
Synergistic cooperative effect of CF3SO2Na and bis(2-butoxyethyl)ether towards selective oxygenation of sulfides with molecular oxygen under visible-light irradiation†
Kai-Jian
Liu
a,
Zheng
Wang
a,
Ling-Hui
Lu
a,
Jin-Yang
Chen
 b,
Fei
Zeng
a,
Ying-Wu
Lin
b,
Zhong
Cao
c,
Xianyong
Yu
d and
Wei-Min
He
*a
b,
Fei
Zeng
a,
Ying-Wu
Lin
b,
Zhong
Cao
c,
Xianyong
Yu
d and
Wei-Min
He
*a
aDepartment of Chemistry and Bioengineering, Hunan University of Science and Engineering, Yongzhou 425100, China. E-mail: weiminhe2016@yeah.net
bSchool of Chemistry and Chemical Engineering, University of South China, Hengyang, 421001, China
cHunan Provincial Key Laboratory of Materials Protection for Electric Power and Transportation, Changsha University of Science and Technology, Changsha, 410114, China
dSchool of Chemistry and Chemical Engineering, Hunan University of Science and Technology, Xiangtan 411201, China
First published on 26th November 2020
Abstract
A safe, practical and eco-friendly method for the switchable synthesis of sulfoxides and sulfones through visible-light-initiated oxygenation of sulfides at ambient temperature under transition-metal-, additives-free and minimal solvent conditions. The synergistic catalytic efforts between CF3SO2Na and 2-butoxyethyl ether represents the key promoting factor for the reaction.
Introduction
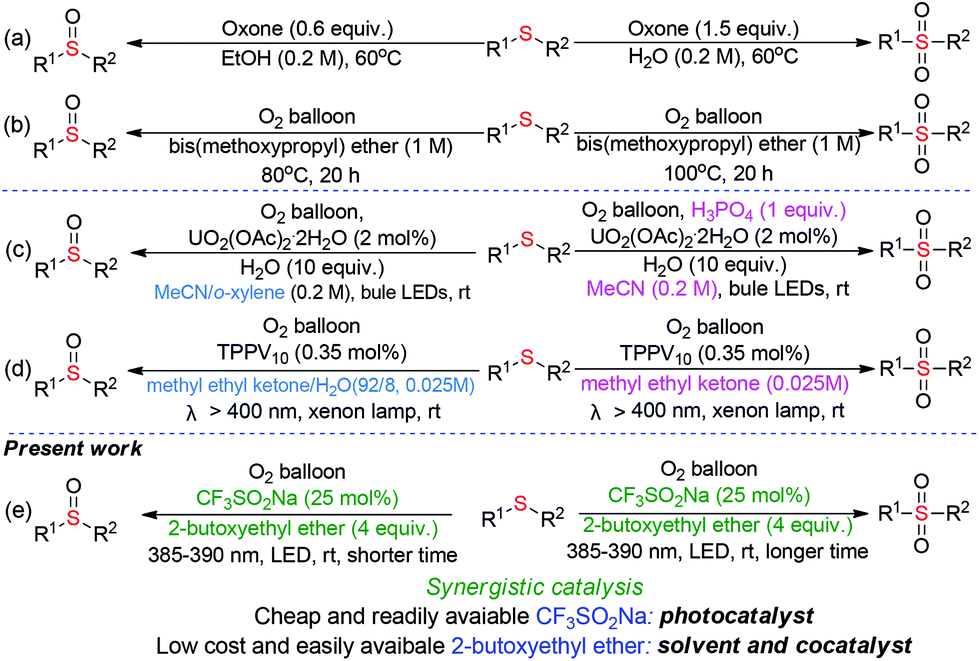
Sulfur-containing molecules perform significant roles in fine chemical, pharmaceutical, and material science, given their diverse oxidation-states.1 Among these S-containing compounds, sulfoxides and sulfones are indispensable motifs in synthetic pharmaceuticals, natural products, and advanced organic materials.2 In the fine chemical and pharmaceutical industry, the selectivity-switchable synthesis of distinct high-value chemicals from the same starting material would be ideal in terms of manufacturing cost and operational simplicity.During the past years, considerable efforts have been made for developing of switchable synthesis of sulfoxides and sulfones through the selective oxidation of abundant and easily available sulfides. In 2012, Liang-Nian He's group pioneered the selective oxygenation of sulfides with stoichiometric amounts of oxone as the oxidant under heating conditions (Scheme 1a).3 Liu and our group independently realized the temperature-controlled selective oxygenation of sulfides using molecular oxygen (1 atm) with ether solvent as the promoter (Scheme 1b).4 However, the usage of O2 in the presence of flammable organic solvent at high temperature results in a potentially inflammable and explosive mixture, which greatly limits their industrial application. Visible-light-induced oxidation with O2 as the sole oxidant has become a prominent sector of synthetic chemistry over the last years as it offers eco-friendly alternatives to the conventional thermal heating process.5 In 2019, Jiang and coworkers disclosed the firstly visible-light-induced UO2(OAc)2·2H2O-catalyzed selective oxygenation of sulfides with O2 in the presence of H2O and H3PO4 as the additives (Scheme 1c).6 Very recently, Kosuke Suzuki and Kazuya Yamaguchi reported the visible-light induced tetraphenylphosphonium decavanadate (TPPV10)-catalyzed switchable oxygenation of sulfides.7 However, expensive uranyl salts or commercially unavailable TPPV10 nano-catalyst was necessary in these photocatalytic processes, which not only increases manufacturing cost but also causes the issue of transition-metal residue in the final products. Moreover, the switching selectivity in the above reactions is fully dependent on different reaction mediums, resulting in the increasing of operating difficulty and production cost. From an environmental and economical point of view, a novel method for the visible-light induced switchable oxygenation of sulfides with O2 in the presence of low cost and readily available photocatalyst under transition-metal- and additives-free conditions would be highly desirable, especially in the pharmacy industry.
 | ||
| Scheme 1 Selective oxidation of sulfides. | ||
We were aware that the cheap and easily available sodium trifluoromethanesulfinate was used as a photocatalyst for the visible-light-induced aerobic oxidation of C–H bonds8 and the ether solvent was employed as a promoter/catalyst for the thermal oxidation with O2. However, to the best of our knowledge, there has been no report about the visible-light-induced oxidation synergistically catalyzed by CF3SO2Na and ether. Under the guidance of green chemistry principles,9 we herein disclose a safe, practical and sustainable protocol for the switchable preparation of sulfoxides and sulfones through visible-light initiated CF3SO2Na/2-butoxyethyl ether synergistic catalyzed oxygenation of sulfides under transition-metal-, additives-free and minimal solvent conditions (Scheme 1e). The synergistic combination of CF3SO2Na and 2-butoxyethyl ether in a relay aerobic oxidation mechanism allows sulfides to undergo deep oxidation.
Results and discussion
We first investigated the visible-light-induced oxygenation of methyl(p-tolyl)sulfane (1a) as the model reaction (Table 1). Conducting the oxygenation of 1a with CF3SO2Na (25 mol%) as a photocatalyst and molecular oxygen as an oxidant in MeCN under the irradiation of LEDs (375–380 nm, 10 W) at ambient temperature for 24 hours delivered the desired sulfoxide (2a) in 19% GC yield (Table 1, entry 1). The effect of the visible light source on the model reaction was next investigated (entries 2–5). Under the irradiation wavelength of 385–390 nm, a 68% GC yield of 2a was obtained after 24 hours (entry 3). As anticipated, the oxygenation efficiency was heavily depended on the nature of the reaction medium (entries 6–12). The ether solvents (entries 9–12) gave better results than non-ether solvents (entries 3 and 6–8). Performing the oxygenation reaction with bis(2-butoxyethyl)ether ether (BE) as the solvent provided 2a in 89% GC yield and the sulfone 3a in 6% GC yield based on 95% conversion of 1a (entry 12). To our delight but unexpectedly, reducing the loading of solvent obviously improved the oxidation capacity of the present reaction system (entries 13–15). A quantitative yield of 3a was obtained by decreasing the amount of BE from 1 mL (about 12 equiv.) to 4 equiv. (entry 14). However, further reducing the loading of BE to 2 equiv. led to a lower yield of 3a (entry 15). As expected, the reaction time exerted a crucial role on the oxygenation outcome (entries 14 vs. 16–18). A 92% yield of 2a and 8% yield of 3a were observed by shorting the oxidation time from 24 hours to 6 hours (entry 17). Both lower conversion of 1a and lower yield of 2a were obtained in the absence of CF3SO2Na or BE (entries 17 vs. 19–21), revealing that the BE has synergetic effect with CF3SO2Na and enhanced oxidation capacity of the photocatalytic system. No oxidation reaction occurred without visible-light irradiation, and the substrate 1a was completely recovered (entry 22).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Light source | Solvent | Time | Conv. | Yieldb | |
| 2a | 3a | |||||
| a Conditions: Methyl 4-tolyl sulfide (1a) (1 mmol), CF3SO2Na (25 mol%), solvent, O2 balloon. b The yields were estimated by GC-MS. c Without CF3SO2Na. d Under dark conditions; BE: 2-butoxyethyl ether; EE: 2-ethoxyethyl ether. | ||||||
| 1 | 375–380 nm | MeCN (1 mL) | 24 h | 19% | 19% | 0 |
| 2 | 380–385 nm | MeCN (1 mL) | 24 h | 21% | 21% | 0 |
| 3 | 385–390 nm | MeCN (1 mL) | 24 h | 68% | 68% | 0 |
| 4 | 390–395 nm | MeCN (1 mL) | 24 h | 62% | 62% | 0 |
| 5 | 395–400 nm | MeCN (1 mL) | 24 h | 49% | 49% | 0 |
| 6 | 385–390 nm | DMF (1 mL) | 24 h | 15% | 15% | 0 |
| 7 | 385–390 nm | MeOH (1 mL) | 24 h | 34% | 34% | 0 |
| 8 | 385–390 nm | DCE (1 mL) | 24 h | 47% | 47% | 0 |
| 9 | 385–390 nm | THF (1 mL) | 24 h | 82% | 80% | 2% |
| 10 | 385–390 nm | ME (1 mL) | 24 h | 88% | 85% | 3% |
| 11 | 385–390 nm | EE (1 mL) | 24 h | 89% | 84% | 5% |
| 12 | 385–390 nm | BE (1 mL) | 24 h | 95% | 89% | 6% |
| 13 | 385–390 nm | BE (8 equiv.) | 24 h | 98% | 41% | 57% |
| 14 | 385–390 nm | BE (4 equiv.) | 24 h | 100% | 0 | 100% |
| 15 | 385–390 nm | BE (2 equiv.) | 24 h | 99% | 27% | 72% |
| 16 | 385–390 nm | BE (4 equiv.) | 12 h | 100% | 47% | 53% |
| 17 | 385–390 nm | BE (4 equiv.) | 6 h | 100% | 92% | 8% |
| 18 | 385–390 nm | BE (4 equiv.) | 4 h | 38% | 37% | 1% |
| 19c | 385–390 nm | BE (4 equiv.) | 6 h | 4% | 4% | 0 |
| 20 | 385–390 nm | MeCN (4 equiv.) | 6 h | 12% | 12% | 0 |
| 21 | 385–390 nm | — | 6 h | 10% | 10% | 0 |
| 22d | — | BE (4 equiv.) | 24 h | 0 | 0 | 0 |
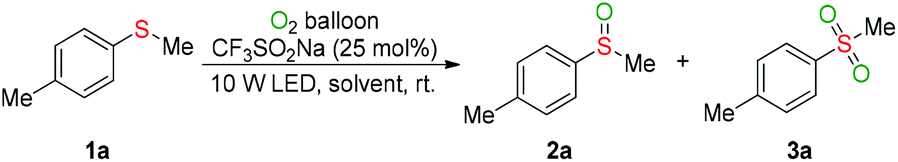
With the optimal conditions, the generality of the selective oxygenation of sulfides 1 to sulfoxides 2 was firstly investigated (Table 2). As seen clearly, phenylthiomethanes with various synthetically important functional groups underwent the visible-light-induced oxygenation efficiently and delivered the desired products (2a–2m) in excellent yields. Neither the electronic effect nor steric hindrance effect of the substituent group on the phenyl ring of substrate 1 had a considerable influence on the oxidative process. It was worth noting that sulfide (1n) is readily oxidized into 1,4-bis(methylsulfinyl)benzene (2n) in 81% yield and the S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond did not have a significant influence during the double oxidative process. To our satisfaction, the methyl sulfide substrates with naphthalene or heteroaromatic ring were also tolerated in the reaction system, which significantly expanded the scope of the oxidation (2o–2s). The phenyl sulfide with various chain lengths, isomeric structures, and functional groups underwent the transformation to generate the sulfoxides in good yields (2t–2w). The dibenzo[b,d]thiophene bearing a sulfur atom in the cyclic framework gave the product (2x) in 78% yield. Given that the thianthrene bearing two sulfur atoms, we were pleased to find that only mono-oxygenation product (2y) was produced in good yield. Moreover, a series of diaryl sulfides with electron-rich and electron-poor groups were well tolerated, delivering the corresponding sulfoxide products (2z–2ag) in good to excellent yields.
O bond did not have a significant influence during the double oxidative process. To our satisfaction, the methyl sulfide substrates with naphthalene or heteroaromatic ring were also tolerated in the reaction system, which significantly expanded the scope of the oxidation (2o–2s). The phenyl sulfide with various chain lengths, isomeric structures, and functional groups underwent the transformation to generate the sulfoxides in good yields (2t–2w). The dibenzo[b,d]thiophene bearing a sulfur atom in the cyclic framework gave the product (2x) in 78% yield. Given that the thianthrene bearing two sulfur atoms, we were pleased to find that only mono-oxygenation product (2y) was produced in good yield. Moreover, a series of diaryl sulfides with electron-rich and electron-poor groups were well tolerated, delivering the corresponding sulfoxide products (2z–2ag) in good to excellent yields.
| a Conditions: Sulfides 1 (1.0 mmol), CF3SO2Na (25 mol%), BE (4.0 mmol), O2 balloon, room temperature. |
|---|
|
Next, the universality of the deep oxygenation of sulfides to sulfones 3 was explored. A wide range of phenylthiomethanes with electron-neutral, -donating, and -withdrawing groups were well compatible under the optimized conditions regardless of their electronic and steric properties (3a–3l). Both polycyclic and quinoline substrates underwent oxygenation reactions smoothly to provide the sulfone products 3m and 3n in 92% and 88% yields, respectively. Phenyl ethyl sulfide was also applicable in the current sulfonylation and provided the corresponding product 3o in 84% yield. Moreover, good to excellent yields of diaryl sulfones (3q–3w) were obtained for the photocatalytic oxygenation of various diaryl sulfides. The dibenzothiophene also proceeded efficiently to give the sulfone 3x in 72% yield. No reaction occurred when dialkyl sulfides, such as dibenzyl sulfide and propyl sulfide, were used as substrates. GC-MS analysis of the reaction mixture showed that a small quantity of sulfone product was formed during the oxidation of diaryl sulfide to diaryl sulfoxide. For example, the oxidation of diaryl sulfide 1af for 24 hours provided 77% GC yield of sulfoxide 2af and 9% GC yield of sulfone 3v.
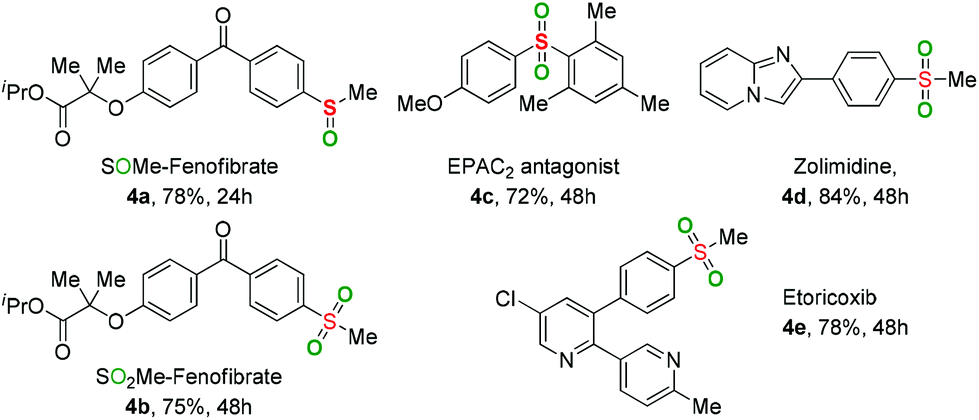
To further verify the applicability of the present oxidation, some sulfoxide- and sulfone-containing pharmaceuticals and their derivatives were prepared through the developed method (Table 3). Both the introduction of –SOMe and –SO2Me motifs into Fenofibrate (a widely used lipid regulator) were smoothly achieved to provide the desired products 4a and 4b in good yields. The sulfone-containing drugs, including EPAC2 antagonist 4c, Zolimidine 4d (anti-ulcer agent), and Etoricoxib 4e (antirheumatoid arthritis drug and COX-2 selective inhibitor) could be easily constructed via the current reaction.
| a Conditions: Sulfides 1 (1.0 mmol), CF3SO2Na (25 mol%), BE (4.0 mmol), O2 balloon, room temperature. |
|---|
|
The effect of visible light irradiation time on the conversion of sulfide 1a was explored. As shown in Fig. 1, in the initial oxygenation stage of 6 hours, both 92% GC yield of sulfoxide 2a and 8% GC yield of sulfone 3a were obtained with full conversion of the sulfide 1a. About at 11 hours, equal amount of 2a and 3a were detected. As the lighting time up to 24 hours, 2a was completely converted into 3a. These results suggested that the 1a was firstly converted into 2a and then the generated 2a was transformed into 3a in the oxidative process.
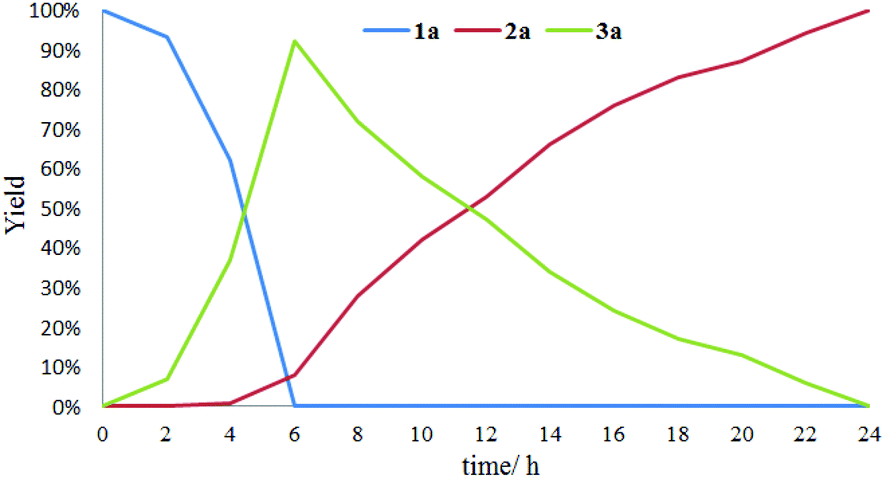 | ||
| Fig. 1 Time course for the oxygenation of 1a. | ||
The light on–off experiments of the oxygenation of sulfide 1a to sulfoxide 2a and oxygenation of 2a to sulfone 3a suggested that both the transformations were inhibited in the absence of visible-light irradiation (Fig. 2a and b). These results demonstrated that the light-dependency of the current oxidative process.
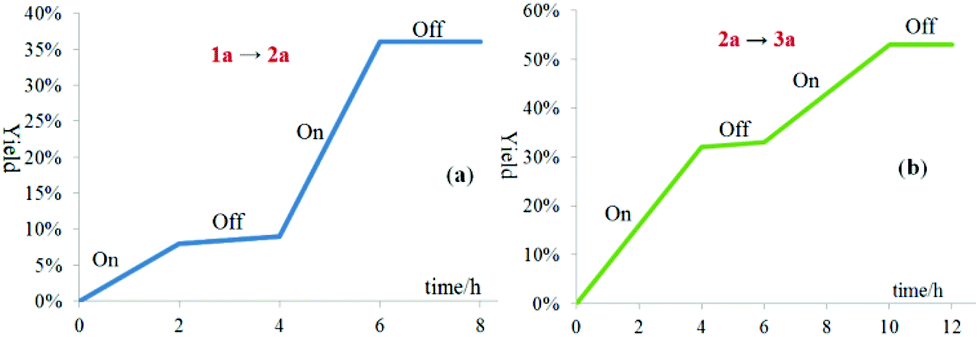 | ||
| Fig. 2 Visible-light irradiation on/off experiments. | ||
In order to gain a better understanding of the visible-light-induced oxidation of sulfides, some control experiments were carried out. The reaction was suppressed by adding TEMPO or BHT as the radical scavenger, confirming that radical species should be involved in this process (Scheme 2a and b). When 1,4-dimethoxybenzene, a well-known sulfide cation radical scavenger, was added into the reaction, only a trace amount of 2a was observed (Scheme 2c), suggesting that the sulfide cation radical might be a key intermediate for the transformation of sulfides.10 Furthermore, the formation of superoxide anion radical was conformed by adding benzoquinone as a superoxide radical scavenger11 (Scheme 2d). Performing the oxidation of sulfoxide 2a with CF3SO2Na as the photocatalyst and O2 as the oxidant in MeCN under the irradiation of LED (385–390 nm) for 24 hours gave the desired sulfone 3a in 21% GC yield (Scheme 2e). Approximately 4% of BE was lost during the oxidative process, suggesting the BE might act an catalyst in the present raction.12 The ultraviolet–visible absorption-spectra showed that only the oxidized CF3SO2Na could absorb visible light and serve as a photocatalyst in the present oxygenation reaction (Fig. S1, ESI†).13
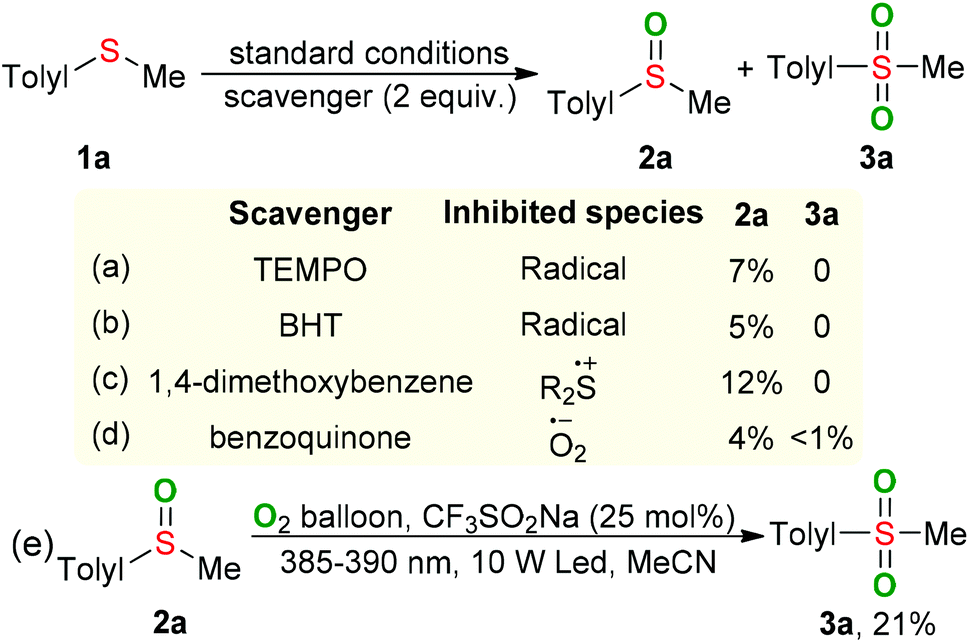 | ||
| Scheme 2 Control experiments. | ||
Based on the above results and related literature,4,6,8 we proposed a plausible mechanism for the photocatalytic selective oxygenation of sulfides (Scheme 3). Initially, the visible-light-induced oxidation of CF3SO2Na with ground-state triplet molecular oxygen generated a penta-coordinate sulfide (C1), which was further excited by light irradiation to produce the excited-state C1*. The excited-state C1* then underwent a single electron transfer (SET) with substrate 1 to generate a sulfide cation radical (S1) along with the formation of ground-state C1. Meanwhile, excited-state C1* also reacted with O2 to form an active superoxide anion radical with the concomitant production of radical C1˙. The subsequent coupling of the intermediate S1 with the superoxide anion radical gave the persulfoxide intermediate (S2), which was subsequently caught by another molecular of substrate 1 to form the sulfoxide 2. The radical C1˙ abstracted hydrogen from the 2-butoxyethyl ether (BE) to generate a carbon-centered radical BE1 along with an intermediate C1–H, which was converted into intermediate C1. Simultaneously, the radical BE1 was further oxidized to generate a peroxide BE2, which oxygenated the sulfoxide 2 into sulfone product 3 and regenerated the BE.
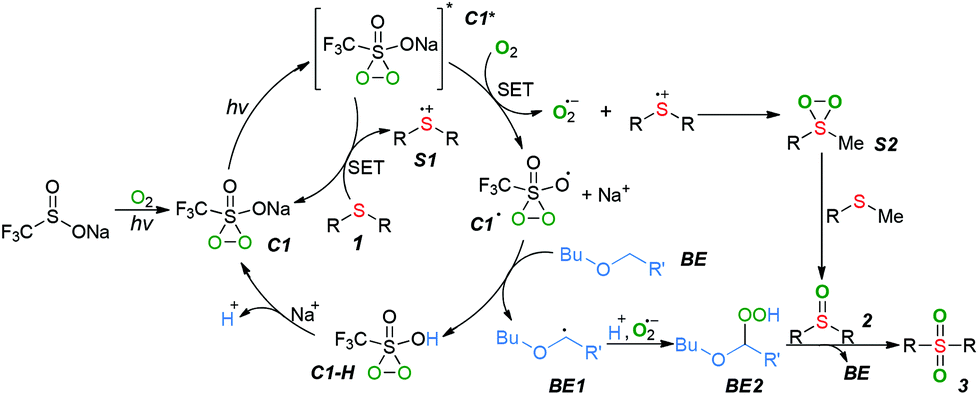 | ||
| Scheme 3 Possible mechanism. | ||
Conclusions
In conclusion, we have successfully demonstrated a safe, practical and eco-friendly photocatalytic method for the switchable synthesis of sulfoxides and sulfones under transition-metal-, additives-free and minimal solvent conditions. With cheap and easily available CF3SO2Na as a photocatalyst, 2-butoxyethyl ether (4 equiv.) as a solvent and a co-catalyst, various sulfides were efficiently and selectively oxidized into sulfoxides (34 examples, 70–95%) and sulfones (28 examples, 72–98%) through the adjustment of reaction time. In sharp contrast to the previous photocatalytic strategies, the present strategy not only avoiding toxic or commercially unavailable photocatalysts, but also significantly simplifying operational procedures and reducing manufacturing cost. Mechanistic studies indicated that the synergistic catalytic efforts between CF3SO2Na and 2-butoxyethyl ether represent the key promoting factor for the oxidation reaction. The protocol will open a new avenue for developing the practical visible-light-initiated oxidation and may also find broad applicability in synthetic chemistry and pharmaceutical chemistry.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the Hunan Provincial Natural Science Foundation of China (No. 2019JJ40090 and 2019JJ20008).Notes and references
- (a) E. A. Ilardi, E. Vitaku and J. T. Njardarson, J. Med. Chem., 2014, 57, 2832 CrossRef CAS; (b) F. Minghao, T. Bingqing, H. L. Steven and J. Xuefeng, Curr. Top. Med. Chem., 2016, 16, 1200 CrossRef; (c) P. Sun, D. Yang, W. Wei, M. Jiang, Z. Wang, L. Zhang, H. Zhang, Z. Zhang, Y. Wang and H. Wang, Green Chem., 2017, 19, 4785 RSC; (d) K. A. Scott and J. T. Njardarson, Top. Curr. Chem., 2018, 376, 5 CrossRef; (e) G. Li, Q. Yan, Z. Gan, Q. Li, X. Dou and D. Yang, Org. Lett., 2019, 21, 7938 CrossRef CAS; (f) G. Li, Z. Gan, K. Kong, X. Dou and D. Yang, Adv. Synth. Catal., 2019, 361, 1808 CrossRef CAS; (g) X.-M. Xu, D.-M. Chen and Z.-L. Wang, Chin. Chem. Lett., 2020, 31, 49 CrossRef CAS; (h) N. Wang, P. Saidhareddy and X. Jiang, Nat. Prod. Rep., 2020, 37, 246 RSC; (i) Y. Wang, Y. Li and X. Jiang, Chem. – Asian J., 2018, 13, 2208 CrossRef CAS.
- (a) P. Rey and L. Tarrago, Antioxidants, 2018, 7, 114 CrossRef; (b) T. Jia, M. Wang and J. Liao, Top. Curr. Chem., 2019, 377, 8 CrossRef; (c) Q. Liu, L. Wang, H. Yue, J.-S. Li, Z. Luo and W. Wei, Green Chem., 2019, 21, 1609 RSC; (d) L.-H. Lu, Z. Wang, W. Xia, P. Cheng, B. Zhang, Z. Cao and W.-M. He, Chin. Chem. Lett., 2019, 30, 1237 CrossRef CAS; (e) X. Gong, M. Yang, J.-B. Liu, F.-S. He, X. Fan and J. Wu, Green Chem., 2020, 22, 1906 RSC; (f) T.-S. Zhang, W.-J. Hao, R. Wang, S.-C. Wang, S.-J. Tu and B. Jiang, Green Chem., 2020, 22, 4259 RSC; (g) Y. Mei, L. Zhao, Q. Liu, S. Ruan, L. Wang and P. Li, Green Chem., 2020, 22, 2270 RSC; (h) S. Chen, Y. Li, M. Wang and X. Jiang, Green Chem., 2020, 22, 322 RSC.
- B. Yu, A.-H. Liu, L.-N. He, B. Li, Z.-F. Diao and Y.-N. Li, Green Chem., 2012, 14, 957 RSC.
- (a) Z. Cheng, P. Sun, A. Tang, W. Jin and C. Liu, Org. Lett., 2019, 21, 8925 CrossRef CAS; (b) K.-J. Liu, J.-H. Deng, J. Yang, S.-F. Gong, Y.-W. Lin, J.-Y. He, Z. Cao and W.-M. He, Green Chem., 2020, 22, 433 RSC.
- (a) W.-L. Lei, B. Yang, Q.-B. Zhang, P.-F. Yuan, L.-Z. Wu and Q. Liu, Green Chem., 2018, 20, 5479 RSC; (b) G. Li, Q. Yan, X. Gong, X. Dou and D. Yang, ACS Sustainable Chem. Eng., 2019, 7, 14009 CrossRef CAS; (c) X. Mi, Y. Kong, J. Zhang, C. Pi and X. Cui, Chin. Chem. Lett., 2019, 30, 2295 CrossRef CAS; (d) Y. Chen, L.-Q. Lu, D.-G. Yu, C.-J. Zhu and W.-J. Xiao, Sci. China: Chem., 2019, 62, 24 CrossRef CAS; (e) Z. Wang, X. Ji, J. Zhao and H. Huang, Green Chem., 2019, 21, 5512 RSC; (f) Y. Lv, P. Bao, H. Yue, J.-S. Li and W. Wei, Green Chem., 2019, 21, 6051 RSC; (g) Q. Yu, Y. Zhang and J.-P. Wan, Green Chem., 2019, 21, 3436 RSC; (h) D.-Q. Dong, L.-X. Li, G.-H. Li, Q. Deng, Z.-L. Wang and S. Long, Chin. J. Catal., 2019, 40, 1494 CrossRef CAS; (i) K.-J. Liu, T.-Y. Zeng, J.-L. Zeng, S.-F. Gong, J.-Y. He, Y.-W. Lin, J.-X. Tan, Z. Cao and W.-M. He, Chin. Chem. Lett., 2019, 30, 2304 CrossRef CAS; (j) Z. Wang and W.-M. He, Chin. J. Org. Chem., 2019, 39, 3594 CrossRef CAS; (k) Z. Gan, G. Li, Q. Yan, W. Deng, Y.-Y. Jiang and D. Yang, Green Chem., 2020, 22, 2956 RSC; (l) Z. Wang, Q. Liu, X. Ji, G.-J. Deng and H. Huang, ACS Catal., 2020, 10, 154 CrossRef CAS; (m) W.-B. He, L.-Q. Gao, X.-J. Chen, Z.-L. Wu, Y. Huang, Z. Cao, X.-H. Xu and W.-M. He, Chin. Chem. Lett., 2020, 31, 1895 CrossRef CAS; (n) T. Han, Y. Jiang, X. Ji, G.-J. Deng and H. Huang, Org. Chem. Front., 2020, 7, 1671 RSC; (o) P. Bao, F. Liu, Y. Lv, H. Yue, J.-S. Li and W. Wei, Org. Chem. Front., 2020, 7, 492 RSC; (p) L.-Y. Xie, Y.-S. Liu, H.-R. Ding, S.-F. Gong, J.-X. Tan, J.-Y. He, Z. Cao and W.-M. He, Chin. J. Catal., 2020, 41, 1168 CrossRef CAS; (q) L. Wang, M. Zhang, Y. Zhang, Q. Liu, X. Zhao, J.-S. Li, Z. Luo and W. Wei, Chin. Chem. Lett., 2020, 31, 67 CrossRef CAS; (r) S. He, X. Chen, F. Zeng, P. Lu, Y. Peng, L. Qu and B. Yu, Chin. Chem. Lett., 2020, 31, 1863 CrossRef CAS; (s) S. Peng, Y.-W. Lin and W.-M. He, Chin. J. Org. Chem., 2020, 40, 541 CrossRef; (t) J. Shi and W. Wei, Chin. J. Org. Chem., 2020, 40, 2170 CrossRef; (u) B. Huang, Y. Li, C. Yang and W. Xia, Green Chem., 2020, 22, 2804 RSC.
- Y. Li, S. A.-e.-A. Rizvi, D. Hu, D. Sun, A. Gao, Y. Zhou, J. Li and X. Jiang, Angew. Chem., Int. Ed., 2019, 58, 13499 CrossRef CAS.
- C. Li, N. Mizuno, K. Murata, K. Ishii, T. Suenobu, K. Yamaguchi and K. Suzuki, Green Chem., 2020, 22, 3896 RSC.
- X. Zhu, Y. Liu, C. Liu, H. Yang and H. Fu, Green Chem., 2020, 22, 4357 RSC.
- (a) Z. Cao, Q. Zhu, Y.-W. Lin and W.-M. He, Chin. Chem. Lett., 2019, 30, 2132 CrossRef CAS; (b) L.-Y. Xie, T.-G. Fang, J.-X. Tan, B. Zhang, Z. Cao, L.-H. Yang and W.-M. He, Green Chem., 2019, 21, 3858 RSC; (c) L.-Y. Xie, S. Peng, T.-G. Fan, Y.-F. Liu, M. Sun, L.-L. Jiang, X.-X. Wang, Z. Cao and W.-M. He, Sci. China: Chem., 2019, 62, 460 CrossRef CAS; (d) W.-H. Bao, Z. Wang, X. Tang, Y.-F. Zhang, J.-X. Tan, Q. Zhu, Z. Cao, Y.-W. Lin and W.-M. He, Chin. Chem. Lett., 2019, 30, 2259 CrossRef CAS; (e) S. Peng, Y.-X. Song, J.-Y. He, S.-S. Tang, J.-X. Tan, Z. Cao, Y.-W. Lin and W.-M. He, Chin. Chem. Lett., 2019, 30, 2287 CrossRef CAS.
- S. M. Bonesi, I. Manet, M. Freccero, M. Fagnoni and A. Albini, Chem. – Eur. J., 2006, 12, 4844 CrossRef CAS.
- E. Baciocchi, T. D. Giacco, F. Elisei, M. F. Gerini, M. Guerra, A. Lapi and P. Liberali, J. Am. Chem. Soc., 2003, 125, 16444 CrossRef CAS.
- K.-J. Liu, J.-H. Deng, T.-Y. Zeng, X.-J. Chen, Y. Huang, Z. Cao, Y.-W. Lin and W.-M. He, Chin. Chem. Lett., 2020, 31, 1868 CrossRef CAS.
- (a) Y. Li, T. Mou, L. Lu and X. Jiang, Chem. Commun., 2019, 55, 14299 RSC; (b) L. Lu, Y. Li and X. Jiang, Green Chem., 2020, 22, 5989 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc02663h |
| This journal is © The Royal Society of Chemistry 2021 |