
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCan determination of extractable organofluorine (EOF) be standardized? First interlaboratory comparisons of EOF and fluorine mass balance in sludge and water matrices†
Anna
Kärrman
 *a,
Leo W. Y.
Yeung
a,
Kyra M.
Spaan
b,
Frank Thomas
Lange
c,
Minh Anh
Nguyen
d,
Merle
Plassmann
b,
Cynthia A.
de Wit
b,
Marco
Scheurer
c,
Raed
Awad
d and
Jonathan P.
Benskin
*b
*a,
Leo W. Y.
Yeung
a,
Kyra M.
Spaan
b,
Frank Thomas
Lange
c,
Minh Anh
Nguyen
d,
Merle
Plassmann
b,
Cynthia A.
de Wit
b,
Marco
Scheurer
c,
Raed
Awad
d and
Jonathan P.
Benskin
*b
aMan-Technology-Environment Research Centre (MTM), Örebro University, Sweden. E-mail: Anna.Karrman@oru.se
bDepartment of Environmental Science, Stockholm University, Sweden. E-mail: Jon.Benskin@aces.su.se
cTZW: DVGW-Technologiezentrum Wasser, Germany
dIVL Swedish Environmental Research Institute, Sweden
First published on 10th September 2021
Abstract
The high proportion of unidentified extractable organofluorine (EOF) observed globally in humans and the environment indicates widespread occurrence of unknown per- and polyfluoroalkyl substances (PFAS). However, efforts to standardize or assess the reproducibility of EOF methods are currently lacking. Here we present the first EOF interlaboratory comparison in water and sludge. Three participants (four organizations) analyzed unfortified and PFAS-fortified ultrapure water, two unfortified groundwater samples, unfortified wastewater treatment plant effluent and sludge, and an unfortified groundwater extract. Participants adopted common sample handling strategies and target lists for EOF mass balance but used in-house combustion ion-chromatography (CIC) and liquid chromatography-tandem mass spectrometry (LC-MS/MS) methods. EOF accuracy ranged from 85–101% and 76–109% for the 60 and 334 ng L−1 fluorine (F) – fortified water samples, respectively, with between-laboratory variation of 9–19%, and within-laboratory variation of 3–27%. In unfortified sludge and aqueous samples, between-laboratory variation ranged from 21–37%. The contribution from sum concentrations of 16 individual PFAS (∑PFAS-16) to EOF ranged from 2.2–60% but extended analysis showed that other targets were prevalent, in particular ultra-short-chain perfluoroalkyl acids (e.g. trifluoroacetic acid) in aqueous samples and perfluoroalkyl acid-precursors (e.g. polyfluoroalkyl phosphate diesters) in sludge. The EOF-CIC method demonstrated promising accuracy, robustness and reporting limits but poor extraction efficiency was observed for some targets (e.g. trifluoroacetic acid).
Environmental significancePer- and polyfluoroalkyl substances (PFAS) occur as a complex mixture in the environment. Typically, targeted analytical methods are used to measure a small subset of PFAS, thereby underestimating the burden to the environment. To address this problem, extractable organofluorine (EOF)-based mass balance approaches have been developed, which aim to capture all EOF in a sample, while quantifying the proportion of organofluorine attributed to known PFAS. However, despite increasing interest, there has been relatively little standardization of EOF methods, leading to questions surrounding reproducibility of data. This work describes the first interlaboratory comparison of EOF in water and sludge matrices. The results demonstrate reproducibility in EOF determination, while confirming a large proportion of unidentified EOF, which would otherwise be missed when using strictly targeted analytical approaches. |
Introduction
Per- and polyfluoroalkyl substances (PFAS) represent a class of >4500 substances containing at least one perfluorocarbon moiety (i.e. –CnF2n–).1 Environmental monitoring of PFAS generally focuses on a suite of ∼30 PFAS, mostly consisting of perfluoroalkyl carboxylic and sulfonic acids (PFCAs and PFSAs, respectively)2 which underestimates the total PFAS burden in the environment. To address this problem, several analytical approaches have emerged which aim to capture the large number and diversity of PFAS. One of these, the so-called “fluorine mass balance” approach, represents a promising method to estimate the fraction of organofluorine from unknown PFAS. Fluorine mass balance studies consist of a combination of measurements of different forms of fluorine, expressed as “fluorine equivalents” (i.e. mass of fluorine per mass of sample [e.g. ng g−1 F] or mass of fluorine per volume of sample [e.g. ng L−1 F]). Total fluorine (TF) refers to the total amount of organic and inorganic fluorine measured directly in a sample (i.e. no sample extraction), while extractable organofluorine (EOF) refers to isolating organofluorine from the sample matrix, while removing inorganic fluorine (typically fluoride or its complexes with Al3+)3,4 from the extract. The sum of “known” and “unknown” EOF can be measured using fluorine-specific approaches such as combustion ion chromatography (CIC) while PFAS that make up the “known” EOF can be measured by targeted approaches (e.g. liquid chromatography-tandem mass spectrometry; LC-MS/MS) and then converted to fluorine-equivalent concentrations. EOF mass balance studies have been carried out on a wide range of matrices, including water,5,6 sediment,7 sludge,8 biological samples,9–15 and consumer products,16,17 all of which report a large proportion of samples containing unidentified EOF.The EOF approach has some advantages over other methods for capturing the large number and diversity of PFAS. For example, unlike the total oxidizable precursors (TOP) assay,18 which is designed to capture perfluoroalkyl acid precursors, EOF analysis captures all organofluorine substances extracted from a sample. Moreover, compared to compound-specific approaches such as non-target analysis,19–21 the EOF approach is not reliant on individual chemical standards. Quantification of any organofluorine substance or the combination of multiple organofluorine substances can be performed using any fluorine-containing standard.
Despite increasing interest, there has been relatively little standardization of EOF methods, leading to questions surrounding reproducibility of data. The few known intercomparison studies involving TF or EOF have focused on comparing different instrumental platforms17,22 and extraction methods.15,22 Round-robin testing and standardization of adsorbable organic fluorine (AOF) was also attempted in the mid-1980s and 1990s, but these efforts were largely unsuccessful,23 likely due to an absence of commercial CIC systems at the time. Compared to EOF (which involves combustion of an extract) AOF involves adsorption of organofluorine on activated carbon followed by combustion of the sorbent. Most recently, a standardized method for determination of AOF was published as a draft in November 2020 (DIN 38409-59 draft), but this approach has not yet been approved.24
In January 2021, the European Commission put into force the revised Drinking Water Directive (DWD), to be implemented by member states within two years.25 A new addition to the DWD is the group approach for PFAS, intended to cover all possible PFAS with a limit of 500 ng L−1 “PFAS Total” in drinking water. This approach is intended to serve as a complement to the 100 ng L−1 limit based on the sum of 20 individual PFAS, as soon as the required monitoring method for PFAS Total becomes available. The method, which is not yet established, should have a measurement uncertainty of <50% and a limit of quantification of max. 30% of the parametric value, i.e. 150 ng L−1. EOF represents a promising metric for capturing PFAS Total, but to date no work has been carried out to standardize or assess the reproducibility of EOF methods.
In addition to European-based regulatory initiatives, total fluorine methods were included as part of the national PFAS management plans in the US26 and Australia/New Zealand,27 highlighting the broad international interest in these approaches. To address the growing international demand for standardization of total PFAS or total fluorine-based methods, the present work aimed to carry out the first interlaboratory comparison of EOF. The main objectives were to (a) assess the accuracy and/or reproducibility of EOF and F-mass balance measurements in sludge and various aqueous matrices using identical extraction methods; (b) evaluate whether method detection limits are suitable for use of EOF in regulatory applications; and finally, (c) identify areas for further improvement in EOF determination.
Materials and methods
Experimental design
The interlaboratory comparison consisted of 3 participants: Örebro University (ORU), Stockholm University (SU), and the Swedish Environmental Research Institute together with the German Water Centre (IVL and TZW, respectively). Common sample handling procedures and targets were adopted by all labs (see Sample handling section) and all participants used a combination of CIC and LC-MS/MS. Isotopically-labelled (IL) standards were added after extraction (necessary for EOF mass balance experiments) and the choice of IL standards and other in-house QC parameters were left up to individual labs.Samples were analyzed between October and December 2020 and included ultrapure water (unfortified [UP0], and fortified at 60.2 [UP60] and 334.4 ng L−1 F [UP334] with a mixture of PFAS), two samples of groundwater (GWlow, GWhigh; both unfortified but known to contain highly contrasting PFAS concentrations), samples of wastewater treatment plant effluent and sludge (both pooled, unfortified), and a pooled groundwater extract (GWext; details in Table 1). All samples were analyzed in triplicate unless noted otherwise. Details on sample preparation and fortification of individual PFAS are provided in the ESI.†
| Abbrev. | Sample | Amount extracted | EOF interlaboratory performance | |
|---|---|---|---|---|
| Accuracya (range, n = 3) | Precisionb (CV%, n = 3) | |||
| na: not applicable. a Recovery of fortified concentration. b Coefficient of variation (CV) between laboratories. c Normalized difference between two laboratories. | ||||
| UP0 | Unfortified ultrapure water | 500 mL | na | na |
| UP60 | Ultrapure water fortified to 60.2 ng L−1 F equivalent to 91 ng L−1 ∑PFAS-6 | 500 mL | 85–101% | 9% |
| UP334 | Ultrapure water fortified to 334.4 ng L−1 F equivalent to 504 ng L−1 ∑PFAS-6 | 500 mL | 76–109% | 19% |
| GWlow | Low-level groundwater (unfortified) | 500 mL | na | 36% |
| GWhigh | High-level groundwater (unfortified) | 500 mL | na | 25% |
| Effluent | Pooled effluent (unfortified) | 250 mL | na | 27% |
| Sludge | Pooled sludge (unfortified) | 0.5 g | na | 43% |
| GWext | Pooled groundwater extract (unfortified) | Direct analysis | na | 2.4%c |
Sample handling
Instrumental analysis
Sample extracts were analyzed for EOF by CIC and for 16 target PFAS (“PFAS-16”; Table S1†) by LC-MS/MS using established methods.9,15,28,29 Briefly, TZW and SU both utilized Thermo-Mitsubishi CICs while ORU employed a Metrohm CIC, all of which were operated under hydropyrolytic conditions, wherein organofluorine is converted to fluoride and then measured by conductivity detection. For LC-MS/MS analysis, ORU and SU used Waters Xevo TQ-S instruments coupled to Acquity UPLCs, while IVL used an AB Sciex API4000 coupled to a Shimadzu Prominence UPLC system. All labs performed analysis in negative ionization, multiple reaction monitoring mode. For determination of EOF mass balance, recovery correction was not performed on either the EOF or target PFAS dataset. In other words, IL-standards added immediately prior to LC-MS/MS analysis are only used to correct target PFAS data for matrix effects, not procedural losses. In an attempt to close the EOF mass balance, ORU also analyzed 14 additional PFAS not included in the interlaboratory comparison. This included trifluoroacetic acid (TFAA), perfluoropropanoic acid (PFPrA) and trifluoromethane sulfonic acid (TFMS) for the aqueous matrices and polyfluoroalkyl phosphate di-esters (diPAPs) and perfluorooctane sulfonamide derivatives for sludge (Table S1†). Details of all methods and instrumentation are provided in the ESI (Tables S2 and S3†).Quality control
The following quality control procedures were included alongside the interlaboratory comparison (see ESI† for details):• Procedural blanks, consisting of reagents but no sample matrix, were analyzed for both EOF and target PFAS.
• Evaluation of removal of inorganic fluorine was done by spike/recovery experiments with NaF performed by SU and ORU.
• To assess accuracy and precision of non-recovery corrected PFAS concentrations, each lab analyzed replicate spiked samples and/or certified reference material and/or in-house reference material. These data were used for internal quality control purposes and were not reported as part of the interlaboratory comparison (see ESI† for the individual labs' quality control results).
• To assess PFAS extraction efficiencies, recovery-corrected PFAS-16 concentrations + the additional 14 PFAS were determined on separate portions of sample which were fortified with internal standard prior to extraction. These results were compared to non-recovery corrected concentrations.
Data handling
All participants performed a blank subtraction of their EOF data using the average of the procedural (reagent) blank. None of the participants performed a blank subtraction on target PFAS data or corrected data for apparent recoveries. To assess the contribution of target PFAS to EOF, target PFAS concentrations were converted to fluorine equivalent concentrations (CF_PFAS; ng g−1 F or ng L−1 F) according to eqn (1):| CF_PFAS = CPFAS × nF × AWF/MWPFAS | (1) |
| EOF mass balance (%) = ∑PFAS-16/EOF × 100% | (2) |
Target PFAS reporting limits were derived from the average plus 3 standard deviations of the procedural blanks or the lowest calibration point. EOF reporting limits were based on the average plus 3 standard deviations of the procedural (reagent) blanks. Concentrations below EOF reporting limits are provided to facilitate interlaboratory comparison. In all cases, error is reported as standard deviation.
Results and discussion
Overviews of the EOF and PFAS-16 results are given in Fig. 1, Table S4 and S5.† All participants reported data for all samples, except the groundwater extract. Due to limited extract availability from IVL, TZW reported n = 2 replicates for ultrapure water and effluent samples, n = 1 replicate for groundwater, sludge, and groundwater extract, and the laboratory procedural blank.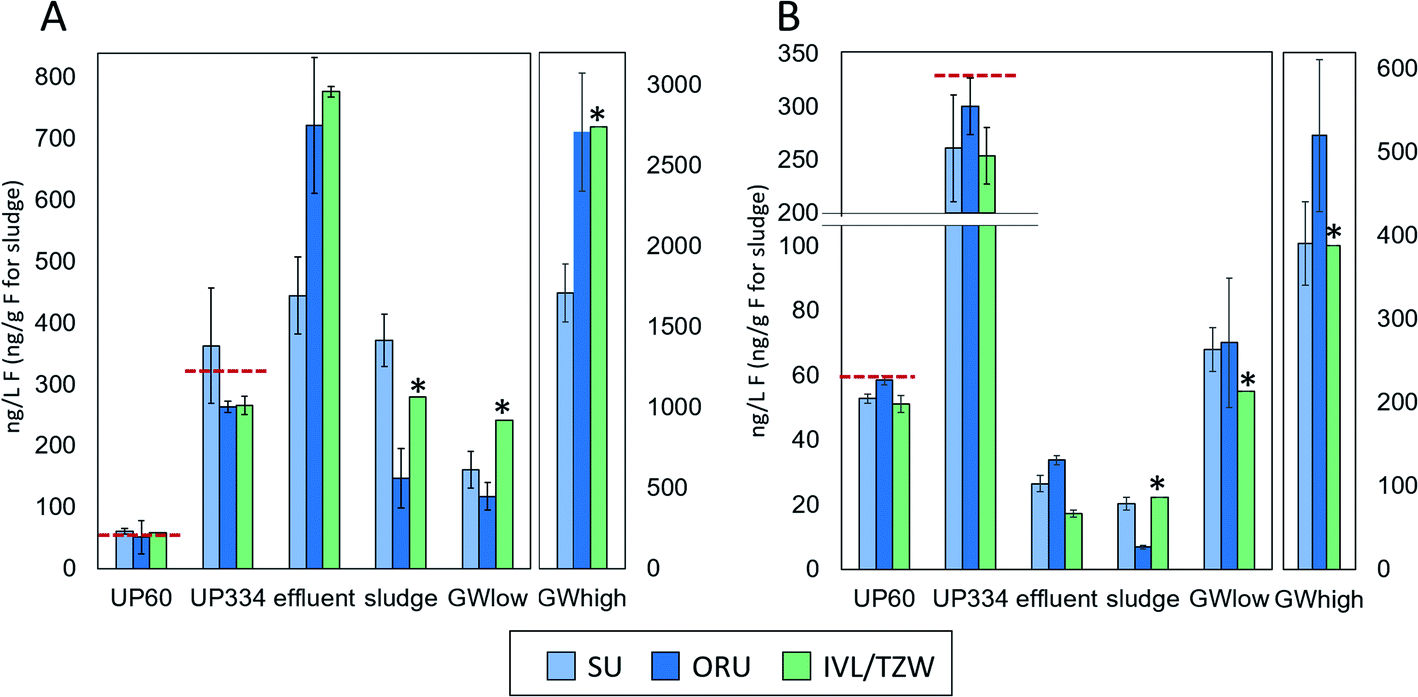 | ||
| Fig. 1 Concentrations (ng L−1 F for aqueous samples and ng g−1 F for sludge) of EOF (A), ∑PFAS-16 (B) in interlaboratory comparison samples. Error bars represent standard deviation (n = 3 for EOF and ∑PFAS-16). The dashed red lines indicate the expected concentrations. * indicates that only a single measurement was performed. | ||
Groundwater extract and (un)fortified ultrapure water
EOF measurements performed on the groundwater extract were reported by ORU and TZW (but not SU) and showed highly consistent results (738 and 756 ng mL−1 F, respectively), indicating good reproducibility of the instrumental analysis methods. In unfortified ultrapure water, EOF concentrations after procedural blank subtraction were negligible, with concentrations <LOD-4.5 ng L−1 F reported by the 3 participants. ∑PFAS-16 concentrations were also low, ranging from <LOD-0.1 ng L−1 F. Accuracy for EOF concentration in ultrapure water fortified to 60.2 ng L−1 F was 85–101% and interlab precision, expressed as coefficient of variation (CV) was 9% (intralab CV = 8–52%, Table S4†). Similar results were obtained for ultrapure water fortified to 334.4 ng L−1 F, with accuracy 76–109% and interlab precision CV 19% (intralab CV = 4–26%). The EOF mass balance for these samples was reasonably consistent across participants with 72–114% identified for UP60 and ∼87–114% identified for UP334. Recovery experiments with NaF added to ultrapure water showed that >96–99% of inorganic fluorine was removed during the sample preparation (see ESI† Quality control for details).Groundwater
PFAS concentrations in GWlow and GWhigh were expected to differ by approximately an order of magnitude (Table 1). For GWlow, SU, ORU, and IVL/TZW reported EOF concentrations of 161, 118, and 242 ng L−1 F resulting in interlab precision of CV 36% (intralab CV = 12.1–12.7%; Table S4†), and ∑PFAS-16 concentrations of 68, 70, and 55 ng L−1 F, respectively (Fig. 1). Taken together, 42, 60, and 23% of the EOF was accounted for by known PFAS, respectively (Fig. 2). In GWhigh, SU reported lower EOF concentrations (1710 ng L−1 F; CV = 10%) compared to ORU (2710 ng L−1 F; CV = 13%) and IVL/TZW (2740 ng L−1 F), while ORU reported higher ∑PFAS-16 concentrations (520 ng L−1 F) compared to SU and IVL (391 and 389 ng L−1 F; Fig. 1). Nevertheless, the interlab precision was 25% and the EOF mass balance was relatively consistent, with 23, 19, and 14% accounted for by known PFAS, respectively (Fig. 2). The variation seen in PFAS-16 concentrations is likely attributable to the fact that internal standards were not fortified prior to extraction. The lower EOF concentrations produced by SU in GWhigh may be due to filtering groundwater samples prior to extraction (not done by ORU and IVL for groundwater), and which may have resulted in loss of some unidentified PFAS due to sorption to filters30 or suspended solids.31 The target PFAS results, on the other hand, did not follow the same pattern as EOF and seems therefore unaffected by the filtration difference.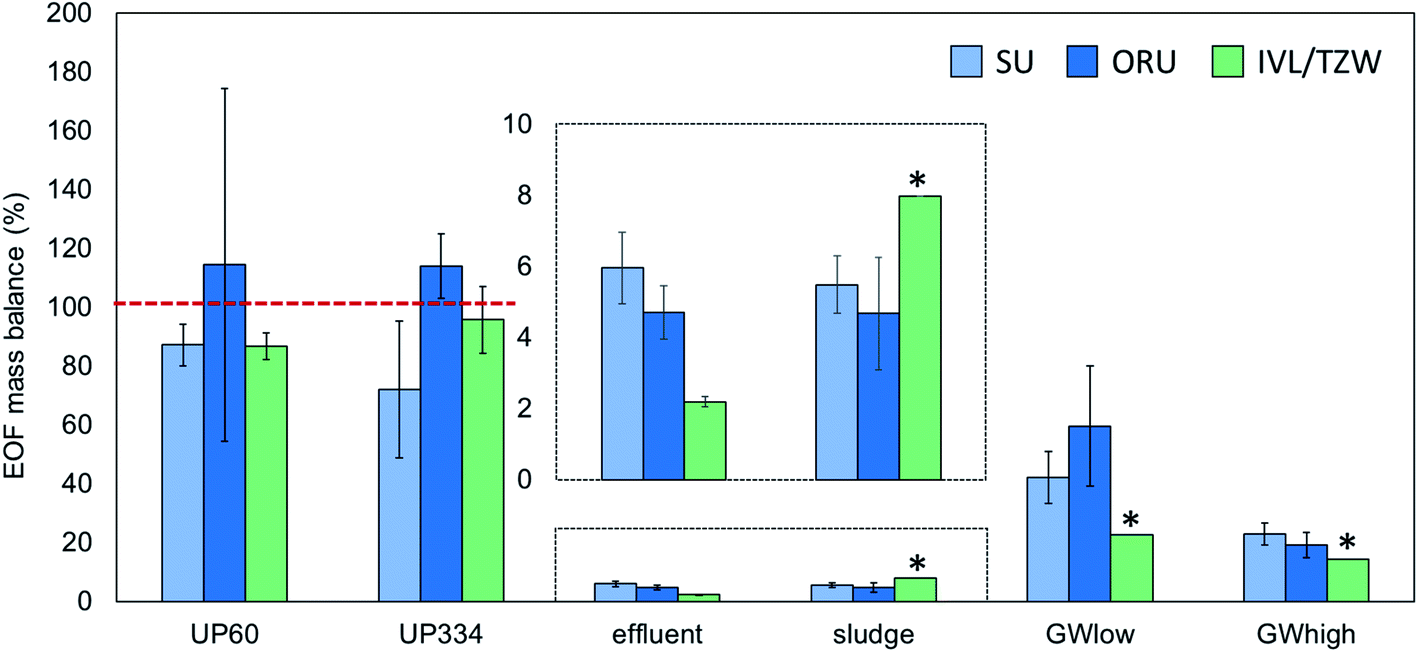 | ||
| Fig. 2 EOF mass balance (%) in interlaboratory comparison samples. Error bars represent pooled standard deviation (n = 3 for EOF and ∑PFAS-16). The dashed red lines indicate the expected EOF mass balance. * indicates that only a single measurement was performed. | ||
Sludge and effluent
EOF concentrations in effluent ranged from 445–785 ng L−1 F with an interlab precision of CV 27% (intralab CV = 12–14%) and ∑PFAS-16 of 17–34 ng L−1 F, resulting in 2.2–6.0% of the EOF attributed to known PFAS (Tables S4 and S5†). In comparison, sludge EOF ranged from 148–372 ng g−1 F (intralab CV = 10–55%), resulting in interlab variation of 43%. Reported ∑PFAS-16 concentrations ranged from 148–372 ng g−1 F resulting in 4.7–8.0% of the EOF accounted for by known PFAS (Fig. 1 and 2). The variability in target concentrations between labs is not surprising considering that IL standards were only added after extraction, thus prohibiting the compensation for extraction losses that is usually done in target quantification. Sludge also tends to be a more heterogeneous matrix that also can be an explanation for higher variability in EOF and PFAS concentrations. Nevertheless, these results show that, provided the losses of known PFAS are proportional to EOF, good consistency in EOF mass balance can be achieved. Recovery experiments with added NaF to sludge showed that >96–99% of inorganic fluorine was removed during the sample preparation (see ESI† Quality control for details).Background level and method reporting limits
CIC instrumental background, evaluated by empty boat combustions, was low and similar across all labs despite differences in manufacturer and/or instrumental parameters. EOF concentrations in UP0 ranged from 13 to 85 ng L−1 F (all participants), similar to in-house procedural blanks for all matrices (42–94 ng L−1 F), indicating that the ultrapure water used here does not contain unknown fluorine that is retained by the extraction sorbent (Table S6†). The EOF reporting limits based on the signal and variation observed in UP0 and water procedural blanks for the three laboratories were 151, 124, and 13 ng L−1 F (average signal plus 3 standard deviations). The reason for the large variation between laboratories is not known and further work is needed to better understand contamination sources and lower the EOF background level.Blank levels have implications for the determination of samples with low concentrations of EOF. While the accuracy of UP60 was very good (85–101%), the concentrations were below method reporting limits for two laboratories. Nevertheless, reporting limits (22–232 ng L−1 when converted to PFOA equivalents) are in the range of the requirements for the proposed DWD limit of 500 ng L−1 PFAS Total in drinking water.25
Extended PFAS target list and extraction efficiency
Analysis of additional targets by ORU in the aqueous samples revealed that ultra-short-chain PFAS made a significant contribution when calculating recovery-corrected PFAS concentrations (Fig. S1 and Table S7†). TFAA was quantified using labeled internal standards (spiked prior to extraction) in effluent (700 ng L−1), and groundwater (GWlow 480 ng L−1 and GWhigh 1960 ng L−1), together with PFPrA and TFMS. Collectively, ultra-short chain PFAS accounted for 46–87% of the 23 quantified PFAS in aqueous samples (Fig. 2 and Table S7†). However, the extraction efficiency was poor for ultra-short chain PFAS and only 1% TFA, 6% TFMS, and 8% PFPrA was recovered. In comparison, the recoveries were good for PFAS-16 (68–101% recovery). Losses of very polar PFAS during extraction might be due to the additional washing step added to remove inorganic fluoride. Therefore, despite their prevalence in water samples, the apparent contribution (i.e. not accounting for procedural loss) of the ultra-short-chain PFAS to measured EOF were only 4.8%, 9.6%, and 0.64% for GWlow, GWhigh and effluent samples, respectively.In sludge, the additional targets analyzed by ORU which were not included in the interlaboratory comparison (e.g. diPAPs) accounted for a significant proportion of EOF (Fig. S2 and Table S7†), consistent with prior reports.8 However, the extended analysis only contributed an additional 45.5 ng g−1 F in sludge, leaving 69% of the measured EOF in sludge unidentified.
Conclusions
Further validation of EOF-CIC is needed and important aspects for are summarized as follows:• Clearly, the greatest drawback from the current study is the small number of laboratories involved, resulting in limited statistical analysis. Future studies with increased numbers of participating laboratories will provide greater confidence in EOF measurements and insight into method limitations.
• CIC-instrumental background was low and consistent, and no systematic differences between laboratories, that could be expected due to e.g. varying calibration routines, were observed. However, differences in procedural blank contamination were observed between labs. The reasons behind this are unclear. Efforts to reduce background F levels and improve reproducibility in blank levels are needed.
• Extraction methods used here were effective for capturing priority PFAS and removing inorganic fluorine, but showed poor extraction efficiency for very polar PFAS (e.g. TFAA) in aqueous matrices. Considering the large variation in PFAS classes, one EOF method will not fit all and the sum parameter will be set by the procedure used. In relation to a PFAS Total assessment, a compromise of modifying protocols to capture the largest number of PFAS in different matrices while ensuring good performance for prioritized substances might be required.
• Method development and quality control of unknown PFAS is inherently difficult since their properties and behavior are unknown. Monitoring possible extraction losses and matrix effects of EOF is therefore challenging. Additional studies are needed to assess the suitability of the method and for example the extent of loss of unknown PFAS from filter sorption.
• Sodium fluoride was used to assess removal efficiency of inorganic fluoride, but a larger concentration range as well as other species of inorganic fluorine known to exist should be evaluated.
Despite the aforementioned areas for future improvement, considering the wide variation in concentrations (∼60 to ∼2500 ng L−1 F) and matrices investigated here, CIC-based EOF measurements revealed good robustness between laboratories, and in all cases achieved measurement uncertainty of <50%, as required by the proposed DWD PFAS Total method.25 This is particularly impressive considering that each lab used their own in-house CIC methods, including calibration using injection of NaF (i.e. no combustion), combustion of PFOS, or combustion of NaF. This indicates that combustion of PFOS is efficient. An efficient combustion, although with varying response for different PFAS and NaF, has been reported in a separate study.32 Comparable results between labs are also promising considering that EOF concentrations cannot be corrected for procedural losses. The small difference between CIC results for the groundwater extract also indicates that the sample treatment procedure will be the most challenging part in standardizing a EOF method. A future comparison study involving a larger number of participants using entirely in-house protocols would shed more light on the current compliance in EOF analysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Anton Ribbenstedt (SU), Fabian Brännström (ORU) and Carl Lemon (ORU) are thanked for assistance with sample preparation and extractions and Birgit Körner (TZW) for CIC measurements. Robin Vestergren (Swedish Chemicals Agency) is thanked for feedback on the experimental design. This study was supported financially by the Swedish Chemicals Agency. Additionally, K. M. Spaan acknowledges funding from the Swedish Research Council FORMAS (Grant 2018-00801). A pre-print version of this article was published as a government report.33References
- OECD, Toward a New Comprehensive Global Database of Per and Polyfluoroalkyl Substances (PFASs), Ser. Risk Manag., 2018, 39, 1–24 Search PubMed.
- A. O. De Silva, J. M. Armitage, T. A. Bruton, C. Dassuncao, W. Heiger-Bernays, X. C. Hu, A. Kärrman, B. Kelly, C. Ng, A. Robuck, M. Sun, T. F. Webster and E. M. Sunderland, PFAS Exposure Pathways for Humans and Wildlife: A Synthesis of Current Knowledge and Key Gaps in Understanding, Environ. Toxicol. Chem., 2021, 40(3), 631–657, DOI:10.1002/etc.4935.
- International Programme on Chemical Safety, Fluorides, World Health Organization, 2002, https://apps.who.int/iris/handle/10665/42415 Search PubMed.
- M. Frankowski and A. Zioła-Frankowska, Speciation Analysis of Aluminium and Aluminium Fluoride Complexes by HPIC-UVVIS, Talanta, 2010, 82(5), 1763–1769, DOI:10.1016/j.talanta.2010.07.066.
- Y. Miyake, N. Yamashita, P. Rostkowski, M. K. So, S. Taniyasu, P. K. S. Lam and K. Kannan, Determination of Trace Levels of Total Fluorine in Water Using Combustion Ion Chromatography for Fluorine: A Mass Balance Approach to Determine Individual Perfluorinated Chemicals in Water, J. Chromatogr. A, 2007, 1143(1–2), 98–104, DOI:10.1016/j.chroma.2006.12.071.
- B. J. Ruyle, H. M. Pickard, D. R. LeBlanc, A. K. Tokranov, C. P. Thackray, X. C. Hu, C. D. Vecitis and E. M. Sunderland, Isolating the AFFF Signature in Coastal Watersheds Using Oxidizable PFAS Precursors and Unexplained Organofluorine, Environ. Sci. Technol., 2021, 55(6), 3686–3695, DOI:10.1021/acs.est.0c07296.
- L. W. Y. Yeung, A. O. De Silva, E. I. H. Loi, C. H. Marvin, S. Taniyasu, N. Yamashita, S. A. Mabury, D. C. G. Muir and P. K. S. Lam, Perfluoroalkyl Substances and Extractable Organic Fluorine in Surface Sediments and Cores from Lake Ontario, Environ. Int., 2013, 59, 389–397, DOI:10.1016/j.envint.2013.06.026.
- L. Yeung, U. Eriksson and A. Kärrman, Time Trend of Unidentified Poly- and Perfluoroalkyalkyl Substances in Sludge from Wastewater Treatment Plants in Sweden, Swedish Environmental Protection Agency, 2017, http://naturvardsverket.diva-portal.org/smash/get/diva2:1138488/FULLTEXT01.pdf Search PubMed.
- K. M. Spaan, C. Van Noordenburg, M. M. Plassmann, L. Schultes, S. Shaw, M. Berger, M. P. Heide-Jørgensen, A. Rosing-Asvid, S. M. Granquist and R. Dietz, Fluorine Mass Balance and Suspect Screening in Marine Mammals from the Northern Hemisphere, Environ. Sci. Technol., 2020, 54(7), 4046–4058, DOI:10.1021/acs.est.9b06773.
- L. Schultes, C. van Noordenburg, K. M. Spaan, M. M. Plassmann, M. Simon, A. Roos and J. P. Benskin, High Concentrations of Unidentified Extractable Organofluorine Observed in Blubber from a Greenland Killer Whale (Orcinus Orca), Environ. Sci. Technol. Lett., 2020, 7(12), 909–915, DOI:10.1021/acs.estlett.0c00661.
- A. Kärrman, T. Wang, R. Kallenborn, A. M. Langseter, S. M. Grønhovd, E. M. Ræder, J. L. Lyche, L. Yeung, F. Chen, U. Eriksson, R. Aro and F. Fredriksson, PFASs in the Nordic Environment: Screening of Poly-And Perfluoroalkyl Substances (PFASs) and Extractable Organic Fluorine (EOF) in the Nordic Environment, Nordic Council of Ministers, 2019, DOI: DOI:10.6027/TN2019-515.
- L. T. Miaz, M. M. Plassmann, I. Gyllenhammar, A. Bignert, O. Sandblom, S. Lignell, A. Glynn and J. P. Benskin, Temporal Trends of Suspect- and Target-per/Polyfluoroalkyl Substances (PFAS), Extractable Organic Fluorine (EOF) and Total Fluorine (TF) in Pooled Serum from First-Time Mothers in Uppsala, Sweden, 1996–2017, Environ. Sci.: Processes Impacts, 2020, 22(4), 1071–1083, 10.1039/C9EM00502A.
- A. Koch, A. Kärrman, L. W. Y. Yeung, M. Jonsson, L. Ahrens and T. Wang, Point Source Characterization of Per- and Polyfluoroalkyl Substances (PFASs) and Extractable Organofluorine (EOF) in Freshwater and Aquatic Invertebrates, Environ. Sci.: Processes Impacts, 2019, 21(11), 1887–1898, 10.1039/c9em00281b.
- L. W. Y. Yeung, Y. Miyake, S. Taniyasu, Y. Wang, H. Yu, M. K. So, G. Jiang, Y. Wu, J. Li, J. P. Giesy, N. Yamashita and P. K. S. Lam, Perfluorinated Compounds and Total and Extractable Organic Fluorine in Human Blood Samples from China, Environ. Sci. Technol., 2008, 42(21), 8140–8145, DOI:10.1021/es800631n.
- A.-M. Kaiser, R. Aro, A. Kärrman, S. Weiss, C. Hartmann, M. Uhl, M. Forsthuber, C. Gundacker and L. W. Y. Yeung, Comparison of Extraction Methods for Per- and Polyfluoroalkyl Substances (PFAS) in Human Serum and Placenta Samples-Insights into Extractable Organic Fluorine (EOF), Anal. Bioanal. Chem., 2021, 413(3), 865–876, DOI:10.1007/s00216-020-03041-5.
- L. Schultes, R. Vestergren, K. Volkova, E. Westberg, T. Jacobson and J. P. Benskin, Per- and Polyfluoroalkyl Substances and Fluorine Mass Balance in Cosmetic Products from the Swedish Market: Implications for Environmental Emissions and Human Exposure, Environ. Sci.: Processes Impacts, 2018, 20(12), 1680–1690, 10.1039/C8EM00368H.
- L. Schultes, G. F. Peaslee, J. D. Brockman, A. Majumdar, S. R. McGuinness, J. T. Wilkinson, O. Sandblom, R. A. Ngwenyama and J. P. Benskin, Total Fluorine Measurements in Food Packaging: How Do Current Methods Perform?, Environ. Sci. Technol. Lett., 2019, 6(2), 73–78, DOI:10.1021/acs.estlett.8b00700.
- E. F. Houtz and D. L. Sedlak, Oxidative Conversion as a Means of Detecting Precursors to Perfluoroalkyl Acids in Urban Runoff, Environ. Sci. Technol., 2012, 46(17), 9342–9349, DOI:10.1021/es302274g.
- P. Jacob, K. A. Barzen-Hanson and D. E. Helbling, Target and Nontarget Analysis of Per- and Polyfluoralkyl Substances in Wastewater from Electronics Fabrication Facilities, Environ. Sci. Technol., 2021, 55(4), 2346–2356, DOI:10.1021/acs.est.0c06690.
- K. A. Barzen-Hanson, S. C. Roberts, S. Choyke, K. Oetjen, A. McAlees, N. Riddell, R. McCrindle, P. L. Ferguson, C. P. Higgins and J. A. Field, Discovery of 40 Classes of Per- and Polyfluoroalkyl Substances in Historical Aqueous Film-Forming Foams (AFFFs) and AFFF-Impacted Groundwater, Environ. Sci. Technol., 2017, 51(4), 2047–2057, DOI:10.1021/acs.est.6b05843.
- J. P. McCord, M. J. Strynar, J. W. Washington, E. L. Bergman and S. M. Goodrow, Emerging Chlorinated Polyfluorinated Polyether Compounds Impacting the Waters of Southwestern New Jersey Identified by Use of Nontargeted Analysis, Environ. Sci. Technol. Lett., 2020, 7(12), 903–908, DOI:10.1021/acs.estlett.0c00640.
- L. Gehrenkemper, F. Simon, P. Roesch, E. Fischer, M. von der Au, J. Pfeifer, A. Cossmer, P. Wittwer, C. Vogel, F.-G. Simon and B. Meermann, Determination of Organically Bound Fluorine Sum Parameters in River Water Samples-Comparison of Combustion Ion Chromatography (CIC) and High Resolution-Continuum Source-Graphite Furnace Molecular Absorption Spectrometry (HR-CS-GFMAS), Anal. Bioanal. Chem., 2021, 413(1), 103–115, DOI:10.1007/s00216-020-03010-y.
- A. Wagner, B. Raue, H.-J. Brauch, E. Worch and F. T. Lange, Determination of Adsorbable Organic Fluorine from Aqueous Environmental Samples by Adsorption to Polystyrene-Divinylbenzene Based Activated Carbon and Combustion Ion Chromatography, J. Chromatogr. A, 2013, 1295, 82–89, DOI:10.1016/j.chroma.2013.04.051.
- German Standard Methods for the Examination of Water, Waste Water and Sludge – Parameters Characterizing Effects and Substances (Group H) – Part 59: Determination of Adsorbable Organically Bound Fluorine, Chlorine, Bromine and Iodine (AOF, AOCl, AOBr, AOI) Using Combustion and Subsequent Ion Chromatographic Measurement (In German). November 11, 2020, DOI:10.31030/3191652.
- Directive (EU) 2020/2184 of the European Parliament and of the Council of 16 December 2020 on the Quality of Water Intended for Human Consumption, L4352020, Official Journal of the European Union, https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=OJ:L:2020:435:TOC Search PubMed.
- US Environmental Protection Agency, EPA's Per- and Polyfluoroalkyl Substances (PFAS) Action Plan, EPA 823R18004, 2019, https://www.epa.gov/sites/production/files/2019-02/documents/pfas_action_plan_021319_508compliant_1.pdf Search PubMed.
- Heads of EPA Australia and New Zealand, PFAS National Environmental Management Plan Version 2.0, 2020, https://www.environment.gov.au/system/files/resources/2fadf1bc-b0b6-44cb-a192-78c522d5ec3f/files/pfas-nemp-2.pdf Search PubMed.
- G. Giovanoulis, M. A. Nguyen, M. Arwidsson, S. Langer, R. Vestergren and A. Lagerqvist, Reduction of Hazardous Chemicals in Swedish Preschool Dust through Article Substitution Actions, Environ. Int., 2019, 130, 104921, DOI:10.1016/j.envint.2019.104921.
- F. T. Lange, B. Körner and J. Müller, Development of a Fluorine-specific Group Parameter EOF for Soil and Other Solid Matrices, project no. L7515008, 2017, in German, from https://pudi.lubw.de/detailseite/-/publication/40334 , accessed 31 Mar 2021 Search PubMed.
- B. Chandramouli, J. P. Benskin, M. C. Hamilton and J. R. Cosgrove, Sorption of Per- and Polyfluoroalkyl Substances (PFASs) on Filter Media: Implications for Phase Partitioning Studies, Environ. Toxicol. Chem., 2015, 34(1), 30–36, DOI:10.1002/etc.2751.
- P. Zhao, X. Xia, J. Dong, N. Xia, X. Jiang, Y. Li and Y. Zhu, Short- and Long-Chain Perfluoroalkyl Substances in the Water, Suspended Particulate Matter, and Surface Sediment of a Turbid River, Sci. Total Environ., 2016, 568, 57–65, DOI:10.1016/j.scitotenv.2016.05.221.
- R. Aro, U. Eriksson, A. Kärrman, I. Reber and L. W. Y. Yeung, Combustion Ion Chromatography for Fluorine Analysis in Environmental Samples, iScience, 2021, 24(9), 102968 CrossRef PubMed.
- KemI, Interlaboratory Comparison of Extractable Organofluorine (EOF) Analysis of Water, Effluent and Sludge, 2021, https://www.kemi.se/download/18.f1b904217860f8d6f035d/1616769307985/PM-5-21-Interlaboratory-Comparison-of-Extractable-Organofluorine-EOF.pdf Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1em00224d |
| This journal is © The Royal Society of Chemistry 2021 |