
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCO2 valorisation towards alcohols by Cu-based electrocatalysts: challenges and perspectives†
Hilmar
Guzmán
 ab,
Nunzio
Russo
a and
Simelys
Hernández
*ab
ab,
Nunzio
Russo
a and
Simelys
Hernández
*ab
aCREST group, Department of Applied Science and Technology (DISAT), Politecnico di Torino, C.so Duca degli Abruzzi, 24, 10129, Turin, Italy. E-mail: simelys.hernandez@polito.it
bCenter for Sustainable Future Technologies, Istituto Italiano di Tecnologia @ Polito, Via Livorno, 60, 10144, Turin, Italy
First published on 11th February 2021
Abstract
Developing efficient technologies to decrease CO2 emissions and dealing with climate change issues are among the most critical challenges in worldwide research. This review discusses the most recent advances on the electrochemical transformation of CO2 to alcohols, mainly methanol, ethanol and n-propanol, as a promising way to produce renewable liquid fuels. The main focus is given to copper-based electrocatalyst with different structures (Cu nanoparticles, oxide-derived Cu, and Cu composites) because Cu is up to now the heterogeneous catalyst with the most relevant activity for producing valuable C1+ hydrocarbons and alcohols via CO2 co-electrolysis. Several factors that impact the reaction activity and selectivities, such as the catalyst morphology, composition, surface structure, electrolyte effects and the electrocatalytic cell design (including liquid-phase and catholyte-free systems) are considered and analysed. This review reports an overview of the state-of-the-art with the most recent investigation highlights. It aims to provide guidance on the best experimental practices, new research directions, and strategies to develop efficient electrocatalysts. An outlook about the main challenges to be still resolved for a future practical application of this technology is also provided, toward a future based on sustainability and independence from fossil fuels.
1 Introduction
Since the industrial revolution, anthropogenic activities have impacted the planet's carbon cycle by the emissions of large amounts of greenhouse gases (GHGs), shifting the equilibrium of human history. The carbon cycle itself is resilient and has feedback cycles that would allow it to return to an equilibrium, but it would take thousands of years.1,2 The increase of GHGs concentrations in the atmosphere involves substantial risks, like warming, glaciers melting, sea-level rise, rapid ecosystem changes, ocean acidification, and extreme weather events.3 Thus, reducing the atmospheric burden of CO2 (>410 ppm in 2019), which is the most prevalent and persistent GHG, is essential for limiting those risks. However, the continuous increase of CO2 emissions is caused by the increasing energy demand, that currently depends on 85% on the use and burning of fossil fuels.4 Hence, developing efficient technologies and new approaches to decrease CO2 emissions and deal with climate change issues are among the most important current challenges in worldwide research.5 CO2 capture and its utilization as raw material, to synthesize high added-value products, is a promising approach to Tackle Global Warming and introduce carbon into a circular economy loop.CO2 is a fully oxidized, chemically inert and very stable molecule from both thermodynamic (ΔG° = −400 kJ mol−1) and kinetic points of view. Thus, its conversion to any organic molecule is difficult because it involves a change in the carbon oxidation state. In general, it is an endergonic and endothermic process; consequently, it requires substantial energy input. There are three different kinds of processes in which the CO2 can be used: physical, chemical and biological; the first regards the technological utilization of CO2 without its transformation, while the others are related to its conversion. The chemical ones can be divided into mineralization, photochemical, electrochemical and thermochemical processes. The last two could be considered the most promising approaches for producing fuels and chemicals from CO2. However, optimized reaction conditions and catalysts with high activity and stability are still needed.6
Among the different possible products, alcohol production from CO2 can lead to a faster transition towards a low C-based economy since they can be stored as liquids at ambient conditions and are compatible with the current energy infrastructure. Methanol and other high-octane alcohols (>C2), like ethanol and propanol, are of particular interest. They can be used as platform chemicals and are emerging as clean and sustainable transportation fuels.7,8 In particular, methanol has been proposed as an alternative energy carrier concerning the hydrogen economy, and is also an intermediate for some bulk chemicals. It could replace gasoline or diesel since it has a high-octane rating (22 MJ kg−1 and almost half of the volumetric energy density of common fuels).9 Ethanol and n-propanol production by electrosynthesis are also of interest because these alcohols can be directly used as fuels in internal combustion engines.10 Indeed, ethanol and n-propanol have an energy density (28 and 35 MJ kg−1, respectively) comparable to conventional fuels (i.e. 46.4 MJ kg−1 for gasoline and 45.6 MJ kg−1 for diesel). Currently, ethanol is extensively used as a fuel additive, but its direct use as fuel or blended with gasoline is increasing.11 Instead, despite its attractive features, n-propanol is not often used as a direct fuel for petrol engines, but as a solvent12 and as a source of hydrogen in some types of a fuel cell.13
The use of alcohols could decrease the CO2 tailpipe emissions due to their lower carbon-to-hydrogen ratio and improved engine efficiency concerning gasoline and other fossil-based fuels. Besides, the use of CO2 as a raw material represents an eco-friendly an economic opportunity to produce C1+ alcohols since the CO2 cost (<80 € per ton) is much lower than that of the oil-cost (∼500 $ per ton). Carbon can be reused and introduced into a circular economy loop, to penetrate the industrial sector (transport or/and chemicals), thus reducing CO2 emissions and the dependence of external energy suppliers. Hence, in perspective, these green fuels could replace fossil fuels and mitigate global warming at the same time.
Regarding the electrochemical pathway, renewable electricity can be directly used for the water splitting to generate the protons (H+) required for the CO2 hydrogenation to use liquid fuels such as methanol, ethanol and n-propanol.14–16 However, the electrocatalytic CO2 reduction (EC CO2R) is often focused on simple 2e− reactions, such as CO17 or formic acid18 formation. Although syngas (CO and H2) production is valuable, because it can be used as fuel or feedstock for current technologies for chemicals production,2 the main disadvantage is its production at low-pressure, which cause high-compression costs of the final product. Consequently, the scientific community is currently addressing the more challenging reactions leading to produce alcohols and other hydrocarbons. The performance of CO2 transformation into fuels or chemicals at Cu-based electrodes is among the best that has ever been achieved for CO2 electroreduction to >C1+ products.15 Compared with other technologies for CO2 mitigation, such as carbon sequestration, the electrochemical CO2 conversion is an environmentally sustainable option that would allow closing the C-loop by generating useful products and storing renewable electricity sources. Besides, this process can be carried out under mild conditions like atmospheric temperature and pressure.19 However, the EC CO2R involves complex and multistep mechanisms, including the adsorption and transformation of the reactants and intermediates at the surface of the electrocatalyst via shuttling of electrons (e−) and protons (H+). The mechanism depends on several factors such as the catalyst material, the kind of electrochemical cell, the electrode configurations and the reaction media (i.e. aqueous electrolyte, gaseous phase or aprotic solvents).
This work presents a critical review of the synthesis of alcohols through the electrochemical conversion of CO2. It discusses the most recent developments that would bring to an efficient and industrially relevant process. The EC CO2R is an interesting alternative considering both environmental and economic points of view. However, as aforementioned the conversion of CO2 to alcohols is a very daring task. The main challenge for implementing this technology at industry level is finding the suitable electrocatalyst and optimized process conditions that would result in the selective production of a single product, with a high conversion and production rate. Herein, the more significant works that have been published for the EC CO2R in liquid- and solvent-free media will be discussed. The focus is on Cu-based electrode materials since cooper is the only catalyst that has produced alcohols and other hydrocarbons with a relevant activity. Moreover, after analysing the scientific works done so far, special attention will be placed on the CO2 conversion in catholyte-free systems. Those are the most promising current configurations of electrochemical cells that could bring to a practical application of this technology in the short- to mid-term, to pursue a fossil-fuel-free world.20 In this regard, it is believed that the current knowledge of thermocatalytic CO2 conversion could be useful. It could provide a bridge for making faster progress in developing new efficient CO2R electrocatalysts. Therefore, a brief discussion on the most relevant achievements in CO2 hydrogenation for methanol production at high temperatures and pressures is also pointed out.
2 CO2 conversion: thermocatalytic vs. electrocatalytic routes
2.1 Useful knowledge from the thermocatalytic CO2 reduction
A considerable amount of literature has been published on thermocatalytic CO2 conversion, including the Sabatier and Fisher–Tropsch processes. This kind of technology is well known for producing synthetic fuels and can be easily scaled-up at an industrial level.21–24 In this process, hydrogen reacts with CO2via hydrogenation reaction to generate methanol and water at high reaction temperatures and pressures (≥220 °C and ≥2 MPa H2), due to low kinetics under milder conditions. To be sustainable and independent from fossil fuels, the H2 can be produced by water electrolysis with renewable energy.25–28 Currently, industrial methanol production is made by syngas (H2/CO mixtures) hydrogenation, although the attempts to perform it with CO2 started from the early 1960s. It has been demonstrated that adding a small quantity of CO2 to the syngas could improve the reaction yield.29 Currently, the CO2-to-methanol conversion by the thermochemical process has reached an adequate development level. It is a promising route to produce low-C fuels, although it has not already earned an industrial scale because of economic reasons.30,31 Since 2009, the methanol installed production capacity (∼100 million tons per year) is rising around 10% annually, and only 2% utilize CO2 as feedstock.32,33The main reactions involved in this process are three: CO2 and CO hydrogenation to methanol and the Reverse Water Gas Shift (RWGS) reaction, as reported in eqn (1), (2) and (3), respectively.34
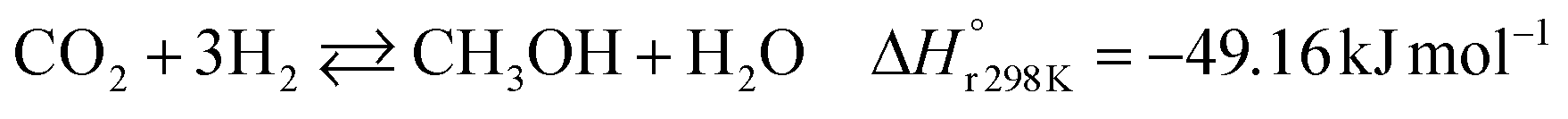 | (1) |
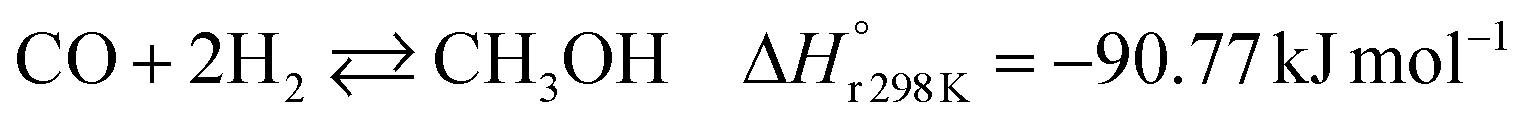 | (2) |
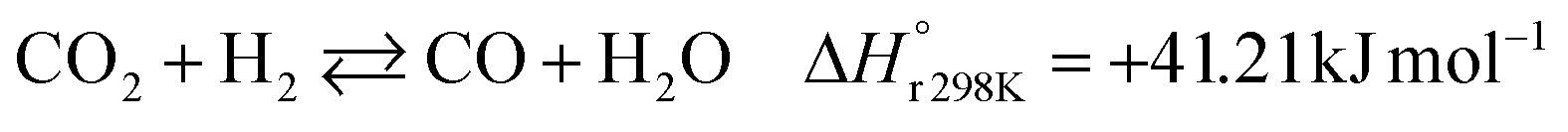 | (3) |
The main challenges of this process are still the low selectivity and conversion efficiency of the currently used catalyst, under mild conditions, being Cu-based materials the most promising ones. The yield towards methanol production of some relevant Cu-based materials reported in the literature are illustrated in Fig. 1 and Table S1 (see ESI†). As shown in Fig. 1, amphoteric metal oxides (i.e. SiO2, Al2O3, TiO2, ZnO, ZrO2)23,35–38 and some promoters (i.e. Ga)22 have been investigated as Cu supports for improving the performance of the catalyst. ZnO and ZrO2 are usually used as supports because they ensure a fair distribution of the active phase. In contrast, Al2O3 is used as both support and promoter because it enhances the thermal stability and specific surface area of the catalyst. In addition, ZnO can preserve the catalyst from poisoning phenomena as it can absorb impurities present in the reagent mixture while keeping the active catalyst sites (normally Cu). Besides, catalyst-containing ZrO2 are more stable and have a high tolerance for water.38,39 Several authors claim that carbon dioxide is absorbed on ZnO and ZrO2, while Cu adsorbs dissociated hydrogen.35,36 On the other hand, SiO2 is a support that improves the performance of the catalyst by increasing its specific surface area, it also attributes basicity to the surface and increases the methanol selectivity. However, it has the disadvantage of a high water adsorption capacity that is a strong inhibitor for the process because it induces the reverse reaction in eqn (1) and (3).36 The effect of TiO2 can be attributed to the formation of a more significant number of basic sites and smaller crystals with a greater specific surface area.23 Instead, Ga is a promoter for the Cu catalyst, which allows the formation of Cu1+, contributes to the methanol selectivity and gives good resistance to sintering phenomena.38 According to the literature, the optimal working conditions have been observed in the 220–270 °C and 2–8 MPa range of temperature and pressure, obtaining CO2 conversion values per pass in the reactor lower than 20%.
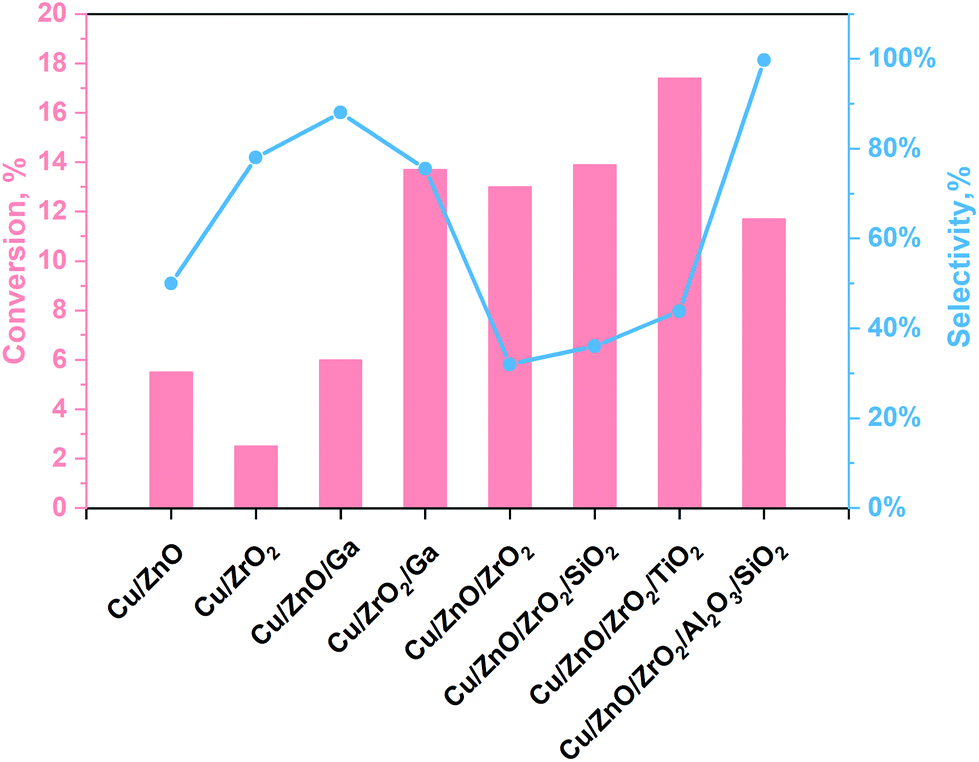 | ||
| Fig. 1 Outstanding results following TC reactions for methanol production from our work40 with data taken from ref. 23 and 35–38. | ||
Some reports have recently provided an outlook of future opportunities for hydrogenation of CO2 into C2+ compounds.41–43 As mentioned above, C2+ oxygenate products are desirable since they have the undoubted advantage of a higher energy density than methanol. However, no commercial implementation has yet been reached. The catalysts used for hydrogenating CO2 in this context are Cu, Fe, and Co-based materials or hybrid catalytic systems with these active sites but also containing noble metals and/or metal oxides with oxygen vacancies. Nonetheless, enhancement of this process by process conditions optimization and novel catalysts development remains an ongoing challenge due to high C–C coupling barriers.38,44 It has been found that such C–C coupling mechanisms could happen via CO or CH3OH intermediates. The former involves the initial generation of CO via RWGS reaction and then the CO intermediate hydrogenation via Fisher–Tropsh-like synthesis (eqn (2)). This pathway proceeds through two reaction mechanisms, namely a redox process (hydrogen acts as a reductant, and does not participate in the formation of intermediates) and/or an associative route (decomposition of intermediates species derived from the association of hydrogen with CO2); while the later combines the CH3OH synthesis (eqn (1)) with the methanol-to-hydrocarbon process.44 For the synthesis of methanol, many studies support the formation of formate species (HCOO*) as the first hydrogenated specie in the CO2 reaction mechanism. Indeed, Grabow and Mavrikakis,45 have performed detailed DFT calculations for demonstrating that the CO2 hydrogenation goes through the formate route on commercial Cu/ZnO/Al2O3. They showed that the hydrogenation of formate (HCOO*) leads to the formation of formic acid (HCOOH*) rather than dioxymethylene (H2COO*), which has been previously suggested. Then, HCOOH* is further hydrogenated to form CH3O2*, which is successively transformed to CH2O* by splitting of its OH group. In the final step, the hydrogenation of CH2O* would yield methoxy (CH3O*) and, successively, to CH3OH (see Fig. 2). However, the activation mechanism and C–C coupling steps already require further research.
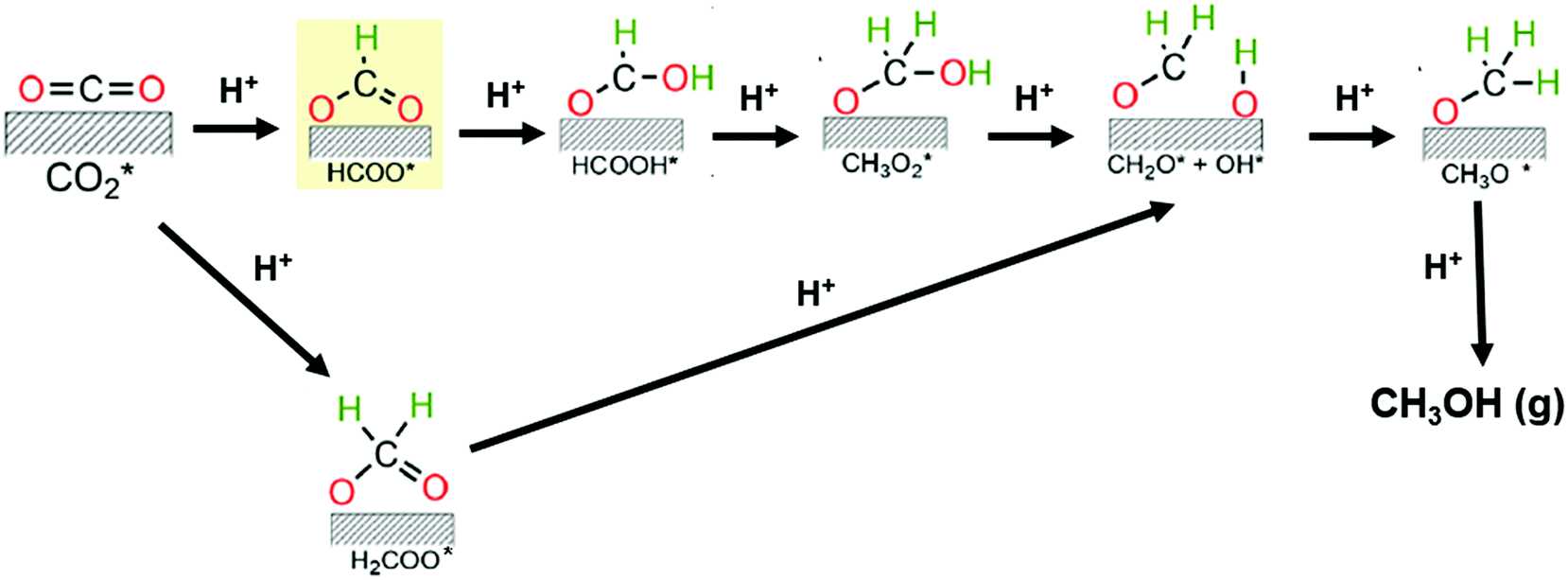 | ||
| Fig. 2 Proposed reaction mechanisms for the CO2 hydrogenation toward methanol. Adapted from ref. 45 and 46. | ||
Even if the thermochemical route for hydrogenation of CO2 is at an advanced level of development, it has some limitations as listed below:39,47
• The formation of valuable chemicals is carried out through multistep process units, commonly starting from fossil fuel-based feedstocks, which involve high operating costs and low energy efficiency.
• Most developed catalysts have a poor stability and/or can be poisoned because of their high H2O sensitivity.
• This process requires a high energy demand: high temperatures (>250 °C) and pressures (>20 bar), which leads to high operational costs and environmental impact. Nevertheless, the more renewable energy sources are employed in the process (e.g. hydrogen, electricity), the lower is the costs and environmental impact of this technology.48
Notwithstanding the limitations, the knowledge advances made on this field could help to develop the EC CO2R process, to find new catalysts formulations or improved operative conditions, as it will be discussed later (see section 4).
2.2 Challenges of the electrocatalytic CO2 reduction
Low temperature electrochemical CO2 conversion is an attractive way to improve the yield to alcohols like methanol or C2+ products. However, it is an ongoing challenge due to the complexity of reaction mechanisms with multiple steps and the high C–C coupling barriers (as it will be discussed below in more detail). The main advantage of the EC CO2R process is the direct use of renewable electricity and water, as electrons (e−) and proton (H+) sources, respectively, to convert CO2 into chemicals or fuels that are traditionally produced from petroleum. Thus, although the EC CO2R is at a lower development level than the conventional thermochemical processes, it can be beneficial in terms of economics and environmental impact, because milder reaction conditions can be used (i.e. room temperature and pressure). Besides, electricity could be obtained from renewable electricity sources to drive the process.Nonetheless, further efforts are still needed to reach relevant electrocatalytic performances of EC CO2R systems towards high volumetric- and mass-energy density alcohols. If the total generated current density of the CO2 co-electrolysis cell exceed a certain threshold (200–400 mA cm−2), contemporaneously, other critical parameters required to establish this technology at a large scale should be achieved, such as a high Faradaic efficiency (FE >70%) and a low cathodic overpotential of <0.5 V.49,50 As will be understood, the electrochemical CO2 reduction is a multidisciplinary problem since several factors must work in synergy to drive the reaction in the desired direction, which represents a challenge for the development of this technology. As shown in Fig. 3, these factors involve the optimization of the reaction medium, operating conditions, electrochemical reactor and nature of the electrocatalyst.
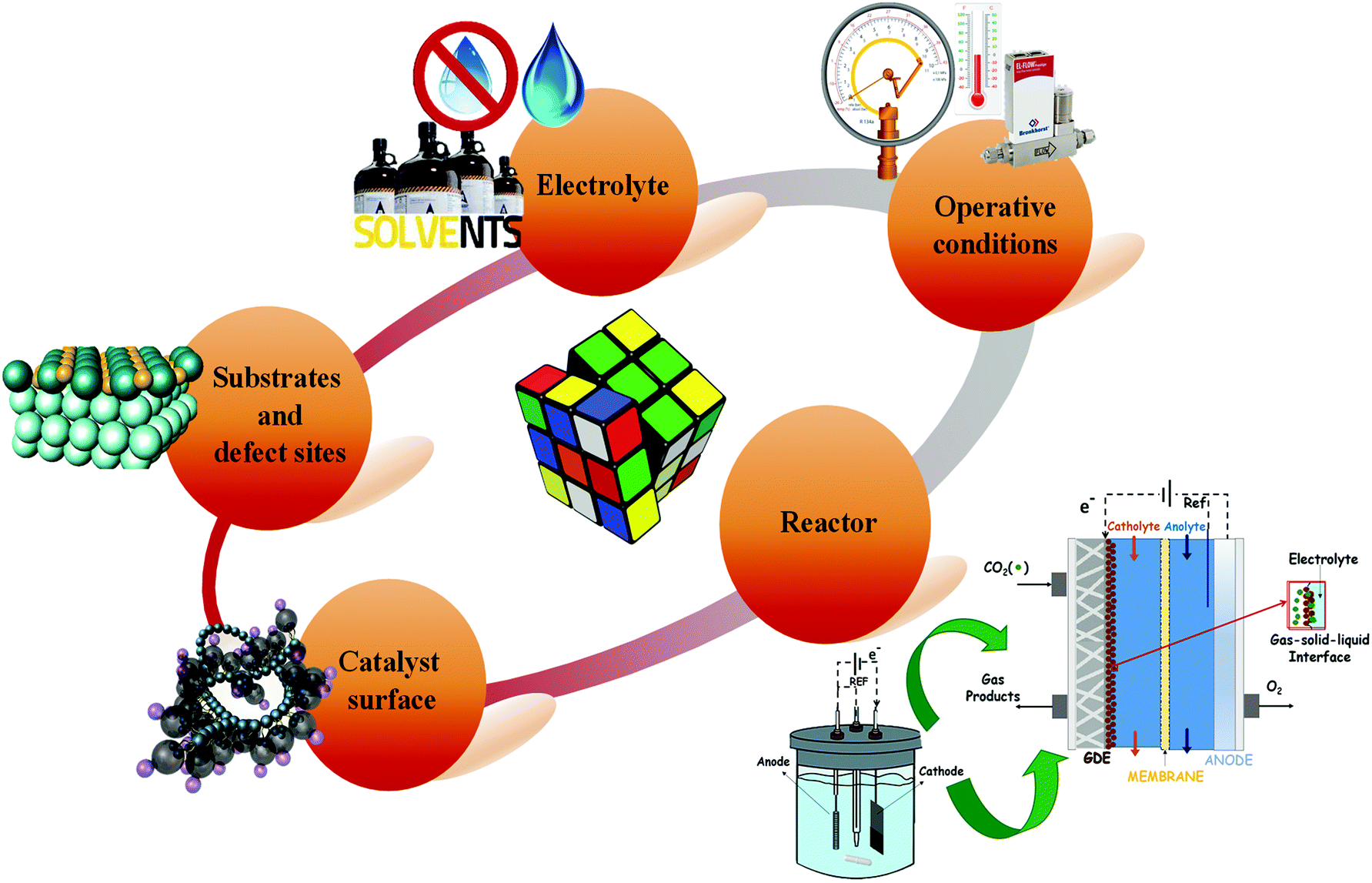 | ||
| Fig. 3 Factors influencing the electrochemical CO2 conversion performance. | ||
Although the future for the CO2-based electrosynthesis processes is promising, most current research works have focused on catalysts development (e.g. metal electrodes, nano-particulate catalysts). Simultaneously, few studies related to the overall electrochemical system design and a direct comparison (with the same catalysts materials) between batch operations and continuous operations, different electrolytes, membranes, etc. When motivation is solely on catalyst selectivity, it can lose sight of the parameters mentioned above, which are required for scaling-up this technology at an industrial level.28 As will be seen in the next sections, recently there are much more outstanding realistic and practical results achieved by electrochemical processes; as well as, a diversity of electrochemical reactors has been developed to achieve the scale-up of this process.51–53
2.3 Thermodynamics, kinetics and reaction mechanism aspects of the EC CO2R to alcohols
Carbon dioxide is a highly stable molecule with a free Gibbs formation energy (ΔG°, at standard conditions) of −400 kJ mol−1. Therefore, a large amount of energy is needed to convert CO2 into added-value products. Table 1 shows a list of the standard reduction potentials (at 1 bar, 25 °C, pH = 0) for EC CO2R to commonly reported electrochemical products.| Cathodic half-cell reaction | E°, V vs. NHE at pH = 0a |
|---|---|
| a All of the EC CO2R standard potentials (E°) here reported were calculated via Gibbs free energy of reaction values taken from ref. 1. b The E° value for the of the formation of the CO2˙− radical is the only exception, which was calculated from the Nernst equation with E° (at pH = 7) = −1.90 V vs. NHE, as reported in ref. 54 and 55. | |
| 4H+ + 4e− → 2H2 | 0.000 |
| CO2 + 2H+ + 2e− → CO + H2O | −0.105 |
| CO2 + 2H+ + 2e− → HCOOH | −0.169 |
| CO2 + 4H+ + 4e− → HCHO + H2O | −0.141 |
| CO2 + 6H+ + 6e− → CH3OH + H2O | 0.017 |
| CO2 + 8H+ + 8e− → CH4 + 2H2O | 0.169 |
| 2CO2 + 10H+ + 10e− ⇄ CH3CHO + 3H2O | 0.050 |
| 2CO2 + 12H+ + 12e− ⇄ C2H5OH + 3H2O | 0.084 |
| 2CO2 + 12H+ + 12e− → C2H4 + 4H2O | 0.079 |
| 2CO2 + 14H+ + 14e− → C2H6 + 4H2O | 0.142 |
| 3CO2 + 16H+ + 16e− ⇄ CH3(CO)CH3 + 5H2O | −0.140 |
| 3CO2 + 18H+ + 18e− → C3H7OH + 5H2O | 0.099 |
| CO2 + e− → CO2˙− | −1.486b |
As mentioned in Table 1, the reported reactions are related to the cathodic half-cell reactions. The overall water splitting and CO2R reactions also involves the Oxygen Evolution Reaction (OER): 2H2O → O2 + 4e− + 4H+, which take place at the anode side (E° = 1.23 V vs. NHE). This means that the numbers of electrons and protons in each reaction (cathodic and anodic) must be equal. It is essential to mention that several research studies have reported the standard potentials for the common CO2 reduction products experimentally found.56–58 Nevertheless, they present variations from each other due to the use of different standard states (for instance, aprotic-instead of aqueous-solvents) and/or different potential scales.
In general, a more positive E° indicates that the reaction is thermodynamically favourable, bearing in mind the theoretical relation: ΔG° = −nFE°, where n is the number of electrons transferred during the redox reaction, and F is the Faraday constant. Therefore, electroreduction products like alcohols, whose E° is more positive (see Table 1), should actually be thermodynamically more favourable than CO or HCOOH. Nonetheless, CO or HCOOH are generally more easily obtained. This behaviour can be ascribed to the fast kinetics of the reactions with a lower number of exchanged protons and electrons (2H+ and 2e−), as well as to the more complex mechanism of the reactions with a higher number of proton-coupled-electron-transfer (PCET) processes. In addition, early researches found that the rate-determining step (RDS) for most of the EC CO2R products is the formation of the CO2˙− radical. As evidenced in Table 1, the standard potential for producing this radical specie is relatively high with respect to the other products. Thus, the major causes of the usually reported high cell overpotentials (the difference between the theoretical thermodynamic potential for the desired product and the real applied potential) are: (i) the high energy needed for the formation of this RDS radical, (ii) the kinetic barriers of multi-PCET, (iii) ohmic losses due to ionic conductivity of the electrolyte, and (iv) mass transport restrictions in the electrode–electrolyte interface.59 For this reason, it is difficult to suppress the hydrogen evolution reaction (HER), which is kinetically more favourable.
Most of researchers often are focused in the electrocatalytic CO2 reduction reaction with the simplest 2e− transfer, such as CO and formic acid (or formate salts) production, for which the best performance has been obtained in literature up to now. In this regard, CO (or syngas) production in Ag-based Membrane Electrode Assembly (MEA) or Gas Diffusion Electrode (GDE) systems have achieved a FE of 70%–95% and current densities (j) of 50 to 300 mA cm−2 with a good stability (>550 h).17,57,60 On the other hand, formate has been produced in a Pd–Pb catalyst in an H-cell configuration, reaching 80% of FE but at a lower current density (11 mA cm−2) and stability (228 h).61 Regarding these products, attempts to demonstrate the CO2 electroreduction technology been already done. Indeed, Avantium recently patented a catalyst composed of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 weight ratio of bismuth to indium, which offers improved catalytic properties for the formation of formate from carbon dioxide, achieving FE = 95% up to 200 mA cm−2.18 From a current techno-economic point of view, these two products are economically viable products with net present values of $13.5 million and $39.4 million (for CO and formic acid, respectively).50
:1 weight ratio of bismuth to indium, which offers improved catalytic properties for the formation of formate from carbon dioxide, achieving FE = 95% up to 200 mA cm−2.18 From a current techno-economic point of view, these two products are economically viable products with net present values of $13.5 million and $39.4 million (for CO and formic acid, respectively).50
It is also important to mention that ethylene is the major product obtained in literature from CO2 electroreduction on Cu-based catalysts. This has been the main C2 product obtained with high FE yields (<70%) and with high stability of up to 150 h.62 Meanwhile, C2+ alcohols like ethanol and n-propanol can become more profitable if acceptable electrocatalytic performances are reached because they have a much higher market potential, since the capital and operating costs of ethylene under an optimistic case are relatively high due to the large amount of electricity needed per kg of product.
To render profitable the production of alcohols from EC CO2R, improved catalytic performances are needed. The catalyst should be active, selective and stable. Specifically, early DFT calculations have reported that there are 2 uphill reaction mechanisms that limit the activity and selectivity of a copper catalyst to alcohols (see Fig. 4). First, the formation of *COOH through CO2 activation and hydrogenation is the RDS, in this case, leading to the production of *CO. Second, the hydrogenation of the so formed *CO is the Selectivity Determining Step (SDS).56,63–66 As shown in Fig. 4(a), there are two possible SDS: formation of *CHO (path 1) or *COH (path 2), being the generation of CH4 more prone than methanol (through path 1), since it is more feasible the hydrogenation in solution than on the catalyst surface. This is due to the weak binding energy (EB) of Cu for H and to the moderate EB of Cu for O or OH.65,67 Therefore, it represents a challenge the tuning of the adjacent chemical environment around Cu atoms and of the binding strengths to the desired intermediates on the catalyst surface (e.g. weaken EB of OH groups), in order to favour the alcohol product without affecting the other steps in the catalytic system. Fig. 4(b) shows the reaction mechanism for the formation of ethanol and n-propanol, which share common intermediates along their reaction pathways, being the *CO–CO coupling the RDS for the formation of this kind of oxygenates. The C–C coupling leads to the formation of *CH2CHO and then to the SDS, that is, the direct conversion of the *CH2CHO intermediate to ethylene (C2H4) or subsequent additional steps for its conversion to ethanol; while the energy needed to generate C2H4 is lower than the energy required to produce ethanol. On the other hand, 1-propanol could be generated via CO insertion on stabilized C2 intermediates.
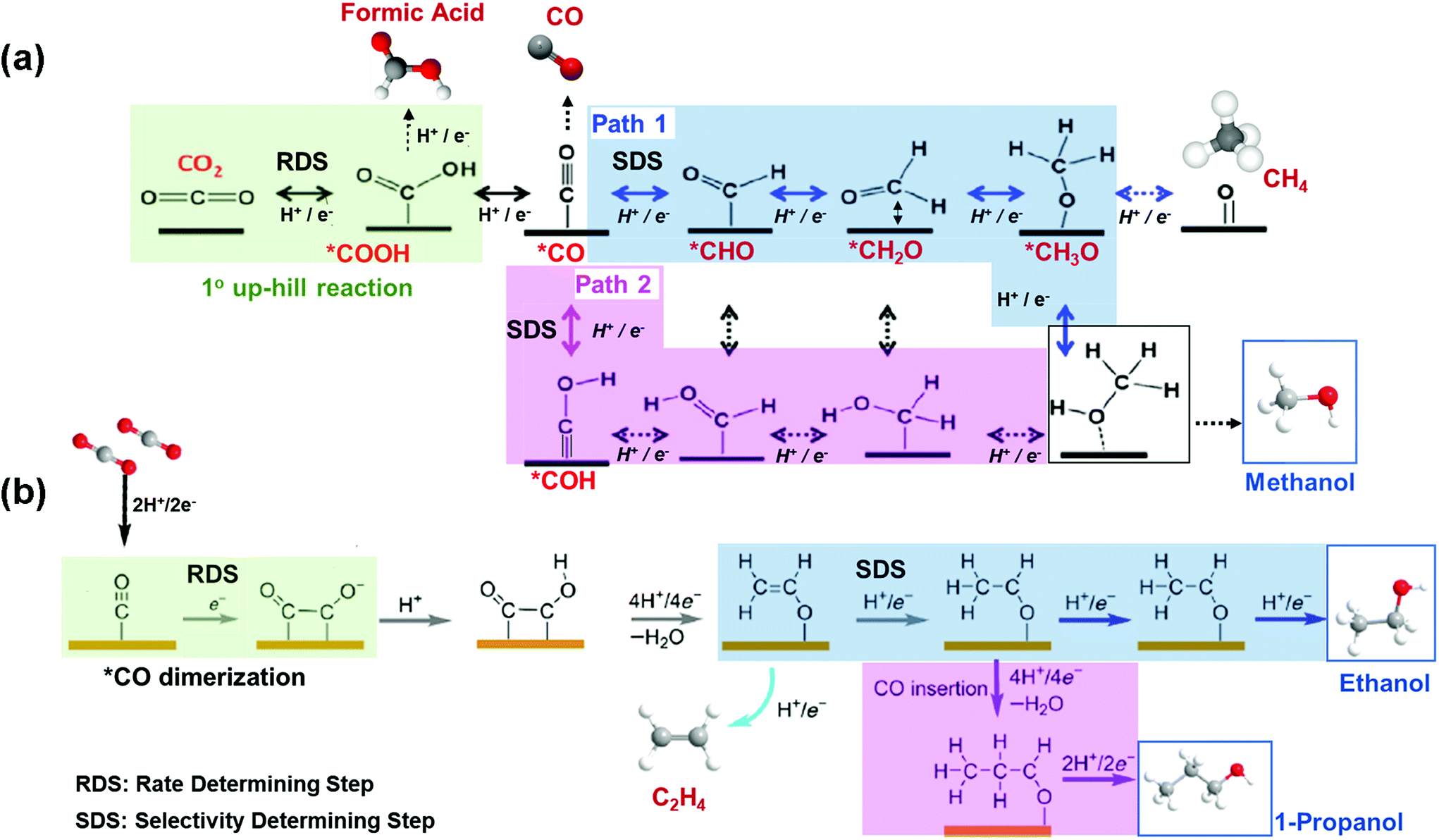 | ||
| Fig. 4 Competing reaction pathways for CO2RR to (a) methanol vs. formic acid, CO and methane, and (b) C2+ alcohols vs. ethylene. Adapted from ref. 63 and 56, respectively. | ||
As shown above, the electrochemical reaction mechanism is essentially constituted by a shuttling of electrons and protons, but it also involves a more complex process requiring adsorption and transformation of the reactants, through intermediates, on the electrocatalyst surface. The process efficiency heavily depends on the reaction conditions, and the low solubility of CO2 (∼38 mM at 20 °C and ambient pressure in water) in the reaction medium is also a controlling factor of the effects on mass transfer limitations. Thus, the conversion of CO2 in the gas phase would avoid the CO2 solubility issue of the liquid phase systems and the necessity of using high pressures (to increase the CO2 solubility in liquid electrolytes). It would also reduce downstream separation costs due to the high product concentrations and, in principle, should reduce side-reactions like H2 production. Nonetheless, although the gas-phase CO2 conversion has gained more attention in the past years, most of the scientific research reported until now on the electrochemical CO2 reduction has been done in aqueous environments. Sections 3.1 and 3.2 summarize the most recent works for the electrochemical CO2 reduction to alcohols in liquid and gas-phase systems, respectively. It will be reported how some of the challenges mentioned above have been addressed and what of them still remains unresolved. Strategies to exploit the opportunities of electrochemistry in the conversion of CO2 and to overcome the weaknesses of this technology will be also pointed out in sections 4.
3 Advances in knowledge of CO2 conversion via electrocatalytic routes
3.1 Electrochemical CO2 reduction: liquid-phase electrolytic solution
The possibility of reducing CO2 to valuable products has been a rapidly expanding field of research in recent years.68,69 Much work has been done on Cu, oxidized Cu and Cu-based materials to understand their activity selectivity and stability.70,71 This is because Cu-based materials are generally considered the only ones that catalyse the electrochemical CO2 reduction to hydrocarbons and oxygenates in relevant quantities. However, high overpotentials, a low selectivity to a single product and competitive side reactions (like the HER) persist. The subsequent discussion of this review is focussed on Cu and Cu-based electrocatalysts, and their ability to catalyse the EC CO2R towards methanol, ethanol and n-propanol as liquid products.Most of the results reported in the literature for alcohols production have been obtained in typical three-electrode systems as those shown in Fig. 5: (a) an undivided electrochemical cell, (b) an H-type cell, (c) a two compartments cell, or (d) a three compartments cell. Instead, in some of the works, the electrocatalysts (cathode and anode) have been assembled with a membrane into a MEA, which is the best way to reduce the gap between these two electrodes, as reported in Fig. 5(e).
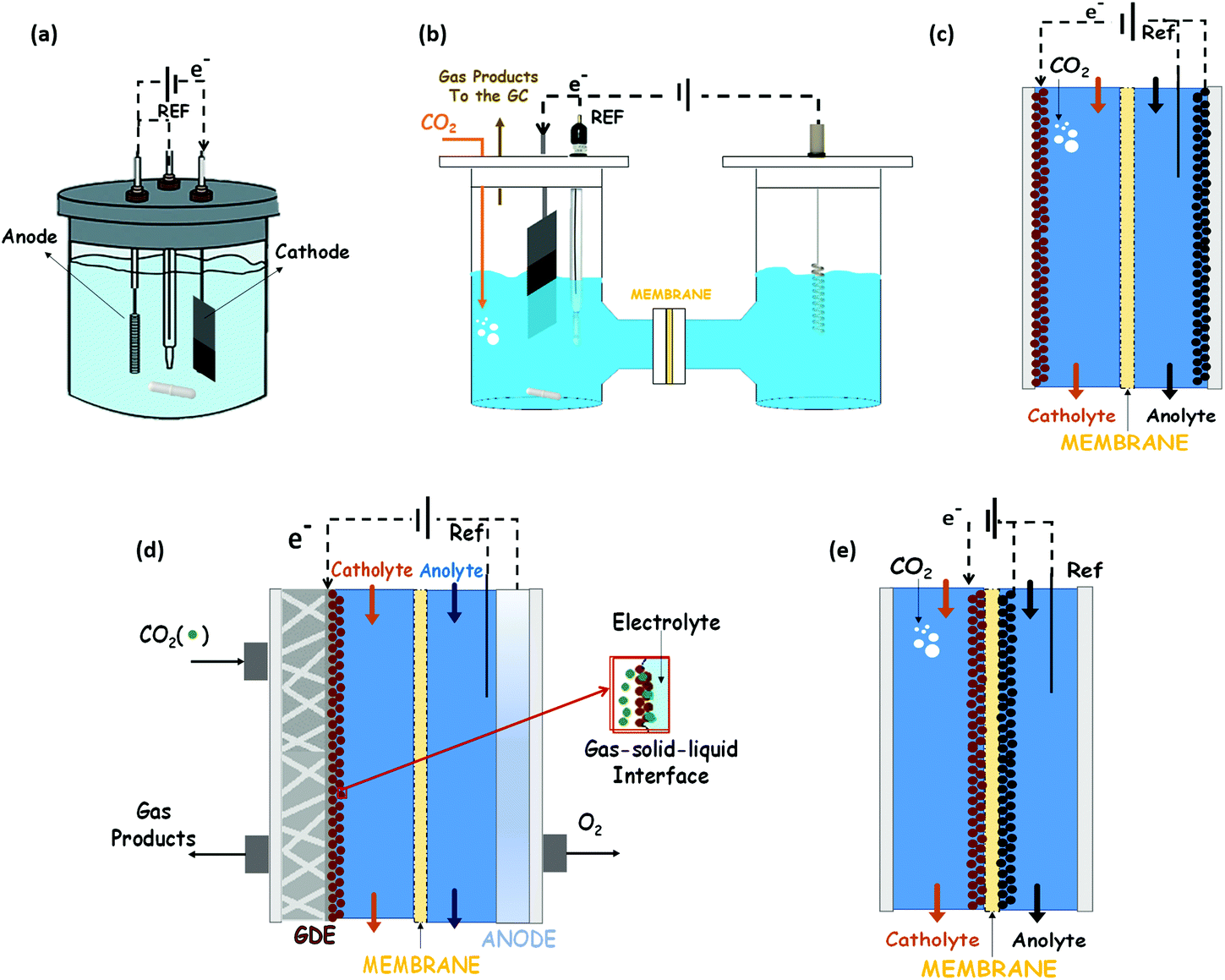 | ||
| Fig. 5 Schematic concepts of (a) traditional 3-electrode cell, (b) H-type cell, (c) two compartments cell, (d) Gas Diffusion Electrode cell and (e) Membrane Electrode Assembly for electrochemical CO2 reduction reactions. | ||
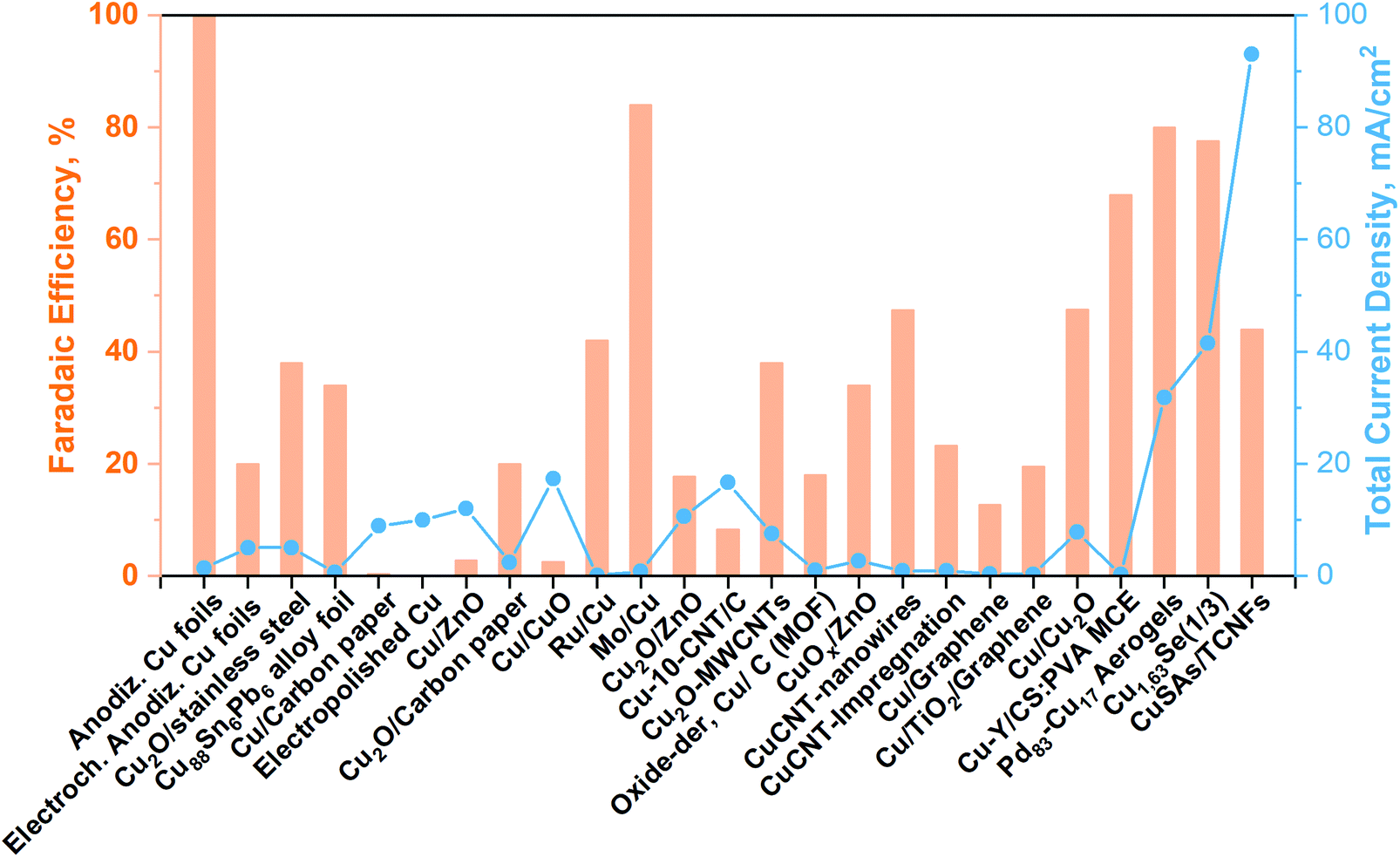 | ||
| Fig. 6 Cu-Based electrocatalyst towards methanol production from CO2 in a liquid electrolyte (batch conditions). Modified from our work40 with updated data taken from ref. 71–83. | ||
As can be seen from Fig. 6, until now, the synthesized Pd83Cu17 (see Fig. 6a) aerogel83 and Cu1.63Se(1/3) (see Fig. 6b) nanocatalyst (∼50 nm)73 are the only materials that have displayed contemporaneously a high current density (32 and 42 mA cm−2, respectively) and a high FE to methanol (80% and 78%, respectively). However, these great performances have been achieved with the use of expensive ionic liquids (i.e. [Bmim]BF4 or [Bmim]PF6) as cathodic electrolytes and CO2 bubbling (<10 mL min−1) in both cases, while a 0.5 M H2SO4 aqueous solution was used as the anodic electrolyte. It is also worth to notice that F-containing ionic liquids can also generate HF during the co-electrolysis process, which also could cause hazardous and safety issues in a large-scale application.
Yang H. et al.74 achieved noticeable performance in this kind of cell by using stable and efficient Cu single atoms decorated through-hole carbon nanofibers (CuSAs/TCNFs), which were directly used as cathode without any binder and vehicle. The morphology of this electrocatalyst is shown in Fig. 7(c). They used a 0.1 M KHCO3 cathodic electrolyte that was continuously purged with CO2 gas for carrying out the co-electrolysis reaction. The systematic fashion of the porous structure expose Cu single atoms on the surface and helps the diffusion of CO2. The CuSAs/TCNFs membrane presented high stability for more than 50 h with the best current density (93 mA cm−2) obtained until now and 44% of FE for methanol production. Textural parameters such as morphology, porous structure and particle size (∼50 nm) or nanowire (or -fibres) diameter (6–700 nm), as well as the possible cooperative effect between the metals (e.g. Pd and Cu; Cu and Se), made the difference in the catalytic performance of these three catalysts. Despite these results, the H-type electrochemical cells have not achieved relevant production rates yet.
 | ||
| Fig. 7 (a) TEM and EDX mapping images (scale bar: 20 nm) of Pd83Cu17 aerogel. Reproduced from ref. 83 with permission of Angew. Chem. (b) TEM image of the Cu1.63Se(1/3) nanocatalysts and the inset is the corresponding elemental mapping (scale bar = 100 nm). Reproduced from ref. 73 with permission of Nature Communications. (c) High resolution-TEM images of CuSAs/TCNFs; the inset of (c) shows the SAED pattern and EDX mapping of a single CuSAs/TCNFs nanofiber. Reproduced from ref. 74 with permission of American Chemical Society. | ||
Electrocatalysts immobilized on a porous and conductive substrate (typically carbon paper or carbon cloth as shown in Fig. 5(d)) have been studied to enhance mass transport within the cell and reduce ohmic losses and pursue high current densities to achieve industrially reasonable production rates. The immobilization of electrocatalysts on these substrates to make GDEs improves the performance of the process by increasing the current density for target chemical products, because of the increase of the CO2 concentration on the catalyst surface. This kind of configuration was initially developed for water electrolysis and fuel cell systems. It provides a triple-phase boundary of CO2 gas, catalyst surface and electrolyte. Thus, this cell configuration (usually known as a gas-phase reactor, because of the gaseous CO2 reactant) has shown an outstanding performance in contrast to CO2-saturated electrolytic solution.20,69 It is important to note that the MEA configuration (Fig. 5(e)) can also be prepared with only one of the electrocatalyst of the system (only the cathode or only the anode attached to the membrane). Indeed, in the GDE configuration shown in Fig. 5d the membrane form a MEA with the anodic electrocatalyst.
Within this context, Albo J. et al.84 developed Cu2O and Cu2O/ZnO GDEs to produce methanol from liquid-phase EC CO2R by using a filter-press electrochemical cell. The goal was to reduce mass transfer limitations founded in their previous work,75 where a two-compartments configuration such as that shown in Fig. 5(c) was used. The use of a three-phases system significantly increased the catalysts performance by reducing mass transfer limitation issues. The maximum achieved faradaic efficiency (at −10 mA cm−2) to methanol was of 42% and 28% for their Cu2O and Cu2O/ZnO-GDE electrodes, respectively, by supplying a CO2 gas flow at 200 mL min−1 through the GDE. The electrolyte solution (0.5 M KHCO3) was previously saturated with CO2. They stated that the inclusion of ZnO might increase the catalytic activity since it is known to strengthen and stabilize Cu and CO2 link in the hydrogenation reaction, improving the selectivity to alcohols.75,85–88 This is related to the thermocatalytic CO2 hydrogenation mechanism with analogous catalysts. However, it was detected the deactivation of the catalysts surface that they allege to be probably linked to the detachment of the catalytic particles during operation, rather than be due to catalyst poisoning. Indeed, the Cu2O/ZnO-GDE electrode was stable over 20 h, where peeling off the catalyst from the carbon paper was observed in a lower quantity compared to the Cu2O-GDE surface. Finally, they carried out an evaluation of some key parameters on the EC CO2R: (i) FEs did not show an increment at higher current densities (ranging from 5 to 40 mA cm−2); the FE to methanol displayed the maximum values at 10 mA cm−2; (ii) a raise in the electrolyte flow rate did not produce a significant change on the methanol production rate; (iii) there was an optimal CO2 gas flow rate to avoid the deactivation of the catalyst (detachment of particles from carbon paper).
On the other hand, some researchers have paid great attention to metal–organic frameworks (MOF), due to their high surface area and unique structure. In this regard, Albo J. et al.89 tested different Cu(II) and Bi(III)-based metal–organic framework blends (HKUST-1 and CAU-17, respectively) to study the synergic effect of Cu and Bi in the electroreduction of CO2 in a filter-press configuration cell with a CO2 flow rate of 200 mL min−1. These materials have been deposited on a carbon paper to form GDEs, whereas the anode used was a platinized titanium electrode. The continuous electroreduction was carried out in a 0.5 M KHCO3 electrolyte. Their results denoted an alcohols selectivity dependence on both current densities and bismuth content: the maximum FE to alcohols was of 36.9% (8.6% for methanol and 28.3% for ethanol), achieved for a blend with a bismuth content of 12% (CuBi12) and at j = 20 mA cm−2. Moreover, they found out that the reaction is more selective towards methanol at 10 mA cm−2, while ethanol is the dominant product at 20 mA cm−2.
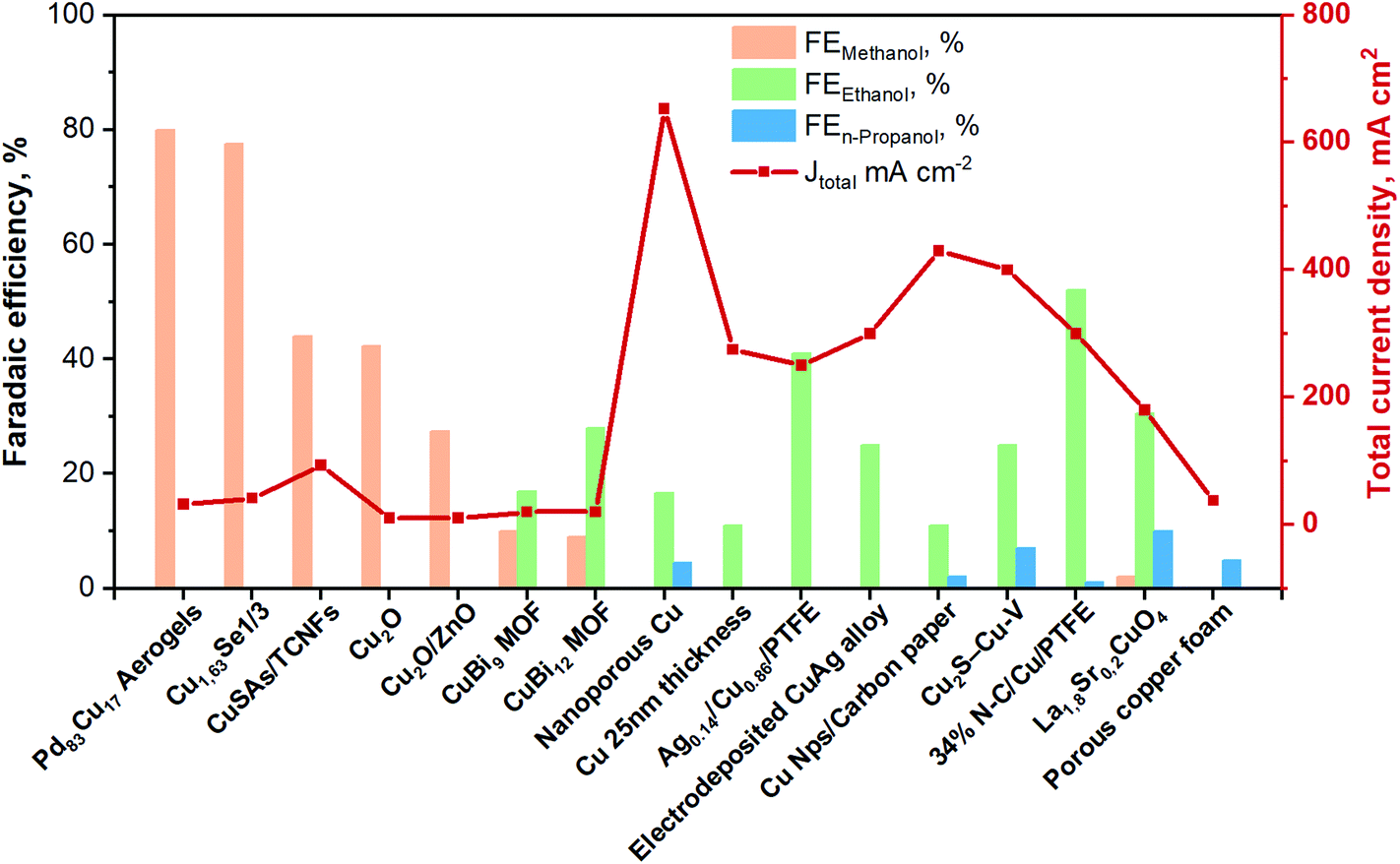 | ||
| Fig. 8 Faradaic efficiencies and total current densities of the best liquid-phase EC CO2R electrocatalysts towards ethanol and n-propanol production in contonuous-flow system. Authors with data taken from ref. 62, 84 and 89–102. | ||
Among the best results for C2+ alcohols production, there is an example of a photo-electrocatalytic material for the CO2 reduction. Indeed, Homayoni H. et al.103 showed that solar illumination of a hybrid CuO/Cu2O nanorod arrays supported on Cu foil (photocathode) can be used to photo-generate alcohols in a continuous-flow photoelectrochemical (PEC) microreactor. A proton exchange membrane separated the cathode and anode compartments in the flow cell, and the supporting electrolyte was 0.1 M sodium bicarbonate. The performance of this Cu-based photocathode was >5 times higher than in several batch systems. The primary products were methanol (FE 4%), ethanol (FE 52%) and isopropanol (FE 40%) with a total photocurrent density of 20 mA cm−2 for 2 h. The high surface-area-to-volume ratio resulting from the narrow reaction channels resulted in an enhanced photocurrent and faradaic efficiency. If the utilized incident light was sunlight, this process would represent a sustainable route to combine renewable energy with CO2 storage in these fuels/chemicals. However, current Cu2O-based photoelectrocatalysts materials for EC CO2R still suffer from poor stability (mainly due to the catalysts transformation and the reduction of Cu1+ to Cu0). Further efforts are needed to enhance and maintain their current densities to a level with practical significance.
Regarding the use of multiple-metallic materials, Cu-based perovskite catalysts of the type: A1.8A′0.2CuO4 (A = La, Pr, and Gd; A′ = Sr and Th) outstands, among others.100 It is important to know that multiple- and bi-metallic materials have been explored in order to improve the selectivity of the reaction for a specific product. These electrocatalysts were deposited on a GDE and tested in 0.5 M KOH aqueous solution under ambient conditions. The cumulative faradaic efficiencies of methanol, ethanol and n-propanol reached 40% in La1.8Sr0.2CuO4 at a constant j of 180 mA cm−2. The preparation of bimetallic system has also been a key tactic to improve the catalytic performance of the EC CO2R process.99 Indeed, one of the best performing Cu-based catalyst systems is a CuAg-wire alloy (containing 6% Ag) electrodeposited on carbon paper (GDE). It has attained a high CO2 reduction with a total current density of 300 mA cm−2 and a FE of around 25% to ethanol in an electrochemical flow cell reactor. The electrolyte was 1 M KOH, and the CO2 flow rate was 7 mL min−1. In that research work, in situ Raman analysis indicated that the high formation of C2+ products is due to two main reasons. First, the stabilization of the Cu1+ overlayer. Second, the presence of high local CO (as a *CO intermediate) owing to the added Ag.98
Similarly, Y. Li and co-workers97 have also shown that adding a second metal with a weaker bonding ability to carbon than Cu (i.e., Ag sites in Cu(111)) led to a shift sideways the ethylene pathway, thus increasing the selectivity for ethanol. Therefore, their Ag0.14/Cu0.86 catalyst deposited on a polytetrafluoroethylene (PTFE) substrate achieved a FE of 41% towards ethanol with a total current density of 250 mA cm−2 at −0.67 V vs. RHE. The electrolyte used was 1 M KOH and an anion exchange membrane for separating the electrodes in a flow-cell system. The X-ray absorption near edge structure (XANES) analysis showed that under applied negative potential, only Cu0 is observed, where mostly CO2 activation occurs. Taking DFT calculations and in situ Raman analysis, they concluded that the Ag/Cu alloy catalyst destabilize the ethylene reaction path favouring ethanol formation. Chen C. et al.104 stated that adding Ag to a Cu–Ag tandem catalyst, a CO-enriched local environment is generated. These conditions can enhance C2+ formation from CO2. The tandem strategy consisted in the increase of the production rate of CO from CO2 on Ag and its subsequent coupling on Cu.
It has been demonstrated that with Cu oxide-derived materials, the interface between Cu1+ and Cu0 contributes to dimerization of *CO, consequently to the production of C2+ products. However, oxidized Cuδ+ species are not stable at high negative applied potentials, which represents a challenge for the performance of the process.105 Therefore, to stabilize these species, Chen C. et al.92 proposed incorporating a heteroatom like boron (B) into the surface of the Cu-based catalyst to affect the electronic configuration of adjacent atoms and reduce the barrier of the *CO dimerization. The FE towards ethanol reached 20%, with a high current density of 33.4 mA cm−2 at low potentials in 0.1 M KHCO3 electrolyte (H-type cell configuration).
In this framework, different researchers have investigated the cooperative effect of highly textured N-doped materials. In a just-published work, Wang X. et al.90 reported a 34% nitrogen–doped carbon (N–C) layer on a Cu surface supported on PTFE (see Fig. 9) with an increased selectivity to ethanol, reaching 52% of FE at a total current density of around 300 mA cm−2. They demonstrated through DFT calculations that the improved performance of this material is due to the suppression of the deoxygenation process, because of the strong electron-donating ability of the confining N–C layer. Thus, C–O bond-breaking from the key intermediate: HOCCH* is prevented. They also registered in situ Raman spectra to explore the interactions between the adsorption of carbonaceous intermediates and the active catalytic surface. The results indicated that the potential for *CO formation on the nitrogen-doped carbon layer on a Cu surface is lower than that on bare Cu. The electrochemical measurements were carried out in a flow cell (three-electrode system) with three compartments. An anion exchange membrane was used to separate the cathodic and anodic chambers containing 1 M KOH. CO2 gas was continuously supplied to the gas chamber located at the backside of the cathode at the rate of 50 mL min−1. Moreover, Chen C. et al.95 have synthesized a composite composed of N-doped graphene quantum dots (NGQ) on CuO-derived Cu nanorods (NGQ/Cu-nr). This dual active sites catalyst achieved a FE of ∼43% for ethanol and ∼0.7% for n-propanol with a total current density of ∼280 mA cm−2. Theoretical studies reveal that this catalyst can stabilize the CH2CHO intermediate for enhancing the production of alcohols through carbon protonation. Moreover, through the implementation of situ Raman spectroscopy, it was demonstrated that adsorbed *CO on Cu was not changed by the presence of N-doped graphene. Therefore, the improvement in alcohols production is due to a combined effect between NGQ and Cu-nr substrate. In another work, Song Y. et al.,106 proposed a metal-free porous and conductive matrix containing N-pyridinic groups with micro/mesopores for improving mass transport of reactants and products. The high e− density in N-pyridinic groups was exploited as an enhancer of the CO* dimerization, enabling the efficient production of ethanol with a high FE of 77% at −0.56 V vs. RHE, but the absolute current density obtained was very low (0.5 mA cm−2), although it was relatively constant during the test duration of 24 h.
 | ||
| Fig. 9 Structural and compositional analyses of the 34% N–C/Cu catalyst on PTFE. (a) Low-magnification SEM image of the 34% N–C/Cu catalyst on PTFE. (b) EDX elemental mapping of Cu, N and C for the 34% N–C/Cu catalyst on PTFE. (c) Scheme of the cross-sectional structure of a N C/Cu/PTFE nanofibre. The white, orange and green layers represent PTFE, Cu and N–C, respectively. Reproduced from ref. 90 with permission of nature energy. | ||
Xu H. et al.93 have also used carbon as conductive support to prepare well-dispersed Cu atoms by an amalgamated Cu–Li method. They stated that the initial high dispersion of the active site was the responsible for achieving a CO2-to-ethanol FE of ∼90% (1.23 mA cm−2) at −0.7 V versus RHE and with a Cu nominal loading of 0.4 wt% (Cu/C-0.4). XANES analysis reveals that Cu0 with a minor component of Cu1+ was present during the testing. Moreover, operando X-ray absorption spectroscopy analysis demonstrated a dynamic and reversible transformation from atomically dispersed Cu atoms to Cun clusters (n = 3 and 4) active sites under the reaction conditions. The electrochemical measurements were carried out in a three-electrodes electrochemical cell using a rotating disk electrode (RDE) as a working electrode.
Regarding the use of porous PTFE layers, Luo M. et al.94 have synthesized and investigated a hybrid cerium hydroxide-doped-Cu/PTFE sample (denoted Ce(OH)x/Cu/PTFE), which allows a tuning of the adsorption of hydrogen on Cu. The CO2RR performance was assessed in a flow cell set-up with 1 M KOH solution as electrolyte. In situ Raman measurements were performed to reveal the reaction intermediates during CO2 reduction. The results revealed that the hydroxide modification did not influence the adsorption of carbonaceous species. Instead, the surperficial Had favoured the formation of ethanol by tackling the Cu–C bind of the *HCCOH intermediate, reaching a FE of 43% for ethanol at an operating current density of 300 mA cm−2.
Some recent studies have shown Cu-based materials with other non-metal dopants different than nitrogen, such as sulphur (e.g. Cu2S–Cu–V).91 Computational calculations suggested that S-enriched Cu and surface vacancies can shift the selectivity away from ethylene towards multi-carbon alcohols.91 The Cu2S–Cu–V nanoparticles with a copper surface shell and a copper sulfide core (see Fig. 10) achieved a total current density of 400 mA cm−2, 25% FE for ethanol, and 7% for n-propanol. The tests were performed in a flow-cell system using a gas-diffusion electrode (1 M KOH electrolyte and a CO2 flow rate of 50 mL min−1).
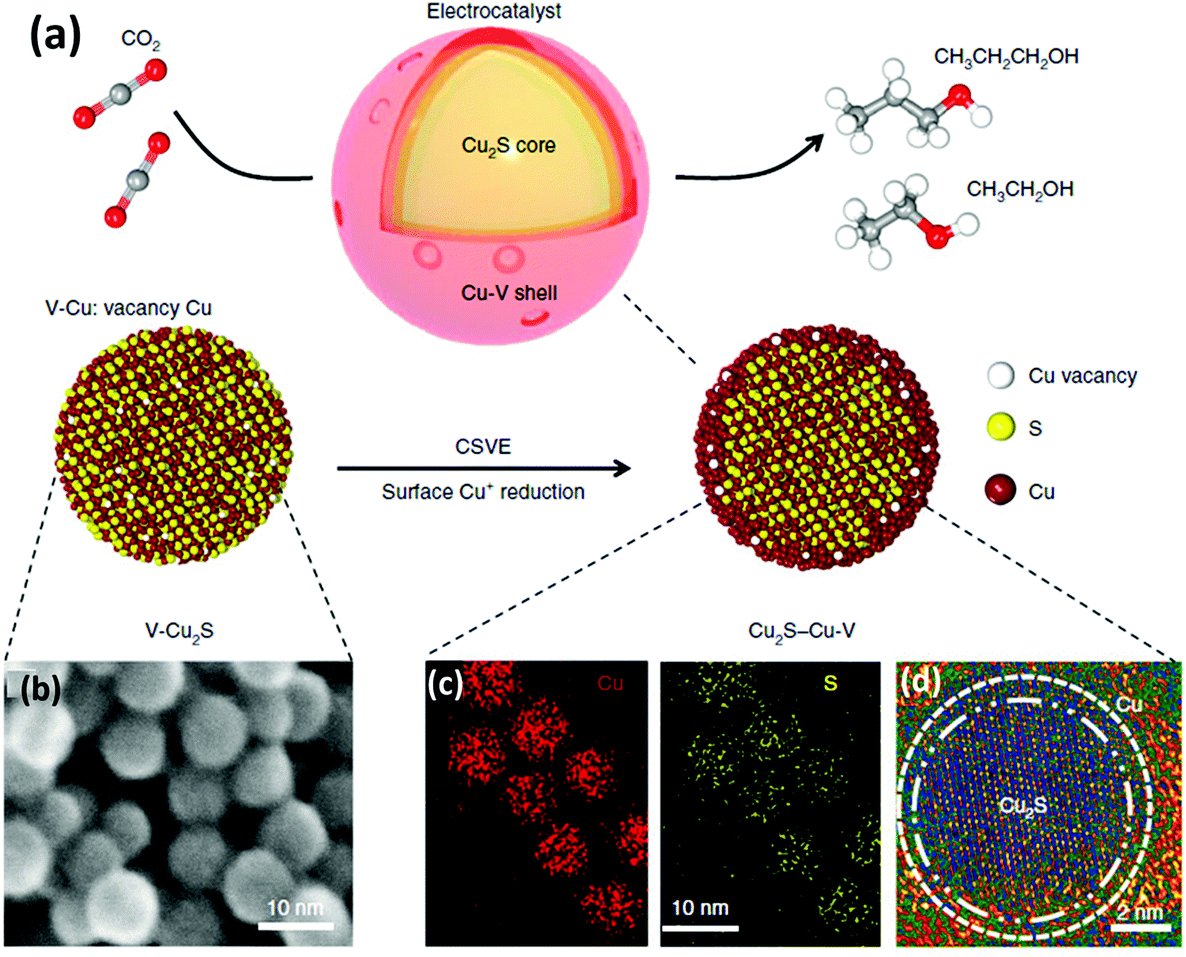 | ||
| Fig. 10 Catalyst design and structural characterization. (a) Schematic illustration of Cu2S–Cu-V electrocatalyst design for production of multi-carbon alcohols from CO2 reduction. (b) TEM showing the uniform size. (d) EDS mapping showing the homogeneous distribution of Cu and S. (d) high-resolution TEM. Reproduced from ref. 91 with permission of nature catalysis. | ||
Cu nanoparticles have also attracted great attention because of their high surface to volume ratio.107 For instance, Dinh C. et al.62 have studied the CO2 reduction reaction activities of Cu nanoparticles (<50 nm) deposited by drop-casting in a GDE and tested in a flow cell reactor under too alkaline conditions (10 M KOH) at 50 mL min−1 of CO2. Using a catalyst thickness of 25 nm, they obtained 11% FE for ethanol at a total current density of 275 mA cm−2. The high pH conditions promoted an improved diffusion of CO2 across the gas–liquid interface reducing ohmic overpotentials. On the other hand, in situ X-ray absorption spectroscopy (XAS) revealed that Cu becomes oxidized when immersed in the alkaline electrolyte. Under the reaction conditions, the surface is primarily Cu0 and remains consistent over the range of potentials and concentrations of interest. Ma S. et al.102 also synthesized Cu nanoparticles with excellent textural characteristics (high surface roughness), which were used to convert CO2 (at 7 mL min−1) in an alkaline flow electrolyser (GDE configuration) in 0.1 M KOH. They reported high conversion of CO2 to ethanol (11% FE) at a total current density of 430 mA cm−2.
Offering improvements, Lv J. et al.96 synthesized nanoporous Cu catalyst by in situ electrochemical reduction of porous CuO and carried out CO2 co-electrolysis with tests in a three-compartments GDE-microcell with a gas flow of 10 ml min−1. Different polymeric and liquid electrolytes were tested: an anion exchange membrane was used with KOH and KHCO3 electrolytes, and a proton exchange membrane was used with KCl and K2SO4 electrolytes, maintaining constant the K+ concentration at 1 M. This configuration permitted for CO2 to be abundantly fed to the catalyst surface, at the electrode–electrolyte interface. It allowed the investigation of CO2 electrolysis at very high current densities, achieving an overall current density of 653 mA cm−2, with 17% FE ethanol and 4.5% FE n-propanol, at an applied potential of −0.67 V vs. RHE. As for the previously presented works, the results reported in this investigation revealed that the production of long-chain of chemicals or fuels (i.e. ≥C2) during the electrocatalytic reduction of CO2 is favoured at high local pH values.
Production of n-propanol from CO2 is very promising considering its higher energy density (35 MJ kg−1) than other alcohols (e.g. methanol 22 MJ kg−1 and ethanol 28 MJ kg−1). Still, it is a challenging product to pursue. In this case, the development of highly selective electrocatalysts is a prerequisite for suppressing ethylene formation and leading CO insertion (as shown in Fig. 4b). Many efforts have been made to produce n-propanol on copper surfaces, but most of them have been inefficient because of the simultaneous formation of many other compounds. For example, Hori Y. et al.108 reported that n-propanol was produced by CO2 reduction on Cu(100) surfaces of Cu single-crystal, achieving a FEn-propanol = 1.5% and FEethanol = 9.7% with a total current density of 5 mA cm−2 in a traditional undivided 3-electrode cell with 0.1 M KHCO3 as the electrolyte solution. They stated that the Cu(100) surface features favoured the stabilization and subsequent coupling of carbon reaction intermediates. In a more recent work, Kim D. et al.109 prepared a densely packed Cu nanoparticle (6.7 nm) ensembles, which re-arranged during catalysis into cube-like particles (×22.5 = 47.7 μg of Cu nanoparticles loaded carbon paper). They reported n-propanol production with a FE of 5.9% at −0.81 V vs. RHE (FEethanol = 13.3%) and a total current density of 12.7 mA cm−2 by using a two compartments configuration separated by an anion exchange membrane with 0.1 M KHCO3. Ren D. et al.110 prepared Cu nanocrystals with defect sites by electroreduction of a Cu2O/Cu(OH)2 films and n-propanol (FE = 11%) was detected at −0.95 V vs. RHE, with a total current density of around 16.4 mA cm−2. They used a two-compartments cell containing 0.1 M KHCO3 both anode and cathode sides, which were separated by an anion-exchange membrane. CO2 was bubbled into the electrolyte at a rate of 20 cm3 min−1 during the 60 minutes experiment. In a subsequent work, Geioushy R. et al.111 continued their attention to the production of n-propanol synthetizing Graphene (GN)/ZnO/Cu2O composites. They evidenced that there is a synergic effect of ZnO/Cu2O, as ZnO is proposed to stabilize Cu1+ (ref. 75, 78 and 85–87) and that Graphene acts as a good support material that contributes to enhance the selectivity towards C3 products. Electrocatalyst with a ZnO/Cu2O weight ratio of 2:1 obtained a FE for n-propanol of 30%. That is the highest value reported in the literature up to now for this product. It was obtained at −0.9 V versus Ag/AgCl with a total current density of 8 mA cm−2. Their electrochemical measurements were performed in a two-compartment cell separated by a glass frit, and CO2 was bubbled continuously in the electrolyte (0.5 M NaHCO3) during the reduction process. N-doped graphene has also been used to support Cu nanoparticles (8 ± 4 nm) coated on TiO2 nano-blocks. A 0.2 M KI aqueous solution was used as the electrolyte in the cathode (GDE-cell configuration) and anode compartments. This electrocatalyst showed a low faradaic efficiency towards n-propanol (3.30%) but a very high FE for the electroreduction of CO2 into methanol and ethanol (19.5% and 43.6%, respectively) that are remarkable values in view of producing an alcohol mixture.80 On the other hand, a polycrystalline Cu foil modified with an electrochemically deposited film of aryl-phenylpyridinium reached 11.8% of FE to n-propanol with a total current density of 1.1 mA cm−2 at −1.1 V vs. RHE. A CO2-saturated electrolyte (0.1 M KHCO3) was used in a two-compartments cell with an anion exchange membrane.112 A very recent study reported the use of highly porous urea-modified copper foam catalysts for the production of n-propanol with a FE up to 4.93% at −0.83 V vs. RHE and with a stable j of 38 mA cm−2 for up to 2 hours. They used a custom-made H-cell with 0.1 M KHCO3 and a proton exchange membrane to separate the electrodes. After electrolysis, a re-arrangement of the catalyst into a dendritic morphology was observed.101
It is worth to mention that, in the work reporting the best-performing copper-based catalysts, high alcohols selectivity have been obtained by using alkaline electrolytes (mainly, KHCO3 or KOH). The best cell configuration has been a continuous flow cell (using or not a GDE). Indeed, the choice of the electrolyte has strong effects on the performance of the electrocatalytic CO2 reduction (current density, selectivity and stability).96,113–116 Besides, it influences the production of H2 from water electrolysis.2 Also, in this case, suppressing the side competitive HER has been observed to be a key factor to improve the EC CO2R performance.
To summarize, Fig. 11 brings together the best scientific works (in batch and in continuous-flow) that have been discussed above. It demonstrates a great diversity of Cu-based electrocatalysts that have been studied in the presence of liquid electrolyte solutions and provides knowledge about the performances for methanol, ethanol and n-propanol production that has been achieved with such systems to the present date. As can be seen, the best methanol performance has been ∼40% of FE with a partial current density of about 30 mA cm−2. Instead, the best ethanol performance has been a FE <50% and a partial current density of around 150 mA cm−2. In contrast, in the case of n-propanol the best FE reached are <10% with a Jn-PrOH <30 mA cm−2. The whole products distribution is listed in Table S3 (ESI†). From this Table, it is possible to appreciate that the selectivity towards products more reduced than C1 (i.e. ethanol) appears to have a correlation with the formation of CO. It seems that the catalyst should be active enough for the CO formation, but should also have a high binding energy towards the formation of *CO intermediate for producing C2+ products. As mentioned in section 2.3, the catalysts have to reduce the barrier of the *CO dimerization to enhance the production of ethanol and n-propanol. In fact, the liquid-phase EC CO2R electrocatalysts with the highest FE towards ethanol (>40%) present the lowest FE towards CO (<3%). Other works aiming to produce ethanol and n-propanol (although with lower performances) are listed in Table S4 (ESI†) to provide a broader literature overview. These latter works include the use of polycrystalline electrodes,108,115 the change of the catalytic layer thickness/roughness and modification of the Cu morphology in a systematic fashion,99,109,110,117–119 the use of graphene and metal oxide as supports.111,120–123
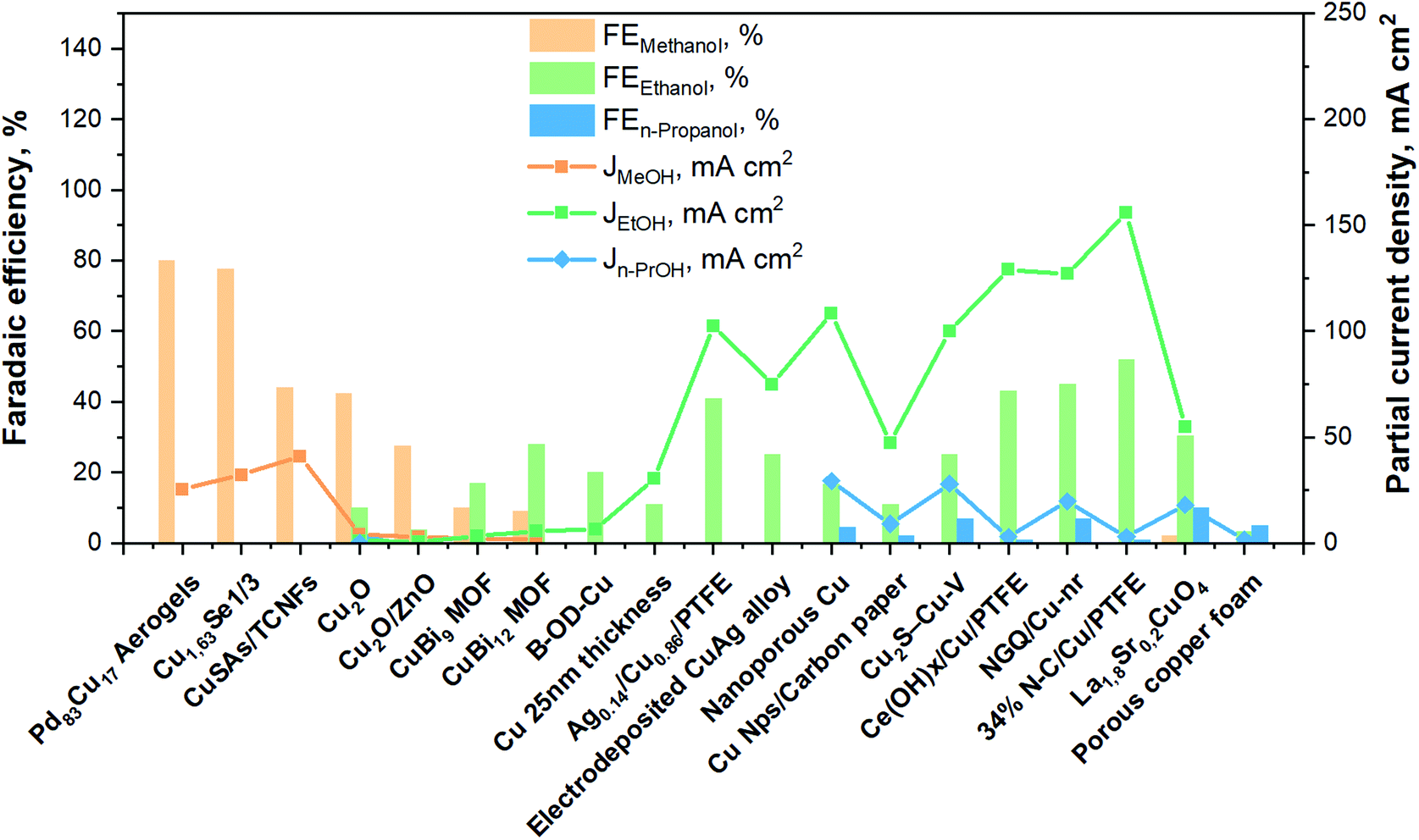 | ||
| Fig. 11 Faradaic efficiencies and partial current densities of the best liquid-phase EC CO2R electrocatalysts towards methanol, ethanol and n-propanol production. Authors with data taken from ref. 62, 73, 74, 83, 84, 90–98, 100–102 and 124. | ||
It is possible to affirm that the presence of the liquid electrolyte influences the CO2 electroreduction process because it can be useful to regulate the local pH and, consequently, the selectivity of the reaction. However, the following fundamental questions still remain: Does the liquid electrolyte reduces the activation barrier for CO2R or does it only resolve the problem of the competition between CO2RR and the thermodynamically preferred HER? Moreover, it is also important to consider another engineering/practical questions like: What could be the shortcomings of recovering the reaction products from the liquid electrolyte? How much energy has to be used for it?
The last questions are critical since alcohols must reach >99% of purity to be commercialized. Therefore, the processes for products purification from the output of the electrochemical cell play a key role in the overall cost of the full alcohols production chain. Distillation units are commonly used for alcohols/water mixtures separation, which usually requires a high investment, but also energy consumption and can highly influence the total energy demand and operative costs of the electrochemical technology. From our recently published work on technoeconomical and life cycle assessment of scaled-up methanol production by both electrocatalytic and thermocatalytic CO2 reduction to methanol, it came out that in the case of EC CO2R in aqueous media, considering a FE of 90% (at 100 mA cm−2) and a methanol productivity in the range of 1 to 106 kg h−1, the distillation columns and vessels constitute at least the 37.4% of the plant capital cost and the utilities account for about 79.8% of the operative costs.48 Moreover, due to the low solubility of CO2 in water, high pressures or costly ionic liquids are required to increase the performance. Therefore, at the moment, the liquid phase electrochemical conversion of CO2 could have low possibilities for fast achieving a practical success. This open the way to new developments for the implementation of different cell configurations like the solvent-less EC CO2R, which currently are at a proof-of-concept stage.40
3.2 Electrochemical CO2 reduction: catholyte-free configuration
A recent approach that overcomes some of the weaknesses mentioned above exploits a catholyte-free electrocatalytic cell design, which was firstly reported by Centi G. et al.129–132 The use of this kind of system for the CO2 reduction to alcohols is attractive because it avoids problems of solubility of CO2 in the liquid phase electrolytes and it is not necessary to use expensive and energy-intensive processes to recover the products from the liquid phase (such as distillation processes). Another advantage of this kind of configuration is the possibility to operate under mild temperatures (T < 150 °C) that can enhance the reaction kinetics and reduce mass transport limitations in the system.Very few reports directly use gaseous or humidified CO2 electrolysis, which emphasizes the challenge of controlling electrolyte-free electrochemical reactions. A complete overview of this kind of system is provided. This represents a strategy for increasing the current densities obtained until now. Fig. 12 shows the two more commonly used electrocatalytic cells in this kind of systems. The solvent-less electrocatalytic cell configuration consists of two chambers separated by a proton exchange membrane (Nafion in most cases).129–132 Gaseous CO2 (H2O-humidified or not) is fed to the cathode, and the anode chamber is filled with a liquid electrolyte solution (or with H2O-humidified N2) for providing the protons needed for the reduction reaction. The H+ then diffuse from the anode through the membrane to the cathode electrode (see Fig. 12a). Usually, the O2 evolution reaction is carried out at the anode over noble metal catalysts (i.e. Pt or IrO2).
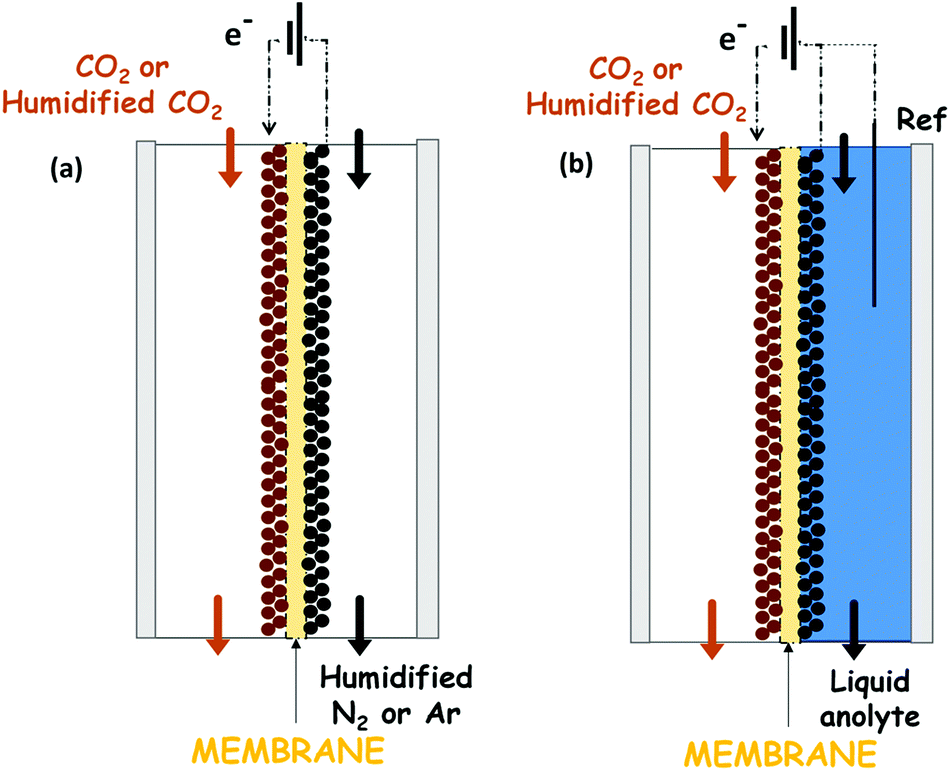 | ||
| Fig. 12 Representation of the most popular catholyte-free configuration, in continuous flow, for the electrochemical CO2 reduction: (a) solvent-less cell configuration, (b) liquid anolyte cell configuration. | ||
To provide an overview about the applicability of this kind of systems for the production of oxygenate fuels like alcohols, the best total current densities and relative FE (relative = excluding the FE of hydrogen) are gathered in Fig. 13. More detailed information is also provided in Table S5 in the ESI.† Furthermore, the total products distribution obtained with the best catholyte-free EC CO2R electrocatalysts is given in The electrocatalysts tested up to now for the CO2 reduction in these systems are based on metal nanoparticles, i.e. Fe, Pt,129–132,141 Co131 and Cu131 (with 10–20 wt% of the metal and about 0.5 mg cm−2), supported on carbon nanotubes (CNTs). In most of these systems the FE towards H2 is very high (>80%) and, therefore, the FE for the CO2 conversion is reported as a relative FE (i.e. selectivity values for CO2 conversion without considering the HER). In the case of Fe/CNTs electrocatalyst, the best relative FE of CO2 conversion for ethanol and methanol reached ∼70% and 21%, respectively, measured at a constant current of −1.41 mA cm−2 and 60 °C (on an electrode of about 3 cm in diameter). On the contrary, the electrocatalyst based on Pt forms isopropanol as the main product, with a relative FE of 34%. The Pt-based electrocatalysts have been also able to form C1+ products after the addition of a CO2-capturing component (a MOF), because of an enhanced surface concentration of CO2, being methanol the main product (60% at −5 mA cm−2). Conversely, Cu/CNTs and Co/CNTs generated lower amounts of hydrocarbons/organics. It is important to mention that the main disadvantage of the use of CNTs as electrocatalyst support is the difficulty in controlling the localization of the metal nanoparticles at the inner or outer surface of the CNTs. In regard to CNT as support of Cu-based catalyst, Jiménez C. et al.142 prepared a Cu/CNT catalyst (particle size between 2 and 5 nm) using supercritical fluid deposition. It was assembled into a polymer-exchange-membrane (PEM)-type electrochemical cell. In this work, CO has been the main reaction product, followed by small amounts of formic acid and methane.
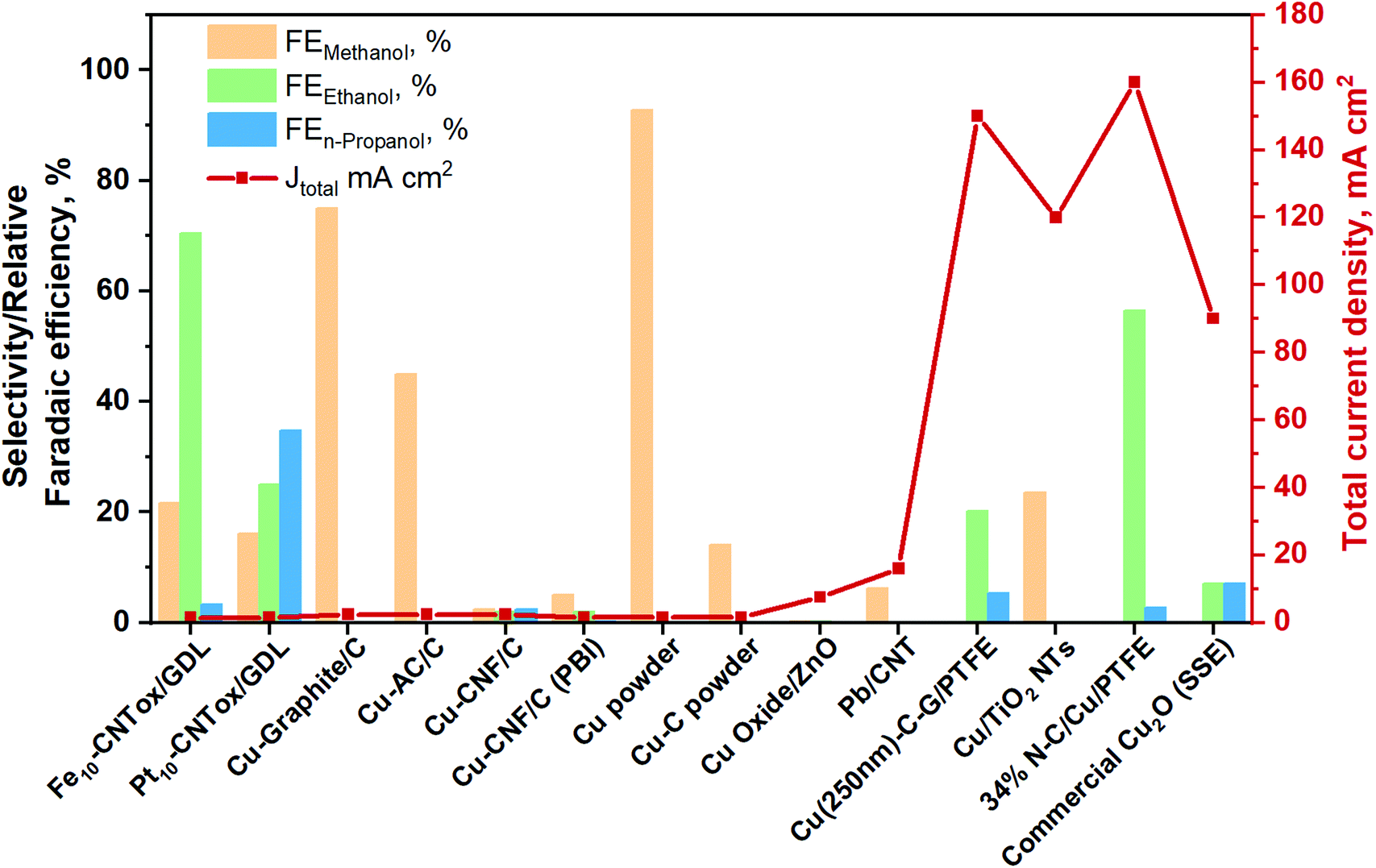 | ||
| Fig. 13 Relative faradaic efficiencies and total current densities of the best catholyte-free EC CO2R electrocatalysts towards methanol, ethanol and n-propanol production. Authors with data taken from ref. 131 and 133–140. | ||
A few years ago, Gutiérrez-Guerra N. et al.136 developed MEAs consisting of different Cu-based cathodic catalysts. Sterion was used as a proton exchange membrane and IrO2 as the anode of the cell. The gas-phase electrocatalytic conversion of CO2 was carried out at temperatures of up to 90 °C. They found that the electrocatalytic activity and selectivity of the catalyst is widely influenced by the nature of the carbon support (i.e. carbon nanofiber (CNF), graphite (G) and activated carbon (AC)). Thus, the Cu–G electrode of 12.56 cm2 reached the best CO2 conversion selectivity (relative FE) of 60 to 75% at −0.8 to −2.4 mA cm−2, respectively, towards methanol.
On the other hand, the Cu–AC and Cu–CNF electrodes were able to catalyse the reaction mainly towards acetaldehyde, both achieving around 60% of selectivity. In particular, the Cu–G cathodic electrode presented a higher particle size value (96 nm) than the other cathodic-catalysts: Cu–CNF and Cu–AC. The Cu–AC showed the lowest particle size value (40 nm), resulting in high catalytic activity and high dispersion of Cu particles on the high surface area of AC support, despite its lower electrical conductivity.
The frequently used proton exchange membranes (like Nafion or Sterion) cannot operate at temperatures higher than 90 °C. In this regard, in a subsequent work, Gutiérrez-Guerra N. et al.137 used H3PO4-doped polybenzimidazole polymer electrolyte membrane (PBI) to carry out the gas EC CO2R at a temperature of up to 110 °C, hence improving the kinetics of the Cu cathodic catalyst supported on CNFs. They exploited again IrO2 as the anodic catalyst because of its superior ability towards the OER. It is worth noting that in this case the main product obtained was acetaldehyde, with a relative FE of 85% and a current density of around −0.8 mA cm−2, similar to in their previous work. By increasing the applied current density (−1.6 mA cm−2) the selectivity of the reaction shifted to lighter and more saturated compounds, presumably due to an increase in protons transfer through the membrane. This Cu–CNF cathodic catalyst produced acetaldehyde, methyl formate, CO, methanol and ethanol with relative FEs of about 70, 20, 3, 3 and 2%, respectively. However, as in the other cases, HER was the main cathodic process. A year later, Gutiérrez-Guerra N. et al.143 tested Cu, and Cu–C materials sputtered on carbon paper in the same electrocatalytic cell. Their results revealed an increase in the production rate of methanol, acetaldehyde and methane by increasing the current densities from 0.8 to 2.4 mA cm−2. Moreover, pure Cu electrocatalyst presented a lower CuO/Cu ratio in the bulk than the Cu–C electrode, as well as lower particles size and more exposed planes and defects. Thus, the authors stated that the higher activity for methanol production using pure Cu is ascribed to these features. Furthermore, they demonstrated that an increase in temperature enhanced the reaction kinetics and in turns the CO2 consumption. The obtained FE towards hydrocarbons and oxygenates in this kind of systems are below 10%, since the HER was favoured being H2 the main product. Within this context, by using Pb nanoparticles on carbon nanotubes under similar conditions, García J. et al.138 observed that low temperatures (60 °C) favoured formic acid production. However, methanol production rate increases with temperature (up to reach 40 at 80 °C), whereas it does not seem to be affected by changes in the current density or the CO2 flowrate.
Pérez-Rodríguez S. et al.144 evaluated the electrochemical reduction of CO2 in a fuel cell (FC)-type reactor by using a Nafion 117 membrane. Fe and Pt deposited in commercial carbon black as support (Vulcan XC-72R) were used as cathodic catalysts. Their performance was evaluated in both acid media (0.5 M H2SO4) and in the gas phase, while pure H2 is used in the anode side. The relative humidity was fixed at 50% for both anode and cathode, and lower currents (e.g. −0.02 mA mg−1) were obtained in gas phase than in the liquid acid phase (e.g. −0.04 mA mg−1). The low performance of these materials under gas-phase conditions was ascribed to the suppression of the HER with respect to the acidic media. As expected, Pt-based electrodes catalysed mainly the formation of molecular hydrogen. In contrast, a Fe-based working electrode promotes the CO2 reduction to hydrocarbons and alcohols, according to the products detected by differential electrochemical mass spectrometry (DEMS) from CO2 reduction in the liquid phase (FEs not reported).
Lately, Merino-Garcia I. et al.145,146 also tried to control electrolyte-free electrochemical CO2 reduction reactions. They performed gas-phase CO2 electroreduction in two-compartment cells by using commercial Cu nanoparticles (25 nm, 40–60 nm and 60–80 nm) on porous carbon paper as a support, which was used as working electrode assembled with a Nafion membrane. Ethylene and methane were the main obtained hydrocarbons. The best performance to ethylene that has been reached in a gas-phase CO2 electrolyser was obtained in this study (FE = 92.8% at 7.5 mA cm−2) by using the Cu25 (Cu nanoparticles of 25 nm) catalyst. They studied the effect of the humidified CO2 feed stream, which did not improve the CH4 productivity and selectivity, but a high H+ transport capacity through the Nafion membrane was evidenced. Subsequently, on the basis of the literature,84 they addressed the dispersion of Cu oxides on ZnO in a half-MEA configuration, working with a 0.1 M KHCO3 aqueous anolyte. The results showed a synergic effect between Cu oxides and ZnO, demonstrated by a lower Tafel slope. They obtained for both Cu oxides/ZnO-GDE and Cu25-GDE a similar FE to ethylene (i.e. ∼91–92% at 7.5 mA cm−2, respectively), being more stable the catalyst containing ZnO as support.147
Metal oxide electrocatalysts have the merits of being promising materials due to their high selectivity and high energy efficiency in liquid phase electrolytes.16 Hossain S. et al.140 carried out the electrocatalytic CO2 conversion to methanol with a 10% Cu-based catalyst supported on TiO2 nanotubes. Pt–Ru/C was used on the anode side with liquid water. They achieved the highest total current density (120 mA cm−2) that has been achieved with methanol production in recent years in a MEA cell configuration, by using a solid polymer electrolyte membrane and a CO2 flow rate of 20 mL min−1.14 Nevertheless, the maximum achieved FE to methanol was only of 4%. The role of TiO2 can be on enhancing the CO2 adsorption and stabilization of CO˙− radical, improving the electrocatalyst surface area and the reaction stability.148
On the other hand, Gabardo C. et al.139 prepared and tested Cu nanoparticles (250 nm) sputtered on a porous PTFE support (the electrocatalyst was then airbrushed with carbon nanoparticles and graphite). The electrochemical CO2 reduction in the gas phase was conducted in a MEA configuration by using an anion exchange membrane and an IrO2/Ti anode immersed in 0.1 M KHCO3 (see Fig. 14). This MEA co-electrolyser operates at industrially relevant current densities, while simultaneously achieving a high selectivity toward multi-carbon products (accumulative FE of 80% towards C2+ products). The authors demonstrated that increasing the operating temperature (up to 40 °C) and decreasing the CO2 flow rate, concentrated liquid (4wt% ethanol) and gas product (30% ethylene) streams can be obtained. A stable total current density >200 mA cm−2 was achieved at −3.9 V for more than 24 h, which is two orders of magnitude greater than the previous results discussed above.
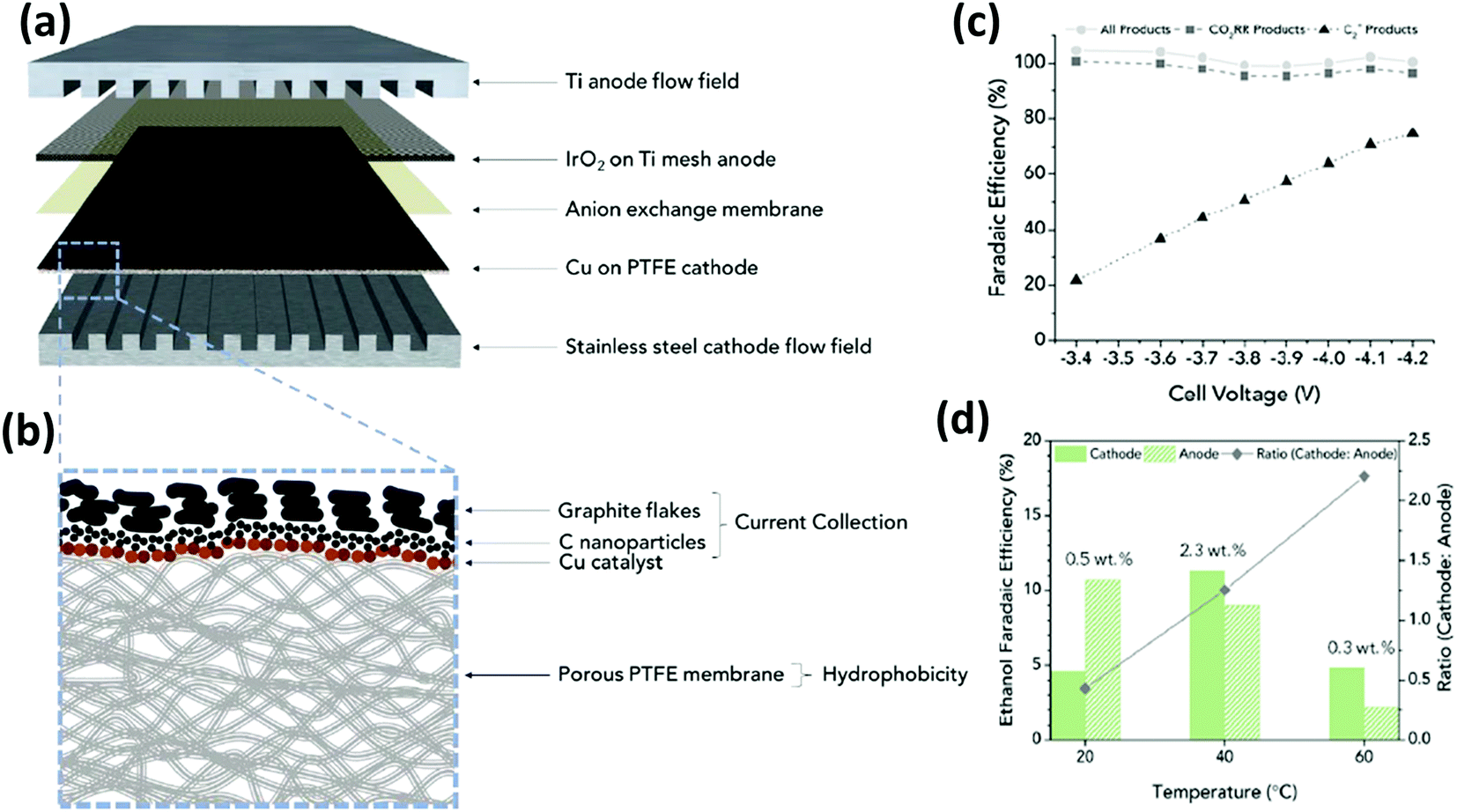 | ||
| Fig. 14 Exploded view of MEA layers inside the electrolyzer (a) where the hatched line box is the Cu on PTFE cathode with the cross-section structure is displayed in (b); (c) faradaic efficiencies of all products (H2 and EC CO2R) and C2+ products and (d) amounts and ratios of FEethanol that are recovered from the cathode and anode streams at each temperature. Reproduced from ref. 139 with permission of Joule. | ||
Inspired by solid-state batteries, Xia C. et al.135 have proposed a solid-state electrolyte (SSE) for a CO2 reduction reaction system. This work aimed to produce HCOOH using a two-dimensional Bi catalyst. However, an electrolyte-free oxygenate solution was also obtained using commercial Cu2O nanoparticles. This solution contained 4.6 mM ethanol, 3.4 mM n-propanol, among other liquid products. They implemented an in operando technique (XAS) in order to study the electronic structure of the Bi catalyst under electrochemical CO2 reduction conditions. The in situ tests were carried out in a traditional H-cell filled with CO2-saturated 0.5 M KHCO3 electrolyte. The results indicated that the active phase was metallic Bi under the negative applied potentials.
In a previously discussed work,90 the 34% N-doped C layer on a Cu surface supported on PTFE was also tested in a MEA catholyte-less system. IrOx/Ti was used as the anode in 0.2 M KHCO3. After operating the MEA system under a full-cell voltage of −3.67 V for 15 h, with a total current density of about 160 mA cm−2, the system retained its ethanol FE of 52%. These results demonstrate the importance of controlling the hydrophobicity of the electrocatalyst surface (provided in this case by the PTFE electrode support) to improve the triple-phase boundary favouring the CO2R at high rates against the HER.
The catholyte-free electrochemical conversion of CO2 system is an attractive alternative to avoid the drawbacks of the liquid-electrolyte based systems. However, further efforts are needed to improve its performance. From the Table S6 (ESI†), it can seen that there is a correlation between CO and ethanol faradaic efficiency, as for the liquid-phase systems. The electrocatalysts with a lower FE to CO (and provably higher *CO binding energy) are those able to enhance C–C coupling for producing ethanol. However, the reaction mechanisms of EC CO2R in the gas phase are not totally similar to those of the same catalysts working in liquid electrolytes, in which most of the investigations have been done. Indeed, Cu, which is the most promising material reported in the literature to give rise to C–C bond formation in EC CO2R in the liquid phase, does not show such good behaviour under gas-phase conditions. In this regard, Genovese C. et al.131 have hypothesized that CO2 dissociates to CO and chemisorbed O, which induces oxidation of the catalyst surface decreasing its activity. Therefore, the further conversion of CO intermediate may also proceed through C–O break dissociation, instead of through CO chemisorption/hydrogenation. On the contrary to metallic Cu, it has been seen that Cu1+ and Cu0 species improve the selectivity to alcohols in the gas phase.136,143
Finally, it is worthy of notice that there is only one catalyst (34% N–C/Cu/PTFE) able to operate at a relevant current density, while simultaneously achieving a high relative FE towards ethanol, with a good stability of 15 h (ref. 90) (see Fig. 12). This catalyst also showed the best performance for alcohols in liquid catholyte solutions, highlighting the fundamental role of the catalyst material on the CO2 reduction selectivity.
From the current state-of-the-art (SoA) of both catholyte and catholyte-free electrochemical conversion of CO2 to alcohols, we understood that the major bottlenecks for the practical implementation of the electrochemical conversion of CO2 are:
• There are few examples of electrocatalysts showing a high FE for single alcohol whereas operating at the same time at relevant current densities, and their long-term stability has not been already proved.
• High overpotentials are usually required (cell potentials >3 V).
• The best alcohols production performance has been achieved in liquid-phase electrolyte configurations, which entails high costs for downstream products separation.
• The competing HER is still predominant in almost all the catholyte-free electrocatalytic systems.
4 Investigation highlights and future perspectives
Among the different metal catalysts reported for the electrochemical CO2 conversion, Cu-based materials are the most promising, abundant, cheap and selective for the production of alcohols (see Fig. 11 and 13). What is more, novel catalysts designs composed of copper with another metal (e.g. alloys, intermetallic compounds)73,83,99,100,124 or metal oxides (as support)75,78,80,84,111,121 have got more attention. It could be due to their possible cooperative and synergistic effect, which could improve the overall performance of the process. The tuning of the material morphology, particle size, textural features and exposed facets are key parameters that usually confer enhanced catalytic activity. In this regard, recent studies have developed porous electrode morphologies to enhance mass transport and, consequently, catalytic activity.96,98,101,106 A large specific surface area and a high dispersion of active sites are essential to increase the catalyst activity. For this reason, catalysts nanostructuring have been widely studied.149 Nevertheless, attention has to be paid to the final size of the metal particles, since it is a determining aspect for the productivity and selectivity to fuels or chemicals. It is generally observed that hydrocarbons and multicarbon oxygenates are favoured on Cu particles higher than 15 nm, whereas CO and H2 are favoured on smaller ones (<3–6 nm).79,149–153 The different selectivity between the particle sizes could be related to the presence or not of specific crystal orientation surface, corners, edges and defects in the electrocatalysts that facilitates the HER and *CO desorption from the catalyst surface.116,154 It is also well known that the Cu crystalline orientation play a key role in determining the products selectivity.155 From theoretical calculations, the ideal electrocatalyst surface for alcohols (i.e. CH3OH) production must have an optimal CO binding energy (−0.67 eV), a weak H adsorption energy to suppress the HER, and weak binding energy for OH species. One fundamental problem of pure metal catalysts is the scaling relations among the adsorption energies of different CO2R intermediates.156 Recently, few examples of Cu alloying (e.g. Cu–Au NPs, oxide-derived Cu4Zn NPs) have been proposed for tuning the EC CO2R activity and selectivity bringing a double gain by overpotential reduction and selectivity enhancement.63,99,157The role of Cu-facets on the EC CO2R reaction mechanism and selectivity has been widely investigated. There are several literature works on experimental and theoretical investigations reviewing these aspects.15,56,158 Therefore, it is out of the scope of this work. Though, it is worth to mention that generally: Cu(100) and stepped (211) facets favour C–C coupling via *CO dimerization and further hydrogenation to ethylene or ethanol, as compared to Cu(111); Cu(110) and Cu(511) promote the production of ethanol, acetate and acetaldehyde, which has been confirmed by DFT simulation.15,56 In the same way, DFT models have also confirmed that the use of concentrated hydroxides (OH−) could further decrease the energy barrier for CO dimerization, thus promoting the C2+ products.56
Regarding the oxidation state of copper, to date, Cu0 and Cu□+ (and/or their interfaces) have been identified in several studies as the copper active sites, but it is still under debate.159–163 It has been reported that Cu1+ or a mix between Cu1+ and Cu2+ promotes methanol production.105,150,164 On the other hand, mechanistic studies of Cu oxide-derived materials revealed that the interface between Cu1+ and Cu0 contributes to the dimerization of *CO species on the electrode surface to induce C–C coupling and generate C2+ products, like ethanol and n-propanol. It is believed that the interface between Cu1+ and Cu0 leads to electrostatic interactions with the adsorbed intermediates since the C atom of *CO at Cu1+ is positively charged, whereas that of *CO at Cu0 is negatively charged.105 In addition, Cu-doping with heteroatoms like B also induces the formation and stabilization of Cu1+/Cu interfaces, because Cu atoms adjacent to B atoms are more positively charged, and this has been demonstrated to increase the energy barrier of the RDS: *CO + *H → *CHO and reduce the barrier of the *CO dimerization, inducing a high activity for EC CO2R towards C2 products.92,160 Within this context, new strategies for N- or S-doped copper80,90,91,106,165 have been exploited to study the synergistic effect between generated defects and interfaces. In this regard, as previously discussed, systematic in situ and in operando characterizations have been developed and used in some research works to identify and monitor the copper actives sites under reaction conditions.62,71,78,90,93–95,97,98,135,166 However, the stability of Cu□+ is of special concern under electrochemical reduction conditions; e.g. time-resolved in situ soft X-ray absorption spectroscopy studies demonstrated that the initial transition step from Cu2+ to Cu+ is very quick, while the further electrochemical reduction of Cu+ to Cu0 is much slower.167 The active sites changes also induce a restructuration of the catalyst morphology, as has been observed by liquid cell transmission electron microscopy under operando electrochemical conditions.168 Thus, the synthesis of electrocatalyst with stable Cu□+ sites still remains a challenge.56 As it has been observed for the Cu-oxides/ZnO systems,75,78,84,87,111,147 we believe that the dispersion of Cu□+ species into other metal oxide substrates (TiO2, Al2O3, ZrO2, etc.)16,49 can be a promising strategy for this purpose, which can take inspiration from the different catalytic systems developed for the thermocatalytic CO2 hydrogenation (see section 2.1). For instance, high selectivity to methanol was achieved by this process using Cu/Zn/Ga/SiO2 (99.5%) and Au/Zn/ZrO2 (100%) catalysts, by using pressurized H2 at mild temperatures (270 °C–220 °C, respectively).38 Therefore, ignoring the electrons source and exploiting the electrochemically produced H+ and H2 species, the CO2 reduction reaction can take place according to similar reaction mechanisms and can follow similar kinetic laws than the thermocatalytic CO2 hydrogenation, at properly tuned operative conditions.169 Thus, this can help in making faster progress in the development of new electrocatalysts materials.
Likewise, the nature of the conductive support plays an important role in enhancing the catalytic activity at the gas-solid interphase and, thus, the productivity of the EC CO2R. Therefore, carbonaceous materials are preferred due to their high-adjustable surface state and good conductivity; they have also been tested for their intrinsic catalytic activity. Different investigations have employed carbon nanotubes, carbon nanofibers, graphite, graphene and activated carbon. By using carbon-based electrodes, it is possible to exploit the confinement effect170 inside their nanopores to create a virtual higher pressure of the reactant (CO2) at the electrocatalyst surface. In turns, the higher surface CO2 concentration could enhance the formation of the C–C bond.171 Moreover, doping carbonaceous materials with heteroatoms of different electronegativities helps to stop its charge neutrality, inducing a redistribution and creating active sites. This method can significantly modify the microstructure of the C-based material, increasing its surface area and structural defects, thus defining the acid/base and hydrophilic/hydrophobic character of the carbon surface. This can determine the CO2 adsorption behaviour and the catalytic and electrochemical properties. For instance, CNT-catalysts functionalized with oxygen (i.e. CNTox) containing carbonyl groups demonstrated a higher selectivity than the non-doped CNT for the formation of ethanol.129–132,141 However, the yield achieved at present with pure C-based catalysts is very low, as can be seen in Fig. 13.
Different types of electrocatalytic reactors have been used for the EC CO2R (see Fig. 5 and 12). The obtained results have demonstrated that the use of a continuous-flow electrocatalytic cell with a suitable design have multiple benefits in comparison with batch systems for scaling-up this process. For instance, the use of a GDE cell has the effect of increasing the current density during the electrochemical reactions by enabling prolonged contact between CO2 and catalytic sites.69 Nevertheless, one disadvantage of this kind of setup is that, when operating with liquid electrolytes as catholyte, the continuous flow removes the products or intermediates from the electrode surface, leading to relatively short residence times. This also depends on the cell design and size, but it severely influences both the faradaic efficiency and products distribution.107 A further key parameter to improve the performance of the EC process is the proper selection of the membrane material, to optimize both mass/charge transport and lifetime. Nonetheless, no matter which type of ion-selective membrane is used (e.g. anionic or cationic), the liquid products can crossover from the catholyte to the anolyte, being then re-oxidized. Moreover, the volatile alcohol products such as ethanol have also experienced evaporation through the gas diffusion layer.139,172 Within this context, bipolar membranes (BPM) is an alternative to avoid the products crossover, but they can promote higher overpotentials. BPM consist of anion- and cation exchange membranes that are laminated together, typically with a catalyst that promotes the auto dissociation of water at the interface. Besides, BPM-based electrolysis cells can maintain constant pH on the two sides by the selective transport of H+ and OH− ions to the cathode and anode, respectively, which could be beneficial for the long-term stability of the electrochemical CO2 conversion system.
A GDE system is able to reduce/eliminate the concentration polarization in the bulk electrolyte (as it occurs in H-type cells). These kinds of systems have reached industrially-relevant total current densities, although the selectivity towards a single product is not satisfactory so far (see Fig. 11). Therefore, the high costs required for the separation of alcohol products from the catholyte outlet stream makes this a still not suitable option to establish this technology at an industrial level. Another promising alternative consists in a typical PEM or AEM (anion exchange membrane) electrolyser design using a MEA. The most common design is composed of a cathode compartment with only gas (or humidified gas) and an anode compartment with a liquid phase anolyte. However, as shown in Fig. 12, electrolyte-free (or zero-gap) designs with humidified N2 or Ar in the anode have also been studied. The MEA-based design stands up between the alternatives to produce more concentrated products, avoiding mixtures with solvents difficult to separate like water, and lowering ohmic losses that are associated to high applied potentials and operative costs. It also does not need to work at high temperatures (as in the case of solid oxide electrolysers). Nevertheless, further efforts are also needed to increase the production rates using MEAs in EC CO2R electrolysers (see Fig. 13).
It is important to highlight that the greatest efforts for electrocatalyst development have been focussed on achieving a high selectivity. It means that required conditions an efficient CO2 reduction can lose sight, thus obtaining materials that are not stable to operate for more than 24 hours (e.g. for ethylene production139). Activity degradation is a consequence of the electrocatalyst instability. In this regard, the formation and deposition of carbonates at the surface of the catalyst has been found to be responsible for EC CO2R activity decay when using liquid electrolytes (e.g. in KHCO3). It reduces the hydrophobicity and blocks the catalyst layer surface, eventually hindering the CO2 flow through the GDL to the catalyst surface and the electrolyte in the case of GDE cells. If cathodic electrodes are not suitably porous, the proper diffusion of gaseous products back through the GDE represents a major challenge (mainly when the cell is operated at high current densities), since bubbles accumulation could also contribute to the physical catalyst degradation/detachment over time.57,96 Therefore, the stability of electrocatalysts is a predominant obstacle to be overcome for a large-scale industrial application. In this framework, efforts have been made to evaluate the stability of GDE-based systems. Those strategies include different methods for the catalyst layer deposition, the study of the effect of the binder (e.g. Nafion) loading in the catalytic ink, and non-conventional gas diffusion layers substrate (e.g. PTFE mats). It has been proved that a proper catalyst deposition method is airbrushing,75,171,173 since it minimizes the agglomeration of the catalyst in the carbon support facilitating the formation of well-defined electrodes. Additionally, PTFE porous layer seems to be less susceptive to degradation over time since the stable hydrophobic gas diffusion layer prevents electrode flooding.62,90,97,139
The conversion of CO2 to any organic molecule requires substantial energy input to overcome the substantial thermodynamic and kinetic barriers. Most of the reported works have been done at ambient temperature and pressure. However, in order to overcome those barriers and increase the reaction rate, in some works of EC CO2R in gas phase the cells were tested in the range 90–110 °C.137,143
The local reaction environment is another crucial factor. Some studies report that a moderate CO2 concentration close to the catalyst surface could lead to an optimum amount of *CO2 and *CO, which may be ideal for C–C coupling towards multicarbon products (i.e. ethanol, n-propanol), while a low concentration of these species favours the competitive HER, and a high CO2 concentration could promote C1 products (i.e. methanol).125,171 In the same way, the reaction pathways towards C2+ products is favoured at relatively high local pH, which is promoted by the presence of OH− ions as mentioned above.56,116,128 Accordingly, more attention should be paid to modelling and computational calculations for predicting the reaction environment that promotes the selective conversion of CO2 into the desired products. In addition, the implementation of in situ/operando characterization techniques can provide key insights concerning the catalyst surface (i.e. structure, composition, oxidation state and reaction intermediates).150
Fig. 15 demonstrates that the faradaic efficiencies and partial current densities for ethanol, n-propanol and methanol have increased steadily over the past 30 years. Partial current densities for ethanol have also increased to ∼150 mA cm−2 as a result of the implementation of GDE or/and MEA cell configurations, which overcome the CO2 solubility issues in aqueous electrolytes. Although the highest faradaic efficiencies have been reached for methanol, the partial current densities are far from an industrial application. In the case of n-propanol, the FE and partial current densities are the less performing results. However, it has been found from a general techno-economic analysis that the production of ethanol and n-propanol from EC CO2R is promising under optimistic conditions (e.g. 300 mA cm−2 and 0.5 V overpotential at 70% faradaic efficiency). At the same time, methanol is the least favourite product from that study since it was the only product with a negative Net Present Value (NPV) using an optimistic case assumption. That is probably because the market value of methanol (0.58$ per kg) is so low that profitability is impossible regardless of process performance, besides its capital and operating costs.50 However, we recently demonstrated that the methanol production at industrial scale (including downstream processing units) is economically competitive in a EC CO2R system achieving 90% of FE and a total of 100 mA cm−2, at productivities greater than 3.3 kg h−1, if an effective allocation of the product in a real market scenario is considered.48 However, to make this process more sustainable than the current methods used to produce methanol the current density must achieve at least 200 mA cm−2, which results in a reduction of 68% of the carbon footprint of this process (reaching up to 2.72 kgCO2 eq. per kgCH3OH).
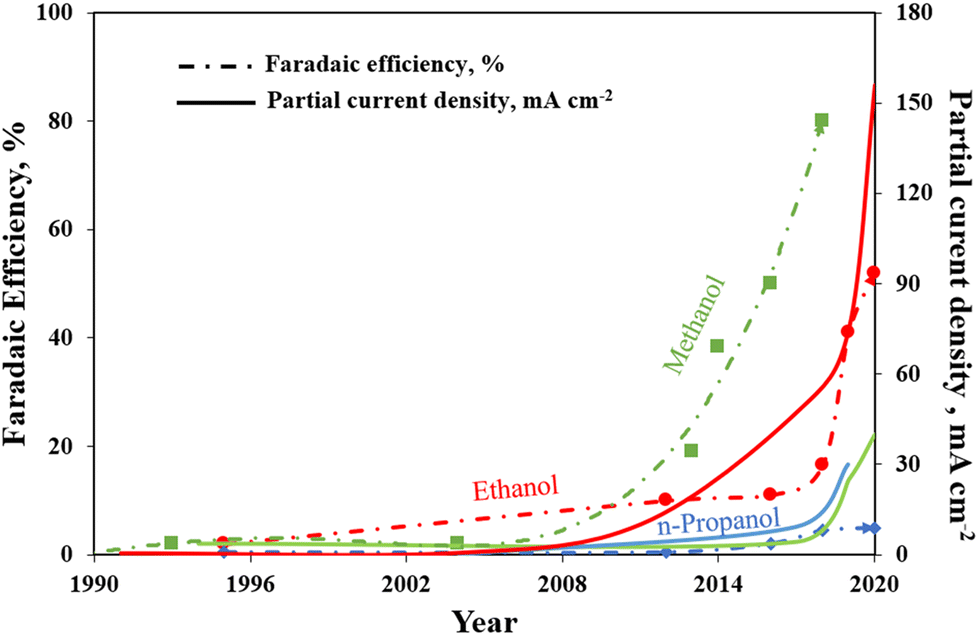 | ||
| Fig. 15 Highest reported faradaic efficiencies (dashed line) and partial current densities (continuous line) for ethanol (red circles), n-propanol (blue diamonds) and methanol (green) over the past three decades. | ||
5 Conclusions
The management of GHGs emissions is one of the most challenging environmental problems to face in the current century. A feasible option to reduce CO2 content in the atmosphere and tackle the environmental problem from GHGs emitted by human activities is to transform it into valuable fuels or chemicals by the electrocatalytic reduction of CO2. This paper reviewed the recent considerable progress in electrochemical CO2 reduction to methanol, ethanol and n-propanol on Cu-based catalysts and the key strategies used to tune its activity and selectivity electrolyte effect, cell configuration, engineering and surface modification of electrocatalysts.From the current state-of-the-art of both catholyte and catholyte-free systems, we realize that the morphology of the nanostructured and composite electrocatalysts (doped or not) did not show high selectivity for a single product (alcohol) while operating at a high current density. The implementation of GDE or/and MEA cell configurations allow tackling the CO2 solubility issues in aqueous electrolytes, reaching high production rates. Although the solvent-less EC CO2R cell design is a promising approach to producing alcohols not dissolved in liquid electrolytes, and increased dominance of the H2 evolution side reaction in almost all the current works limits the practical application of those systems. Thus, until now, the best EC CO2R performance towards alcohols production has been achieved in liquid-phase electrocatalytic configuration, which entails high costs for downstream products separation.
Electrochemical CO2 reduction processes still need further experimental analysis and theoretical efforts to: (i) improve the design of the electrocatalytic cell, (ii) optimize the catalytic sites activity, selectivity and stability, and (iii) engineering the electrocatalyst, to enhance the charge and mass transport within the system, for reducing ohmic losses and pursuing high current densities (>200 mA cm−2) towards industrially relevant rates. Even if many challenges remain, it is assumed that with further investigation, the perspective of implementing CO2 electrolysis for producing fossil-free fuels and chemicals at an industry level could be realised soon.
Abbreviations
| AC | Activated carbon |
| AEM | Anion exchange membrane |
| BE | Binding energy |
| CNF | Carbon nanofiber |
| CNT | Carbon nanotubes |
| CO2R | CO2 reduction |
| Cu | Copper |
| DEMS | Differential electrochemical mass spectrometry |
| DFT | Density functional theory |
| E° | Standard reduction potential |
| EC | Electrochemical |
| EDX | Energy-dispersive X-ray spectroscopy |
| EtOH | Ethanol |
| FC | Fuel cell |
| G | Graphite |
| GDE | Gas diffusion electrode |
| GHG | Greenhouse gas |
| GN | Graphene |
| HER | Hydrogen evolution reaction |
| MEA | Membrane electrode assembly |
| MeOH | Methanol |
| MOF | Metal–organic framework |
| NHE | Normal hydrogen electrode |
| NPV | Net present value |
| OER | Oxygen evolution reaction |
| PBI | Polybenzimidazole polymer electrolyte membrane |
| PCET | Proton-coupled-electron-transfer |
| PEC | Photoelectrochemical |
| PEM | Polymer exchange membrane |
| PrOH | Propanol |
| PTFE | Polytetrafluoroethylene (Teflon) |
| RDS | Rate determining step |
| RE | Reference electrode |
| RWGS | Reverse water gas shift |
| SAED | Selected area (electron) diffraction |
| SDS | Selectivity determining step |
| SEM | Scanning electroscope microscope |
| SI | Supporting information |
| Syngas | CO and H2 |
| TC | Thermochemical |
| TEM | Transmission electron microscopy |
| TRL | Technology readiness level |
| WE | Working electrode |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been performed with the financial support of Eni SpA and the R&D Program Energy Transition (Cattura e Utilizzo CO2), the European Union’s Horizon 2020 Research and Innovation Action program under the Project SunCoChem (Grant Agreement No 862192) and the ERC@Polito CO2 Synthesis project granted by Politecnico di Torino.References
- I. Ganesh, Renewable Sustainable Energy Rev., 2016, 59, 1269–1297 CrossRef CAS.
- S. Hernández, M. A. Farkhondehfal, S. Francesc, M. Makkee, G. Saracco and N. Russo, Green Chem., 2017, 19, 2326–2346 RSC.
- T. F. Stocker, D. Qin, G. K. Plattner, M. M. B. Tignor, S. K. Allen, J. Boschung, A. Nauels, Y. Xia, V. Bex and P. M. Midgley, Climate change 2013 the physical science basis: Working Group I contribution to the fifth assessment report of the intergovernmental panel on climate change, New York, 2013 Search PubMed.
- L. E. Singer and D. Peterson, International Energy Outlook 2016, Washington, DC 20585, 2016 Search PubMed.
- Strategic Energy Technology Plan, https://ec.europa.eu/energy/topics/technology-and-innovation/strategic-energy-technology-plan_en, (accessed 28 May 2020).
- I. Ganesh, Renewable Sustainable Energy Rev., 2014, 31, 221–257 CrossRef CAS.
- Methanex, The Power of Agility, http://www.methanex.com/about-methanol/methanol-vehicle-fuel, (accessed 9 October 2018).
- X. Zhao, M. Yin, L. Ma, L. Liang, C. Liu, J. Liao, T. Lu and W. Xing, Energy Environ. Sci., 2011, 4, 2736–2753 RSC.
- G. A. Olah, Angew. Chem., Int. Ed., 2005, 44, 2636–2639 CrossRef CAS.
- J. P. Soto Veiga and T. L. Romanelli, J. Cleaner Prod., 2020, 260, 121092 CrossRef.
- S. Madiwale, K. Alagu and V. Bhojwani, Therm. Sci., 2020, 24, 27–36 CrossRef.
- J. C. Thermodynamics, A. Mehrdad and M. Hajikarimi, J. Chem. Thermodyn., 2020, 142, 105972 CrossRef.
- B. C. Ong, S. K. Kamarudin and S. Basri, Int. J. Hydrogen Energy, 2017, 42, 10142–10157 CrossRef CAS.
- Y. Liu, F. Li, X. Zhang and X. Ji, Curr. Opin. Green Sustain. Chem., 2020, 23, 10–17 CrossRef.
- T. K. Todorova, M. W. Schreiber and M. Fontecave, ACS Catal., 2020, 10, 1754–1768 CrossRef CAS.
- G. Liu, T. Tran-Phu, H. Chen and A. Tricoli, Adv. Sustain. Syst., 2018, 2, 1800028 CrossRef.
- C. T. Dinh, F. P. García De Arquer, D. Sinton and E. H. Sargent, ACS Energy Lett., 2018, 3, 2835–2840 CrossRef CAS.
- R. Parajuli, D. Ansovini, M. F. Philips and K. J. P. Schouten, WO 2019/141827 Catalyst system for catalysed electrochemical reactions and preparation hereof, applications and uses thereof, 2019 Search PubMed.
- A. Dominguez-Ramos, B. Singh, X. Zhang, E. G. Hertwich and A. Irabien, J. Cleaner Prod., 2015, 104, 148–155 CrossRef CAS.
- J. T. Song, H. Song, B. Kim and J. Oh, Catalysts, 2019, 9, 224 CrossRef.
- J. Wu, M. Saito, M. Takeuchi and T. Watanabe, Appl. Catal., A, 2001, 218, 235–240 CrossRef CAS.
- F. Studt, I. Sharafutdinov, F. Abild-Pedersen, C. F. Elkjær, J. S. Hummelshøj, S. Dahl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2014, 6, 320–324 CrossRef CAS.
- J. Xiao, D. Mao, X. Guo and J. Yu, Appl. Surf. Sci., 2015, 338, 146–153 CrossRef CAS.
- E. A. Morosanu, F. Salomone, R. Pirone and S. Bensaid, Catalysts, 2020, 10, 283 CrossRef CAS.
- M. Pérez-Fortes, J. C. Schöneberger, A. Boulamanti and E. Tzimas, Appl. Energy, 2016, 161, 718–732 CrossRef.
- B. M. Tackett, E. Gomez and J. G. Chen, Nat. Catal., 2019, 2, 381–386 CrossRef CAS.
- H. W. Haitham Al-Kalbani, J. Xuan and S. García, Appl. Energy, 2016, 165, 1–13 CrossRef.
- J. B. Vennekoetter, R. Sengpiel and M. Wessling, Chem. Eng. J., 2019, 364, 89–101 CrossRef CAS.
- L. Samiee and S. Gandzha, Rev. Chem. Eng. DOI:10.1515/revce-2019-0012.
- D. Bellotti, M. Rivarolo and L. Magistri, Energy Procedia, 2019, 158, 4721–4728 CrossRef CAS.
- Z. Qin, G. Zhai, X. Wu, Y. Yu and Z. Zhang, Energy Convers. Manag., 2016, 124, 168–179 CrossRef CAS.
- F. Ausfelder, A. Bazzanella, H. VanBracki, R. Wilde, C. Beckmann, R. Mills, E. Rightor, C. Tam, N. Trudeau and P. Botschek, Technology Roadmap Energy and GHG Reductions in the the Chemical Industry via Catalytic Processes, 2013 Search PubMed.
- E. Alper and O. Yuksel Orhan, Petroleum, 2017, 3, 109–126 CrossRef.
- A. A. Kiss, J. J. Pragt, H. J. Vos, G. Bargeman and M. T. de Groot, Chem. Eng. J., 2016, 284, 260–269 CrossRef CAS.
- T. Witoon, N. Kachaban, W. Donphai, P. Kidkhunthod, K. Faungnawakij, M. Chareonpanich and J. Limtrakul, Energy Convers. Manag., 2016, 118, 21–31 CrossRef CAS.
- T. Phongamwong, U. Chantaprasertporn, T. Witoon, T. Numpilai, Y. Poo-arporn, W. Limphirat, W. Donphai, P. Dittanet, M. Chareonpanich and J. Limtrakul, Chem. Eng. J., 2017, 316, 692–703 CrossRef CAS.
- J. Toyir, R. Miloua, N. E. Elkadri, M. Nawdali, H. Toufik, F. Miloua and M. Saito, Phys. Procedia, 2009, 2, 1075–1079 CrossRef CAS.
- S. Saeidi, N. A. S. Amin and M. R. Rahimpour, J. CO2 Util., 2014, 5, 66–81 CrossRef CAS.
- R. P. Ye, L. Lin, Q. Li, Z. Zhou, T. Wang, C. K. Russell, H. Adidharma, Z. Xu, Y. G. Yao and M. Fan, Catal. Sci. Technol., 2018, 8, 3428–3449 RSC.
- H. Guzmán, M. A. Farkhondehfal, K. R. Tolod, N. Russo and S. Hernández, in Solar Hydrogen Production Processes, Systems and Technologies, Elsevier Inc., 2019, p. 560 Search PubMed.
- P. Kangvansura, L. M. Chew, W. Saengsui, P. Santawaja, Y. Poo-arporn, M. Muhler, H. Schulz and A. Worayingyong, Catal. Today, 2016, 275, 59–65 CrossRef CAS.
- M. K. Gnanamani, G. Jacobs, R. A. Keogh, W. D. Shafer, D. E. Sparks, S. D. Hopps, G. A. Thomas and B. H. Davis, Appl. Catal., A, 2015, 499, 39–46 CrossRef CAS.
- Y. Yang, T. Lin, X. Qi, F. Yu, Y. An, Z. Li, Y. Dai, L. Zhong, H. Wang and Y. Sun, Appl. Catal., A, 2018, 549, 179–187 CrossRef CAS.
- Y. N. Gao, S. Liu, Z. Zhao, H. C. Tao and Z. Y. Sun, Wuli Huaxue Xuebao/Acta Phys. – Chim. Sin., 2018, 34, 858–872 CAS.
- L. C. Grabow and M. Mavrikakis, ACS Catal., 2011, 1, 365–384 CrossRef CAS.
- A. Álvarez, A. Bansode, A. Urakawa, A. V. Bavykina, T. A. Wezendonk, M. Makkee, J. Gascon and F. Kapteijn, Chem. Rev., 2017, 117, 9804–9838 CrossRef.
- A. Zachopoulos and E. Heracleous, J. CO2 Util., 2017, 21, 360–367 CrossRef CAS.
- H. Guzmán, F. Salomone, E. Batuecas, T. Tommasi, N. Russo, S. Bensaid and S. Hernández, Chem. Eng. J., 2020, 127973 CrossRef.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS.
- M. Jouny, W. Luc and F. Jiao, Ind. Eng. Chem. Res., 2018, 57, 2165–2177 CrossRef CAS.
- CELBICON project, https://www.celbicon.org/, (accessed 28 March 2020).
- RECODE project, https://www.recodeh2020.eu/, (accessed 28 March 2020).
- OCEAN project, https://www.spire2030.eu/ocean, (accessed 28 March 2020).
- F. R. Keene, C. Creutz and N. Sutin, Coord. Chem. Rev., 1985, 64, 247–260 CrossRef CAS.
- H. A. Schwarz and R. W. Dodson, J. Phys. Chem., 1989, 93, 409–414 CrossRef CAS.
- L. Fan, C. Xia, F. Yang, J. Wang, H. Wang and Y. Lu, Sci. Adv., 2020, 6, 1–18 Search PubMed.
- U. O. Nwabara, E. R. Cofell, S. Verma, E. Negro and P. J. A. Kenis, ChemSusChem, 2020, 13, 855–875 CrossRef CAS.
- W. Zhang, Y. Hu, L. Ma, G. Zhu, Y. Wang, X. Xue, R. Chen, S. Yang and Z. Jin, Adv. Sci., 2018, 5, 1700275 CrossRef.
- P. Akhter, M. A. Farkhondehfal, S. Hernández, M. Hussain, A. Fina, G. Saracco, A. U. Khan and N. Russo, J. Environ. Chem. Eng., 2016, 4, 3934–3953 CrossRef CAS.
- R. B. Kutz, Q. Chen, H. Yang, S. D. Sajjad, Z. Liu and I. R. Masel, Energy Technol., 2017, 5, 929–936 CrossRef CAS.
- X. Lu, Y. Wu, X. Yuan and H. Wang, Angew. Chem., 2019, 131, 4071–4075 CrossRef.
- C. T. Dinh, T. Burdyny, G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. Pelayo García De Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS.
- X. Nie, W. Luo, M. J. Janik and A. Asthagiri, J. Catal., 2014, 312, 108–122 CrossRef CAS.
- H. A. Hansen, C. Shi, A. C. Lausche, A. A. Peterson and J. K. Nørskov, Phys. Chem. Chem. Phys., 2016, 18, 9194–9201 RSC.
- S. Back, H. Kim and Y. Jung, ACS Catal., 2015, 5, 965–971 CrossRef CAS.
- S. P. Liu, M. Zhao, W. Gao, Q. Jiang and T. Jacob, Electrocatalysis, 2017, 8, 647–656 CrossRef CAS.
- A. Vasileff, C. Xu, Y. Jiao, Y. Zheng and S. Z. Qiao, Chem, 2018, 4, 1809–1831 CAS.
- A. Irabien, M. Alvarez-Guerra, J. Albo and A. Dominguez-Ramos, Electrochemical conversion of CO2 to value-added products, Elsevier Inc., 2018 Search PubMed.
- R. Lin, J. Guo, X. Li, P. Patel and A. Seifitokaldani, Catalysts, 2020, 10, 473 CrossRef CAS.
- J. D. Goodpaster, A. T. Bell and M. Head-Gordon, J. Phys. Chem. Lett., 2016, 7, 1471–1477 CrossRef CAS.
- K. Zhao, Y. Liu, X. Quan, S. Chen and H. Yu, ACS Appl. Mater. Interfaces, 2017, 9, 5302–5311 CrossRef CAS.
- J. Albo, M. Alvarez-Guerra, P. Castaño and A. Irabien, Green Chem., 2015, 17, 2304–2324 RSC.
- D. Yang, Q. Zhu, C. Chen, H. Liu, Z. Liu, Z. Zhao, X. Zhang, S. Liu and B. Han, Nat. Commun., 2019, 10, 1–9 CrossRef.
- H. Yang, Y. Wu, G. Li, Q. Lin, Q. Hu, Q. Zhang, J. Liu and C. He, J. Am. Chem. Soc., 2019, 141, 12717–12723 CrossRef CAS.
- J. Albo, A. Sáez, J. Solla-Gullón, V. Montiel and A. Irabien, Appl. Catal., B, 2015, 176–177, 709–717 CrossRef CAS.
- C. Genovese, C. Ampelli, S. Perathoner and G. Centi, Green Chem., 2017, 19, 2406–2415 RSC.
- M. I. Malik, Z. O. Malaibari, M. Atieh and B. Abussaud, Chem. Eng. Sci., 2016, 152, 468–477 CrossRef.
- M. T. H. Le, Louisiana State University and Agricultural and Mechanical College, 2011 Search PubMed.
- B. C. Marepally, C. Ampelli, C. Genovese, F. Tavella, L. Veyre, E. A. Quadrelli, S. Perathoner and G. Centi, J. CO2 Util., 2017, 21, 534–542 CrossRef CAS.
- J. Yuan, M. P. Yang, Q. L. Hu, S. M. Li, H. Wang and J. X. Lu, J. CO2 Util., 2018, 24, 334–340 CrossRef CAS.
- J. Hazarika and M. S. Manna, Electrochim. Acta, 2019, 328, 135053 CrossRef CAS.
- A. Marcos-Madrazo, C. Casado-Coterillo and Á. Irabien, ChemElectroChem, 2019, 6, 5273–5282 CrossRef CAS.
- L. Lu, X. Sun, J. Ma, D. Yang, H. Wu, B. Zhang, J. Zhang and B. Han, Angew. Chem., Int. Ed., 2018, 57, 14149–14153 CrossRef CAS.
- J. Albo and A. Irabien, J. Catal., 2016, 343, 232–239 CrossRef CAS.
- M. Aresta, A. Dibenedetto and E. Quaranta, J. Catal., 2016, 343, 2–45 CrossRef CAS.
- E. Andrews, M. Ren, F. Wang, Z. Zhang, P. Sprunger, R. Kurtz and J. Flake, J. Electrochem. Soc., 2013, 160, 841–846 CrossRef.
- M. S. Spencer, Top. Catal., 1999, 8, 259–266 CrossRef CAS.
- M. Le, M. Ren, Z. Zhang, P. T. Sprunger, R. L. Kurtz and J. C. Flake, J. Electrochem. Soc., 2011, 158, E45 CrossRef CAS.
- J. Albo, M. Perfecto-Irigaray, G. Beobide and A. Irabien, J. CO2 Util., 2019, 33, 157–165 CrossRef CAS.
- X. Wang, Z. Wang, F. P. G. De Arquer, C. Dinh, A. Ozden, Y. C. Li, D. Nam, J. Li, Y. Liu, J. Wicks, Z. Chen, M. Chi, B. Chen, Y. Wang, J. Tam, J. Y. Howe, A. Proppe, P. Todorović, F. Li, T. Zhuang, C. M. Gabardo, A. R. Kirmani, C. Mccallum, S. Hung, Y. Lum, M. Luo, Y. Min, A. Xu, C. P. O. Brien, B. Stephen, B. Sun, A. H. Ip, L. J. Richter, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Energy, 2020, 5, 478–486 CrossRef CAS.
- T. Zhuang, Z. Liang, A. Seifitokaldani, Y. Li, P. De Luna, T. Burdyny, F. Che, F. Meng, Y. Min, R. Quintero-bermudez, C. T. Dinh, Y. Pang, M. Zhong, B. Zhang, J. Li, P. Chen, X. Zheng, H. Liang, W. Ge, B. Ye, D. Sinton, S. Yu and E. H. Sargent, Nat. Catal., 2018, 1, 421–428 CrossRef CAS.
- C. Chen, X. Sun, L. Lu, D. Yang, J. Ma, Q. Zhu, Q. Qian and B. Han, Green Chem., 2018, 20, 4579–4583 RSC.
- H. Xu, D. Rebollar, H. He, L. Chong, Y. Liu, C. Liu, C. J. Sun, T. Li, J. V. Muntean, R. E. Winans, D. J. Liu and T. Xu, Nat. Energy, 2020, 5, 623–632 CrossRef CAS.
- M. Luo, Z. Wang, Y. C. Li, J. Li, F. Li, Y. Lum, D. H. Nam, B. Chen, J. Wicks, A. Xu, T. Zhuang, W. R. Leow, X. Wang, C. T. Dinh, Y. Wang, Y. Wang, D. Sinton and E. H. Sargent, Nat. Commun., 2019, 10, 1–7 CrossRef.
- C. Chen, X. Yan, S. Liu, Y. Wu, Q. Wan, X. Sun, Q. Zhu, H. Liu, J. Ma, L. Zheng, H. Wu and B. Han, Angew. Chem., 2020, 100049, 16601–16606 Search PubMed.
- J. J. Lv, M. Jouny, W. Luc, W. Zhu, J. J. Zhu and F. Jiao, Adv. Mater., 2018, 30, 1–8 Search PubMed.
- Y. C. Li, Z. Wang, T. Yuan, D. H. Nam, M. Luo, J. Wicks, B. Chen, J. Li, F. Li, F. P. G. De Arquer, Y. Wang, C. T. Dinh, O. Voznyy, D. Sinton and E. H. Sargent, J. Am. Chem. Soc., 2019, 141, 8584–8591 CrossRef CAS.
- T. T. H. Hoang, S. Verma, S. Ma, T. T. Fister, J. Timoshenko, A. I. Frenkel, P. J. A. Kenis and A. A. Gewirth, J. Am. Chem. Soc., 2018, 140, 5791–5797 CrossRef CAS.
- F. Jia, X. Yu and L. Zhang, J. Power Sources, 2014, 252, 85–89 CrossRef CAS.
- M. Schwartz, R. L. Cook, V. M. Kehae, R. C. MacDuff, J. Patel and A. F. Sammells, J. Electrochem. Soc., 1993, 140, 614 CrossRef CAS.
- J. A. Rudd, E. Kazimierska, L. B. Hamdy, O. J. E. Bain, S. Ahn, A. R. Barron and E. Andreoli, ChemRxiv, DOI: DOI:10.26434/chemrxiv.12022623.v1.
- S. Ma, M. Sadakiyo, R. Luo, M. Heima, M. Yamauchi and P. J. A. Kenis, J. Power Sources, 2016, 301, 219–228 CrossRef CAS.
- H. Homayoni, W. Chanmanee, N. R. de Tacconi, B. H. Dennis and K. Rajeshwar, J. Electrochem. Soc., 2015, 162, E115–E122 CrossRef CAS.
- C. Chen, Y. Li, S. Yu, S. Louisia, J. Jin, M. Li, M. B. Ross and P. Yang, Joule, 2020, 4, 1688–1699 CrossRef CAS.
- H. Xiao, W. A. Goddard, T. Cheng and Y. Liu, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6685–6688 CrossRef CAS.
- Y. Song, W. Chen, C. Zhao, S. Li, W. Wei and Y. Sun, Angew. Chem., Int. Ed., 2017, 56, 10840–10844 CrossRef CAS.
- D. M. Weekes, D. A. Salvatore, A. Reyes, A. Huang and C. P. Berlinguette, Acc. Chem. Res., 2018, 51, 910–918 CrossRef CAS.
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, J. Mol. Catal. A: Chem., 2003, 199, 39–47 CrossRef CAS.
- D. Kim, C. S. Kley, Y. Li and P. Yang, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 10560–10565 CrossRef CAS.
- D. Ren, N. T. Wong, A. D. Handoko, Y. Huang and B. S. Yeo, J. Phys. Chem. Lett., 2016, 7, 20–24 CrossRef CAS.
- R. A. Geioushy, M. M. Khaled, K. Alhooshani, A. S. Hakeem and A. Rinaldi, Electrochim. Acta, 2017, 245, 456–462 CrossRef CAS.
- Z. Han, R. Kortlever, H. Y. Chen, J. C. Peters and T. Agapie, ACS Cent. Sci., 2017, 3, 853–859 CrossRef CAS.
- P. P. Sharma and X. D. Zhou, Wiley Online Libr., 2017, 6, 1–21 CAS.
- H. Zhong, K. Fujii, Y. Nakano and F. Jin, J. Phys. Chem. C, 2015, 119, 55–61 CrossRef CAS.
- Y. Hori, A. Murata and R. Takahashi, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 2309–2326 RSC.
- R. Kas, R. Kortlever, H. Yilmaz, M. T. M. Koper and G. Mul, ChemElectroChem, 2015, 2, 354–358 CrossRef CAS.
- D. Ren, Y. Deng, A. D. Handoko, C. S. Chen, S. Malkhandi and B. S. Yeo, ACS Catal., 2015, 5, 2814–2821 CrossRef CAS.
- F. Y. Zhang, T. Sheng, N. Tian, L. Liu, C. Xiao, B. A. Lu, B. B. Xu, Z. Y. Zhou and S. G. Sun, Chem. Commun., 2017, 53, 8085–8088 RSC.
- M. Rahaman, A. Dutta, A. Zanetti and P. Broekmann, ACS Catal., 2017, 7, 7946–7956 CrossRef CAS.
- R. A. Geioushy, M. M. Khaled, A. S. Hakeem, K. Alhooshani and C. Basheer, J. Electroanal. Chem., 2017, 785, 138–143 CrossRef CAS.
- J. Yuan, J. J. Zhang, M. P. Yang, W. J. Meng, H. Wang and J. X. Lu, Catalysts, 2018, 8, 171 CrossRef CAS.
- Y. Song, R. Peng, D. K. Hensley, P. V. Bonnesen, L. Liang, Z. Wu, H. M. Meyer, M. Chi, C. Ma, B. G. Sumpter and A. J. Rondinone, ChemistrySelect, 2016, 1, 6055–6061 CrossRef CAS.
- J. Yuan, M. P. Yang, Q. L. Hu, S. M. Li, H. Wang and J. X. Lu, J. CO2 Util., 2018, 24, 334–340 CrossRef CAS.
- J. Albo, M. Perfecto-Irigaray, G. Beobide and A. Irabien, J. CO2 Util., 2019, 33, 157–165 CrossRef CAS.
- F. L. P. Veenstra, N. Ackerl, A. J. Martín and J. Pérez-Ramírez, Chem, 2020, 6, 1–16 Search PubMed.
- L. Mandal, K. R. Yang, M. R. Motapothula, D. Ren, P. Lobaccaro, A. Patra, M. Sherburne, V. S. Batista, B. S. Yeo, J. W. Ager, J. Martin and T. Venkatesan, ACS Appl. Mater. Interfaces, 2018, 10, 8574–8584 CrossRef CAS.
- F. Dattila, R. Garclá-Muelas and N. López, ACS Energy Lett., 2020, 5, 3176–3184 CrossRef CAS.
- Y. Lum, B. Yue, P. Lobaccaro, A. T. Bell and J. W. Ager, J. Phys. Chem. C, 2017, 121, 14191–14203 CrossRef CAS.
- G. Centi, S. Perathoner, G. Winè and M. Gangeri, Green Chem., 2007, 9, 671–678 RSC.
- M. Gangeri, S. Perathoner, S. Caudo, G. Centi, J. Amadou, D. Bégin, C. Pham-Huu, M. J. Ledoux, J. P. Tessonnier, D. S. Su and R. Schlögl, Catal. Today, 2009, 143, 57–63 CrossRef CAS.
- C. Genovese, C. Ampelli, S. Perathoner and G. Centi, J. Catal., 2013, 308, 237–249 CrossRef CAS.
- C. Genovese, C. Ampelli, S. Perathoner and G. Centi, J. Energy Chem., 2013, 22, 202–213 CrossRef CAS.
- N. Gutiérrez-Guerra, J. A. González, J. C. Serrano-Ruiz, E. López-Fernández, J. L. Valverde and A. de Lucas-Consuegra, J. Energy Chem., 2019, 46–53 CrossRef.
- I. Merino-Garcia, J. Albo, J. Solla-Gullón, V. Montiel and A. Irabien, J. CO2 Util., 2019, 31, 135–142 CrossRef CAS.
- C. Xia, P. Zhu, Q. Jiang, Y. Pan, W. Liang, E. Stavitsk, H. N. Alshareef and H. Wang, Nat. Energy, 2019, 4, 776–785 CrossRef CAS.
- N. Gutiérrez-Guerra, L. Moreno-López, J. C. Serrano-Ruiz, J. L. Valverde and A. de Lucas-Consuegra, Appl. Catal., B, 2016, 188, 272–282 CrossRef.
- N. Gutiérrez-Guerra, J. L. Valverde, A. Romero, J. C. Serrano-Ruiz and A. de Lucas-Consuegra, Electrochem. Commun., 2017, 81, 128–131 CrossRef.
- J. García, C. Jiménez, F. Martínez, R. Camarillo and J. Rincón, J. Catal., 2018, 367, 72–80 CrossRef.
- C. M. Gabardo, P. Colin, O. Brien, P. Jonathan, J. Li, H. Edward, D. Sinton, C. P. O. Brien, J. P. Edwards, C. Mccallum, Y. Xu, C. Dinh, J. Li, E. H. Sargent and D. Sinton, Joule, 2019, 3, 2777–2791 CrossRef CAS.
- S. K. S. Hossain, J. Saleem, S. U. Rahman, S. M. J. Zaidi, G. McKay and C. K. Cheng, Catalysts, 2019, 9, 1–19 Search PubMed.
- B. C. Marepally, C. Ampelli, C. Genovese, T. Saboo, S. Perathoner, F. M. Wisser, L. Veyre, J. Canivet, E. A. Quadrelli and G. Centi, ChemSusChem, 2017, 10, 4442–4446 CrossRef CAS.
- C. Jiménez, J. García, F. Martínez, R. Camarillo and J. Rincón, Electrochim. Acta, 2020, 337, 135663 CrossRef.
- N. Gutiérrez-Guerra, J. A. González, J. C. Serrano-Ruiz, E. López-Fernández, J. L. Valverde and A. de Lucas-Consuegra, J. Energy Chem., 2019, 31, 46–53 CrossRef.
- S. Pérez-Rodríguez, F. Barreras, E. Pastor and M. J. Lázaro, Int. J. Hydrogen Energy, 2016, 41, 19756–19765 CrossRef.
- I. Merino-Garcia, J. Albo and A. Irabien, Energy Technol., 2017, 5, 922–928 CrossRef CAS.
- I. Merino-Garcia, J. Albo and A. Irabien, Nanotechnology, 2018, 29, 14001 CrossRef CAS.
- I. Merino-Garcia, J. Albo, J. Solla-Gullón, V. Montiel and A. Irabien, J. CO2 Util., 2019, 31, 135–142 CrossRef CAS.
- M. Rattalino, M. Makkee, A. Lamberti, A. Chiodoni, K. Bejtka, A. Sacco, F. C. Pirri and N. Russo, Int. J. Hydrogen Energy, 2019, 45, 26458–26471 Search PubMed.
- D. Gao, R. M. Arán-Ais, H. S. Jeon and B. Roldan Cuenya, Nat. Catal., 2019, 2, 198–210 CrossRef CAS.
- J. J. Velasco-Vélez, T. Jones, D. Gao, E. Carbonio, R. Arrigo, C. J. Hsu, Y. C. Huang, C. L. Dong, J. M. Chen, J. F. Lee, P. Strasser, B. Roldan Cuenya, R. Schlögl, A. Knop-Gericke and C. H. Chuang, ACS Sustainable Chem. Eng., 2019, 7, 1485–1492 CrossRef.
- A. Karelovic and P. Ruiz, Catal. Sci. Technol., 2015, 5, 869–881 RSC.
- F. Arena, K. Barbera, G. Italiano, G. Bonura, L. Spadaro and F. Frusteri, J. Catal., 2007, 249, 185–194 CrossRef CAS.
- I. Hjorth, M. Nord, M. Rønning, J. Yang and D. Chen, Catal. Today, 2020, 357, 311–321 CrossRef CAS.
- O. A. Baturina, Q. Lu, M. A. Padilla, L. Xin, W. Li, A. Serov, K. Artyushkova, P. Atanassov, F. Xu, A. Epshteyn, T. Brintlinger, M. Schuette and G. E. Collins, ACS Catal., 2014, 4, 3682–3695 CrossRef CAS.
- W. J. Durand, A. A. Peterson, F. Studt, F. Abild-Pedersen and J. K. Nørskov, Surf. Sci., 2011, 605, 1354–1359 CrossRef CAS.
- Y. Li and Q. Sun, Adv. Energy Mater., 2016, 6, 1–19 Search PubMed.
- D. Ren, B. S. H. Ang and B. S. Yeo, ACS Catal., 2016, 6, 8239–8247 CrossRef CAS.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- T. T. H. Hoang, S. Ma, J. I. Gold, P. J. A. Kenis and A. A. Gewirth, ACS Catal., 2017, 7, 3313–3321 CrossRef CAS.
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. De Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero-Bermudez, T. K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974–980 CrossRef CAS.
- K. Jiang, R. B. Sandberg, A. J. Akey, X. Liu, D. C. Bell, J. K. Nørskov, K. Chan and H. Wang, Nat. Catal., 2018, 1, 111–119 CrossRef CAS.
- Z. Q. Liang, T. T. Zhuang, A. Seifitokaldani, J. Li, C. W. Huang, C. S. Tan, Y. Li, P. De Luna, C. T. Dinh, Y. Hu, Q. Xiao, P. L. Hsieh, Y. Wang, F. Li, R. Quintero-Bermudez, Y. Zhou, P. Chen, Y. Pang, S. C. Lo, L. J. Chen, H. Tan, Z. Xu, S. Zhao, D. Sinton and E. H. Sargent, Nat. Commun., 2018, 9, 1–8 CrossRef CAS.
- H. Jung, S. Y. Lee, C. W. Lee, M. K. Cho, D. H. Won, C. Kim, H. S. Oh, B. K. Min and Y. J. Hwang, J. Am. Chem. Soc., 2019, 141, 4624–4633 CrossRef CAS.
- T. Qin, Y. Qian, F. Zhang and B. L. Lin, Chin. Chem. Lett., 2019, 30, 314–318 CrossRef CAS.
- G. Centi, S. Perathoner, G. Winè and M. Gangeri, Green Chem., 2007, 9, 671–678 RSC.
- T. T. Zhuang, Y. Pang, Z. Q. Liang, Z. Wang, Y. Li, C. S. Tan, J. Li, C. T. Dinh, P. De Luna, P. L. Hsieh, T. Burdyny, H. H. Li, M. Liu, Y. Wang, F. Li, A. Proppe, A. Johnston, D. H. Nam, Z. Y. Wu, Y. R. Zheng, A. H. Ip, H. Tan, L. J. Chen, S. H. Yu, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Catal., 2018, 1, 946–951 CrossRef CAS.
- P. De Luna, R. Quintero-Bermudez, C. T. DInh, M. B. Ross, O. S. Bushuyev, P. Todorović, T. Regier, S. O. Kelley, P. Yang and E. H. Sargent, Nat. Catal., 2018, 1, 103–110 CrossRef CAS.
- R. M. Arán-Ais, R. Rizo, P. Grosse, G. Algara-Siller, K. Dembélé, M. Plodinec, T. Lunkenbein, S. W. Chee and B. R. Cuenya, Nat. Commun., 2020, 11, 1–8 Search PubMed.
- G. Horányi, Catal. Today, 1994, 19, 285–311 CrossRef.
- S. Pariente, P. Trens, F. Fajula, F. Di Renzo and N. Tanchoux, Appl. Catal., A, 2006, 307, 51–57 CrossRef CAS.
- Y. C. Tan, K. Berm, H. Song, J. Oh, Y. C. Tan, K. B. Lee, H. Song and J. Oh, Joule, 2020, 4, 1104–1120 CrossRef CAS.
- J. Zhang, W. Luo and A. Züttel, J. Catal., 2020, 385, 140–145 CrossRef CAS.
- H. R. Q. Jhong, F. R. Brushett and P. J. A. Kenis, Adv. Energy Mater., 2013, 3, 589–599 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc03334k |
| This journal is © The Royal Society of Chemistry 2021 |