
Reduction of silver ions to form silver nanoparticles by redox-active organic molecules: coupled impact of the redox state and environmental factors†
Feng
Dong
 ab,
Chao
Wu
c,
Ai-Jun
Miao
*c and
Ke
Pan
*a
ab,
Chao
Wu
c,
Ai-Jun
Miao
*c and
Ke
Pan
*a
aShenzhen Key Laboratory of Marine Microbiome Engineering, Institute for Advanced Study, Shenzhen University, Shenzhen, 518060, China. E-mail: panke@szu.edu.cn
bKey Laboratory of Optoelectronic Devices and Systems of Ministry of Education and Guangdong Province, College of Optoelectronic Engineering, Shenzhen University, Shenzhen, 518060, China
cState Key Laboratory of Pollution Control and Resource Reuse, School of the Environment, Nanjing University, Nanjing, Jiangsu Province 210046, China. E-mail: miaoaj@nju.edu.cn
First published on 9th December 2020
Abstract
The release of silver ions (Ag+) is an important process in determining the environmental fate and toxic effects of silver nanoparticles (AgNPs), but the released Ag+ can be reduced to form new AgNPs in environments. Here the reduction of Ag+ by redox-active organic molecules to form AgNPs under different lighting and oxygen conditions was investigated using anthraquinonoe-2,6-disulphonate (AQDS) and nicotinamide adenine dinucleotide (NAD+) as representatives. In anaerobic environments, oxidized AQDS (AQDSox) and NAD+ failed to reduce Ag+ to form AgNPs, whereas reduced AQDS (AQDSred) and NAD+ (NADH) produced a concentration-dependent AgNP production pattern, underlining a key role of the redox state in Ag+ reduction by organics. The presence of O2 diminished the reducing capacity of NADH and AQDSred, probably due to their oxidation by O2. However, under UV light, the AgNP formation rate was dramatically enhanced in AQDS, independent of O2. Our results revealed that the Ag+ photoreduction was mainly attributed to the photoexcited AQDS, not the radical intermediates. For the complex organic humic acid (HA), and consistent with the phenomena in AQDS, the reduced HA (HAred) reduced Ag+ at a much faster rate than the native HA (HAox) in anaerobic and dark environments, and the amounts of AgNPs produced by HAred decreased in oxic environments. UV illumination enabled significant Ag+ reduction in both HAox and HAred, and O2 promoted the AgNP formation. Our study systematically revealed different Ag+-reduction pathways to form AgNPs, with an organic's redox state and environmental factors having a combined effect.
Environmental significanceThe mechanism of reduction of silver ions (Ag+) to form silver nanoparticles (AgNPs) is poorly understood under environmentally relevant conditions. Our work shows that the redox-active organic's redox state couples with light and O2 to influence Ag+ reduction and AgNP formation kinetics. We find considerable Ag+ reduction by reduced organics, but not by oxidized ones under dark conditions, where O2 diminishes the AgNP formation. In contrast, significant Ag+ reduction by organics is obtained under UV irradiation regardless of their redox state, and O2 has a limited influence. The results demonstrate different pathways in Ag+ reduction to form AgNPs by environmental organics. Our findings are meaningful for evaluating the environmental fate and risks of AgNPs. |
1 Introduction
Many fields have used silver nanoparticles (AgNPs) as an antimicrobial agent.1,2 However, there is increasing concern about the ecological risk of AgNPs due to their high toxicity towards a variety of environmental organisms.3–7 The speciation of AgNPs strongly affects their toxicity, with the bioavailability of Ag+ playing a crucial role.8,9 The dissolution of AgNPs to release Ag+ is a thermodynamically favorable process in natural environments.10 Given the standard reduction potential of +799 mV for Ag+/Ag, the released Ag+ has a strong tendency to be reduced to metallic Ag. In contrast to dissolution, the reduction of Ag+ results in the decline of free-Ag+ concentration, thereby decreasing the toxicity.11In aquatic environments, dissolved organic matter (DOM) such as humic acid (HA) can reduce Ag+ to form colloidal Ag.12 Solar irradiation enhances Ag+ reduction due to the superoxide anion radical (O2˙−) produced by the reaction between photo-excited DOM and O2.13–15 But similar light-driven AgNP formation by Suwannee River HA has also been observed under O2-free conditions where O2˙− production was absent.16 Moreover, Wimmer and co-workers recently detected ng L−1 quantities of AgNPs in the deep water layer of natural lakes, where sunlight intensity is low.17 Here, DOM-mediated Ag+ reduction was found without light irradiation in a natural environment, complicating our understanding of AgNP generation. At present, the electron-transfer pathway from DOM to Ag+ to generate AgNPs remains ambiguous under environmentally relevant conditions. Generally, radical intermediates such as O2˙− can be produced by photoactivated organics to reduce Ag+ under light radiation, or DOM may directly transfer electrons to Ag+ to produce AgNPs, but the reducing capacity varies greatly among different types of DOM due to their heterogenous and complex chemical composition.
The production of AgNPs via Ag+ reduction has also been frequently observed in biological systems. Previous studies have documented biogenic AgNPs in bacteria and fungi, which formed via an O2˙− production mechanism.18–20 Microbial Ag+ reduction also readily occurs in anoxic and anaerobic environments.11,21,22 Indeed, many other metal ions and metal oxides, including Fe3+, SeO42−, and TeO42−, have reportedly been reduced by anaerobic bacteria or methanogenic archaea into zerovalent nanoparticles.23–25 Considering that these metals have relatively high reduction potentials (>400 mV), from a thermodynamic point of view, it is not surprising that they could serve as electron acceptors to facilitate the reduction. The biotic metal ion reduction was ascribed to the reducing molecules or groups in the biological metabolites.25–27 In fact, the microbial products derived from metabolism and plant/animal degradation might contain a variety of redox-active substances such as humics, phenazines, quinines, flavins, and cytochromes, which can significantly influence the geochemical transformation of many elements.28–30 For example, saprophytic bacteria (Shewanella spp.) transported intracellular electrons to exterior HA and flavin mononucleotides that further donated electrons to iron minerals dissolving the minerals as well as to Hg(II) forming mercury particles.31–33 We assume that these redox-active substances are involved in the extracellular AgNP production. Silver nanoparticles have also been found inside cells,34 with the Ag+ reduction documented to be closely correlated with cytosolic electron transport chain (ETC) components, including nicotinamide adenine dinucleotide phosphate,35 nitrate reductase36,37 and Mtr–Omc cross-membrane electron transport machinery.22,30 However, our mechanistic understanding of the relationship between the ETC and AgNP formation is very limited. It should be noted that the redox-active substances in the ETC undergo multiple redox cycles in a series of electron transfer processes that are accompanied by a cyclic change of the reduced and oxidized states. While many electron transporters are sensitive to light,29 the influence of the redox state on Ag+ reduction in different light radiation environments remains an open question.
To elucidate the role of redox-active organics in Ag+ reduction and the corresponding AgNP production, we used disodium anthraquinone-2,6-disulfouate (AQDS), which is a typical HA analogue, and nicotinamide adenine dinucleotide (NAD) as the representative of environmental and biological electron transporters, respectively. We examined the AgNP formation kinetics of the organics under different concentrations and redox states. The reductions of Ag+ by oxidized and reduced HA were compared in the presence and absence of oxygen under darkness, visible and UV light conditions. We aim to elucidate the potentially different pathways of Ag+ reduction by environmental organic molecules. A generalized model was proposed by exploring the influence of environmental factors on AgNP production. Our results demonstrated that an organic's redox state combines with oxygen and light radiation to control Ag+ reduction, providing a new insight into the biogeochemical cycle of AgNPs in natural environments.
2 Materials and methods
2.1 Chemicals
Silver nitrate (AgNO3), sodium borohydride (NaBH4), sodium citrate, bovine serum albumin (BSA), nitrotetrazolium blue chloride, HA (organic mixture extracted from a natural environment), palladium catalyzer (10 wt% loaded on activated carbon; Pd/C), tris(hydroxymethyl)aminomethane (Tris), dimethyl sulfoxide (DMSO), dipotassium hydrogen phosphate (K2HPO4), potassium dihydrogen phosphate (KH2PO4) and superoxide dismutase (SOD) were purchased from Sigma-Aldrich. Nicotinamide adenine dinucleotide disodium salt (NAD+), the reduced form of nicotinamide adenine dinucleotide (NADH), AQDS, hydrogen peroxide (H2O2), acetic acid, acetylacetone, and ammonium acetate were purchased from Macklin (Shanghai Macklin Biochemical Co., Ltd., Shanghai, China).2.2 Ag+ reduction by NADH and AQDS
To avoid the influence of O2, AgNO3 was reacted with NADH solutions under anaerobic conditions. Although the environmentally relevant concentration of Ag+ or AgNPs was in the low range (ng L−1),38,39 for the sake of feasibility and quick measurement, UV-vis spectrometry that measured the surface plasmon resonance absorbance of AgNPs was used in order to quantify the AgNP formation kinetics at high-time resolution, and relatively high concentrations of Ag+ and organics were used to obtain a sufficient amount of AgNPs formed from the Ag+ reduction. The two solutions were mixed in a serum bottle (100 mL volume) with shaking at 30 °C. The anaerobic conditions were obtained by flushing nitrogen gas into the solution and headspace for at least 20 min, followed by sealing the bottle with a rubber stopper. To investigate the concentration effect, the AgNO3 and NADH concentrations varied from 0–2 and 0–4 mM, respectively. To analyze the AgNP formation kinetics, the UV-vis absorption spectra (UVAS) of the mixture were measured as a function of time (UV-2600, Shimadzu). The reduction of Ag+ by AQDS was performed with the same procedure. Citrate and NaBH4 were used as the controls since they had different Ag+ reducing capabilities and were commonly used for AgNP synthesis in the laboratory. The hydrodynamic size of the generated AgNPs was measured by a dynamic light scattering method (DLS; Zetasizer Nano, Malvern Instruments). Their morphology was analyzed by transmission electron microscopy (TEM) combined with energy dispersive X-ray spectrometry (EDS) for elemental analysis (JEOL, JEM2100F).2.3 Effects of the NAD and AQDS redox state on Ag+ reduction
To investigate the influence of the redox state on Ag+ reduction, the reductions of Ag+ by NAD+ and NADH or oxidized and reduced AQDS were compared under anaerobic conditions. The purchased AQDS was already in an oxidized state (AQDSox). The reduced AQDS (AQDSred) was produced by a chemical reduction method.40 Briefly, N2 was flushed into the AQDSox solution (30 mL) and headspace for 30 min to remove the oxygen from an anaerobic bottle (100 mL volume). Hydrogen gas (H2), produced from the hydrolysis of 100 mM NaBH4 (1 mg mL−1 Pd/C as the catalyst), was then passed through the AQDSox solution for 1 h. After sealing the bottle, the reduction continued for another 20 h at 30 °C in an incubator with shaking. The AQDSred solution was obtained by filtering the reaction mixture through a 0.2 μm membrane to remove the Pd/C catalyst and then flushing with N2 to remove residual H2.An experiment with partially oxidized NADH or AQDSred was also performed. The reoxidation of NADH and AQDSred was achieved by exposing them to air for 15 h at 30 °C in the dark. The oxidation was quenched by flushing N2 into the solution to remove the unreacted O2; NAD+ and AQDSox were used as controls. The same procedures were used for the Ag+ reduction experiments in NADH and AQDS under different redox states.
2.4 Photoreduction of Ag+ into silver nanoparticles
To investigate the influence of light on Ag+ reduction, 0.1 mM AgNO3 was reduced with 0.5 mM NAD+ or AQDSox (10 mL total volume) in quartz vials (20 mL) without adjustment of pH in the dark and under light irradiation at 30 °C. A control experiment was performed by using sole AgNO3 solution (0.1 mM). The pH of 0.5 mM AQDSox and NAD+ solution was 6.2 and 7.3, respectively. The vials were wrapped with multiple layers of aluminum foil to achieve a dark environment. For the photoreduction, low pressure mercury vapor (8 W) and light-emitting-diode light (4 W) lamps were used to simulate the UV and visible light of solar irradiation, respectively. The spectral irradiance of the two lamps and UV-vis absorption spectra of AgNO3 and organics are shown in Fig. S1 (ESI†). In addition, the AgNP generation kinetics in aerobic and anaerobic environments under different light conditions were compared to examine the effect of O2. For the aerobic reactions, the vials were exposed to air without sealing. For the anaerobic reactions, the vials were flushed with N2 for at least 15 min and sealed with a rubber stopper. To measure the AgNP-formation kinetics, the UVAS of the samples were determined at designated time intervals.2.5 Analyzing the role of radical species in Ag+ reduction
The hydroxyl radical (˙OH) production from irradiating AQDSox with UV light was analyzed by a colorimetric method using Tris and Nash's reagent and measuring the absorbance at 412 nm.41 To examine the involvement of ˙OH in Ag+ reduction, we measured AgNP formation in the presence of DMSO, which is an ˙OH scavenger.42 In this experiment, AgNO3 (0.1 mM) was mixed with AQDSox (0.5 mM) in the presence of 0–50 mM DMSO under anaerobic conditions. The control was 0.5 mM AgNO3 mixed with 50 mM DMSO in the absence of AQDS. In addition, we measured the reduction of 0.1 mM Ag+ under UV light radiation in the presence of H2O2 (0–10 mM). To examine whether O2˙− participated in generating AgNPs, 0.1 mM Ag+ was reduced with 0.5 mM AQDSox under UV irradiation in the presence of 0–200 kU L−1 of SOD under oxic conditions. The AgNP formation kinetics were analyzed as described above. As a control, AgNO3 (0.1 mM) was mixed with 200 kU L−1 of SOD in the absence of AQDSox.2.6 Reduction of Ag+ by HA
We investigated the reduction of 0.1 mM Ag+ by native or reduced HA under light illumination. The purchased HA had been exposed to air for a very long time, and thus was assumed to be in an oxidized state (HAox).43,44 The reduced HA (HAred) was prepared by the Pd/C catalyzed reduction method discussed above. To study the effect of the redox state, we first analyzed the AgNP-formation kinetics by reducing 0.1 or 0.5 mM AgNO3 with 100 mg L−1 of HAox or HAred in quartz vials in the dark. By using relatively higher concentrations of AgNO3 and HA than those in natural environments, it was more feasible to measure the AgNP formation kinetics with UV-vis absorption spectrometry at high time-resolution. To investigate the influence of light illumination and O2, we reacted 0.5 mM AgNO3 with 100 mg L−1 of HAox or HAred (pH, 6.5–7.5) in the dark and under visible and UV light irradiation conditions, with and without the presence of air. The temperature was set at 30 °C. The AgNP-formation kinetics were determined from the UVAS. Because HA showed a considerable adsorption spectral overlap with the AgNPs, the spectrum of the AgNPs formed was obtained by subtracting the HA spectrum from that of the mixture.2.7 Statistical analysis
The software OriginPro (v. 9.0) was used for the plotting, while PASW Statistics (v. 18.0) was used to conduct the statistical analysis. For the statistical significance analysis, *, **, and *** denote p values of <0.05, <0.01, and <0.001, respectively.3 Results and discussion
3.1 AgNP formation kinetics
To avoid O2 interference, the reactions between Ag+ and NADH were performed under anaerobic conditions. When reducing 0.1 mM Ag+ with 0.5 mM NADH, the noticeable absorbance between 400 and 550 nm (where NAD+ and NADH displayed negligible absorption) implied the formation of AgNPs (Fig. S2a†). The UVAS curve featuring the localized surface plasmon resonance (LSPR) of the AgNPs was obtained by subtracting the absorbance of NADH from that of the mixture (Fig. S2b†). The declining absorbance at 323 nm, resulting from NADH oxidation to NAD+, was accompanied by an increase in the peak absorbance value (Apeak) of the LSPR band, demonstrating the direct reduction of Ag+ by NADH. The Apeak of the AgNP's UVAS curve is well correlated with AgNP concentration when the nanoparticle's morphology and surrounding environment do not significantly change.45,46 For convenience, it was used to quantify the generated AgNPs. During the reaction, the Apeak increased slowly during the first day, followed by a dramatic rise, and leveling off after 4 days, implying an apparent AgNP growth process (Fig. S2c†). The initially slow change in the Apeak was presumably due to the small size and low concentration of freshly generated AgNPs, which the UVAS could not readily detect. We therefore used the Apeak during the rapid-growth phase to calculate the AgNP-formation rate.Fig. 1a shows that the AgNP-formation rate generally increased with Ag+ concentration. At Ag+ concentrations of 0–0.2 mM, all Ag+ appeared to be reduced by 0.5 mM NADH. The Apeak of the matured AgNPs was proportional to the initially added Ag+ concentration, and their normalized LSPR bands were quite similar, suggesting that AgNP formation was consistent across different Ag+ concentrations (Fig. S3†). At high Ag+ concentrations (≥0.5 mM), we observed the gradual decrease of absorbance of finally formed AgNPs at around 400 nm, a red-shift of the absorption spectra, a broadening of the UVAS bands, and the evolving color of the corresponding AgNP suspension (Fig. 1b). In response to the increasing Ag+ concentration, the hydrodynamic size gradually increased from 10 to over 250 nm (Fig. 1c), probably due to the aggregation of the AgNPs47 or the formation of large AgNPs.48 The generated nanoparticles were almost spherical (Fig. 1d), and the EDS results revealed apparent intensities at 2.98 and 22.2 keV (kilo-electron-volts), which correspond to Ag, confirming AgNP generation.30
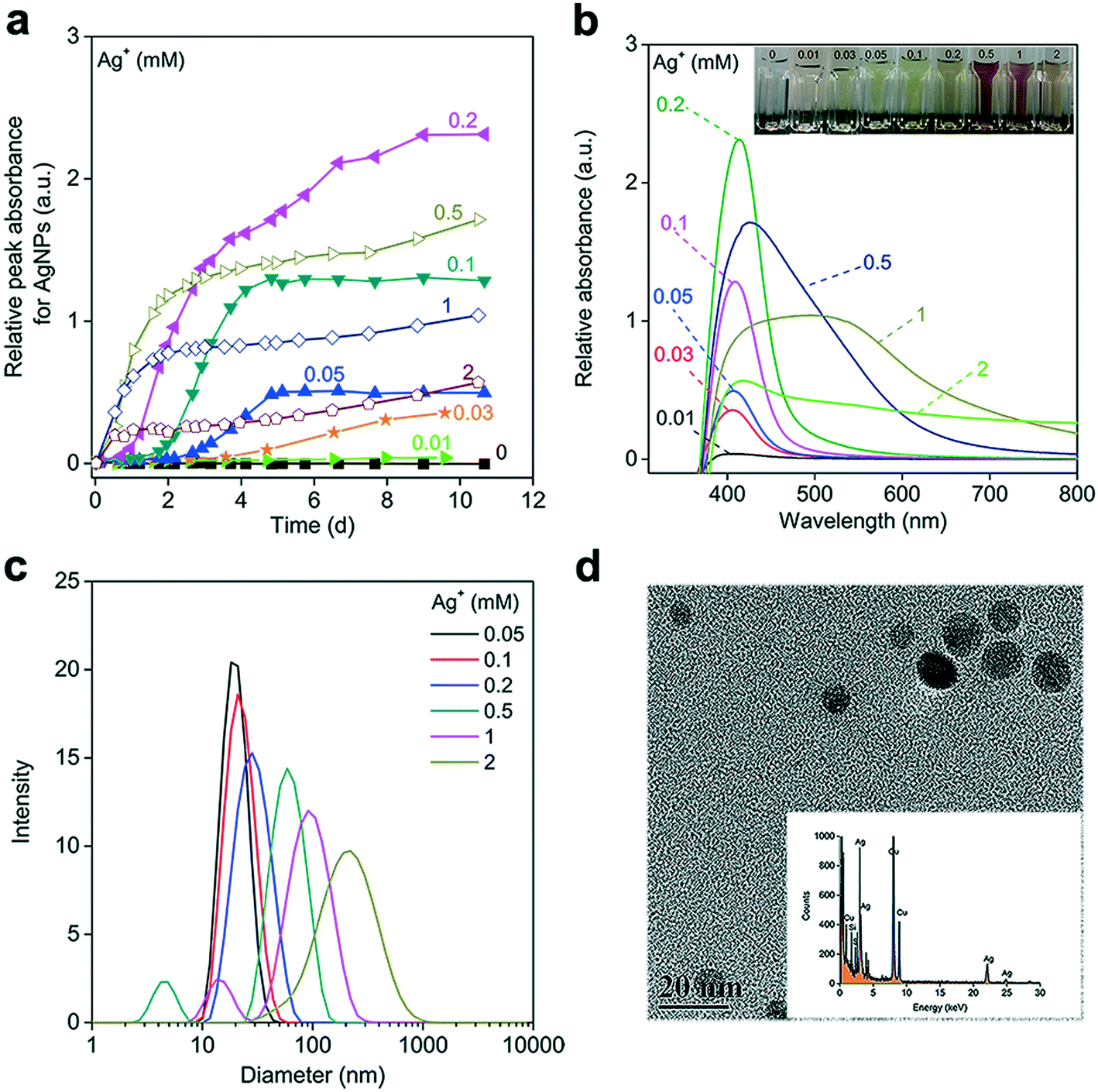 | ||
| Fig. 1 Formation of silver nanoparticles (AgNPs) by reducing varying concentrations of Ag+ (0–2 mM) with 0.5 mM NADH under anaerobic conditions. (a) AgNP formation kinetics. The relative peak absorbance of the AgNPs was the peak value of the UV-vis absorption spectra (UVAS) of the AgNPs, obtained by subtracting the spectrum of NADH from that of the final mixed products. a.u. refers to arbitrary units of absorbance. (b) UVAS of the AgNPs formed by ∼10 days. The inset shows the corresponding formed AgNP suspensions with their evolving colors. (c) Hydrodynamic size of the formed AgNPs as measured by a dynamic light scattering (DLS) method. AgNP concentrations <0.05 mM were below the detection limit of DLS. (d) Representative transmission electron microscopy (TEM) image of the AgNPs generated by reducing 0.1 mM Ag+ with 1 mM NADH under anaerobic conditions. The inset plots the EDS spectrum of the AgNPs formed. | ||
Increasing the NADH concentration promoted AgNP generation (Fig. 2a). At NADH concentrations >0.5 mM, the complete reduction of 0.1 mM Ag+ was achieved with nearly the same amount of AgNPs produced (Apeak = 1.28 ± 0.04) in 7 days. But as the NADH concentration increased from 2 to 4 mM, the generation of apparent AgNPs within the UVAS detection limit (Apeak > 0.01) was gradually delayed, suggesting that very high NADH concentrations may not favor the initial formation and growth of AgNPs. We verified this phenomenon with citrate, which is an important component of the respiration chain that produces NADH for the ETC, as well as a reductant for AgNP synthesis in the laboratory.49 At an Ag+ concentration of 0.1 mM, AgNPs could barely form in the presence of citrate, probably because citrate is not a strong reducing agent, exhibiting much weaker electron donating activity than NADH. When we increased the Ag+ concentration to 1 mM, a consistent trend was observed, but the concentration effect was more pronounced (Fig. 2b). The citrate-reduced AgNPs were not stable and tended to aggregate, as demonstrated by the reduced Apeak in the latter phase. Notably, a very high concentration of citrate (≥30 mM) completely prevented AgNP formation (Fig. 2c), probably due to the strong Ag–citrate complexation, as observed in a previous study.50
 | ||
| Fig. 2 Influence of the reducing organics' concentrations on silver nanoparticle (AgNP) formation. (a) Kinetics of AgNP formation when reducing 0.1 mM Ag+ with 0.1–4 mM NADH under anaerobic conditions. (b) Kinetics of AgNP formation when reducing 1 mM Ag+ with 1–40 mM citrate under anaerobic conditions. (c) AgNP-formation rates, calculated by linear regression of the relative peak absorbance of the formed AgNPs against time during the rapid-growth phases in panels a and b. Error bar represents the standard error of the slope. The pH of NADH (0.1–4 mM) and citrate solution (1–40 mM) was 5.7–8.2 and 6.9–7.5, respectively. | ||
Taken together, our data indicated that the concentrations of both the reducing organics and Ag+ significantly influenced the kinetics of AgNP formation, which involves nucleation, growth, and maturation processes.51 The reduction of Ag+ generated AgNP nuclei that were the basic building units for silver nanocrystals.52 Sufficient Ag+ and NADH might be required to produce a large number of Ag nuclei and subsequent small AgNP clusters. Aggregative growth, with small nanoparticles directly attaching to one another to form larger particles, has been reported to be a critical mechanism governing AgNP maturation, when the small AgNP clusters have overcome the aggregation energy barrier to form mature AgNPs through coalescence.51–53 The presence of very high concentrations of the reducing agent may give rise to excessive interaction between the organics and primarily formed AgNP clusters. This may reduce the collision rates or increase the surface energy barrier for effective attachment, which would account for the delayed maturation and reduced AgNP growth rate.
3.2 Effects of the organic redox state on the reduction of Ag+
To elucidate the influence of the redox state on Ag+ reduction, the AgNP-formation kinetics of reduced and oxidized organic molecules were compared under anaerobic conditions to avoid the interference of O2. Fig. 3 shows that neither NAD+ nor AQDSox was able to reduce Ag+ to form AgNPs. In contrast, upon mixing Ag+ with AQDSred, AgNPs quickly generated at a rate similar to that in a NaBH4 solution, suggesting that the reduced AQDS had a very strong Ag+ reduction activity (Fig. S4a and b†). The amount of AgNPs formed by AQDSred increased linearly with Ag+ concentration (Fig. S4c†). Pre-exposure to air resulted in an apparent delay in AgNP production in reoxidized NADH (Fig. 3a) and a decreased amount of AgNPs formed in reoxidized AQDSred (Fig. 3b). Previous work has shown that the reduced moieties of dissolved and particulate organic matter undergo oxidation by O2 in oxic environments, which may involve the transfer of electrons from reduced organics/quinones to O2 and the release of protons.54,55 We expected that exposure to air would cause a fraction of the NADH or AQDSred to be oxidized by O2 to NAD+ or AQDSox (which showed no Ag+ reducing capability) and lead to decreased reducing power.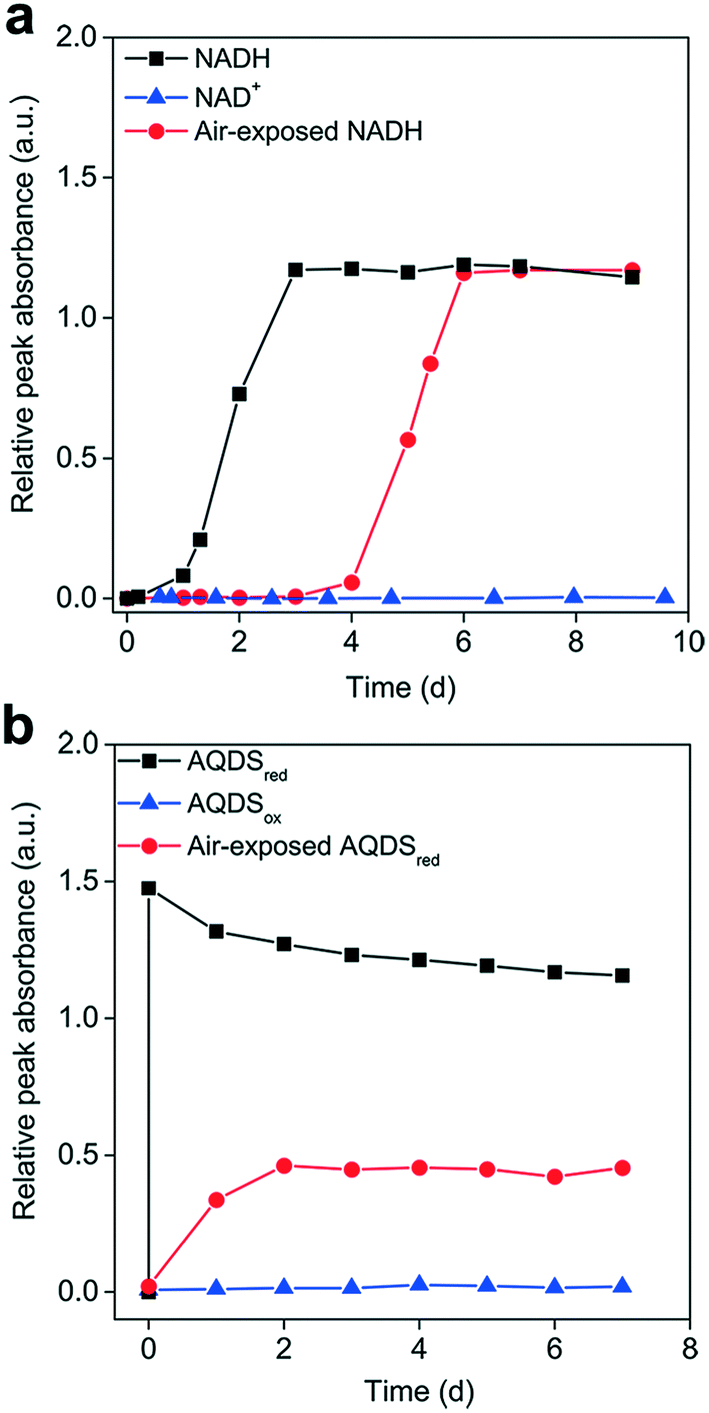 | ||
| Fig. 3 Influence of the organic's redox state on Ag+ reduction to generate silver nanoparticles (AgNPs). AgNP-formation kinetics when reducing 0.1 mM Ag+ with (a) 0.5 mM NADH, NAD+, and air-exposed NADH; or (b) 0.5 mM AQDSred, AQDSox, and air-exposed AQDSred under anaerobic conditions. Air exposure for 15 h may allow for the partial oxidation of NADH to NAD+ and AQDSred to AQDSox. | ||
Although identical amounts of AgNPs were formed in the reoxidized NADH and primary NADH, the AgNP formation process was significantly delayed in the reoxidized NADH. We hypothesized that the NAD+ produced competes with NADH for Ag+ binding, thus limiting the availability of free Ag+ for NADH reduction. Amino acids with a high affinity to silver have been shown to inhibit the AgNP formation mediated by photo-reduced tyrosine and c-cytochrome in peptides, which was attributed to silver–amino acid complexation.56,57 We tested our hypothesis by reducing Ag+ with NADH in the presence of BSA, which can complex with Ag+ but not reduce it. Our results showed a concentration-dependent delay in AgNP production in the presence of BSA (Fig. S5†), likely due to Ag+–BSA interactions preventing Ag+ reduction by NADH, thus supporting our hypothesis.
Earlier work has shown that chemically-reduced HA reduces Ag+ and forms AgNPs at a rate much faster than untreated HA.58 A similar phenomenon was also reported for the reduction of Hg2+ to Hg0 by microbially-reduced HA.32 Our results were consistent with these observations, highlighting the important role of the reduced state in Ag+ reduction. In general, an anoxic/anaerobic environment enables redox-active organics such as humics and AQDS in a reduced state,54 as they can generally serve as electron acceptors for microbial respiration in anaerobic consortia.59,60 It was expected that these microbially-reduced organics would transfer electrons to Ag+ to support AgNP production. In contrast, in oxic environments, not only would the organics mostly be in an oxidized state due to oxidation by O2, but also the O2 molecules would compete with Ag+ for the limited electron-donating capacity of the redox-active organics,54,55 consequently restricting AgNP formation.
3.3 Influence of light irradiation on Ag+ reduction
While the redox-active organics are expected to be present in an oxidized state after long-term exposure to dissolved O2, they also receive solar irradiation in surface waters and soils. Therefore, we explored the Ag+ reduction behavior of NAD+ and AQDSox under simulated visible and UV light. Fig. 4a shows that UV irradiation, but not darkness or visible light, drove the Ag+ reduction. The roughly similar levels of AgNP formation rates in the presence and absence of NAD+ might suggest that NAD+ had a limited influence on Ag+ reduction. However, Ag+ was rapidly reduced by AQDSox under UV illumination, which was independent of oxygen. The UV light catalyzed AgNP-formation rate mediated by AQDSox was more than one order of magnitude greater than that mediated by NAD+ and the control. The photochemical characteristics of organic substances are reportedly associated with their molecular structure and corresponding light absorption.61 The discrepancy between AQDSox and NAD+ may be due to the different optical properties of quininone and nicotinamide/adenine groups under the lighting conditions used, which needs further investigation. | ||
| Fig. 4 Effects of oxygen and light on Ag+ reduction by organics. (a) Silver nanoparticle (AgNP)-formation rate when reducing 0.1 mM AgNO3 with 0.5 mM NAD+ or AQDSox under dark, visible light, or UV light conditions in the presence and absence of air. Reducing sole AgNO3 (0.1 mM) was performed in the absence of organics as a control. (b) Hydroxyl radical (˙OH) production from exposing 0.5 mM AQDSox to UV irradiation in an anaerobic environment. The ˙OH produced was quantified with a colorimetric method using a Nash agent; the absorbance was measured at 412 nm and increased with Tris concentration. (c) Impact of DMSO on AgNP formation by reducing Ag+ (0.1 mM) with AQDSox (0.5 mM) under UV light irradiation in an anaerobic environment. | ||
Many DOM can be promoted to the excited singlet state (1DOM*) upon absorbing solar energy, which undergoes intersystem crossing to the excited triplet state (3DOM*).62 The formed 3DOM* is an important photoactivated component mediating the interaction between DOM and surrounding environments. It can react with O2, H2O and other chemicals to produce radicals and superoxide, such as O2˙−, ˙OH and H2O2.63–65 The photochemical behaviors of AQDS are consistent with that of DOM to some extent.66,67 With regard to the influence of O2, our results showed that AQDSox exhibited apparent Ag+ photoreduction activity in oxic environments. It has been reported that O2˙− mediates the Ag+ reduction by DOM in sunlit waters, with photo-excited DOM transferring electrons to the oxygen molecule to produce O2˙− that participates in Ag+ reduction.14,15 The AgNPs, particularly those that are small, tend to dissolve in oxygenated water.68 That AgNP production under aerobic conditions is faster than that under anaerobic conditions may reflect O2˙−-photoreduction outcompeting dissolution and vice versa. The discrepancy between the aerobic and anaerobic AgNP-formation rates was not as large in AQDSox, suggesting that O2˙− was not an essential factor in these Ag+ photoreduction processes. This would agree with earlier work reporting O2-independent Ag+ reduction by HA under simulated solar irradiation.16,69
As an O2˙− mechanism can be excluded in the absence of O2, we explored other pathways in the anaerobic Ag+ reduction process. Apart from O2˙−, other radical species such as ˙OH are also ubiquitously observed in aquatic environments under solar radiation,70 where the aromatic and less saturated components of DOM play a positive role.71 Indeed, minor concentrations of ˙OH were detected over time when irradiating AQDSox with UV light over time in anaerobic environments (Fig. 4b). Consistent with a recent work showing ˙OH production from photosensitized NAD+,41 the ˙OH production rate increased with Tris and AQDS concentration. These results suggested that the photoexcited AQDS might react with surrounding chemicals to produce hydroxyl radicals during the Ag+ reduction process. Photo-excited organics, particularly quinone, can act as both a strong reductant and an oxidant.62 In oxic environments, AQDS can be promoted from the ground state into the excited state upon UV light radiation, which can reduce O2 to produce O2˙−.66 Frequently, photo-induced ˙OH generation involves the dismutation of O2˙− to produce H2O2, followed by photolysis of H2O2 to produce ˙OH in aquatic environments.72,73 This can be ruled out in anaerobic environments, where O2˙− is not expected to be produced. The photo-induced ˙OH production pathway was still not well understood, and it may be produced through photo-activated organics abstracting an H atom from H2O or OH−.74 More types of aromatic and oxygenated DOM were reported to be more efficient in producing˙OH,71 strongly suggesting the involvement of AQDS in ˙OH photoproduction.75
To further elucidate the potential role of photoexcited AQDS and radical intermediates in AgNP production, we first performed a control experiment by investigating the photoreduction of Ag+ by AQDS in the presence of an ˙OH scavenger. Adding DMSO that can react with ˙OH significantly suppressed AgNP production in a dose-dependent pattern (Fig. 4c). The concentrations of DMSO >5 mM completely inhibited AgNP formation. However, given the redox potential of ˙OH/H2O of +2.73 V,76,77 it was unlikely for ˙OH to act as a reductant to directly reduce Ag+. Since the DOM in the excited state was able to oxidize various organic molecules,62 the inhibitory effect of DMSO may be ascribed to their interaction with photoactivated AQDS,66 diminishing the Ag+ reduction. Additionally, we investigated the AgNP formation process in the presence of H2O2 instead of AQDS. It is known that H2O2 is a strong ˙OH producer under UV irradiation.74 We did not observe considerable AgNP production in the presence of different concentrations of H2O2 (0.1–10 mM) under UV light in anaerobic environments (Fig. S6†), excluding the direct involvement of ˙OH in Ag+ photoreduction. In addition, we found that the addition of SOD, which catalyzes O2˙− disproportionation, failed to diminish Ag+ reduction, but to some extent, it had a positive influence on the AgNP generation (Fig. S7†). Collectively, these results suggest that the radical intermediates including ˙OH and O2˙− were not involved in the Ag+ reduction in anaerobic environments. It was more likely that the photoexcited AQDS directly participated in the light-driven AgNP production. It has reported that the direct Ag+ reduction process involved the binding of Ag+ to DOM.16,58 Recently, a ligand-to-metal charge transfer (LMCT) mechanism was found to partly account for the direct reduction of Fe3+ by organics without the involvement of radical intermediates.78 Given a similar redox potential of Ag+/Ag0 to Fe3+/Fe2+, the LMCT process may be an important contribution to the photoactivated AQDS-mediated Ag+ reduction.
3.4 Reduction of Ag+ by oxidized versus reduced HA
We explored the Ag+ reduction in natural organics by using HA as a representative. It is known that HA can serve as an electron shuttle, mediating the interactions between microbes and environments.79,80 We found that AgNPs were gradually formed in both native HA (HAox) and chemically reduced HA (HAred) in dark and O2-free environments. The LSPR spectra featuring these AgNPs formed can be found in Fig. S8.† The AgNPs were spherical in shape, as can be observed in the TEM images, and EDS confirmed them to be elemental Ag (Fig. S9†). Increasing the Ag+ concentration facilitated AgNP formation, which was in line with the concentration-dependent Ag+-reduction profiles of AQDSred and NADH. The native HA was expected to be in a largely oxidized state, as it had been exposed to air for a long time; it induced only very slow AgNP formation (Fig. 5a). In contrast, AgNPs quickly formed in HAred on the first day (phase I), accounting for approximately one third of the total AgNPs formed over 7 days, which was followed by a slow growth process (phase II). The HAred produced several-fold amounts of AgNPs and enabled faster AgNP-formation rates than HAox, confirming the major contribution of the reduced state to Ag+ reduction in the dark (Fig. 5a and b), which was consistent with the phenomena in NAD+ and AQDS. | ||
| Fig. 5 Influence of the redox state on Ag+ reduction by humic acid (HA) in anaerobic environments. (a) Formation of silver nanoparticles (AgNPs) as a function of time when reducing 0.1 or 0.5 mM Ag+ with 100 mg L−1 native HA (HAox) or chemically reduced HA (HAred). (b) AgNP-formation rate, calculated as the slope of the line obtained by linear regression of the relative peak absorbance of the formed AgNPs against time during the steady-growth phase (phase II), shown in panel (a). Error bars refer to standard errors of the slopes. | ||
Humic acid comprises a pool of organic components. The dramatic AgNP generation observed in phase I was in accordance with the swift Ag+ reduction in AQDSred. This may indicate that a fraction of HAred, likely linked to quinone moieties,81 has strong reducing powers and can quickly transfer electrons to Ag+ at first contact. This phenomenon matched well with the previously reported strong electron donating capacity of other chemically and microbially reduced HAs.40,82 Besides, HA contains diverse functional molecules, such as phenol and carbonyl, which can also be the electron donor for Ag+ reduction.83,84 The subsequent relatively slow AgNP formation may involve some relatively low redox-active components, including carboxyl, as indicated by the weak Ag+ reducing capacity of citrate.
3.5 Influence of light and oxygen on Ag+ reduction by HA
The Ag+ reduction in oxidized and reduced HA was further examined under different O2 and light conditions. The generated AgNP profiles were in the order of dark < visible light < UV light in terms of the final production and formation rate, in line with the trend observed in AQDSox (Fig. 6a and b). In the dark and under visible light, AgNPs were produced at a greater rate in HAred than in HAox regardless of the presence of O2, suggesting a pathway for direct Ag+ reduction other than O2-related intermediate radicals (Fig. 6a). | ||
| Fig. 6 Influence of light and oxygen on the reduction of Ag+ by humic acid (HA). (a) Silver nanoparticle (AgNP)-formation kinetics of reducing 0.1 mM Ag+ with 100 mg L−1 native HA (HAox) or chemically reduced HA (HAred) in the presence (open) or absence (solid) of air in the dark and under visible and UV light irradiation. The data points for the same HA redox state were identically shaped in each plot. (b) AgNP-formation rates, calculated as the slope of the line obtained by linear regression of the relative peak absorbance of the formed AgNPs against time during the steady-growth phase (phase II, refer to Fig. 5a) in panel (a). Error bars denote the standard errors of the slopes. a.u., arbitrary units. | ||
Exposure to air decreased the amount of AgNPs formed in the dark and under visible light. In HAox, the slight decline in aerobic AgNP formation probably resulted from the relatively quick AgNP dissolution in comparison to the slow Ag+ reduction. The negative effect of O2 was more pronounced in HAred, especially during phase I, when the amount of AgNPs formed under aerobic conditions was approximately one third of that in anaerobic environments (Fig. 6a). This result was consistent with the O2 effect in AQDSred, probably because O2 competed with Ag+ for the electron-donating capacity; alternatively, reduced HA may lose its electron-donating capacity upon O2-oxidation.55 Strikingly, for HAred, the presence of O2 imposed no significant influence on the Ag+ reduction during phase II (Fig. 6b), suggesting the involvement of other reducing fractions. In fact, not all reduced HA moieties can be reoxidized by O2,44 and these moieties may still preserve the reducing power to slowly reduce Ag+ in the presence of O2.
The reduction of Ag+ was remarkably enhanced under UV light under both oxic and anoxic conditions, and it was an order of magnitude greater than that in the dark and under visible light (Fig. 6a and b). In both HAox and HAred, the presence of O2 significantly promoted AgNP production, indicating the involvement of both O2˙− and photoexcited HA in Ag+ reduction in oxic environments versus sole photoexcited HA in anaerobic environments. During the first 5 h, slightly higher amounts of AgNPs were observed from HAred than from HAox (Fig. 6a), which was particularly obvious in anaerobic environments. This might imply that direct and photocatalytic reduction were collectively responsible for the initial AgNP formation in HAred. However, during the following phase, HAox displayed a faster Ag+ reduction rate, and a larger amount of AgNPs finally formed in HAox than in HAred (Fig. 6b), demonstrating the major contribution of photoactivated organics to forming AgNPs under UV light radiation in this phase. The photoactivity of HA is closely correlated with its molecular composition.85,86 The quantum yield of radicals under light radiation is closely linked to less saturated components.71 The smaller amount of AgNPs finally formed in HAred may be partially attributed to the decreased radical production due to the degradation of light-sensitive unsaturated moieties while preparing the chemical reduction.
4 Conclusions
This work systematically revealed the kinetics of Ag+ reduction by typical redox-active organic molecules to generate AgNPs. Our results show that reduced AQDS is a very strong Ag+ reducer, followed by NADH. At relatively low levels, the AgNP-formation rate generally increased proportionally with the Ag+ or organic concentration. Yet the presence of very high concentrations of organics had a negative influence on AgNP formation. This inhibitory effect was more pronounced for the organics with weak reducing capacity such as citrate.The organics' redox state coupled with environmental factors, including O2 and light, significantly impacted the AgNP-generation processes. Our results indicate that NAD+ and AQDS in a reduced state are capable of direct Ag+ reduction and AgNP generation in O2-free and dark environments, while those in an oxidized state are not. The presence of O2 strongly diminished their Ag+ reduction power, probably because O2 oxidized the reduced organics or competed for the electron donating capacity. This was also true for natural organics such as HA, in which the quinone moiety is believed to be a dominant redox-active component. The quicker electron transfer from reduced HA to Ag+ enables much faster AgNP formation in comparison to the slow Ag+-reduction process for native HA. A fraction of the reduced HA was likely oxidized by O2 in oxic environments, decreasing their electron donating capacity. Nevertheless, light radiation, particularly UV light, enabled Ag+ reduction mediated by oxidized AQDS and HA in an O2-independent manner. Under UV light, photoexcited AQDS or HA contributed significantly more to AgNP production than did O2˙−. In the presence of O2, the UV-irradiated organics probably combined with the O2˙−-pathway to collectively contribute to Ag+ reduction, as evidenced by the faster AgNP formation rates observed in oxic environments than in anoxic environments. Our findings have revealed different Ag+-reduction pathways forming AgNPs in redox-active organics, with dual impacts from the organic's redox state and environmental factors. This indicates potentially sustainable AgNP production, accompanied by the cyclic oxidation–reduction of redox-active organics, in natural environments.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the Natural Science Foundation of China (41676095, 21822605, 21677068 and 41907347), the Fundamental Research Funds for the Central Universities (021114380148), the China Postdoctoral Science Foundation (2020M672807) and the Shenzhen Science and Technology Innovation Commission of China (JCYJ20180507182227257, KQTD20180412181334790). We acknowledge Na Zhang for the help with graphical artwork. We thank Dongge Li at Shanghai Jiao Tong University for the TEM characterization of nanoparticles.References
- Y. H. Joe, D. H. Park and J. Hwang, Evaluation of Ag nanoparticle coated air filter against aerosolized virus: anti-viral efficiency with dust loading, J. Hazard. Mater., 2016, 301, 547–553 CrossRef CAS PubMed.
- G. A. Sotiriou and S. E. Pratsinis, Engineering nanosilver as an antibacterial, biosensor and bioimaging material, Curr. Opin. Chem. Eng., 2011, 1, 3–10 CrossRef CAS PubMed.
- C. S. Ward, J.-F. Pan, B. P. Colman, Z. Wang, C. A. Gwin, T. C. Williams, A. Ardis, C. K. Gunsch and D. E. Hunt, Conserved microbial toxicity responses for acute and chronic silver nanoparticle treatments in wetland mesocosms, Environ. Sci. Technol., 2019, 53, 3268–3276 CrossRef CAS PubMed.
- J. N. Smith, D. G. Thomas, H. Jolley, V. K. Kodali, M. H. Littke, P. Munusamy, D. R. Baer, M. J. Gaffrey, B. D. Thrall and J. G. Teeguarden, All that is silver is not toxic: silver ion and particle kinetics reveals the role of silver ion aging and dosimetry on the toxicity of silver nanoparticles, Part. Fibre Toxicol., 2018, 15, 47 CrossRef CAS PubMed.
- J. D. Martin, P. C. Frost, H. Hintelmann, K. Newman, M. J. Paterson, L. Hayhurst, M. D. Rennie, M. A. Xenopoulos, V. Yargeau and C. D. Metcalfe, Accumulation of silver in yellow perch (Perca flavescens) and northern pike (Esox lucius) from a lake dosed with nanosilver, Environ. Sci. Technol., 2018, 52, 11114–11122 CrossRef CAS PubMed.
- L. Yuan, C. J. Richardson, M. Ho, C. W. Willis, B. P. Colman and M. R. Wiesner, Stress responses of aquatic plants to silver nanoparticles, Environ. Sci. Technol., 2018, 52, 2558–2565 CrossRef CAS PubMed.
- A.-J. Miao, K. A. Schwehr, C. Xu, S.-J. Zhang, Z. Luo, A. Quigg and P. H. Santschi, The algal toxicity of silver engineered nanoparticles and detoxification by exopolymeric substances, Environ. Pollut., 2009, 157, 3034–3041 CrossRef CAS PubMed.
- F. Dong, N. F. Mohd Zaidi, E. Valsami-Jones and J.-U. Kreft, Time-resolved toxicity study reveals the dynamic interactions between uncoated silver nanoparticles and bacteria, Nanotoxicology, 2017, 11, 637–646 CrossRef CAS PubMed.
- L. Zhang and W.-X. Wang, Dominant role of silver ions in silver nanoparticle toxicity to a unicellular alga: evidence from luminogen imaging, Environ. Sci. Technol., 2019, 53, 494–502 CrossRef CAS PubMed.
- C. Levard, E. M. Hotze, G. V. Lowry and G. E. Brown, Environmental transformations of silver nanoparticles: impact on stability and toxicity, Environ. Sci. Technol., 2012, 46, 6900–6914 CrossRef CAS PubMed.
- F. Dong and Y. Zhou, Differential transformation and antibacterial effects of silver nanoparticles in aerobic and anaerobic environment, Nanotoxicology, 2019, 13, 339–353 CrossRef CAS PubMed.
- N. Akaighe, R. I. MacCuspie, D. A. Navarro, D. S. Aga, S. Banerjee, M. Sohn and V. K. Sharma, Humic acid-induced silver nanoparticle formation under environmentally relevant conditions, Environ. Sci. Technol., 2011, 45, 3895–3901 CrossRef CAS PubMed.
- H. Liu, X. Gu, C. Wei, H. Fu, P. J. J. Alvarez, Q. Li, S. Zheng, X. Qu and D. Zhu, Threshold concentrations of silver ions exist for the sunlight-induced formation of silver nanoparticles in the presence of natural organic matter, Environ. Sci. Technol., 2018, 52, 4040–4050 CrossRef CAS PubMed.
- Y. Yin, J. Liu and G. Jiang, Sunlight-induced reduction of ionic Ag and Au to metallic nanoparticles by dissolved organic matter, ACS Nano, 2012, 6, 7910–7919 CrossRef CAS PubMed.
- A. M. Jones, S. Garg, D. He, A. N. Pham and T. D. Waite, Superoxide-mediated formation and charging of silver nanoparticles, Environ. Sci. Technol., 2011, 45, 1428–1434 CrossRef CAS PubMed.
- W.-C. Hou, B. Stuart, R. Howes and R. G. Zepp, Sunlight-driven reduction of silver ions by natural organic matter: formation and transformation of silver nanoparticles, Environ. Sci. Technol., 2013, 47, 7713–7721 CrossRef CAS PubMed.
- A. Wimmer, A. Kalinnik and M. Schuster, New insights into the formation of silver-based nanoparticles under natural and semi-natural conditions, Water Res., 2018, 141, 227–234 CrossRef CAS PubMed.
- A. R. Badireddy, J. Farner Budarz, S. M. Marinakos, S. Chellam and M. R. Wiesner, Formation of silver nanoparticles in visible light-illuminated waters: mechanism and possible impacts on the persistence of AgNPs and bacterial lysis, Environ. Eng. Sci., 2014, 31, 338–349 CrossRef CAS.
- X. Zhang, C.-W. Yang, H.-Q. Yu and G.-P. Sheng, Light-induced reduction of silver ions to silver nanoparticles in aquatic environments by microbial extracellular polymeric substances (EPS), Water Res., 2016, 106, 242–248 CrossRef CAS PubMed.
- Y. Yin, X. Yang, L. Hu, Z. Tan, L. Zhao, Z. Zhang, J. Liu and G. Jiang, Superoxide-mediated extracellular biosynthesis of silver nanoparticles by the fungus Fusarium oxysporum, Environ. Sci. Technol. Lett., 2016, 3, 160–165 CrossRef CAS.
- N. Yin, R. Gao, B. Knowles, J. Wang, P. Wang, G. Sun and Y. Cui, Formation of silver nanoparticles by human gut microbiota, Sci. Total Environ., 2019, 651, 1489–1494 CrossRef CAS PubMed.
- N. C. Kiat, S. Krishnakumar, L. Xin, M. Munusamy, J. Lianghui, Y. Liang, T. Chuyang, S. Hao, K. Staffan and C. Bin, Influence of outer membrane c-type cytochromes on particle size and activity of extracellular nanoparticles produced by Shewanella oneidensis, Biotechnol. Bioeng., 2013, 110, 1831–1837 CrossRef PubMed.
- A. S. Eswayah, T. J. Smith and P. H. E. Gardiner, Microbial transformations of selenium species of relevance to bioremediation, Appl. Environ. Microbiol., 2016, 82, 4848–4859 CrossRef CAS PubMed.
- A. Ramos-Ruiz, J. A. Field, J. V. Wilkening and R. Sierra-Alvarez, Recovery of elemental tellurium nanoparticles by the reduction of tellurium oxyanions in a methanogenic microbial consortium, Environ. Sci. Technol., 2016, 50, 1492–1500 CrossRef CAS PubMed.
- H. Shang, M. Daye, O. Sivan, C. S. Borlina, N. Tamura, B. P. Weiss and T. Bosak, Formation of zerovalent iron in iron-reducing cultures of Methanosarcina barkeri, Environ. Sci. Technol., 2020, 54, 7354–7365 CrossRef CAS PubMed.
- F. Kang, P. J. Alvarez and D. Zhu, Microbial extracellular polymeric substances reduce Ag+ to silver nanoparticles and antagonize bactericidal activity, Environ. Sci. Technol., 2014, 48, 316–322 CrossRef CAS PubMed.
- A. Manceau, K. L. Nagy, M. A. Marcus, M. Lanson, N. Geoffroy, T. Jacquet and T. Kirpichtchikova, Formation of metallic copper nanoparticles at the soil−root interface, Environ. Sci. Technol., 2008, 42, 1766–1772 CrossRef CAS PubMed.
- X. Liu, L. Shi and J.-D. Gu, Microbial electrocatalysis: redox mediators responsible for extracellular electron transfer, Biotechnol. Adv., 2018, 36, 1815–1827 CrossRef CAS PubMed.
- N. R. Glasser, S. H. Saunders and D. K. Newman, The colorful world of extracellular electron shuttles, Annu. Rev. Microbiol., 2017, 71, 731–751 CrossRef CAS PubMed.
- N. Law, S. Ansari, F. R. Livens, J. C. Renshaw and J. R. Lloyd, Formation of nanoscale elemental silver particles via enzymatic reduction by Geobacter sulfurreducens, Appl. Environ. Microbiol., 2008, 74, 7090–7093 CrossRef CAS PubMed.
- S. H. Light, L. Su, R. Rivera-Lugo, J. A. Cornejo, A. Louie, A. T. Iavarone, C. M. Ajo-Franklin and D. A. Portnoy, A flavin-based extracellular electron transfer mechanism in diverse Gram-positive bacteria, Nature, 2018, 562, 140–144 CrossRef CAS PubMed.
- B. Gu, Y. Bian, C. L. Miller, W. Dong, X. Jiang and L. Liang, Mercury reduction and complexation by natural organic matter in anoxic environments, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 1479–1483 CrossRef CAS PubMed.
- Y. Bai, A. Mellage, O. A. Cirpka, T. Sun, L. T. Angenent, S. B. Haderlein and A. Kappler, AQDS and redox-active NOM enables microbial Fe(III)-mineral reduction at cm-scales, Environ. Sci. Technol., 2020, 54, 4131–4139 CrossRef CAS PubMed.
- T. Klaus, R. Joerger, E. Olsson and C. G. Granqvist, Silver-based crystalline nanoparticles, microbially fabricated, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 13611–13614 CrossRef CAS PubMed.
- S. Hietzschold, A. Walter, C. Davis, A. A. Taylor and L. Sepunaru, Does nitrate reductase play a role in silver nanoparticle synthesis? Evidence for NADPH as the sole reducing agent, ACS Sustainable Chem. Eng., 2019, 7, 8070–8076 CrossRef CAS.
- S. Anil Kumar, M. K. Abyaneh, S. W. Gosavi, S. K. Kulkarni, R. Pasricha, A. Ahmad and M. I. Khan, Nitrate reductase-mediated synthesis of silver nanoparticles from AgNO3, Biotechnol. Lett., 2007, 29, 439–445 CrossRef CAS PubMed.
- J. Ali, N. Ali, L. Wang, H. Waseem and G. Pan, Revisiting the mechanistic pathways for bacterial mediated synthesis of noble metal nanoparticles, J. Microbiol. Methods, 2019, 159, 18–25 CrossRef CAS PubMed.
- L.-S. Wen, P. H. Santschi, G. A. Gill, C. L. Paternostro and R. D. Lehman, Colloidal and particulate silver in river and estuarine waters of Texas, Environ. Sci. Technol., 1997, 31, 723–731 CrossRef CAS.
- A. Wimmer, A. A. Markus and M. Schuster, Silver nanoparticle levels in river water: real environmental measurements and modeling approaches—a comparative study, Environ. Sci. Technol. Lett., 2019, 6, 353–358 CrossRef CAS.
- J. Jiang and A. Kappler, Kinetics of microbial and chemical reduction of humic substances: implications for electron shuttling, Environ. Sci. Technol., 2008, 42, 3563–3569 CrossRef CAS PubMed.
- J. Kim, S. H. Lee, F. Tieves, C. E. Paul, F. Hollmann and C. B. Park, Nicotinamide adenine dinucleotide as a photocatalyst, Sci. Adv., 2019, 5, eaax0501 CrossRef CAS PubMed.
- C. D. Georgiou, H. J. Sun, C. P. McKay, K. Grintzalis, I. Papapostolou, D. Zisimopoulos, K. Panagiotidis, G. Zhang, E. Koutsopoulou, G. E. Christidis and I. Margiolaki, Evidence for photochemical production of reactive oxygen species in desert soils, Nat. Commun., 2015, 6, 7100 CrossRef CAS PubMed.
- M. P. Lau, M. Hupfer and H.-P. Grossart, Reduction-oxidation cycles of organic matter increase bacterial activity in the pelagic oxycline, Environ. Microbiol. Rep., 2017, 9, 257–267 CrossRef CAS PubMed.
- M. Aeschbacher, M. Sander and R. P. Schwarzenbach, Novel electrochemical approach to assess the redox properties of humic substances, Environ. Sci. Technol., 2010, 44, 87–93 CrossRef CAS PubMed.
- W. Haiss, N. T. K. Thanh, J. Aveyard and D. G. Fernig, Determination of size and concentration of gold nanoparticles from UV−Vis spectra, Anal. Chem., 2007, 79, 4215–4221 CrossRef CAS PubMed.
- M. Sikder, J. R. Lead, G. T. Chandler and M. Baalousha, A rapid approach for measuring silver nanoparticle concentration and dissolution in seawater by UV–Vis, Sci. Total Environ., 2018, 618, 597–607 CrossRef CAS PubMed.
- F. Dong, E. Valsami-Jones and J.-U. Kreft, New, rapid method to measure dissolved silver concentration in silver nanoparticle suspensions by aggregation combined with centrifugation, J. Nanopart. Res., 2016, 18, 1–12 CrossRef CAS.
- N. G. Bastús, J. Piella and V. Puntes, Quantifying the sensitivity of multipolar (dipolar, quadrupolar, and octapolar) surface plasmon resonances in silver nanoparticles: the effect of size, composition, and surface coating, Langmuir, 2016, 32, 290–300 CrossRef PubMed.
- N. G. Bastús, F. Merkoçi, J. Piella and V. Puntes, Synthesis of highly monodisperse citrate-stabilized silver nanoparticles of up to 200 nm: kinetic control and catalytic properties, Chem. Mater., 2014, 26, 2836–2846 CrossRef.
- X. C. Jiang, C. Y. Chen, W. M. Chen and A. B. Yu, Role of citric acid in the formation of silver nanoplates through a synergistic reduction approach, Langmuir, 2010, 26, 4400–4408 CrossRef CAS PubMed.
- F. Wang, V. N. Richards, S. P. Shields and W. E. Buhro, Kinetics and mechanisms of aggregative nanocrystal growth, Chem. Mater., 2013, 26, 5–21 CrossRef.
- J. Polte, X. Tuaev, M. Wuithschick, A. Fischer, A. F. Thuenemann, K. Rademann, R. Kraehnert and F. Emmerling, Formation mechanism of colloidal silver nanoparticles: analogies and differences to the growth of gold nanoparticles, ACS Nano, 2012, 6, 5791–5802 CrossRef CAS PubMed.
- T. J. Woehl, C. Park, J. E. Evans, I. Arslan, W. D. Ristenpart and N. D. Browning, Direct observation of aggregative nanoparticle growth: kinetic modeling of the size distribution and growth rate, Nano Lett., 2014, 14, 373–378 CrossRef CAS PubMed.
- N. Walpen, M. P. Lau, A. Fiskal, G. J. Getzinger, S. A. Meyer, T. F. Nelson, M. A. Lever, M. H. Schroth and M. Sander, Oxidation of reduced peat particulate organic matter by dissolved oxygen: quantification of apparent rate constants in the field, Environ. Sci. Technol., 2018, 52, 11151–11160 CrossRef CAS PubMed.
- F. Maurer, I. Christl and R. Kretzschmar, Reduction and reoxidation of humic acid: influence on spectroscopic properties and proton binding, Environ. Sci. Technol., 2010, 44, 5787–5792 CrossRef CAS PubMed.
- S. I. Vasylevskyi, S. Kracht, P. Corcosa, K. M. Fromm, B. Giese and M. Füeg, Formation of silver nanoparticles by electron transfer in peptides and c-cytochromes, Angew. Chem., Int. Ed., 2017, 56, 5926–5930 CrossRef CAS PubMed.
- S. Kracht, M. Messerer, M. Lang, S. Eckhardt, M. Lauz, B. Grobéty, K. M. Fromm and B. Giese, Electron transfer in peptides: on the formation of silver nanoparticles, Angew. Chem., Int. Ed., 2015, 54, 2912–2916 CrossRef CAS PubMed.
- F. Maurer, I. Christl, M. Hoffmann and R. Kretzschmar, Reduction and reoxidation of humic acid: influence on speciation of cadmium and silver, Environ. Sci. Technol., 2012, 46, 8808–8816 CrossRef CAS PubMed.
- D. R. Lovley, J. D. Coates, E. L. Blunt-Harris, E. J. P. Phillips and J. C. Woodward, Humic substances as electron acceptors for microbial respiration, Nature, 1996, 382, 445 CrossRef CAS.
- F. J. Cervantes, F. A. M. D. Bok, T. Duong-Dac, A. J. M. Stams, G. Lettinga and J. A. Field, Reduction of humic substances by halorespiring, sulphate-reducing and methanogenic microorganisms, Environ. Microbiol., 2002, 4, 51–57 CrossRef CAS PubMed.
- G. McKay, Emerging investigator series: critical review of photophysical models for the optical and photochemical properties of dissolved organic matter, Environ. Sci.: Processes Impacts, 2020, 22, 1139–1165 RSC.
- K. McNeill and S. Canonica, Triplet state dissolved organic matter in aquatic photochemistry: reaction mechanisms, substrate scope, and photophysical properties, Environ. Sci.: Processes Impacts, 2016, 18, 1381–1399 RSC.
- S. Garg, A. L. Rose and T. D. Waite, Photochemical production of superoxide and hydrogen peroxide from natural organic matter, Geochim. Cosmochim. Acta, 2011, 75, 4310–4320 CrossRef CAS.
- X. Yuan, P. S. Nico, X. Huang, T. Liu, C. Ulrich, K. H. Williams and J. A. Davis, Production of hydrogen peroxide in groundwater at Rifle, Colorado, Environ. Sci. Technol., 2017, 51, 7881–7891 CrossRef CAS PubMed.
- D. Zhang, S. Yan and W. Song, Photochemically induced formation of reactive oxygen species (ROS) from effluent organic matter, Environ. Sci. Technol., 2014, 48, 12645–12653 CrossRef CAS PubMed.
- S. Garg, A. L. Rose and T. D. Waite, Production of reactive oxygen species on photolysis of dilute aqueous quinone solutions, Photochem. Photobiol., 2007, 83, 904–913 CrossRef CAS PubMed.
- Y. Zhang, R. Del Vecchio and N. V. Blough, Investigating the mechanism of hydrogen peroxide photoproduction by humic substances, Environ. Sci. Technol., 2012, 46, 11836–11843 CrossRef CAS PubMed.
- G. A. Sotiriou, A. Meyer, J. T. N. Knijnenburg, S. Panke and S. E. Pratsinis, Quantifying the origin of released Ag+ ions from nanosilver, Langmuir, 2012, 28, 15929–15936 CrossRef CAS PubMed.
- N. F. Adegboyega, V. K. Sharma, L. Cizmas and C. M. Sayes, UV light induces Ag nanoparticle formation: roles of natural organic matter, iron, and oxygen, Environ. Chem. Lett., 2016, 14, 353–357 CrossRef CAS.
- Y. Chen, R. M. Hozalski, L. G. Olmanson, B. P. Page, J. C. Finlay, P. L. Brezonik and W. A. Arnold, Prediction of photochemically produced reactive intermediates in surface waters via satellite remote sensing, Environ. Sci. Technol., 2020, 54, 6671–6681 CrossRef CAS PubMed.
- S. M. Berg, Q. T. Whiting, J. A. Herrli, R. Winkels, K. H. Wammer and C. K. Remucal, The role of dissolved organic matter composition in determining photochemical reactivity at the molecular level, Environ. Sci. Technol., 2019, 53, 11725–11734 CrossRef CAS PubMed.
- S. E. Page, W. A. Arnold and K. McNeill, Assessing the contribution of free hydroxyl radical in organic matter-sensitized photohydroxylation reactions, Environ. Sci. Technol., 2011, 45, 2818–2825 CrossRef CAS PubMed.
- S. E. Page, M. Sander, W. A. Arnold and K. McNeill, Hydroxyl radical formation upon oxidation of reduced humic acids by oxygen in the dark, Environ. Sci. Technol., 2012, 46, 1590–1597 CrossRef CAS PubMed.
- S. Gligorovski, R. Strekowski, S. Barbati and D. Vione, Environmental implications of hydroxyl radicals (˙OH), Chem. Rev., 2015, 115, 13051–13092 CrossRef CAS.
- A. E. Alegría, A. Ferrer, G. Santiago, E. Sepúlveda and W. Flores, Photochemistry of water-soluble quinones. Production of the hydroxyl radical, singlet oxygen and the superoxide ion, J. Photochem. Photobiol., A, 1999, 127, 57–65 CrossRef.
- S. Siahrostami, G.-L. Li, V. Viswanathan and J. K. Nørskov, One- or two-electron water oxidation, hydroxyl radical, or H2O2 evolution, J. Phys. Chem. Lett., 2017, 8, 1157–1160 CrossRef CAS PubMed.
- Y. Sheng, I. A. Abreu, D. E. Cabelli, M. J. Maroney, A.-F. Miller, M. Teixeira and J. S. Valentine, Superoxide dismutases and superoxide reductases, Chem. Rev., 2014, 114, 3854–3918 CrossRef CAS PubMed.
- G. Xing, S. Garg and T. D. Waite, Is superoxide-mediated Fe(III) reduction important in sunlit surface waters?, Environ. Sci. Technol., 2019, 53, 13179–13190 CrossRef CAS PubMed.
- A. Sundman, J. M. Byrne, I. Bauer, N. Menguy and A. Kappler, Interactions between magnetite and humic substances: redox reactions and dissolution processes, Geochem. Trans., 2017, 18, 6 CrossRef.
- L. Klüpfel, A. Piepenbrock, A. Kappler and M. Sander, Humic substances as fully regenerable electron acceptors in recurrently anoxic environments, Nat. Geosci., 2014, 7, 195 CrossRef.
- M. Aeschbacher, D. Vergari, R. P. Schwarzenbach and M. Sander, Electrochemical analysis of proton and electron transfer equilibria of the reducible moieties in humic acids, Environ. Sci. Technol., 2011, 45, 8385–8394 CrossRef CAS.
- N. Walpen, G. J. Getzinger, M. H. Schroth and M. Sander, Electron-donating phenolic and electron-accepting quinone moieties in peat dissolved organic matter: quantities and redox transformations in the context of peat biogeochemistry, Environ. Sci. Technol., 2018, 52, 5236–5245 CrossRef CAS PubMed.
- D. Leyva, R. Jaffe and F. Fernandez-Lima, Structural characterization of dissolved organic matter at the chemical formula level using TIMS-FT-ICR MS/MS, Anal. Chem., 2020, 92, 11960–11966 CrossRef CAS PubMed.
- Y. Wang and J. Ma, Quantitative determination of redox-active carbonyls of natural dissolved organic matter, Water Res., 2020, 185, 116142 CrossRef CAS PubMed.
- A. C. Maizel, J. Li and C. K. Remucal, Relationships between dissolved organic matter composition and photochemistry in lakes of diverse trophic status, Environ. Sci. Technol., 2017, 51, 9624–9632 CrossRef CAS PubMed.
- J. Lv, R. Han, Z. Huang, L. Luo, D. Cao and S. Zhang, Relationship between molecular components and reducing capacities of humic substances, ACS Earth Space Chem., 2018, 2, 330–339 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0en00820f |
| This journal is © The Royal Society of Chemistry 2021 |