
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolid–electrolyte-interphase design in constrained ensemble for solid-state batteries†
William
Fitzhugh‡
,
Xi
Chen‡
 ,
Yichao
Wang
,
Luhan
Ye
and
Xin
Li
*
,
Yichao
Wang
,
Luhan
Ye
and
Xin
Li
*
John A. Paulson School of Engineering and Applied Sciences, Harvard University, Cambridge, Massachusetts 02138, USA. E-mail: lixin@seas.harvard.edu; Tel: +1 617 496 3075
First published on 20th July 2021
Abstract
Solid-state-batteries (SSBs) represent one of the most promising directions in the energy-storage field. The development of SSBs, however, is currently limited by the complex [electro-]chemical reactions that inevitably occur at the interface of solid-state electrolyte (SSE) particles. Moreover, given the material complexity of such systems, there is no straightforward methodology for addressing these interface instabilities. In this work, a combined high-throughput ab initio computation and machine learning approach is used to study and design solid-state solid–electrolyte-interphase (SEI) with tunable electrochemical stabilities using our unique constrained ensemble description. Machine learning reveals that the ability of a solid-state SEI to be stabilized by the mechanical constriction effect is a nonconvex and nonlinear, but deterministic none-the-less, function of composition. The power of this approach is demonstrated using the interface of glass and ceramic sulfide families of solid-electrolytes. Finally, it is experimentally verified that the designed interfaces, in fact, decompose and electrochemically passivate based on our predictions.
Broader contextSolid-state batteries are widely seen as the next great step in energy storage technology to displace lithium-ion batteries as the gold standard of secondary batteries. However, the development of solid state batteries has remained slow, largely due to complicated [electro-]chemical reactions that occur at interfaces between solid phases. Thus, there is an urgent need to develop a more effective approach to handle this major design hurdle. In this work, we introduce a comprehensive method to systematically study and, ultimately, design solid–electrolyte-interphases for such an all-solid electrochemical system. Within the general framework of mechanical constriction design, a new concept of critical effective modulus of interphase is proposed and quantified. Any local mechanical constriction level below this critical value will allow decompositions to happen. The decomposition, however, will fill local voids and cracks, and eventually increase the local mechanical constriction beyond the critical value to suppress further decompositions through metastability. Therefore, from this unique perspective of interphase electrochemical evolution, designing the interphase with a reduced critical effective modulus becomes an effective approach to increase dynamic interface stabilities. The method can be applied to the interface design of broad types of material systems, including glass, ceramic, polymer and their composites. |
The rapid charging, high energy density, and non-flammability of SSBs1–8 are critical for the ongoing push to achieve 100% vehicle electrification – a necessity for meaningful progress on climate change. Despite this promise, there remain many technical barriers in the way of broadly adoptable SSBs. One of the greatest of which is the electrochemical instability often found at the various interfaces in SSB cells.9–13
In our previous work, we have detailed how the fundamental thermodynamics of materials deviate in SSBs as compared to conventional liquid-cells.11–13 In short, liquid-cells operate in accordance with the well-studied state variables of pressure (p), temperature (T), and voltage (ϕ). In other words, the operator of a conventional cell sets these state variables experimentally and the material phases equilibrate (by, for example, [de]lithiation). Thus, the conventional liquid-cell can be well described by the pTϕ thermodynamic ensemble, which we term the “unconstrained” ensemble for contrast with the “constrained” ensemble for all-solid-state systems, described below.
Our previous work has shown that, unlike liquid systems, pressure is not a valid state variable in SSBs. In other words, the experimenter does not have direct control of the pressure within a working cell. When chemical decompositions occur within SSBs, strong local pressure gradients can form as the pressure is a function of the phases which are present locally. In liquid-cells, the liquid–electrolyte would flow to equilibrate this local pressure in response to any external pressure applied at the cell level. This cannot occur in an SSB, where the SSE maintains a pressure gradient. Thus, an operator has no direct control of the local pressure within an SSB. As a result, the electrochemical stability of solid-electrolytes varies considerably depending on cell and material design.
To describe these profound thermodynamic differences in SSBs, we introduce the concept of “constrained” ensemble, where the pressure is no-longer a state variable but is a function of the mole-fraction of local phases ({x}i). There are two critical insights for understanding this change of state variables. The first is the observation that many solid-electrolytes tend to decompose to products that are larger in total volume that the original electrolyte. The second is that, unlike liquid batteries, this volume expansion induced by decomposition has a profound impact on the underlying thermodynamics of the system.
With regards to the volume expansion, consider the electrolyte Li10GeP2S12 (LGPS). When LGPS chemically decays, it forms the products Li4GeS4 (LGS) and Li3PS4 (LPS). These products maintain a volume that is 2% larger than the original LGPS. Moreover, the volume expansion from electrochemical decompositions of LGPS when charged vs. lithium metal is even larger. In fact, the volume increases can reach values in excess of 30% at the complete oxidation (at high voltage) and reduction (at low voltage) limits.11–13 This increase in volume is referred to as the reaction strain (or reaction dilation) and is denoted εRXN. It is important to note that the reaction strain is a stress-free strain, meaning that unlike conventional strain, it does not result from an applied stress – it is solely the result of the [electro-]chemical decomposition that forms products having a different volume than the initial reactant (i.e. the electrolyte).
However, under an applied mechanical constriction, like the typical conditions of SSBs, the reaction strain can result in local stresses. That is to say, although the reaction strain is not caused by mechanical stress, it can cause new local mechanical stresses to form within the battery. These local stresses are the result of trying to fit a larger phase (the decomposed products) into the space that was previously occupied by a smaller phase (the electrolyte). This phenomenon is generalized by the formula p = KeffεRXN, where the pressure (p) is the average compressive stress on a solid volume unit and Keff is the local effective mechanical modulus. This formula quantifies that for a solid system, characterized by the modulus Keff, to accommodate some expanded decomposition products, a local pressure of p must be generated. The reaction strain is directly a function of the mole fraction of the present phases. For example, when the respective mole fractions of LGPS, LGS, and LPS are 1, 0, 0, then the reaction strain is 0%. Conversely, when the respective mole fractions are 0, 1/3, 2/3, the reaction strain is 2%. As a result of pressure being a function of εRXN and εRXN being a function of the present mole fractions, it is seen that pressure is now no-longer a state variable but instead a function of the present mole fractions.
This dependency of the mechanical stress distribution on the local mole fractions is what causes such strong changes in the thermodynamics of solid-state vs. liquid batteries. To illustrate this, consider a thought experiment in which a small volume fraction of the total electrolyte, located at ![[r with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0072_20d1.gif) 0, has decomposed. The decomposed [volume] fraction (xD) is now a piecewise function of the form:
0, has decomposed. The decomposed [volume] fraction (xD) is now a piecewise function of the form:
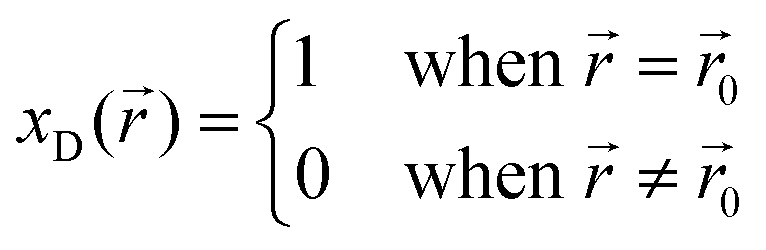 0 and zero everywhere else. As a result, the pressure caused by this reaction strain will also be non-zero only in the neighborhood of 0. In a liquid battery, such a pressure gradient would not be in equilibrium as fluids must have equal pressures at all point. As a result, the decomposed products would flow within the liquid electrolytes until the pressure comes to equilibrium with the environment (i.e. 1 atm for non-pressurized battery cells). In a solid-state system, however, this flow of the decomposed products will not occur because solids can maintain pressure gradients. The result is that a fluid battery will maintain a constant pressure equal to the environment regardless of the decomposed fractions whereas a solid-state battery can potentially have significant localized pressure that is not controlled by the external environment. That is, pressure is a valid state variable of liquid batteries but not one of solid-state batteries.
0 and zero everywhere else. As a result, the pressure caused by this reaction strain will also be non-zero only in the neighborhood of 0. In a liquid battery, such a pressure gradient would not be in equilibrium as fluids must have equal pressures at all point. As a result, the decomposed products would flow within the liquid electrolytes until the pressure comes to equilibrium with the environment (i.e. 1 atm for non-pressurized battery cells). In a solid-state system, however, this flow of the decomposed products will not occur because solids can maintain pressure gradients. The result is that a fluid battery will maintain a constant pressure equal to the environment regardless of the decomposed fractions whereas a solid-state battery can potentially have significant localized pressure that is not controlled by the external environment. That is, pressure is a valid state variable of liquid batteries but not one of solid-state batteries.
Consideration of the functional form of p({x}i) has shown to have profound impact on the intrinsic electrochemical stability of ceramic-sulfide SSEs. When the functional form is taken to be constant (i.e. isobaric conditions), the traditional pTϕ (grand canonical) ensemble is recovered, and the electrochemical stability is seen to be on the order of 1.7–2.1 V vs. lithium for LGPS.14,15 In contrast, when the pressure is taken to be proportional to the local strain, the (meta)stability window can greatly expand to approximately 1–4 V,1,12,13 and kinetic stability can reach down to 0 V and up to 10 V.1,4
We then for the first time apply the constrained ensemble to explicitly treat the calculation of interface stability, and articulate quantitatively one most critical aspect regarding interface stabilities in SSBs in electrochemical cycling, through the implementation of high-throughput ab initio computations and machine learning. We find that when the effective local modulus at the interface (Keff) is above some critical threshold decided by interface materials (denoted Kcrit), the interface will remain stable throughout cycling. When the effective modulus is lower than this threshold, the interface is unstable and will grow with cycling and thus limit the battery performance. Finally, we utilize this computational approach to develop a method for engineering solid–electrolyte-interphases (SEI's) which are stabilized through electrochemical evolution within a prescribed voltage window.
Modelling the interfaces within the constrained ensemble
The free energy picture of interface decomposition is depicted in Fig. 1(a). The interface is initially composed of two phases (α and β) and is seen to decompose with a reaction energy magnitude of Ghullvia some reaction: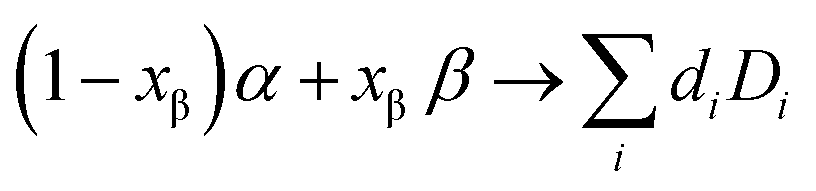 | (1) |
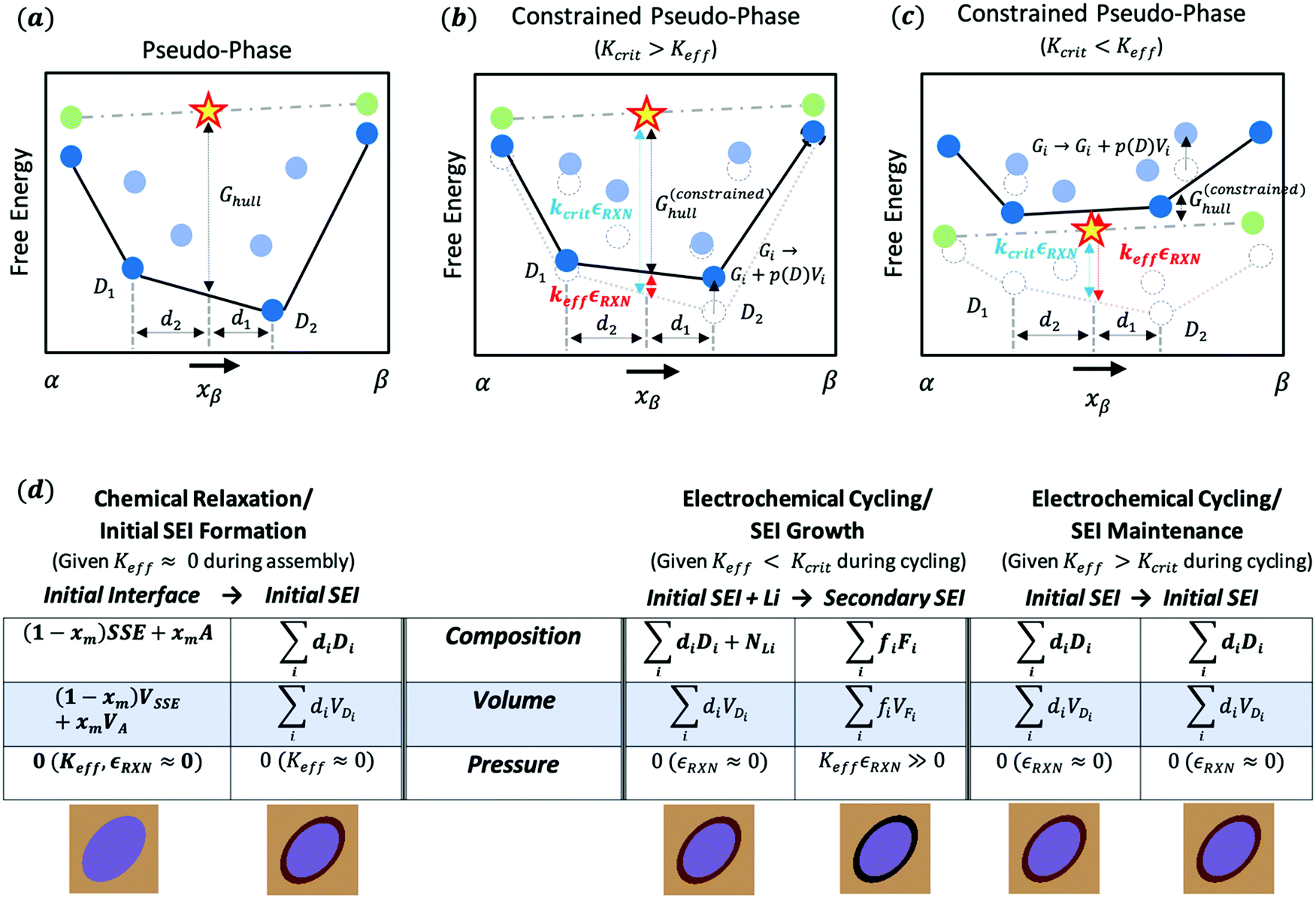 | ||
| Fig. 1 Understanding the critical effective modulus Kcrit within the constrained ensemble description. (a) Convex hull illustration for pseudo-phase models of the solid SEI. The yellow star represents the interphase composition (x-axis) and energy (y-axis) of the two reactive phases represented by green dots. The dark blue dots represent phases that are thermodynamically favorable and the black line represents a linear combination of these phases (i.e. the convex hull). The phase labels D1 and D2 and the lengths d2 and d1 represent the well-known “lever rule” for interpreting phase diagrams. The height of the interphase (yellow star) over the convex hull, Ghull, measures the magnitude of the decomposition energy. For a more detailed understanding of convex-hull pseudo-phase methods, the reader is directed to ref. 9 and 10. (b) An illustration of how a weak mechanical constriction (Keff < Kcrit) impacts the interphase convex hull. In this case, each of the potential decomposition phases experiences an increase in free energy corresponding to the pressurization of the phase equal to the generate pressure times the phase's volume. Under these weak constriction conditions the shift in energy is slight and doesn’t impact which phases are thermodynamically preferred. (c) Same as (b) but now with a strong mechanical constriction (Keff > Kcrit). As such, the interphase is now lower in free energy than the prior decomposition products. (d) Comparison of the different chemical and electrochemical reactions that can occur during battery assembly (left) and cycling (right) within the constrained ensemble framework. Generally, mechanical constriction cannot be applied throughout the entire assembly process, so initial SEI formation will likely occur in an unconstrained ensemble. In contrast, during battery cycling the battery may or may not be constrained, which can change the nature of the SEI to propagate and grow with cycling. The illustrations below the table in (d) illustrate how the electrolyte (light brown) and active material (blue) react to form a stable or unstable SEI depending on mechanical constriction. The initial SEI is in dark brown. If the battery is cycled with weak constriction (Keff < Kcrit), this initial SEI will further react to a new SEI (black) and likely grow. If the battery is cycled with a strong constriction (Keff > Kcrit), the initial SEI will remain in place. | ||
In liquid-cells (pTϕ ensemble), both the initial interface and decomposed interface exist at the same pressure, which is controlled by the operator. However, analogously to the effects of constraint on bulk SSE's, the pseudo-phase decomposition in solid-state reactions takes the form:
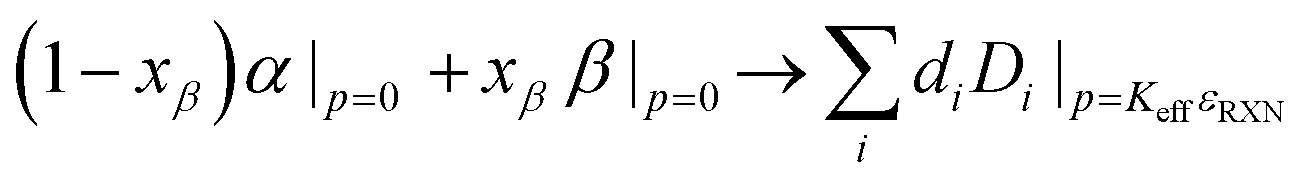 | (2) |
| ΔG0RXN + KcritεRXNV0 = 0 | (3) |
Deviation from grand canonical behavior is possible after the battery cell is fully assembled, at which point mechanical constriction may (or may not) be applied. In the absence of mechanical constriction, or when the local mechanical constriction is weak (Keff ≪ Kcrit), the initially formed SEI will be able to further decay once the battery is assembled and the system becomes open to lithium. This is represented in Fig. 1(d) as SEI growth. As the battery is cycled, the voltage will likely pass in and out of the SEI's electrochemical stability window, causing the initial SEI to continuously decay into secondary SEIs allowing continued growth. In contrast, if the local mechanical constriction is sufficiently rigid (Keff > Kcrit), either initially or after certain cycling steps, the following cycling of the voltage in and out of the initial SEI's stability window will not lead to further decompositions. This process is described as “SEI maintenance” through the local stability gained from the electrochemical evolution that could contain processes of both the initial decomposition (Keff < Kcrit) and the further stabilization (Keff > Kcrit).
Towards SEI maintenance: Kcrit distribution and elemental correlation
From a functional perspective, given the inevitable formation of an initial solid–electrolyte-interphase (SEI) during battery assembly, the goal of solid-state battery design should be to focus on designing the initial SEI's such that they don’t grow during cycling. Fig. 2 illustrates a computational process to determine such ideal initial SEI's. In short, an ensemble of 50 randomly initialized decision trees is trained to predict Kcrit of the ground state phase at any input composition. The ensemble is then used to fine-tune the composition of known parent glass phase sulfides with good lithium-ion conductivity. The resulting child compositions should be minimally changed from the parent compositions but modulated to decrease the interface Kcrit to as low of values as is possible. In this paper, we illustrate the computational method using only the interface between various glass coatings and four well known ceramic-sulfides. However, such a process is readily generalizable to the interface between the glass coatings and the active electrode materials, between ceramic electrolyte and uncoated electrode materials, as well as coatings beyond glasses, i.e., (nano)crystalline or polymer phases.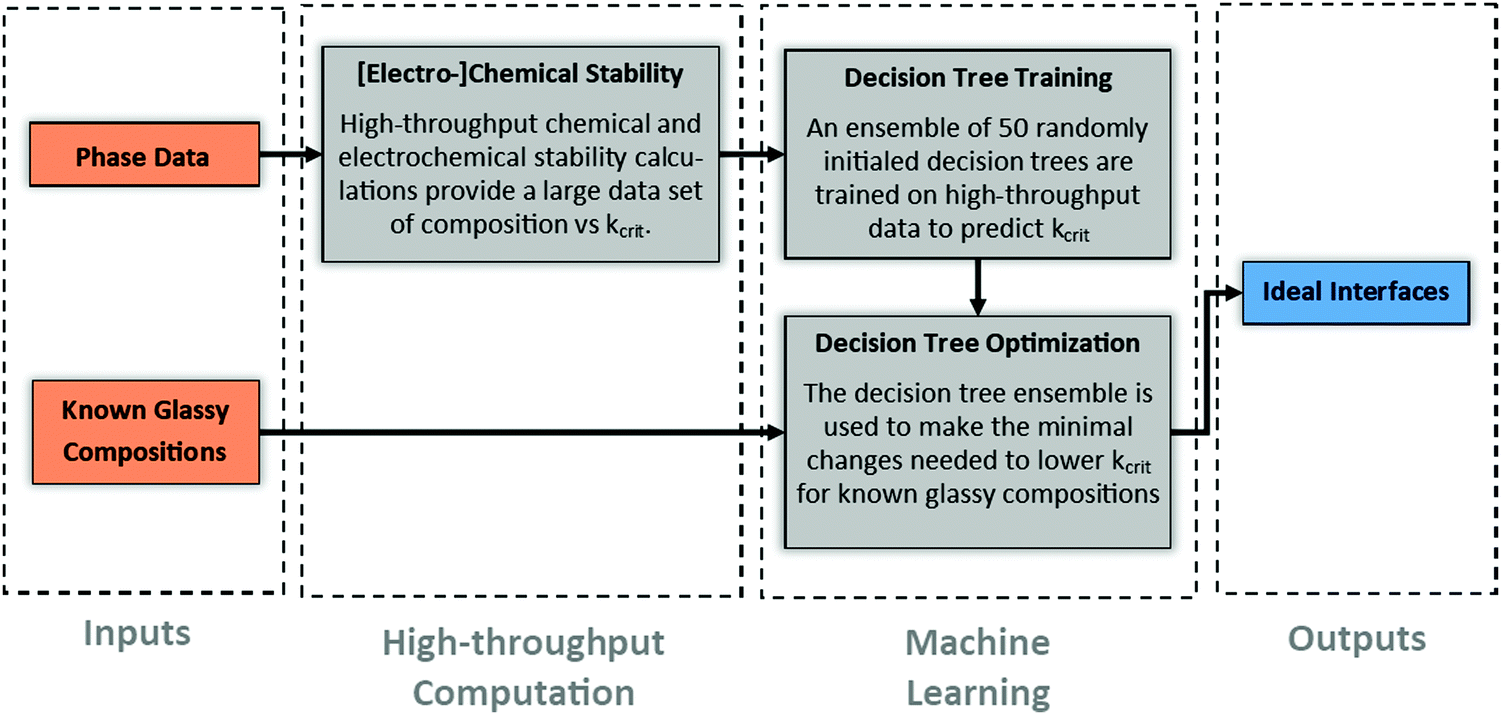 | ||
| Fig. 2 Overview of computational approach. High throughput computation is performed to obtain datapoints on how Kcrit varies with composition. These datapoints are then leveraged to train an ensemble of 50 decision trees to predict Kcrit based on composition. Finally, using known glassy compounds with good lithium-ion conductivity, the decision tree is used to make minimal compositional changes to lower Kcrit. The resulting compounds should have significantly improved response to mechanical constriction. These represent ideal interfaces for SSEs as they are both ionically conductive and mechanically metastable. | ||
Over 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 SEI's were simulated using pseudo-binary calculation of over 20000 electronic insulators from the Materials Project with four well known ceramic-sulfide solid electrolytes – Li10GeP2S12 (LGPS), Li10SiP2S12 (LSPS), Li7P3S11 (LPS), and Na7P3S11 (NPS). Each data point consisted of the initial SEI composition, formed by simulating a chemical reaction between the SSE and the coating, and the Kcrit corresponding to stabilizing the initial SEI at a cathode voltage (e.g., from 2 V to 4 V) vs. lithium metal. In other words, Kcrit indicates how much local mechanical constriction is required to stop the initial SEI from decomposing when cycled to the cathode voltage region. A lower Kcrit thus also suggests that the interface is more likely to be stabilized in further cycling steps even if the initial local Keff is less than Kcrit, as the electrochemical evolution will more easily increase the local Keff beyond Kcrit.
000 SEI's were simulated using pseudo-binary calculation of over 20000 electronic insulators from the Materials Project with four well known ceramic-sulfide solid electrolytes – Li10GeP2S12 (LGPS), Li10SiP2S12 (LSPS), Li7P3S11 (LPS), and Na7P3S11 (NPS). Each data point consisted of the initial SEI composition, formed by simulating a chemical reaction between the SSE and the coating, and the Kcrit corresponding to stabilizing the initial SEI at a cathode voltage (e.g., from 2 V to 4 V) vs. lithium metal. In other words, Kcrit indicates how much local mechanical constriction is required to stop the initial SEI from decomposing when cycled to the cathode voltage region. A lower Kcrit thus also suggests that the interface is more likely to be stabilized in further cycling steps even if the initial local Keff is less than Kcrit, as the electrochemical evolution will more easily increase the local Keff beyond Kcrit.
Fig. 3(a) shows the distribution of Kcrit determined for interfaces of LPS paired with the various coating materials. The distribution has a mean, median, and standard deviation of 18.5, 14.8, and 13.0 GPa, respectively. The large standard deviation indicates that the value of Kcrit of an initial interface can vary considerably with composition and, thus, allows for substantial engineering to optimize the interface's susceptibility to mechanical constriction within the constrained ensemble framework. Similarly, Fig. S1(a–c) (ESI†) show the Kcrit distribution for interfaces with LGPS, LSPS, and NPS. Fig. S1(d) (ESI†) depicts these statistical figures of merit (mean, median, standard deviation) for each SSE interface distribution. It remains true for each SSE that the distribution is quite broad, implying there is ample opportunity for engineering ideal interfaces with low Kcrit values.
 | ||
| Fig. 3 Statistical behavior of interface Kcrit at 4 V. (a) The distribution of Kcrit for binary pseudo-phases where one of the interfacing materials is LPS. (b) The correlation of elemental composition and Kcrit for all pseudo-phases considered (LPS, LGPS, LSPS, NPS). Red elements are those that lower Kcrit [ideal], whereas those elements that are blue raise Kcrit. | ||
The correlation between the elemental composition of SEIs and Kcrit is depicted in Fig. 3(b). Those atomic species with negative correlation (red) tend to lower Kcrit as their atomic fraction is increased. Conversely, those atomic species with positive correlation (blue) tend to raise Kcrit. The highly electronegative oxygen, fluorine, and chlorine are seen to be amongst the most beneficial atomic species for maintaining a low Kcrit at the cathode voltage. This is likely attributed to these anions having high voltage redox potential. In other words, SEIs with high levels of oxygen, fluorine, and chlorine are less prone to delithiation in the considered voltage ranges. Additionally, V, Mo, and W are also seen to improve the stability of high voltage SEIs, presumably indicating that these atomic species are also unlikely to undergo reactions below 4 V.
Designing SEIs with low Kcrit
In light of these results, it is apparent that initial SEIs could be designed to minimize Kcrit and hence decrease growth of the SEI upon cycling. In evaluating interfaces that are likely to occur in solid-state cathode, such as between LPS and common cathode materials, Fig. S2 (ESI†) shows that the Kcrit tends to be non-linear, oscillatory, with multiple local minima vs. composition. This highly nonlinear relationship between Kcrit and composition suggests that optimizing Kcrit directly is difficult, considering that the composition space is very high dimensional.To computationally capture the nonlinear effects of composition on Kcrit, we utilized an ensemble model of decision trees. The model was trained on the previously determined SEI data to predict Kcrit. The conditional statements of each node are learned by the tree to most accurately reflect the underlying composition dependence of Kcrit. The minimum unit of an example tree section illustrated in Fig. 4(a), together with the tree with more layers and branches depicted in Fig. S3 (ESI†), confirms the importance of electronegative anions for maintaining a low Kcrit. For example, the left most branch at each layer implements a conditional statement on O or Cl. If the material has a low composition of those anions, the tree is with a higher predicted value of Kcrit. Similarly, the right most branch states that if the material has a high composition of Li, F, Al, the tree is with a lower predicted value of Kcrit. While each node corresponds to the evaluation of a single element, each element may be evaluated many times throughout the tree. Thus, each tree represents an approximate mapping of the Kcrit manifold. The ensemble then performs a weighted average of the predicted manifolds for the final value, i.e., to predict a value for a given composition and a weighted average then gives a final prediction for the input composition. As shown in Fig. 4(b), which compares the predicted and actual Kcrit values for a test set of 10000 SEIs, this approach can readily capture the complicated compositional dependence, especially in the lower range of Kcrit. The root mean square error (RMSE) for this test set is 2.59 GPa.
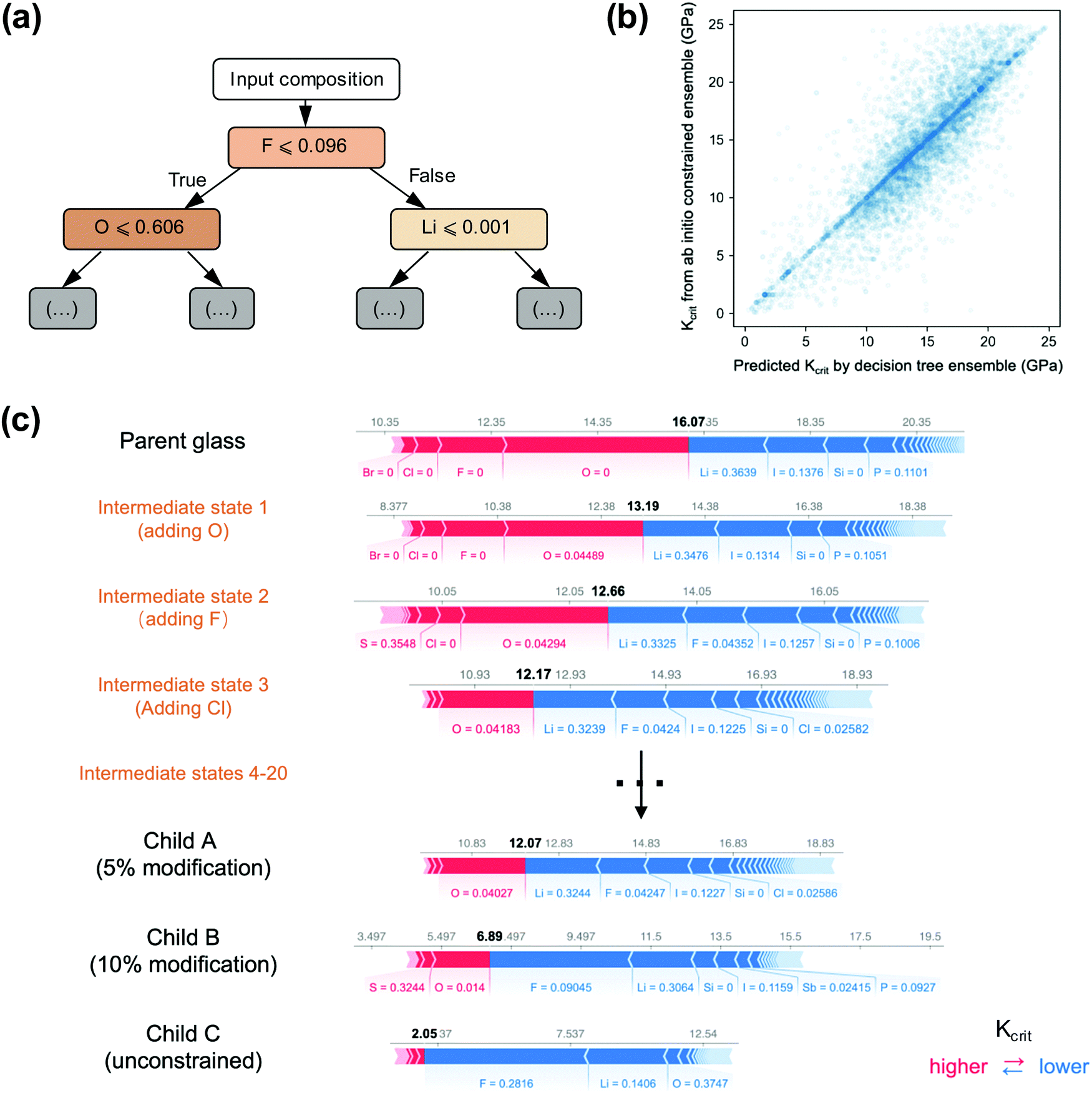 | ||
| Fig. 4 Decision tree method and results for composition design. (a) Illustration of the structure and mechanism of decision trees. The input composition is parsed through a hierarchy of criteria on the composition and generates output (see Fig. S3 for larger illustration, ESI†). (b) Comparison of the predicted Kcrit by the decision tree and the calculated Kcrit at the interface to LSPS at 4 V from our constrained ensemble ab initio platform, with a root-mean-square error (RMSE) of 2.59 GPa. (c) Optimization from (top to bottom) parent glassy material to child A (5% compositional change) to child B (10% compositional change) to child C (unlimited compositional change). The individual influence of each element is calculated by the SHAP model. Three intermediate states during the optimization process illustrate how the O, F, and Cl elements are introduced into the optimized Child A material as suggested by the SHAP model. | ||
Using the trained decision tree ensemble, we can modify the composition of known lithium-ion conductors to decrease the Kcrit and improve the SEI stability. To help us understand the role of each element on interface Kcrit, we applied a feature-attribution model SHAP, or Shapley additive explanations,17 to our trained ensemble to extract the influence of the composition of each element on the Kcrit. More technical details about the optimization method is described in the Methods. We tested various materials with changing levels of restriction on the elemental composition to better understand how Kcrit will change based on the doping amount. Table S1 (ESI†) shows compositional optimization of 10 well known sulfide glass SSE's and the corresponding child phases where the composition has been changed to lower the interface Kcrit to LSPS. Child A, B, and C correspond to maximum allowed changes in the stoichiometry of each element as 0.05, 0.1, or no constraint, respectively. Note that in practice it is preferable to make the minimum possible compositional changes to lower the Kcrit, as the ionic conductivity will likely fall quickly as composition is deviated. In Table S1 (ESI†), the known sulfide glasses have Kcrit values that are characteristically on the order of 15–17 GPa from our prediction. These values decrease to 10–13 GPa in child A phases, 6–9 GPa in child B phases, and 1.5–2 GPa in child C phases. This Kcrit decrease is expected to remarkably improve interface stability in actual solid-state battery cells, as the local mechanical constriction Keff is not necessarily at a sufficiently high level, depending on materials and battery assembly approaches.
We now use the first parent compound and its child compositions in Table S1 (ESI†) as an example to illustrate how our platform finds new compositions with lowered Kcrit. In Fig. 4c, the length of each bar (in the unit of GPa) approximates how much the corresponding element increases (red) or decreases (blue) the Kcrit. The SHAP model analyzed that for the parent glass of Li0.364P0.110S0.388I0.138, the absence of O, F and Cl are dominantly responsible for the relatively high Kcrit, as the first three long red bars are with O = 0, F = 0 and Cl = 0 in Fig. 4c, indicating that the presence of O, F and Cl in the compound will reduce Kcrit. Meanwhile, the blue bars for existing elements of Li, I, P in the parent glass indicate that having them as existing nonzero compositions is not in conflict with the optimization goal of reducing Kcrit.
It's natural to understand that the optimization effort should be given to the features with red bars, since the length of red bar also roughly indicates how much we can further decrease the Kcrit if we fully optimize the composition of the corresponding elements starting from a given composition. We take an approach to efficiently optimize Kcrit by focusing on the element corresponding to either the longest red bar at each step, or the 2nd longest red bar if the longest bar element has just been updated in the previous step (i.e., a pseudo-longest red bar). The actual optimization process of Kcrit is also influenced by the concurrent composition of other elements brought by changing the composition of one element. For example, the child A, B, C compositions bring down the Kcrit from the 16.07 GPa (parent glass) to 12.07 GPa (Child A), 6.89 (Child B), and 2.05 GPa (Child C) with an increasing modification range of compositions. F, O and Cl are first introduced to Child A composition of Li0.324P0.098S0.346I0.138Cl0.026F0.042O0.040, because these three elements all appear as either the longest or the pseudo-longest red bar in the initial intermediate optimization steps from the Parent toward the Child A composition (Fig. 4c), with Li, P, S, I compositions being forced to make slight concurrent decreases. The algorithm is very efficient in finding the right composition, as the intermediate compositions after the first 3 intermediate steps are very close to the Child A composition, with the remaining 17 intermediate steps just as a fine-tune of the composition. Through the composition optimization from Parent to Child A, B, C compounds, the total length of red bars shrinks and the Kcrit decreases generally. At Child C with unconstrained compositional change, the red bars are very short, suggesting a very limited remaining room to further decrease Kcrit through compositional modification. The general trend of adding more F, O, Cl, Br for reduced Kcrit as predicted by the SHAP model is also in good agreement with the elemental correlation map (Fig. 3(b)).
Using the predicted Kcrit of SEI at 4 V, we further predict the 4 V interface (to LGPS) decomposition energy for the SEI under various levels of composition modification (Fig. 5(a)). Similar to Kcrit, the interface decomposition energy also tends to decrease with the level of modification. A Keff of 5 GPa can further lower the interface decomposition energies from that at 0 GPa while keeping the similar decreasing trend with composition modification. A Keff of 10 GPa can stabilize child materials at the 4 V interface to LGPS with a composition modification larger than 2.5%, for which Keff > Kcrit is satisfied due to the reduced Kcrit by composition modifications (Fig. 5b). With a 15 GPa Keff, all the materials, including the parent material will be stabilized. The trends described in Fig. 5a and b are qualitatively the same for interfaces calculated at 2 V and 3 V (Fig. S4, ESI†), except for some quantitative changes by voltage. Note that the Kcrit at 2 V are all below 4.6 GPa and that at 3 V are all above 4.9 GPa at all modifications.
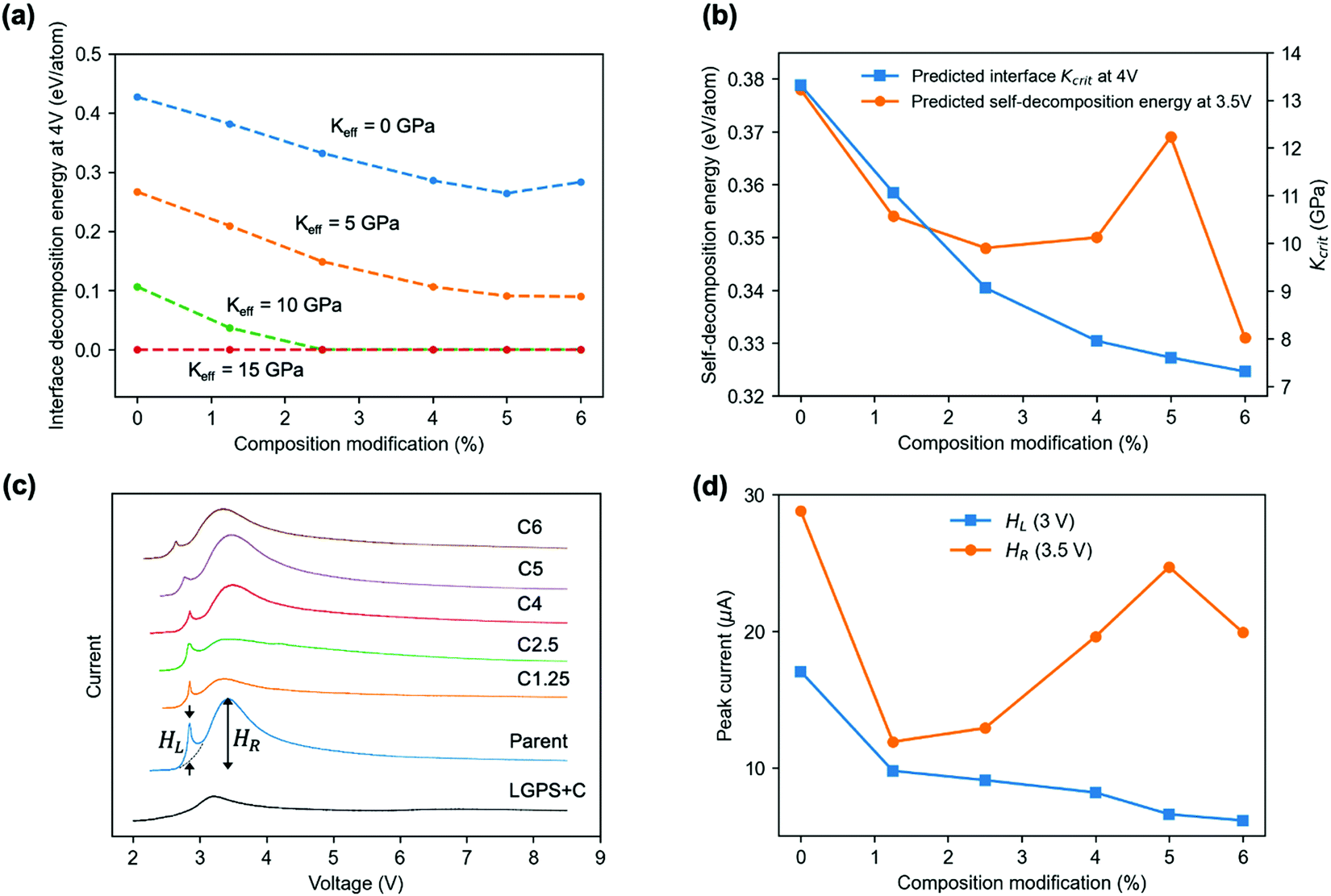 | ||
| Fig. 5 Material modification and experimental validation based on parent glass Li0.364P0.11S0.388I0.138 (Table S2, ESI†). (a) Predicted interface decomposition energy at 4 V for the parent (0%) and modified glass coatings (1.25, 2.5, 4, 5, 6%) to LGPS electrolyte at different Keff. The x% composition modification suggested by our SHAP model means the stoichiometry of each element is allowed to change up to a number of x/100, regardless of the initial amount of this element. (b) Predicted Kcrit of coating materials at the interface to LGPS electrolyte (blue) and the self-decomposition energy of coating materials at Keff = 0 GPa (orange) vs. composition modification level. (c) CV test of the synthesized parent glass and five modified child glasses, labelled as Cx, where x corresponds to the modification level in (a). HL and HR mark the peak height for the decomposition current at around 3 V (pre-peak) and 3.5 V (broad peak), respectively. (d) Pre-peak (HL) and broad peak (HR) decomposition currents vs. composition modification level. | ||
Testing the model by experiment
To further test the predictability of our model, we synthetized the parent and predicted child glasses of the first row of Table S2 (ESI†) and performed cyclic voltammetry (CV) test based on batteries of normalized cathode weight, area and composition, as shown in Fig. 5(c) (Methods). Two decomposition current peaks labelled as HL and HR are observed at 3 V and 3.5 V, respectively. The pre-peak HL is found to decrease monotonically with modification, while the broad peak HR is oscillatory (Fig. 5(d)). We found that the predicted self-decomposition energy at 3.5 V at Keff = 0 GPa in Fig. 5(b) exhibits a very similar trend to the measured HR curve, suggesting that the change of broad peak intensity around 3.5 V with composition modification should be mainly caused by the self-decomposition current of the glasses in the initial charge, where the decomposition happens at the glass region near the many inevitable pores in the cold pressed pellet,18 where Keff = 0 GPa. Note that without glasses, the control cathode of LGPS mixed only with amorphous conductive carbon shows a broad peak of decomposition current below 9 μA in the CV test, which defines the background contribution from the LGPS self-decomposition to the measured current that is independent from the modification to the glasses. Thus, the pre-peak HL has to be contributed by the interface decomposition between LGPS and glass at a weak local mechanical constriction in the initial charge, consistent with the general decreasing trend for both curves of the measured HL (Fig. 5d) and the predicted interface decomposition energy at a low Keff (Fig. 5a and Fig. S4, ESI†) versus modifications.The fact that all the HL pre-peaks are around 3 V suggests that the local Keff at the interface in the initial charge is around 4.6 to 4.9 GPa so that it is higher than all the Kcrit at 2 V (Fig. S4a, ESI†) but lower than all the Kcrit at 3 V (Fig. S4b, ESI†). Such an initial interface Keff is decided by the mechanical property of the two materials that form the interface (glass sulfide and LGPS here) and the processing and testing procedures (hand mixing, cold initial and stack press here) that define the interface contact. Only when the voltage is charged approaching 3 V, the local Keff starts to be smaller than Kcrit at the interface, as Kcrit increases with charge (comparing Fig. 5b and Fig. S4ab, ESI†), so that the interface decomposition happens. However, after the initial charge, the decomposition current almost vanishes in discharge and the following CV cycles below 4 V (Fig. S5, ESI†), suggesting that the local Keff of the glass–LGPS mixture upon the initial self and interface decompositions is increased beyond the Kcrit at all modifications both at the interface (Kcrit < 14 GPa, Fig. 5b) and inside the glass (Kselfcrit < 6 GPa, Fig. S6, ESI†). Therefore, it is through these local decompositions in the initial charge that the order of Keff and Kcrit is switched at some low Keff regions, leading to the largely suppressed decomposition in further cycling, where the local Keff is increased beyond 14 GPa throughout the charged pellet and Keff > Kcrit is generally satisfied below 4 V. Note that through the comparison between computation and experiment an upper limit of Keff value around 15 to 20 GPa is also estimated for crystalline phase of LSPS12 and LGPS.1,4,13 Such a Keff is largely decided by the mechanical modulus of the electrolyte materials, which is similar for crystalline19 and amorphous20 sulfides.
We have shown that within the constrained ensemble that is unique to solid-state batteries, the use of randomly initialized decision tree ensembles has significant potential for optimizing SEIs at multiple solid–solid interfaces. Fine-tuning the SEI composition based on the decision tree results has been able to reduce the Kcrit significantly. Although the examples given here are tested at an experimental condition with strong mechanical constriction (see Methods) so that the decomposition current is largely inhibited after the initial charge regardless of the composition, in practice, the composition modification strategy for the electrolyte and coating materials could greatly reduce the burden of void elimination procedures during battery assembly and micro-crack prevention approaches during battery test, favoring the ideal manufacturing conditions at weak mechanical constriction. As a result, the method opens the door to realizable solid-state battery design and production based on mechanical constriction induced metastability.
Methods
Experiment
The amorphous solid electrolytes (P1-C6) are synthesized by high energy ball milling at 350 rpm. Stoichiometric amounts of Li2S (>99.9% purity, Alfa Aesar), P2S5 (>99% purity, Sigma Aldrich), LiI (>99% purity, Sigma Aldrich), LiF(>99.99% purity, Sigma Aldrich), and Li2O (>97%, Sigma Aldrich) were milled for 20 hours. A total of 1 mg LGPS–amorphous SE–carbon composite cathode (weight ratio = 45:45:10) was used as the cathode. The composite was mixed and rolled into a thin film by a dry method with the addition of 5% PTFE. The diameter of the cathode film is 0.49 cm2. The area and weight of all batteries were kept constant for all CV measurements. 120 mg LGPS was applied as the electrolyte while Li/graphite (capacity ratio = 2.5:1) was applied as an anode. The battery of Li/graphite–LPSCl–LGPS-cathode was pressed in a homemade pressurized cell, where LPSCl represents Li5.5PS4.5Cl1.5 that is used to guarantee a stable anode interface following our recent approach.21 The batteries were initially pressed at 467 MPa while the operating pressure was kept at 250 MPa during battery test to ensure the mechanical constriction. In the CV test, a voltage ranges from the open circuit voltage to 8.5 V was applied with the scan rate of 0.1 mV s−1. The control sample labelled as LGPS + C in Fig. 5 is constructed with the similar procedure but using only 0.5 mg of LGPS + C (weight ratio = 90:10) as cathode, without mixing with the amorphous glass.
High-throughput computation
Thermodynamical chemical decomposition reactions between materials (DFT data obtained from Materials Project) and an electrolyte were first calculated to generate an interphase for each material/electrolyte combination by constructing phase diagram using the Python Materials Genomics (pymatgen) library22 with the binary-search algorithm.9 The electrochemical stability of each interphase is then calculated from its grand potential phase diagram to obtain decomposition energy and reaction strain for calculating Kcrit by eqn (2) and (3). All ab initio calculations of phase data were obtained following the Material Project calculation guidelines in the Vienna ab initio Software Package (VASP). The mechanical constriction induced metastability calculations were performed on the computational platform we developed following the perturbation or Lagrangian optimization methods.11–13Decision tree learning
A decision tree consists of hierarchical computation (decision) nodes. A typical decision tree is depicted in Fig. S3 (ESI†). The input data to the decision trees is in the form (X,y) = ({x1,x2,…,xn},y) where xi are the features and y is a target value. The decision tree can perform both the regression and classification tasks, depending on whether the nature of target variable y being continuous or a finite number of classes. Starting with the input features, each node of the tree applies a conditional statement on the value of a feature, then moves to a subsequent node based on the truth of that statement. The optimization of the tree includes choosing both the feature and threshold for the criteria for each node that overall best splits the set of items. Instead of measuring the error, better metrics such as the cross entropy and the Gini index are generally used to measure the goodness of the choice of criteria and data split.23We use an ensemble model of individual decision trees, the Extremely Randomized Tree model.24 In such models, a number of N trees are initialized simultaneously (N = 30 in our setting). Each tree in the ensemble is fed with training data sampled from the training set. A random subset of candidate features is used, from which thresholds are drawn at random for each candidate feature, and the best of these randomly generated thresholds is picked as the splitting rule.
Feature attribution and composition optimization
SHAP, or Shapley additive explanations, is a game theoretic approach to explain the output of any machine learning model.17 It connects optimal credit allocation with local explanations using the classic Shapley values from game theory and their related extensions and obtain the influence of the individual features on the output. In our case, we apply it to extract how each elemental composition of the SEI influences the Kcrit.We compute the SHAP values and choose the element with the most positive SHAP value, i.e., the presence of which has the most positive influence on the Kcrit. We then optimize the composition of this chosen element under the given constraint. With the composition optimized, we compute the SHAP value again and optimize the next element with the most positive SHAP value. Due to the composition constraint, it is possible that the element that is just updated still has the highest SHAP value. In this case, we move to the element with the second-highest SHAP value (pseudo-longest red bar in Fig. 4c) in order to proceed the optimization. The optimization process ended until the Kcrit cannot be further reduced.
Conflicts of interest
The authors declare no competing financial or non-financial interests.Acknowledgements
The work was supported by Harvard Climate Change Solutions Fund, Dean's Competitive Fund for Promising Scholarship at Harvard University, Harvard Data Science Initiative Competitive Research Fund, Nissan USA and LG Energy Solution, Ltd. Computational work was supported by computational resources from the Harvard Odyssey cluster and the national Extreme Science and Engineering Discovery Environment (XSEDE).References
- L. Ye, et al., Toward Higher Voltage Solid-State Batteries by Metastability and Kinetic Stability Design, Adv. Energy Mater., 2020, 10, 2001569 CrossRef CAS.
- Y. Kato, et al., High-power all-solid-state batteries using sulfide superionic conductors, Nat. Energy, 2016, 1, 16030 CrossRef CAS.
- Y. G. Lee, et al., High-energy long-cycling all-solid-state lithium metal batteries enabled by silver–carbon composite anodes, Nat. Energy, 2020, 5, 299–308 CrossRef CAS.
- Y. Su, et al., A more stable lithium anode by mechanical constriction for solid state batteries, Energy Environ. Sci., 2020, 13, 908–916 RSC.
- Z. Gao, et al., Promises, Challenges, and Recent Progress of Inorganic Solid-State Electrolytes for All-Solid-State Lithium Batteries, Adv. Mater., 2018, 30, 1705702 CrossRef PubMed.
- J. Janek and W. G. Zeier, A solid future for battery development, Nat. Energy, 2016, 1, 1–4 Search PubMed.
- N. Kamaya, et al., A lithium superionic conductor, Nat. Mater., 2011, 10, 682–686 CrossRef CAS PubMed.
- P. Adeli, et al., Boosting Solid-State Diffusivity and Conductivity in Lithium Superionic Argyrodites by Halide Substitution, Angew. Chem., Int. Ed., 2019, 58, 8681–8686 CrossRef CAS PubMed.
- W. Fitzhugh, et al., A High-Throughput Search for Functionally Stable Interfaces in Sulfide Solid-State Lithium Ion Conductors, Adv. Energy Mater., 2019, 9, 1900807 CrossRef.
- Y. Zhu, X. He and Y. Mo, First principles study on electrochemical and chemical stability of solid electrolyte–electrode interfaces in all-solid-state Li-ion batteries, J. Mater. Chem. A, 2016, 4, 3253–3266 RSC.
- W. Fitzhugh, L. Ye and X. Li, The effects of mechanical constriction on the operation of sulfide based solid-state batteries, J. Mater. Chem. A, 2019, 7, 23604–23627 RSC.
- F. Wu, W. Fitzhugh, L. Ye, J. Ning and X. Li, Advanced sulfide solid electrolyte by core-shell structural design, Nat. Commun., 2018, 9, 4037 CrossRef PubMed.
- W. Fitzhugh, F. Wu, L. Ye, H. Su and X. Li, Strain-Stabilized Ceramic-Sulfide Electrolytes, Small, 2019, 15, 1901470 CrossRef PubMed.
- S. P. Ong, et al., Phase stability, electrochemical stability and ionic conductivity of the Li10±1MP2X12 (M = Ge, Si, Sn, Al or P, and X = O, S or Se) family of superionic conductors, Energy Environ. Sci., 2013, 6, 148–156 RSC.
- F. Han, Y. Zhu, X. He, Y. Mo and C. Wang, Electrochemical Stability of Li10GeP2S12 and Li7La3Zr2O12 Solid Electrolytes, Adv. Energy Mater., 2016, 6, 1501590 CrossRef.
- A. M. Nolan, Y. Zhu, X. He, Q. Bai and Y. Mo, Computation-Accelerated Design of Materials and Interfaces for All-Solid-State Lithium-Ion Batteries, Joule, 2018, 2, 2016–2046 CrossRef CAS.
- S. M. Lundberg, P. G. Allen and S.-I. Lee, A Unified Approach to Interpreting Model Predictions, Arxiv: 1705.07874.
- A. Sakuda, A. Hayashi and M. Tatsumisago, Sulfide solid electrolyte with favorable mechanical property for all-solid-state lithium battery, Sci. Rep., 2013, 3, 1–5 Search PubMed.
- Z. Deng, Z. Wang, I.-H. Chu, J. Luo and S. P. Ong, Elastic Properties of Alkali Superionic Conductor Electrolytes from First Principles Calculations, J. Electrochem. Soc., 2016, 163, A67–A74 CrossRef CAS.
- F. P. McGrogan, et al., Compliant Yet Brittle Mechanical Behavior of Li2S-P2S5 Lithium-Ion-Conducting Solid Electrolyte, Adv. Energy Mater., 2017, 7, 1602011 CrossRef.
- L. Ye and X. Li, A dynamic stability design strategy for lithium metal solid state batteries, Nature, 2021, 593, 218–222 CrossRef CAS PubMed.
- S. P. Ong, et al., Python Materials Genomics (pymatgen): A robust, open-source python library for materials analysis, Comput. Mater. Sci., 2013, 68, 314–319 CrossRef CAS.
- C. M. Bishop, Pattern recognition and machine learning, Springer, 2006 Search PubMed.
- P. Geurts, D. Ernst and L. Wehenkel, Extremely randomized trees, Mach. Learn., 2006, 63, 3–42 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ee00754h |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2021 |