrossRef CAS.
- G. Cera, T. Haven and L. Ackermann, Chem. – Eur. J., 2017, 23, 3577–3582 CrossRef CAS.
- T. Matsubara, S. Asako, L. Ilies and E. Nakamura, J. Am. Chem. Soc., 2014, 136, 646–649 CrossRef CAS.
- (a) L. Ilies, Y. Itabashi, R. Shang and E. Nakamura, ACS Catal., 2017, 7, 89–92 CrossRef CAS; (b) J. Loup, D. Zell, J. C. A. Oliveira, D. Stalke and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 14197–14201 CrossRef CAS; (c) M. Y. Wong, T. Yamakawa and N. Yoshikai, Org. Lett., 2015, 17, 442–445 CrossRef CAS; (d) A. Matsumoto, L. Ilies and E. Nakamura, J. Am. Chem. Soc., 2011, 133, 6557–6559 CrossRef CAS; (e) L. Ilies, S. Asako and E. Nakamura, J. Am. Chem. Soc., 2011, 133, 7672–7675 CrossRef CAS.
- (a) F. Ferlin, P. M. L. Navarro, Y. Gu, D. Lanari and L. Vaccaro, Green Chem., 2020, 22, 397–403 RSC; (b) F. Ferlin, A. Marini, N. Ascani, L. Ackermann, D. Lanari and L. Vaccaro, ChemCatChem, 2020, 12, 449–454 CrossRef CAS; (c) F. Ferlin, S. R. Yetra, S. Warratz, L. Vaccaro and L. Ackermann, Chem. – Eur. J., 2019, 25, 11427–11431 CrossRef CAS; (d) F. Ferlin, L. Luciani, S. Santoro, A. Marrocchi, D. Lanari, A. Bechtoldt, L. Ackermann and L. Vaccaro, Green Chem., 2018, 20, 2888–2893 RSC; (e) A. Bechtoldt, M. E. Baumert, L. Vaccaro and L. Ackermann, Green Chem., 2018, 20, 398–402 RSC; (f) F. Ferlin, S. Santoro, L. Ackermann and L. Vaccaro, Green Chem., 2017, 19, 2510–2514 RSC; (g) D. Rasina, A. Kahler-Quesada, S. Ziarelli, S. Warratz, H. Cao, S. Santoro, L. Ackermann and L. Vaccaro, Green Chem., 2016, 18, 5025–5030 RSC.
- See the ESI† for further details on optimization and experimental procedures.
- E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066–3072 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc03351k |
| This journal is © The Royal Society of Chemistry 2021 |