
Albumin protein coronas render nanoparticles surface active: consonant interactions at air–water and at lipid monolayer interfaces†
Nasim
Ganji
 and
Geoffrey D.
Bothun
*
and
Geoffrey D.
Bothun
*
Department of Chemical Engineering, University of Rhode Island, 303 Fascitelli Engineering Building, 2 East Alumni Ave, Kingston, RI 02881, USA. E-mail: gbothun@uri.edu; Tel: +1 401 874 9518
First published on 4th January 2021
Abstract
Protein coronas are known to alter the physicochemical properties, colloidal stability, and biological fate of nanoparticles. Using human serum albumin (HSA) and polystyrene nanoparticles (NPs) with anionic or cationic surface chemistries, we show that protein coronas also govern the surface activity of PS nanoparticles as well as their interactions with a model red blood cell (RBC) lipid monolayer. The adsorption kinetics of bare nanoparticles (no corona) and nanoparticles with a hard corona (HC) at an air–water interface were well-described theoretically, which revealed that the adsorption energy was greater with the corona due to hydrophobic interactions that were enhanced with protein restructuring. Corona complexation increased the concentration of nanoparticles at the interface and led to the formation of interfacial aggregates. Despite clear differences in monolayer structure, the compressibility of PS–HC monolayers was similar to free HSA, indicating that conformational changes associated with the protein were not restricted in a hard corona. The intrinsic behavior of the proteins driving the surface activity and compressibility of the complexes at an air–water interface was also observed at an air–lipid (RBC)–water interface. In this case the lipid monolayer acted as a barrier and reduced the interface concentration of bare nanoparticles. However, with a corona the nanoparticles penetrated into the monolayer and led to the formation of NP–HC–lipid ‘pillars’ that extended into air. Our results suggest that nanoparticle surface activity, and changes in surface activity due to corona formation, are insightful parameters to predicting nanoparticle–membrane interactions, complementing the conventional view that electrostatic forces are dominant.
Environmental significanceThe environmental concentration of polymeric particles continues to increase due to the significant amount of disposed plastic waste that degrades over time into polymeric nanoparticles, which have been shown to translocate across living cells to the lymphatic and circulatory systems and accumulate in secondary organs. The adverse effects associated with interactions between polymeric nanoparticles and environmental or biological systems may be analogous to those generally observed for engineered nanoparticles where formed coronas are known to govern their fate. We show that albumin coronas govern the interfacial behavior of polystyrene nanoparticles, with the corona masking the anionic or cationic charge of the particles and protein restructuring yielding an additional driving force for corona-modified nanoparticles to adhere to interfaces. |
Introduction
The environmental concentration of polymeric particles continues to increase due to the significant amount of disposed plastic waste.1–3 Plastics weather and degrade over time into micro- (<5 mm) and nano- (<100 nm) plastics,4–7 which pose a threat both to environmental and human health.6–14 Small plastic particles are ingested by organisms that are at the bottom of the food-chain and may bioaccumulate.1 It is estimated that humans consume 74![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to 120000 microplastic particles on average per year through ingestion and inhalation.15 The potential adverse health effects associated with these materials is analogous to those observed with engineered nanoparticles (ENPs).16–18 Toxicological studies conducted in vitro and in vivo have demonstrated that polymeric ENPs can translocate across living cells to the lymphatic and/or circulatory system,19,20 accumulate in secondary organs,21 and adversely impact the immune system.22–24
000 to 120000 microplastic particles on average per year through ingestion and inhalation.15 The potential adverse health effects associated with these materials is analogous to those observed with engineered nanoparticles (ENPs).16–18 Toxicological studies conducted in vitro and in vivo have demonstrated that polymeric ENPs can translocate across living cells to the lymphatic and/or circulatory system,19,20 accumulate in secondary organs,21 and adversely impact the immune system.22–24
Nanoparticle cellular uptake begins with particle adhesion to the cell surface and subsequent interactions with lipids and other components of the cell membrane. The interfacial and biophysical forces that modulate this process can be examined using lipid bilayers or monolayers as model cell membranes.25–34 Two main advantages of model membranes are that (1) the lipid composition and structure can be precisely controlled, thereby capturing essential biophysical aspects of cell membranes, and (2) the membrane organization and disruption can be measured directly using techniques that are not amenable to living cells.18 While model systems have been used extensively to study ENP–membrane interactions,16,18,35 few studies that have examined the effect of a biomolecular corona on these interactions. The biomolecular corona ultimately determines the biological identity of an ENP.36–42
Upon encountering biological fluids such as blood, nanoparticles are covered by biomolecules, notably proteins, that form a corona.41,42 The corona is composed of a tightly bound, but not completely irreversibly adsorbed layer of biomolecules (the “hard” corona or HC) that is surrounded by a more loosely bound and rapidly exchanging layer of biomolecules (the “soft” corona or SC).43 The formation of a corona has been reported for several nanoparticles, including polystyrene.36,44–47 The amount, composition, and orientation of biomolecules present in the corona strongly influence NPs adsorption, distribution, and elimination in biological systems, and governs their interactions with cellular membranes.48–50 Despite the importance of the biomolecular corona in dominating nanoparticle interactions at biological interfaces, the influence of protein corona formation on nanoparticle behavior at biological membranes has only recently begun to receive attention.51
Within the context of nanoparticle–membrane interactions, some studies have demonstrated increased cellular uptake for serum incubated ENPs relative to serum-free conditions,52–54 while other studies have shown reduced adhesion and uptake after incubation in serum.55–60 For instance, Lesniak et al.60 have examined the adhesion of polystyrene and silica NPs to A549 (lung cancer) cell membranes and have shown that the presence of biomolecular corona strongly reduces nanoparticle adhesion (and uptake) by weakening nonspecific interactions between NPs and the cell membrane. In contrast, Chithrani et al.54 have reported a greater uptake of gold NPs by HeLa (cervical cancer) cells when a serum protein corona is present. Detailed studies are needed to determine the surface activity of native and corona-modified nanoparticles, how this activity governs interactions with lipid membrane interfaces, and how surface and interfacial activity relate to the composition and physicochemical properties of the NPs and the formed corona.
We propose that the surface activity of corona-modified nanoparticles (i.e. at the air–water interface), which is governed by the amphiphilicity of the corona coating and the ability of the corona proteins to adsorb and undergo conformational changes at the interface, is directly related to the extent of nanoparticle adsorption at lipid interfaces. This would provide a new parameter, particle surface activity, to determine a priori the potential interactions with biological membranes. To test this hypothesis, we have examined the response of a human red blood cell (RBC) model membrane, deployed as a lipid monolayer, to the adhesion of polystyrene (PS) nanoparticles with anionic or cationic surface chemistries, and modified with a human serum albumin (HSA) corona. The Langmuir technique, combined with fluorescence and Brewster angle microscopy, was used to measure the kinetics of PS NP adhesion and the monolayer response, and to identify the properties of the particles and coronas that contribute to the activity at air–water and air–lipid–water interfaces.
Experimental
Materials
All materials were used as received unless otherwise noted. 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE), egg sphingomyelin (SM), and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (Liss Rhod PE) were purchased from Avanti Polar Lipids (Alabaster, AL). Unmodified (PS) and carboxylate-modified fluorescent polystyrene (PS-COOH) NPs were purchased form Polysciences Inc. (Warrington, PA). Amine-modified fluorescent polystyrene (PS-NH) NPs and human serum albumin (HSA, lyophilized powder, essentially fatty acid free) were purchased from Sigma Aldrich. NPs were washed before monolayer experiments by centrifugation and rinsing in order to remove impurities or any excess surfactant used during the polymerization process. Phosphate buffer saline (PBS, pH 7.4) was purchased from Fisher Scientific (Waltham, MA). Chloroform (CHCl3, >99.8%), acetone (C3H6O, >99.5%), and ethanol (C2H6O, >99.5%) from Fisher Scientific (Waltham, MA) were used as solvents for making stock solutions of the lipids and cleaning the Langmuir trough. Deionized (DI) ultra-filtered water was obtained from a Millipore Direct-Q3 UV purification system (Billerica, MA) at 18.2 mΩ resistance and pH 6.5.The model monolayer was composed of lipids naturally occurring in the outer layer of human erythrocytes;61–65 POPC:POPE:SM at the molar ratio 44.9:12:43.1, respectively. A small quantity (1 mol%) of rhodamine-conjugated phosphatidylethanolamine (PE) lipid was added to this mixture as a fluorescent probe.
Formation of nanoparticle–hard corona complexes
NP–HC complexes were prepared following the procedure reported for carboxylate-modified PS NPs by Di Silvio et al.66,67 NP solutions were added to 1.5 mL microcentrifuge tubes to attain final NP concentrations of 1 mg mL−1. HSA (5% in PBS) was added to the microcentrifuge tubes, and the tubes were incubated at 37 °C for 1 h. The tubes were subsequently centrifuged three times (18000g, 4 °C) with a PBS solution wash between each centrifugation step. The sedimented NPs were re-dispersed in PBS to isolate the NP–HC complexes.
Characterization of nanoparticles and nanoparticle–hard corona complexes
NPs and NP–HC complexes were characterized using transmission electron microscopy (TEM; JEOL JEM-2100F) operating at 200 kV and a Malvern Zetasizer Nano ZSX for their core radius, and hydrodynamic radius and zeta (ζ) potentials, respectively. The average size of PS NPs was determined by analyzing multiple TEM images with ImageJ software (n > 50).68 To measure the average ζ-potentials and hydrodynamic diameter (dh) of the NPs, the as-received particles were diluted in PBS and analyzed at 25 °C. The values reported are based on triplicate measurements of three different samples. Adsorption of HSA on PS NPs were visualized by performing negative-staining TEM.69–71 One drop of the diluted NP–HC solution was placed on a carbon coated grid and blotted with filter paper, after which a small aliquot of 2% uranyl acetate was placed on the grid and was dried thoroughly at room temperature before imaging.NP–HC complexes were analyzed further using thermogravimetric analysis (TGA; TA Q500, New Castle, DE) for their protein content. The amount of HSA adsorbed on the NPs was determined by measuring the weight loss of the NP–HC complexes in the range of 200–550 °C due to protein degradation,72 and subtracting it from the weight loss of the NPs over the same temperature range. Heating was performed in a platinum crucible under a nitrogen flow (60 mL min−1) at a rate of 10 °C min−1 up to 1000 °C.
Monolayer surface pressure measurements
Monolayers experiments were conducted at 23 °C as previously described.30 Monolayers were prepared in Teflon® Langmuir–Blodgett trough (KN2002, KSV NIMA, Biolin Scientific Inc., Linthicum Heights, MD) filled with PBS by spreading dissolved lipids in chloroform at the air–water interface and allowing 45 min for the chloroform to evaporate. Isotherms were generated for a single compression/expansion cycle at a barrier rate of 2 cm2 min−1 and the interfacial tension (γ) or surface pressure (π = γ0 − γ, where γ0 = 72.5 mN m−1) was measured using paper Wilhelmy plates. The total area of the trough during this cycle ranged from roughly 70–240 cm2. After recording an isotherm, the trough was set to maintain a constant initial surface pressure (π0 = 30 mN m−1). Once the monolayer stabilized and π0 remained constant, the barrier positions were fixed at the corresponding interfacial area and HSA (22.75 mg L−1), NPs (10 mg L−1), or NP–HC (10 mg L−1) complexes were added to the subphase by injecting them behind the barriers without disrupting the monolayer. The NP and NP–HC subphase concentrations correspond to the amount needed to provide excess surface coverage based on the PS NP cross sectional area at a monolayer surface area of 240 cm2. To determine the adsorption kinetics of NPs and NP–HC complexes at the lipid–water interface, dynamic changes in γ, expressed as γ − γ0, were monitored over 400–600 min. The same monolayer experiments were conducted in the absence of lipid monolayers to determine the surface activity and adsorption kinetics of NPs and NP–HC complexes at the air–water interface. Sample volumes of 2 mL were removed from the Langmuir trough subphase at the end of the monolayer experiments to determine the concentration of PS NPs by UV-vis spectroscopy (Jasco, Tokyo, Japan) based on the maximum peak height at a wavelength of 240 nm after baseline subtraction. All experiments were conducted at least in duplicate.The morphology of the monolayers was visualized using fluorescence and Brewster angle microscopy. For fluorescence microscopy, the Langmuir films were transferred to plasma cleaned glass slides using the Langmuir–Blodgett (LB) deposition technique at constant surface pressures of 10, 20, or 30 mN m−1 at a deposition rate of 0.5 mm min−1.73 A CytoViva microscope equipped with a Dual Mode Fluorescent Module was used to obtain fluorescent images of the deposited film. Brewster angle microscopy (BAM) was used to enable real-time observation of monolayers at the air–water interface in a Langmuir trough. BAM provides information on homogeneity, phase behavior and the film morphology by detecting changes in the refractive index of the water surface in the presence of surfactants or surface-active molecules.
Results and discussion
Characterization of nanoparticles and nanoparticle–corona complexes
The average diameter (d) of the unmodified, carboxylate- and amine-modified PS NPs was 98 ± 9 nm based on TEM analysis (Fig. S1†). Similar hydrodynamic diameters (dh) were measured for all three NPs (Fig. 1A), which was consistent with the average particle diameter measured by TEM. The carboxylate PS-COOH NPs and the unmodified PS NPs were negatively charged, with the surface charge on the unmodified particles due to an anionic surfactant coating (Fig. 1B). The amine PS-NH NPs were cationic owing to the secondary amine groups. | ||
| Fig. 1 (A) Average hydrodynamic diameters (dh), (B) the ζ-potential of NPs and NP–HC complexes, and (C) the increase in NP dh upon adsorption of HSA (inset: representative micrograph of PS–HC complexes; HSA is negatively stained). Samples were prepared in pH 7.4 PBS and the reported values are based on triplicate measurements of three different samples. (D) the calculated amounts of HSA comprising the protein corona for the different NP–HC complexes (inset: schematic of the dimensions of the HSA as an equilateral triangular prism74). Error bars correspond to one standard deviation for triplicate experiments. | ||
The changes in ζ-potential and dh of the particles (Fig. 1A and B) with a protein corona provided direct evidence of the complexation of PS NPs by HSA. Fig. 1C shows the increase of PS NPs hydrodynamic diameter upon HSA incubation for concentrations ranging from 0.1 to 600 μM HSA. From these results a concentration of 300 μM HSA was assumed to saturate the NP surface and form a close-packed protein monolayer.69 The increase in dh due to corona formation was ∼20 nm and was common to all three PS NPs corresponding to a shell thickness of ∼10 nm (Fig. 1A), which was similar to the z-averaged hydrodynamic diameter measured for HSA (10.7 ± 2.3 nm). An HSA shell thickness of 7 ± 1 nm for PS–HC complexes was further shown by negative-staining TEM imaging (Fig. 1C, inset). Upon protein complexation, the ζ-potential of the particles became either negative, in the case of amine-modified PS NPs, or less negative for unmodified and carboxylate-modified PS NPs, approaching the value measured for HSA in PBS (−9.9 ± 1.2 mV) (Fig. 1B). These data indicate that NPs form complexes with HSA, and that complexation occurred for all three PS nanoparticles with different surface chemistries.
Considering the dimensions of HSA (76 × 76 × 28 Å3)74 and the nanoparticle surface area (πd2h), we estimated that 1.0 × 103 to 1.5 × 103 HSA molecules (based on flat or edge-on binding configurations, respectively) are required to form a close-packed monolayer of protein corona. This was confirmed by TGA where 1.0 × 103 to 1.1 × 103 HSA per NP were measured at saturation (Fig. 1D), which is in good agreement with our calculations for HSA binding.
Insight into the mechanism of HSA binding and corona formation can be gained from the protein surface charge distribution and the Debye screening length (κ−1) at physiological salt concentration in PBS. The structure of HSA at pH 7.4 was determined computationally and shows the coexistence of acidic (blue; aspartic acid, glutamic acid) and basic (red; arginine, histidine, lysine) amino acid residues on the protein surface (Fig. S2†). Hydrophobic (silver) and polar (yellow) residues are also shown. Despite the net negative charge of HSA, positive and negative amino acids are distributed on its surface and provide local binding sites for opposite charges. Furthermore, κ−1 is approximately 0.8 nm (150 mM monovalent ions), which means that the proteins and NPs come into close contact before experience electrostatic interactions. At this length scale van der Waals attraction was assumed to be the driving force for HSA adsorption, aided by the mixed charge distribution on the protein surface.
Dynamic interfacial tension of nanoparticles and nanoparticle–corona complexes at the air–water interface
Dynamic adsorption behavior can be described by a three-stage process as depicted in Fig. 2A–C, respectively. During stage 1, γ decreases slowly due to the adsorption of individual particles to a pristine interface. As the excess surface concentration of NPs increases during stage 2, γ decreases more rapidly. As t → ∞ during stage 3 the interface approaches maximum coverage and the rate of NP surface adsorption decreases due to a steric barrier. During this stage γ plateaus, reflecting a pseudo-equilibrium condition.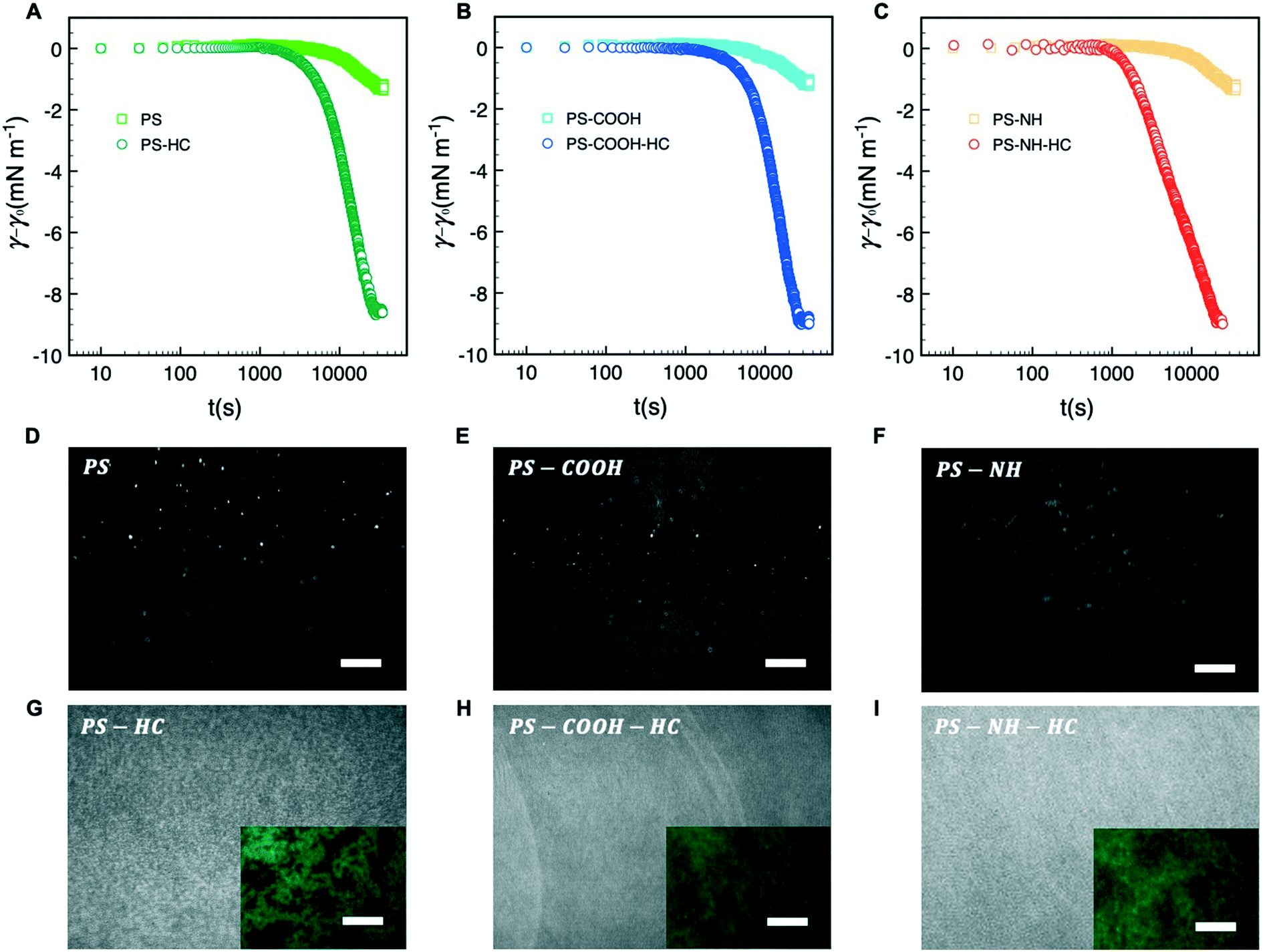 | ||
| Fig. 2 Dynamic changes in surface tension for (A) PS, (B) PS-COOH, and (C) PS-NH nanoparticles before and after complexation with HSA plotted in a semi-logarithm scale. Representative fluorescence microscopy (scale bars = 20 μm) and BAM images (scale bars = 300 μm) are shown for (D) PS, (E) PS-COOH, and (F) PS-NH NP and (G) PS–HC, (H) PS-COOH-HC, and (I) PS-NH-HC complexes at the air–water interface at pseudo-equilibrium conditions (t → ∞). | ||
As shown in Fig. 2A–C, bare NPs were not appreciably surface active with a maximum change in interfacial tension of approximately −1.5 mN m−1 that we attribute to particle adsorption at the interface. HSA corona complexation rendered the NPs surface active due to hydrophobic interactions at the air–water arising from the hydrophobic amino acid residues of the proteins interface. Similar surface activities, or reductions in interfacial tension, were observed for all three NP–HC complexes and the difference between the pseudo-equilibrium interfacial tension at the end of stage 3 with and without a corona was approximately −7 mN m−1.
The morphology and packing of NP and NP–HC Langmuir films at the interface were examined at the pseudo-equilibrium condition (stage 3) using fluorescence microscopy and BAM. Bare NPs adsorbed at the interface and formed fractal aggregates on the micrometer scale and larger due to attractive van der Waals and capillary interactions75 (Fig. 2D–F; S3A and B†). For NP–HC complexes, the formed monolayers were thicker based on BAM reflectance (Fig. 2G–I) and comprised of denser aggregates that we attribute to a combination of capillary interactions and interparticle attraction driven by the hydrophobic interactions between coronas with restructured proteins at the air–water interface (Fig. 2G–I inset; S3C and B†).
The subphase concentrations of PS NPs were analyzed by UV-vis spectroscopy at the end of stage 3 to further quantify the extent of NP and NP–HC adsorption at the air–water interface. The NP surface concentrations, Γ, were determined by mass balance as Γ = (ci − ceq)bV(VNPρPSA)−1 where (ci − ceq)b is the change in bulk PS concentration from initial (ci) to pseudo-equilibrium (ceq), VNP is the mean PS NP volume, ρPS is the density of polystyrene, and V and A are the trough volume and area, respectively. For bare NPs, the surface concentrations reflect maximum fractional surface coverages (Θ∞) from 0.34 to 0.48. The presence of a protein corona increased NP adsorption, and the surface excess concentration for all three types of NP–HC complexes was similar at Γ+corona ≈ 15 × 1013 NP m−2 (Fig. 3). At this surface concentration Θ∞ ≈ 0.9. The increase in adsorption due to corona complexation was approximately 45% for PS-NH and PS-COOH, and 62% for unmodified PS. While the value of Θ∞ ≈ 0.9 is near that for a hexagonally packed monolayer of spheres (0.91),76 the microscopy analysis shows that the PS–HC layers were comprised of interfacial aggregates rather than a continuous monolayer.
 | ||
| Fig. 3 Surface concentration (Γ, NP m−2) of PS nanoparticles with and without a formed corona (− corona and + corona, respectively) (A) at the air–water interface and (B) at the air–lipid–water interface. Results in (B) show the change in concentration, Γ+RBC/Γ−RBC, when the RBC lipid layer is present relative to an air–water interface. Error bars represent one standard deviation of triplicate experiments. | ||
Adsorption kinetics of nanoparticles and nanoparticle–corona complexes at the air–water interface
The presence of a hard corona on PS nanoparticles leads to similar apparent surface activities (Fig. 2A–C) and similar interfacial structures (Fig. 2G–I), consistent with the view that formed protein coronas govern the physicochemical properties of nanoparticles. To determine if corona formation also leads to similar kinetics of NP adsorption, the dynamic interfacial tension results were analyzed using the model of Ward and Tordai.80 The following asymptotic equations have been employed to interpret data from the early (t → 0) and late (t → ∞) times of NP adsorption.At early times (stage 1), an individual NP that is adsorbing to the interface encounters a bare interface leading to a change in γ. Assuming there is no barrier to adsorption at this stage, particle diffusion to the interface is the rate-limiting step and the diffusion-controlled Ward and Tordai mechanism can be applied.77 Bizmark et al.78 modified the Ward and Tordai model to account for NPs larger than 10 nm with adsorption trapping energy exceeding 103kBT
 | (1) |
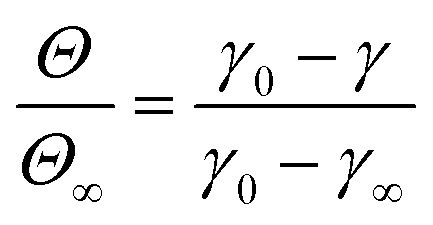 | (2) |
The stage 1 region in Fig. 2A–C were defined by the range Θ = 0 to 0.3,79 providing a basis for calculating γ at Θ = 0.3 using eqn (2). Considering the stage 1 adsorption energy as 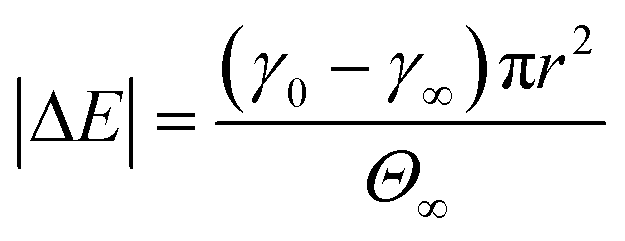 , effective diffusion coefficients, D, were determined based on eqn (1) by linear regressions of γ − γ0vs. t0.5. Table 1 reports the values of D for NPs and NP–HC complexes, and compares them to diffusion coefficients predicted by the Stoke–Einstein equation
, effective diffusion coefficients, D, were determined based on eqn (1) by linear regressions of γ − γ0vs. t0.5. Table 1 reports the values of D for NPs and NP–HC complexes, and compares them to diffusion coefficients predicted by the Stoke–Einstein equation 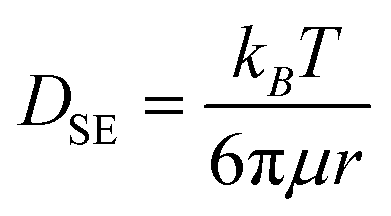 , where r is the hydrodynamic radii of the particles and μ is the viscosity of water at room temperature. Values of D and DSE differ by just 30 to 47%, indicating that eqn (1) is valid during the early times adsorption of particles from the bulk to the air–water interface.
, where r is the hydrodynamic radii of the particles and μ is the viscosity of water at room temperature. Values of D and DSE differ by just 30 to 47%, indicating that eqn (1) is valid during the early times adsorption of particles from the bulk to the air–water interface.
| D SE (10−12 m2 s−1) | D (10−12 m2 s−1) | Stage 1 (diffusion-controlled) | Stage 2 (protein restructuring) | Stage 3 (barrier-controlled) | |
|---|---|---|---|---|---|
| |ΔES1| (104kBT, J) | |ΔES2|/|ΔES1| | k a (10−6 m s−1) | |||
| a Standard deviation in DSE based measured standard deviations of hydrodynamic radii. b Unit of 100kBT. | |||||
| PS | 4.91 ± 0.12 | 7.92 ± 0.78 | 1.35 ± 0.02 | – | – |
| PS-COOH | 4.73 ± 0.29 | 6.78 ± 0.15 | 1.81 ± 0.05 | – | – |
| PS-NH | 4.63 ± 0.35 | 6.25 ± 0.18 | 2.61 ± 0.02 | – | – |
| PS–HC | 3.99 ± 0.30 | 3.78 ± 0.37 | 8.11 ± 0.19 | 6.86 | 1.70 ± 0.19 |
| PS-COOH-HC | 3.71 ± 0.27 | 3.31 ± 0.18 | 6.13 ± 0.24 | 7.43 | 1.51 ± 0.93 |
| PS-NH-HC | 3.54 ± 0.16 | 4.54 ± 0.93 | 8.99 ± 1.03 | 11.39 | 5.83 ± 0.42 |
| HSA | 66.0 ± 6.1 | – | 14.64 ± 0.95 | 2.16 | – |
Using DSE values, eqn (1) was then used to calculate the stage 1 adsorption energy, |ΔES1|. As shown in Table 1, the magnitude of the adsorption energy correlated with the ζ-potential of the NPs where |ΔES1| increased as the ζ-potential became less negative (or more positive). This result is consistent with anionic NPs being electrostatically repelled from the air–water interface, which has been shown to have a negative surface potential.79,80
Unlike NP adsorption, two distinct stages with clearly different slopes were observed for NP–HC complexes when γ − γ0vs. t0.5 was plotted from Θ = 0 to 0.3 (Fig. S4A and B†). This is consistent with recent work by Tian et al.81 for the adsorption kinetics of poly(ethylene oxide) (PEO)-modified polystyrene NPs to air–water interfaces. As shown in Fig. 2, although the transition between these two stages occurs at an earlier time for PS-NH-HC compared to PS–HC and PS-COOH-HC, no statistically significant difference in interfacial tension is observed at this transition point between the three types of NP–HC complexes. For stage 1, we calculated the diffusion coefficients and adsorption energies of NP–HC complexes as we did for bare NPs; using the slope of γ − γ0vs. t0.5 according eqn (1). Values for D and DSE are remarkably similar and confirm the effects of adsorbed coronas on increasing the hydrodynamic diameters and reducing diffusivity compared to the bare NPs. The effect of the corona can also be observed in |ΔES1|, with significantly greater values being measured for NP–HC complexes due to hydrophobic interactions at the air–water interface (i.e. greater thermodynamic driving force for particle trapping at the interface). The greatest driving force is observed for the particles where the corona is formed on cationic amine-modified PS, which also has a ζ-potential closest to zero.
At the end of stage 1 the surface coverage, Θ, was less than 0.05 for the NP–HCs. Hence, for stage 2 we inferred that particle adsorption is also diffusion-controlled and that the Stokes–Einstein equation can be applied to estimate the diffusion coefficients of the NP–HCs. The adsorption energy during stage 2, |ΔES2|, was calculated according to eqn (1) (Table 1; reported as |ΔES2|/|ΔES1|) and was approximately 7- to 11-fold greater than for stage 1. The observed two-stage transition for NP–HC complexes is attributed to initial particle diffusion (stage 1) followed by protein restructuring (stage 2) at the air–water interface.78 Protein restructuring, exposing hydrophobic residues at the air–water interface as the protein unfolds, led to significant reductions in interfacial tension accompanied by high adsorption energies.
As the interface becomes saturated (t → ∞ and Θ > 0.75),78 the presence of adsorbed particles hinders additional particle attachment. Stage 3 adsorption kinetics can be described by introducing a blocking function to the long-time Ward and Tordai approximation to account for the adsorption barrier at high NP surface coverage82
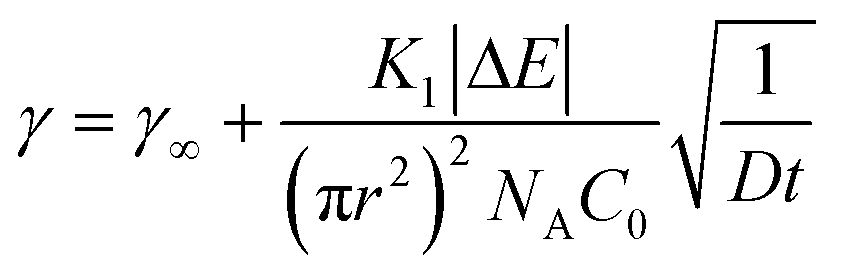 | (3) |
 | (4) |
![[k with combining low line]](https://www.rsc.org/images/entities/i_char_006b_0332.gif)
![[a with combining low line]](https://www.rsc.org/images/entities/char_0061_0332.gif) is the dimensionless adsorption constant. The adsorption constant, ka, can be determined as, ka = DNAC0πr2.
is the dimensionless adsorption constant. The adsorption constant, ka, can be determined as, ka = DNAC0πr2.
For bare PS NPs Θ∞ ≤ 0.48, indicating that adsorbing particles did not experience a crowded interface and the adsorption is diffusion-controlled at any time during the process. For NP–HC complexes, ka was calculated from the gradient of γ − γ0vs. t−0.5 (shown for PS-COOH-HC in Fig. S4C†). The values of ka for all NP–HC complexes are listed in Table 1. PS-NH-HC complexes have a greater adsorption constant compared to unmodified and carboxylate-modified NP–HC complexes. The greater value of ka for PS-NH-HC denotes a faster rate of adsorption in stage 3, which is consistent with greater adsorption energies for particle attachment (|ΔES1|) and protein restructuring (|ΔES2|) for the amine-modified PS compared to the anionic nanoparticles.
Surface pressure–area isotherms of nanoparticles and nanoparticle–corona complexes at the air–water interface
The compressibility and structure of the interfacial layers were further examined through surface pressure–area (π–A) isotherms and BAM. HSA alone shows the characteristic sigmoidal shape with a steep increase in π (reduction in γ) upon initial compression as restructured proteins pack at the interface, followed by a transition near π = 12 mN m−1 where hydrophilic residues are expelled from the interface due to steric hinderance (Fig. 4A).83 Additional compression yields a second transition near π = 20 mN m−1 as the reconfigured proteins continue to pack. A maximum surface compressional modulus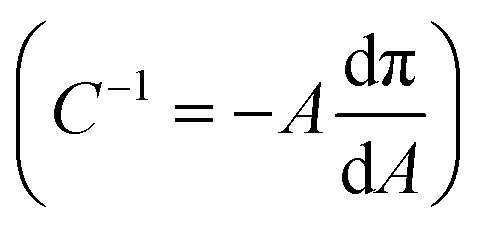 , or resistance to packing, is observed at 40 mN m−1. The NP–HC complexes exhibit strikingly similar behavior; both transitions are observed and shape of the C−1vs. A curves are nearly superimposable for PS-COOH-HC at the point of inflection (Fig. 4A; position of maximum C−1). PS–HC exhibited similar behavior (Fig. S5A1†). This similarity is not attributed to unbound HSA in NP–HC samples as we confirmed that there was no measurable unbound HSA after the separation step during corona formation, consistent with previous work showing that protein coronas are stable, exhibiting little protein desorption.69 These results demonstrate that HSA bound within a corona complex behaves similarly to unbound HSA at the air–water interface, which infers that the free energy change associated with surface activity and corresponding conformational changes competes with the attractive protein–particle interactions that lead to corona formation. Complexes formed with cationic PS NPs, PS-NH-HC (Fig. S5B1†), deviated slightly and exhibited a C−1 that was 10 mN m−1 lower than HSA or the other NP–HC complexes. This reflects a lower resistance to compression that may be due to a greater preference for the proteins to remain in the NP-bound state as indicated by the stronger binding and restructuring energies determined from the kinetic analysis. The corona proteins (negatively charged) may also have resisted reconfiguration due to stronger electrostatic interactions with the positively charged particle surface.
, or resistance to packing, is observed at 40 mN m−1. The NP–HC complexes exhibit strikingly similar behavior; both transitions are observed and shape of the C−1vs. A curves are nearly superimposable for PS-COOH-HC at the point of inflection (Fig. 4A; position of maximum C−1). PS–HC exhibited similar behavior (Fig. S5A1†). This similarity is not attributed to unbound HSA in NP–HC samples as we confirmed that there was no measurable unbound HSA after the separation step during corona formation, consistent with previous work showing that protein coronas are stable, exhibiting little protein desorption.69 These results demonstrate that HSA bound within a corona complex behaves similarly to unbound HSA at the air–water interface, which infers that the free energy change associated with surface activity and corresponding conformational changes competes with the attractive protein–particle interactions that lead to corona formation. Complexes formed with cationic PS NPs, PS-NH-HC (Fig. S5B1†), deviated slightly and exhibited a C−1 that was 10 mN m−1 lower than HSA or the other NP–HC complexes. This reflects a lower resistance to compression that may be due to a greater preference for the proteins to remain in the NP-bound state as indicated by the stronger binding and restructuring energies determined from the kinetic analysis. The corona proteins (negatively charged) may also have resisted reconfiguration due to stronger electrostatic interactions with the positively charged particle surface.
 | ||
| Fig. 4 Surface pressure–area isotherms (π–A) and corresponding surface compressional moduli (C−1) for HSA and the PS-COOH hard corona complexes (PS-COOH-HC) at the air–water interface. The open squares shown on the isotherms denote the conditions for BAM imaging shown in B. BAM images for HSA alone are shown in C. Scale bars in BAM images represent 300 μm. | ||
BAM analysis of the films show thick NP–HC monolayers, based on reflectance, and a variety of lateral and clustered phases that span the compression range (Fig. 4B; S5A2 and B2†). These phases are not observed for HSA alone (Fig. 4C), though small differences in HSA film thickness were evident. At no point in the NP–HC isotherms were void spaces (black regions void of reflectance) observed, indicating the coexistence of dense and lean NP–HC regions. The exception to this is for PS-COOH-HC at high compression (π = 21.5 mN m−1; Fig. 4B) where a defect is seen in the layer. It is interesting to note that the apparent thickness (reflectance) of the NP–HC layers did not change significantly with compression as shown for HSA. To accommodate the NP–HC layers with compression some of the particles must have been displaced into the aqueous phase without forming wrinkles, at least at the length scales observable by BAM imaging.
Nanoparticle and nanoparticle–corona complexes at the air–lipid–water interface
We now compare the behavior of NPs and NP–HC complexes at an air–water interface to when a model RBC lipid monolayer is present. Dynamic changes in interfacial tension were determined as γ − γL, where γL is the initial interfacial tension at air–lipid–water interface (γL = 42.5 mN m−1 corresponding to an initial surface pressure, π = γ0 − γL, of 30 mN m−1). At this surface pressure the compressed monolayer has an average area per lipid, A, of 63 Å2 per molecule with coexisting liquid-expanded (LE; rich in POPC and POPE lipids) and liquid-condensed (LC; rich in SM lipid) phases, as previously observed63 (Fig. S6†). By compressing the monolayer to π = 30 mN m−1 we can achieve an area per lipid representative of lipid packing within a cell membrane.Interfacial tensions are often reported to decrease with time as proteins or protein–nanoparticle complexes penetrate into a lipid monolayer and fill void space between lipids at the air–water interface.84 We observed this for initial surface pressures less than 10 mN m−1 where A ≥ 82 Å2 per molecule (Fig. S6†). However, increases in interfacial tension were observed for HSA (Fig. S7;† shown as decreases in π) and for NP and NP–HC complexes (Fig. 5A1–C1) at an initial surface pressure of 30 mN m−1. We measured the surface concentrations of PS and PS–HC complexes that produced the increases in interfacial tension. Based on the relative concentrations with and without an RBC monolayer present, Γ+RBC/Γ−RBC (Fig. 3B), the monolayer reduced the amount of bare PS nanoparticles at the air–lipid–water interface by 80% (PS, PS-COOH) and 50% (PS-NH), which correlates to a Θ∞ of approximately 0.07 (PS), 0.1 (PS-COOH), and 0.24 (PS-NH). Comparatively, particles with protein coronas showed a high degree of surface coverage similar to when no lipid monolayer was present.
 | ||
| Fig. 5 Dynamic changes in interfacial tension for (A1) PS, (B1) PS-COOH, and (C1) PS-NH nanoparticles before and after complexation with HSA, plotted in a semi-logarithm scale. Corresponding BAM images are shown in A2–C2 at early (103 s) and late (104 s) times during the adsorption process. Scale bars in A2–C2 represent 300 μm. | ||
BAM imaging was conducted at early (103 s) and late (104 s) times as the NPs and NP–HC complexes adsorbed to and interacted with the lipid monolayer (Fig. 5A2–C2). Coexisting LE–LC phases are observed at early times for NPs with and without formed coronas. Bare NPs accumulate at the interface and appear to thicken LC domains (some free, lighter grey LC domains are still observed). For anionic PS and PS-COOH, binding is consistent with electrostatic and charge–dipole interactions with LC domains, where the lipid dipole moment extends perpendicular to the air–water interface with the positively charged choline headgroup extending into the aqueous subphase.32,85–87 For cationic PS-NH, large fractal aggregates of NPs are observed, reflecting the greater measured surface coverage, Θ∞ (Fig. 5C2). Unlike anionic NPs, cationic NPs have been shown to preferential bind to LE phases where the lipid headgroup dipole is parallel to the interface and the negatively charged phosphate group is accessible.87 Therefore, we attribute the structures formed with PS-NH to NP binding primarily to the LE phase followed by NP aggregation. Free LC domains remain present throughout the process.
The presence of a HC, where hydrophobic interactions between HSA and zwitterionic lipids are dominant, led to the formation of unique structures composed of clustered LC domains at early times, and a thick interfacial layer similar to HSA at the air–lipid–water interface at pseudo-equilibrium. Interactions between HSA and zwitterionic lipid monolayers have been shown to decrease with increasing lipid packing (lower area per lipid),88 suggesting that the NP–HC complexes preferentially bind to the LE phase. Given the high surface coverage of the corona complexes at the air–lipid–water interface and the observed aggregation behavior at the air–water interface (Fig. 2), we ascribe the clustering of LC domains to these domains being excluded from LE domains with bound aggregates of NP–HC complexes.
The significant difference in surface coverage between bare and corona-complexed NPs, and distinct differences in interfacial structure, suggests that different mechanisms are at play. Bare NPs appear to have preferentially bound to LC or LE phases, adsorbing lipids and partially removing them from the interface, while NP–HC complexes and the lipids remained an integral part of the interfacial layer. Di Silvio et al.67 showed that bare 100 nm PS-COOH NPs disrupted a zwitterionic supported lipid bilayer by lipid extraction. Bilayer disruption was observed when the particles were coated with a soft protein corona of fetal bovine serum, but not a hard corona. This was attributed to the weakly bound soft corona proteins (or free proteins) acting in concert with the NPs on the membrane surface. Our results demonstrating that the NP–HC complexes disrupt monolayers and that HC proteins drive this disruption may reflect the different protein used (HSA vs. FBS) and the more fluid nature of a monolayer. Out of plane distortions are more restricted in a supported lipid monolayer that is adsorbed onto a solid surface.
Surface pressure–area isotherms were generated under compression for the lipid monolayers with NP–HC complexes (Fig. 6A). The RBC monolayer exhibited a continuous increase in π with decreasing area, the nucleation of LC domains between 10 to 15 mN m−1 (bright spots at π ≥ 15 mN m−1 in BAM images; Fig. 6C), and the growth of the LC domains up to 35 mN m−1. With the addition of HSA, the isotherm resembles that of HSA alone at the air–water interface, with hydrophilic residues expelled from the interface at a slightly higher surface pressure (π = 15 mN m−1 compared to 12 mN m−1). LC domains were not observed, and the structure of the layer was again visually similar to HSA alone based on reflectance (Fig. 4C and 6D). These results indicate that the interface was comprised of HSA–lipid complexes, with these complexes being “squeezed out” of the monolayer at high surface pressures. The reversibility of the compression–expansion isotherms (data not shown) suggests that the displaced complexes remain near the interface and re-adsorbed at low surface pressures, consistent with previous results for bovine serum albumin and zwitterionic dipalmitoylphosphatidyl ethanolamine.89
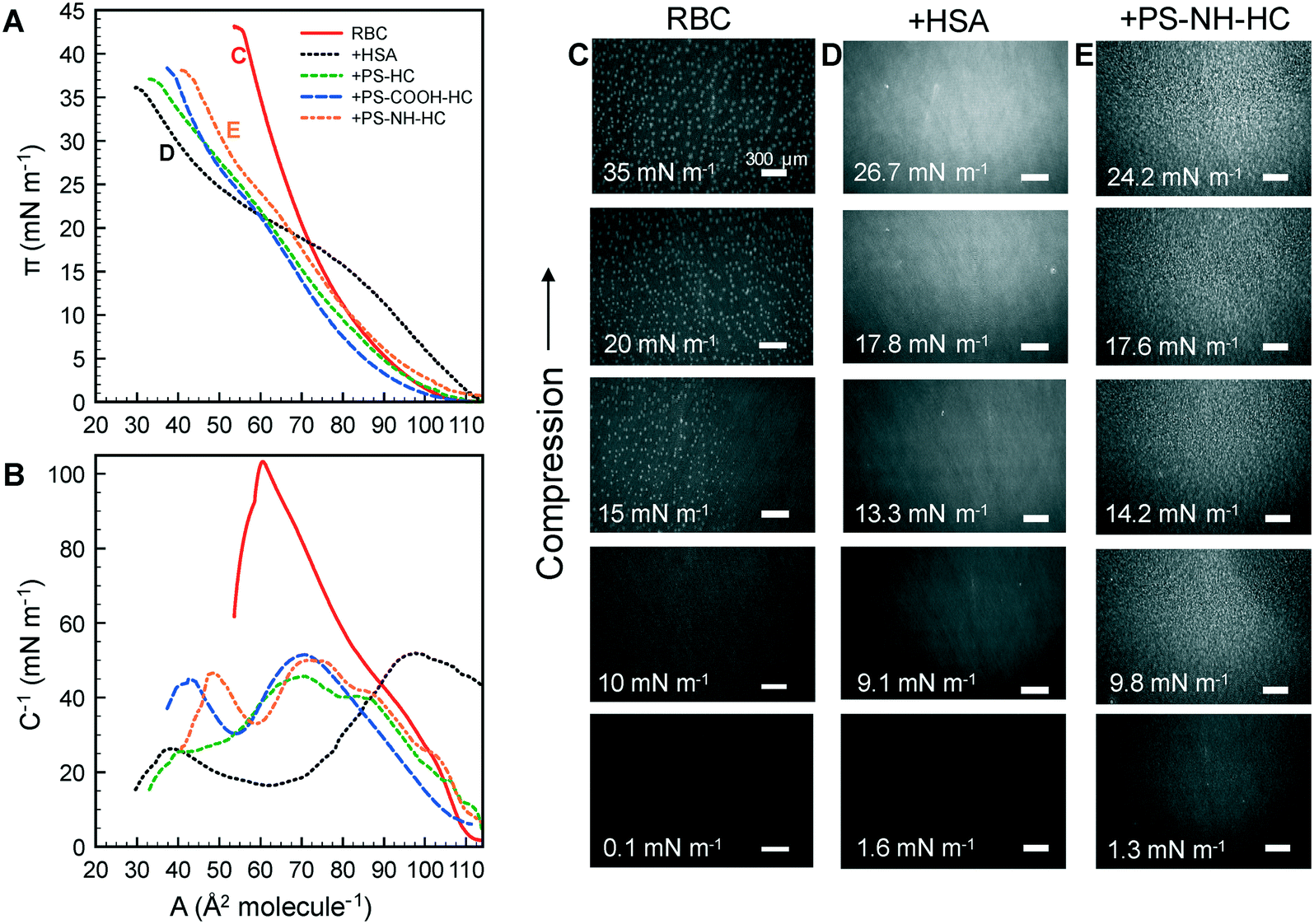 | ||
| Fig. 6 Surface pressure–area isotherms (A) and surface compressional moduli (B) of the RBC monolayer alone and exposed to HSA and the NP–HC complexes. Corresponding BAM images are shown for (B and C) the RBC monolayer exposed to (D) HSA or (E) PS-NH-HC. Scale bars in C–E represent 300 μm. | ||
With NP–HC complexes the intrinsic behavior of the proteins driving the surface activity and compressibility of the complexes at an air–water interface is also observed at an air–lipid–water interface. Transitions in surface pressure between 20–25 mN m−1 due to hydrophilic residues being expelled from the interface are observed for NP–HC complexes. The conformational changes in corona proteins at the air–water interface also occur at the air–lipid–water interface. Maximum C−1 values were 103 mN m−1 for the lipid monolayer, consistent with previous results for a model RBS outer membrane leaflet,61 52 mN m−1 when exposed to HSA, and ranged from 46–51 mN m−1 for mixed layers of lipid + NP complexes. The presence of lipid added additional compressibility compared to the air–water interface, however the values were similar to HSA alone.
BAM images for PS-NH-HC are shown in Fig. 6E (PS–HC and PS-COOH-HC led to the same structures and are not shown). Small, bright spots are observed at π ≥ 9.8 mN m−1 and become larger with compression. This is attributed to the increasing displacement of lipid/PS complexes into the air, whereas HSA–lipid complexes are displaced into the aqueous phase. The displacement of lipid/PS complexes above the interface may be due to adsorbed lipids rendering them more hydrophobic. Therefore, the increases in dynamic interfacial tension observed in Fig. 5 are likely due to the formation of lipid/PS complex and the displacement behavior rather than lipid condensation, which has been previously reported for charged nanoparticles interacting with zwitterionic lipid monolayers or bilayers.32,85–87
Conclusions
Surface pressure measurements were coupled with fluorescence and Brewster angle microscopy to investigate the effects of a serum protein corona on the intrinsic surface activity of charged polystyrene nanoparticles and on the interactions with a model human red blood cell (RBC) lipid monolayer. We show that ‘classic’ theoretical models can capture the adsorption kinetics of bare and corona-complexed NPs, and the additional particle trapping energies associated with protein restructuring, which was confirmed experimentally. The ability for proteins within a corona to restructuring infers an additional driving force for nanoparticle–corona complexes to adhere to biological interfaces. This was further observed with the lipid monolayer present for all NP–HC complexes examined, suggesting that the protein corona controlled monolayer adhesion. Interestingly, the lipid monolayer prevented the majority of the bare nanoparticles from adsorbing at the interface (compared to an air–water interface), but was able to accommodate most of the NP–HC complexes because of their appreciable surface activity. In addition to the surface activity, this may reflect the affinity of HSA for the lipids – HSA binds and transports fatty acids through the bloodstream. We should point out that, while other studies with serum coronas have shown similar features,50 this work represents an initial demonstration of the interfacial interactions that occur when nanoparticle–HSA corona complexes interact with a model lipid monolayer. We expect that the surface activity-based approach may translate more broadly to other nanoparticle–protein corona complexes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by the National Science Foundation (NSF) under Grant No. CBET-1055652. Additional resources were provided by the Rhode Island Center for Nanoscience and Nanotechnology, which is supported by the Rhode Island NSF Experim Track-1 Experimental Program to Stimulate Competitive Research (EPSCoR) under Grant No. OIA-1655221. We thank Dr. Irene Andreu in the Department of Chemical Engineering at URI for assisting with TEM imaging.References
- S. Kihara, N. J. van der Heijden, C. K. Seal, J. P. Mata, A. E. Whitten, I. Köper and D. J. McGillivray, Soft and Hard Interactions between Polystyrene Nanoplastics and Human Serum Albumin Protein Corona, Bioconjugate Chem., 2019, 30(4), 1067–1076 CrossRef CAS
.
- S. Lambert and M. Wagner, Characterisation of nanoplastics during the degradation of polystyrene, Chemosphere, 2016, 145, 265–268 CrossRef CAS
- A. Cózar, F. Echevarría, J. I. González-Gordillo, X. Irigoien, B. Ubeda, S. Hernández-León, A. T. Palma, S. Navarro, J. García-de-Lomas, A. Ruiz, M. L. Fernández-de-Puelles and C. M. Duarte, Plastic debris in the open ocean, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 10239–10244 CrossRef
- H. Bouwmeester, P. C. H. Hollman and R. J. B. Peters, Potential Health Impact of Environmentally Released Micro- and Nanoplastics in the Human Food Production Chain: Experiences from Nanotoxicology, Environ. Sci. Technol., 2015, 49, 8932–8947 CrossRef CAS
- A. Rajeev, V. Erapalapati, N. Madhavan and M. G. Basavaraj, Conversion of expanded polystyrene waste to nanoparticles via nanoprecipitation, J. Appl. Polym. Sci., 2016, 133, 2–6 CrossRef
- J. Costa, P. S. M. Santos, A. C. Duarte and T. Rocha-santos, (Nano) plastics in the environment – Sources, fates and effects, Sci. Total Environ., 2016, 566–567, 15–26 CrossRef
- A. L. Dawson, S. Kawaguchi, C. K. King, K. A. Townsend, R. King, W. M. Huston and S. M. Bengtson Nash, Turning microplastics into nanoplastics through digestive fragmentation by Antarctic krill, Nat. Commun., 2018, 9, 1001 CrossRef
- Y. Chae and Y. J. An, Current research trends on plastic pollution and ecological impacts on the soil ecosystem: A review, Environ. Pollut., 2018, 240, 387–395 CrossRef CAS
- M. Carbery, W. Andrew O'connor, T. Palanisami, W. O'connor and P. Thavamani, Trophic transfer of microplastics and mixed contaminants in the marine food web and implications for human health, Environ. Int., 2018, 115, 400–409 CrossRef
- M. Smith, D. C. Love, C. M. Rochman and R. A. Neff, Microplastics in Seafood and the Implications for Human Health, Curr. Environ. Health Rep., 2018, 5, 375–386 CrossRef CAS
- A. D. Vethaak and H. A. Leslie, Plastic Debris is a Human Health Issue, Environ. Sci. Technol., 2016, 50, 6825–6826 CrossRef CAS
- R. H. Waring, R. M. Harris and S. C. Mitchell, Plastic contamination of the food chain: A threat to human health?, Maturitas, 2018, 115, 64–68 CrossRef CAS
- M. Revel, A. Châtel and C. Mouneyrac, Micro(nano)plastics: a threat to human health?, Curr. Opin. Environ. Sci. Health, 2018, 1, 17–23 CrossRef
- R. R. Hurley and L. Nizzetto, Fate and occurrence of micro(nano)plastics in soils: Knowledge gaps and possible risks, Curr. Opin. Environ. Sci. Health, 2018, 1, 6–11 CrossRef
- K. D. Cox, G. A. Covernton, H. L. Davies, J. F. Dower, F. Juanes and S. E. Dudas, Human Consumption of Microplastics, Environ. Sci. Technol., 2019, 53, 7068–7074 CrossRef CAS
- A. M. Farnoud and S. Nazemidashtarjandi, Emerging investigator series: interactions of engineered nanomaterials with the cell plasma membrane; what have we learned from membrane models?, Environ. Sci.: Nano, 2019, 6, 13–40 RSC
- C. Loos, T. Syrovets, A. Musyanovych, V. Mailänder, K. Landfester, G. Ulrich Nienhaus and T. Simmet, Functionalized polystyrene nanoparticles as a platform for studying bio-nano interactions, Beilstein J. Nanotechnol., 2014, 5, 2403–2412 CrossRef
- E. Guzmán and E. Santini, Lung surfactant-particles at fluid interfaces for toxicity assessments, Curr. Opin. Colloid Interface Sci., 2019, 39, 24–39 CrossRef
- A. des Rieux, E. G. E. Ragnarsson, E. Gullberg, V. Préat, Y.-J. Schneider and P. Artursson, Transport of nanoparticles across an in vitro model of the human intestinal follicle associated epithelium, Eur. J. Pharm. Sci., 2005, 25(4–5), 455–465 CrossRef CAS
- J. Cabellos, C. Delpivo, E. Fernández-Rosas, S. Vázquez-Campos and G. Janer, Contribution of M-cells and other experimental variables in the translocation of TiO2 nanoparticles across in vitro intestinal models, NanoImpact, 2017, 5, 51–60 CrossRef
- K. E. Carr, S. H. Smyth, M. T. McCullough, J. F. Morris and S. M. Moyes, Morphological aspects of interactions between microparticles and mammalian cells: intestinal uptake and onward movement, Prog. Histochem. Cytochem., 2012, 46, 185–252 CrossRef
- G. F. Schirinzi, I. Pérez-Pomeda, J. Sanchís, C. Rossini, M. Farré and D. Barceló, Cytotoxic effects of commonly used nanomaterials and microplastics on cerebral and epithelial human cells, Environ. Res., 2017, 159, 579–587 CrossRef CAS
- D. M. Brown, M. R. Wilson, W. MacNee, V. Stone and K. Donaldson, Size-dependent proinflammatory effects of ultrafine polystyrene particles: A role for surface area and oxidative stress in the enhanced activity of ultrafines, Toxicol. Appl. Pharmacol., 2001, 175, 191–199 CrossRef CAS
- H. Makkar, S. K. Verma, P. K. Panda, N. Pramanik, E. Jha and M. Suar, Molecular insight to size and dose-dependent cellular toxicity exhibited by a green synthesized bioceramic nanohybrid with macrophages for dental applications, Toxicol. Res., 2018, 7(5), 959–969 CrossRef CAS
- A. K. Sachan, R. K. Harishchandra, C. Bantz, M. Maskos, R. Reichelt and H. J. Galla, High-resolution investigation of nanoparticle interaction with a model pulmonary surfactant monolayer, ACS Nano, 2012, 6(2), 1677–1687 CrossRef CAS
- R. K. Harishchandra, A. K. Sachan, A. Kerth, G. Lentzen, T. Neuhaus and H. J. Galla, Compatible solutes: Ectoine and hydroxyectoine improve functional nanostructures in artificial lung surfactants, Biochim. Biophys. Acta, Biomembr., 2011, 1808, 2830–2840 CrossRef CAS
- S. Nazemidashtarjandi and A. M. Farnoud, Membrane outer leaflet is the primary regulator of membrane damage induced by silica nanoparticles in vesicles and erythrocytes, Environ. Sci.: Nano, 2019, 6, 1219–1232 RSC
- Y. Chen and G. D. Bothun, Lipid-Assisted formation and dispersion of aqueous and bilayer-embedded Nano-C 6o, Langmuir, 2009, 25, 4875–4879 CrossRef CAS
- A. Xi and G. D. Bothun, Centrifugation-based assay for examining nanoparticle-lipid membrane binding and disruption, Analyst, 2014, 139, 973–981 RSC
- N. Ganji, I. A. Khan and G. D. Bothun, Surface Activity of Poly(ethylene glycol)-Coated Silver Nanoparticles in the Presence of a Lipid Monolayer, Langmuir, 2018, 34, 2039–2045 CrossRef CAS
- N. M. Anaya, F. Faghihzadeh, N. Ganji, G. Bothun and V. Oyanedel-Craver, Comparative study between chemostat and batch reactors to quantify membrane permeability changes on bacteria exposed to silver nanoparticles, Sci. Total Environ., 2016, 565, 841–848 CrossRef CAS
- G. D. Bothun, N. Ganji, I. A. Khan, A. Xi and C. Bobba, Anionic and Cationic Silver Nanoparticle Binding Restructures Net-Anionic PC/PG Monolayers with Saturated or Unsaturated Lipids, Langmuir, 2017, 33, 353–360 CrossRef CAS
- E. Guzmán, M. Ferrari, E. Santini, L. Liggieri and F. Ravera, Effect of silica nanoparticles on the interfacial properties of a canonical lipid mixture, Colloids Surf., B, 2015, 136, 971–980 CrossRef
- E. Guzmán, L. Liggieri, E. Santini, M. Ferrari and F. Ravera, Influence of Silica Nanoparticles on Thermodynamic and Structural Properties of DPPC – Palmitic Acid Langmuir Monolayers, Colloids Surf., A, 2012, 413, 280–287 CrossRef
- K. L. Chen and G. D. Bothun, Nanoparticles Meet Cell Membranes: Probing Nonspecific Interactions using Model Membranes, Environ. Sci. Technol., 2014, 48, 873–880 CrossRef CAS
- Q. Wang, M. Lim, X. Liu, Z. Wang and K. L. Chen, Influence of Solution Chemistry and Soft Protein Coronas on the Interactions of Silver Nanoparticles with Model Biological Membranes, Environ. Sci. Technol., 2016, 50, 2301–2309 CrossRef CAS
- S. Schöttler, K. Klein, K. Landfester and V. Mailänder, Protein source and choice of anticoagulant decisively affect nanoparticle protein corona and cellular uptake, Nanoscale, 2016, 8, 5526–5536 RSC
- C. Corbo, R. Molinaro, A. Parodi, N. E. Toledano Furman, F. Salvatore and E. Tasciotti, The impact of nanoparticle protein corona on cytotoxicity, Nanomedicine, 2016, 11(1), 81–100 CrossRef CAS
- R. D. Vinluan, III and J. Zheng, Serum protein adsorption and excretion pathways of metal nanoparticles, Nanomedicine, 2015, 10(17), 2781–2794 CrossRef
- G. Hu, B. Jiao, X. Shi, R. P. Valle, Q. Fan and Y. Y. Zuo, Physicochemical properties of nanoparticles regulate translocation across pulmonary surfactant monolayer and formation of lipoprotein corona, ACS Nano, 2013, 7, 10525–10533 CrossRef CAS
- M. P. Monopoli, C. Åberg, A. Salvati and K. A. Dawson, Biomolecular coronas provide the biological identity of nanosized materials, Nat. Nanotechnol., 2012, 7, 779–786 CrossRef CAS
- D. Docter, D. Westmeier, M. Markiewicz, S. Stolte, S. K. Knauer and R. H. Stauber, The nanoparticle biomolecule corona: lessons learned - challenge accepted?, Chem. Soc. Rev., 2015, 44, 6094–6121 RSC
- V. H. Nguyen and B. J. Lee, Protein corona: A new approach for nanomedicine design, Int. J. Nanomed., 2017, 12, 3137–3151 CrossRef CAS
- S. Milani, F. Baldelli Bombelli, A. S. Pitek, K. A. Dawson and J. Rädler, Reversible versus Irreversible Binding of Transferrin to Polystyrene Nanoparticles: Soft and Hard Corona, ACS Nano, 2012, 6, 2532–2541 CrossRef CAS
- K. A. Dawson, A. Lesniak, F. Fenaroli, M. P. Monopoli, A. Christoffer and A. Salvati, Effects of the Presence or Absence of a Protein Corona on Silica Nanoparticle Uptake and Impact on Cells, ACS Nano, 2012, 6, 5845–5847 CrossRef
- J. R. Lawrence, G. D. W. Swerhone, J. J. Dynes, A. P. Hitchcock and D. R. Korber, Complex organic corona formation on carbon nanotubes reduces microbial toxicity by suppressing reactive oxygen species production, Environ. Sci.: Nano, 2016, 3, 181–189 RSC
- E. Casals, T. Pfaller, A. Duschl, G. J. Oostingh and V. Puntes, Time evolution of the nanoparticle protein corona, ACS Nano, 2010, 4, 3623–3632 CrossRef CAS
- E. S. Melby, S. E. Lohse, J. E. Park, A. M. Vartanian, R. A. Putans, H. B. Abbott, R. J. Hamers, C. J. Murphy and J. A. Pedersen, Cascading Effects of Nanoparticle Coatings: Surface Functionalization Dictates the Assemblage of Complexed Proteins and Subsequent Interaction with Model Cell Membranes, ACS Nano, 2017, 11, 5489–5499 CrossRef CAS
- S. Dominguez-Medina, L. Kisley, L. J. Tauzin, A. Hoggard, B. Shuang, A. S. D. S. Indrasekara, S. Chen, L.-Y. Wang, P. J. Derry, A. Liopo, E. R. Zubarev, C. F. Landes and S. Link, Adsorption and Unfolding of a Single Protein Triggers Nanoparticle Aggregation, ACS Nano, 2016, 10, 2103–2112 CrossRef CAS
- S. Dominguez-Medina, J. Blankenburg, J. Olson, C. F. Landes and S. Link, Adsorption of a protein monolayer via hydrophobic interactions prevents nanoparticle aggregation
under harsh environmental conditions, ACS Sustainable Chem. Eng., 2013, 1, 833–842 CrossRef CAS
- P. C. Ke, S. Lin, W. J. Parak, T. P. Davis and F. Caruso, A Decade of the Protein Corona, ACS Nano, 2017, 11, 11773–11776 CrossRef CAS
- K. Serre, L. Giraudo, L. Leserman and P. Machy, Liposomes targeted to Fc receptors for antigen presentation by dendritic cells in vitro and in vivo, Methods Enzymol., 2003, 373, 100–118 CAS
- S. Nagayama, K. ichi Ogawara, Y. Fukuoka, K. Higaki and T. Kimura, Time-dependent changes in opsonin amount associated on nanoparticles alter their hepatic uptake characteristics, Int. J. Pharm., 2007, 342, 215–221 CrossRef CAS
- B. D. Chithrani, A. A. Ghazani and W. C. W. Chan, Determining the size and shape dependence of gold nanoparticle uptake into mammalian cells, Nano Lett., 2006, 6, 662–668 CrossRef CAS
- D. Guarnieri, A. Guaccio, S. Fusco and P. A. Netti, Effect of serum proteins on polystyrene nanoparticle uptake and intracellular trafficking in endothelial cells, J. Nanopart. Res., 2011, 13, 4295–4309 CrossRef CAS
- Y. Zhu, W. Li, Q. Li, Y. Li, Y. Li, X. Zhang and Q. Huang, Effects of serum proteins on intracellular uptake and cytotoxicity of carbon nanoparticles, Carbon, 2009, 47(5), 1351–1358 CrossRef CAS
- X. Jiang, S. Weise, M. Hafner, C. Rocker, F. Zhang, W. J. Parak and G. Ulrich Nienhaus, Quantitative analysis of the protein corona on FePt nanoparticles formed by transferrin binding, J. R. Soc., Interface, 2010, 7, S5–S13 CAS
- P. C. Patel, D. A. Giljohann, W. L. Daniel, D. Zheng, A. E. Prigodich and C. A. Mirkin, Scavenger Receptors Mediate Cellular Uptake of Polyvalent Oligonucleotide-Functionalized Gold Nanoparticles, Bioconjugate Chem., 2010, 21(12), 2250–2256 CrossRef CAS
- I. Stayton, J. Winiarz, K. Shannon and Y. Ma, Study of uptake and loss of silica nanoparticles in living human lung epithelial cells at single cell level, Anal. Bioanal. Chem., 2009, 394, 1595–1608 CrossRef CAS
- A. Lesniak, A. Salvati, M. J. Santos-Martinez, M. W. Radomski, K. A. Dawson and C. Åberg, Nanoparticle Adhesion to the Cell Membrane and Its Effect on Nanoparticle Uptake Efficiency, J. Am. Chem. Soc., 2013, 135, 1438–1444 CrossRef CAS
- M. Arczewska, G. Czernel and M. Gagoś, Effect of the amphotericin B and its copper complex on a model of the outer leaflet of human erythrocyte membrane, J. Phys. Chem. B, 2016, 120, 11191–11204 CrossRef CAS
- K. H
![[a with combining cedilla]](https://www.rsc.org/images/entities/char_0061_0327.gif) c-Wydro and P. Dynarowicz-Łtka, Externalization of phosphatidylserine from inner to outer layer may alter the effect of plant sterols on human erythrocyte membrane - The Langmuir monolayer studies, Biochim. Biophys. Acta, Biomembr., 2012, 1818, 2184–2191 CrossRef
c-Wydro and P. Dynarowicz-Łtka, Externalization of phosphatidylserine from inner to outer layer may alter the effect of plant sterols on human erythrocyte membrane - The Langmuir monolayer studies, Biochim. Biophys. Acta, Biomembr., 2012, 1818, 2184–2191 CrossRef - P. Wydro, The influence of cholesterol on multicomponent Langmuir monolayers imitating outer and inner leaflet of human erythrocyte membrane, Colloids Surf., B, 2013, 103, 67–74 CrossRef CAS
- J. A. Virtanen, K. H. Cheng and P. Somerharju, Phospholipid composition of the mammalian red cell membrane can be rationalized by a superlattice model, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4964–4969 CrossRef CAS
-
Y. Yawata, Cell Membrane: The Reb Blood Cell as a Model, Wiley, 2003 Search PubMed
- D. Di Silvio, M. Maccarini, R. Parker, A. Mackie, G. Fragneto and F. Baldelli Bombelli, The effect of the protein corona on the interaction between nanoparticles and lipid bilayers, J. Colloid Interface Sci., 2017, 504, 741–750 CrossRef CAS
- D. Di Silvio, N. Rigby, B. Bajka, A. Mayes, A. Mackie and F. Baldelli Bombelli, Technical tip: high-resolution isolation of nanoparticle-protein corona complexes from physiological fluids, Nanoscale, 2015, 7, 11980–11990 RSC
- J. B. Sheffield, An introduction to ImageJ: A useful tool for biological image processing and analysis, Microsc. Microanal., 2008, 14, 898–899 CrossRef
- S. Yu, A. Perálvarez-Marín, C. Minelli, J. Faraudo, A. Roig and A. Laromaine, Albumin-coated SPIONs: An experimental and theoretical evaluation of protein conformation, binding affinity and competition with serum proteins, Nanoscale, 2016, 8, 14393–14405 RSC
- M. Kokkinopoulou, J. Simon, K. Landfester, V. Mailänder and I. Lieberwirth, Visualization of the protein corona: Towards a biomolecular understanding of nanoparticle-cell-interactions, Nanoscale, 2017, 9, 8858–8870 RSC
- P. Renz, M. Kokkinopoulou, K. Landfester and I. Lieberwirth, Imaging of Polymeric Nanoparticles: Hard Challenge for Soft Objects, Macromol. Chem. Phys., 2016, 217, 1879–1885 CrossRef CAS
- A. Adamiano, I. G. Lesci, D. Fabbri and N. Roveri, Adsorption of bovine serum albumin onto synthetic Fe-doped geomimetic chrysotile, J. R. Soc., Interface, 2015, 12, 20150186 CrossRef
- L. F. Chi, M. Anders, H. Fuchs, R. R. Johnston and H. Ringsdorf, Domain Structures in Langmuir-Blodgett Films Investigated by Atomic Force Microscopy, Science, 1993, 259, 213–216 CrossRef CAS
- X. Min and C. Daniel, Atomic structure and chemistry of humam serum albumin, Nature, 2015, 360, 40–46 Search PubMed
- V. Garbin, J. C. Crocker and K. J. Stebe, Nanoparticles at fluid interfaces: Exploiting capping ligands to control adsorption, stability and dynamics, J. Colloid Interface Sci., 2012, 387, 1–11 CrossRef CAS
- K. Du, E. Glogowski, T. Emrick, T. P. Russell and A. D. Dinsmore, Adsorption energy of nano- and microparticles at liquid-liquid interfaces, Langmuir, 2010, 26, 12518–12522 CrossRef CAS
- A. F. H. Ward and L. Tordai, Time-Dependence of Boundary Tensions of Solutions I. The Role of Diffusion in Time-Effects, J. Chem. Phys., 1946, 14, 453–461 CrossRef CAS
- N. Bizmark, M. A. Ioannidis and D. E. Henneke, Irreversible adsorption-driven assembly of nanoparticles at fluid interfaces revealed by a dynamic surface tension probe, Langmuir, 2014, 30, 710–717 CrossRef CAS
- M. Flury and S. Aramrak, Role of air-water interfaces in colloid transport in porous media: A review, Water Resour. Res., 2017, 53, 5247–5275 CrossRef
- M. Manciu and E. Ruckenstein, Ions near the air/water interface: I. Compatibility of zeta potential and surface tension experiments, Colloids Surf., A, 2012, 400, 27–35 CrossRef CAS
- C. Tian, J. Feng, H. J. Cho, S. S. Datta and R. K. Prud'Homme, Adsorption and Denaturation of Structured Polymeric Nanoparticles at an Interface, Nano Lett., 2018, 18, 4854–4860 CrossRef CAS
- Z. Adamczyk, Kinetics of diffusion-controlled adsorption of colloid particles and proteins, J. Colloid Interface Sci., 2000, 229, 477–489 CrossRef CAS
- J. Sánchez-González, J. Ruiz-García and M. Gálvez-Ruiz, Langmuir–Blodgett films of biopolymers: a method to obtain protein multilayers, J. Colloid Interface Sci., 2003, 267, 286–293 CrossRef
- S. M. Lystvet, S. Volden, Ø. Halskau and W. R. Glomm, Immobilization onto gold nanoparticles alters α-lactalbumin interaction with pure and mixed phospholipid monolayers, Soft Matter, 2011, 7, 11501–11509 RSC
- A. C. Mensch, J. T. Buchman, C. L. Haynes, J. A. Pedersen and R. J. Hamers, Quaternary Amine-Terminated Quantum Dots Induce Structural Changes to Supported Lipid Bilayers, Langmuir, 2018, 34, 12369–12378 CrossRef CAS
- A. M. Farnoud and J. Fiegel, Interaction of dipalmitoyl phosphatidylcholine monolayers with a particle-laden subphase, J. Phys. Chem. B, 2013, 117, 12124–12134 CrossRef CAS
- B. Wang, L. Zhang, C. B. Sung and S. Granick, Nanoparticle-induced surface reconstruction of phospholipid membranes, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18171–18175 CrossRef CAS
- X. Wang, Y. Zhang, J. Wu, M. Wang, G. Cui, J. Li and G. Brezesinski, Dynamical and morphological studies on the adsorption and penetration of human serum albumin into phospholipid monolayers at the air/water interface, Colloids Surf., B, 2002, 23, 339–347 CrossRef CAS
- N. C. de Souza, W. Caetano, R. Itri, C. A. Rodrigues, O. N. Oliveira, J. A. Giacometti and M. Ferreira, Interaction of small amounts of bovine serum albumin with phospholipid monolayers investigated by surface pressure and atomic force microscopy, J. Colloid Interface Sci., 2006, 297, 546–553 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0en00934b |
| This journal is © The Royal Society of Chemistry 2021 |