
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Two-photon, visible light water splitting at a molecular ruthenium complex†
Jacob
Schneidewind
 *a,
Miguel A.
Argüello Cordero
b,
Henrik
Junge
a,
Stefan
Lochbrunner
b and
Matthias
Beller
a
*a,
Miguel A.
Argüello Cordero
b,
Henrik
Junge
a,
Stefan
Lochbrunner
b and
Matthias
Beller
a
aLeibniz-Institut für Katalyse e.V., Albert-Einstein-Str. 29a, Rostock 18059, Germany. E-mail: Jacob.Schneidewind@catalysis.de
bInstitute for Physics and Department of Life, Light and Matter, University of Rostock, 18051 Rostock, Germany
First published on 18th June 2021
Abstract
Water splitting to give molecular oxygen and hydrogen or the corresponding protons and electrons is a fundamental four-electron redox process, which forms the basis of photosynthesis and is a promising approach to convert solar into chemical energy. Artificial water splitting systems have struggled with orchestrating the kinetically complex absorption of four photons as well as the difficult utilization of visible light. Based on a detailed kinetic, spectroscopic and computational study of Milstein's ruthenium complex, we report a new mechanistic paradigm for water splitting, which requires only two photons and offers a new method to extend the range of usable wavelengths far into the visible region. We show that two-photon water splitting is enabled by absorption of the first, shorter wavelength photon, which produces an intermediate capable of absorbing the second, longer wavelength photon (up to 630 nm). The second absorption then causes O–O bond formation and liberation of O2. Theoretical modelling shows that two-photon water splitting can be used to achieve a maximum solar-to-hydrogen efficiency of 18.8%, which could be increased further to 28.6% through photochemical instead of thermal H2 release. It is therefore possible to exceed the maximum efficiency of dual absorber systems while only requiring a single catalyst. Due to the lower kinetic complexity, intrinsic utilization of a wide wavelength range and high-performance potential, we believe that this mechanism will inspire the development of a new class of water splitting systems that go beyond the reaction blueprint of photosynthesis.
Broader contextGreen hydrogen will play an integral part in the global transition to renewable energy, which is being recognized by an increasing number of countries that are adopting green hydrogen strategies. Economic production of green hydrogen, however, remains challenging. Existing approaches such as using renewable electricity to power water electrolysis are not currently competitive with hydrogen from fossil sources. Photocatalytic water splitting could offer a pathway for low-cost green hydrogen production: in this process, a photocatalyst mediates water splitting by directly utilizing solar energy. Yet, the development of suitable photocatalysts has been hampered by two important challenges: (1) so far, catalysts have required the absorption of at least four photons per reaction, creating a high degree of complexity; (2) It has been challenging to effectively utilize visible light, which is necessary to obtain practical solar-to-hydrogen efficiencies. Herein, we report a new mechanistic paradigm for water splitting, which reduces the number of required photons to two and offers a new method to extend the range of usable wavelengths far into the visible region. These insights will form the basis to develop a new class of photocatalysts that can overcome existing challenges and could pave the way for low-cost green hydrogen production. |
Water splitting is a four-electron redox process, which is accomplished in natural photosynthesis by consecutive absorption of four photons in photosystem II (Kok's cycle).1,2 The kinetic complexity of orchestrating these four photon absorption and redox events is solved in nature through the so-called Z scheme, a complex chain of light-driven redox reactions enabling an efficient flow of electrons through the responsible enzymes.3 Artificial water splitting systems are attractive means of converting solar into chemical energy and could play a pivotal role in a global transition towards the use of renewable energy.4 Photocatalysts for water splitting have largely adopted a similar approach to natural photosynthesis, relying on absorption of four photons to accomplish water splitting.5 This is because most systems are based on semiconductors, for which absorption of a photon produces one electron/hole pair (except for processes like singlet fission6,7) and thus, four of these events are required to complete the reaction.5 Due to the absence of an equally elegant electron transfer system like the Z scheme, however, artificial systems have struggled to deal with this kinetic complexity, resulting in systems which are not yet productive enough to compete with other approaches for solar energy conversion.4 Progress has been made to address this challenge by using alternative reaction pathways,8,9 although these can present other hurdles (see Fig. 1).
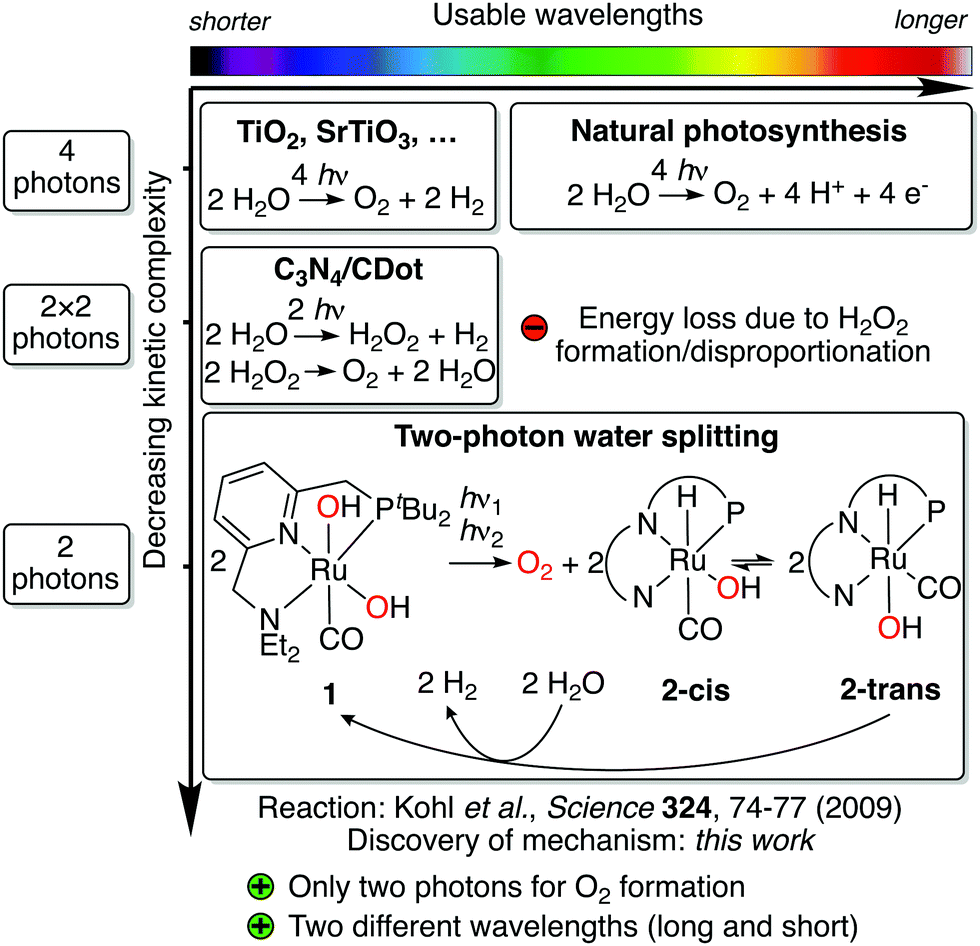 | ||
| Fig. 1 Overview of approaches to light-driven water splitting, arranged by usable wavelength range (top scale) and kinetic complexity (side scale). Note that wavelength ranges are only approximate. Simplified representation of the pincer ligand is shown for 2-cis and 2-trans. | ||
Another longstanding challenge for artificial water splitting systems has been the efficient utilization of visible light, which is necessary for the productive use of sunlight. Most artificial systems, however, have been limited to UV and blue light.10 While in nature this is also addressed using the Z scheme, advances for artificial systems include the combination of smaller band gap semiconductors with low overpotential catalysts,11,12 artificial Z schemes,3 photon upconversion,13,14 and the combination of different photoanodes and cathodes.15 A challenge shared by these approaches is that electrons and holes are generated with the same mechanism for each of the four redox steps. While each step might have a different redox potential,16 the respective electron–hole generation mechanism thus has to match the largest of all potentials, therefore setting the minimum energy requirement for all photon absorptions.
Here, we report the discovery of a new mechanistic paradigm for water splitting, which addresses these challenges. It is based on three key aspects:
(1) The first mechanism for overall water splitting which requires only two instead of four photons. Hence, kinetic complexity is significantly reduced compared to the reaction blueprint of photosynthesis.
(2) A new way for water splitting to extend the range of usable wavelengths far into the visible region through sequential photon absorption by two different reaction species. Each species absorbs photons at different wavelengths, thus tailoring photon absorption for each redox event. This allows for productive utilization of light with wavelengths up to 630 nm and is a method for visible light utilization that is distinct from all described above. Theoretical modelling shows that a maximum solar-to-hydrogen efficiency of up to 18.8% can thus be achieved, which could be increased further to 28.6% via photochemical instead of thermal H2 release. This would be higher than the limit of dual absorber systems while requiring only a single catalyst.
(3) An unprecedented O–O bond formation mechanism proceeding via light-induced oxo–hydroxo coupling. This new mechanism is in contrast to established water nucleophilic attack or dinuclear oxo coupling mechanisms.17
Related two-photon mechanisms have previously been reported in the context of photochromism,18,19 organic photoredox reactions,20,21 and OH˙ generation.22–24 Evidence for these mechanisms was garnered through square rate/photon flux relationships,18,19,21 dual irradiation and wavelength dependency experiments,18,19,21 independent generation of the second photon absorbing intermediate,19–23 single and two pulse transient absorption spectroscopy,18,19,21–23 irradiation with ultrashort pulses,18 kinetic modelling,18,19,22,23 and quantum chemical studies.19,24 Compared to water splitting, the previously studied reactions are relatively simple transformations (single bond breaking events). This work is, to the best of our knowledge, the first description of two-photon chemistry in the context of a highly complex transformation such as water splitting.
The new mechanism was discovered through a detailed kinetic, spectroscopic and computational study of a previously reported reaction involving Milstein's ruthenium complex.25 In their seminal study in 2009, Milstein and coworkers reported that broadband irradiation of ruthenium dihydroxo complex 1 led to evolution of O2 and concomitant formation of hydrido hydroxo complex 2-trans (see Fig. 1).25 Heating 2-trans in water led to evolution of H2 and regeneration of 1, closing the quasi-catalytic cycle. Due to the different reaction conditions for both steps and moderate yields, the reaction is not catalytic, but it contains the core reactivity of an overall water splitting process. In contrast to most artificial water splitting systems, it is a circular sequence of organometallic reaction steps involving a single type of complex. Since the H2 release step is redox neutral, all electron transfer events for water splitting occur during irradiation of 1, the reaction that is subject of this study. Through elegant isotope labeling and trapping experiments, Milstein and coworkers showed that O–O bond formation occurs intramolecularly and that O2 is formed in its triplet ground state.25 While originally it was proposed that irradiation of 1 produces H2O2, which then disproportionates to form O2 and water,25 subsequent theoretical results have shown that this pathway is unlikely.26 Hence, this unusual water splitting reaction has eluded understanding for the past eleven years.
Results
We started our investigation by also first looking at the possibility of a H2O2 disproportionation pathway. Experimentally, we found that H2O2 disproportionation catalyzed by 1 in the dark is too slow to explain the observed O2 formation (see ESI,† Section 5). Irradiation might accelerate the H2O2 disproportionation rate but is difficult to study experimentally for this system (see ESI,† Section 5.2). We therefore studied possible H2O2 pathways theoretically, finding that H2O2 formation from 1 is not energetically feasible (see ESI,† Section 11.2.8), which is in agreement with the previous findings.26To elucidate the mechanism, we then turned to investigating the physico-chemical behavior of 1. Synthesis and characterization of 1 gave results consistent with Milstein and coworkers’ structural assignment of 1 being a cis-dihydroxo complex (see ESI,† Section 3.1.1). The lowest energy absorption feature is a metal-to-ligand charge-transfer (MLCT) band from 350–400 nm (see Fig. 2 and ESI,† Section 11.2.3) and weak fluorescence in water can be observed at 500 nm when 1 is irradiated at 370 nm (see Fig. 2). Irradiation of 1 for two days in water using a broadband quartz-tungsten-halogen light source (320–1000 nm) leads to formation of 2-cis in ca. 20% yield (concentration–time profile see ESI,† Fig. S4.1–1 and Section 4.1.2 for structural assignment).
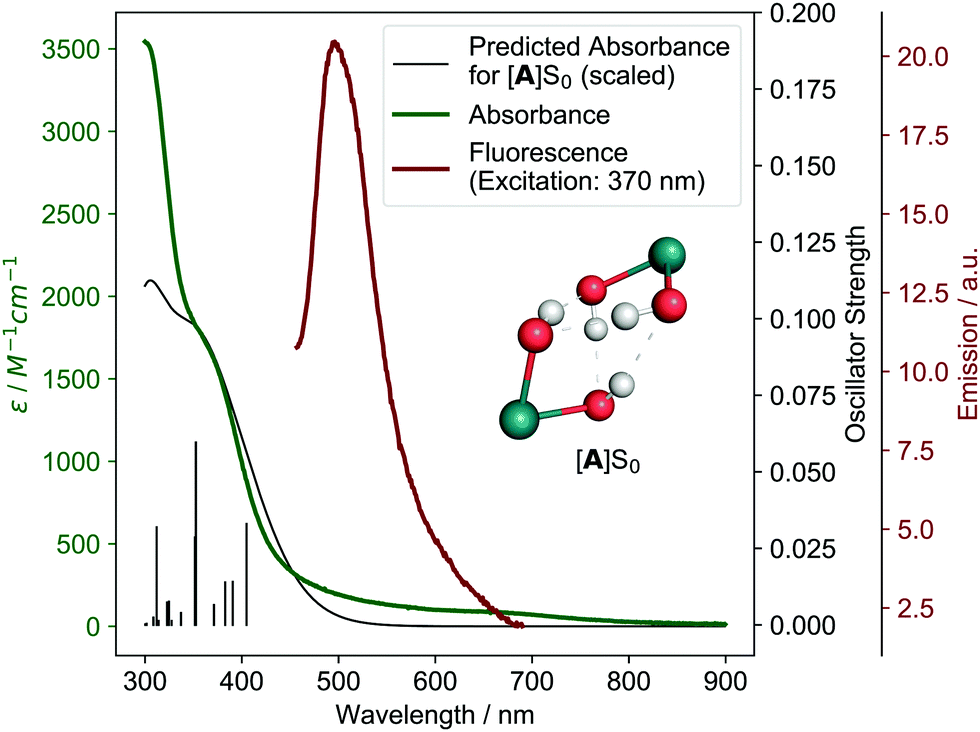 | ||
| Fig. 2 Experimental (green) and theoretical (black) absorbance of 1 (theoretical model [A]S0, see below) as well experimental fluorescence (red). Individual theoretical transitions are shown as vertical lines. Inlet shows structure of [A]S0 with other ligands omitted for clarity. | ||
2-cis refers to the isomer of 2 in which hydrido and hydroxo ligands are in a cis configuration and it isomerizes to form the more stable 2-trans over time in the dark (details see ESI,† Section 4.1.2). Just like Milstein an co-workers,25 we found that 2-trans does not show significant photoreactivity, although it can photochemically isomerize back to 2-cis in the presence of O2 (see ESI,† Section 4.1.2 and Fig. S11.2-17 for UV/vis absorption of 2-trans). Heating of 2-trans in water leads to regeneration of 1 (see ESI,† Section 3.1.4). This is consistent with the findings of Milstein and co-workers, who reported that H2 is thermally released in this regeneration step, closing the water splitting cycle.25 However, formation of side products prevents quantitative H2 release for this step (see ESI,† Fig. S7.1.9).
During irradiation of 1, reversible formation of a side product, named Oxo Dimer, is also observed, which is tentatively assigned to an oxo-bridged dimer that slowly hydrolyses back to 1 (see ESI,† Section 4.1.2). Using in situ O2 detection, formation of O2 can be unambiguously detected in both the liquid and the gas phase (see ESI,† Sections 4.2 and 4.3) during irradiation of 1. Kinetic modeling of the concentration–time profile for the formation of 2-cis and the initial rate of O2 formation in the liquid phase show that both occur at a comparable rate, indicating that they are part of the same reaction (see ESI,† Section 4.4). Notably, the formed O2 appears to be consumed again during irradiation, likely due to photochemical oxidation of 1 (see ESI,† Section 4.3.2). A lower yield of 2 (20% vs. 45%), formation of the Oxo Dimer side product and consumption of O2 slightly differ from the original results.25 Nevertheless, the core water splitting reactivity is identical and these differences do not impact the water splitting mechanistic investigation herein.
Kinetic studies
Next, we performed kinetic studies based on initial rates of O2 formation in the liquid phase as well as NMR measurements of 2-cis formation. Performing the reaction in D2O instead of H2O shows that there is only a small H/D kinetic isotope effect (1.18, see ESI,† Section 4.2.4). Furthermore, the reaction rate does not increase with increasing temperature (see ESI,† Section 4.2.4). We then studied how the initial rate of O2 formation varies with photon flux density: for these experiments, irradiation was performed using a 320–500 nm filtered light source and we found that the rate increases non-linearly with increasing photon flux densities. Notably, the relationship can be described well using a square dependence (see Fig. 3A), indicating that two photons are absorbed during the reaction.18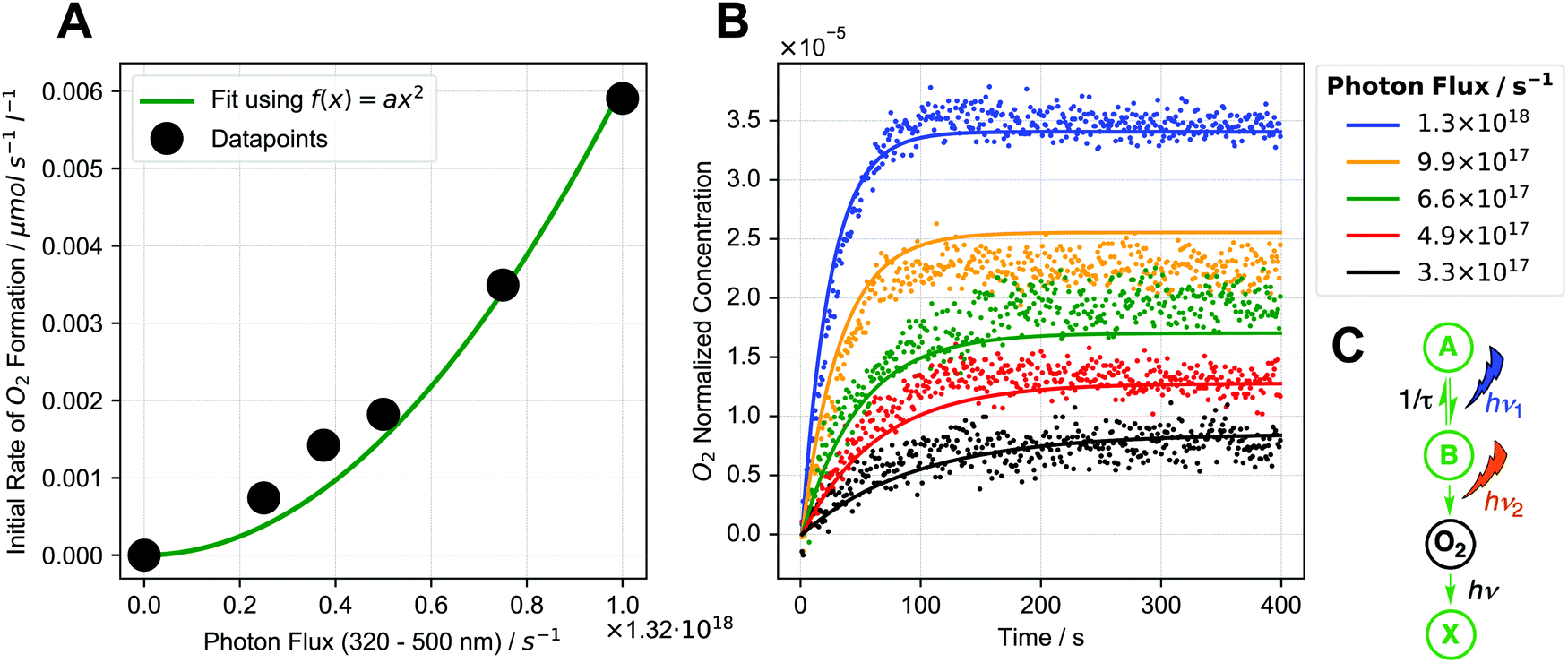 | ||
| Fig. 3 (A) Dependence of initial rate of O2 formation on photon flux (black dots) along with square fit (green). (B) Global fit of photochemical kinetic model (C) to O2 formation data for different photon fluxes (dots: experimental data, lines: fits of kinetic model). Global fit means that one set of parameters is used for all data sets. (C) Schematic representation of photochemical kinetic model. Details see ESI,† Sections 4.2.3 and 10. | ||
Based on this observation, there are two feasible reaction pathways:
(1) Two equivalents of 1 separately absorb one photon each, generating two equivalents of an intermediate, which then react in a bimolecular reaction (akin to triplet–triplet annihilation13) Each photon is absorbed by starting complex 1 in this case.
or
(2) 1 absorbs a photon, generating an intermediate, which then absorbs a second photon. The two photons are absorbed by different species in this case (the first by 1, the second by the intermediate).
Simultaneous two-photon excitation can be excluded as the photon flux densities used in our experiments are too low to observe this effect.18,27,28
To gain further insight, we developed a photochemical kinetic model to describe the initial rate/photon flux data (details see ESI,† Section 10). Fig. 3B shows a global fit of the sequential two-photon mechanism (pathway 2, see Fig. 3C) to experimental O2 formation data for different photon fluxes, showing reasonably good agreement. This model also reproduces the square initial rate/photon flux relationship (see ESI,† Fig. S10.3–1B), which occurs when intermediate B is transient and decays back to the starting complex. The square relationship results from the two sequential photon absorption steps, because an increase in photon flux increases the rate of B formation (first absorption step) while also increasing the chance of B absorbing a photon before it decays (second absorption step).18,21 A sequential two-photon mechanism is therefore consistent with the kinetic data. Mechanisms with more than two photon absorptions were also modelled but were found to be unlikely (see ESI,† Section 10.5). When analyzing the initial rate/photon flux data with our photochemical kinetic model alone, however, a bimolecular mechanism (pathway 1, see ESI,† Section 10.5.1) could be just as valid as the sequential two-photon mechanism.
To determine the operative pathway, we therefore studied the effects of using different wavelengths for irradiation by using either one or two different light sources simultaneously. When 1 is irradiated using a 320–400 nm filtered light source, 2-cis is formed in 6% yield after 17 h. Expectedly, when irradiation is performed using a 495–1000 nm filtered light source, no 2-cis is formed (as 1 does not significantly absorb >495 nm light). Remarkably, when both the 320–400 nm and 495–1000 nm light source are used simultaneously, 2-cis yield increases to 10% (see Fig. 4A).
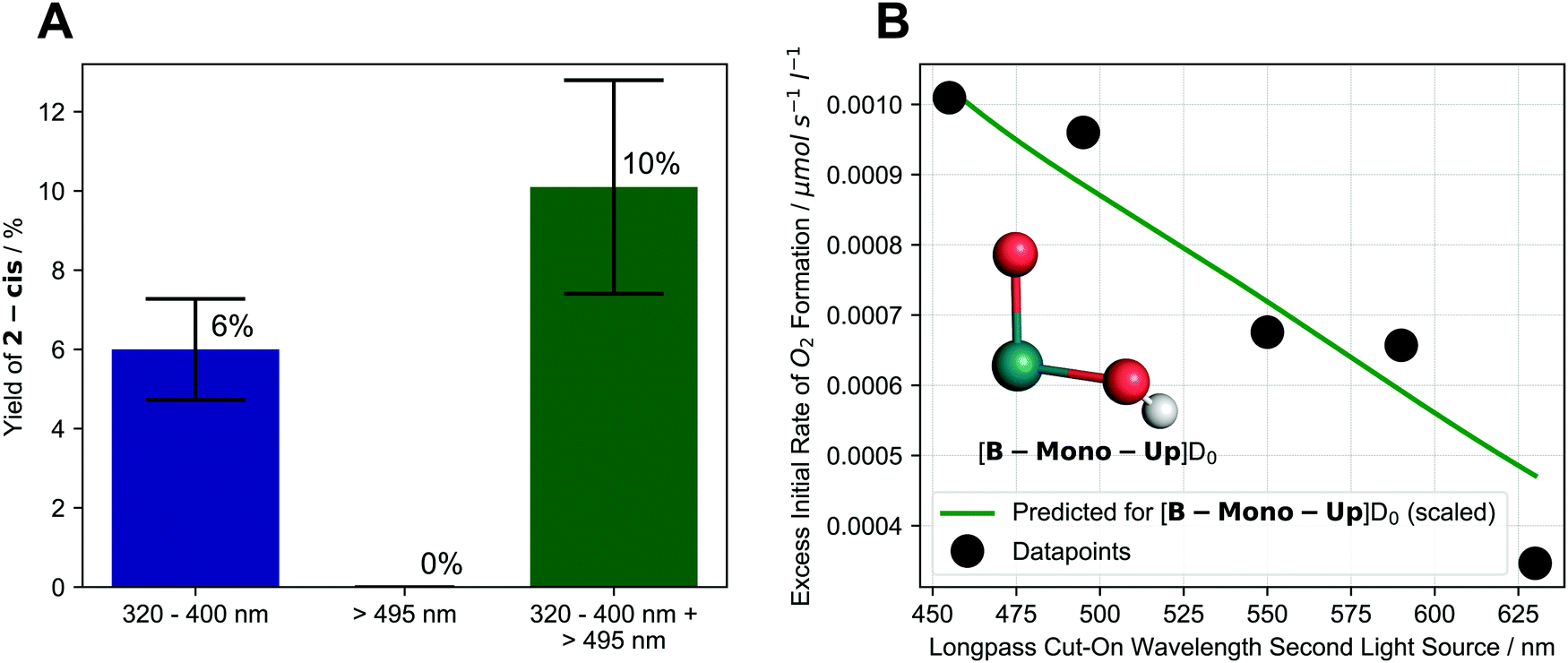 | ||
| Fig. 4 (A) Effect of different irradiation wavelengths and dual irradiation on formation of 2-cis after 17 h of irradiation. Colored bars and text indicate average values while black error bars indicate upper and lower limit of experimental values; (B) dependence of excess initial rate of O2 formation on longpass cut-on wavelength of second light source (first light source: 320–400 nm) along with scaled, predicted behavior for [B-Mono-Up]D0 (see below). Inlet shows structure of [B-Mono-Up]D0 with other ligands omitted for clarity. Details see ESI,† Sections 4.1.4, 4.2.5, 11.4.1. | ||
These results show that using light, which is not significantly absorbed by the starting complex 1, can enhance the water splitting reaction in a synergistic way. In turn, this suggests that excitation of 1 by a 320–400 nm photon leads to formation of an intermediate, which absorbs a second, longer wavelength photon to complete the water splitting reaction.
To investigate this effect more closely we performed the following series of dual irradiation experiments: a solution of 1 was simultaneously irradiated with a 320–400 nm filtered light source as well as a second light source with different longpass filters, resulting in X–1000 nm wavelength intervals (455–1000 nm, 495–1000 nm, etc.) for the second light source. For each longpass filter, the excess initial rate of O2 formation was determined, which was calculated by subtracting the initial rates of single light source irradiation (320–400 nm or X–1000 nm) from the dual irradiation rate (details see ESI,† Section 4.2.5).
In this experiment, 320–400 nm irradiation excites 1 and generates the intermediate, while the effect of longpass filters on excess initial rate effectively probes the absorption behavior of the intermediate for water splitting. Fig. 4B shows that already starting at 630 nm, an excess initial rate can be observed, which increases roughly linearly all the way to 455 nm. This indicates that an intermediate, which can absorb 455–630 nm photons, is responsible for the second photon absorption during water splitting. The experimental longpass filter/excess rate relationship is in good agreement with a theoretical prediction for the computationally identified intermediate ([B-Mono-Up]D0, see Fig. 4B green line, below, and ESI,† Section 11.4.1). The dual irradiation results are consistent with the predictions of the photochemical kinetic model for the sequential two-photon mechanism (see ESI,† Section 10.4).
We also performed time-resolved UV/vis spectroscopy in an attempt to gain another spectro-kinetic perspective but no useful information could be extracted in this case (details see ESI,† Section 8.3).
Ultrafast pump–probe spectroscopy
To investigate the dynamics of 1 after excitation, we performed ultrafast pump–probe spectroscopy, exciting 1 with 400 nm laser pulses (details see ESI,† Section 9). In fact, two transient species could be detected, having lifetimes of τ = 6 ps and τ = 150 ps, respectively. Decay associated spectra (DAS) indicate that the τ = 6 ps species corresponds to an emissive singlet state of 1, explaining the complex's weak fluorescence. DAS for the τ = 150 ps species agree well with the predicted DAS for an isomer of the computationally identified intermediate, referred to as [B-trans]T0 (see ESI,† Fig. S9.2-1). Using the photochemical kinetic model (see ESI,† Section 10.3), we can estimate that the lifetime of the second photon absorbing intermediate has to be at least 10 ns to explain the observed rates. Therefore, the τ = 150 ps species is likely not the corresponding intermediate, although it might partially isomerize to it below the detection limit (see ESI,† Section 9.2). Direct detection of the intermediate might thus require higher sensitivity measurements, which should be the goal for future studies of this system.Computational studies
To connect the described experimental insights and develop a holistic mechanistic understanding, we performed single- (density functional theory, DFT) as well as multi-configurational (complete active space self-consistent field, CASSCF) computations. Regarding previous theoretical studies, only Chen and Fang investigated a mechanism for O2 formation that does not proceed via H2O2 disproportionation.26 The authors proposed a hydrogen-bonded dimer of 1, which upon excitation undergoes intermolecular proton transfer followed by O–O bond formation. While the O–O bond formation step is energetically infeasible in this mechanism (barrier over 200 kJ mol−1), the first reaction step offers an interesting starting point for our study. In the following, the electronic state (spin and energy level) is indicated for each intermediate behind square brackets.Start for our mechanism (see Fig. 5A) is also a hydrogen-bonded dimer of 1, named [A]S0, for which time-dependent (TD)-DFT calculations reproduce the experimental UV/vis spectrum of 1 well (see Fig. 2). Excitation of the MLCT band at 350–400 nm (see ESI,† Section 11.2.3 for natural transition orbitals) leads to initial population of one of the six lowest excited singlet states, [A]S1–6, with an energy of 238–293 kJ mol−1 (relative to [A]S0) based on absorption and fluorescence (500 nm) wavelengths. This species was also observed in pump–probe spectroscopy. Intersystem crossing combined with a proton-coupled electron transfer (PCET) leads to formation of [B]T0 (219.7 kJ mol−1 relative to [A]S0). PCET of excited metal complexes has ample precedent.29 In [B]T0, the left ruthenium center has been formally oxidized to Ru(III) (although spin density is largely on the oxo ligand), while the right one has been reduced to Ru(I), leading to a charge-transfer state (see ESI,† Fig. S11.2-12 for spin density). This means that the first photon absorption has transferred the first of four electrons.
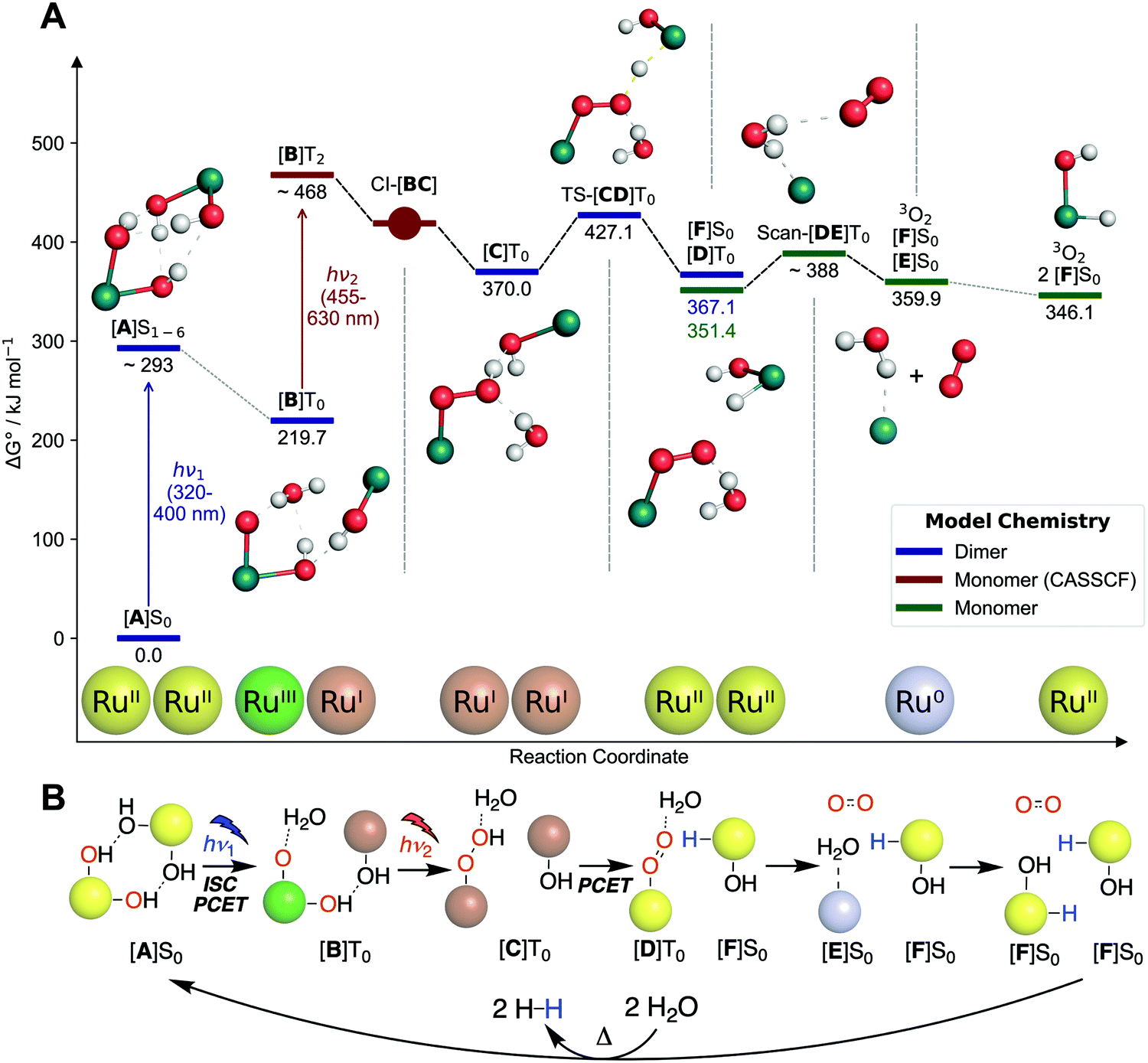 | ||
| Fig. 5 (A) DFT computed free energy profile. Energies of excited species ([A]S1–6 and [B]T2) are only approximate. Franck–Condon geometry is shown for [A]S1–6. Abbreviations: “CI” conical intersections, “TS” transition state, “Scan” relaxed potential energy surface scan. (B) Schematic representation of mechanism shown in (A). Colored spheres indicate ruthenium centers with corresponding formal oxidation states (Ru0 = grey, RuI = brown, RuII = yellow, RuIII = green). Abbreviations: “ISC” intersystem crossing, “PCET” proton-coupled electron transfer. Shown oxidation states are only formal to visualize electron flow. Structures are shown without other ligands for clarity. | ||
[B]T0 subsequently absorbs the second photon. Since O–O bond formation occurs intramolecularly,25 one can expect that the second photon absorption is centered on the left part of the dimer, [B-Mono]D0, containing a hydroxo and oxo ligand. Indeed, the TD-DFT UV/vis spectrum for [B-Mono-Up]D0 (conformational isomer of [B-Mono]D0, see ESI,† Section 11.4.1) agrees well with the dual irradiation data (see Fig. 4, details see ESI,† Section 11.4.1). Hence, photon absorption by [B-Mono]D0 was studied using CASSCF (see Fig. 6). Absorption of a 455–630 nm photon (transition wavelength CASSCF: 482 nm, [B]T0 TD-DFT: 539 nm, see ESI,† Section 11.3.1) leads to population of the D2 state (248 kJ mol−1 in energy relative to [B-Mono]D0, see Fig. 6). For this transition, electron density is transferred from the hydroxo ligand and ruthenium center to the oxo ligand (see ESI,† Fig. S11.3-4). Energetically downhill, at 211 kJ mol−1 relative energy, the D2/D1 minimum energy conical intersection (MECI) was located (peaked, bifurcating30). At this point, the O–O distance has shortened from 2.8 to 2.0 Å and depopulation of the O–O σ* orbital occurs for the D1 state (see ESI,† Fig. S11.3-5), instead populating a Ru–CO π* orbital, which leads to a bent CO coordination. Further downhill at 154 kJ mol−1, the D1/D0 MECI was located (sloped, single path30), at which the O–O bond is almost completely formed (1.6 Å). Here, depopulation of the Ru–CO π* orbital occurs for D0, instead populating the Ru(dz2) orbital (see ESI,† Fig. S11.3-6). Finally, at 125 kJ mol−1 relative energy, the O–O bond is fully formed in hydroperoxo complex [C-Mono]D0. Overall, this unprecedented reaction can be seen as a photoinduced oxo–hydroxo reductive elimination to enable a highly endothermic O–O bond formation. After formation of [B-Mono]D2 by photon absorption, there is no energetic barrier for this reaction step. Formally, two electrons are transferred to the left ruthenium center in the process, reducing it from Ru(III) to Ru(I). Hence, the second photon absorption transfers the second and third out of four electrons.
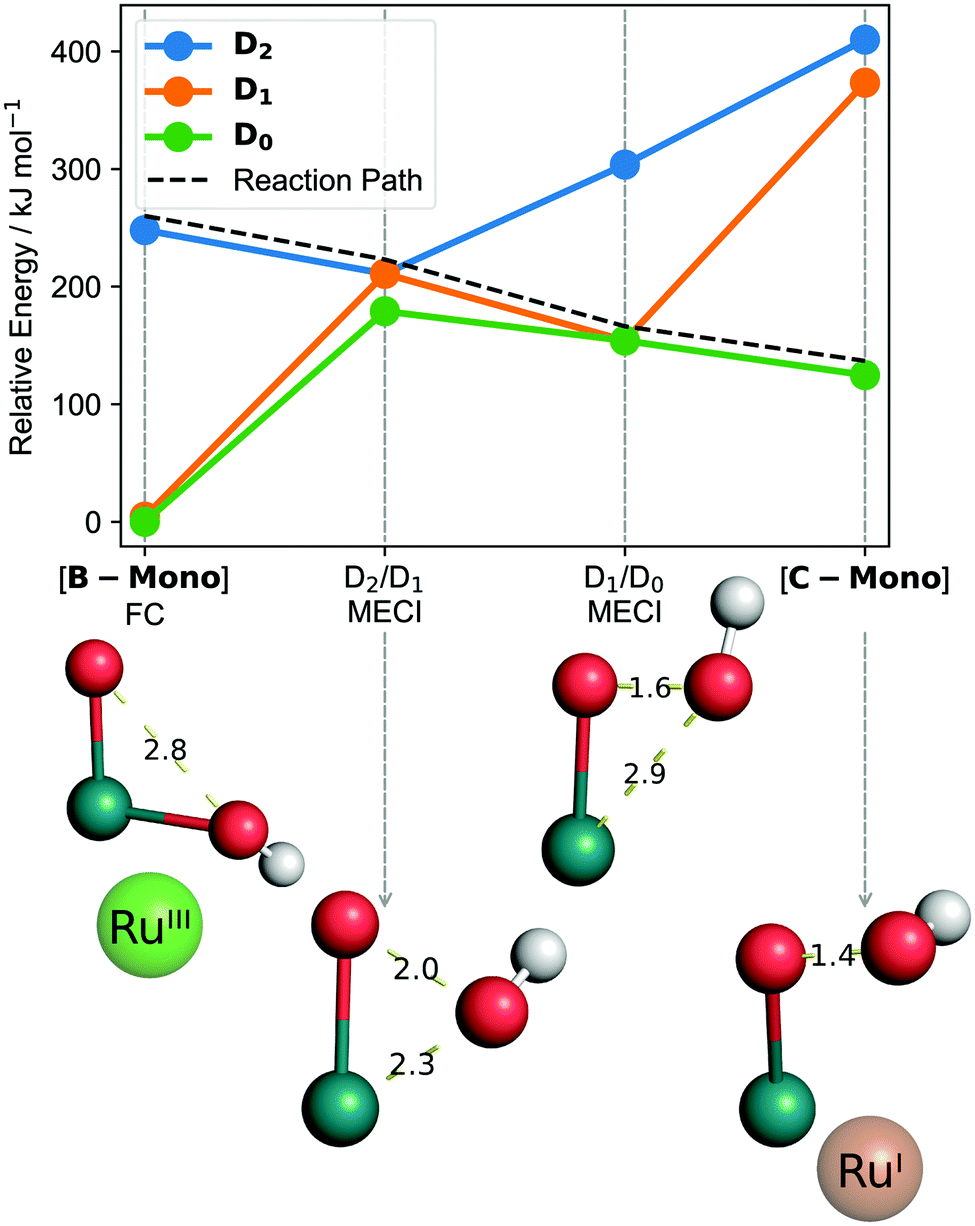 | ||
| Fig. 6 CASSCF computed reaction from [B-Mono] to [C-Mono] including minimum energy conical intersections (MECI). Bond distances are in Ångström. Calculations in the gas phase, for solution phase results see ESI,† Fig. S11.3-1. “FC” refers to Franck–Condon geometry. Shown oxidation states are only formal to visualize electron flow. Structures are shown without other ligands for clarity. | ||
The remaining reaction steps were calculated using DFT in the triplet ground state. In the dimeric model [C]T0 (spin density see ESI,† Fig. S11.2-13), the fourth and final electron transfer can take place via a PCET from the hydroperoxo ligand to the right Ru(I) center. This forms a superoxo ligand (bond distance of 1.3 Å) on the left ruthenium ([D]T0, spin density see ESI,† Fig. S11.2-14), while generating the first hydrido hydroxo product complex [F]S0 on the right. This step has a modest barrier of 57.1 kJ mol−1. Likely this barrier can be overcome with residual vibrational energy from the second photon absorption. Furthermore, it can likely explain the small amount of H2O2 formation observed by Milstein and co-workers: in a small fraction of all reactions, hydrolysis of [C]T0 might occur, liberating H2O2 instead of directly forming the superoxo ligand.
At this point the hydrogen-bonded dimer [D]T0 [F]S0 likely dissociates, as the monomer parts are 15.7 kJ mol−1 more stable than the dimeric structure. The superoxo ligand in [D]T0 is then displaced by water, leading to liberation of 3O2 and formation of Ru(0) complex [E]S0 (oxidation states of superoxo complexes are ill defined, so the conversion of Ru(II) to Ru(0) during superoxo dissociation is just a formal reduction). Finally, in a formal oxidative addition of water, [E]S0 can be converted to a second equivalent of [F]S0.31–33 The ligand configuration in [F]S0 is different from 2-cis. However, experimentally it was found that independent of water splitting, complex 2 isomerizes during irradiation to form 2-cis (see ESI,† Section 4.1.2).
To close the reaction sequence for overall water splitting, redox neutral protonation of the hydride ligands in two [F]S0 equivalents leads to liberation of two H2 molecules and regeneration of [A]S0,31–34 which is endothermic by 72 kJ mol−1 per [F]S0 (see ESI,† Table S11.2-7). The mechanism of H2 formation has previously been thoroughly investigated using computational methods.31–34 It proceeds via the following steps:
(1) A proton attacks the hydride ligand to from a dihydrogen ligand.
(2) The dihydrogen ligand dissociates, producing H2 and a five-coordinate Ru(II) hydroxo complex intermediate.
(3) Hydroxide coordination to the five-coordinate intermediate produces dihydroxo complex [A-Mono]S0 (which can subsequently form the hydrogen bridged dimer [A]S0).
This process may be assisted by metal–ligand cooperation with the pincer ligand.34 Before hydride protonation, [F]S0 might isomerize to the more stable trans isomer ([F-trans]S0 analogues to 2-trans), although this does not make a large energetic difference (see ESI,† Section 11.2.9). For the thermodynamic considerations herein, we assume H2 release from [F]S0.
We can summarize that the proposed water splitting mechanism rests on the following pieces of evidence: firstly, experiments indicate that 2-cis and O2 are formed at a comparable rate (see ESI,† Section 4.4) and both show the unusual effect that their formation is synergistically enhanced by light which is not absorbed by starting complex 1 (see Fig. 4). We consider this strong evidence that 2-cis and O2 are the two products of the water splitting reaction and other (unknown) processes for the formation of 2-cis or O2 do not appear to play a significant role. Hence, both can be used as indicators to study the water splitting reaction.
Secondly, the two-photon nature of the water splitting reaction was elucidated using six different methods inspired by those previously used:18–24
(1) A square relationship between initial rate of O2 formation and photon flux (see Fig. 3A) indicates that two photons are involved in the reaction.18,19,21
(2) Dual irradiation experiments revealed that light, which is not absorbed by starting complex 1, enhances formation of 2-cis (see Fig. 4A). This is consistent with a sequential two-photon mechanism in which an intermediate absorbs the second photon that cannot be absorbed by the starting material.18
(3) Wavelength dependent dual irradiation experiments of O2 formation revealed the absorption behavior of the second photon absorbing intermediate (see Fig. 4B). The obtained data is consistent with the computational prediction for intermediate [B-Mono-Up]D0.
(4) Ultrafast pump–probe spectroscopy allowed for observation of an isomer ([B-trans]T0) of the proposed intermediate ([B]T0, see ESI,† Section 9.2).
(5) A kinetic model for two-photon water splitting was developed which is consistent with experimental O2 formation data (see Fig. 3B), the observed square initial rate/photon flux relationship (see ESI,† Section 10.2) and dual irradiation data (see ESI,† Section 10.4).
(6) Single- and multi-configurational computations provided a complete mechanistic description (see Fig. 5 and 6). The computations are consistent with the experimental absorption behaviors of both complex 1 (see Fig. 1) and the second photon absorbing intermediate (see Fig. 4B).
Combined, these experimental, kinetic and computational pieces of evidence, which are all consistent with one another, strongly support the two-photon water splitting paradigm proposed herein.
Modelling solar-to-hydrogen efficiency
In the studied system, two-photon water splitting only proceeds with a low yield (20% of 2-cis after 2 days of irradiation, 320–500 nm quantum yield up to 1.4 × 10−4%, see ESI,† Section 4.2.3), which is likely due to a short lifetime of [B]T0 (see ESI,† Section 10.3 and 10.4). For a low lifetime of [B]T0, the intermediate's probability of absorbing a photon before decaying back to [A]S0 is also low, rendering the reaction less efficient. To some extent, this deficiency is not surprising since the structure of 1 has in no way been optimized to facilitate photochemical reactions or stabilize charge transfer states. This model system may therefore serve primarily as a proof-of-concept for two-photon water splitting. However, increasing the lifetime of [B]T0 would linearly increase the quantum yield (see ESI,† Section 4.2.3). Lifetimes on the order of milliseconds, which have been experimentally demonstrated for charge-transfer states,35,36 would be sufficient for the intermediate to absorb a photon before decaying most of the time (see ESI,† Section 11.5.2.1). A major target moving forward should therefore be to improve the lifetime of [B]T0.To gain an understanding for the potential of this mechanism to efficiently convert solar energy, we developed a model to calculate maximum theoretical solar-to-hydrogen (STH) efficiencies for various water splitting mechanisms, based on the work of Bolton et al.37 and Seitz et al.38 (details see ESI,† Section 11.5). Assuming full photon absorption, 100% quantum efficiency and a sufficiently long lifetime of [B]T0 (on the order of milliseconds, see ESI,† Section 11.5.2.1), we found that the maximum STH efficiency for [A]S0 → 2[F]S0 + 3O2 is 13.27%. This efficiency is achieved when the longest absorption wavelength for [A]S0 is 455 nm, while the longest absorption wavelength for [B]T0 is 517 nm (see ESI,† Fig. S11.5-1), which are relatively close to the current values (ca. 400 and 630 nm). To complete the water splitting reaction, two equivalents of H2 have to be released from two equivalents of [F]S0. If this thermal reaction is driven using solar energy (the operating temperature for solar water splitting reactors has been estimated to be 60 °C39), an additional STH efficiency of 5.52% is obtained, giving a total STH efficiency of 18.79% (details see ESI,† Section 11.5.2.1). This efficiency is higher than what can be achieved using normal single absorber photocatalysts that require absorption of four photons (18.11%, see ESI,† Table S11.5-1), despite relying on just one catalyst and requiring absorption of only two photons. It is also encouraging in terms of practical applicability, since a STH efficiency of 10% has been identified as a threshold for commercial viability of solar water splitting.39
Notably, the maximum theoretical STH efficiency can be further improved if hydrogen is not thermally released from [F]S0 but rather photochemically. Photochemical H2 release from transition metal monohydrides has previously been reported.40 If each equivalent of [F]S0 absorbs one photon to release H2, the total maximum STH efficiency is 28.65% (see ESI,† Table S11.5-3 and Fig. S11.5-2). In this case, the optimal longest absorption wavelengths for [A]S0, [B]T0 and [F]S0 are 491, 584 and 748 nm, respectively. This efficiency is higher than what can be achieved for dual absorber systems such as two-absorber Z schemes (27.72%, see ESI,† Table S11.5-2), while still relying on just one catalyst. In total, four photons are needed for this reaction sequence (two for [A]S0 → 2[F]S0 + 3O2 and two for 2[F]S0 + 2H2O → [A]S0 + 2H2). Since the redox neutral, photochemical reaction of [F]S0 would be independent from that of [A]S0, this would not increase the kinetic complexity of two-photon water splitting itself. We can therefore see that high STH efficiencies can theoretically be achieved using two-photon water splitting. Important factors to enable this are a long lifetime of [B]T0, well matched absorption wavelengths of [A]S0 and [B]T0, high quantum efficiencies and photochemical H2 release from [F]S0.
As a whole, this highlights a new and complementary way to think about solar water splitting systems: conventionally, a photoabsorber is used, which generates charge carriers that are transported to the active sites of hydrogen and oxygen evolution. Hence, there is a separation between light absorption and chemical reactivity. In this new paradigm, water splitting can be seen as a sequence of photochemical reactions, generating different light absorbing intermediates at each step to complete the reaction. This means that there is a direct interplay between light absorption and chemical reactivity. It remains to be seen whether practical systems can be designed based on the proof-of-concept system studied herein. However, the modelling of STH efficiencies shows that theoretically, efficient solar energy utilization can be achieved with simple systems (single catalyst) and low kinetic complexity (between a total of two and four photon absorptions).
Conclusion
In summary, we describe a new mechanistic paradigm for overall water splitting, which requires absorption of only two photons and presents a new method to extend the range of usable wavelengths far into the visible region. Kinetic results show that consecutive, non-linear absorption of a 320–400 nm photon followed by a 455–630 nm photon occurs. These are absorbed by two different species. Computational investigations, supported by ultra-fast spectroscopy, revealed that the first photon absorption causes a spin–flip and the first proton-coupled electron transfer, resulting in a charge-transfer state, which is capable of absorbing the second photon. The second photon absorption enables a highly endothermic O–O bond forming reaction by transferring the second and third electrons. This results in a reaction product that can undergo the fourth and final electron transfer without additional energy input. While for this particular model system the reaction does not proceed with high yield or efficiency, theoretical modelling shows that a maximum solar-to-hydrogen efficiency of up to 18.8% can be achieved, which could be increased further to 28.6% through photochemical instead of thermal H2 release. Therefore, we expect that the detailed mechanistic insight laid out herein can inspire a new class of artificial water splitting systems which take advantage of:(1) the low kinetic complexity of requiring only two photons,
(2) the intrinsic ability of this mechanism to utilize a wide wavelength range and
(3) the high-performance potential.
We envision that two-photon water splitting can be used as the turnover enabling step to develop the first homogeneous photocatalysts for overall water splitting. Furthermore, it is possible that defined active sites in heterogeneous materials (e.g. metal–organic frameworks or hybrid structures41) can be constructed that facilitate two-photon water splitting, giving rise to a new class of visible light absorbing heterogeneous water splitting catalysts. Ultimately, this could enable efficient and economical water splitting systems that go beyond the reaction blueprint of natural photosynthesis.
Funding
Financial support by Fonds der Chemischen Industrie (Kekulé-Stipendium no. 102151 for J. S.) and the Deutsche Forschungsgemeinschaft (Priority Program SPP 2102, project LO 714/11-1) is gratefully acknowledged.Author contributions
J. S. conceived and coordinated the project, performed all experimental (except ultrafast pump–probe spectroscopy) and computational work, wrote analysis software, performed data analysis, curated data for supporting information and public repository, and wrote the manuscript. M. A. A. C. and S. L. planned ultrafast pump–probe spectroscopy experiments, M. A. A. C. performed pump–probe spectroscopy experiments and analyzed resulting data. H. J. and M. B. provided experimental infrastructure and resources as well as funding. All authors discussed the results and revised the manuscript.Data availability
Original data supporting the results of this study is available at: https://github.com/jschneidewind/Water-SplittingCode availability
Code developed for this study is available at: https://github.com/jschneidewind/Water-SplittingConflicts of interest
The authors have no competing interests.Acknowledgements
Hrishi Olickel is gratefully acknowledged for providing computational resources and valuable discussions. Maximilian Marx (LIKAT) is acknowledged for crystallization attempts of 1, IR measurements and valuable discussions. Prof. David Milstein (Weizmann Institute of Science) and Dr Stephan Kohl (TU Berlin) are acknowledged for providing original data and valuable discussions. Dr Wolfgang Baumann and Susann Buchholz (both LIKAT) are acknowledged for performing NMR measurements. Dr Jabor Rabeah (LIKAT) is acknowledged for performing EPR measurements. Peter Kucmierczyk (Evonik), Dr Nils Rockstroh, Astrid Lehmann, Andreas Hutter, Matthias Auer and Anja Kammer (all LIKAT) are acknowledged for technical support. Prof. Robert H. Morris (University of Toronto) is acknowledged for enabling early experimental work.References
- B. Kok, B. Forbush and M. McGloin, Photochem. Photobiol., 1970, 11, 457–475 Search PubMed.
- J. Kern, R. Chatterjee, I. D. Young, F. D. Fuller, L. Lassalle, M. Ibrahim, S. Gul, T. Fransson, A. S. Brewster, R. Alonso-Mori, R. Hussein, M. Zhang, L. Douthit, C. de Lichtenberg, M. H. Cheah, D. Shevela, J. Wersig, I. Seuffert, D. Sokaras, E. Pastor, C. Weninger, T. Kroll, R. G. Sierra, P. Aller, A. Butryn, A. M. Orville, M. Liang, A. Batyuk, J. E. Koglin, S. Carbajo, S. Boutet, N. W. Moriarty, J. M. Holton, H. Dobbek, P. D. Adams, U. Bergmann, N. K. Sauter, A. Zouni, J. Messinger, J. Yano and V. K. Yachandra, Nature, 2018, 563, 421–425 Search PubMed.
- Y. Wang, H. Suzuki, J. Xie, O. Tomita, D. J. Martin, M. Higashi, D. Kong, R. Abe and J. Tang, Chem. Rev., 2018, 118, 5201–5241 Search PubMed.
- R. J. Detz, J. N. H. Reek and B. C. C. van der Zwaan, Energy Environ. Sci., 2018, 11, 1653–1669 Search PubMed.
- T. Hisatomi and K. Domen, Nat. Catal., 2019, 1 Search PubMed.
- M. Einzinger, T. Wu, J. F. Kompalla, H. L. Smith, C. F. Perkinson, L. Nienhaus, S. Wieghold, D. N. Congreve, A. Kahn, M. G. Bawendi and M. A. Baldo, Nature, 2019, 571, 90–94 Search PubMed.
- M. S. Martinez, A. J. Nozik and M. C. Beard, J. Chem. Phys., 2019, 151, 114111 Search PubMed.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S.-T. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970–974 Search PubMed.
- F. Kuttassery, S. Mathew, S. Sagawa, S. N. Remello, A. Thomas, D. Yamamoto, S. Onuki, Y. Nabetani, H. Tachibana and H. Inoue, ChemSusChem, 2017, 10, 1909–1915 Search PubMed.
- T. Takata and K. Domen, ACS Energy Lett., 2019, 4, 542–549 Search PubMed.
- Q. Wang, M. Nakabayashi, T. Hisatomi, S. Sun, S. Akiyama, Z. Wang, Z. Pan, X. Xiao, T. Watanabe, T. Yamada, N. Shibata, T. Takata and K. Domen, Nat. Mater., 2019, 18, 827–832 Search PubMed.
- Z. Wang, Y. Inoue, T. Hisatomi, R. Ishikawa, Q. Wang, T. Takata, S. Chen, N. Shibata, Y. Ikuhara and K. Domen, Nat. Catal., 2018, 1, 756 Search PubMed.
- T. Yu, Y. Liu, Y. Zeng, J. Chen, G. Yang and Y. Li, Chem. – Eur. J., 2019, 25, 16270–16276 Search PubMed.
- M. Zhang, Y. Lin, T. J. Mullen, W. Lin, L.-D. Sun, C.-H. Yan, T. E. Patten, D. Wang and G. Liu, J. Phys. Chem. Lett., 2012, 3, 3188–3192 Search PubMed.
- B. Seger, I. E. Castelli, P. C. K. Vesborg, K. W. Jacobsen, O. Hansen and I. Chorkendorff, Energy Environ. Sci., 2014, 7, 2397–2413 Search PubMed.
- L.-P. Wang and T. Van Voorhis, J. Phys. Chem. Lett., 2011, 2, 2200–2204 Search PubMed.
- M. D. Kärkäs, O. Verho, E. V. Johnston and B. Åkermark, Chem. Rev., 2014, 114, 11863–12001 Search PubMed.
- K. Mutoh, Y. Nakagawa, A. Sakamoto, Y. Kobayashi and J. Abe, J. Am. Chem. Soc., 2015, 137, 5674–5677 Search PubMed.
- Y. Kobayashi, K. Mutoh and J. Abe, J. Photochem. Photobiol., C, 2018, 34, 2–28 Search PubMed.
- I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725–728 Search PubMed.
- F. Glaser, C. Kerzig and O. S. Wenger, Angew. Chem., Int. Ed., 2020, 59, 10266–10284 Search PubMed.
- M. Goez, M. Schiewek and M. H. O. Musa, Angew. Chem., Int. Ed., 2002, 41, 1535–1538 Search PubMed.
- M. Goez, D. von Ramin-Marro, M. H. Othman Musa and M. Schiewek, J. Phys. Chem. A, 2004, 108, 1090–1100 Search PubMed.
- W. Domcke, J. Ehrmaier and A. L. Sobolewski, ChemPhotoChem, 2019, 3, 10–23 Search PubMed.
- S. W. Kohl, L. Weiner, L. Schwartsburd, L. Konstantinovski, L. J. W. Shimon, Y. Ben-David, M. A. Iron and D. Milstein, Science, 2009, 324, 74–77 Search PubMed.
- Y. Chen and W.-H. Fang, J. Phys. Chem. A, 2010, 114, 10334–10338 Search PubMed.
- R.-Q. Li, C. Zhang, B.-R. Xie, W.-Y. Yu, W.-X. Qiu, H. Cheng and X.-Z. Zhang, Biomaterials, 2019, 194, 84–93 Search PubMed.
- G. Langer, K.-D. Bouchal, H. Grün, P. Burgholzer and T. Berer, Opt. Express, 2013, 21, 22410–22422 Search PubMed.
- O. S. Wenger, Acc. Chem. Res., 2013, 46, 1517–1526 Search PubMed.
- I. F. Galván, M. G. Delcey, T. B. Pedersen, F. Aquilante and R. Lindh, J. Chem. Theory Comput., 2016, 12, 3636–3653 Search PubMed.
- X. Yang and M. B. Hall, J. Am. Chem. Soc., 2010, 132, 120–130 Search PubMed.
- K. S. Sandhya and C. H. Suresh, Organometallics, 2011, 30, 3888–3891 Search PubMed.
- C. Ma, S. Piccinin and S. Fabris, ACS Catal., 2012, 2, 1500–1506 Search PubMed.
- H. Li and M. B. Hall, ACS Catal., 2015, 5, 1895–1913 Search PubMed.
- Z. Lin, R. Kabe, K. Wang and C. Adachi, Nat. Commun., 2020, 11, 191 Search PubMed.
- Y. Han, L. Dobeck, A. Gong, F. Meng, C. W. Spangler and L. H. Spangler, Chem. Commun., 2005, 1067–1069 Search PubMed.
- J. R. Bolton, S. J. Strickler and J. S. Connolly, Nature, 1985, 316, 495–500 Search PubMed.
- L. C. Seitz, Z. Chen, A. J. Forman, B. A. Pinaud, J. D. Benck and T. F. Jaramillo, ChemSusChem, 2014, 7, 1372–1385 Search PubMed.
- B. A. Pinaud, J. D. Benck, L. C. Seitz, A. J. Forman, Z. Chen, T. G. Deutsch, B. D. James, K. N. Baum, G. N. Baum, S. Ardo, H. Wang, E. Miller and T. F. Jaramillo, Energy Environ. Sci., 2013, 6, 1983–2002 Search PubMed.
- S. M. Barrett, C. L. Pitman, A. G. Walden and A. J. M. Miller, J. Am. Chem. Soc., 2014, 136, 14718–14721 Search PubMed.
- L. Luo and P. A. Maggard, Cryst. Growth Des., 2013, 13, 5282–5288 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Methods and materials, Fig. S3.1-1–S11.6-32, Tables S4.1-1–S11.5-4. See DOI: 10.1039/d1ee01053k |
| This journal is © The Royal Society of Chemistry 2021 |