
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct dimethyl carbonate synthesis from CO2 and methanol catalyzed by CeO2 and assisted by 2-cyanopyridine: a cradle-to-gate greenhouse gas emission study†
Hajime
Ohno
 a,
Mahdi
Ikhlayel
a,
Masazumi
Tamura
b,
Kenji
Nakao
c,
Kimihito
Suzuki
c,
Kentaro
Morita
d,
Yuzuru
Kato
d,
Keiichi
Tomishige
*e and
Yasuhiro
Fukushima
*a
a,
Mahdi
Ikhlayel
a,
Masazumi
Tamura
b,
Kenji
Nakao
c,
Kimihito
Suzuki
c,
Kentaro
Morita
d,
Yuzuru
Kato
d,
Keiichi
Tomishige
*e and
Yasuhiro
Fukushima
*a
aDepartment of Chemical Engineering, Graduate School of Engineering, Tohoku University, 6-6-07, Aramaki Aza Aoba, Aoba-ku, Sendai, Miyagi 980-8579, Japan. E-mail: fuku@sis.che.tohoku.ac.jp
bResearch Center for Artificial Photosynthesis, Osaka City University, 3-3-138, Sugimoto, Sumiyoshi-ku, Osaka, 558-8585, Japan
cNippon Steel Corporation, 20-1 Shintomi, Futtsu, Chiba 293-8511, Japan
dNippon Steel Engineering Co., Ltd., 46-59, Nakabaru, Tobata-ku, Kitakyushu-shi, Fukuoka 804-8505, Japan
eDepartment of Applied Chemistry, Graduate School of Engineering, Tohoku University, 6-6-07, Aramaki Aza Aoba, Aoba-ku, Sendai, Miyagi 980-8579, Japan. E-mail: tomi@erec.che.tohoku.ac.jp
First published on 8th December 2020
Abstract
The direct synthesis of dimethyl carbonate (DMC) from CO2 and methanol is an attractive alternative route utilizing CO2 instead of toxic phosgene as a carbonate source. The route is thermodynamically difficult because of the equilibrium limitation of the reaction 2CH3OH + CO2 → (CH3O)2O + H2O. In addition, the azeotrope formed by DMC and methanol makes the separation of DMC from unreacted methanol complex and energy intensive. The use of CeO2 and 2-cyanopyridine as a catalyst and dehydration agent solved both the equilibrium constraint and the separation challenge. In this study, the direct DMC synthesis from CO2 and methanol over CeO2 with 2-cyanopyridine was evaluated in terms of greenhouse gas (GHG) emission with the aid of process simulation. It was validated that the cradle-to-gate greenhouse gas emission attributed to the product of this system (0.39 kg-CO2-eq per kg-DMC) becomes much lower than that in conventional commercialized processes. The heat exchange in the process reduced the emission further to 0.34 kg-CO2-eq per kg-DMC. Among the items associated with emissions, methanol consumption shared the largest part (0.63 kg-CO2-eq per kg-DMC), while the converted CO2 was regarded as an important offset (−0.49 kg-CO2-eq per kg-DMC). It is due to the use of the typical methanol production from natural gas (0.88 kg-CO2-eq per kg-methanol). It suggests that if the methanol production with its associated GHG emission accounting for less than 0.41 or 0.34 kg-CO2-eq per kg-methanol is applicable for with or without heat-exchanging cases, the presented process achieves negative emission. Furthermore, based on the results, the requirements for the practical process implementation are discussed by comparing the lifecycle GHG emission results with other DMC synthesis routes.
Introduction
The reduction of atmospheric CO2 concentration by carbon capture and utilization (CCU) is recognized as one of the technological solutions to mitigate climate change.1–3 In this regard, where and how CO2 should be captured, and what should be produced by utilizing CO2 as a feedstock are of considerable interest in various fields.3–8 As emphasized in previous studies,3,6–9 the chemical industry plays a significant role in addressing the excess CO2 emissions by converting CO2 into a feedstock for the production of various chemicals and decoupling it from fossil resources.There are two types of chemical CO2 conversion to feedstocks: reductive and non-reductive. The reductive conversion was considered to utilize CO2 for the synthesis of an energy source (e.g., methane and methanol) and CO for the further synthesis of feedstocks.8,9 However, its contribution to net CO2 emission reduction highly depends on the availability of energy sources generated by a low-carbon technology because huge energy is required to reduce the C atom in CO2 which is thermodynamically stable.3,7–9 In contrast, the non-reductive conversion into carbonates, carbamates, and ureas utilizing the carbonyl group present in CO2 is less energy-intensive by conserving the oxidation state of the C atom.3,6,8,10 As a result of energy saving, the non-reductive conversion has an advantage in net CO2 emission reduction against the reductive conversion.3,8,10
As one of the non-reductive CO2 conversions, the synthesis of dimethyl carbonate (DMC) has attracted attention. DMC is recognized as a green reagent for methylation and carbonylation, substituting toxic dimethyl sulfate and phosgene.11 DMC is an alternative carbonyl source to phosgene for producing diphenyl carbonate which is a precursor of mass-consumed polycarbonates.12
Historically, DMC itself has been produced from phosgene although the replacement of this route has been proceeding.6,11,13 Accordingly, many DMC synthesis routes have been investigated so far13 including CO2-utilizing routes which satisfy multiple principles of green chemistry.14 Among the proposed routes, those starting from CO (e.g., Bayer, EniChem, and Ube industries) and the transesterification of a cyclic carbonate synthesized from CO2 (e.g., the Asahi Kasei process) are commercialized.6,13 If CO could be obtained from CO2, the former routes can also be regarded as a synthesis of DMC from CO2.6 However, owing to the considerable energy inputs required to synthesize precursors such as CO and cyclic carbonates from CO2, the merit of these processes to reduce CO2 emission may be limited.
In contrast, the direct DMC synthesis from CO2 and methanol (MeOH) is a promising route that is based on a much simpler reaction (eqn (1)) requiring no precursor. However, it is restricted by the thermodynamic stability of CO2.15,16
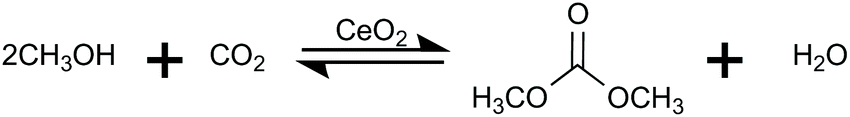 | (1) |
Various catalysts, such as ZrO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 17–19 and Cu–Ni based20–23 catalysts, have been explored for the direct DMC synthesis to address this limitation.15,24 Additionally, the introduction of dehydration agents24–28 and utilization of reactive distillation27 were also suggested to shift the reaction equilibrium. However, there is still a drawback in separating DMC from the reaction effluent because of the azeotrope formation between unreacted MeOH and DMC. Therefore, high MeOH-based conversions are needed for direct DMC synthesis.10,24
17–19 and Cu–Ni based20–23 catalysts, have been explored for the direct DMC synthesis to address this limitation.15,24 Additionally, the introduction of dehydration agents24–28 and utilization of reactive distillation27 were also suggested to shift the reaction equilibrium. However, there is still a drawback in separating DMC from the reaction effluent because of the azeotrope formation between unreacted MeOH and DMC. Therefore, high MeOH-based conversions are needed for direct DMC synthesis.10,24
In addition to the abovementioned catalysts, there have been a lot of recent reports on DMC synthesis from CO2 and methanol using CeO2-based catalysts, which were reported in our previous studies.29–32 We reported that the combination of DMC synthesis from CO2 and methanol with the hydration of nitriles, where both the reactions are catalyzed by CeO2, was effective in the enhancement of MeOH conversion into DMC,33–35 and 2-cyanopyridine (2-CP) was found to be one of the most effective nitriles.24,25 In addition to methanol, CeO2 catalyzes the reaction of CO2 with a variety of alcohols and amines to give the corresponding carbonates,36,37 carbamates,38,39 ureas40,41 and so on.42 The combination of nitrile hydration and the reactions with severe equilibrium limitation was found to be effective for high conversion of the target products.43–50
Honda et al. (2013)25 effectively utilized CeO2 and 2-CP as a catalyst and dehydration agent, respectively, to directly synthesize DMC. This system achieved a 96% MeOH conversion into DMC by the efficient and quick removal of water that was generated together with DMC during the reaction (eqn (1)) of hydrating 2-CP into 2-picolinamide (2-PA, eqn (2)).24 Both reactions are catalyzed by CeO2.
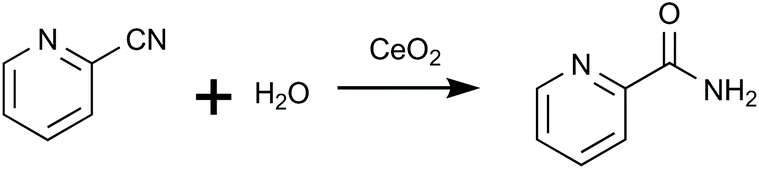 | (2) |
Owing to the high conversion, the energy required to extract DMC was lower compared to the direct DMC synthesis with other catalytic systems. In addition, the continuous synthesis process51–53 and the potential to regenerate 2-CP by dehydrating 2-PA (the reverse reaction of eqn (2))25,54 were also suggested. Although this system is highly attractive in terms of system-wide, net CO2 emission reduction, it has not been systematically evaluated yet.
The direct DMC synthesis was evaluated by Yu et al. (2018)55 and Wu and Chien (2019)56 who conducted techno-economic assessments using process simulations to quantify the gate-to-gate CO2 emissions from the processes. In both pioneering studies, ethylene oxide (EO) was used as the dehydration agent with the Cu–Ni bimetallic nanocomposite catalyst, resulting in a low gate-to-gate CO2 emission (0.049 to −0.232 kg-CO2 per kg-DMC) from the process. Because CO2 was used as a feedstock, the net emission could be negative. Yu et al. (2020)57 synthesized diethyl carbonate by reacting CO2 with 2-CP catalyzed by CeO2. They revealed the effectiveness of 2-CP as a dehydration agent as well as the importance of the 2-CP regeneration process in reducing CO2 emissions. These results demonstrate the great potential to directly synthesize DMC with dehydration agents to lower CO2 emissions in DMC as well as polycarbonate production.
In this study, CO2 was used to directly produce DMC with 2-CP catalyzed by CeO2. The CO2 utilization was systematically investigated by evaluating the lifecycle greenhouse gas emission (GHG) supported by process simulations. By simulating the process, including the reaction, separation, dehydration agent regeneration, and recycling loops, the energy demand for the DMC synthesis process was validated and the GHG emissions associated with the process was evaluated. Furthermore, the GHG emissions to produce feedstocks for the synthesis were also included in the evaluation to extend the scope to the cradle-to-gate domain. Finally, the results of the evaluation were compared with those of other DMC synthesis routes (the oxidative carbonylation of MeOH and the transesterification of ethylene carbonate (EC)) and the direct DMC synthesis process proposed by Wu and Chien (2019)56 by utilizing the break-even analysis as a method of sensitivity analysis.
Methodology and data
Process development: simulation and optimization
The flowsheet of the direct DMC synthesis route over CeO2 with 2-cyanopyridine was developed based on a patent by Shinkai et al. (2017),54 our earlier publications,24,25 and the studies from another research group.51,52 The simulation was performed in Aspen Plus V11.0, and the UNIQUAC method is used for the thermodynamic model applied in this study. The components used in this work consisted of MeOH, CO2, DMC, water, 2-CP, and 2-PA. There was only limited information of basic physical and chemical properties for 2-CP and 2-PA available in the ASPEN database. The missing properties were estimated using ASPEN Plus based on group contribution methods (GCMs) (e.g., the Joback method58 was referred to in the base case of this study). Furthermore, the binary interactions between components were also estimated with the UNIQUAC method.The direct DMC synthesis reaction modeled in this study was based on the kinetics of eqn (1) and (2), both catalyzed by CeO2 and established by Honda et al.24 The reaction provided in eqn (1) effectively occurs because the reaction displayed in eqn (2) shifts the equilibrium of eqn (1) to the carbonate side by removing water with a high reaction rate.24 As a result, 2-PA and DMC are formed. In this study, the 2-CP regeneration by dehydrating 2-PA (i.e., the reverse reaction of eqn (2)) was also simulated to minimize the operating cost and waste generation.57 This reaction is simulated as an equilibrium reaction based on the properties estimated by the UNIQUAC method because of the lack of reaction kinetics, although the use of several catalysts was reported.25,54,59 Side reactions and byproducts were not considered in this study because of the high DMC selectivity in the overall reaction of eqn (1) and (2) (>99%)51 and 2-CP in the 2-PA dehydration.25
The process flow diagram (PFD) of the simulated base case process is shown in Fig. 1. The process consists of a reactor (continuous stirred-tank reactor: CSTR), two distillation columns including a column for reactive distillation, a flash tank, a valve, and a couple of pumps, compressors, coolers, mixers, and splitters.
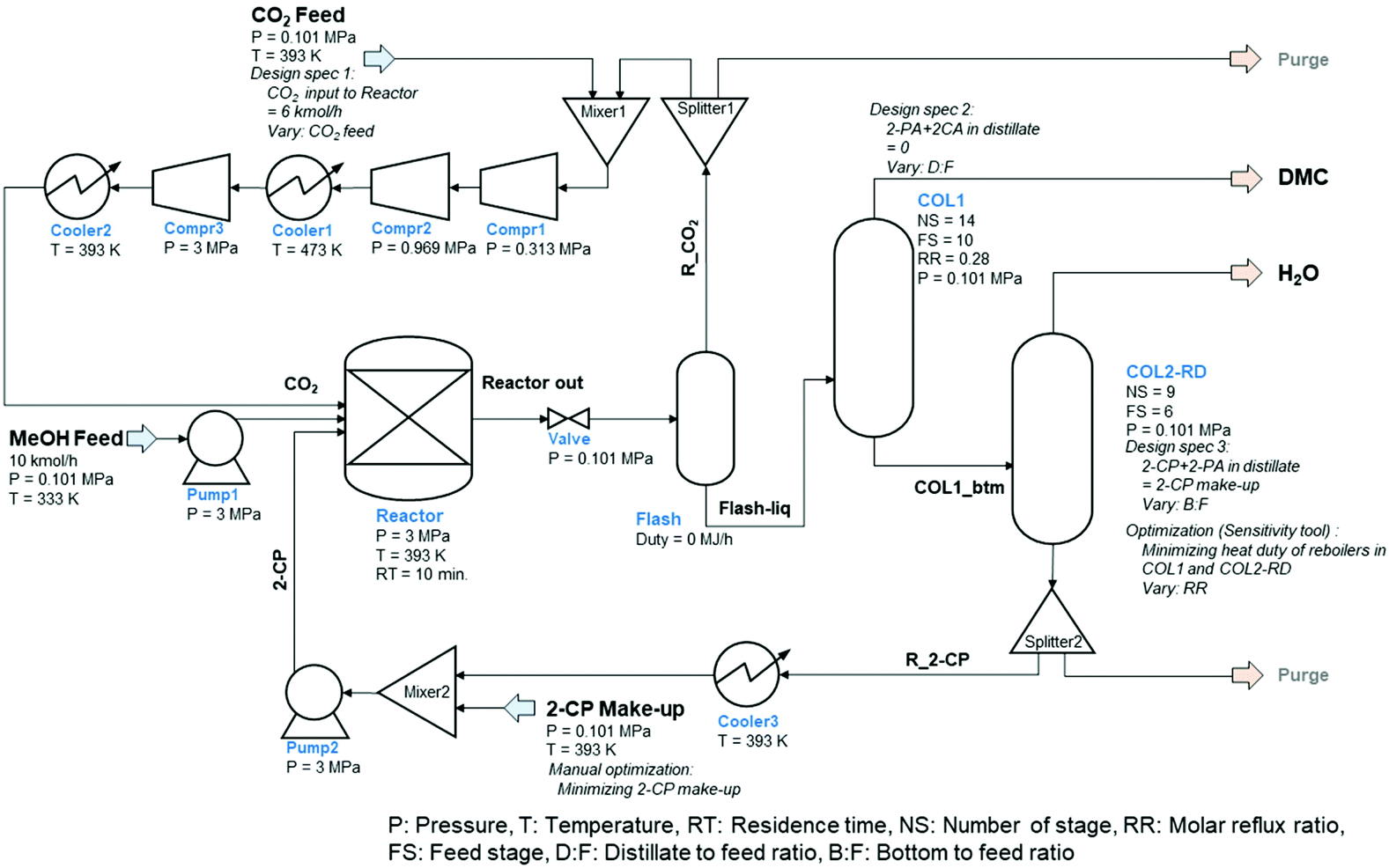 | ||
| Fig. 1 Process flow diagram of the direct DMC synthesis with 2-cyanopyridine catalyzed by CeO2 (base case). | ||
MeOH was fed in the liquid phase at 10 kmol h−1, 0.101 MPa, and 333 K. After pressurizing, MeOH is transferred to the reactor together with CO2, 2-CP, and recycled fractions resulting in the reactions provided in eqn (1) and (2). The reaction temperature, pressure, and residence time were set according to the optimized conditions for the continuous process (393 K, 3 MPa, and 10 min).51 The reaction is a vapor–liquid phase reaction under these conditions. The required catalyst loading for the 10 kmol h−1 MeOH feed was 440 kg CeO2,51 although it was not an input into the simulation because the catalyst effect was already reflected in the reaction kinetics. The reactor effluent, which is in the liquid phase, connects to the pressure release valve reducing the pressure to ambient (i.e., 0.101 MPa). After reducing the pressure, unreacted CO2, MeOH, and a part of the DMC were vaporized and separated for recycling by flash operation under adiabatic conditions. The liquid phase mainly containing DMC and 2-PA was sent to the first column (COL1), which was modeled by RadFrac to extract DMC as a distillate. The required conditions, number of stages, feed stage, and reflux ratio were determined using the DSTWU (Winn–Underwood–Gilliland shortcut design and calculation) model in advance of the simulation. The distillate-to-feed ratio was modified by the design-specification function in ASPEN Plus during the entire simulation to maintain the mass balance in the process including the recycling loops. The bottom stream from COL1 was sent to the reactive distillation column (COL2-RD) to regenerate 2-CP by dehydrating 2-PA (i.e., the reverse reaction of eqn (2)). The specifications (spec) of the column were roughly determined by DSTWU and adjusted in detail by utilizing the design-spec and the sensitivity analysis functions. The distillate of COL2-RD mostly consisted of water that was eliminated from the process. The regenerated 2-CP was extracted from the bottom of COL2-RD to be recycled back to the reactor through Splitter2, Cooler2, Mixer2, and Pump2 to adjust the temperature and pressure to that of the MeOH feed. The make-up of 2-CP was added from Mixer2 to maintain the mass balance of 2-CP in the process. With respect to another recycling loop in the process, the vapor fraction from Flash was mixed with the CO2 feed provided under normal pressure. To gradually pressurize the stream to 3 MPa, three compressors (Compr1–3) were introduced. The number of compressors dictated the pressure change ratio between two compressors to be less than 5.60 Considering the heat resistance of the compressors, Cooler1 was installed between Comp2 and 3 to maintain the temperature of the compressed streams below 473 K because the temperature of the stream increased based on the compression. The stream was cooled to 393 K by Cooler2. The flow rates of the CO2 feed and 2-CP make-up were determined by the design-spec function and manual optimization. Because there were several specifications determined by the design-spec function, the optimization function in ASPEN Plus hardly converged to minimize the flow rate of the 2-CP feed within the iteration limit of the Wegstein method. Consequently, the minimum make-up of 2-CP was determined by manually reducing the value in a stepwise manner.
Although two purge streams were included in the process, they were not active because there was no accumulation of inert components in the process. Considering the side reactions and by-products24 in the process, the two purge streams to be split from the recycling streams will play an important role in maintaining the process mass balance. However, this is not the case in this study. Moreover, pressure drops in each unit were not considered throughout the simulation to simplify the process.
The choice of the GCM affects the conversion of the reverse reaction of eqn (2). In the base case, the standard Gibbs free energy of formation 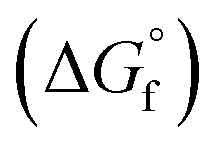 of 2-CP and 2-PA was estimated by the Joback method.58 Based on the GCM, the reverse reaction of eqn (2) proceeds spontaneously because of the negative Gibbs free energy change in the reaction. Furthermore, the reaction hardly proceeds with the use of the Benson method61 for
of 2-CP and 2-PA was estimated by the Joback method.58 Based on the GCM, the reverse reaction of eqn (2) proceeds spontaneously because of the negative Gibbs free energy change in the reaction. Furthermore, the reaction hardly proceeds with the use of the Benson method61 for 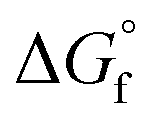 of both 2-CP and 2-PA. Considering this variety, the 2-CP regeneration process based on the different GCM was also simulated in addition to the base case shown in Fig. 1. The process and results are described in the ESI.†
of both 2-CP and 2-PA. Considering this variety, the 2-CP regeneration process based on the different GCM was also simulated in addition to the base case shown in Fig. 1. The process and results are described in the ESI.†
Lifecycle GHG emission accounting
The direct DMC synthesis process was evaluated by particularly focusing on GHG emission quantified as the CO2 equivalent value (kg-CO2-eq). The scope of the evaluation is cradle-to-gate, which includes the emission in the production of feedstocks consumed in the process as well as that induced by the consumption of energy in the DMC synthesis process. The functional unit was a 1 kg-DMC production.The cradle-to-gate emission Etotal (kg-CO2-eq per kg-DMC) was calculated as follows:
| Etotal = Efeedstock + Eenergy + Eothers, | (3) |
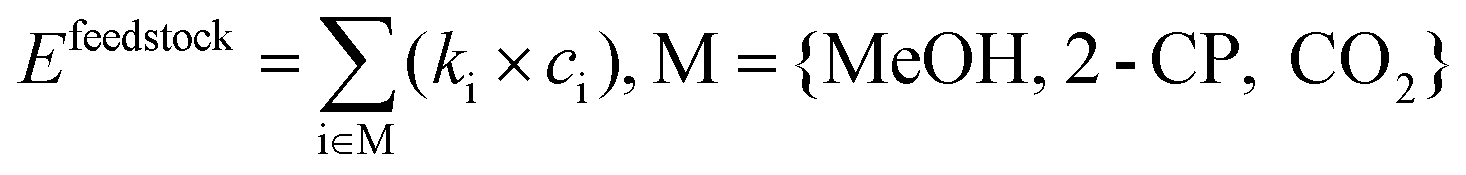 | (4) |
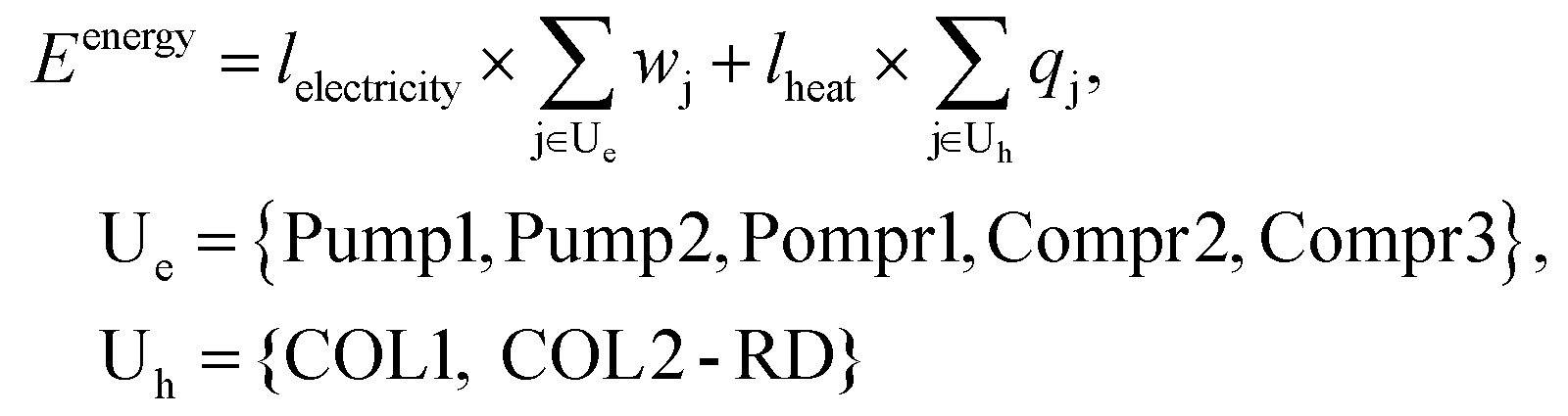 | (5) |
Break-even analysis
Based on the Etotal analysis, the performance of the direct DMC synthesis was compared with those of already commercialized processes included in the lifecycle assessment (LCA) database (Ecoinvent version 3.563 and the extension database for GaBi ts “Ia: organic intermediates”64) and previous studies on other direct synthesis processes.56 The referenced processes correspond to the transesterification of cyclic carbonates63 (for example, Asahi-chem. process6), oxidative carbonylation of MeOH64 (e.g., EniChem process11), and direct synthesis with ethylene oxide (EO) as a dehydration agent.56 Although there are several studies on conducting LCA of the transesterification of cyclic carbonates,16,65 oxidative carbonylation,16,66 and also other DMC synthesis routes,67 the values from the databases were used for more clearer comparison. In terms of the system boundary, the databases’ values consider the emissions in transportation and construction processes although these are not considered in the studied process due to the lack of consideration of the process scale and the period of the production. Because the contributions from transportation and construction in the databases’ values tend to be negligible, it remains a future direction of this study. The difference from the reference process ΔE was formulated as follows:| ΔE = Eref − Etotal, | (6) |
As comprehensively summarized by von der Assen et al. (2016), the efficiency of carbon capture varies with the CO2 source and the available capturing technology.5 Furthermore, due to the lack of kinetic data for the reverse reaction of eqn (2), the reaction in COL2-RD was simulated in this study as an equilibrium reaction. It achieved almost 100% conversion and separation at normal pressure in accordance with the nature of the equilibrium reaction in the simulator, although pioneering studies reported technical difficulties in the 2-PA dehydration, such as the low reaction rate (0.12 mmol h−1 gcat−1, catalyzed by Na2/SiO2)25 and the need for a reactive distillation with solvent under low pressure (502 K, 54.3 kPa catalyzed by Cs2O/SiO2).54 The details of the simulated reaction in COL2-RD are provided in the ESI.†
Accordingly, the emission inventory of captured CO2ki (i = CO2) and the energy consumption in COL2-RD (i.e., heat duty of the reboiler) qj (j = COL2-RD) were set as variables because uncertainties of these parameters and results were likely to be high. Consequently, eqn (3) can be reformulated as:
| Etotal = Efix + kici + lheatqj, (i = CO2, j = COL2-RD) | (7) |
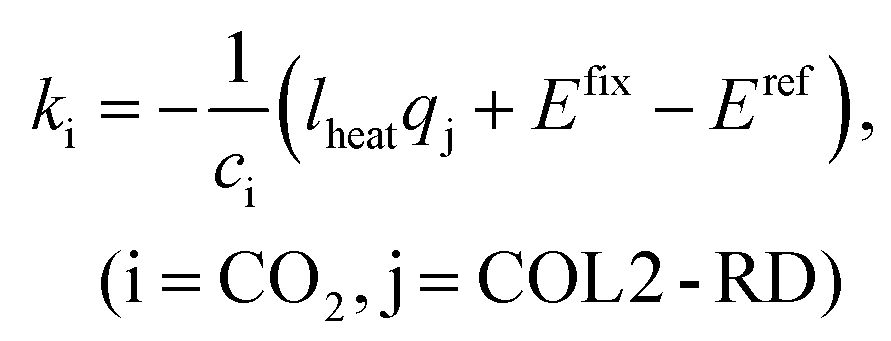 | (8) |
Data
The data used as GHG emission inventories of feedstocks and energy supplies were referred from Ecoinvent version 3.563 and Professional Database62 provided for GaBi ts version 9.2.68 The inventory for electric power generation was determined based on the Japanese grid mix in 2017.69 The inventories were assessed based on the CML 2001 method.70 The inventories used in the evaluation are given in the ESI.†
The lifecycle inventory for 2-CP production could not be obtained from any database or literature. Thus, it was estimated using a molecular structure-based model: Finechem.71,72 Although the use of this method may introduce a large uncertainty to the result,73 it is useful to estimate the embodied GHG emission of the target chemical based on its molecular structure by applying an artificial neural network.71–73 Details of this method and the validation of its result are described in the ESI.†
Results and discussion
Process overview: streams and unit operations
The results of the simulation in terms of streams are summarized in Table 1. In the simulated process, 5.01 kmol h−1 of product stream was obtained including 99.7% DMC with a small amount of residual CO2 and MeOH against 10 kmol h−1 of MeOH feed. The DMC yield based on MeOH feed was 99.4%. Despite a high conversion in the reactor (>99.9%), 0.028 kmol h−1 DMC was lost together with the water removed from COL2-RD. This was related to the design specifications for COL1 to not contaminate the product stream with 2-PA and 2-CP.| Stream name | MeOH feed | CO2 Feed | 2-CP make-up | 2-CP | CO2 | Reactor_out | R_CO2 | Flash_liq | DMC | COL1_btm | Water | R_2CP |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| From | Pump2 | Cooler1 | Reactor | Flash | Flash | COL1 | COL1 | COL2-RD | Splitter2 | |||
| To | Pump1 | Mixer1 | Mixer2 | Reactor | Reactor | Valve | Splitter1 | COL1 | COL2-RD | Cooler3 | ||
| Phase | Liquid | Vapor | Liquid | Liquid | Mixed | Liquid | Vapor | Liquid | Liquid | Liquid | Liquid | Liquid |
| Temperature (K) | 333 | 393 | 393 | 394 | 393 | 393 | 370 | 370 | 341 | 515 | 488 | 488 |
| Pressure (MPa) | 0.101 | 0.101 | 0.101 | 3.00 | 3.00 | 3.000 | 0.101 | 0.101 | 0.101 | 0.101 | 0.101 | 0.101 |
| Molar vapor fraction | 0.00 | 1.00 | 0.00 | 0.00 | 0.80 | 0.00 | 1.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Mole flow (kmol h−1) | 10.00 | 5.03 | 0.02 | 5.01 | 7.74 | 12.75 | 2.70 | 10.04 | 5.01 | 5.04 | 5.05 | 4.99 |
| Mass flow (kg h−1) | 320.42 | 221.45 | 2.08 | 521.72 | 423.13 | 1265.27 | 201.68 | 1063.59 | 449.30 | 614.29 | 94.64 | 519.64 |
| Mole flows (kmol h −1 ) | ||||||||||||
| MeOH | 10.000 | 0.000 | 0.000 | 0.000 | 0.001 | 0.002 | 0.001 | 0.001 | 0.001 | 0.000 | 0.000 | 0.000 |
| CO2 | 0.000 | 5.032 | 0.000 | 0.000 | 5.996 | 0.997 | 0.964 | 0.033 | 0.033 | 0.000 | 0.000 | 0.000 |
| DMC | 0.000 | 0.000 | 0.000 | 0.000 | 1.657 | 6.656 | 1.657 | 4.999 | 4.971 | 0.028 | 0.028 | 0.000 |
| 2-PA | 0.000 | 0.000 | 0.000 | 0.000 | 0.081 | 5.081 | 0.081 | 4.999 | 0.000 | 4.999 | 0.000 | 0.000 |
| 2-CP | 0.000 | 0.000 | 0.020 | 5.011 | 0.000 | 0.012 | 0.000 | 0.012 | 0.000 | 0.012 | 0.020 | 4.991 |
| Water | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 4.999 | 0.000 |
| Mass fractions | ||||||||||||
| MeOH | 1.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| CO2 | 0.000 | 1.000 | 0.000 | 0.000 | 0.624 | 0.035 | 0.210 | 0.001 | 0.003 | 0.000 | 0.000 | 0.000 |
| DMC | 0.000 | 0.000 | 0.000 | 0.000 | 0.353 | 0.474 | 0.740 | 0.423 | 0.997 | 0.004 | 0.026 | 0.000 |
| 2-PA | 0.000 | 0.000 | 0.000 | 0.000 | 0.023 | 0.490 | 0.049 | 0.574 | 0.000 | 0.994 | 0.000 | 0.000 |
| 2-CP | 0.000 | 0.000 | 1.000 | 1.000 | 0.000 | 0.001 | 0.000 | 0.001 | 0.000 | 0.002 | 0.022 | 1.000 |
| Water | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.952 | 0.000 |
The CO2 feed flow rate was calculated by the design-spec function. Because the conversion of the reaction (eqn (1)) in the reactor depends on the CO2 flow rate into the reactor (Fig. S4†), the CO2 feed rate into the process was designed to keep the CO2 input to the reactor higher than the stoichiometry (6 kmol h−1) through the recycling loop. As a result, 0.032 kmol h−1 more CO2 was fed into the process. This excess CO2 amount into the reactor effectively reduced impurities in DMC extracted from the top of COL1. Although the reaction conversion in the reactor is quite high (>99%) under the specified conditions, residual MeOH is contained in the reactor effluent (Reactor_out). By vaporizing and separating dissolved CO2via the valve and the flash together with a part of the MeOH towards recycling, the MeOH content in the distillate of COL1 was halved (0.001 kmol h−1).
Furthermore, a high yield was achieved by a mere 0.02 kmol h−1 make-up of 2-CP, corresponding to 0.4% of the stoichiometry of the overall reaction of eqn (1) and (2). It results from the almost complete regeneration and separation of 2-CP in COL2-RD. The output of water from the top of COL2-RD contained 0.02 kmol h−1 of 2-CP, and thus the mass balance between the input and output of 2-CP was closed. For this mass balance, the bottom to feed ratio of COL2-RD was designed to be 0.991 by the design-spec function.
Fig. 2 shows (a) the key parameters used and designed for the columns (COL1 and COL2-RD), and (b) and (c) are the temperature profiles (the plot of the number of stages vs. temperature) with the component compositions in the columns. COL1 effectively separated DMC into the distillate and 2-PA into bottoms based on their large difference in boiling point (363 K and 558 K). The temperature was stable between stage numbers 2 to 6 because of the small compositional changes in the stages. However, to achieve a high DMC purity without 2-PA and 2-CP contamination, this number of stages was required for the distillate to feed ratio of 0.498 designed by the design-spec function. DSTWU for COL1 suggested ten to twenty stages to make the separation feasible. To achieve a distillate purity of higher than 99.5% without 2-PA and 2-CP contamination, at least fourteen stages were required according to the RadFrac model. Therefore, the number of stages determined in this study was fourteen. The total of COL2-RD stages is nine. In COL2-RD, the reaction was set to start at stage number 5 and end at stage number 9. As shown in Table S2 and Fig. S1,† the 2-PA dehydration reaction progressed spontaneously based on the 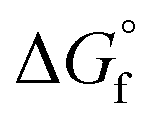 of 2-CP and 2-PA estimated by Joback’ GCM58 converting 2-PA immediately into 2-CP after entering COL2-RD on stage number 6. Notably, such a quick reaction seems to be detached from reality as mentioned in section S1.† The reflux ratio of COL2-RD was determined by the sensitivity analysis tool instead of the optimization tool because of simulation convergence issues. The sensitivity analysis was conducted by varying the reflux ratio of COL2-RD to minimize the energy consumption in the column reboiler within the converging range of the reflux ratio. Increasing the reflux ratio in COL2-RD increases the heat duty of the reboiler. In contrast, when the reflux ratio was low, the simulation was not converged because more than 0.02 kmol h−1 of 2-CP was removed with water from the top. Therefore, the sensitivity analysis explored the minimum converging reflux ratio by focusing on the energy consumption in the column. As a result of the analysis, the lowest reflux ratio that could be converged was 0.9. Consequently, COL2-RD separated 2-CP as bottoms in 100% purity and almost all water (≈5.00 kmol h−1) generated as a result of the reaction (eqn (1)) was removed with the distillate.
of 2-CP and 2-PA estimated by Joback’ GCM58 converting 2-PA immediately into 2-CP after entering COL2-RD on stage number 6. Notably, such a quick reaction seems to be detached from reality as mentioned in section S1.† The reflux ratio of COL2-RD was determined by the sensitivity analysis tool instead of the optimization tool because of simulation convergence issues. The sensitivity analysis was conducted by varying the reflux ratio of COL2-RD to minimize the energy consumption in the column reboiler within the converging range of the reflux ratio. Increasing the reflux ratio in COL2-RD increases the heat duty of the reboiler. In contrast, when the reflux ratio was low, the simulation was not converged because more than 0.02 kmol h−1 of 2-CP was removed with water from the top. Therefore, the sensitivity analysis explored the minimum converging reflux ratio by focusing on the energy consumption in the column. As a result of the analysis, the lowest reflux ratio that could be converged was 0.9. Consequently, COL2-RD separated 2-CP as bottoms in 100% purity and almost all water (≈5.00 kmol h−1) generated as a result of the reaction (eqn (1)) was removed with the distillate.
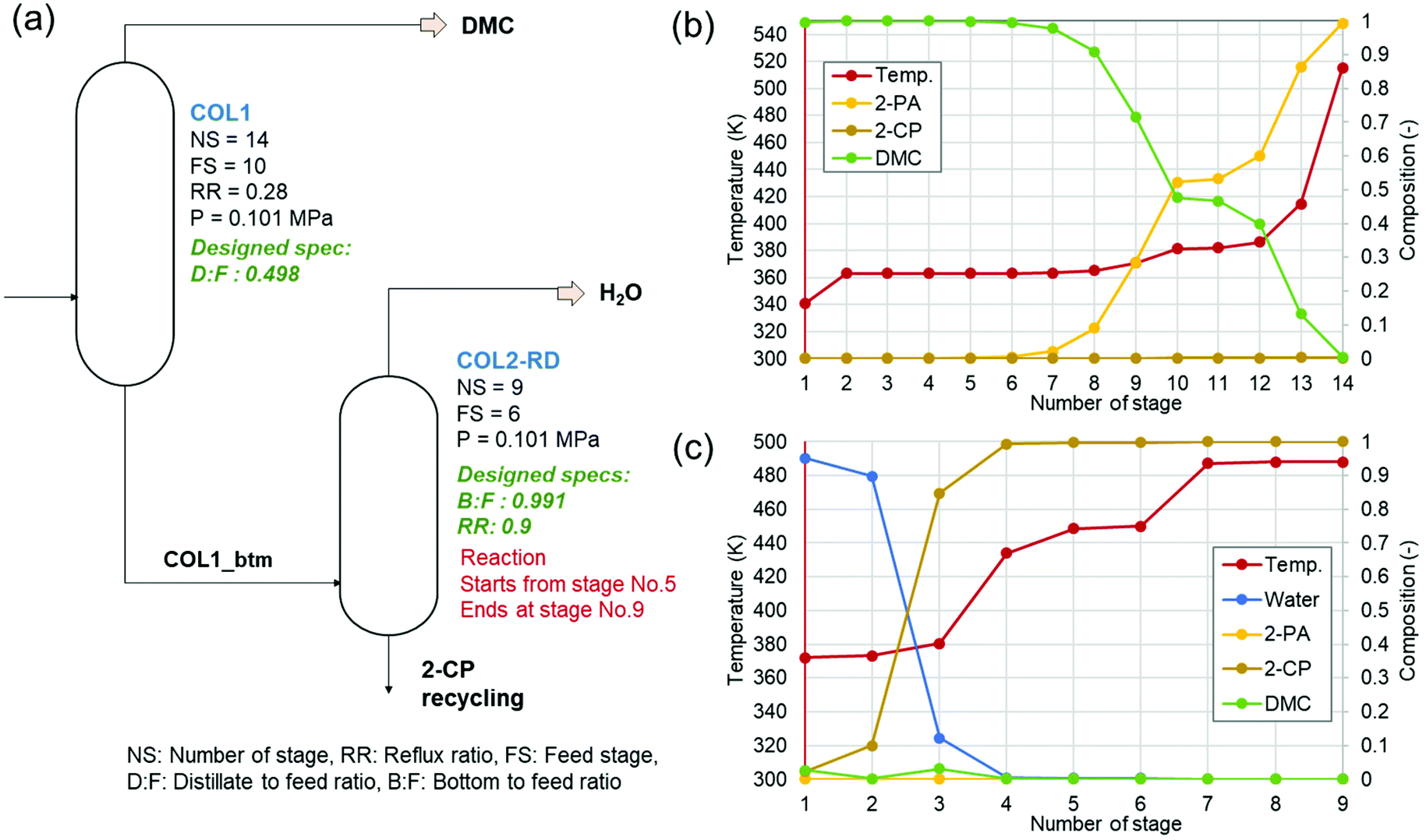 | ||
| Fig. 2 Designs and profiles of columns. (a) Key parameters and designed specifications (written with green), (b) temperature and composition by stages in COL1, and (c) the same for COL2-RD. | ||
Energy consumption
Table 2 summarizes the energy consumption of each process unit. Because the reaction occurring in the reactor was exothermic, the reactor required cooling. Furthermore, cooling was also required for the recycling loops to limit the temperature difference of the streams before the reactor (393 K). The temperature increase in the CO2 recycling loop resulted from the compression (562 K for Compr2, and 569 K for Compr3) and high temperature after the separation of 2-CP in COL2-RD (488 K). For the column condensers cooling was also needed to condense vaporized components at the top. These cooling demands were met by air cooling. The reboilers need to be heated to vaporize the lower boiling point components in the columns. Although the number of stages of COL2-RD (nine stages) was lower than that of COL1 (fourteen stages), COL2-RD had a higher heating demand than COL1 because of its higher reflux ratio of 0.9 compared to 0.28 in COL1. These heating demands were assumed to be supplied by high-pressure steam through the combustion of natural gas. The electricity demands for the pumps were not that high compared to the compressors because there was less volume change in the compression of the liquid compared to the vapor. The compressors required almost even work owing to the gradual compression through the three compressors.| Base case | Heat-exchanging | Energy sources | |
|---|---|---|---|
| a Heat supply by the steam produced by natural gas combustion.63 | |||
| Heat demanding units (MJ h −1 ) | |||
| Reactor | −282.33 | −282.33 | Air |
| Cooler1 (temperature change) | −47.35 (562 to 473 K) | −47.35 (562 to 473 K) | Air |
| Cooler2 (temperature change) | −137.79 (571 to 393 K) | −17.68 (410 to 393 K) | Air |
| Cooler3 (temperature change) | −205.46 (488 to 393 K) | −19.09 (402 to 393 K) | Air |
| COL1_condenser | −238.98 | −238.98 | Air |
| COL2-RD_condenser | −394.71 | −394.71 | Air |
| COL1_reboiler | 446.96 | 141.79 | Steama |
| COL2-RD_reboiler | 602.62 | 602.62 | Steama |
| Electricity demanding units (kW) | |||
| Pump1 | 1.16 | 1.16 | Grid power |
| Pump2 | 0.45 | 0.45 | Grid power |
| Compr1 | 11.55 | 11.55 | Grid power |
| Compr2 | 14.05 | 14.05 | Grid power |
| Compr3 | 14.62 | 14.24 | Grid power |
Focusing on the large temperature change in the coolers, the possibility of heat exchange was considered to reduce the external heating demands in the columns. The locations for the four heat exchangers (HEX1–4) were suggested by the ASPEN energy analyzer. Fig. 3 shows the suggested heat-exchanging streams setting the minimum temperature difference (ΔTmin) to 10 K, which is the lowest value for typical chemical processes.74 The stream between Compr3 and Cooler2 (compressed CO2 feed stream) as well as the stream between Splitter2 and Cooler3 (2-CP recycling) were selected as hot process streams for the exchange. The stream before COL1 (Flash_liq) and the reboiler of COL1 were chosen as cold sides. Each hot process stream supplies heat to both cold process streams. Among these heat exchanges, the combination of Splitter2-to-Cooler3 and Flash_liq represented the ΔTmin point (Fig. S5†). Splitter2-to-Cooler3 provided 186.4 MJ h−1 for the cold process streams utilizing the high enthalpy of the 2-CP recycling stream. Because of these heat exchanges, 305.2 MJ h−1 of heating demand in the COL1_reboiler could be reduced. The energy consumptions for each unit in the heat-exchanging case are summarized in Table 2. Based on this heat exchange, the heats of reaction in the reactor and COL2-RD were not considered suitable for the exchange. By utilizing the excess heat from the reactor, a considerably higher reduction in heating demand would be achieved. In addition to the exchange of the process heat, the introduction of an external heat source such as waste heat from other industries would also reduce the energy consumption in this process.
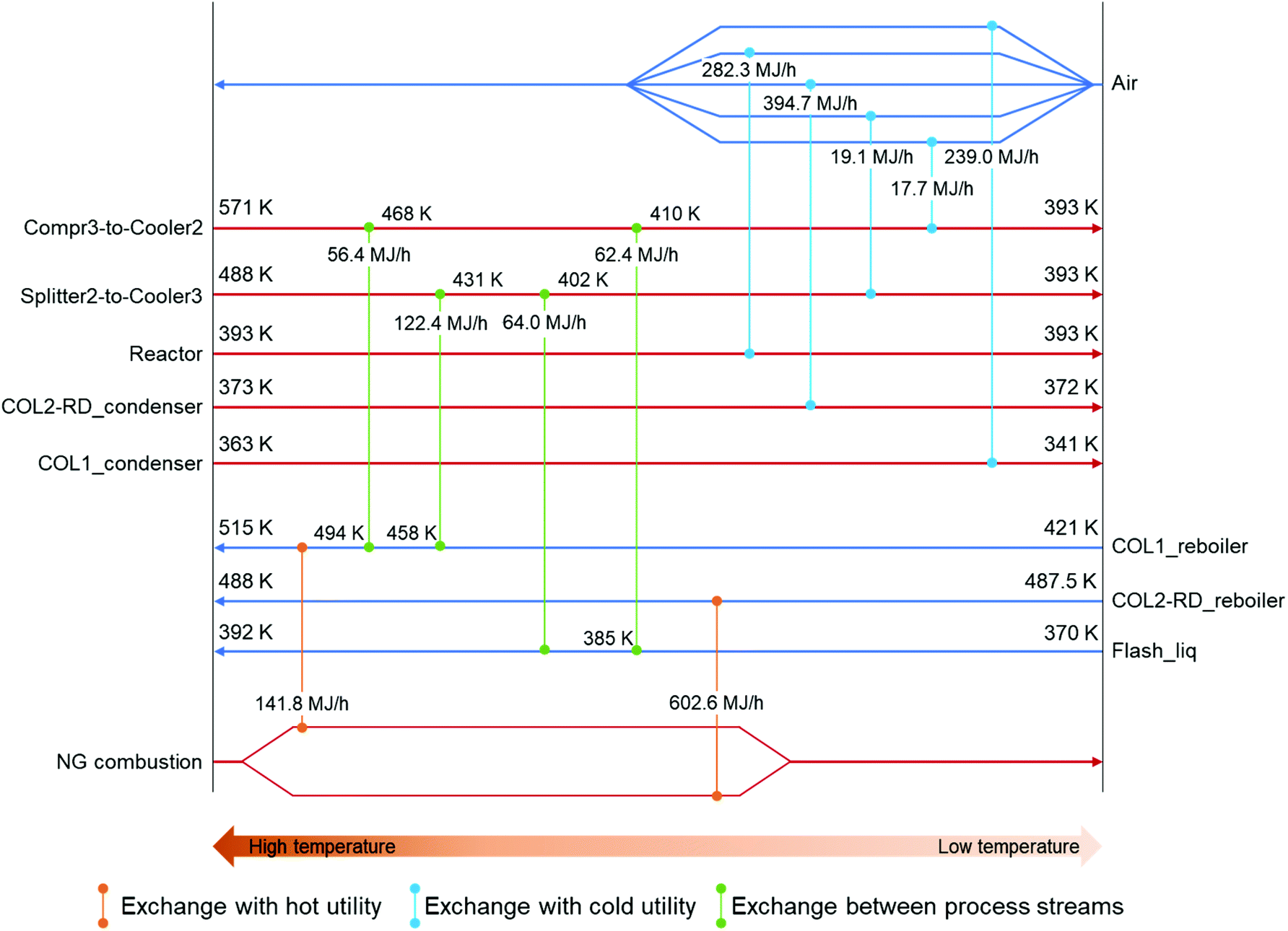 | ||
| Fig. 3 Combination of heat-exchanging streams. Red and blue horizontal arrows represent hot and cold process streams, respectively. The stream names are located on either side according to the stream type. The vertical lines with circle connectors indicate combinations of heat exchange. Temperature changes after the exchanges are indicated right after the connection on the horizontal lines. Exchanged heat was indicated on each vertical line. | ||
Lifecycle GHG emission of the process and comparison with other processes
Based on the energy consumption results in the process, the GHG emissions of the process were evaluated (Table 3). Owing to the use of CO2 as a feedstock, the cradle-to-gate GHG emissions of this 1 kg-DMC process remained low, around 0.34 to 0.39 kg-CO2-eq. The difference between the base and heat-exchanging case is only the emission associated with the energy consumption in COL1. The GHG emissions of the commercialized processes in the databases were 2.12 and 3.43 kg-CO2-eq per kg-DMC for the cyclic carbonate route63 and the oxidative carbonylation route,64 respectively. Compared to these processes, the direct route examined in this study achieves much lower GHG emissions.| Emission category | GHG emission (kg-CO2-eq per kg-DMC) | ||
|---|---|---|---|
| Base case | Heat-exchanging | ||
| Feedstock production | MeOH | 0.63 | 0.63 |
| CO2 | −0.49 | −0.49 | |
| 2-CP | 0.03 | 0.03 | |
| Energy consumption | COL1_reboiler | 0.07 | 0.02 |
| COL2-RD_reboiler | 0.09 | 0.09 | |
| Electricity | 0.06 | 0.06 | |
| Others | Wastewater treatment | 0.01 | 0.01 |
| Total | 0.39 | 0.35 | |
The contribution of MeOH to the total process emissions was the largest. In this study, the lifecycle inventory for MeOH production from natural gas was referred to as the typical conventional MeOH production route.63 Since there are other innovative routes for MeOH production, such as the low-temperature syngas conversion,75 utilization of captured CO2 as a feedstock,76 and the bio-based approach,77 this emission could be reduced. If the emission associated with MeOH production could be decreased to less than 0.24 or 0.29 kg-CO2-eq per kg-DMC in the base or the heat-exchanging case, respectively, this proposed DMC synthesis process would become a negative emission process. To achieve this, MeOH needs to be produced by a technology whose GHG emissions are less than 0.34 or 0.41 kg-CO2-eq per kg-MeOH, respectively. Moreover, it has been reported that CeO2 can also catalyze the reaction of ethanol,24,31,34,78 and 1-propanol,24,35 which can be derived from biomass,79 with CO2 and an appropriate dehydrating agent, can promote the formation of the corresponding carbonates. Therefore, methanol can be replaced by ethanol or 1-propanol as a greener substance, although further investigation on the process using ethanol and 1-propanol is necessary.
Owing to the 2-CP recycling via the reactive 2-PA distillation, the emission associated with 2-CP was sufficiently low, while the embodied GHG emission of 2-CP was assumed to be high (6.2 kg-CO2-eq per kg-2-CP) by the Finechem approach71,72 (Table S4†). If the 2-CP recycling is not applied in the process, 7.16 kg-CO2-eq per kg-DMC would be emitted associated with the use of 2-CP according to the stoichiometry, whereas COL2-RD contributed only 0.09 kg-CO2-eq per kg-DMC to recycle 2-CP. Thus, the recycling of 2-CP is crucial for this process.
With respect to the energy-related emissions, the COL2-RD reboiler was marked as critical, although the contribution did not differ much from other factors. However, this result for the COL2-RD reboiler should be noted as a “singular point”, which could only be achieved in the simulation. Considering the drawbacks of the 2-PA dehydration reported in previous studies,25,54 the further increase in GHG emissions implementing the 2-PA dehydration is easily supposed. By referring to Benson's GCM,61 the equilibrium conversion of 2-PA dehydration at normal pressure was much lower than that of the base case as simulated by Joback's GCM58 (Fig. S1†). Accordingly, an alternative 2-CP regeneration process including a reactor for the 2-PA dehydration, and two distillation columns separating 2-CP, 2-PA, and water at reduced pressure (0.01 MPa) was simulated (Fig. S6†). This process contributes 0.21 kg-CO2-eq per kg-DMC instead of using COL2-RD in the base case. With this process, the excess heat of 2-CP recycling stream becomes no longer utilizable to supply heat to the COL1_reboiler due to a lower temperature (429 K) than the base case (488 K). Consequently, the total GHG emissions of the proposed DMC synthesis were 0.48 and 0.51 kg-CO2-eq per kg-DMC for the cases with and without heat exchange, respectively.
The break-even lines are shown in Fig. 4(a) comparing commercialized63,64 and innovative DMC synthesis technologies56 with the process simulated in this study and the additional requirement to provide zero gate-to-gate emission. In addition, the plots for CO2 capture yields by various CO2 sources were also added, according to von der Assen et al. (2016).5 These yields reflect the energy demands and transportation embedded into the captured amount of CO2 from various CO2 sources located in Europe. Although the location of the present process is assumed to be in Japan, the values of CO2 capture yields: −kCO2 in Europe are useful benchmarks for evaluating the process. The green dots represent the heat-exchanging cases. The comparison with our process referring to the heat-exchanging case was demonstrated using break-even lines. Besides, Fig. 4(b) presents the GHG emission breakdowns by emission contributors in each case corresponding to the typical condition plotted on the break-even lines in Fig. 4(a). Based on Fig. 4(b), the margin for 2-CP regeneration and CO2 capture yields can be observed from the GHG emission point of view.
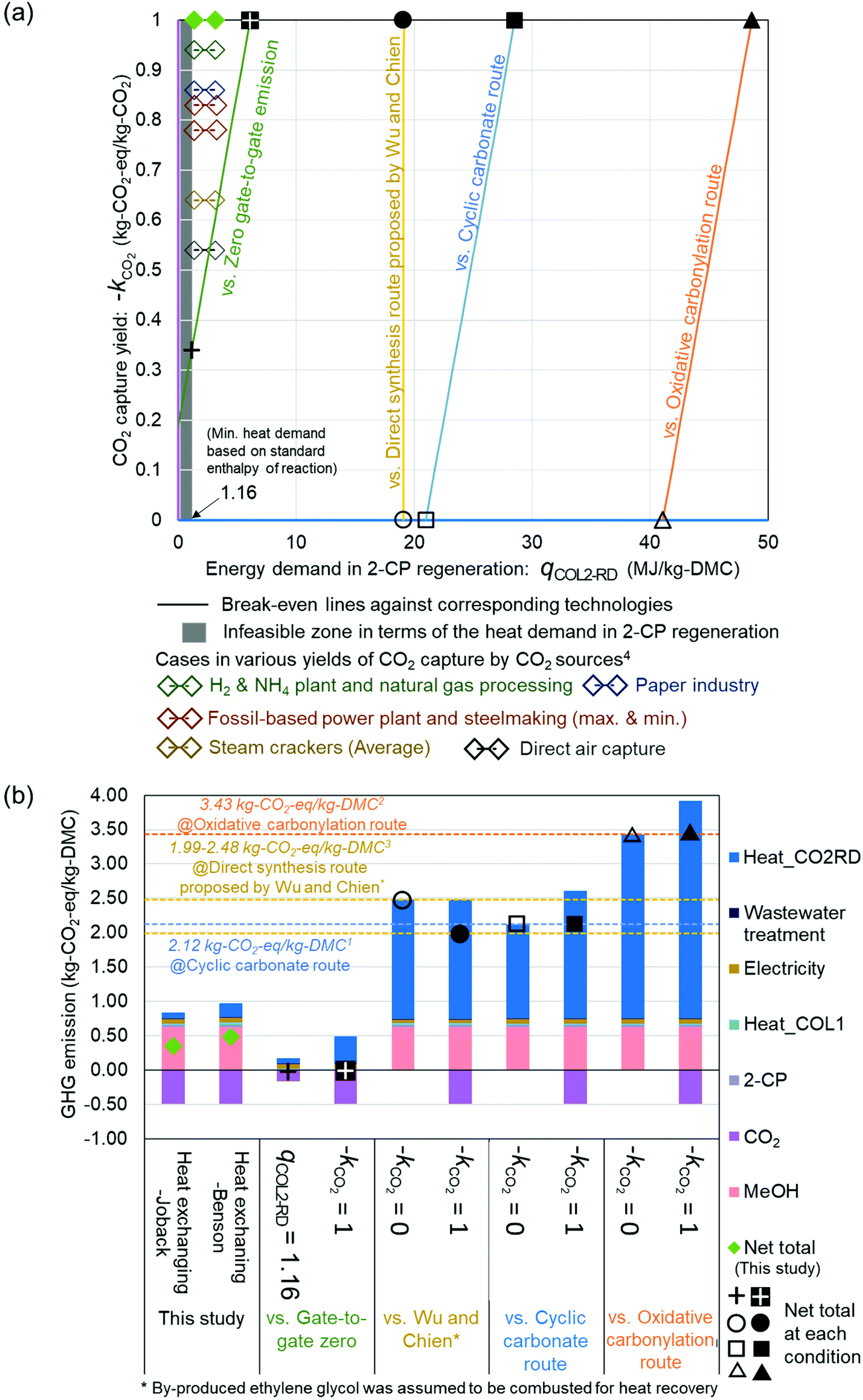 | ||
| Fig. 4 (a) Break-even lines against commercialized63,64 and innovative56 technologies. The x and y-axes represent the energy demand of COL2-RD: qCOL2-RD and CO2 capture yield: −kCO2, respectively. The colors of axes correspond to the color of bars for Heat_COL2-RD and CO2 in (b). When the intersection of the value of the x-axis and y-axis is on a break-even line, the GHG emission for the 1 kg-DMC production over the CeO2 catalyst with 2-CP is identical to the corresponding technology. The heat demand in 2-CP regeneration (x-axis) cannot be less than 1.16 MJ per kg-DMC according to the standard enthalpy of the reaction: 2-PA → 2-CP + H2O. (b) GHG emission per kg DMC production of the studied process as well as the typical cases corresponding to the plot on the break-even lines. The breakdown is colored by emission contributors. 1Ecoinvent version 3.5,632Extension database for GaBi ts “Ia: organic intermediates”,643Calculated based on Wu and Chien (2019),564von der Assen et al. (2016).5 | ||
The simulated case was plotted on the left side of all break-even lines owing to its advantageous assumptions on −kCO2 and the 2-CP regeneration reaction. Even under the thermodynamically more challenging assumption that the regeneration reaction is based on Benson's GCM61 as noted in the ESI (Fig. S1†), the energy demand in 2-CP regeneration (3.15 MJ per kg-DMC) remained on the left side of the break-even lines. To achieve zero gate-to-gate emission in the process, the requirements for −kCO2 and the energy demand in 2-CP regeneration were much stricter compared to other processes: above 0.35 kg-CO2-eq per kg-CO2 and less than 6.2 MJ per kg-DMC, respectively. Against the commercialized processes, there is enough margin for qCOL2-RD. If the ideal CO2 yield (−kCO2 = 1) could be utilized, 2-CP regeneration can consume 28.6 and 48.7 MJ per kg-DMC in this proposed process with lower GHG emissions than the commercialized DMC production via the cyclic carbonate (2.12 kg-CO2-eq per kg-DMC) and oxidative carbonylation route (3.43 kg-CO2-eq per kg-DMC), respectively (Fig. 4(b)). Even if the process utilizes CO2 without any emission reduction effect with respect to CCU (i.e., −kCO2 = 0), qCOL2-RD still maintains a large margin against commercial processes.
In contrast, the direct synthesis route using EO as a dehydrating agent, reported by Wu and Chien (2019),56 is highly competitive to the present process. The comparison of their process with the heat-exchanging case was conducted by consistently referring to the heat and electricity supply inventories used by Wu and Chien (2019).56 Applying their inventories to our process, the total process emission was 0.37 kg-CO2-eq per kg-DMC. Wu and Chien reported that the minimum gate-to-gate GHG emission in the optimized process was negative (−0.23 kg-CO2-eq per kg-DMC). Extending their scope simply to the cradle-to-gate case by additionally considering the embodied GHG emissions of the consumed feedstocks (MeOH and EO), the total emission of the process was 1.37 kg-CO2-eq per kg-DMC without taking the byproduct (0.61 kg of ethylene glycol (EG)) into account. If EG could be sold and substitute the conventional EG production, the total emission would be even much lower (0.17 kg-CO2-eq per kg-DMC), offsetting GHG emissions associated with EG produced as a byproduct. Because the contribution of COL2-RD to the total emission (0.12 kg-CO2-eq per kg-DMC with Wu and Chien's inventory) was lower than the difference between the total emission of our process (0.37 kg-CO2-eq per kg-DMC) and selling EG (0.17 kg-CO2-eq per kg-DMC), our process could not compete with the optimized process of Wu and Chien in terms of GHG emission if EG is sold. However, it would still have some advantages in case EG could not be sold. In the case of not selling EG, it was assumed that the generated EG was combusted as a heat source in the process. Based on the EG combustion, 0.87 kg-CO2-eq per kg-DMC of additional emission occurred while 9.14 MJ per kg-DMC was obtained. By utilizing the obtained heat, the heat demand in the process was balanced and the total emission became 1.99 kg-CO2-eq per kg-DMC and 2.48 kg-CO2-eq per kg-DMC for not selling EG in the case of −kCO2 = 1 and 0, respectively. The break-even line against the no-EG selling case is shown in Fig. 4. The line became vertical because both the compared processes were CCU processes that depend on the carbon capture yield. The value of qCOL2-RD in our process should be below 19.1 MJ per kg-DMC to make the GHG emission of our process less than that reported by Wu and Chien without selling EG.
Conclusion
The direct DMC synthesis process catalyzed by CeO2 using 2-CP was simulated, and its cradle-to-gate GHG emission per kg DMC production was evaluated. Owing to the high conversion, the energy consumption in the reaction step could be less than that of the direct synthesis process proposed by Wu and Chien (2019).56 In Wu and Chien's process, they need a reactive distillation column along with the reactor to separate the recycling stream (i.e., unreacted MeOH and CO2) from the EG and DMC mixture. The reactive distillation column consumes 2.5 MJ per kg-DMC in their system, while our process does not consume energy in the reaction step. Although the GHG emissions of their process decreased below that of our process by selling the byproduct EG, the investment cost of our process could be lower. For example, the present process includes two columns having 14 and 9 stages, respectively, while the process of Wu and Chien needs four columns with 49, 13, 18, and 3 stages, including a reactive distillation column connected to the main reactor for the reaction (eqn (1)). In addition, due to the flammable and explosive nature of EO, the necessity to safely handle the EO in their process was pointed out.56 The use of 2-CP, which is solid (melting point: 302 K) under standard conditions (298 K, 0.101 MPa), would also be advantageous at this point. To verify our process advantages, further investigation into the economic aspects of the process is necessary.Based on the break-even analysis, the direction of the development of the 2-CP regeneration process is suggested. To improve the GHG emission in our process over other processes, except for the EG-selling case of the Wu and Chien process, 19.1 MJ per kg-DMC of qCOL2-RD is allowed in our process. Our process consumes 1.35 to 3.16 MJ per kg-DMC for 2-CP regeneration using the estimation of 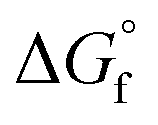 by the GCM of Joback58 and Benson.61 Because the Gibbs free energy change in the regeneration reaction based on
by the GCM of Joback58 and Benson.61 Because the Gibbs free energy change in the regeneration reaction based on 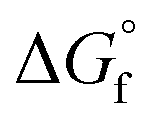 of both 2-CP and 2-PA estimated by Benson's method was the highest among the
of both 2-CP and 2-PA estimated by Benson's method was the highest among the 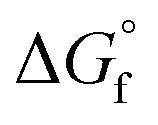 combinations estimated by different GCMs, there is scope to theoretically decrease the GHG emission below that in other DMC synthesis processes. Previously, the 2-PA dehydration was experimentally studied under low pressure with a catalyst and solvent (502 K, 54.3 kPa catalyzed by Cs2O/SiO2).54 To apply such a low-pressure dehydration of 2-PA and a separation of 2-CP from the solvent in the DMC synthesis, it is required to further investigate the operating conditions.
combinations estimated by different GCMs, there is scope to theoretically decrease the GHG emission below that in other DMC synthesis processes. Previously, the 2-PA dehydration was experimentally studied under low pressure with a catalyst and solvent (502 K, 54.3 kPa catalyzed by Cs2O/SiO2).54 To apply such a low-pressure dehydration of 2-PA and a separation of 2-CP from the solvent in the DMC synthesis, it is required to further investigate the operating conditions.
The energy requirement for catalyst reactivation needs to be considered to implement the process. As reported in previous experimental studies, CeO2 gradually deactivates during operation.24,52 The catalyst deactivated after 35 h of continuous synthesis can be reactivated by washing it with methanol at 393 K for 1 h and calcining it with dry air at 573 K for 12 h.52 Accordingly, by introducing multiple fixed bed reactors and switching them for reactivation, the process can be continuously operated. The number of reactors and switching schedules as well as the energy consumption for calcination of the catalyst should be optimized by an economic analysis.
The comprehensive development of a polycarbonate synthesis process (i.e., the major use of DMC) that includes the direct DMC synthesis utilizing CO2 as a feedstock will be investigated in the future. The use of DMC as a carbonyl source substitutes the use of toxic phosgene in PC production.12 In addition to carbonates, carbamate and urea synthesis from CO2 with the same catalytic system has also been established.32,48–50 Such CO2 conversions to chemicals for durable material production can be regarded by society as CO2 sequestration. The decarbonization in durable material production using chemicals from CO2 will be an environmentally friendly way to advance a step further toward a sustainable and greener process development.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This paper is based on the results obtained from a ‘NEDO Feasibility Study Program’ commissioned by the New Energy and Industrial Technology Development Organization (NEDO).References
- J. Rockström, O. Gaffney, J. Rogelj, M. Meinshausen, N. Nakicenovic and H. J. Schellnhuber, Science, 2017, 355, 1269–1271 CrossRef.
- M. Bui, C. S. Adjiman, A. Bardow, E. J. Anthony, A. Boston, S. Brown, P. S. Fennell, S. Fuss, A. Galindo, L. A. Hackett, J. P. Hallett, H. J. Herzog, G. Jackson, J. Kemper, S. Krevor, G. C. Maitland, M. Matuszewski, I. S. Metcalfe, C. Petit, G. Puxty, J. Reimer, D. M. Reiner, E. S. Rubin, S. A. Scott, N. Shah, B. Smit, J. P. M. Trusler, P. Webley, J. Wilcox and N. Mac Dowell, Energy Environ. Sci., 2018, 11, 1062–1176 RSC.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS.
- N. Mac Dowell, P. S. Fennell, N. Shah and G. C. Maitland, Nat. Clim. Change, 2017, 7, 243–249 CrossRef CAS.
- N. von der Assen, L. J. Muller, A. Steingrube, P. Voll and A. Bardow, Environ. Sci. Technol., 2016, 50, 1093–1101 CrossRef CAS.
- J. Artz, T. E. Muller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS.
- A. Katelhon, R. Meys, S. Deutz, S. Suh and A. Bardow, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 11187–11194 CrossRef CAS.
- M. Aresta, A. Dibenedetto and E. Quaranta, J. Catal., 2016, 343, 2–45 CrossRef CAS.
- A. Al-Mamoori, A. Krishnamurthy, A. A. Rownaghi and F. Rezaei, Energy Technol., 2017, 5, 834–849 CrossRef.
- M. Aresta, A. Dibenedetto and A. Dutta, Catal. Today, 2017, 281, 345–351 CrossRef CAS.
- P. Tundo and M. Selva, Acc. Chem. Res., 2002, 35, 706–716 CrossRef CAS.
- S. Fukuoka, I. Fukawa, T. Adachi, H. Fujita, N. Sugiyama and T. Sawa, Org. Process Res. Dev., 2019, 23, 145–169 CrossRef CAS.
- H.-Z. Tan, Z.-Q. Wang, Z.-N. Xu, J. Sun, Y.-P. Xu, Q.-S. Chen, Y. Chen and G.-C. Guo, Catal. Today, 2018, 316, 2–12 CrossRef CAS.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- B. A. V. Santos, V. M. T. M. Silva, J. M. Loureiro and A. E. Rodrigues, ChemBioEng Rev., 2014, 1, 214–229 CrossRef CAS.
- P. Kongpanna, V. Pavarajarn, R. Gani and S. Assabumrungrat, Chem. Eng. Res. Des., 2015, 93, 496–510 CrossRef CAS.
- K. T. Jung and A. T. Bell, J. Catal., 2001, 204, 339–347 CrossRef CAS.
- K. Tomishige, Y. Ikeda, T. Sakaihori and K. Fujimoto, J. Catal., 2000, 192, 355–362 CrossRef CAS.
- K. Tomishige, T. Sakaihori, Y. Ikeda and K. Fujimoto, Catal. Lett., 1999, 58, 225–229 CrossRef CAS.
- J. Bian, M. Xiao, S. J. Wang, Y. X. Lu and Y. Z. Meng, J. Colloid Interface Sci., 2009, 334, 50–57 CrossRef CAS.
- J. Bian, M. Xiao, S.-J. Wang, Y.-X. Lu and Y.-Z. Meng, Appl. Surf. Sci., 2009, 255, 7188–7196 CrossRef CAS.
- X. J. Wang, M. Xiao, S. J. Wang, Y. X. Lu and Y. Z. Meng, J. Mol. Catal. A: Chem., 2007, 278, 92–96 CrossRef CAS.
- X. L. Wu, Y. Z. Meng, M. Xiao and Y. X. Lu, J. Mol. Catal. A: Chem., 2006, 249, 93–97 CrossRef CAS.
- M. Honda, M. Tamura, Y. Nakagawa, K. Nakao, K. Suzuki and K. Tomishige, J. Catal., 2014, 318, 95–107 CrossRef CAS.
- M. Honda, M. Tamura, Y. Nakagawa, S. Sonehara, K. Suzuki, K. Fujimoto and K. Tomishige, ChemSusChem, 2013, 6, 1341–1344 CrossRef CAS.
- V. Eta, P. Mäki-Arvela, J. Wärn, T. Salmi, J. P. Mikkola and D. Y. Murzin, Appl. Catal., A, 2011, 404, 39–46 CAS.
- X. Hu, H. Cheng, X. Kang, L. Chen, X. Yuan and Z. Qi, Chem. Eng. Process., 2018, 129, 109–117 CrossRef CAS.
- T. Sakakura, J.-C. Choi, Y. Saito, T. Masuda, T. Sako and T. Oriyama, J. Org. Chem., 1999, 64, 4506–4508 CrossRef CAS.
- K. Tomishige, Y. Furusawa, Y. Ikeda, M. Asadullah and K. Fujimoto, Catal. Lett., 2001, 76, 71–74 CrossRef CAS.
- K. Tomishige and K. Kunimori, Appl. Catal., A, 2002, 237, 103–109 CrossRef CAS.
- Y. Yoshida, Y. Arai, S. Kado, K. Kunimori and K. Tomishige, Catal. Today, 2006, 115, 95–101 CrossRef CAS.
- K. Tomishige, Y. Gu, T. Chang, M. Tamura and Y. Nakagawa, Mater. Today Sustainability., 2020, 9, 100035 CrossRef.
- M. Honda, A. Suzuki, B. Noorjahan, K. Fujimoto, K. Suzuki and K. Tomishige, Chem. Commun., 2009, 4596–4598 RSC.
- M. Honda, S. Kuno, N. Begum, K.-i. Fujimoto, K. Suzuki, Y. Nakagawa and K. Tomishige, Appl. Catal., A, 2010, 384, 165–170 CrossRef CAS.
- M. Honda, S. Kuno, S. Sonehara, K.-i. Fujimoto, K. Suzuki, Y. Nakagawa and K. Tomishige, ChemCatChem, 2011, 3, 365–370 CrossRef CAS.
- K. Tomishige, H. Yasuda, Y. Yoshida, M. Nurunnabi, B. Li and K. Kunimori, Catal. Lett., 2004, 95, 45–49 CrossRef CAS.
- K. Tomishige, H. Yasuda, Y. Yoshida, M. Nurunnabi, B. T. Li and K. Kunimori, Green Chem., 2004, 6, 206–214 RSC.
- M. Honda, S. Sonehara, H. Yasuda, Y. Nakagawa and K. Tomishige, Green Chem., 2011, 13, 3406–3413 RSC.
- M. Tamura, M. Honda, K. Noro, Y. Nakagawa and K. Tomishige, J. Catal., 2013, 305, 191–203 CrossRef CAS.
- M. Tamura, K. Noro, M. Honda, Y. Nakagawa and K. Tomishige, Green Chem., 2013, 15, 1567–1577 RSC.
- M. Tamura, K. Ito, Y. Nakagawa and K. Tomishige, J. Catal., 2016, 343, 75–85 CrossRef CAS.
- M. Tamura, M. Honda, Y. Nakagawa and K. Tomishige, J. Chem. Technol. Biotechnol., 2014, 89, 19–33 CrossRef CAS.
- M. Honda, M. Tamura, K. Nakao, K. Suzuki, Y. Nakagawa and K. Tomishige, ACS Catal., 2014, 4, 1893–1896 CrossRef CAS.
- M. Tamura, K. Ito, M. Honda, Y. Nakagawa, H. Sugimoto and K. Tomishige, Sci. Rep., 2016, 6, 24038 CrossRef.
- Y. Gu, K. Matsuda, A. Nakayama, M. Tamura, Y. Nakagawa and K. Tomishige, ACS Sustainable Chem. Eng., 2019, 7, 6304–6315 CrossRef CAS.
- Y. Gu, A. Miura, M. Tamura, Y. Nakagawa and K. Tomishige, ACS Sustainable Chem. Eng., 2019, 7, 16795–16802 CrossRef CAS.
- M. Tamura, A. Miura, M. Honda, Y. Gu, Y. Nakagawa and K. Tomishige, ChemCatChem, 2018, 10, 4821–4825 CrossRef.
- M. Honda, M. Tamura, Y. Nakagawa and K. Tomishige, Catal. Sci. Technol., 2014, 4, 2830–2845 RSC.
- K. Tomishige, M. Tamura and Y. Nakagawa, Chem. Rec., 2019, 19, 1354–1379 CrossRef CAS.
- K. Tomishige, Y. Gu, Y. Nakagawa and M. Tamura, Front. Energy Res., 2020, 8, 117 CrossRef.
- A. Bansode and A. Urakawa, ACS Catal., 2014, 4, 3877–3880 CrossRef CAS.
- D. Stoian, A. Bansode, F. Medina and A. Urakawa, Catal. Today, 2017, 283, 2–10 CrossRef CAS.
- D. Stoian, F. Medina and A. Urakawa, ACS Catal., 2018, 8, 3181–3193 CrossRef CAS.
- Y. Shinkai, H. Liu, H. Harada, S. Isahaya, K. Tomishige, Y. Nakagawa, M. Tamura, K. Suzuki and Y. Namiki, WO2017221908A1, 2017.
- B.-Y. Yu, M.-K. Chen and I. L. Chien, Ind. Eng. Chem. Res., 2018, 57, 639–652 CrossRef CAS.
- T.-W. Wu and I. L. Chien, Ind. Eng. Chem. Res., 2019, 59, 1234–1248 CrossRef.
- B.-Y. Yu, P.-J. Wu, C.-C. Tsai and S.-T. Lin, J. CO2 Util., 2020, 41, 101254 CrossRef CAS.
- K. G. Joback and R. C. Reid, Chem. Eng. Commun., 1987, 57, 233–243 CrossRef CAS.
- Y. Li, Y. Zhao, S. Wang and X. Ma, Chin. Chem. Lett., 2019, 30, 494–498 CrossRef CAS.
- L. T. Biegler, I. E. Grossmann and A. W. Wasterberg, Systematic Methods of Chemical Process Design, Pearson, 1997 Search PubMed.
- S. W. Benson, F. R. Cruickshank, D. M. Golden, G. R. Haugen, H. E. O'Neal, A. S. Rodgers, R. Shaw and R. Walsh, Chem. Rev., 1969, 69, 279–324 CrossRef CAS.
- Sphera, Professional Database 2020, http://www.gabi-software.com/support/gabi/gabi-6-lci-documentation/professional-database/, (accessed August 19th, 2020).
- G. Wernet, C. Bauer, B. Steubing, J. Reinhard, E. Moreno-Ruiz and B. Weidema, Int. J. Life Cycle Assess., 2016, 21, 1218–1230 CrossRef.
- Sphera, Extension database Ia: Intermediates organic, http://www.gabi-software.com/support/gabi/gabi-database-2019-lci-documentation/extension-database-ia-intermediates-organic/, (accessed August 19th, 2020).
- J. G. M.-S. Monteiro, O. Q. F. Araújo and J. L. de Medeiros, Clean Technol. Environ. Policy, 2009, 11, 459–472 CrossRef CAS.
- I. Garcia-Herrero, R. M. Cuellar-Franca, V. M. Enriquez-Gutierrez, M. Alvarez-Guerra, A. Irabien and A. Azapagic, ACS Sustainable Chem. Eng., 2016, 4, 2088–2097 CrossRef CAS.
- M. Aresta and M. Galatola, J. Cleaner Prod., 1999, 7, 181–193 CrossRef.
- Sphera, Gabi ts, http://www.gabi-software.com/software/, (accessed July 10th, 2020).
- APEC Energy Working Group: Expert Group on Energy Data and Analysis, Electricity Power Generation Table, https://www.egeda.ewg.apec.org/egeda/database_info/electricity_select_form2.html, (accessed July 10th, 2020).
- H.-J. Althaus, C. Bauer, G. Doka, R. Dones, R. Frischknecht, S. Hellweg, S. Humbert, T. K. Niels Jungbluth, Y. Loerincik, M. Margni and T. Nemecek, Implementation of Life Cycle Impact Assessment Methods, 2010 Search PubMed https://www.ecoinvent.org/files/201007_hischier_weidema_implementation_of_lcia_methods.pdf .
- G. Wernet, S. Hellweg, U. Fischer, S. Papadokonstantakis and K. Hungerbühler, Environ. Sci. Technol., 2008, 42, 6717–6722 CrossRef CAS.
- G. Wernet, S. Papadokonstantakis, S. Hellweg and K. Hungerbühler, Green Chem., 2009, 11, 1826–1831 RSC.
- A. G. Parvatker and M. J. Eckelman, ACS Sustainable Chem. Eng., 2018, 7, 350–367 CrossRef.
- H. Tatsumi and K. Matsuda, Pinch Technology (in Japanese), The Energy Conservation Center, Japan, Tokyo, Japan, 2002 Search PubMed.
- A. Yoko, Y. Fukushima, T. Shimizu, Y. Kikuchi, T. Shimizu, A. Guzman-Urbina, K. Ouchi, H. Hirai, G. Seong, T. Tomai and T. Adschiri, Chem. Eng. Process, 2019, 142, 107531 CrossRef CAS.
- M. Pérez-Fortes, J. C. Schöneberger, A. Boulamanti and E. Tzimas, Appl. Energy, 2016, 161, 718–732 CrossRef.
- L. Bonfim-Rocha, M. L. Gimenes, S. H. Bernardo de Faria, R. O. Silva and L. J. Esteller, J. Cleaner Prod., 2018, 187, 1043–1056 CrossRef.
- T. Chang, M. Tamura, Y. Nakagawa, N. Fukaya, J.-C. Choi, T. Mishima, S. Matsumoto, S. Hamura and K. Tomishige, Green Chem., 2020, 22, 7321–7327 RSC.
- Y. Amada, S. Koso, Y. Nakagawa and K. Tomishige, ChemSusChem, 2010, 3, 728–736 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc03349a |
| This journal is © The Royal Society of Chemistry 2021 |