
Carbon dioxide derived carbonized polymer dots for multicolor light-emitting diodes
Bin
Liu
 *ab,
Bo
Chu
b,
Ya-Ling
Wang
a,
Lan-Fang
Hu
b,
Shengliang
Hu
a and
Xing-Hong
Zhang
*b
*ab,
Bo
Chu
b,
Ya-Ling
Wang
a,
Lan-Fang
Hu
b,
Shengliang
Hu
a and
Xing-Hong
Zhang
*b
aSchool of Energy and Power Engineering, North University of China, Taiyuan 030051, P. R. China. E-mail: liubin@nuc.edu.cn
bMOE Key Laboratory of Macromolecular Synthesis and Functionalization, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou 310027, P. R. China. E-mail: xhzhang@zju.edu.cn
First published on 18th November 2020
Abstract
Carbonized polymer dots (CPDs) have received great interest from researchers in recent years. However, the solid-state photoluminescence quenching of CPDs is a big challenge owing to the resonance energy transfer or π–π* interaction. Herein, we report a new type of carbon dioxide (CO2) derived CPD exhibiting quantum yields of 46.2% in solution and 11.3% as a solid. These CPDs were prepared using ethylenediamine and trimethylolpropane tri(cyclic carbonate)ether synthesized from the reaction of carbon dioxide (CO2) and trimethylolpropane triglycidyl ether as precursors in ethanol. Gel permeation chromatography results revealed that the formation of CPDs involved the polymerization of small molecules and then the transformation of the polymers to CPDs. The solid-state fluorescence is attributed to the self-passivation of poly(hydroxyurethane) chains on the surface of the carbon core. Particularly, the synthesis of CPDs consumed nearly 26.7 wt% CO2, thus providing a new method for fixing CO2 to functional carbon materials. We have also demonstrated the application of CO2-derived CPDs by the fabrication of multicolor light-emitting diodes (LEDs), and a warm white LED is obtained when CPDs are applied as single phosphors by changing the amount of CPDs and the type of chip.
Introduction
Carbon dioxide (CO2), an essential component of greenhouse gases, has caused a remarkable increase in the atmospheric temperature and an abnormal change in the global climate. According to the CO2·earth website, the average concentration of CO2 increased from 338 ppm in 1980 to 416 ppm in 2020, and the global surface temperature increased by one degree relative to pre-industrial times.1 The number has been rising year after year, threatening the future of mankind. Viewed from another perspective, CO2 is a nontoxic, naturally abundant, cheap and low-cost one-carbon building block. A great deal of work has been done on the conversion of CO2 into small organic compounds2–4 and polymers.5–9 The fixation of CO2 into useful or value-added materials can also effectively utilize CO2 and create economic benefits at the same time, but it is still rather limited.10,11Recently, carbonized polymer dots (CPDs), as emerging and green environment protecting materials with high carbon content, have attracted considerable research interest due to their special structures, tunable photoelectric properties, high photo-stability, and low toxicity. These merits make them excellent materials for optoelectronic device construction, catalysis, ion detection and so on.12–18 Different from any other kinds of carbon dots (CDs), polymer chains are attached on the surface of CPDs, providing the possibilities of solid-state luminescence. However, most CPDs still suffer from self-quenching and low quantum yield (QY),19,20 greatly hindering their development and application. In general, the low QY of CPDs is attributed to the presence of sub-fluorophores (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, CO, NO, etc.), resulting in very weak photoluminescence (PL).20 Some research groups have taken measures to solve this issue. Yang and coworkers20,21 used a crosslinking method to enhance the PL intensity and promote the crosslink enhanced emission (CEE) effect. Dai and coworkers22 developed a general and facile “ship-in-a-bottle” strategy to prepare polymer dots with a QY of 17.3% in solution. For the self-quenching of CPDs, Liu and coworkers23 prepared a kind of CPD with abundant surface PVA chains to resist self-quenching. Yang and coworkers14 reported self-quenching-resistant solid-state fluorescent CPDs with a solid-state QY of 8.5% from maleic acid and ethylenediamine (EDA). Therefore, it is of great significance to obtain CPDs with a high QY and self-quenching-resistant properties simultaneously for their applications.
N, CO, NO, etc.), resulting in very weak photoluminescence (PL).20 Some research groups have taken measures to solve this issue. Yang and coworkers20,21 used a crosslinking method to enhance the PL intensity and promote the crosslink enhanced emission (CEE) effect. Dai and coworkers22 developed a general and facile “ship-in-a-bottle” strategy to prepare polymer dots with a QY of 17.3% in solution. For the self-quenching of CPDs, Liu and coworkers23 prepared a kind of CPD with abundant surface PVA chains to resist self-quenching. Yang and coworkers14 reported self-quenching-resistant solid-state fluorescent CPDs with a solid-state QY of 8.5% from maleic acid and ethylenediamine (EDA). Therefore, it is of great significance to obtain CPDs with a high QY and self-quenching-resistant properties simultaneously for their applications.
CPDs can often be synthesized from oxygen-rich precursors;12,24–26 therefore, we were interested in exploring the utilization of CO2 for preparing CPDs. Herein, a solvothermal treatment of EDA and trimethylolpropane tri(cyclic carbonate)ether (TPTE) was performed, following which CPDs were obtained. TPTE was synthesized by trimethylolpropane triglycidyl ether and CO2, thus achieving the transition from CO2 to nanocarbons. The CPDs showing an average diameter of 4.5 nm exhibited a strong blue solution fluorescence and a blue-green solid-state fluorescence. The QYs of the CPDs could reach up to 46.2% in ethanol and 11.3% in the solid state, respectively. These values were higher than those for most CPDs reported to date.20,21,27–30 The dynamic process was investigated in detail through tuning different reaction temperatures to illuminate the formation mechanisms of the CPDs. Furthermore, when the CPDs were applied as single phosphors integrated with a 420 nm InGaN chip, a warm white light-emitting diode (LED) with color coordinates of (0.376, 0.288), high color rendering index (CRI) of 85, and low correlated color temperature (CCT) of 3161 K was fabricated.
Results and discussion
The CPDs were prepared via the solvothermal treatment of a mixture containing TPTE and EDA at 230 °C for 12 h in ethanol, as described in the Experimental section. The five-membered cyclic carbonate groups of TPTE can react with amines to produce carbamates and hydroxyl groups under mild conditions (∼95 °C, no catalysts).31 TPTE was synthesized by the cycloaddition reaction from trimethylolpropane triglycidyl ether and CO2, realizing the fixation of CO2 with 30.4 wt% relative to TPTE.32,33 In general, five-membered cyclic carbonates can be prepared from the reaction of CO2 with diols, allyl alcohols or propargylic alcohol derivatives.34–36 However, the cycloaddition reaction of epoxides and CO2 is the most effective method to prepare multifunctional cyclic carbonates and avoid the generation of by-products,37 maximizing the utilization of CO2. Ultimately, 26.7 wt% CO2 was converted into CPDs since no decarburization process occurred; this realized the transition from CO2 to nanocarbons (Scheme 1). Therefore, the idea of CO2-immobilizing nanocarbons is put forward.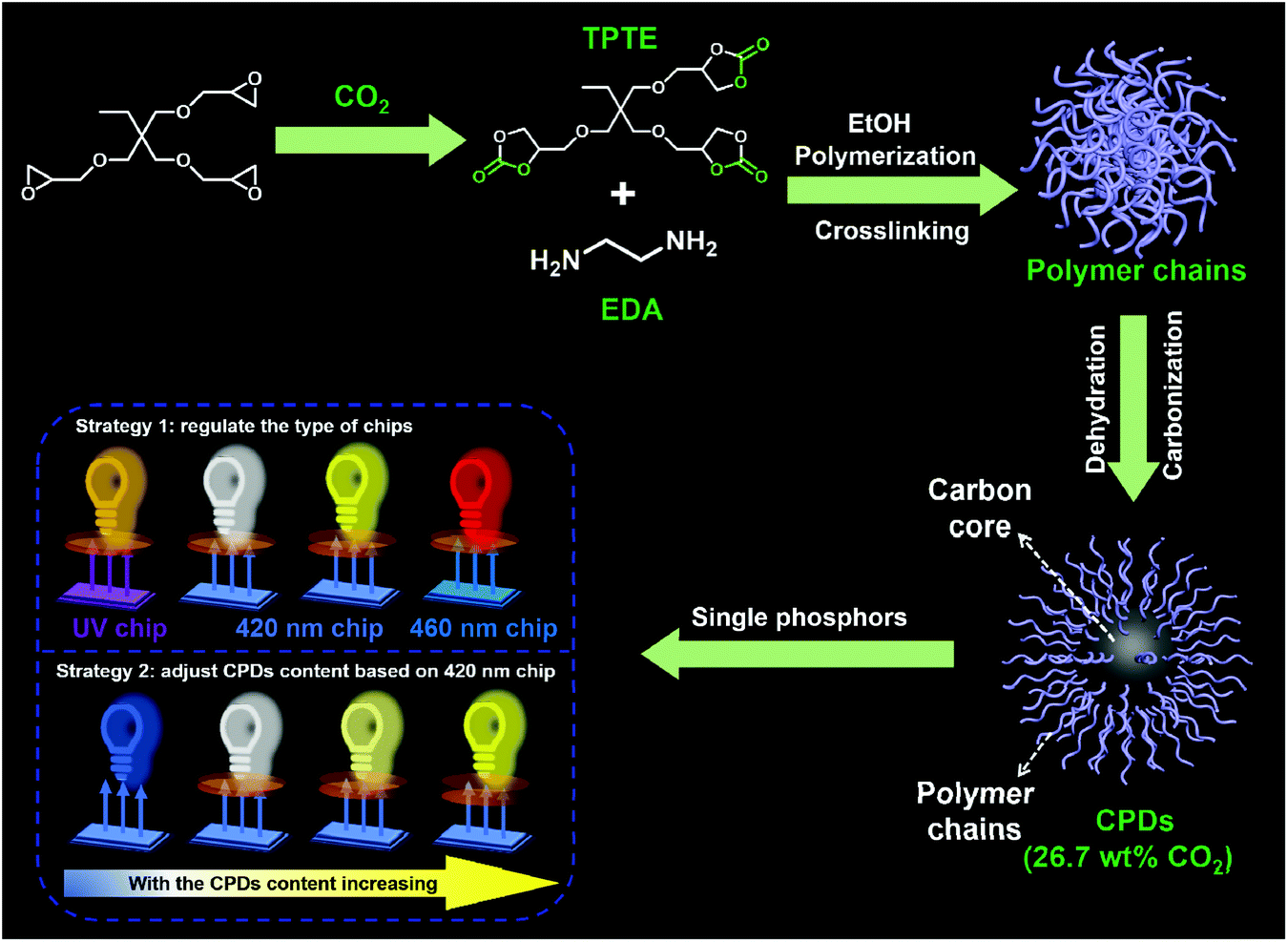 | ||
| Scheme 1 Schematic representation of the formation of CPDs and their application in multicolor LEDs. | ||
As proposed by Yang and co-workers, the formation process of most CPDs using polymers as precursors usually undergoes crosslinking, dehydration, and carbonization, and the CEE effect can improve the QY of CPDs.20,21 However, when small molecules are used as precursors, it becomes difficult to confirm the occurrence of polymerization during the preparation process of CPDs. Therefore, to demonstrate the polymerization process during the formation of CPDs and further prepare CPDs with a high QY, TPTE and EDA with equivalent functional groups were used as precursors to prepare CPDs in ethanol. The solvothermal process was performed at 140, 170, 200, and 230 °C, respectively, to study the formation mechanism of CPDs.
Transmission electron microscopy (TEM), gel permeation chromatography (GPC), and Fourier transform infrared spectroscopy (FT-IR) were performed to determine the formation process and structural change of the CPDs. As shown in Fig. 1, the diameter of the particles decreased clearly from ∼50 nm to ∼4.5 nm, and the number of particles increased significantly as the temperature increased from 140 to 230 °C. Most importantly, lattice fringes in the dots appeared when the temperature increased to 230 °C (Fig. 1e). High-resolution TEM (HRTEM) measurements revealed that the CPDs were graphitic in nature, with a lattice fringe spacing of 0.21 nm coincident with the (100) facet of graphite (inset in Fig. 1e). The results indicate that a high temperature is necessary for the formation of CPDs and its graphite structure.38 Thus, the smaller size and more particles at high temperatures were attributed to the high carbonization and graphitization degrees.
 | ||
| Fig. 1 TEM images of the products obtained at (a) 140, (b) 170, (c) and (d) 200, and (e) 230 °C in ethanol, respectively. The insets are the size distribution histogram (upper right) and the HRTEM (lower right) image of CPDs obtained at 230 °C. | ||
The data from some characterization methods, including NMR, XRD and FT-IR, cannot be used to directly explain the formation process of CPDs. Therefore, gel permeation chromatography (GPC) was selected to study the formation mechanism of CPDs. The separation process of GPC is based on size exclusion; thus GPC is also known as size exclusive chromatography (SEC), and the polymers are separated from each other based on their hydrodynamic volumes. As shown in Fig. 2a, the GPC spectra showed that the carbonized product consisted of two parts: a polymer and a spherical nanoparticle. Owing to the fact that CPDs are spherical, the hydrodynamic radius was smaller than that of the polymer and the elution time was the slowest in the GPC spectra. The polymers possessed large hydrodynamic volumes and eluted earlier at 17.1 min than the spherical nanoparticles, which eluted at between 17.5 and 17.9 min. The peak at 17.1 min decreased and disappeared with increasing the reaction temperature, indicating the decreasing content of soluble polymers. When the reaction temperature reached 230 °C, only one peak appeared, indicating that the polymer had completely disappeared and was converted into CPDs with a particle size of 4.5 nm. The enlarged GPC spectra between 17.7 and 18.2 min showed that the diameter of the nanoparticles gradually decreased as the reaction temperature increased (inset in Fig. 2a), which was consistent with the TEM results. The above results provide direct evidence for the formation process of CPDs. In the FT-IR spectra (Fig. 2b), some partially residual cyclic carbonates (1790 cm−1) still existed when the temperature was below 230 °C. Common stretching vibrations of O–H/N–H at ∼3338 cm−1, C–H at 2965, 2922, and 2878 cm−1, CO of carbamate at 1690 cm−1, and C–OH at 1108 and 1048 cm−1 were all observed. The surface groups of CPDs were also further investigated by XPS analysis (Fig. 2c–f). A full scan XPS analysis revealed the presence of carbon, nitrogen, and oxygen (Fig. 2c). The high-resolution C 1s band was deconvoluted into five peaks at 284.6, 285.4, 286.0, 286.8, and 288.0 eV, corresponding to the CC/C–C, C–N, C–O/C–OH, O–CO, and NH–CO groups, respectively (Fig. 2d).39–41 The high-resolution N 1s spectrum revealed the presence of OC–N (399.8 eV) and C–NH (401.6 eV) (Fig. 2e).42 Oxygen was detected by an O 1s signal composed of two peaks at 531.2 and 532.7 eV (Fig. 2f), indicating the presence of CO and C–O–C/C–OH.43 The surface composition of the CPDs evaluated by the XPS spectra was in agreement with the FT-IR results.
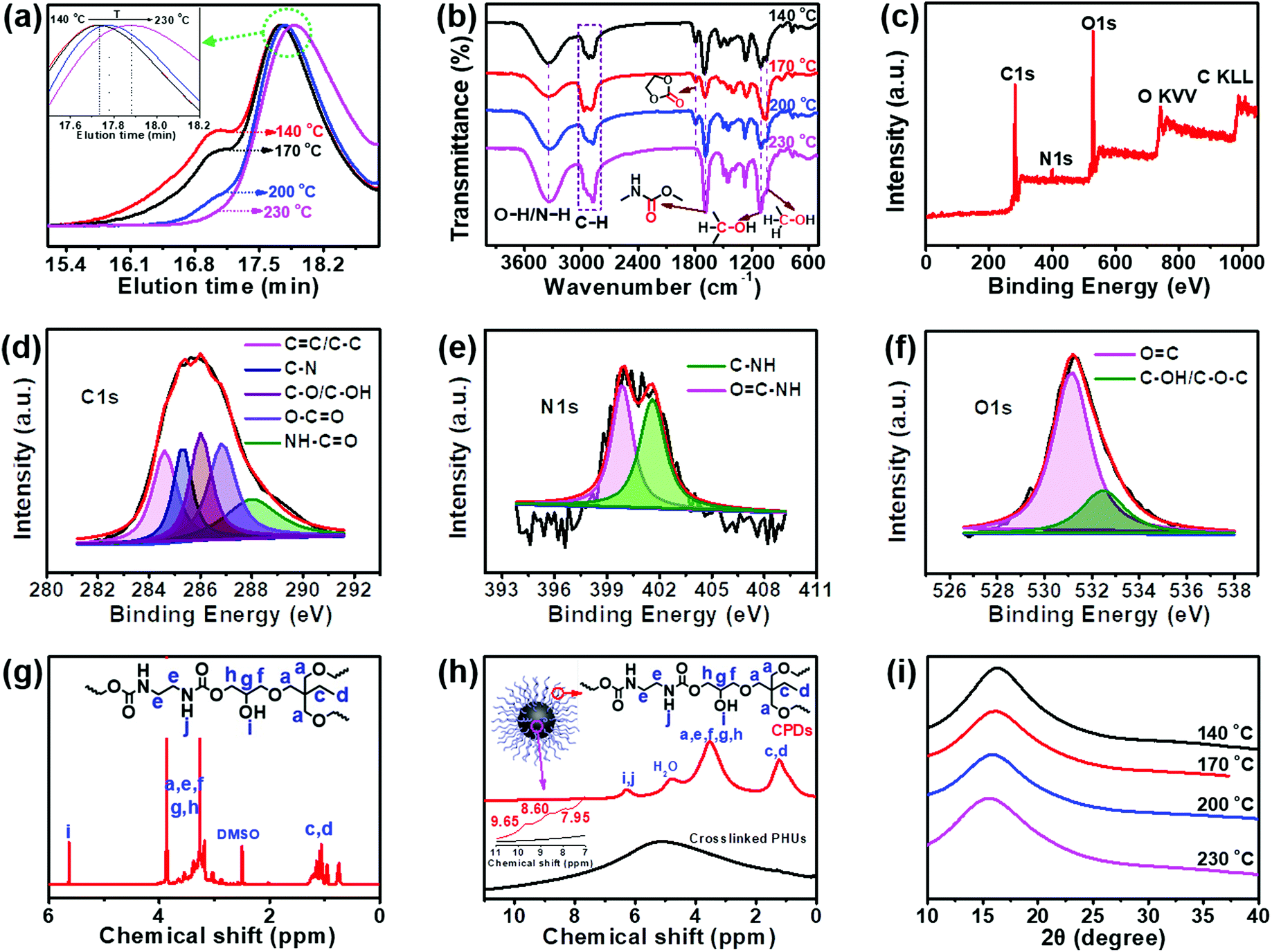 | ||
| Fig. 2 (a) GPC and (b) FT-IR spectra of products prepared at different temperatures. (c) XPS full scan, (d) high resolution C 1s, (e) high resolution N 1s, and (f) high resolution O 1s of CPDs. (g) 1H NMR (DMSO-d6 + D2O) spectra of the CPDs. (h) 1H SSNMR spectra of the CPDs and crosslinked PHUs. (i) WAXS spectra of the CPDs. | ||
The same groups in the CPDs could also be found in the 1H nuclear magnetic resonance (NMR) and 1H solid-state NMR (SSNMR) spectra, as shown in Fig. 2g and h. More importantly, not only the surface groups but also the inner core structures of the CPDs could be obtained from the 1H SSNMR spectra.44 Compared with the 1H SSNMR spectra of the crosslinked PHUs prepared from TPTE and EDA (black curve in Fig. 2h), aromatic protons were detected in the range of 7.0–10.0 ppm in the CPDs (inset in Fig. 2h). Besides, carbamate structures still existed at 230 °C (Fig. 2b, g, and h), demonstrating that hydroxyurethane chains were attached on the inner core structures of the CPDs. The wide-angle X-ray scattering (WAXS) spectra displayed a broad peak centered around 2θ = 15° (Fig. 2i), which is attributed to highly disordered carbon and polymer chains and is similar to those of the products prepared at lower temperatures. Overall, the CPDs consisted of graphitized core and non-graphitized surface poly(hydroxyurethane) chains, as shown in Scheme 1.
All the products prepared at different temperatures emitted bright blue fluorescence in aqueous solutions (inset in Fig. 3a). The absorption spectra exhibited two strong absorption bands in the ultraviolet (UV) region with a tail extending to the visible region (Fig. 3a). The former absorption peak at 267 nm was attributed to the n–π* transitions of the isolated carbonyls in carbamates or the π–π* transitions of the aromatic sp2 domains.45 The latter absorption band located between 327 and 348 nm was assigned to the n–π* transitions of carbonyl clusters (i.e., interactions between carbamate groups) according to previous studies.46–48 The intensity of the absorption peak at 267 nm decreased obviously with an increase in the temperature, confirming that the peak belonged to the n–π* transitions of the isolated carbonyls. However, the latter absorption band showed an obvious red shift from 327 to 348 nm. Meanwhile, a red-shift was also observed in the photoluminescence excitation (PLE) spectra, and the maximum peaks were very close to the UV-vis absorption peaks; this indicated that the fluorescence emission mainly originated from the n–π* transitions of the carbonyl clusters. Molecular-state and core-state fluorescence are two widely proposed luminescence mechanisms.49 Here, the emission of the carbonyl clusters of the CPDs belonged to the category of the molecular-state luminescence mechanism.
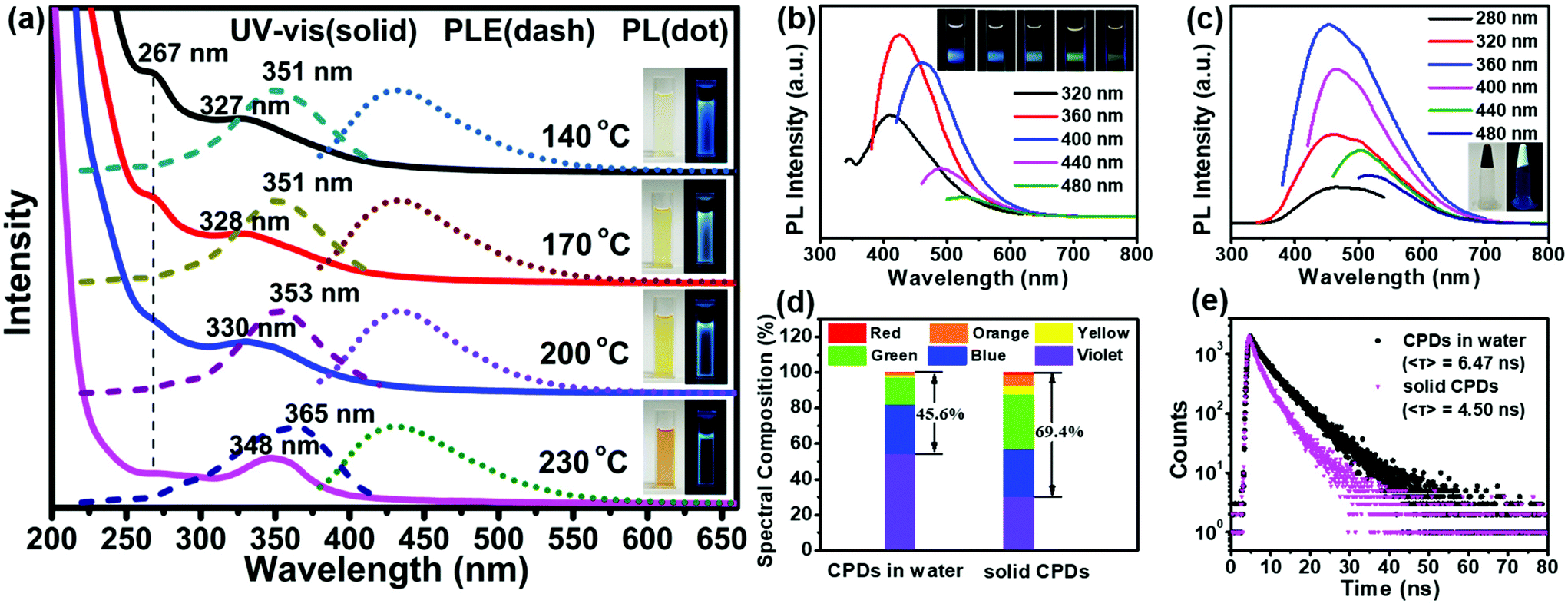 | ||
| Fig. 3 (a) UV-vis absorption, PL and PLE spectra of products prepared at different temperatures. (b) and (c) PL spectra of the CPDs in solution (b) and solid-state form (c) under different excitation wavelengths. (d) and (e) Blue-to-red spectral composition (d) and time-resolved fluorescence decay curves (e) (λex = 405 nm) of the CPDs in solution and solid-state form. Insets: (a) and (c) Photographs of product in solution (a) and solid CPDs (c) in daylight (left) and under UV light (right, λex = 365 nm), respectively; (b) photographs of CPDs in solution under different excitation wavelengths. | ||
Under 360 nm irradiation, the CPD solution exhibited a strong blue luminescence centered at 425 nm and excitation-dependent PL characteristics (Fig. 3b). In addition, the maximum PL peaks did not change with the reaction temperature (Fig. 3a). The QY of the CPDs in solution could reach up to 46.2%; this is higher than those of the most CPDs reported to date.20,21,27–29 The solid-state CPDs were black colloids, indicating that the carbonization of the CPDs occurred obviously and that residual polymer chains were present. Moreover, the CPDs exhibited an obvious solid-state luminescence with excitation-dependent PL behaviours (Fig. 3c). Most CDs/CPDs showed no fluorescence in the solid state or in a poor solvent owing to the severe aggregation-caused quenching phenomenon. Here, solid-state CPDs with a QY of 11.3% were obtained via a one-step solvothermal method without further passivation and dispersion. This is because the PHUs did not undergo absolute dehydration and carbonization, resulting in a large amount of PHUs remaining on the carbon core surface (Scheme 1). Owing to the self-absorption of the solid CPDs, the emission peak was red-shifted, and the full width at half maximum (FWHM) broadened. Consequently, the blue-to-red spectral composition of the CPDs in the solid state could reach up to 69.4%, which was obviously higher than that (45.6%) in solution (Fig. 3d). A higher value of the blue-to-red spectral composition facilitates the conversion of UV light into visible light, providing possibilities for the application of CPDs in phosphor-converted LEDs. In addition, compared with the average lifetime in solution (6.47 ns), the lower lifetime in solid (4.50 ns) is related to the resonance energy transfer (Fig. 3e).23,50
Of special note, the reaction solvent plays a key role in determining the morphology, structure, carbonization degree and PL property of the final CPDs. Our concurrent study showed that in water, a hyperbranched nanocarbon material was formed using the same precursors45 because the zero ζ-potential induced the self-assembly of CPDs as per the Derjaguin–Landau–Verwey–Overbeek (DLVO) theory. In this work, the utilization of ethanol led to particles with a high ζ-potential (13.3 mV, reaction temperature: 230 °C), inhibiting the self-assembly of CPDs.
Considering the direct solid-state luminescence, broad emission peak, and high blue-to-red spectral composition, the CPDs were applied as single phosphors to fabricate multicolor phosphor-converted LEDs. Usually, multicolor LEDs involve various kinds of phosphors with different emission properties. In particular, the preparation of white LEDs often involves the mixing of a variety of phosphors, such as red, green and yellow phosphors or blue and yellow phosphors,51,52 which may lead to reabsorption and color imbalance, requiring trial and error experiments.53 Therefore, single-phosphor-based LEDs offer obvious advantages in this respect. Firstly, three types of chip (365, 420, and 460 nm) were selected to combine with CPDs. As shown in Fig. 4a, when excited by the UV chips, the excess CPDs absorbed all of the UV light and emitted orange light centered at 620 nm with Commission Internationale de L'Eclairage (CIE) coordinates of (0.582, 0.413) (line A and point A in Fig. 4a and b). Similarly, when 420 and 460 nm blue chips were chosen to excite the CPDs, yellow and red LEDs could be obtained, respectively (line B, C and point B, C in Fig. 4a and b). The corresponding CIE coordinates were located at (0.549, 0.443) and (0.545, 0.309), and digital photographs are inset in Fig. 4a. Besides, the fabrication of multicolor LEDs can also be realized by adjusting the content of solid CPDs (25, 35, 45, and 55 mg). When the CPD content was 25 mg, a warm white LED was achieved with CIE coordinates of (0.376, 0.288), low CCT of 3161 K, and high CRI of 85 (line A′ and point A′ in Fig. 4c and d). The CRI value was higher than those of most single-phosphor-based white LEDs and sufficient to meet the commercial requirements.54–56 The PL intensity of the CPDs markedly was enhanced with increase in the CPD content. Meanwhile, the CRIs (from 85 to 65) and CCTs (from 3161 to 1999 K) of the LEDs decreased obviously with increase in the yellow light proportion. Additionally the CIE coordinates moved toward the yellow region. When the CPD content was sufficient blue light could be absorbed completely and converted into yellow light (line C′, D′ and point C′, D′ in Fig. 4c and d). As a consequence, the change from white to yellow LEDs was realized by regulating the CPD content.
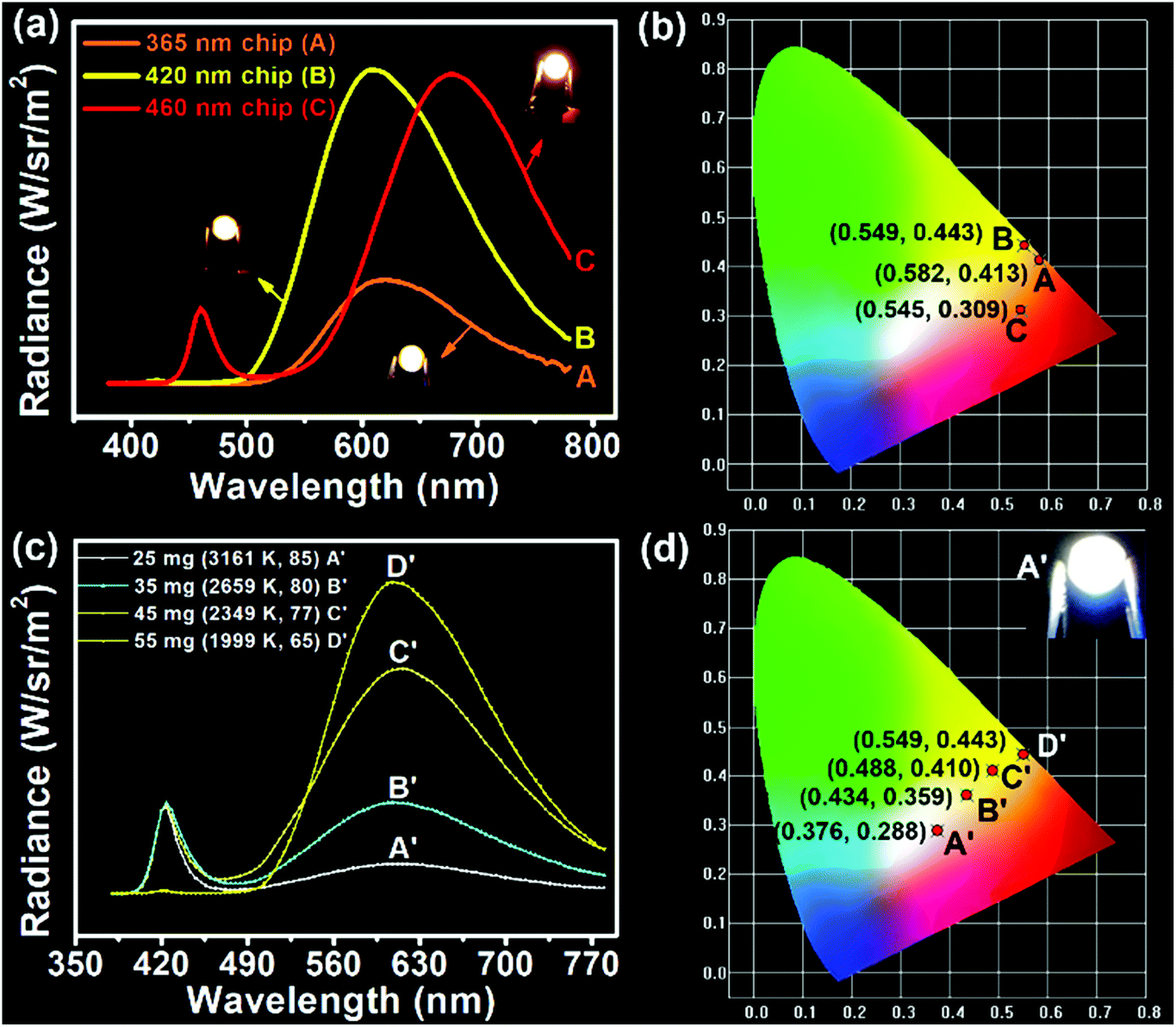 | ||
| Fig. 4 (a) EL spectra and (b) CIE coordinates of LEDs based on different kinds of chips combined with CPDs under 3.0 V. (c) EL spectra and (d) CIE coordinates of the LEDs based on different amounts of CPDs combined with a 420 nm blue chip under 3.0 V. The insets are corresponding digital photographs. | ||
Conclusions
In conclusion, this work provided a facile method to convert CO2 into solid-state luminescent CPDs, which are popular energy conversion materials. The prepared CPDs showed quantum yields of 46.2% in solution and 11.3% in the solid state via the solvothermal treatment of TPTE and EDA; 26.7 wt% CO2 was incorporated. The formation mechanisms of CPDs (i.e., polymerization, crosslinking, and further carbonization) and their structures were supported by GPC data, FT-IR spectra, XPS scan and high-resolution TEM observations. Moreover, the CPDs could be appropriately applied as LED convertors to produce multicolor LEDs by varying the chip type and CPD content, providing a new pathway towards low-cost and high-performance multicolor LEDs.Experimental
Materials and methods
Trimethylolpropane tri(cyclic carbonate)ether (TPTE) was prepared according to our previous literature procedures.45 A zinc–cobalt(III) double metal cyanide complex (Zn–Co(III) DMCC) catalyst was prepared according to a previous report.32 Ethylenediamine (EDA) and ethanol were used as analytical reagents. Carbon dioxide (99.99%) and all the other reagents were commercially available and used as received without further purification.Characterization studies
High-resolution transmission electron microscope (HRTEM) measurements were recorded on a JEM-2100 system equipped with a CCD camera operating at an accelerating voltage of 200 kV. The ζ-potentials were recorded at 25 °C using a Delsa Nano C particle analyzer (Beckman Coulter Ireland Inc.). Gel permeation chromatography (GPC) was carried out using a PL-GPC220 chromatograph (Polymer Laboratories) equipped with an HP 1100 pump at 40 °C with THF as the eluent at a flow rate of 1.0 mL min−1. The sample concentration was 0.3 wt% and the injection volume 50 μL. Fourier transform infrared (FT-IR) spectra were recorded on a Bruker TENSOR 27 spectrometer. Photoluminescence (PL) and photoluminescence excitation (PLE) spectra were recorded on a QM40-LS spectrophotometer using a xenon lamp as an excitation source. The absolute fluorescence quantum yield (QY) was measured on a QM40-LS spectrophotometer with a calibrated integrating sphere. The fluorescence decay curves were measured on a FLS920 transient fluorescence and phosphorescence spectrograph. UV-vis absorption spectra were recorded on a Cary 100 Scan UV-vis spectrophotometer (America). 1H NMR spectra were recorded on a Bruker 400 MHz instrument. 1H Solid-state NMR spectra were recorded at a magnetic field strength of 9.4 T on a Bruker Avance III-HD 400 MHz NMR spectrometer at resonance frequencies of 400.13 MHz. Wide angle X-ray scattering (WAXS) patterns were recorded on a BL16B1 beamline in the Shanghai Synchrotron Radiation Facility. X-ray photoelectron spectroscopy (XPS) was performed on a ESCALAB VG ESCALAB MARK II spectrometer with a mono X-Ray source Mg Kα excitation (1253.6 eV), and the experimental data were deconvoluted by using the Gaussian–Lorentzian mixture peak-fitting software. Spectra Scan PR655 was used to analyze the spectra, CIE coordinates, correlated color temperature, and color rendering index of the LEDs.Synthesis of TPTE
Trimethylolpropane triglycidyl ether (15.0 g, 49.6 mmol), Zn–Co(III) DMCC (29.6 mg) and cetyltrimethylammonium bromide (622.5 mg, 1.7 mmol) were poured into a 100 mL autoclave. Then the autoclave was pressurized to 40 bar pressures with CO2 and heated at 120 °C for 24 h. Afterward, the autoclave was cooled to room temperature, and the pressure was slowly vented. Afterwards, Zn–Co(III) DMCC and cetyltrimethylammonium bromide were removed by using a basic aluminum oxide column.Synthesis of carbon-dioxide-derived CPDs
Carbon-dioxide-derived CPDs were prepared by a solvothermal method with TPTE and EDA as the precursors and ethanol as the solvent. The details are as follows: TPTE (200.0 mg, 0.46 mmol) and EDA (41.5 mg, 0.69 mmol) were dissolved in ethanol and poured into a poly(tetrafluoroethylene)-lined stainless steel autoclave and then heated at 230 °C for 12 h. Afterwards, the resulting solution was filtered with a 0.22 μm syringe filter, and the ethanol was removed by vacuum distillation.Synthesis of crosslinked poly(hydroxyurethane)s (PHUs)
A 0.20 g amount of TPTE (0.46 mmol) and 0.41 g of EDA (0.69 mmol) were added to a 10 mL round bottom flask, heated to 90 °C, and kept for 18 h under an N2 atmosphere.Fabrication of the multicolor LEDs
Multicolor LEDs were fabricated by changing the type of the LED chip and the amount of the CPDs. Three kinds of InGaN based LED chip (365, 420 and 460 nm) were selected. Concentrated CPDs (55 mg CPDs) were added into a cup-shaped optical lens and placed into an air-circulating oven at 60 °C for 2 h to remove the ethanol. Finally, the optical lens was placed on the top of InGaN based LED chips to accomplish the fabrication of LEDs. Similarly, CPDs (25, 35, 45, and 55 mg) were added into a cup-shaped optical lens combined with a 420 nm InGaN blue LED chip, respectively.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support of the National Natural Science Foundation of China (52003254 and 51973190), the Natural Science Foundation of Shanxi Province (201901D211262 and 201901D211282), and the State Key Laboratory of Motor Vehicle Biofuel Technology.Notes and references
- N. P. Gillett, V. K. Arora, G. M. Flato, J. F. Scinocca and K. von Salzen, Geophys. Res. Lett., 2012, 39, L01704 CrossRef.
- N. Yadav, F. Seidi, D. Crespy and V. D'Elia, ChemSusChem, 2019, 12, 724 CrossRef CAS.
- J. Kothandaraman, A. Goeppert, M. Czaun, G. A. Olah and G. K. S. Prakash, J. Am. Chem. Soc., 2016, 138, 778 CrossRef CAS.
- X. M. Hu and K. Daasbjerg, Nature, 2019, 575, 598 CrossRef CAS.
- D. J. Darensbourg and S. J. Wilson, Green Chem., 2012, 14, 2665 RSC.
- H. Cao, Y. S. Qin, C. W. Zhuo, X. H. Wang and F. S. Wang, ACS Catal., 2019, 9, 8669 CrossRef CAS.
- X.-B. Lu and D. J. Darensbourg, Chem. Soc. Rev., 2012, 41, 1462 RSC.
- Y. Li, Y.-Y. Zhang, B. Liu and X.-H. Zhang, Chin. J. Polym. Sci., 2018, 36, 139 CrossRef CAS.
- B. Song, A. Qin and B. Z. Tang, Acta Chim. Sin., 2020, 78, 9 CrossRef.
- R. S. Haszeldine, Science, 2009, 325, 1647 CrossRef CAS.
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434 CrossRef CAS.
- J. Liu, D. Li, K. Zhang, M. Yang, H. Sun and B. Yang, Small, 2018, 14, 1703919 CrossRef.
- S. Zhu, Y. Song, X. Zhao, J. Shao, J. Zhang and B. Yang, Nano Res., 2015, 8, 355 CrossRef CAS.
- J. Shao, S. Zhu, H. Liu, Y. Song, S. Tao and B. Yang, Adv. Sci., 2017, 4, 1700395 CrossRef.
- X. Huang, L. Yang, S. Hao, B. Zheng, L. Yan, F. Qu, A. M. Asiri and X. Sun, Inorg. Chem. Front., 2017, 4, 537 RSC.
- S. Liu, J. Tian, L. Wang, Y. Zhang, X. Qin, Y. Luo, A. M. Asiri, A. O. Al-Youbi and X. Sun, Adv. Mater., 2012, 24, 2037 CrossRef CAS.
- H. Song, X. Liu, B. Wanga, Z. Tang and S. Lu, Sci. Bull., 2019, 64, 1788 CrossRef CAS.
- B. Wang, J. Li, Z. Tang, B. Yang and S. Lu, Sci. Bull., 2019, 64, 1285 CrossRef CAS.
- Y. Deng, X. Chen, F. Wang, X. Zhang, D. Zhao and D. Shen, Nanoscale, 2014, 6, 10388 RSC.
- S. Zhu, Y. Song, J. Shao, X. Zhao and B. Yang, Angew. Chem., Int. Ed., 2015, 54, 14626 CrossRef CAS.
- S. Zhu, L. Wang, N. Zhou, X. Zhao, Y. Song, S. Maharjan, J. Zhang, L. Lu, H. Wang and B. Yang, Chem. Commun., 2014, 50, 13845 RSC.
- Z.-A. Qiao, Q. Huo, M. Chi, G. M. Veith, A. J. Binder and S. Dai, Adv. Mater., 2012, 24, 6017 CrossRef CAS.
- Y. Chen, M. Zheng, Y. Xiao, H. Dong, H. Zhang, J. Zhuang, H. Hu, B. Lei and Y. Liu, Adv. Mater., 2016, 28, 312 CrossRef CAS.
- X. Zhao, J. Li, D. Liu, M. Yang, W. Wang, S. Zhu and B. Yang, iScience, 2020, 23, 100982 CrossRef CAS.
- Z. Wang, Y. Liu, S. Zhen, X. Li, W. Zhang, X. Sun, B. Xu, X. Wang, Z. Gao and X. Meng, Adv. Sci., 2020, 7, 1902688 CrossRef CAS.
- J.-X. Zheng, X.-H. Liu, Y.-Z. Yang, X.-G. Liu and B.-S. Xu, New Carbon Mater., 2018, 33, 276 CrossRef.
- Y. Yang, J. Cui, M. Zheng, C. Hu, S. Tan, Y. Xiao, Q. Yang and Y. Liu, Chem. Commun., 2012, 48, 380 RSC.
- Y. Sun, W. Cao, S. Li, S. Jin, K. Hu, L. Hu, Y. Huang, X. Gao, Y. Wu and X.-J. Liang, Sci. Rep., 2013, 3, 3036 CrossRef.
- B. Sun, B. Zhao, D. Wang, Y. Wang, Q. Tang, S. Zhu, B. Yang and H. Sun, Nanoscale, 2016, 8, 9837 RSC.
- W. Li, Y. Liu, B. Wang, H. Song, Z. Liu, S. Lu and B. Yang, Chin. Chem. Lett., 2019, 30, 2323 CrossRef CAS.
- A. Steblyanko, W. Choi, F. Sanda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2375 CrossRef CAS.
- R.-J. Wei, X.-H. Zhang, B.-Y. Du, Z.-Q. Fan and G.-R. Qi, J. Mol. Catal. A: Chem., 2013, 379, 38 CrossRef CAS.
- B. Liu, Y.-Y. Zhang, X.-H. Zhang, B.-Y. Du and Z.-Q. Fan, Polym. Chem., 2016, 7, 3731 RSC.
- M. Tamura, M. Honda, Y. Nakagawa and K. Tomishige, J. Chem. Technol. Biotechnol., 2014, 89, 19 CrossRef CAS.
- S. Minakata, I. Sasaki and T. Ide, Angew. Chem., Int. Ed., 2010, 49, 1309 CrossRef CAS.
- W. Yamada, Y. Sugawara, H. M. Cheng, T. Ikeno and T. Yamada, Eur. J. Org. Chem., 2007, 2604 CrossRef CAS.
- X. Jiang, F. Gou, F. Chen and H. Jing, Green Chem., 2016, 18, 3567 RSC.
- J. Wang, C.-F. Wang and S. Chen, Angew. Chem., Int. Ed., 2012, 51, 9297 CrossRef CAS.
- C. Shen, J. Wang, Y. Cao and Y. Lu, J. Mater. Chem. C, 2015, 3, 6668 RSC.
- S. Zhu, Q. Meng, L. Wang, J. Zhang, Y. Song, H. Jin, K. Zhang, H. Sun, H. Wang and B. Yang, Angew. Chem., Int. Ed., 2013, 52, 3953 CrossRef CAS.
- L. Bao, C. Liu, Z.-L. Zhang and D.-W. Pang, Adv. Mater., 2015, 27, 1663 CrossRef CAS.
- S. Lu, L. Sui, J. Liu, S. Zhu, A. Chen, M. Jin and B. Yang, Adv. Mater., 2017, 29, 1603443 CrossRef.
- T. Zhang, J. Zhu, Y. Zhai, H. Wang, X. Bai, B. Dong, H. Wang and H. Song, Nanoscale, 2017, 9, 13042 RSC.
- S. Liu and S. P. Armes, Angew. Chem., Int. Ed., 2002, 41, 1413 CrossRef CAS.
- Y.-L. Wang, B. Liu, J.-L. Yang, X.-H. Cao, Y.-Z. Yang, Q. Yang, A. Greiner, J.-T. Xu and X.-H. Zhang, Adv. Opt. Mater., 2019, 7, 1900659 CrossRef.
- S. Kim, S. W. Hwang, M.-K. Kim, D. Y. Shin, D. H. Shin, C. O. Kim, S. B. Yang, J. H. Park, E. Hwang, S.-H. Choi, G. Ko, S. Sim, C. Sone, H. J. Choi, S. Bae and B. H. Hong, ACS Nano, 2012, 6, 8203 CrossRef CAS.
- X. Miao, D. Qu, D. Yang, B. Nie, Y. Zhao, H. Fan and Z. Sun, Adv. Mater., 2018, 30, 1870002 CrossRef.
- B. Liu, Y.-L. Wang, W. Bai, J.-T. Xu, Z.-K. Xu, K. Yang, Y.-Z. Yang, X.-H. Zhang and B.-Y. Du, J. Mater. Chem. C, 2017, 5, 4892 RSC.
- B. E. Kwak, H. J. Yoo and D. H. Kim, Adv. Opt. Mater., 2019, 7, 1900932 CrossRef CAS.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer Science & Business Media, 2007 Search PubMed.
- E. F. Schubert and J. K. Kim, Science, 2005, 308, 1274 CrossRef CAS.
- T. Jun, K. Sim, S. Iimura, M. Sasase, H. Kamioka, J. Kim and H. Hosono, Adv. Mater., 2018, 30, 1804547 CrossRef.
- F. Arcudi, L. Đorđević and M. Prato, Angew. Chem., Int. Ed., 2017, 56, 4170 CrossRef CAS.
- A. Birkel, K. A. Denault, N. C. George, C. E. Doll, B. Hery, A. A. Mikhailovsky, C. S. Birkel, B.-C. Hong and R. Seshadri, Chem. Mater., 2012, 24, 1198 CrossRef CAS.
- B. Liu, H.-Y. Duan, Y.-L. Wang, B.-Y. Du, Q. Yang, J.-T. Xu, Y.-Z. Yang, A. Greiner and X.-H. Zhang, Mater. Horiz., 2018, 5, 932 RSC.
- W.-S. Song and H. Yang, Chem. Mater., 2012, 24, 1961 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |