
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Degradation of perfluorooctane sulfonate via in situ electro-generated ferrate and permanganate oxidants in NOM-rich source waters†
Sean T.
McBeath
 * and
Nigel J. D.
Graham
* and
Nigel J. D.
Graham
Department of Civil & Environmental Engineering, Imperial College London, London SW7 2AZ, UK. E-mail: s.mcbeath17@imperial.ac.uk
First published on 5th August 2021
Abstract
A novel process involving the in situ electrochemical generation of ferrate and permanganate oxidants, in circumneutral conditions, from low concentration aqueous iron (Fe2+) and manganese (Mn2+), is investigated for the treatment of the ubiquitous and highly recalcitrant micro-pollutant, perfluorooctane sulfonate (PFOS). The present study investigated the efficacy of both electro-oxidation (EO), and the simultaneous EO and ferrate/permanganate generation and oxidation, of PFOS as a potential drinking water treatment technology. While permanganate was shown to have little effect on PFOS removal, significantly increased degradation was observed when EO was coupled with ferrate generation and oxidation, significantly exceeding that of solely EO. From an initial concentration of 0.80 μM, final PFOS concentrations of 0.53 (±0.004), 0.43 (±0.01) and 0.27 (±0.01) μM were yielded during 10, 40 and 80 mA cm−2 electrolysis and an initial Fe2+ = 179 μM. In general, PFOS degradation rates increased with both increasing current density and initial Fe2+ concentration. Degradation was observed to follow mixed zero- and pseudo-first-order reaction kinetics for both the EO and simultaneous EO and ferrate oxidation. Finally, PFOS oxidation was not inhibited by the presence of low and high molecular weight organic scavenger species, and high concentrations of natural organic matter (NOM) improved PFOS removal due to hydrophobic interaction. Reduced ferrate species were also observed to increase NOM removal after electrolysis, by iron coagulant formation and subsequent flocculation.
Water impactElectrochemical water treatment technologies present a promising alternative to conventional processes, particularly for small, remote and/or decentralised applications. Using high oxygen overpotential electrodes, an advanced electro-oxidation process facilitates a novel pathway for in situ ferrate and permanganate (“green” oxidants) production from background Fe/Mn in neutral pH conditions, leading to the degradation of PFOS contaminants by simultaneous electro- and chemical oxidation processes. |
1. Introduction
Per- and polyfluoroalkyl substances (PFAS) are a wide class of synthetically made chemicals, characterised by an aliphatic carbon backbone with fluorine atom substitutions on the alkyl chain in place of hydrogen. Perfluorooctane sulfonate (PFOS) is a particularly ubiquitous PFAS species, consisting of an eight carbon chain and hydrophilic sulfonate functional group, previously used in a number of applications including as a mist suppressant agent for carcinogenic aerosols,1 aqueous film-forming foams, surfactants and lubricants,2 as well as various household products such as carpet, clothing and non-stick cookware.3 PFOS is also known to be an especially stable PFAS species due to its long carbon chain and strong hydrophobicity when compared to other common PFAS species like perfluorooctanoic acid (PFOA).4,5 The prevalence of PFOS in natural waters varies largely and is dependent on contaminant source location. In a worldwide survey conducted in 15 countries and 41 cities during 2004–2010, in both industrialised and non-industrialised areas, PFOS levels ranged from trace to 70.1 ng L−1.6 A United States Environmental Protection Agency (US EPA) survey found PFOS contamination ranging from 40–43 ng L−1 as an average in 50 US states in contaminated waters, with individual levels ranging from trace to over 1800 ng L−1.7 Other studies have found PFOS at concentrations of 132 ng L−1 in US surface waters,8 and as high as 0.011–2270 μg L−1 in waters near the release of aqueous film fire-fighting foam.9In general, PFAS are known to be chemically stable and resistant to biodegradation and conventional treatment processes,10–13 leading to the accumulation of perfluorinated compounds in the environment, as well as in wildlife and humans.13,14 Moreover, their toxic nature presents many harmful health effects to humans,15–17 leading many governments to impose health-based notification levels.10 Due to their widespread contamination and recalcitrant nature, some communities may be particularly susceptible to PFOS contamination. In particular, small and remote communities may be especially vulnerable owing to difficulties in implementing sophisticated centralised treatment approaches, due to the lack of economy-of-scale, availability of trained operators, or limitations in supplying the necessary process chemicals.18
The development of efficient decentralised water treatment options has increased in recent years, in part due to their potential applicability for vulnerable communities like small and remote systems.19–21 In particular, electrochemical processes are of especial interest due to their favorable economy-of-scale and ability to eliminate chemical supply chains associated with conventional water treatment unit operations like coagulation and disinfection, with technologies such as electrocoagulation and electrochemical oxidation (electro-oxidation or EO), respectively. The latter technology has the capability of eliminating the chemical supply chain associated with conventional oxidation processes such as those based on chlorine, hydrogen peroxide, permanganate and ferrate. Additionally, through the advancement of powerful high oxygen overpotential electrode materials such as boron-doped diamond (BDD), EO processes have become exceedingly more effective. BDD is reported as having the greatest electrical potential range of water stability (−1.25–2.3 VSHE),22,23 which minimises the anodic oxygen evolution reaction (OER) ([R1]), while facilitating the generation of powerful reactive oxygen species, principally hydroxyl radicals (˙OH) ([R2]):
| 2H2O → O2 + 4H+ + 4e− | ([R1]) |
| BDD + H2O → BDD(˙OH) + H+ + e− | ([R2]) |
The use of BDD for electro-oxidation has been previously explored for a large number of organic micropollutants, including cyclic, heterocyclic and aliphatic compounds (such as pesticides, dyes, pharmaceuticals, phenols, aliphatic acids, humic substances and perfluorinated acids),24–28 including PFOS.29–33 In general, researchers have found that PFOS degradation via BDD-EO is a mass transfer limited process, which proceeds primarily through direct oxidation at the electrode surface,31,33 rather than mediated by ˙OH oxidation. Moreover, batch electrolysis experiments showed that PFOS degradation was unaffected when the ˙OH scavenger tert-butanol was present,32 further indicating that the PFOS oxidation mechanism proceeds by direct electron transfer, as well as suggesting the effectiveness of BDD-EO for PFOS degradation in the presence of low molecular weight (MW) organic scavenger species. More recently, researchers have found that boron doping levels in BDD electrodes did not effect PFOS degradation,29 however modified BDD materials with SnO2–F and BDD/SnO2–F anodes provided improved PFOS degradation by increasing the oxygen evolution potential ([R1]) during electrolysis.30
Because the main mechanism of PFOS degradation during BBD EO has been associated with direct oxidation at the electrode surface, this renders the process a point-source application for treatment (i.e., limited to the electrode surface). However, a potentially powerful aspect of the BDD-EO process, yet to be investigated for PFOS degradation, is the simultaneous direct oxidation and synthesis of in situ chemical oxidants for downstream treatment.34 The generation of in situ oxidants via BDD anodic oxidation has been previously investigated for chemicals such as persulfate (E0 = +1.96 VSHE),35 peroxodiphosphate (E0 = +2.07 VSHE)36 and various chlorine species.37 More recently, the circumneutral generation of powerful iron and manganese-based oxidants, namely ferrate(VI) (Fe6+, FeO42−)38,39 and permanganate(VII) (Mn7+, MnO4−),40 from low concentrations of aqueous Fe2+ and Mn2+ typical of groundwater sources, respectively, have been reported. Both ferrate and permanganate are known to have high redox potentials (E0Fe(VI) = +2.2 VSHE, E0Mn(VII) = +1.5 VSHE), with non-toxic reduction products (e.g., hydrolysis species of Fe3+ and Mn3+), whereby the former has been observed to function as an effective coagulant chemical for further treatment.41 Moreover, ferrate (dosed as potassium ferrate) and its reduced species Fe(V) and Fe(IV) have previously been observed to oxidise PFOS in neutral and alkaline pH conditions, with maximum removals of 34%.42 Furthermore, a synergistic effect with ferrate and zero-valent iron for PFOS degradation has recently been reported in the literature.43 Permanganate has received limited attention as a potential oxidant for PFOS degradation, but has previously been observed to degrade PFOS in high temperature (65 °C) and acidic (pH 4.2) conditions.44 In addition to the largely unknown simultaneous effects of electro-generated oxidant species like ferrate and permanganate on PFOS degradation, the oxidation scavenging effect in the presence of high concentrations of both low and high MW fractions of dissolved organic species, particularly natural organic matter (NOM), has not been thoroughly investigated to date.
The current study has investigated the simultaneous EO of PFOS and generation of ferrate and permanganate from low oxidation state, low concentration iron (Fe2+) and manganese (Mn2+), for enhanced PFOS removal. The study presents a novel reaction pathway for both ferrate and permanganate generation, in circumneutral pH conditions, as well as the first demonstration of such a process for applications concerning the abatement of PFOS in model surface and groundwater sources. The separate effects on PFOS oxidation by EO, EO–ferrate and EO–permanganate systems were investigated. Finally, the scavenging effects of a representative low MW hydrophilic NOM analogue species (resorcinol), as well as real water NOM with a large range of MW fractions, were also considered.
2. Materials & methods
2.1 Electrochemical reactor setup and procedures
Electro-oxidation experiments were conducted using a parallel plate, batch-recycle, flow through reactor. The anolyte water temperature was held constant throughout electrolysis at 21.0 ± 0.8 °C using thermo-regulated glass and an applied thermal control (ATC) Kt recirculating chiller. A single-drive Watson-Marlow 505S peristaltic pump with dual-heads controlled both the anolyte (1000 mL) and catholyte (1000 mL) flow rate at 355 mL min−1. The anolyte solution was stirred throughout electrolysis with a magnetic stirrer at an approximate rate of 60–120 rpm. A thin-film (2–3 μm) monocrystalline BDD anode (NeoCoat®) on a 1 mm silicon substrate, prepared by a chemical vapour deposition process, was used for all experiments. The cathode was an austenitic, face centred cubic crystal stainless steel 304 alloy. Both the anode and cathode had dimensions of 50 × 50 × 1 mm. The anolyte and catholyte were separated by a 10 mm inter-electrode gap and a Nafion-324 perfluorinated proton exchange membrane (Sigma-Aldrich), to eliminate the electrochemical reduction of ferrate and permanganate at the cathode surface. The custom electrochemical cell was fabricated with electrically inert polyvinyl chloride material, with stainless steel hardware and braces for assembly. Inert rubber was used for the o-ring and gaskets in the assembled cell. A Keithley 2460-EC Electrochemistry Lab System potentiostat was used for current/potential control and measurements.2.2 Synthetic water
A phosphate buffer (pH = 7.1, 0.1 M) composed of ultrapure reverse osmosis (RO) water, NaH2PO4 and Na2HPO4 (Fisher Scientific) were used for all experiments. The desired initial Fe2+ and Mn2+ concentrations were attained by addition of FeCl2 (Acros Organics) and MnCl2 (Acros Organics), respectively. Initial Fe2+ concentrations of 179, 54, 18 and 9 μM (10, 3, 1 and 0.5 mg L−1, respectively) and Mn2+ concentrations of 182 and 55 μM (10 and 3 mg L−1) were used for the anolyte solution during ferrate and permanganate tests, respectively. Stock PFOS (Aldrich, ≥98.0%) solutions were prepared at ∼25 mg L−1 in ultrapure RO water and stored in the refrigerator at 4 °C. An initial PFOS concentration of 400 μg L−1 (0.8 μM) was used throughout the study, except for experiments investigating the initial PFOS concentration effect on degradation, where an initial concentration of 4.6 mg L−1 (9.2 μM) was used. Selected conditions were investigated using resorcinol (analytical reagent grade, Fisher Scientific) as a model NOM surrogate, representative of low MW oxidation by-product precursor species. Resorcinol stock solutions were prepared at 20 mg L−1 and stored in the refrigerator at 4 °C to avoid degradation due to increased temperature and light exposure. Finally, the synthetic ‘real’ water experiments were conducted using a RO isolated natural organic matter, collected from the raw water source at the Chellow Heights Water Treatment Plant (United Kingdom). The NOM extract was dissolved in high purity water to reach an initial dissolved organic concentration (DOC) of 3.00 mg L−1, with a corresponding UV-absorbance at 254 nm (UV254) of 0.100 cm−1.2.3 Analytical methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 M ratio, producing a light-absorbing radical cation (ABTS˙+) with a visible UV-absorption maximum at 415 nm. The ABTS method is a sensitive method for ferrate and permanganate quantification, with levels of detection and quantification of 0.1 and 0.4 μM, and 0.01 and 0.03 μM, respectively. Ferrate standards and samples were analysed using a Shimadzu UV-4201PC spectrophotometer. Ferrate and permanganate standards were prepared using potassium ferrate (>99%) (Guangzhou Kexing Chemicals Ltd.) and potassium permanganate (>99%) (Alfa Aesar) in a concentration range of 0–4 mg L−1. Ferrate and permanganate concentrations were then determined from experimental measurements, as follows:
:1 M ratio, producing a light-absorbing radical cation (ABTS˙+) with a visible UV-absorption maximum at 415 nm. The ABTS method is a sensitive method for ferrate and permanganate quantification, with levels of detection and quantification of 0.1 and 0.4 μM, and 0.01 and 0.03 μM, respectively. Ferrate standards and samples were analysed using a Shimadzu UV-4201PC spectrophotometer. Ferrate and permanganate standards were prepared using potassium ferrate (>99%) (Guangzhou Kexing Chemicals Ltd.) and potassium permanganate (>99%) (Alfa Aesar) in a concentration range of 0–4 mg L−1. Ferrate and permanganate concentrations were then determined from experimental measurements, as follows: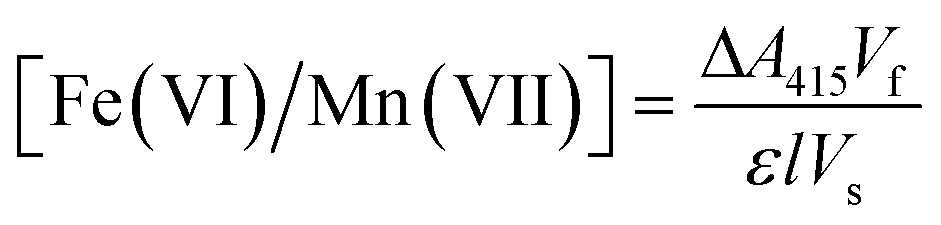 | (1) |
000 M−1 cm−1) and permanganate (140030 M−1 cm−1) standards, the cell path length (1 cm), and the volume of the sample extracted from the anolyte (5–15 mL), respectively. Samples were added to the ABTS solution and immediately analyzed spectrophotometrically to avoid both the degradation of ferrate and permanganate (prior to a subsequent addition to ABTS) and self-decay of ABTS.
3. Results and discussion
3.1 Electrochemical ferrate and permanganate synthesis
Previous studies have demonstrated the circumneutral electrochemical generation of ferrate38 and permanganate40 from low initial concentrations of Fe2+ and Mn2+, respectively. Through the use of radical scavengers and cyclic voltammetry, these previous studies demonstrated that ferrate and permanganate generation was attributed to both ˙OH mediated and direct oxidation at the electrode surface. In the current study, the same initial Mn2+ concentrations (182 and 55 μM) and Fe2+ concentrations (54, 18 and 9 μM), at three current density conditions of 10, 40 and 80 mA cm−2 over 120 minutes of electrolysis, were used for the PFOS degradation experiments; however, an additional Fe2+ initial concentration of 179 μM was investigated. For both ferrate and permanganate synthesis processes, the operating cell potentials were relatively stable throughout the entirety of electrolysis (120 minutes), regardless of the initial iron or manganese concentration, but were dependent on the current density: 6.0–6.6 V, 14.1–14.7 and 18.0–18.6 V during 10, 40 and 80 mA cm−2 operations.It was previously observed that no significant differences in ferrate synthesis were observed during electrolysis at the three current density conditions, despite increased generation of ˙OH as the current density increased, as described elsewhere.38,40 A mathematical model was developed to describe ferrate and permanganate synthesis under mass transport limitations, which agreed well with the experimentally derived data, yielding mass transfer coefficients between 0.24–2.8 × 10−8 m s−1, depending on current density and initial iron/manganese concentration.38,40 These results demonstrated that the rate-limiting step under these low iron concentration conditions was the diffusion of Fe2+ to the BDD electrode surface from the bulk water solution, through the Nernst diffusion layer. After 120 minutes of electrolysis, 3.15 (±0.10), 0.90 (±0.09) and 0.40 (±0.06) μM of ferrate was synthesised under 54, 18 and 9 μM initial Fe2+ conditions, respectively. Similarly, no significant effect on permanganate synthesis was observed due to current density variations during electrolysis in the presence of Mn02+ = 55 μM, yielding a maximum concentration of 0.09 μM after 120 minutes of electrolysis. When the initial manganese concentration was increased to 182 μM, the maximum permanganate concentration generated was 0.926 (±0.01) μM for 80 and 40 mA cm−2 operations.
When the initial Fe2+ concentration was increased to 179 μM, the process was no longer diffusion limited, whereby ferrate generation was observed to be affected by the operating current density. Like previous studies investigating the circumneutral electro-synthesis of ferrate using BDD electrodes and high concentrations of Fe3+ (3–30 mM),47,48 an increase in current density correlated with an increase in ferrate synthesis, reaching maximum concentrations of 28.7 (±4.5), 18.4 (±0.7) and 13.9 (±0.5) μM after 120 minutes during 80, 40 and 10 mA cm−2 electrolysis, respectively (Fig. 1). These levels of ferrate generation at 80, 40 and 10 mA cm−2 correspond to a yield of 16.0, 10.3 and 7.8%, and faradaic efficiencies of 0.069, 0.089 and 0.268%, respectively.
 | ||
| Fig. 1 Variation of electrochemical ferrate generation with current density at initial Fe2+ = 179 μM (pH = 7, T = 21.0 ± 0.8 °C). | ||
The majority of the initial aqueous iron and manganese was not converted to ferrate and permanganate, respectively, with yields of only 4.7–16.0% and 0.01–0.51%, depending on current density and initial iron and manganese concentrations, respectively. The resulting faradaic efficiencies for ferrate and permanganate generation were 2.5–0.27 × 10−3% and 0.4–3.1 × 10−4%, respectively. Although these efficiencies are low, they have been shown previously to be sufficient to produce an increased oxidation of atrazine.49,50 Moreover, strategies to improve the faradaic efficiency of both ferrate and permanganate synthesis have been previously reported and determined via the aforementioned mathematical model, which include increasing reactor residence time, decreasing the inter-electrode gap or increasing the anode area.38,40 For example, if the electrochemical cell was redesigned to accommodate a 100 × 300 mm and a 5 mm inter-electrode gap, ferrate synthesis would double, in half of the time.38
3.2 PFOS degradation
The degradation of PFOS was investigated during EO with, and without, iron and manganese addition, to determine the effect of ferrate and permanganate during the simultaneous EO and oxidant generation process. Experiments were conducted using the same current densities and iron/manganese conditions used in the previously discussed ferrate and permanganate synthesis study. The EO of PFOS has generally been shown to be slow due to the high electronegativity of fluorine atoms bonded to the carbon backbone. Dehalogenation of PFOS by electrochemical reduction at the cathode has been previously observed to increase the susceptibility of subsequent oxidation (at the anode),51–57 however, in this study the objective was to understand the oxidation effect of EO, EO–Fe(VI) and EO–Mn(VII) only, and limit reactions to the anolyte (hence the separated cell).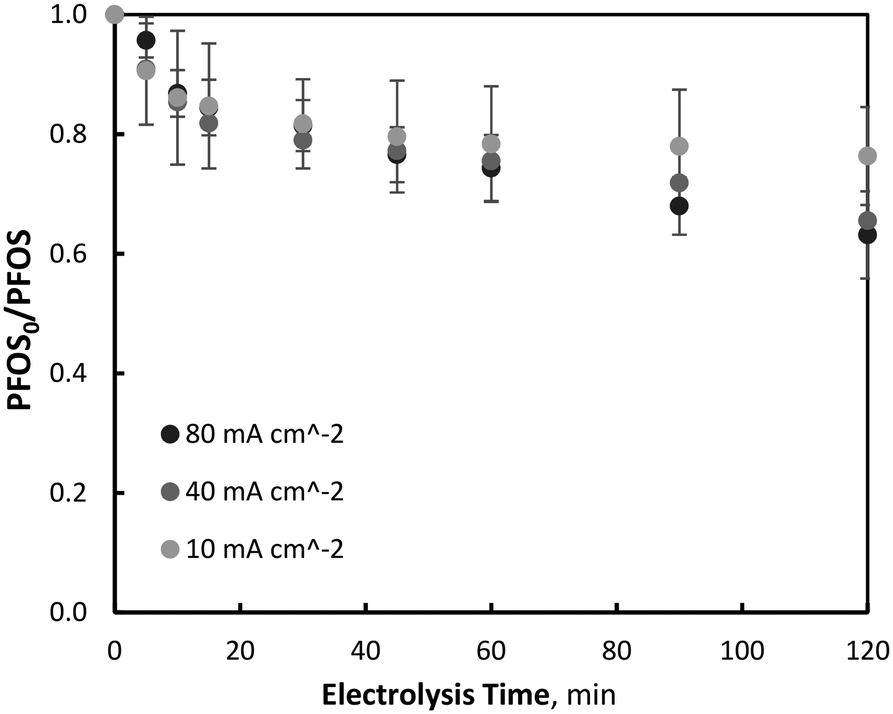 | ||
| Fig. 2 Variation of EO of PFOS with current density (PFOS0 = 400 μg L−1, pH = 7, T = 21.0 ± 0.8 °C). | ||
It was observed that PFOS degradation did not follow pseudo-first-order reaction kinetics throughout the entirety of electrolysis, particularly within the first 15–20 minutes of electrolysis. Beyond this initial period of degradation (15–20 minutes) which was characterised by a linear relationship between PFOS and electrolysis time (zero-order kinetics), pollutant degradation began to follow a first-order reaction kinetic relationship (ln(PFOS0/PFOSt)) with time (Fig. 3). Further tests were repeated with an increased initial PFOS concentration of 4.60 mg L−1, an order of magnitude greater than that used for the previous experiments, to determine whether diffusion limitations were associated with the non-first-order behavior. At the increased initial concentration, after 120 minutes of electrolysis and 10, 40 and 80 mA cm−2 current density operations, PFOS was observed to decrease by 22.0 (±2.0), 32.6 (±1.4) and 35.3 (±2.4) % respectively, which was within the standard deviation of reductions observed at low initial PFOS tests, indicating no significant differences in degradation at the two initial PFOS concentrations. Similar results have been observed previously by other researchers,32 and correspond well to the linear region of the Langmuir–Hinshelwood model for surface chemical reactions.58 Moreover, PFOS degradation did not differ significantly between the three current densities investigated during the initial electrolysis period, suggesting the concentration of ˙OH or available direct oxidation sites on the electrode surface did not affect PFOS degradation early in the process. Because this initial period (0–20 minutes) of degradation is linear with time and not dependent on PFOS or oxidant (˙OH or direct electrode surface sites) concentration, it is consistent with zero-order kinetics, a phenomenon observed in previous PFOS degradation by BDD-EO studies during non-mass transport limited conditions using a rotating disk electrode.31 This mixed reaction kinetic system has been observed previously during BDD-EO with micro-pollutants like sulfamethoxazole (SMX)59 and the abatement of chemical oxygen demand,60 as well as previously described in a kinetic model.61 After 15 minutes of electrolysis characterised by a zero-order reaction rate of k0 = 0.0088 μM min−1, degradation followed pseudo-first order kinetics with reaction rate constants of k1 = 0.0028, 0.0020 and 0.0007 min−1 during 80, 40 and 10 mA cm−2 operations, respectively. An overview of the mixed-order reaction kinetic data is summarised in Table 1. No clear PFOS degradation pathway could be determined or confidently proposed based on the oxidation by-products identified throughout electrolysis, however, the predominant species formed throughout EO were: pentafluoroethane sulfonic acid, perfluoroheptane sulfonic acid (PFHpS), trifluoroacetic acid (TFA), perfluoropentane sulfonic acid (PFPeS), perfluorohexane sulfonic acid (PFHxS), pentafluorobutanesulfonic acid (PFBS) and pentafluoropropionic acid (PFPrA). More information on oxidation by-product formation is provided in the Supplemental Materials.
 | ||
| Fig. 3 Pseudo-first-order degradation analysis of PFOS during EO operations (shaded area indicating zero-first-order kinetic behavior). The corresponding reaction order and coefficient of determination for each EO operation is summarised in Table 1. | ||
| [Fe2+] | i | Reaction type | Zero-order | Pseudo-first-order | ||||
|---|---|---|---|---|---|---|---|---|
| μM | mA cm−2 | k 0, uM min−1 | R 2 | Time, min | k 1, min−1 | R 2 | Time, min | |
| 0.0 | 10 | Mixed | 0.0088 | 0.96 | 0–15 | 0.0007 | 0.99 | 15–120 |
| 40 | 0.0020 | 0.97 | ||||||
| 80 | 0.0028 | 0.99 | ||||||
| 9 | 10 | Mixed | 0.0088 | 0.95 | 0–15 | 0.0013 | 0.96 | 15–120 |
| 40 | 0.0026 | 0.99 | ||||||
| 80 | 0.0053 | 0.98 | ||||||
| 18 | 10 | Mixed | 0.0070 | 0.98 | 0–15 | 0.0018 | 0.97 | 15–120 |
| 40 | 0.0031 | 0.97 | ||||||
| 80 | 0.0061 | 0.98 | ||||||
| 54 | 10 | Mixed | 0.0117 | 1.00 | 0–10 | 0.0023 | 0.97 | 10–120 |
| 40 | 0.0054 | 0.99 | ||||||
| 80 | First | N/A | 0.0086 | 1.00 | 0–120 | |||
| 179 | 10 | Mixed | 0.0125 | 1.00 | 0–10 | 0.0020 | 0.99 | 10–120 |
| 40 | 0.0046 | 0.98 | ||||||
| 80 | First | N/A | 0.0095 | 0.99 | 0–120 | |||
 | ||
| Fig. 4 Pseudo-first-order degradation analysis of PFOS during simultaneous EO and ferrate synthesis operations (PFOS0 = 400 μg L−1, pH = 7, T = 21.0 ± 0.8 °C). | ||
To better understand the greatly increased degradation of PFOS when even a small amount of iron is present in the water matrix, the degradation due to ferrate, independently of EO, was investigated by first electrochemically generating ferrate oxidants and then mixing with PFOS-containing waters. Electrolysis proceeded with an initial Fe2+ concentration of 179 μM and 80 mA cm−2, reaching an initial ferrate concentration of 17.3 μM. A PFOS stock solution was then added to the electrochemically generated ferrate solution to reach an initial PFOS concentration of 0.80 μM. PFOS and ferrate concentrations were monitored over 120 minutes of mixing in a glass beaker, without recirculation through the electrochemical reactor, to understand the independent effect of ferrate degradation on PFOS (i.e., without EO occurring). PFOS was observed to degrade quickly, reaching a final concentration of 0.58 (±0.02) μM within 30 minutes of stirring (Fig. S3†).
From the results, a second-order reaction rate constant of 33.8 (±1.4) M−1 s−1 was observed between the electrochemically-derived ferrate and PFOS (Fig. S4†). From the EO-only and ferrate-only degradation results, a greater PFOS degradation was observed during the simultaneous process (coupled EO and electro-ferrate degradation) compared to that which would be predicted from the yielded first-order reaction rate of the EO system and the second-order reaction rate of the ferrate system. While a 66% reduction in PFOS was observed during the simultaneous EO–Fe(VI) process, a theoretical reduction of 63% would be expected from the yielded k1 and k2. This increased degradation can be associated with a few potential phenomena, including the electrochemical regeneration of ferrate from reduced iron species (i.e., ferrate reduction byproducts, Fe(III)–Fe(V)), as well as oxidation by lower cationic state iron species including Fe(V) and Fe(IV). Previous researchers have also observed efficient degradation of PFOS from lower oxidation state ferrate species, particularly Fe(V) and Fe(IV) species.42 Moreover, the presence of iron in the form of Fe2+ and ZVI has been observed to have synergistic effects on PFOS removal during UV irradiation62 and ferrate oxidation,43 respectively. Although a benefit of ferrate production exists for the overall degradation of PFOS, as the initial iron concentrations increases, both direct oxidation sites and adsorbed ˙OH will be more readily scavenged (by iron cations). This scavenging effect may decrease the availability of oxidation sites (direct surface sites and ˙OH) which may be involved in the degradation of PFOS. However, even at the lowest initial Fe2+ concentration of 9 μM, significantly increased removal of PFOS was observed when compared to the EO-only process.
3.3 Scavenging effect of NOM
The effect of NOM on the EO and ferrate oxidation processes was investigated with the addition of a NOM surrogate, namely, resorcinol (section 3.3.1), and real surface water NOM (section 3.3.2). For the tests with resorcinol, the compound was added to the water matrix at a concentration of 9.1 μM, an order of magnitude greater than PFOS (0.80 μM). For the real water NOM tests, a RO isolated NOM was added to achieve an initial DOC concentration and UV254 absorbance of 3.00 mg L−1 and 0.100 cm−1, respectively.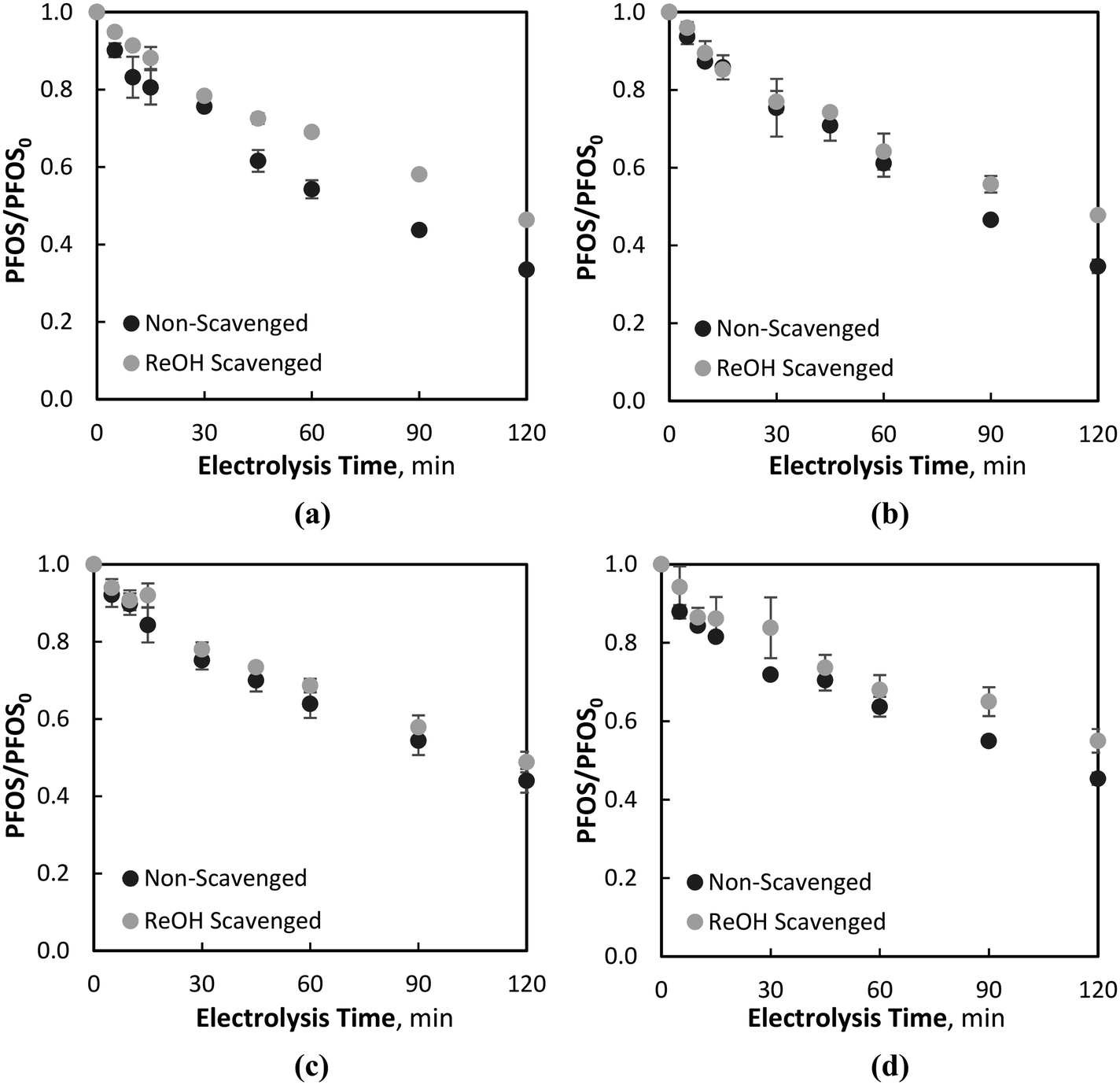 | ||
| Fig. 5 PFOS degradation at 80 mA cm−2 with and without the resorcinol (ReOH) scavenger at: (a) Fe2+ = 179 μM, (b) Fe2+ = 54 μM, (c) Fe2+ = 18 μM, and (d) Fe2+ = 9 μM. | ||
This phenomenon is further evidenced during electrolysis in waters containing no iron (Fe2+ = 0 μM), where no significant differences in PFOS degradation were observed when the resorcinol scavenger was added to the water matrix. Resorcinol has been found previously to be primarily degraded via ˙OH oxidation during BDD-EO66 and was observed to be quickly degraded in control studies containing no PFOS or Fe2+, yielding a pseudo-first-order rate constant of 0.184 min−1. Owing to the high degradation of resorcinol via EO, and relatively low degradation of PFOS in the same system, with and without the presence of the resorcinol scavenger (i.e., no scavenging effect due to resorcinol), it is believed that PFOS degradation is primarily via direct EO, and not ˙OH attack, as previously observed in systems containing the ˙OH scavenger tert-butanol. The recalcitrant nature of PFOS with respect to ˙OH degradation has been previously reported,12,33,67–69 which may indicate no adverse effect on PFOS degradation due to the presence of low MW organic scavengers, which are predominantly oxidised by ˙OH attack.24 During similar studies investigating the degradation of atrazine, a pollutant found to be readily degraded by adsorbed ˙OH on the BDD surface, the presence of resorcinol was found to inhibit the degradation of the target pollutant for both the EO and EO–ferrate systems.50 Moreover, even with ferrate being highly scavenged by the resorcinol, PFOS removal was still observed to be significantly greater for all initial Fe2+ concentrations compared to an EO-only (i.e., Fe2+ = 0 μM) process (Fig. 5). In general, the difference between PFOS degradation during resorcinol scavenged and non-scavenged processes decreased during electrolysis with the lower initial Fe+2 concentrations, highlighting the role of PFOS degradation via ferrate oxidation. While the differences were small during Fe02+ = 18 and 9 μM electrolysis conditions, PFOS degradations during scavenged and non-scavenged electrolysis were still significant.
The tests showed that PFOS removal was not significantly different between all conditions tested, whereby a 67.3 (±0.7)% reduction was observed after 120 minutes of electrolysis, indicating no enhanced treatment due to ferrate production (Fig. S6†). The total organic carbon content was also substantially mineralised, with an average reduction in DOC of 70 (±3) % after 120 minutes of electrolysis (Fig. S7†). Unlike the previous resorcinol conditions, no significant difference in PFOS reduction was observed during EO–ferrate operations in the presence of NOM compared to the absence of NOM, suggesting no ferrate scavenging. However, it is unlikely that no scavenging occurred, as electrochemically generated ferrate oxidation has previously been observed to be adversely affected by NOM.71 Interestingly, PFOS removal during the EO-only process (i.e., Fe2+ = 0 μM) was observed to exceed that which was observed during non-NOM experiments, indicating greater PFOS abatement conditions in a high NOM containing waters. This is likely due to hydrophobic interaction between PFOS and NOM, which has previously been observed to facilitate physical adsorption.72–74 With PFOS–NOM hydrophobic interactions, the transport of PFOS to the site of oxidation at the electrode surface may be increased due to the greater diffusive flux of high concentration NOM to the electrode.
To investigate this further, NOM experiments were repeated at a lower concentration, corresponding to an initial DOC and UV254 of 1.00 mg L−1 and 0.044 cm−1, respectively. Consistent with the previous results, a decrease in PFOS removal occurred during EO, corresponding to the lower NOM concentration. A clear trend of increased PFOS removal was observed with an increase in initial NOM concentration, as seen in Fig. 6. With a decrease in NOM, less PFOS absorption by hydrophobic interaction would be expected and thereby decreased PFOS flux to the electrode surface. Another phenomenon that may account for the increased removal of PFOS during NOM-scavenged experiments is the increased stability of ferrate in the presence of NOM. While ferrate decomposition can increase due to interactions with its reduction by-products (i.e., Fe3+ oxide and hydroxide species), some researchers have observed the complexation of dissolved organic matter with Fe3+ (from reduced ferrate), which consequently increases the stability of ferrate in the presence of the NOM substances. As a result of this increased stability, ferrate was able to interact with target pollutant species for a longer period compared to water matrices not containing dissolved organic matter.75 Moreover, recent studies have suggested that NOM may function as an electron shuttle in the presence of ferrate, whereby oxidized mediators such as phenoxy radicals or benzoquinone-like compounds are generated when the phenolic moieties are oxidized by ferrate, which was observed to improve the degradation of the organic pollutant 4-tert-butylphenol.76
 | ||
| Fig. 6 PFOS removal during EO, with varying initial NOM concentrations. | ||
Furthermore, when iron is present in the water matrix, both low cationic state iron and chemically-reduced ferrate could provide additional PFOS removal due to coagulation effects, if the appropriate iron oxide and hydroxide species are formed, as previously observed.41,77 Therefore, the increased removal of PFOS in the presence of NOM may be associated with a coagulation effect, whereby NOM is removed via iron electrocoagulation, with PFOS absorbed to the formed flocs. To investigate this phenomenon, the same conditions used in the previous NOM experiments were applied (80 mA cm−2, Fe2+ = 54, 9 and 0 μM and DOC0 = 3 mg L−1). After 120 minutes of electrolysis, the electrical current was stopped, and the complete volume of anolyte water was pumped into the anolyte vessel and gently stirred for an additional 120 minutes to facilitate coagulation.
A further reduction in NOM was observed due to coagulation and flocculation effects after the initial DOC mineralisation during electrolysis, with reductions of 28.3 (±4.3) and 16.6 (±9.0) % for Fe02+ = 54 and 9 μM conditions, respectively (Fig. 7). Control studies were also conducted with no iron addition (i.e., Fe02+ = 0 μM) throughout electrolysis and coagulation. As expected, no effect of coagulation was observed, whereby all reductions in DOC occurred during EO.
 | ||
| Fig. 7 DOC reduction during EO (0–120 min) and coagulation/flocculation (120–240 min) with initial Fe2+ concentration of (a) 9 μM, and (b) 54 μM. | ||
The PFOS concentration was also monitored over the 120 minutes of mixing time, to determine whether any removal occurred due to coagulation effects. No additional PFOS removal occurred during this period, and even a small increase in concentration was yielded for both Fe02+ = 54 and 9 μM (Fig. S8†). Previous coagulation studies found that coagulant doses needed to exceed 10 mg L−1 for any significant reductions in PFOS to occur.78 Moreover, PFOS adsorption on NOM and removal by iron coagulation decreased with increasing humic and fulvic acid concentrations, due to both steric hindrance effects and competitive adsorption with NOM molecules, PFOS and the Fe3+ coagulants. This helps to explain why a slight increase in PFOS was observed over the 120 minutes of mixing, as NOM (composed of both humic and fulvic substances) adsorbed to Fe3+ coagulants to form flocs, which was evidenced by the decrease in DOC for both initial Fe2+ concentrations; PFOS would then be competing for adsorption sites with both coagulants and other flocs.
4. Conclusions
In this study, a novel electrochemical oxidation process for the simultaneous circumneutral generation of ferrate and permanganate and degradation of PFOS is presented. The electro-synthesis of ferrate and permanganate was observed to be mass transport limited, with oxidant synthesis increasing with initial Fe2+/Mn2+ concentration. At the highest current density (80 mA cm−2) and initial Fe2+ and Mn2+ concentrations of 179 and 182 μM, the corresponding concentrations of ferrate and permanganate generated were 28.7 and 0.926 μM, respectively.PFOS degradation was best described by mixed-order reaction kinetics, with the first 15–20 minutes of electrolysis following zero-order kinetics, and the remaining 20–120 minutes of treatment well described by pseudo-first-order kinetics with reaction rate constants of 0.0007–0.0028 min−1, as current increased from 10 to 80 mA cm−2. When Fe2+ was added to the water matrix, increased PFOS degradation was observed at all initial concentrations and current densities. Reaction rates increased with increasing initial iron concentration and current density, ranging from 0.0013–0.0095 min−1. Although no significant differences in PFOS degradation were observed during EO with initial concentrations of 4.60 mg L−1 and 400 μg L−1, a further decrease in initial PFOS concentration will be considered for future studies, to better reflect natural pollutant concentrations which are commonly found in surface and groundwater sources. No increased removal of PFOS was observed during simultaneous permanganate synthesis experiments, when compared to the EO-only process. In the presence of naturally extracted NOM, increased reductions in PFOS were observed, suggesting hydrophobic interactions with PFOS and NOM increasing the diffusive mass flux of the target contaminant to the electrode surface.
Given the promising results highlighted, the simultaneous EO and ferrate oxidation process presents a potentially suitable alternative technology for small, remote and/or decentralised treatment applications. While further scale-up tests would be required to demonstrate the technology's viability in practice, these bench-scale tests have demonstrated its potential suitability to treat recalcitrant organic pollutants like PFOS, even in highly challenging, high DOC water matrices.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to acknowledge the support of Imperial College London with a President's Scholarship for S. T. McBeath and the Natural Sciences and Engineering Research Council of Canada (NSERC) [PGSD3-516562-2018]. The authors would also like to thank Professor David Wilkinson (University of British Columbia) for his advisory role throughout the research.References
- M. Pilat and R. Pegnam, Particle emissions from chrome plating, Aerosol Sci. Technol., 2006, 40(8), 639–648 CrossRef CAS.
- C. A. Moody and J. A. Field, Perfluorinated surfactants and the environmental implications of their use in fire-fighting foams, Environ. Sci. Technol., 2000, 34(18), 3864–3870 CrossRef CAS.
- S. Fujii, C. Polprasert, S. Tanaka, N. P. H. Lien and Y. Qiu, New POPs in the water environment: Distribution, bioaccumulation and treatment of perfluorinated compounds - A review paper, J. Water Supply: Res. Technol.--AQUA, 2007, 56(5), 313–326 CrossRef CAS.
- S. Park, L. S. Lee, V. F. Medina, A. Zull and S. Waisner, Heat-activated persulfate oxidation of PFOA, 6:2 fluorotelomer sulfonate, and PFOS under conditions suitable for in-situ groundwater remediation, Chemosphere, 2016, 145, 376–383 CrossRef CAS PubMed.
- H. Hori, A. Yamamoto, E. Hayakawa, S. Taniyasu, N. Yamashita and S. Kutsuna, et al. Efficient decomposition of environmentally persistent perfluorocarboxylic acids by use of persulfate as a photochemical oxidant, Environ. Sci. Technol., 2005, 39(7), 2383–2388 CrossRef CAS PubMed.
- C. Kunacheva, S. Fujii, S. Tanaka, S. T. M. L. Seneviratne, N. P. H. Lien and M. Nozoe, et al. Worldwide surveys of perfluorooctane sulfonate (PFOS) and perfluorooctanoic acid (PFOA) in water environment in recent years, Water Sci. Technol., 2012, 66(12), 2764–2771 CrossRef CAS.
- U. K. Vedagiri, R. H. Anderson, H. M. Loso and C. M. Schwach, Ambient levels of PFOS and PFOA in multiple environmental media, Remediation, 2018, 28(2), 9–51 CrossRef.
- R. Qu, J. Liu, L. Wang and Z. Wang, The toxic effect and bioaccumulation in aquatic oligochaete Limnodrilus hoffmeisteri after combined exposure to cadmium and perfluorooctane sulfonate at different pH values, Chemosphere, 2016, 152, 496–502 CrossRef CAS PubMed.
- J. P. Giesy and K. Kannan, Perfluorochemical Surfactants in the Environment, Environ. Sci. Technol., 2002, 36(7), 147A–152A CrossRef PubMed.
- D. Lu, S. Sha, J. Luo, Z. Huang and X. Zhang Jackie, Treatment train approaches for the remediation of per- and polyfluoroalkyl substances (PFAS): A critical review, J. Hazard. Mater., 2020, 386(December 2019), 121963, DOI:10.1016/j.jhazmat.2019.121963.
- K. H. Kucharzyk, R. Darlington, M. Benotti, R. Deeb and E. Hawley, Novel treatment technologies for PFAS compounds: A critical review, J. Environ. Manage., 2017, 204, 757–764, DOI:10.1016/j.jenvman.2017.08.016.
- B. N. Nzeribe, M. Crimi, S. Mededovic Thagard and T. M. Holsen, Physico-Chemical Processes for the Treatment of Per- And Polyfluoroalkyl Substances (PFAS): A review, Crit. Rev. Environ. Sci. Technol., 2019, 49(10), 866–915 CrossRef.
- M. F. Rahman, S. Peldszus and W. B. Anderson, Behaviour and fate of perfluoroalkyl and polyfluoroalkyl substances (PFASs) in drinking water treatment: A review, Water Res., 2014, 50, 318–340, DOI:10.1016/j.watres.2013.10.045.
- F. Suja, B. K. Pramanik and S. M. Zain, Contamination, bioaccumulation and toxic effects of perfluorinated chemicals (PFCs) in the water environment: A review paper, Water Sci. Technol., 2009, 60(6), 1533–1544 CrossRef CAS PubMed.
- X. Shi, Y. Du, L. PKS, W. RSS and B. Zhou, Developmental toxicity and alteration of gene expression in zebrafish embryos exposed to PFOS, Toxicol. Appl. Pharmacol., 2008, 230(1), 23–32 CrossRef CAS.
- F. Chen, C. Wei, Q. Chen, J. Zhang, L. Wang and Z. Zhou, et al. Internal concentrations of perfluorobutane sulfonate (PFBS) comparable to those of perfluorooctane sulfonate (PFOS) induce reproductive toxicity in Caenorhabditis elegans, Ecotoxicol. Environ. Saf., 2018, 158, 223–229 CrossRef CAS.
- N. Yin, R. Yang, S. Liang, S. Liang, B. Hu and T. Ruan, et al. Evaluation of the early developmental neural toxicity of F-53B, as compared to PFOS, with an in vitro mouse stem cell differentiation model, Chemosphere, 2018, 204, 109–118 CrossRef CAS PubMed.
- G. L. Rupp, The challenges of installing innovative treatment in small water systems, J. Environ. Health, 2001, 64(1), 1–22 Search PubMed.
- H. Haider, R. Sadiq and S. Tesfamariam, Performance indicators for small- and medium-sized water supply systems: a review, Environ. Rev., 2014, 22, 1–40, DOI:10.1139/er-2013-0013.
- B. P. Chaplin, The Prospect of Electrochemical Technologies Advancing Worldwide Water Treatment, Acc. Chem. Res., 2019, 52(3), 596–604 CrossRef CAS PubMed.
- M. A. Hamouda, W. B. Anderson and P. M. Huck, Employing multi-criteria decision analysis to select sustainable point-of-use and point-of-entry water treatment systems, Water Supply., 2012, 12(5), 637–647 CrossRef.
- A. Kraft, Doped diamond: A compact review on a new, versatile electrode material, Int. J. Electrochem. Sci., 2007, 2, 355–385 CAS.
- H. B. Martin, A. Argoitia, U. Landau, A. B. Anderson and J. C. Angus, Hydrogen and oxygen evolution on boron-doped diamond electrodes, J. Electrochem. Soc., 1996, 143(6), 133–136 CrossRef.
- S. T. McBeath, D. P. Wilkinson and N. J. D. Graham, Application of Boron-Doped Diamond Electrodes for the Anodic Oxidation of Pesticide Micropollutants in a Water Treatment Process: A Critical Review, Environ. Sci.: Water Res. Technol., 2019, 5, 2090–2107 RSC.
- Á. Anglada, A. Urtiaga and I. Ortiz, Contributions of electrochemical oxidation to waste-water treatment: Fundamentals and review of applications, J. Chem. Technol. Biotechnol., 2009, 84(12), 1747–1755 CrossRef.
- H. Särkkä, A. Bhatnagar and M. Sillanpää, Recent developments of electro-oxidation in water treatment - A review, J. Electroanal. Chem., 2015, 754, 46–56, DOI:10.1016/j.jelechem.2015.06.016.
- M. Panizza and G. Cerisola, Direct and mediated anodic oxidation of organic pollutants, Chem. Rev., 2009, 109(12), 6541–6569 CrossRef CAS PubMed.
- B. P. Chaplin, Critical review of electrochemical advanced oxidation processes for water treatment applications, Environ. Sci.: Processes Impacts, 2014, 16(6), 1182–1203, 10.1039/C3EM00679D.
- M. Pierpaoli, M. Szopińska, B. K. Wilk, M. Sobaszek, A. Łuczkiewicz and R. Bogdanowicz, et al. Electrochemical oxidation of PFOA and PFOS in landfill leachates at low and highly boron-doped diamond electrodes, J. Hazard. Mater., 2021, 403, 123606 CrossRef CAS PubMed.
- Q. Zhuo, J. Wang, J. Niu, B. Yang and Y. Yang, Electrochemical oxidation of perfluorooctane sulfonate (PFOS) substitute by modified boron doped diamond (BDD) anodes, Chem. Eng. J., 2020, 379(June 2019), 122280, DOI:10.1016/j.cej.2019.122280.
- K. E. Carter and J. Farrell, Oxidative destruction of perfluorooctane sulfonate using boron-doped diamond film electrodes, Environ. Sci. Technol., 2008, 42(16), 6111–6115 CrossRef CAS PubMed.
- C. E. Schaefer, C. Andaya, A. Burant, C. W. Condee, A. Urtiaga and T. J. Strathmann, et al. Electrochemical treatment of perfluorooctanoic acid and perfluorooctane sulfonate: Insights into mechanisms and application to groundwater treatment, Chem. Eng. J., 2017, 317, 424–432, DOI:10.1016/j.cej.2017.02.107.
- Q. Zhuo, S. Deng, B. Yang, J. Huang, B. Wang and T. Zhang, et al. Degradation of perfluorinated compounds on a boron-doped diamond electrode, Electrochim. Acta, 2012, 77, 17–22, DOI:10.1016/j.electacta.2012.04.145.
- C. A. Martínez-Huitle and M. Panizza, Electrochemical oxidation of organic pollutants for wastewater treatment, Curr. Opin. Electrochem., 2018, 11(1), 62–71 CrossRef.
- K. C. de Freitas Araújo, D. R. da Silva, E. V. dos Santos, H. Varela and C. A. Martínez-Huitle, Investigation of persulfate production on BDD anode by understanding the impact of water concentration, J. Electroanal. Chem., 2020, 860, 113927, DOI:10.1016/j.jelechem.2020.113927.
- A. Sánchez, J. Llanos, C. Sáez, P. Cañizares and M. A. Rodrigo, On the applications of peroxodiphosphate produced by BDD-electrolyses, Chem. Eng. J., 2013, 233, 8–13 CrossRef.
- E. Mostafa, P. Reinsberg, S. Garcia-Segura and H. Baltruschat, Chlorine species evolution during electrochlorination on boron-doped diamond anodes: In-situ electrogeneration of Cl2, Cl2O and ClO2, Electrochim. Acta, 2018, 281, 831–840, DOI:10.1016/j.electacta.2018.05.099.
- S. T. McBeath, D. P. Wilkinson and N. J. D. Graham, Exploiting water contaminants: In-situ electrochemical generation of ferrates using ambient raw water iron (Fe2+), J. Environ. Chem. Eng., 2020, 8(4), 103834, DOI:10.1016/j.jece.2020.103834.
- S. T. McBeath, J. T. English, D. P. Wilkinson and N. J. D. Graham, Circumneutral electrosynthesis of ferrate oxidant: An emerging technology for small, remote and decentralised water treatment applications, Curr. Opin. Electrochem., 2021, 27, 100680 CrossRef CAS.
- S. T. McBeath, D. P. Wilkinson and N. J. D. Graham, Advanced electrochemical oxidation for the simultaneous removal of manganese and generation of permanganate oxidant, Environ. Sci.: Water Res. Technol., 2020, 6(9), 2405–2415 RSC.
- J.-Q. Jiang, C. Stanford and M. Petri, Practical application of ferrate(VI) for water and wastewater treatment – Site study's approach, Water-Energy Nexus, 2018, 1(1), 42–46, DOI:10.1016/j.wen.2018.05.001.
- B. J. Yates, R. Darlington, R. Zboril and V. K. Sharma, High-valent iron-based oxidants to treat perfluorooctanesulfonate and perfluorooctanoic acid in water, Environ. Chem. Lett., 2014, 12(3), 413–417 CrossRef CAS.
- S. Deng, Y. Bao, G. Cagnetta, J. Huang and G. Yu, Mechanochemical degradation of perfluorohexane sulfonate: Synergistic effect of ferrate(VI) and zero-valent iron, Environ. Pollut., 2020, 264, 114789, DOI:10.1016/j.envpol.2020.114789.
- C. S. Liu, K. Shih and F. Wang, Oxidative decomposition of perfluorooctanesulfonate in water by permanganate, Sep. Purif. Technol., 2012, 87, 95–100, DOI:10.1016/j.seppur.2011.11.027.
- S. T. McBeath, D. P. Wilkinson and N. J. D. Graham, Analytical quantification of aqueous permanganate: Direct and indirect spectrophotometric determination for water treatment processes, Chemosphere, 2020, 251, 126626, DOI:10.1016/j.chemosphere.2020.126626.
- Y. Lee, J. Yoon and U. Von Gunten, Spectrophotometric determination of ferrate (Fe(VI)) in water by ABTS, Water Res., 2005, 39(10), 1946–1953 CrossRef CAS PubMed.
- M. A. Cataldo-Hernández, R. Govindarajan, A. Bonakdarpour, M. Mohseni and D. P. Wilkinson, Electrosynthesis of ferrate in a batch reactor at neutral conditions for drinking water applications, Can. J. Chem. Eng., 2018, 9999, 1–8 Search PubMed.
- M. A. Cataldo-Hernández, A. Bonakdarpour, J. T. English, M. Mohseni and D. P. Wilkinson, A membrane-based electrochemical flow reactor for generation of ferrates at near neutral pH conditions, React. Chem. Eng., 2019, 4(6), 1116–1125 RSC.
- S. T. McBeath and N. J. D. Graham, In-situ electrochemical generation of permanganate for the treatment of atrazine, Sep. Purif. Technol., 2021, 260, 118252, DOI:10.1016/j.seppur.2020.118252.
- S. T. McBeath and N. J. D. Graham, Simultaneous electrochemical oxidation and ferrate generation for the treatment of atrazine: A novel process for water treatment applications, J. Hazard. Mater., 2021, 411, 125167 CrossRef CAS PubMed.
- C. D. Vecitis, H. Park, J. Cheng, B. T. Mader and M. R. Hoffmann, Treatment technologies for aqueous perfluorooctanesulfonate (PFOS) and perfluorooctanoate (PFOA), Front. Environ. Sci. Eng. China, 2009, 3(2), 129–151 CrossRef CAS.
- G. D. Sayles, G. You, M. Wang and M. J. Kupferle, DDT, DDD, and DDE dechlorination by zero-valent iron, Environ. Sci. Technol., 1997, 31(12), 3448–3454 CrossRef CAS.
- H. K. Yak, B. W. Wenclawiak, I. F. Cheng, J. G. Doyle and C. M. Wai, Reductive dechlorination of polychlorinated biphenyls by zerovalent iron in subcritical water, Environ. Sci. Technol., 1999, 33(8), 1307–1310 CrossRef CAS.
- C. G. Jones, J. Silverman, M. Al-Sheikhly, P. Neta and D. L. Poster, Dechlorination of Polychlorinated Biphenyls in Industrial Transformer Oil by Radiolytic and Photolytic Methods, Environ. Sci. Technol., 2003, 37(24), 5773–5777 CrossRef CAS PubMed.
- D. C. Hinz, C. M. Wai and B. W. Wenclawiak, Remediation of a nonachloro biphenyl congener with zero-valent iron in subcritical water, J. Environ. Monit., 2000, 2(1), 45–48 RSC.
- W. X. Zhang, Nanoscale iron particles for environmental remediation: An overview, J. Nanopart. Res., 2003, 5, 323–332 CrossRef CAS.
- C. B. Wang and W. X. Zhang, Synthesizing nanoscale iron particles for rapid and complete dechlorination of TCE and PCBs, Environ. Sci. Technol., 1997, 31(7), 2154–2156 CrossRef CAS.
- H. S. Fogler, Essentials of Chemical Reaction Engineering, Upper Saddle River, NJ, Prentice Hall, 2014 Search PubMed.
- S. Li, D. Bejan, M. S. McDowell and N. J. Bunce, Mixed first and zero order kinetics in the electrooxidation of sulfamethoxazole at a boron-doped diamond (BDD) anode, J. Appl. Electrochem., 2008, 38(2), 151–159 CrossRef CAS.
- E. Mousset, Y. Pechaud, N. Oturan and M. A. Oturan, Charge transfer/mass transport competition in advanced hybrid electrocatalytic wastewater treatment: Development of a new current efficiency relation, Appl. Catal., B, 2019, 240(August 2018), 102–111, DOI:10.1016/j.apcatb.2018.08.055.
- M. Panizza, P. A. Michaud, G. Cerisola and C. H. Comninellis, Anodic oxidation of 2-naphthol at boron-doped diamond electrodes, J. Electroanal. Chem., 2001, 507(1–2), 206–214 CrossRef CAS.
- L. Jin, P. Zhang, T. Shao and S. Zhao, Ferric ion mediated photodecomposition of aqueous perfluorooctane sulfonate (PFOS) under UV irradiation and its mechanism, J. Hazard. Mater., 2014, 271, 9–15, DOI:10.1016/j.jhazmat.2014.01.061.
- T. Bond, E. H. Goslan, B. Jefferson, F. Roddick, L. Fan and S. A. Parsons, Chemical and biological oxidation of NOM surrogates and effect on HAA formation, Water Res., 2009, 43(10), 2615–2622, DOI:10.1016/j.watres.2009.03.036.
- J.-P. Croue, G. V. Korshin and M. M. Benjamin, Characterization of Natural Organic Matter in Drinking Wate, AWWA Research Foundation and American Water Works Association, 2000 Search PubMed.
- V. K. Sharma, Ferrate(VI) and ferrate(V) oxidation of organic compounds: Kinetics and mechanism, Coord. Chem. Rev., 2013, 257(2), 495–510, DOI:10.1016/j.ccr.2012.04.014.
- B. K. Körbahti and P. Demirbüken, Electrochemical oxidation of resorcinol in aqueous medium using boron-doped diamond anode: Reaction kinetics and process optimization with response surface methodology, Front. Chem., 2017, 5(October), 1–14 Search PubMed.
- H. Moriwaki, Y. Takagi, M. Tanaka, K. Tsuruho, K. Okitsu and Y. Maeda, Sonochemical decomposition of perfluorooctane sulfonate and perfluorooctanoic acid, Environ. Sci. Technol., 2005, 39(9), 3388–3392 CrossRef CAS PubMed.
- X. J. Lyu, W. W. Li, P. K. S. Lam and H. Q. Yu, Insights into perfluorooctane sulfonate photodegradation in a catalyst-free aqueous solution, Sci. Rep., 2015, 5, 1–6 Search PubMed.
- C. D. Vecitis, Y. Wang, J. Cheng, H. Park, B. T. Mader and M. R. Hoffmann, Sonochemical degradation of perfluorooctanesulfonate in aqueous film-forming foams, Environ. Sci. Technol., 2010, 44(1), 432–438 CrossRef CAS PubMed.
- J. K. Edzwald, Coagulation in drinking water treatment: Particles, organics and coagulants, Water Sci. Technol., 1993, 27(11), 21–35 CrossRef CAS.
- K. L. Dubrawski, M. Cataldo, Z. Dubrawski, A. Mazumder, D. P. Wilkinson and M. Mohseni, In-situ electrochemical Fe(VI) for removal of microcystin-LR from drinking water: comparing dosing of the ferrate ion by electrochemical and chemical means, J. Water Health, 2018, 16(3), 414–424 CrossRef CAS PubMed.
- Z. Du, S. Deng, Y. Bei, Q. Huang, B. Wang and J. Huang, et al. Adsorption behavior and mechanism of perfluorinated compounds on various adsorbents-A review, J. Hazard. Mater., 2014, 274, 443–454, DOI:10.1016/j.jhazmat.2014.04.038.
- H. Chen, C. Zhang, Y. Yu and J. Han, Sorption of perfluorooctane sulfonate (PFOS) on marine sediments, Mar. Pollut. Bull., 2012, 64(5), 902–906, DOI:10.1016/j.marpolbul.2012.03.012.
- C. Jia, C. You and G. Pan, Effect of temperature on the sorption and desorption of perfluorooctane sulfonate on humic acid, J. Environ. Sci., 2010, 22(3), 355–361 CrossRef CAS.
- Y. Jiang, J. E. Goodwill, J. E. Tobiason and D. A. Reckhow, Effect of different solutes, natural organic matter, and particulate Fe(III) on ferrate(VI) decomposition in aqueous solutions, Environ. Sci. Technol., 2015, 49(5), 2841–2848 CrossRef CAS PubMed.
- Q. Zheng, N. Wu, R. Qu, G. Albasher, W. Cao and B. Li, et al. Kinetics and reaction pathways for the transformation of 4-tert-butylphenol by ferrate(VI), J. Hazard. Mater., 2021, 401(April 2020), 123405, DOI:10.1016/j.jhazmat.2020.123405.
- K. T. Tien, N. Graham and J. Q. Jiang, Evaluating the coagulation performance of ferrate: A preliminary study, in ACS Symposium Series, 2008 Search PubMed.
- Y. Bao, J. Niu, Z. Xu, D. Gao, J. Shi and X. Sun, et al. Removal of perfluorooctane sulfonate (PFOS) and perfluorooctanoate (PFOA) from water by coagulation: Mechanisms and influencing factors, J. Colloid Interface Sci., 2014, 434, 59–64, DOI:10.1016/j.jcis.2014.07.041.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ew00399b |
| This journal is © The Royal Society of Chemistry 2021 |