
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThio-assisted reductive electrolytic cleavage of lignin β-O-4 models and authentic lignin†
Zhen
Fang
 ab,
Michael G.
Flynn
a,
James E.
Jackson
*c and
Eric L.
Hegg
*ab
ab,
Michael G.
Flynn
a,
James E.
Jackson
*c and
Eric L.
Hegg
*ab
aDepartment of Biochemistry & Molecular Biology, Michigan State University, 603 Wilson Rd, East Lansing, Michigan 48824, USA. E-mail: erichegg@msu.edu
bGreat Lakes Bioenergy Research Center, Michigan State University, 1129 Farm Lane, East Lansing, Michigan 48824, USA
cDepartment of Chemistry, Michigan State University, 578 S Shaw Ln, East Lansing, Michigan 48824, USA. E-mail: jackson@chemistry.msu.edu
First published on 8th December 2020
Abstract
Avoiding the use of expensive catalysts and harsh conditions such as elevated temperatures and high pressures is a critical goal in lignin depolymerization and valorization. In this study, we present a thio-assisted electrocatalytic reductive approach using inexpensive reticulated vitreous carbon (RVC) as the working cathode to cleave the β-O-4-type linkages in keto aryl ethers. In the presence of a pre-electrolyzed disulfide (2,2′-dithiodiethanol) and a radical inhibitor (BHT) at room temperature at a current density of 2.5 mA cm−2, cathodic reduction of nonphenolic β-O-4 dimers afforded over 90% of the corresponding monomeric C–O cleavage products in only 1.5 h. Extended to DDQ-oxidized poplar lignin, this combination of electric current and disulfide, applied over 6 h, released 36 wt% of ethyl acetate soluble fragments and 26 wt% of aqueous soluble fragments, leaving only 38 wt% of insoluble residue. These findings represent a significant improvement over the current alone values (24 wt% ethyl acetate soluble; 22 wt% aqueous soluble; 54 wt% insoluble residue) and represent an important next step in our efforts to develop a mild electrochemical method for reductive lignin deconstruction.
Introduction
Lignin is the most abundant renewable aromatic biopolymer on earth, and it has been recognized as a promising feedstock for fuels and chemicals.1–5 The complex three-dimensional structure and diverse functionalities of lignin, however, complicate the deconstruction of this macromolecule. The alkyl and aryl units of lignin's heterogeneous polymer skeleton are connected via ether or carbon–carbon bonds,1 with the β-O-4 ether linkage being the most abundant, accounting for approximately 50% and 60% of the linkages in softwoods and hardwoods, respectively.1 Efficient cleavage of the β-O-4 linkage is therefore critical for lignin depolymerization, and indeed, aerobic oxidation,6–11 reduction,12–14 and hydrolysis15–17 of this bond have been extensively studied as depolymerization strategies.1,2,4,5An ideal lignin depolymerization process would (i) avoid costly catalysts,18,19 severe conditions such as high pressures/temperatures,12,14,20 and the use of hazardous gaseous reagents such as hydrogen; (ii) produce a minimum amount of chemical waste; and (iii) retain the maximum amount of the feedstock carbon and energy content.21 Compared to conventional reductants and oxidants, electric current is an inherently clean and inexpensive reagent, capable of cleaving lignin model compounds22–24 and depolymerizing real lignin.25 Because only electrons and protons are removed or added during electrolysis, generation of additional pollutants or reagent wastes is largely avoided.26 Moreover, the counter-electrode cell offers the opportunity to perform additional desired organic transformations, or simply to split water. For instance, in a reductive process, the resulting pure O2 byproduct from water-splitting is a “free” non-polluting oxidant which may be discharged or used in other non-electrolytic reactions.27,28 Most existing reports on electrolytic lignin decomposition describe anodic oxidation, beginning with the pioneering studies by Utley et al. where mechanisms of electro-oxidative cleavage of lignin models were explored using nickel anodes in various solvent systems.29 Most related oxidative strategies30,31 have generally required expensive electrode materials such as Au32 or IrO2,33 or the assistance of photo-catalysts.34,35 Some reductive approaches have also used electrodes made of catalytic metals such as RANEY® nickel or Pd.22,23,36 Electrocatalytic hydrogenation/hydrogenolysis (ECH) with such metal electrodes typically leads to saturation of potentially desirable aromatic products, though we have recently identified promising strategies to modulate selectivities.37 These further reactions add to the complexity of the cleavage product streams, complicating isolation, purification, and further direct utilization of targeted products. Thus, the development of a simple, low-cost approach to selectively and efficiently cleave the ether bonds in β-O-4 linkages remains a critical need for effective lignin depolymerization and upgrading to useful products.
We previously reported the successful reductive cleavage of lignin-relevant α-keto aryl ether dimers by various small organic thiols.38,39 This bio-inspired approach formed phenol and acetophenone quantitatively from 2-phenoxyacetophenone after 24 h treatment with 100 eq. β-mercaptoethanol (BME) in refluxing acetonitrile. The resulting disulfide byproduct could theoretically be reduced back to the active thiol by various reductants. Of greatest interest here would be electrochemical reduction, regenerating the thiolate with no reagent byproducts (Scheme 1).40 This approach could potentially lead to a sustainable cycle connecting electrochemical reduction to lignin cleavage via thiols as small diffusible redox carriers (Fig. 1). We now report a thio-assisted electro-reductive approach using inexpensive reticulated vitreous carbon (RVC) as the working cathode to cleave α-oxidized β-O-4-type linkages in model compounds and in lignin samples. This study, intended to extend our biomimetic thiol-mediated reductive cleavage method to electrochemical reduction, reveals additional cleavage processes that are not completely interpretable within the above mechanistic framework, but promise significant value nonetheless.
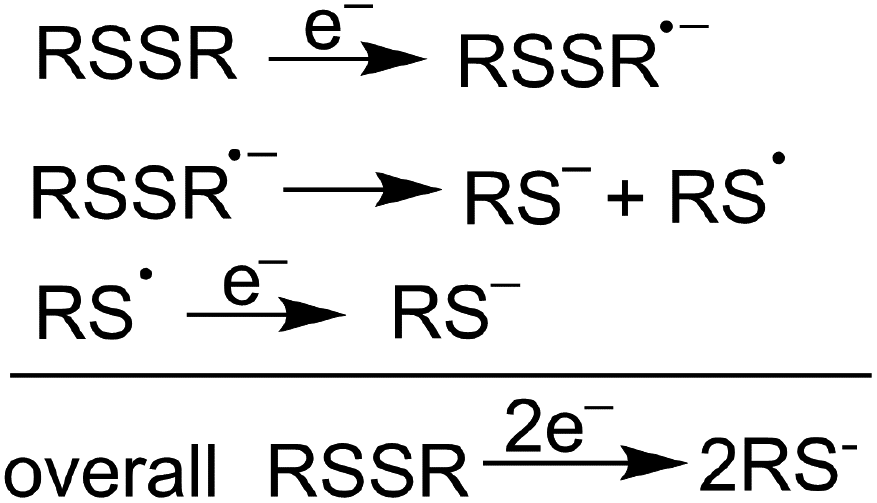 | ||
| Scheme 1 Schematic illustration of disulfide reduction via successive one-electron transfer steps. | ||
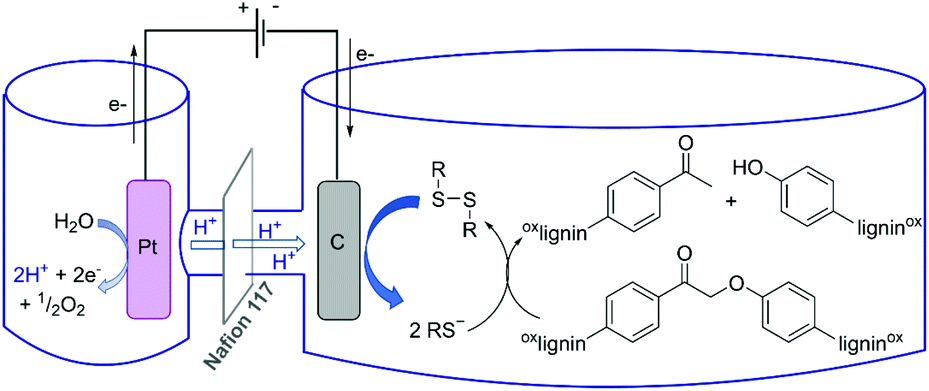 | ||
| Fig. 1 Potential schematic mechanism of lignin deconstruction in a thio-assisted electrolytic system. | ||
Results and discussion
Thio-assisted electrolysis of 2-phenoxyacetophenone
Treatment of 2-phenoxyacetophenone (4.7 mM in 20 mL) with current alone was first performed as a control at room temperature in an H-type electrochemical cell. DMF, the polar aprotic solvent that most effectively promoted keto aryl ether bond cleavage in the previous SN2 mechanism,38 was utilized as the solvent on the cathode side. For most experiments, a constant current of 5 mA was applied, passing through a 2 cm2 reticulated vitreous carbon (RVC) cathode (Table 1), providing an area-based current density of 2.5 mA cm−2; herein these conditions are simply described as “5 mA”. Interestingly, current alone cleaved 2-phenoxyacetophenone (compound I in Table 1) to phenol and acetophenone in 2.5 h (entry 1), although the substrate barely decayed in the first 45 min (Fig. 2a). Pinacol (compound IV in Table 1), known to form from the reductive dimerization of acetophenone,41 was also produced as the reaction progressed. As expected, successful cleavage required the α-keto (oxidized) form of this β-O-4 linkage model; no reaction occurred with the parent alcohol analog under 5 mA electric current alone (Fig. S1†).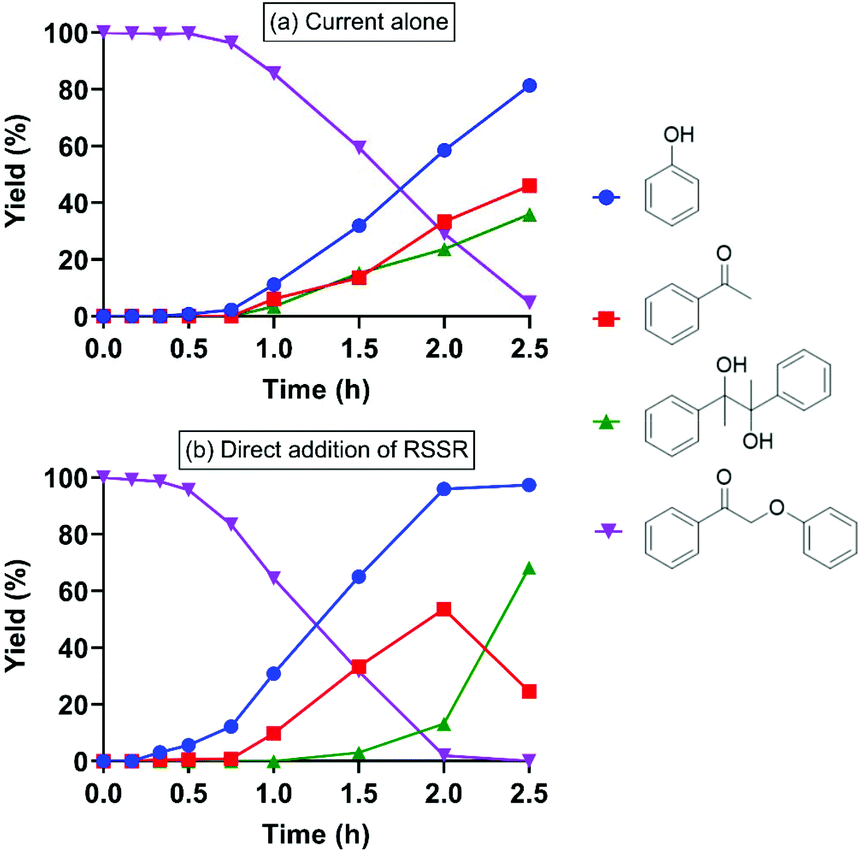 | ||
| Fig. 2 Electrolysis of 2-phenoxyacetophenone (compound I) under 5 mA at room temperature with (a) current alone; and (b) current with direct (i.e., applied simultaneously with) addition of RSSR. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pt wire in pH 8 phosphate buffer
:Pt wire in pH 8 phosphate buffer
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Currenta (5 mA) | RSSR | Pre-electrolysis of RSSR | BHT | N2 atmosphere | Conv. (%) | Yields (%) | Current efficiencyc (%) | ||
| II | III | IV | ||||||||
| a Passage of two equivalents of electrons required approximately 1 h under these conditions. b 1-Phenylethanol (6%) was formed as well. c Pre-electrolysis time was included in the calculation. | ||||||||||
| 1 | Yes | — | — | — | — | 95 | 81 | 46 | 36 | 40 |
| 2 | Yes | Yes | — | — | — | >99 | 97 | 24 | 68 | 53 |
| 3 | Removed after pre-electrolysis of RSSR | Yes | 1 h | — | — | 25 | 16 | — | — | 16 |
| 4 | Yes | Yes | 1 h | — | — | >99 | >99 | 0.3 | 73 | 43 |
| 5 | Yes | Yes | 1 h | 1 eq. | — | >99 | 96 | 15 | 88 | 45 |
| 6 | Yes | Yes | 30 min (with BHT together) | 1 eq. | — | >99 | 96 | 79 | 17 | 35 |
| 7 | Yes | Yes | — | — | Yes | >99 | 84 | 0.8 | 86 | 51 |
| 8 | Yes | Yes | — | 1 eq. | Yes | >99 | 82 | 60 | 8 | 25 |
| 9 | Yes | Yes | 1 h | — | Yes | >99b | 94 | — | 65 | 51 |
| 10 | Yes | Yes | 1 h | 1 eq. | Yes | >99 | 86 | 2 | 60 | 37 |
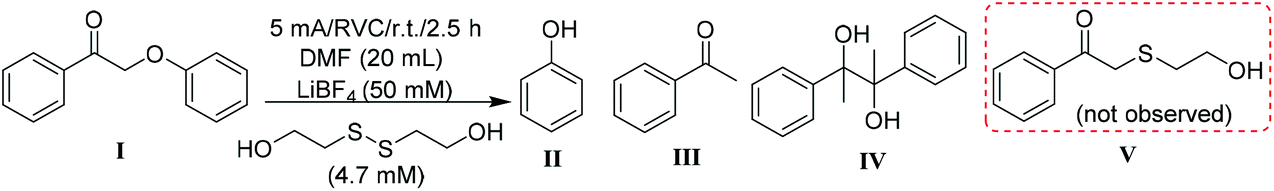
Stoichiometrically, only two equivalents of thiolate are needed to reductively cleave 2-phenoxyacetophenone via a reaction mechanism consisting of an SN2 displacement of phenoxide by thiolate, followed by nucleophilic attack of thiolate on the thioether intermediate (compound V in Table 1) to replace 1-phenylethen-1-olate (the enolate of acetophenone).38 To probe the susceptibility of disulfide to electrolytic cleavage and its effect on the deconstruction of lignin dimer model compounds, 1 equivalent of 2,2′-dithiodiethanol (the disulfide derived from β-mercaptoethanol oxidation) was included in the DMF solution in the cathode cell (entry 2). The 2-phenoxyacetophenone cleavage displayed a reaction profile similar to that in Fig. 2a, with virtually no formation of phenol and acetophenone in the first 1 h (Fig. 2b) at room temperature. In addition to 2,2′-dithiodiethanol, other disulfides including cystine (Fig. S2†) exhibited comparable rates of substrate consumption. The critical nucleophile that cleaved the ether bond in the previous studies,38,39 2-hydroxyethanethiolate (here referred to as RS−), was initially presumed to be the active species in the electrochemically driven reaction. At the 5 mA current used in these experiments, two equivalents of electrons were calculated to pass through the cathode in 1 h, and the RSSR should ideally then be reduced to 2 equivalents of RS−. Therefore, RSSR was pre-electrolyzed in DMF at 5 mA for 1 h to reduce the disulfide, the current was then removed, and the substrate 2-phenoxyacetophenone was added (entry 3). The substrate decayed only very slowly (Fig. 3a) under these conditions, consistent with our previous finding that heat was required to get reasonable reaction rates in the thiolate-enabled dimer cleavage.38 In a parallel control, the combination of β-mercaptoethanol (hereafter referred to as RSH) with either an insoluble (K2CO3) or a soluble (Et3N) base in DMF also showed little or no cleavage of 2-phenoxyacetophenone at room temperature (Fig. S3 and S4†).
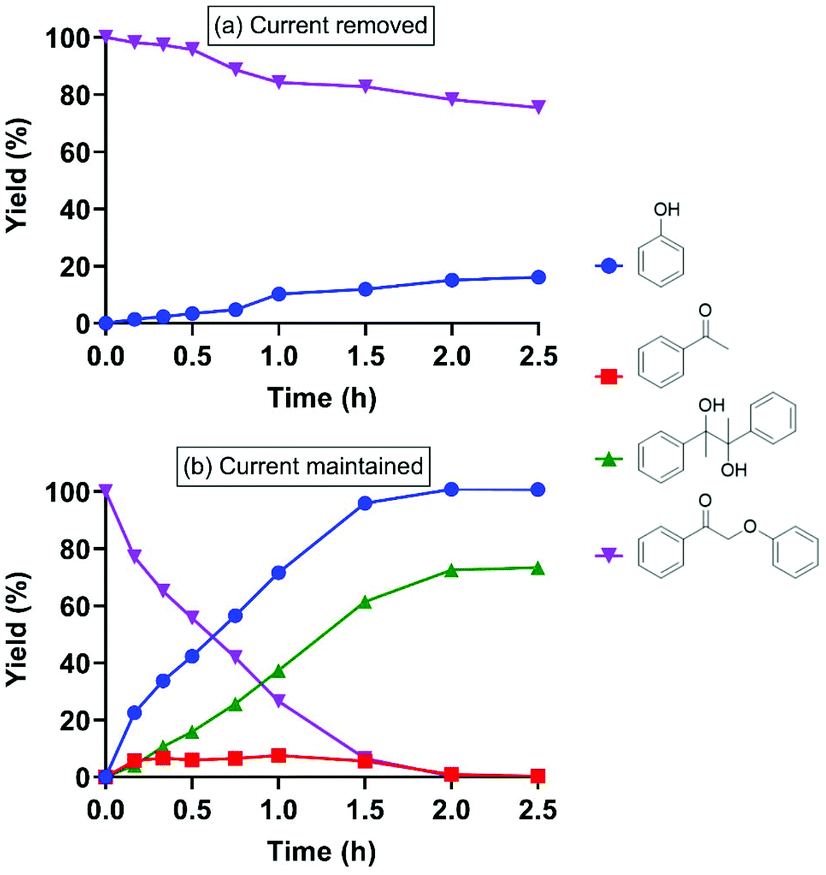 | ||
| Fig. 3 Effect of flowing current (5 mA) on cleavage of 2-phenoxyacetophenone (I) with 1 h pre-electrolyzed RSSR: (a) current was removed after I was added (entry 3); (b) current was maintained throughout the entire reaction (entry 4). | ||
When the current was maintained following pre-electrolysis of RSSR, 2-phenoxyacetophenone decayed significantly faster (Fig. 3b). As expected, cleavage was faster after an hour of pre-electrolysis (i.e., two equivalents of electrons, the quantity expected to fully reduce the disulfide), than with only 30 min of pre-electrolysis (Fig. S5†). Notably, the thioether (compound V in Table 1), the critical diagnostic intermediate in the SN2 mechanism previously studied, was not observed in the cathode cell. One potential explanation is that some species other than simple RS− was involved in attacking the substrate during electrolysis.
Thiyl radical (referred to as RS˙) formed during the cathodic disulfide reduction might contribute to the substrate decomposition. To probe this possibility, the current was turned off once the RSSR was pre-electrolyzed for 1 h, and then the radical trap butylated hydroxytoluene (BHT) was added together with the substrate (current was still absent). Although 2-phenoxyacetophenone was cleaved to a small extent, the reaction ceased after 45 min (Fig. S6†), showing a similar profile to that in Fig. 3a, where BHT was not employed. In addition, when RSSR was pre-electrolyzed for only 30 min, the current removed, BHT added, and the resulting solution stirred for an additional 30 min, no cleavage of substrate was observed (Fig. S7†); however, if no BHT was added, a slow reaction did occur (Fig. S8†). In a parallel control, a thiyl radical generated from RSH and AIBN (azobisisobutyronitrile) yielded essentially no substrate cleavage in the absence of current (Fig. S9†). Thus, as in the previous thio-based cleavage studies, we infer that dimer cleavage does not involve free thiyl radical processes,38 suggesting instead that the electric current forms diffusible electron-carrier species, which activate ether bond cleavage.
In the absence of the radical trap BHT, when RSSR was pre-electrolyzed for 1 h before addition of 2-phenoxyacetophenone substrate, pinacol began forming almost immediately and only small quantities of acetophenone were observed as current flow continued and the starting material was cleaved (Fig. 3b). In contrast, addition of BHT to 1 h pre-electrolyzed RSSR (entry 5) allowed acetophenone to accumulate to 50% yield before it was further reduced and dimerized to pinacol (Fig. 4a).
 | ||
| Fig. 4 Effect of BHT on the electrolysis of 2-phenoxyacetophenone (I) using 5 mA of current. Condition: Prior to the addition of I, (a) BHT was added after RSSR was pre-electrolyzed for 1 h (entry 5); (b) BHT and RSSR were pre-electrolyzed together for 30 min (entry 6). | ||
With these results in mind, BHT and RSSR (1:1 molar ratio) were pre-electrolyzed together for 1 h prior to addition of 2-phenoxyacetophenone and 5 mA of current was maintained throughout the entire reaction. Compared to pre-electrolysis of RSSR alone (for 30 min) in the absence of BHT (Fig. S5†), in which acetophenone only accumulated to 14% yield and up to 70% of pinacol was formed, with the BHT present in the pre-electrolysis mixture (entry 6), 2-phenoxyacetophenone was completely cleaved in 1.5 h with a quantitative yield of phenol and up to 80% of acetophenone (Fig. 4b). In addition, no pinacol was observed until all substrate had been cleaved (Fig. 4b) at 1.5 h. Doubling the quantity of BHT used did not lead to a statistically significant change in the reaction profile (Fig. S10†).
To study further the potential role of a disulfide-derived radical species during substrate cleavage in the cathode cell, the effect of O2, a known radical trap,42 was evaluated. Even when RSSR was not pre-electrolyzed, the substrate decayed much faster under anoxic conditions; under a N2 atmosphere (entry 7), the reaction was nearly complete after 2 h with pinacol and phenol being the major products (Fig. 5a), while 40% of the substrate remained unreacted after 2 h when the same reaction was performed under fully aerobic conditions (Fig. 2b). Moreover, addition of BHT to reactions performed under N2 (entry 8) again inhibited the formation of pinacol (Fig. 5b), but still showed faster cleavage of 2-phenoxyacetophenone than in the open air (Fig. S11†).
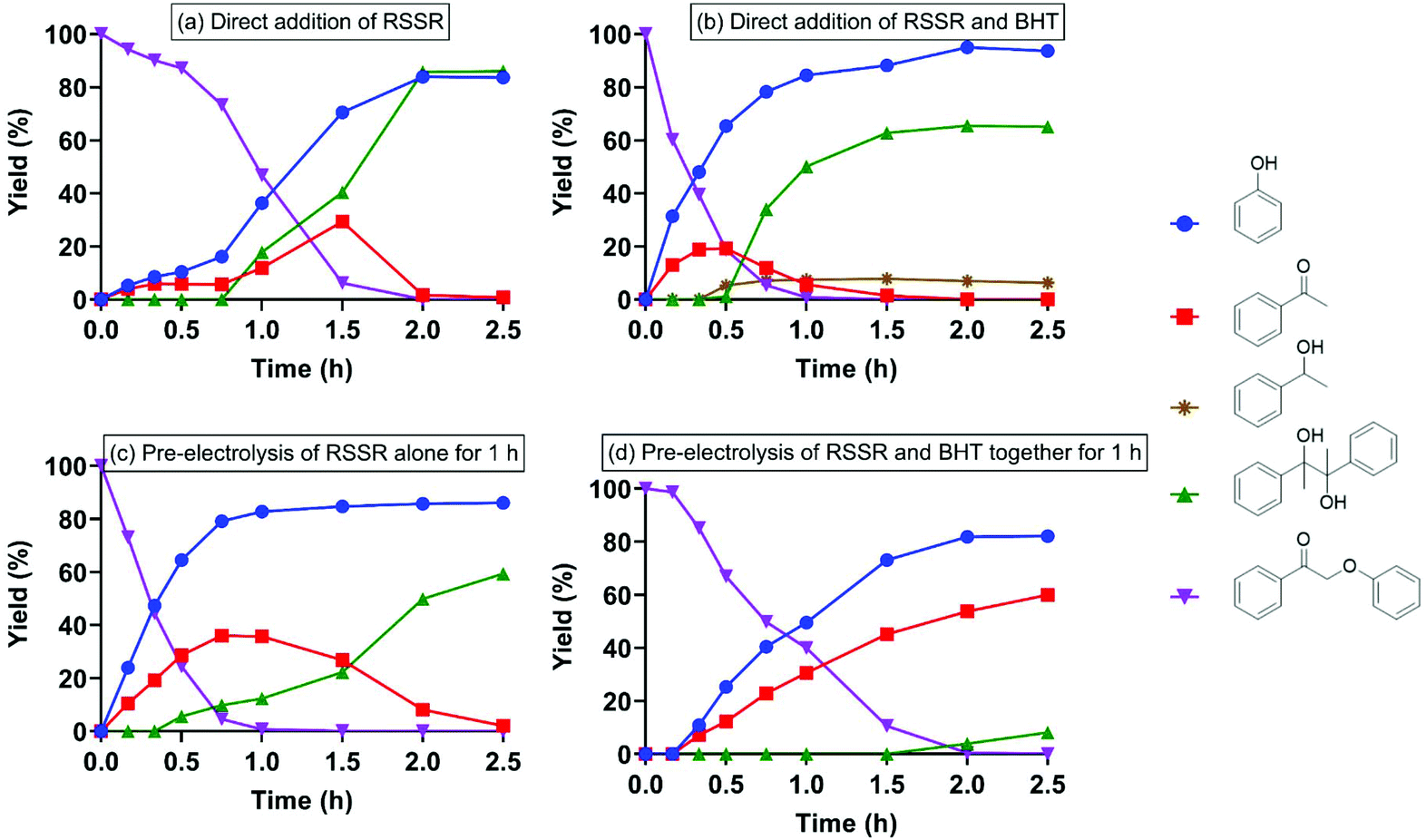 | ||
| Fig. 5 Thio-assisted cathodic electrolysis of 2-phenoxyacetophenone (I) under N2 atmosphere. (a) Direct addition (i.e., applied simultaneously with) of RSSR (entry 7) with I; (b) direct addition of RSSR and BHT with I (entry 8); (c) RSSR alone was pre-electrolyzed for 1 h prior to addition of I (entry 9); (d) RSSR and BHT were pre-electrolyzed together for 1 h prior to addition of I (entry 10). | ||
One pathway considered for the C–O ether bond cleavage in the keto aryl ether substrate was one-electron reduction by an electron transfer agent. In that scenario, the disulfide radical anion (RSSR˙−) might act as an electron carrier. Oxygen could (a) consume an electron from RSSR˙−,43 slowing the rate of 2-phenoxyacetophenone cleavage relative to that under N2 (Scheme 2a), or (b) react with RS˙ or RS− to form the RSO2˙ radical or anion (Scheme 2b).44–46 To test this possibility, RSSR was pre-electrolyzed for 1 h under N2; again, after substrate addition, pinacol and phenol were the major products (entry 9, Fig. 5c). Here, the substrate was completely consumed in only 1 h while previous controls had required at least 1.5 h. Including BHT during RSSR pre-electrolysis under N2 (entry 10) did not slow the 2-phenoxyacetophenone deconstruction (Fig. 5d), indicating again that the cleavage did not solely depend on the thiyl radical and that oxygen competed to abstract the electron from the electron transfer agent, thereby diverting the reaction.
 | ||
| Scheme 2 (a) A proposed mechanism to cleave 2-phenoxyacetophenone via a one-electron reduction pathway; (b) potential mechanism of O2 trapping the thiyl radical. | ||
Importantly, we found that the electrolyte used in this study, LiBF4, was critical in promoting the cleavage of 2-phenoxyacetophenone. Li+, a known ion pairing agent and Lewis acid,47 favored the formation of pinacol (Scheme S1†) and presumably shifted the equilibrium of the cleavage reaction. When NaBF4 was employed as the electrolyte, approximately 50% of the substrate remained unreacted and pinacol was not observed even after 2.5 h (Fig. S12†). Under these same conditions using LiBF4 as the electrolyte, however, the substrate 2-phenoxyacetophenone was nearly completely cleaved in 1.5 h and pinacol was formed (Fig. 3b).
Current efficiencies (CE%) of all reactions were calculated as follows:48
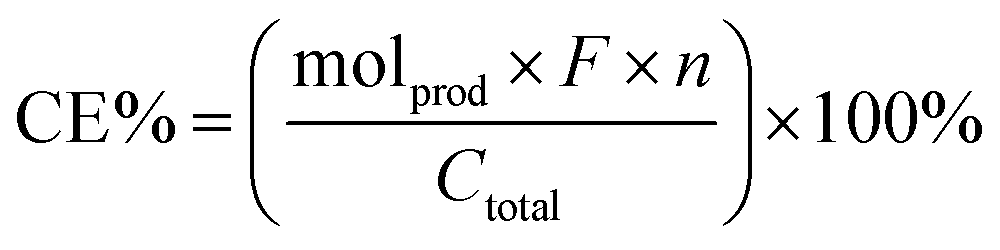 485 C mol−1); n is the number of electrons per reaction and Ctotal is the total charge passed. Overall, entry 2 shows the highest faradaic efficiency (∼53%).
485 C mol−1); n is the number of electrons per reaction and Ctotal is the total charge passed. Overall, entry 2 shows the highest faradaic efficiency (∼53%).
Thio-assisted electrolysis of other β-O-4 model compounds
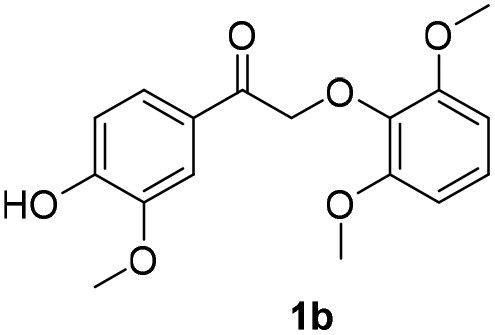
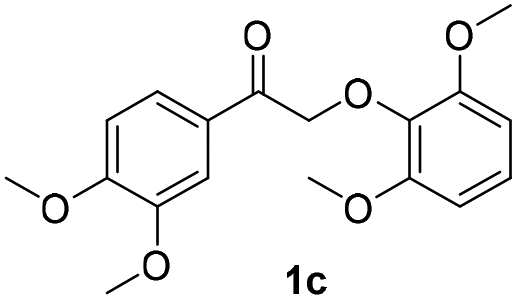
In our previous study, 2-phenoxyacetophenone was reacted with 100 eq. of β-mercaptoethanol (BME) and a large excess of insoluble base (K2CO3) in refluxing acetonitrile (∼85 °C) for 24 h to achieve complete cleavage of substrate.38 In the present work, however, 2-phenoxyacetophenone was completely decomposed in 1.5 h at room temperature in the presence of only 1 eq. of 2,2′-dithiodiethanol (the disulfide form of BME) and 5 mA electric current, producing a quantitative yield of phenol and up to 80% of acetophenone (Fig. 4b). In addition, the formation of pinacol was inhibited by BHT until after 1.5 h.Encouraged by the success in thio-assisted electrolytic cleavage of 2-phenoxyacetophenone, we explored this system with other α-keto β-O-4 dimers (1a–c) more closely related to real lignin. We posited that the presence on the aryl ketone of the electron-donating 4-methoxy or -hydroxy groups commonly found in lignin might inhibit the pinacol-forming reductive dimerization.
The results of these experiments are summarized in Table 2 and Fig. S13–S18.† Electric current (5 mA) alone still cleaved the β-O-4 dimers bearing methoxy substituents. As expected, with pre-electrolyzed 2,2′-dithiodiethanol (1 eq.), the cleavage time was shortened, except in the case of dimer 1b where the substituent para to the ketone moiety is –OH. We speculate that this phenolic –OH in 1b may be deprotonated, forming a quinone methide intermediate which would resist further reduction and cleavage of the ether bond (Scheme S2†). Without addition of the disulfide, an “induction period” (around 45 min) was found in the electrolysis of both dimers 1a and 1c where very limited dimer cleavages occurred (Fig. S13 and S17†), and acetoveratrone was generated only in moderate yields. Pre-electrolysis of the disulfide shortened the induction time by half (Fig. S14 and S18†) and improved the yields of acetoveratrone to 72% (dimer 1a) and 45% (dimer 1c), with quantitative yields of guaiacol (dimer 1a) and 53% syringol (dimer 1c), respectively. Termination of the reaction prior to acetoveratrone decay, which usually occurred after the dimers were consumed (i.e., at 1.5 h for 1a and at 2 h for 1c), would help maximize the monomer yields (Fig. S14 and S18†). As expected, more promising results were observed when BHT and RSSR were pre-electrolyzed together. All the monomeric products were produced in 1.5 h with over 90% yields (Fig. S19 and S20†). Notably, dimer 1a, which required 24 h to produce a quantitative yield of acetoveratrone in the previous study (employing refluxing acetonitrile with 10 eq. β-mercaptoethanol and a large excess of K2CO3),38 decomposed to acetoveratrone (91%) and guaiacol (90%) using 1 eq. of 2,2′-dithiodiethanol at room temperature in only 1.5 h (Fig. S19†) in the current study. Together, the shortened reaction time, lower reaction temperature, and significantly reduced thio-compound loading suggest that electrochemical depolymerization might succeed under practical conditions with authentic lignin.
| Dimer | Current alonea | Current + disulfide and BHTb | ||||
|---|---|---|---|---|---|---|
| Ketone (%) | Phenol (%) | Conv. (%) | Ketone (%) | Phenol (%) | Conv. (%) | |
|
a The reaction was performed in DMF with 5 mA only (no disulfide) at room temperature for 2 h.
b The disulfide (2,2′-dithiodiethanol) and BHT (1:1 molar ratio) were pre-electrolyzed together for 40 min in DMF at 5 mA, and the dimer was then added and the reaction stirred for 1.5 h.
|
||||||
|
37 | 58 | 54 | 91 | 90 | 97 |
|
12 | 22 | 20 | 22 | 28 | 30 |
|
27 | 36 | 33 | 90 | >99 | 96 |
Thio-assisted electrolysis of ligninox
The successful cleavage of the model β-O-4 lignin dimers described above encouraged us to apply the optimized conditions to authentic lignin samples. The lignin used in this study was extracted from hybrid poplar following the previous described copper-catalyzed alkaline peroxide (Cu-AHP) strategy.49 Cu-AHP lignin was oxidized (referred to as ligninox) with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ),10 which was confirmed by 2D HSQC spectra (Fig. S21†). To improve the conductivity of the reaction mixture and the solubility of ligninox, a solvent mixture of pH 8 phosphate buffer/isopropanol (2:1, v/v) was employed and the reaction was performed under a higher current density (10 mA cm−2) for 6 h.
Electric current was found to be critical in this aqueous reaction system as ligninox showed essentially no change in the absence of flowing current. This is consistent with our previous finding that water inhibited the thio-assisted keto aryl ether cleavage. In the previous study, increased thiol loading, elevated temperature and N2 protection were required to break the ether bond.38 In contrast, at room temperature, the combination of electric current and disulfide successfully solubilized 62% of ligninox in only 6 h (Fig. 6) while only 46% of the ligninox was solubilized under analogous conditions in the absence of the disulfide, again demonstrating that the thiol assists in the lignin cleavage process. In the control reaction where no disulfide was added, electric current (20 mA) alone afforded 24 wt% of ethyl acetate soluble (EA soluble) products from ligninox (Fig. 6), including various functionalized S/G/H type phenolic monomers (Fig. 7) along with a series of dimers, trimers, tetramers and other oligomers (Fig. S22†). The addition of RSSR (2,2′-dithiodiethanol) and BHT to the reaction mixture improved the yield of EA soluble products to 36 wt%, with a concomitant decrease in the insoluble material from 54 wt% to 38 wt% (Fig. 6). Although slight differences were observed via LCMS-QTOF in the distribution of products in the EA soluble fractions (Fig. S23†), the profiles were relatively similar, indicating that the addition of the disulfide did not significantly complicate the product streams. Addition of disulfide and BHT increased the amount of aqueous soluble products from 22 wt% to 26 wt%, and analysis by gel permeation chromatography (GPC) indicated that the molecular weight of the major products ranged from approximately 600–1000 Da (Fig. S24†). Overall, at ambient temperature and atmosphere, electroreductive degradation of ligninox solubilized a significant fraction of the lignin, leaving only 38–54 wt% of insoluble residual. Compared to previous work on depolymerization of different types of lignin, i.e. via catalytic electrolysis (77–82 wt% residual),50 aerobic oxidation-hydrolysis (47–78 wt% residual)9 or a two-step oxidative approach (50–91 wt% residual),10 the present study represents an encouraging first step in developing a simple new strategy for lignin depolymerization and valorization.
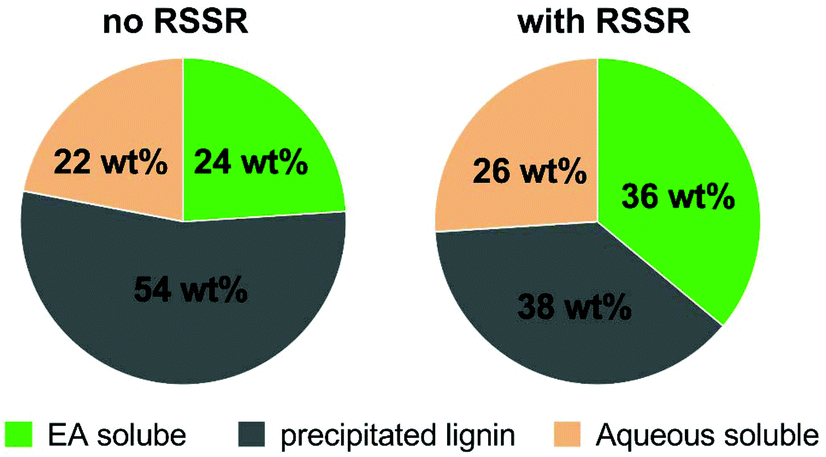 | ||
| Fig. 6 Composition of each fraction from electrolysis of ligninox; wt% of each fraction was calculated based on the weight of ligninox. Note: wt% of aqueous-soluble fraction was determined by 1 − (wt% of EA soluble + wt% of insoluble). | ||
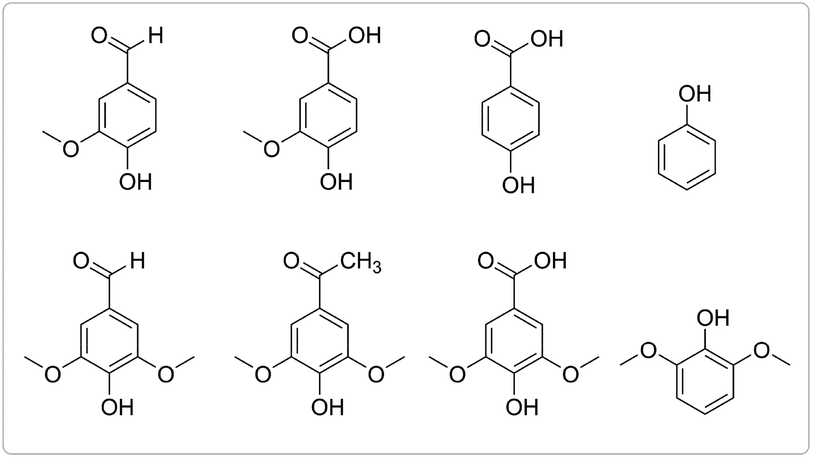 | ||
| Fig. 7 Phenolic monomers from electrolysis of ligninox; products were determined by GC-MS. | ||
Conclusion
Building on our previously reported thiolate-based reductive cleavage of β-O-4 linkages via an SN2 pathway,38,39 this study aimed to electrochemically reduce the disulfide byproduct to achieve an electrocatalytic cycle (Fig. 1). Electrolytic reductive treatment of α-keto β-O-4 model dimers together with disulfides does indeed result in ether cleavage. With pre-electrolyzed disulfide and BHT, this approach cleaved the β-O-4 models and produced the corresponding phenolic and keto monomers with over 90% yields in only 1.5 h. Using only 1 eq. of disulfide at room temperature, this surprisingly mild and rapid cleavage represented an unexpectedly large improvement over our previous mechanistically motivated work in which 10 eq. of thiol in refluxing acetonitrile required 24 h to completely cleave the oxidized β-O-4 dimers. Applied to pre-oxidized authentic lignin, the electrochemical method showed promising levels of cleavage and solubilization. In the electrochemical context, the use of thiols, originally envisioned to serve as small diffusible redox carriers via the thiol/disulfide couple, enables a significantly milder approach to deconstruction of lignin to small-molecule fragments. Though the mechanistic details of these improvements remain incompletely understood, we summarize our findings as follows: (1) pre-reduction of the disulfide forms active species that promote cleavage of 2-phenoxyacetophenone; (2) for complete substrate conversion, electric current must be supplied throughout the reaction time; (3) dimer cleavage is not likely to involve free thiyl radicals; and (4) substrate is cleaved faster under a N2 atmosphere than under air. These results represent a substantial step forward in linking the reducing power of electrical current to the deconstruction and potential valorization of lignin as an organic feedstock.Experimental section
General materials
Acetone (ACS grade), 3,4-dimethoxyacetophenone (98%), 4-hydroxy-3-methoxyacetophenone (acetovanillone, 98%), 2,6-di-tert-butyl-4-methylphenol (BHT, >99% GC), 1-phenylethanol (98%), acetonitrile (99.5+%), dichloromethane (ACS grade), N-bromosuccinimide (NBS, 99%), lithium tetrafluoroborate (LiBF4, 98%), N,N-dimethylformamide (DMF, 99.8%, anhydrous), syringol, 2,3-diphenyl-butane-2,3-diol (pinacol), hexane (>98.5%), and plastic silica gel 60 F-254 plates (for thin layer chromatography, TLC) were purchased from Sigma-Aldrich (St Louis, MO). Acetophenone (98.8%) and triethyl amine were purchased from J.T. Baker (Phillipsburg, NJ), while benzyl chloride and 2-phenoxyacetophenone (>98%) were purchased from TCI, America, Ltd (Portland, OR). Guaiacol (99+%) was purchased from Acros Organics (New Jersey). Tetrahydrofuran (THF, 99+% with 250 ppm BHT) and phenol (99+%) were purchased from Alfa Aesar (Haverhill, MA). Sodium sulfate (anhydrous) and ethyl acetate (99.9%) were purchased from Fisher Chemicals (Pittsburgh, PA), and p-toluenesulfonic acid (TsOH) was purchased from Spectrum (New Brunswick, NJ). Finally, Chloroform-d (CDCl3, 99.8 atom% D) was purchased from Cambridge Isotope Laboratories (Andover, MA).Characterization of products
Column chromatography was performed on Silicycle (Quebec City, Canada) SiliaFlash P60 silica gel (40–63 μm). 1H and 13C NMR spectra were acquired on Agilent 500/54 premium shielded instruments with tetramethylsilane or residual solvent as the internal reference. High performance liquid chromatography (HPLC) was conducted on an Agilent 1260 Infinity equipped with an Agilent G1315D1260 diode array detector VL, monitoring at 250 nm (for experiments related to dimer 1a–c) or 280 nm (for experiments related to 2-phenoxyacetophenone) and recording 190–400 nm. A 10 L sample injection using a Supelco Ascentis Express C18 column (15 cm × 4.6 cm, 2.7 μm, St Louis, MO) at 0.5 mL min−1, with a mobile phase of 35/65 and 50/50 acetonitrile/water (with 0.5 M H2SO4) was applied at 30 °C on analyses of 1a–c and 2-phenoxyacetophenone related experiments, respectively. Yields of products were calibrated using authentic compounds as external standards. Gel permeation chromatography (GPC) was performed using a Waters Ultrahydrogel 250 7.8 × 300 mm column equipped with a Waters Ultrahydrogel 6 × 40 mm guard column at 40 °C with a mobile phase of 5 mM NaOH in 80/20 0.1 M aqueous sodium nitrate/acetonitrile at a flow rate of 0.7 mL min−1. Polystyrene sulfonic acid and poly(ethylene glycol) were employed as external standards. 1H13C-gradient heteronuclear single quantum coherence (HSQC) was performed at ambient temperature on a 500 MHz Bruker NMR spectrometer equipped with a 5 mm iProbe (BBO probe). Spectra were collected by utilizing the Bruker pulse sequence “hsqcedetgpsisp2.3” with an acquisition time of 63.9 ms (F2, 512 complex points for 1H) and 63.9 ms (F1, 1024 increments for the 13C dimension), using a delay of 1.5 s with 48 scans per increment with spectral widths of 8013 Hz (1H) and 20 kHz (13C).General procedure for thio-assisted cathodic electrolysis of dimers
Reactions were conducted at room temperature up to 2.5 h in an H-type electrochemical cell (separated by a Nafion 117 membrane) wherein a platinum wire in 20 mL of pH 8 phosphate buffer was placed in the anode half-cell and the reticulated vitreous carbon (RVC, 2 cm2 immersed) was employed as the cathode. The start of the reaction (t = 0) was defined as the time point when substrate (2-phenoxyacetophenone or dimers 1a–c) was added. (Note: reactions were run in open air unless otherwise noted.)For reactions run without pre-electrolysis, 20 mg of the dimer (2-phenoxyacetophenone or 1a–c), 2,2′-dithiodiethanol (none or 1 eq.) and BHT (none or 1 eq.) were directly added to 20 mL DMF in the cathode half-cell and a 5 mA electric current was applied for the duration of the reaction (the voltage of the system ranged from 5.6–6.0 V in DMF under our conditions). Sample aliquots (250 μL) were taken from the cathode cell at timed intervals and diluted with another 250 μL of acetonitrile before HPLC analysis.
For reactions involving pre-electrolysis of the disulfide (2,2′-dithodiethanol), 2,2′-dithiodiethanol alone was added to 20 mL DMF and the 5 mA electric current was applied for 30 min or 1 h. Then, 20 mg of the dimer (2-phenoxyacetophenone or 1a–c) and BHT (none or 1 eq.) were added in solid form. Sampling for HPLC analysis was performed as above.
For reactions run under a N2 atmosphere: 20 mL of DMF alone was placed in the sealed cathode half-cell and the cell was purged with N2 for 30 min with stirring. Subsequent steps were as described above except that a N2 balloon was connected to the cathode half-cell throughout the reaction. At different time intervals, 500 μL samples were taken from the cathode cell for HPLC analysis.
Synthesis of β-O-4 dimers (1a–c)
Dimers 1a–c were synthesized based on previous reports with only minor modifications.38A mixture of 2-bromo-1-(3,4-dimethoxyphenyl)ethanone (2.54 g, 9.8 mmol), guaiacol (1.21 g, 9.8 mmol), and K2CO3 (1.35 g, 9.8 mmol) in acetone (50 mL) was stirred at room temperature overnight. The solid was removed by vacuum filtration and the solution was extracted with three 20 mL aliquots of DCM. The combined organic layers were separated and dried with anhydrous Na2SO4 and concentrated under vacuum. The crude product was purified by column chromatography on silica gel (hexane/ethyl acetate = 2/1) to produce 1a (2.65 g, 8.8 mmol) as a white solid in 89% yield.
1H NMR (500 MHz, CDCl3): 7.69 (dd, J = 8.4, 2.0 Hz, 1H), 7.61 (d, J = 2.0 Hz, 1H), 7.01–6.94 (m, 1H), 6.94–6.88 (m, 2H), 6.88–6.82 (m, 2H), 5.31 (s, 2H), 3.96 (s, 3H), 3.94 (s, 3H), 3.90 (s, 3H); 13C NMR (126 MHz, CDCl3): 193.2, 153.7, 149.6, 149.1, 147.5, 127.8, 122.7, 122.3, 120.8, 114.6, 112.0, 110.4, 110.1, 71.9, 56.1, 56.0, 55.9. Spectral data are in accordance with those previously reported.38
A solution of N-bromosuccinimide (NBS, 6.96 g, 39 mmol) dissolved in anhydrous acetonitrile (30 mL) was added dropwise with stirring to a solution of the above benzyl protected acetovanillone (10 g, 39 mmol) and p-toluenesulfonic acid (TsOH, 10 g, 58.6 mmol) in anhydrous acetonitrile (70 mL). The mixture was then heated and stirred at 100 °C for 2 h under a N2 atmosphere. After cooling to room temperature, the solvent was evaporated in vacuo. The crude product was redissolved in dichloromethane (DCM, 70 mL) and washed three times with 30 mL aliquots of deionized water. The organic layer was separated and dried with anhydrous Na2SO4 and concentrated under vacuum. Without further purification, the resulting crude protected bromoacetovanillone (5 g, 14.9 mmol) was combined with syringol (2.3 g, 14.9 mmol) and K2CO3 (2.06 g, 14.9 mmol) in acetone (100 mL) and stirred at room temperature overnight. The solid KBr byproduct was removed by vacuum filtration and the solution was extracted with three 30 mL aliquots of DCM. The organic layers were combined and dried with anhydrous Na2SO4 and concentrated under vacuum. The resulting crude protected dimer (2 g) was dissolved in methanol (50 mL), 10% Pd/C (0.2 g) was added, and the mixture was stirred for 3 h at room temperature. The solid catalyst was separated by vacuum filtration and washed with methanol. Then, the filtrate was concentrated under vacuum and the crude product was purified by column chromatography on silica gel (hexane/ethyl acetate = 2/1) to give a yellow solid.
1H NMR (500 MHz, CDCl3): 7.69–7.64 (m, 2H), 7.02 (t, J = 8.4 Hz, 1H), 6.95 (d, J = 8.1 Hz, 1H), 6.59 (d, J = 8.4 Hz, 2H), 5.16 (s, 2H), 3.96 (s, 3H), 3.82 (s, 6H); 13C NMR (126 MHz, CDCl3): 193.5, 153.2, 150.5, 146.6, 136.6, 128.0, 124.0, 123.5, 113.8, 110.4, 105.3, 75.2, 56.1, 56.1. Spectral data are in accordance with those previously reported.38
1H NMR (500 MHz, CDCl3): 7.73 (dd, J = 8.3, 2.0 Hz, 1H), 7.66 (d, J = 2.0 Hz, 1H), 7.02 (t, J = 8.4 Hz, 1H), 6.90 (d, J = 8.4 Hz, 1H), 6.59 (d, J = 8.5 Hz, 2H), 5.16 (s, 2H), 3.95 (d, J = 1.8 Hz, 6H), 3.82 (s, 6H); 13C NMR (126 MHz, CDCl3): 193.7, 153.4, 153.3, 153.2, 148.9, 136.6, 128.3, 124.0, 123.0, 110.6, 110.0, 105.2, 105.2, 75.2, 56.0, 56.0, 56.0, 55.9. Spectral data are in accordance with those previously reported.38
Oxidation of Cu-AHP lignin
Lignin from alkaline pre-extracted hybrid poplar was isolated via the Cu-AHP pretreatment process as previously described using 1 mM of copper and 2 mM of bipyridine.49 Hydrogen peroxide (H2O2, 100 mg per g of biomass) was added in 10 equal aliquots at 1 h intervals while shaking at 30 °C. DDQ-catalyzed oxidation of Cu-AHP lignin was performed following a previous literature report.10 Briefly, 3 g of lignin was solubilized in 1,2-dimethoxyethane/2-ethoxyethanol (v/v = 3:2, 42 mL) and then DDQ (10 wt%) and t-BuONO (10 wt%) were added. The solution was heated at 80 °C overnight under an oxygen atmosphere. The oxidized lignin product (ligninox) was precipitated by addition of 500 mL of diethyl ether, isolated by filtration, washed with additional diethyl ether (200 mL), and dried in vacuo, yielding 2.7 g of light-brown ligninox.
General procedure for thio-assisted electrolysis of ligninox
Reactions were conducted at room temperature in an H-type electrochemical cell (separated by a Nafion 117 membrane) with 20 mL of pH 8 phosphate buffer and 20 mL of 2:1, v/v pH 8 phosphatebuffer/isopropanol in the anodic and cathodic cell, in which a platinum wire was used as the anode and reticulated vitreous carbon (RVC) was employed as the cathode, respectively. A mixture of LiBF4 (100 mg) and BHT (100 mg) was added into the cathodic cell. After 1 h of pre-electrolysis at 20 mA (current density = 10 mA cm−2), ligninox (100 mg) and 2,2′-dithiodiethanol (RSSR, 100 mg) were added to the catholyte and the reaction continued at 20 mA for an additional 6 h. The solvent was removed under a N2 flow, and the crude product was solubilized in 20 mL EtOAc/water (1:1, v/v). The solution was acidified with 60% H2SO4 to pH 2, precipitating insoluble material. The precipitate was filtered, washed with water, dried in vacuo, and the remaining filtrate was extracted three times with 30 mL EtOAc. The organic layers were combined, dried with anhydrous Na2SO4, and concentrated in vacuo. In the parallel control reaction, all set-up and work-up protocols were the same except that the RSSR and BHT components were omitted.
Conflicts of interest
A patent application has been submitted by E. L. Hegg, J. E. Jackson, and G. E. Klinger (Methods for Lignin Depolymerization Using Thiols – WO/2018/195000). As holders of this patent application, we could potentially benefit financially from the technology discussed in this manuscript.Acknowledgements
This material is based upon work supported by the Great Lakes Bioenergy Research Center, U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research under Award Number DE-SC0018409. The authors gratefully acknowledge Casey Johnny and Tony Schilmiller in the Research Technology Support Facility at Michigan State University for the GC-MS and LCMS-TOF analyses. ZF thanks Nicholas Schlecht for help in performing selected reactions and Dr. Md Asmaul Hoque at UW-Madison for running the cyclic voltammetry of 2,2′-dithiodiethanol (Fig. S31†).Notes and references
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110(6), 3552–3599 CrossRef CAS PubMed.
- Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118(2), 614–678 CrossRef CAS PubMed.
- W. Schutyser, T. Renders, S. Van den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47(3), 852–908 RSC.
- B. M. Upton and A. M. Kasko, Chem. Rev., 2016, 116(4), 2275–2306 CrossRef CAS PubMed.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115(21), 11559–11624 CrossRef CAS PubMed.
- I. Hasegawa, Y. Inoue, Y. Muranaka, T. Yasukawa and K. Mae, Energy Fuels, 2011, 25(2), 791–796 CrossRef CAS.
- A. Rahimi, A. Azarpira, H. Kim, J. Ralph and S. S. Stahl, J. Am. Chem. Soc., 2013, 135(17), 6415–6418 CrossRef CAS PubMed.
- A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Nature, 2014, 515(7526), 249–252 CrossRef CAS PubMed.
- A. Das, A. Rahimi, A. Ulbrich, M. Alherech, A. H. Motagamwala, A. Bhalla, L. da Costa Sousa, V. Balan, J. A. Dumesic, E. L. Hegg, B. E. Dale, J. Ralph, J. J. Coon and S. S. Stahl, ACS Sustainable Chem. Eng., 2018, 6(3), 3367–3374 CrossRef CAS.
- Y. Song, A. H. Motagamwala, S. D. Karlen, J. A. Dumesic, J. Ralph, J. K. Mobley and M. Crocker, Green Chem., 2019, 21(14), 3940–3947 RSC.
- O. Musl, M. Holzlechner, S. Winklehner, G. Gübitz, A. Potthast, T. Rosenau and S. Böhmdorfer, ACS Sustainable Chem. Eng., 2019, 7(18), 15163–15172 CrossRef CAS.
- Y. Li, B. Demir, L. M. Vázquez Ramos, M. Chen, J. A. Dumesic and J. Ralph, Green Chem., 2019, 21(13), 3561–3572 RSC.
- E. Feghali, G. Carrot, P. Thuéry, C. Genre and T. Cantat, Energy Environ. Sci., 2015, 8(9), 2734–2743 RSC.
- E. M. Anderson, R. Katahira, M. Reed, M. G. Resch, E. M. Karp, G. T. Beckham and Y. Román-Leshkov, ACS Sustainable Chem. Eng., 2016, 4(12), 6940–6950 CrossRef CAS.
- L. Fan, Y. Zhang, S. Liu, N. Zhou, P. Chen, Y. Cheng, M. Addy, Q. Lu, M. M. Omar, Y. Liu, Y. Wang, L. Dai, E. Anderson, P. Peng, H. Lei and R. Ruan, Bioresour. Technol., 2017, 241, 1118–1126 CrossRef CAS PubMed.
- G. Jiang, D. J. Nowakowski and A. V. Bridgwater, Energy Fuels, 2010, 24(8), 4470–4475 CrossRef CAS.
- P. R. Patwardhan, R. C. Brown and B. H. Shanks, ChemSusChem, 2011, 4(11), 1629–1636 CrossRef CAS PubMed.
- P. J. Deuss and K. Barta, Coord. Chem. Rev., 2016, 306, 510–532 CrossRef CAS.
- S. K. Hanson and R. T. Baker, Acc. Chem. Res., 2015, 48(7), 2037–2048 CrossRef CAS.
- S. Van den Bosch, W. Schutyser, S. F. Koelewijn, T. Renders, C. M. Courtin and B. F. Sels, ChemComm, 2015, 51(67), 13158–13161 RSC.
- C. H. Lam, S. Das, N. C. Erickson, C. D. Hyzer, M. Garedew, J. E. Anderson, T. J. Wallington, M. A. Tamor, J. E. Jackson and C. M. Saffron, Sustainable Energy Fuels, 2017, 1(2), 258–266 RSC.
- B. Mahdavi, A. Lafrance, A. Martel, J. Lessard, H. Ménard and L. Brossard, J. Appl. Electrochem., 1997, 27, 605–611 CrossRef CAS.
- A. Cyr, F. Chiltz, P. Jeanson, A. Martel, L. Brossard, J. Lessard and H. Ménard, Can. J. Chem., 2000, 78(3), 307–315 CrossRef CAS.
- X. Du, H. Zhang, K. P. Sullivan, P. Gogoi and Y. Deng, ChemSusChem, 2020, 13(17), 4318–4343 CrossRef CAS PubMed.
- S. Stiefel, J. Lölsberg, L. Kipshagen, R. Möller-Gulland and M. Wessling, Electrochem. Commun., 2015, 61, 49–52 CrossRef CAS.
- B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Green Chem., 2010, 12(12), 2099–2119 RSC.
- B. H. Nguyen, R. J. Perkins, J. A. Smith and K. D. Moeller, J. Org. Chem., 2015, 80(24), 11953–11962 CrossRef CAS PubMed.
- T. Wu, B. H. Nguyen, M. C. Daugherty and K. D. Moeller, Angew. Chem., Int. Ed., 2019, 58(11), 3562–3565 CrossRef CAS PubMed.
- V. L. Pardini, C. Z. Smith, J. H. P. Utley, R. R. Vargas and H. Viertler, J. Org. Chem., 1991, 56(26), 7305–7313 CrossRef CAS.
- H. Zhu, L. Wang, Y. Chen, G. Li, H. Li, Y. Tang and P. Wan, RSC Adv., 2014, 4(56), 29917–29924 RSC.
- Y.-S. Wang, F. Yang, Z.-H. Liu, L. Yuan and G. Li, Catal. Commun., 2015, 67, 49–53 CrossRef CAS.
- P. Parpot, A. P. Bettencourt, A. M. Carvalho and E. M. Belgsir, J. Appl. Electrochem., 2000, 30(6), 727–731 CrossRef CAS.
- R. Tolba, M. Tian, J. Wen, Z.-H. Jiang and A. Chen, J. Electroanal. Chem., 2010, 649(1–2), 9–15 CrossRef CAS.
- I. Bosque, G. Magallanes, M. Rigoulet, M. D. Karkas and C. R. J. Stephenson, ACS Cent. Sci., 2017, 3(6), 621–628 CrossRef CAS PubMed.
- M. Tian, J. Wen, D. MacDonald, R. M. Asmussen and A. Chen, Electrochem. Commun., 2010, 12(4), 527–530 CrossRef CAS.
- P. Dabo, A. Cyr, J. Lessard, L. Brossard and H. Ménard, Can. J. Chem., 1999, 77, 1225–1229 CAS.
- Y. Zhou, G. E. Klinger, E. L. Hegg, C. M. Saffron and J. E. Jackson, J. Am. Chem. Soc., 2020, 142(8), 4037–4050 CrossRef CAS PubMed.
- G. E. Klinger, Y. Zhou, P. Hao, J. Robbins, J. M. Aquilina, J. E. Jackson and E. L. Hegg, ChemSusChem, 2019, 12(21), 4775–4779 CrossRef CAS PubMed.
- G. E. Klinger, Y. Zhou, J. A. Foote, A. M. Wester, Y. Cui, M. Alherech, S. S. Stahl, J. E. Jackson and E. L. Hegg, ChemSusChem, 2020, 13(17), 4394–4399 CrossRef CAS PubMed.
- S. Antonello, R. Benassi, G. Gavioli, F. Taddei and F. Maran, J. Am. Chem. Soc., 2002, 124(25), 7529–7538 CrossRef CAS PubMed.
- K. Nakahara, K. Naba, T. Saitoh, T. Sugai, R. Obata, S. Nishiyama, Y. Einaga and T. Yamamoto, ChemElectroChem, 2019, 6(16), 4153–4157 CrossRef CAS.
- A. Fava, G. Reichenbach and U. Peron, J. Am. Chem. Soc., 1967, 89(25), 6696–6700 CrossRef CAS.
- S. P. Mezyk and D. A. Armstrong, J. Chem. Soc., Perkin Trans. 2, 1999,(7), 1411–1420 RSC.
- H. Wariishi, K. Valli, V. Renganathan and M. H. Gold, J. Biol. Chem., 1989, 264(24), 14185–14191 CrossRef CAS.
- X. Zhang, N. Zhang, H. P. Schuchmann and C. von Sonntag, J. Phys. Chem., 1994, 98, 6541–6547 CrossRef CAS.
- M. P. Bertrand, Org. Prep. Proced. Int., 1994, 26(3), 257–290 CrossRef CAS.
- C. P. Andrieux, M. Robert and J. M. Saveant, J. Am. Chem. Soc., 1995, 117, 9340–9346 CrossRef CAS.
- C. H. Lam, C. B. Lowe, Z. Li, K. N. Longe, J. T. Rayburn, M. A. Caldwell, C. E. Houdek, J. B. Maguire, C. M. Saffron, D. J. Miller and J. E. Jackson, Green Chem., 2015, 17(1), 601–609 RSC.
- A. Bhalla, N. Bansal, R. J. Stoklosa, M. Fountain, J. Ralph, D. B. Hodge and E. L. Hegg, Biotechnol. Biofuels, 2016, 9, 34 CrossRef.
- X. Du, W. Liu, Z. Zhang, A. Mulyadi, A. Brittain, J. Gong and Y. Deng, ChemSusChem, 2017, 10(5), 847–854 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc03597a |
| This journal is © The Royal Society of Chemistry 2021 |