
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCation ordered Ni-rich layered cathode for ultra-long battery life†
Un-Hyuck
Kim
 a,
Geon-Tae
Park
a,
Patrick
Conlin
b,
Nickolas
Ashburn
b,
Kyeongjae
Cho
b,
Young-Sang
Yu
c,
David A.
Shapiro
c,
Filippo
Maglia
d,
Sung-Jin
Kim
d,
Peter
Lamp
d,
Chong S.
Yoon
*e and
Yang-Kook
Sun
*a
a,
Geon-Tae
Park
a,
Patrick
Conlin
b,
Nickolas
Ashburn
b,
Kyeongjae
Cho
b,
Young-Sang
Yu
c,
David A.
Shapiro
c,
Filippo
Maglia
d,
Sung-Jin
Kim
d,
Peter
Lamp
d,
Chong S.
Yoon
*e and
Yang-Kook
Sun
*a
aDepartment of Energy Engineering, Hanyang University, Seoul 04763, South Korea
bDepartment of Materials Science and Engineering and Department of Physics, University of Texas at Dallas Richardson, TX 75080, USA
cAdvanced Light Source, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA
dBMW Group, Petuelring 130, 80788 München, Germany
eDepartment of Materials Science and Engineering, Hanyang University, Seoul 04763, South Korea
First published on 29th January 2021
Abstract
Fluorine doping of a compositionally graded cathode, with an average concentration of Li[Ni0.80Co0.05Mn0.15]O2, yields a high discharge capacity of 216 mA h g−1 with unprecedented cycling stability by retaining 78% of its initial capacity after 8000 cycles. The cathode is cycled at 100% depth of discharge (DOD), unlike the currently deployed layered cathode whose DOD is limited to 60–80% to compensate for capacity fading and guarantee the required battery life. Additionally, the capacity and cycling stability of the cathode easily surpass those of the existing state-of-the-art batteries, while achieving the energy density goal of 800 W h kg−1cathode for electric vehicles (EV) with ultra-long cycle life. The structural and chemical stabilities of the cathode were provided by the compositional partitioning and unique microstructure of the compositionally graded cathode combined with the ordered site-intermixing of Li and transition metal (TM) ions discovered via transmission electron microscopy. F doping induced the formation of a 2ahex × 2ahex × chex superlattice from ordered Li occupation in TM slabs and vice versa, which has been proven to be essential for suppressing microcrack formation in deeply charged states, while maintaining the structural stability of the cathode during extended cycling. Furthermore, the proposed cathode allows for the recycling of used EV batteries in energy storage systems, thereby alleviating the negative environmental impact by reducing the CO2 emissions and cost associated with disposing of dead batteries.
Broader contextAlthough the global market share of electric vehicles (EVs) is estimated to grow rapidly, the driving range and lifetime of EVs are still inadequate, which is mainly caused by the limitation of their electrical power source, the Li-ion battery (LIB). The battery performance of a LIB is dictated by its cathode because the energy density and the cycling stability of conventional Ni-rich NCM or NCA cathodes are relatively inferior compared to those of the graphite anode. Our work presents a highly stable electrochemical performance of an F-doped compositionally graded Ni-rich cathode at full depth of discharge. The cathode cycled for 8000 cycles in a pouch-type full cell while incurring only 22% loss of the initial capacity after the unprecedented long-cycling. Furthermore, it was shown that F-doping provides an atomic-scale engineered “cation-ordered structure” that stabilizes the structure of the cathode in its deeply delithiated state. The result reported in this paper represents a pioneering work that demonstrates that a Ni-rich NCM cathode with its Ni content higher than 80% can have an ultra-long cycling life while providing a sufficient discharge capacity for EVs. |
Introduction
Since their introduction in 1991, Li-ion batteries (LIBs) have attracted significant attention as the power source for portable electronic devices and home appliances owing to their high energy densities. However, for replacing the internal combustion engines for powering public transportation as a fuel for electric vehicles (EVs), the energy density provided by current state-of-the-art LIBs remains inadequate to meet the driving range threshold such that the consumer's fear of running out of electricity while driving is allayed. In addition to the high energy density required by EVs, long cycle life is also expected due to the relatively long vehicle service life (5–10 years requiring ∼1000 charging–discharging cycles). However, it has been well documented that the high energy density for batteries usually translates to inferior cycling stability, which is particularly true for batteries employing layered oxide cathodes, such as Li[NixCoy(Al or Mn)1−x−y]O2, where Al = nickel cobalt aluminum oxide (NCA) and Mn = nickel cobalt manganese oxide (NCM).1–5 According to the accelerated power fading test of Ni-rich NCA cathodes performed by Argonne National Laboratory, the graphite anode from an aged cell recovered its original capacity, whereas the cathode was permanently damaged and failed to deliver its pristine potential.6 Specifically, the inherent structural instabilities of NCA and NCM cathodes in their deeply delithiated state prevent their long-term cycling. To overcome this problem, the energy density of the NCA cathodes employed in existing EVs is compromised by limiting the depth of discharge (DOD) to 60%, which adds dead weight and increases battery cost.7,8 One effective way of increasing the cycling stability of Ni-rich NCM and NCA cathodes without DOD restriction is the introduction of compositional gradation into individual cathode particles, wherein the Ni fraction continuously decreases from the particle core to the surface for dissipating and relieving the strain resulting from repeated Li insertion/removal and minimizing the surface degradation from the highly reactive Ni4+ species.9,10 Herein, we show that F doping, when combined with the compositional gradation scheme, can extend the cycle life of Ni-rich NCM cathodes to an unprecedented level (>8000 cycles). What makes its long-term cycling stability remarkable is that the cell was operated at 100% DOD, which was considered impossible for Ni-rich NCM or NCA cathodes with Ni fraction >0.8. During the post-mortem structural study of the cycled cathodes, it was discovered that cation mixing, which was long considered to hinder the Li diffusion and needed to be eliminated during cathode synthesis,11–15 in fact, helps to stabilize the delithiated structure and contributes greatly to extending the battery life.Results and discussion
1. Structural characterization
A compositionally graded Li[Ni0.80Co0.05Mn0.15]O2 (denoted as GC80 hereafter), whose performance as a cathode is field-proven, has been deployed in the latest model of e-Niro from Kia; the detailed chemical composition distribution for GC80 is shown in Fig. S1 and Table S1 (ESI†). The GC80 cathode was first coated with 1 mol% NH4F; then, it underwent heat treatment at 400 °C, thereby allowing F to diffuse into the particle interior. Energy-dispersive X-ray spectroscopy (EDS) mapping of a cross-section of the NH4F-coated GC80 cathode before and after heat treatment revealed that F, which was confined to the particle surface as part of the NH4F coating layer, was present throughout the particle interior (Fig. 1a and Fig. S2, ESI†). X-ray diffraction (XRD) patterns indicate that the F-doped GC80 cathodes (1, 2, and 10 mol% F-doped GC80 denoted as F1-, F2-, and F10-GC80, respectively) possessed a hexagonal NaFeO2 structure (space group of R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) without any impurity phase (Fig. S3 and Table S2, ESI†). The calculated a- and c-axis lattice parameters increased monotonically with increasing fractions of F (Fig. 1b).16 The increase in the unit cell dimension likely stems from the partial replacement of Ni3+ (ionic radius = 0.56 Å) with Ni2+ (ionic radius = 0.69 Å),17 as the introduction of F− into the O2− sites in the LiMO2 lattice necessarily produces Ni2+ for maintaining charge neutrality. The abundant presence of Ni2+ was confirmed by the decreasing intensity of I(003)/I(104), which suggested an increasing level of cation mixing. The increased level of cation mixing induced by F doping was directly verified by the Rietveld refinement of the XRD data (Fig. 1c and Table S2, ESI†).
m) without any impurity phase (Fig. S3 and Table S2, ESI†). The calculated a- and c-axis lattice parameters increased monotonically with increasing fractions of F (Fig. 1b).16 The increase in the unit cell dimension likely stems from the partial replacement of Ni3+ (ionic radius = 0.56 Å) with Ni2+ (ionic radius = 0.69 Å),17 as the introduction of F− into the O2− sites in the LiMO2 lattice necessarily produces Ni2+ for maintaining charge neutrality. The abundant presence of Ni2+ was confirmed by the decreasing intensity of I(003)/I(104), which suggested an increasing level of cation mixing. The increased level of cation mixing induced by F doping was directly verified by the Rietveld refinement of the XRD data (Fig. 1c and Table S2, ESI†).
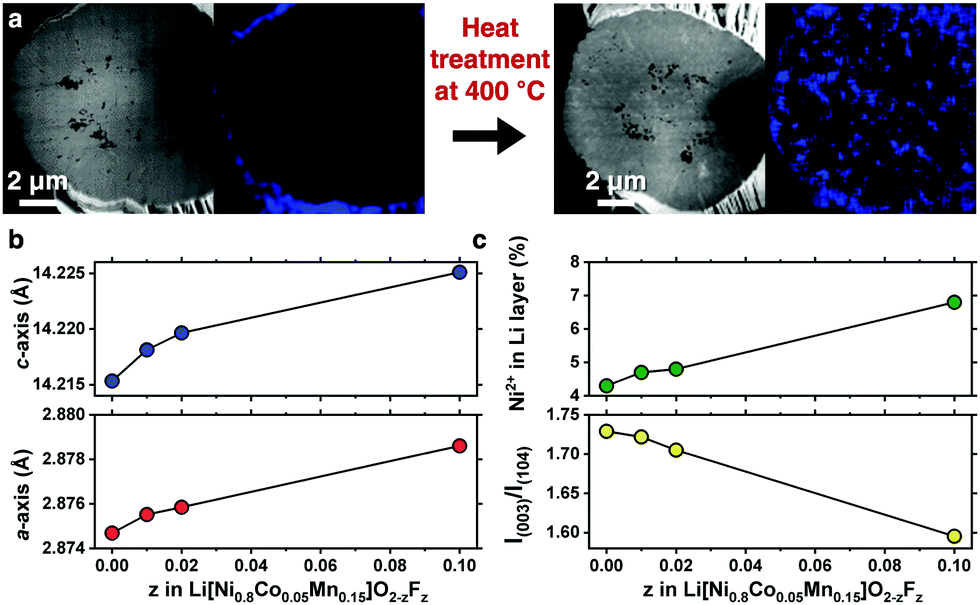 | ||
| Fig. 1 Structural details of the F-doped GC80 cathodes. (a) EDS mapping of F Kα emission for a cross-section of the F-doped GC80 before (left-hand side) and after the heat treatment (right-hand side). (b) Calculated a- and c-axis lattice parameters with increasing amount of F doping. (c) Cation mixing of Ni2+ in the Li layer and intensity ratio of the (003) peak to (104) peak as a function of the F concentration. | ||
2. Electrochemical performance
The fundamental electrochemical performance of the GC80 and F-doped cathodes was characterized as a function of the F fraction in 2032 coin-type half-cells with a Li metal anode, as well as in pouch-type full cells employing graphite as an anode. The initial charge–discharge curves at a 0.1C rate in Fig. 2a show that the discharge capacities delivered by GC80, F0.5-GC80, and F1-GC80 are nearly identical at 215–216 mA h g−1; however, for F2-GC80, the discharge capacity decreases slightly to 210 mA h g−1. However, F doping noticeably improved the cycling stability, as evident from the cycle performance data obtained at a 0.5C rate with a voltage of 2.7–4.3 V (Fig. 2b). The F0.5-, F1-, and F2-GC80 cathodes retained 95.6%, 99.7%, and 104.2% of their initial capacity after 100 cycles, respectively, whereas the pristine GC80 cathode retained 94.2% of its initial capacity. For the F2-GC80 cathode, long stabilization cycles (∼30 cycles), wherein the capacity steadily increased before stabilization, were observed. Hence, based on the cycling performance data shown in Fig. 2b, 1 mol% was chosen as the optimal level of doping for the GC80 cathode. To evaluate the F1-GC80 cathode under high-stress conditions, high temperature and long-term cathode cycling were performed (Fig. 2c–e). At 45 °C, the capacities at 0.1C and 4.3 V delivered by the pristine GC80 and F1-GC80 cathodes were identical (217.7 mA h g−1), but the latter considerably outperformed the former in cycling stability at elevated temperatures, as the capacity retention of the F1-GC80 cathode exceeded 96.5% after 100 cycles. In comparison, the pristine cathode retained 93.8% during the same cycling period. In the pouch-type full cell using graphite as an anode, the initial capacities (194 mA h g−1 between 3.0 and 4.2 V at 0.1C) of the two cathodes were nearly identical owing to their similar average chemical compositions, as seen in the half-cells, but the F1-GC80 cathode exhibited outstanding capacity retention of 91.1% after 1000 cycles when cycled at a high temperature of 45 °C, whereas the GC80 cathode retained only 64.6% of its initial capacity, as presented in Fig. 2e. The highlight of this study is the unprecedented cycling stability demonstrated by the F1-GC80 cathode as the full cell using the F1-GC80 cathode and a graphite anode was cycled for 8000 cycles while still retaining more than 82% of its initial capacity. In contrast, the capacity for the full cell using the pristine GC80 cathode deteriorated markedly beyond 1000 cycles (Fig. 2f). The remarkable cycling stability of the F1-GC80 cathode is further exceptional because the cathode was cycled at its full DOD. To the best of our knowledge, no Ni-rich layered cathodes have demonstrated such cycling stability at 100% DOD. The F1-GC80 cathode, if deployed in an EV, will increase the battery energy density by 170% because the DOD for the current NCA cathodes is maintained at 60%, which will enable the EV to easily surpass the consumer driving range threshold. EV batteries deploying the proposed cathode will provide an exceedingly long battery lifetime of ∼20 years, assuming daily full charging. The battery will certainly outlast the mechanical components and be reclaimed for other uses, reducing the environmental impact from disposing of dead batteries.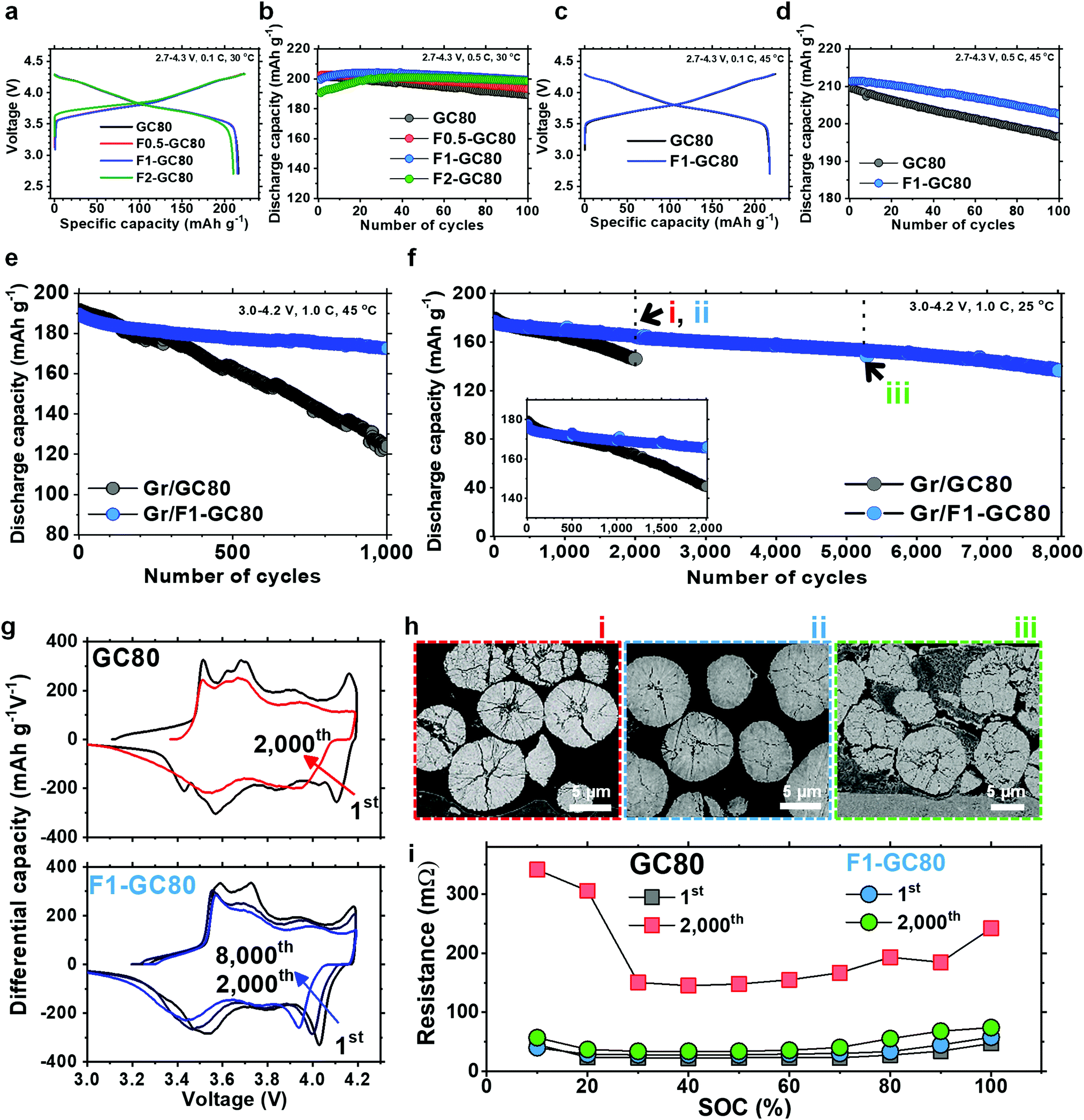 | ||
| Fig. 2 Electrochemical performance of F-doped GC80 cathodes. (a) Initial charge–discharge curves of the F doped GC80 with different F concentrations. (b) Cycling performance of the F doped GC80 cathode in half-cells. (c) Initial charge–discharge curves of the GC80 and F1-GC80 cathodes at 45 °C. (d) Cycling performance of the GC80 and F1-GC80 cathodes at 45 °C. Long term cycling performance of the GC80 and F1-GC80 cathodes using pouch-type full cells at (e) 45 °C and (f) 25 °C. (g) dQ dV−1 curves for the GC80 and F1-GC80 cathodes. (h) Cross-sectional SEM image of the GC80 cathode after 2000 cycles and F1-GC80 cathode after 2000 and 5000 cycles. (i) DC resistance as different states of charge after the first and 2000th cycles measured to characterize the high pulse power. | ||
The cathode will also be an ideal candidate for stationary large-format energy storage systems (ESS), wherein a long cycle lifetime is essential to increase the cost-effectiveness of LIBs. In fact, the calculated correlation between cycle lifetime and DOD for different battery technologies based on the manufacturer's data showed that generic NCA cathodes will last less than 2000 cycles at 100% DOD.7 The cycle lifetime exhibited by the F1-GC80 cathode suggests that the cycling stability of the Ni-rich NCM or NCA cathodes can be extended well beyond commonly accepted limits, provided that the material composition and structure are properly engineered.
The superior structural stability of the F1-GC80 cathode during cycling was confirmed by dQ dV−1 curves obtained by differentiating the respective charge–discharge curves (Fig. 2g). The dQ dV−1 curve for the GC80 cathode exhibits multiple transitions (hexagonal (H1) → monoclinic (M) → hexagonal (H2) → hexagonal (H3)). The last transition, i.e., H2 → H3, near the charge end, causes an abrupt lattice contraction/expansion, which builds up local stress concentration, and eventually leads to the microcracking of the cathode particles.4,18–21 A quick decay of the redox peaks corresponding to the H2 → H3 phase transition, which is evident in the dQ dV−1 curve for the pristine GC80 cathode, is associated with the continuous structural deterioration of the cathode. However, the H2 → H3 redox peaks for the F1-GC80 cathode decay at a much slower rate during the same cycling period, confirming the structural stability of the F-doped cathode. Visual inspection of the cycled cathode particles unequivocally confirmed the superior mechanical stability of the F-doped cathode as the cycled GC80 cathode particles developed numerous microcracks, which nearly fractured some secondary particles into fragments after 2000 cycles. In comparison, no visible cracks, besides some hairline cracks, were visible in the cycled F1-GC80 cathode particles even after 2000 cycles. Visible fragmentation of the cathode particles was observed only after 5000 cycles (Fig. 2h). To assess the dynamic power capability of the F1-GC80, a high pulse power characterization test was employed using the current pulse protocol for discharging and charging provided by the US Department of Energy (Fig. 2i).22,23 The DC resistance, measured as a function of the state of charge (SOC) for both the pristine GC80 and F1-GC80 cathodes, was nearly identical during the first cycle. However, after 2000 cycles, the resistance for the pristine GC80 cathode increased by a factor of 6.7 at 60% SOC, which translated into a severe power loss as shown in Fig. S4 (ESI†) (78.3% power loss), whereas the resistance increase for the F1-GC80 cathode was limited to 1.2 (23.3% power loss).
3. Cation ordering induced by F-doping
To investigate the exceptional cycling stability exhibited by the F1-GC80 cathode, a detailed structural study of the cycled cathodes was conducted using transmission electron microscopy (TEM). Fig. 3a–c compares the selected area electron diffractions (SAEDs) of the [100] zone from the as-prepared F1-GC80 cathode with those of the cathodes charged upto 4.3 V (first cycle) and after 2000 cycles.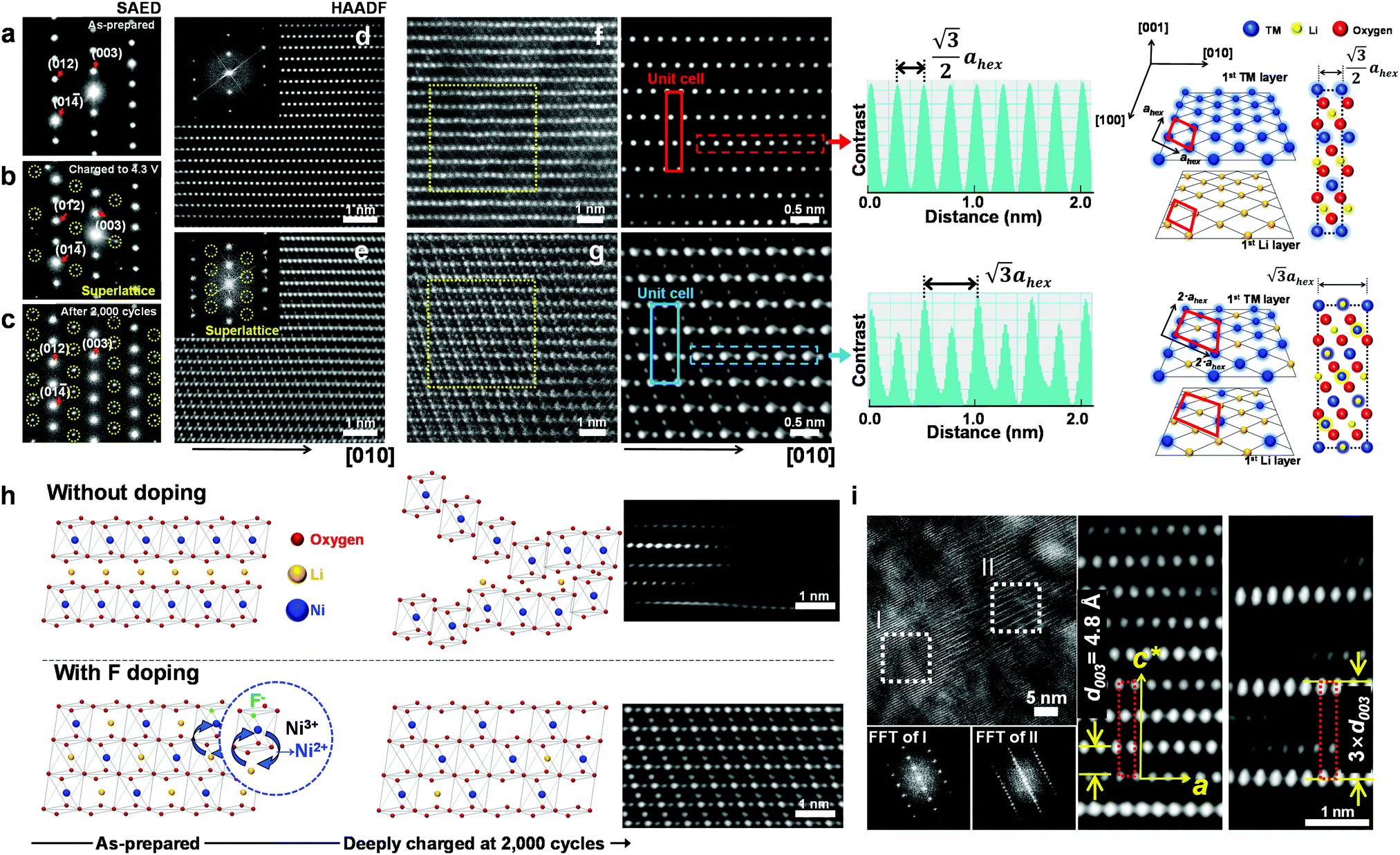 | ||
| Fig. 3 Cation ordering induced by F-doping through the formation of a superlattice. (a–c) [100] zone SAED patterns from the F1-GC80 cathode in the as-prepared state, after charging to 4.3 V, and after 2000 cycles (discharge state) marked by yellow circles in Fig. S5 (ESI†). [100] zone HAADF TEM images with corresponding Fourier-filtered images: (d) normal layered structure of the as-prepared F1-GC80 cathode and (e) cation ordered layer structure of the cycled F1-GC80 cathode (20 cycles). (f and g) Magnified Fourier-filtered [100] zone HAADF TEM images comparing the normal layered structure of the as-prepared F1-GC80 cathode with the cation ordered structure of the 2000 cycled F1-GC80 cathode with corresponding schematic drawings of atomic arrangements (normal vs. ordered structure with a 2ahex × 2ahex × chex supercell). Line scans along the respective TM layers highlight the alternating contrast variation of the cation-ordered structure with a periodicity that is twice as large as that of the normal layered structure. (h) Illustration comparing the relative structural stability of the GC80 and F1-GC80 cathodes in the deeply charged state with corresponding HR-TEM images. (i) [100] zone HR-TEM image of the F1-GC80 charged to 4.3 V and corresponding Fourier filtered transforms of a faulted region with a new stacking sequence (see text). | ||
The extra diffraction spots (circled in yellow) appearing in the charged and cycled cathodes maintain a simple fractional relationship between the lattice spacings, indicating that the extra peaks originated from a superlattice structure, rather than from a secondary phase. The most probable solution for the superlattice is the migration of Ni ions into the Li layer and subsequent ordered occupation of the Li sites, which are mostly vacant in the delithiated state. The abundance of Ni2+ ions caused by substituting O2− ions with F− facilitates the Ni migration into the Li layer due to similar ionic radii of Li+ and Ni2+ ions. During the discharge, these migrated Ni ions apparently remain in the Li sites, as evidenced by the superlattice peaks whose intensity substantially increases after 2000 cycles. The [100] zone high-resolution (HR) TEM image of the as-prepared F1-GC80 in Fig. 3d shows a normal layered structure with a fully occupied Ni and Li layer. Fourier transform (FT) of the HRTEM image of the as-prepared cathode reproduces the [100] zone SAED diffraction. The accompanying FT-filtered image shows no contrast between two transition metal (TM) layers because Li ions, with low electron scattering power, do not generate sufficient contrast for observation. In comparison, the FT of the HRTEM image from the cycled cathode exhibits extra superlattice peaks and the FT-filtered image evidently reveals the presence of TM ions between two TM layers, as shown in Fig. 3e (F1-GC80). A random occupation of Ni ions would not produce superlattice peaks, but only the relative changes in peak intensities. The superlattice peaks signify that the Ni ions migrate into the Li layer in a precisely ordered pattern.
High-angle annular dark-field (HAADF) images obtained using Cs-corrected optics verify the ordered occupation of Ni ions in the Li layer and Li ions in the TM layer. The FT-filtered HAADF image of a normal layered lattice in the [100] zone reveals clearly resolved TM ions with identical contrast of periodicity of 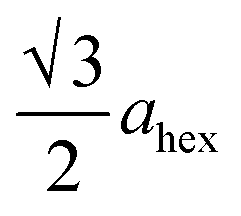 , as shown in Fig. 3f, whereas the corresponding HAADF image from the cation-ordered region from the F1-GC80 cathode cycled for 2000 cycles shows the alternating contrast in along the TM layer with a periodicity of
, as shown in Fig. 3f, whereas the corresponding HAADF image from the cation-ordered region from the F1-GC80 cathode cycled for 2000 cycles shows the alternating contrast in along the TM layer with a periodicity of 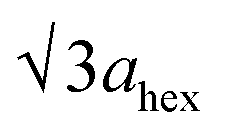 , resulting from the Li ions occupying alternate TM sites to form a supercell with 2ahex × 2ahex × chex in Fig. 3g. Cation mixing, i.e., random presence of TM ions in the Li layer, in a layered structure is believed to impede Li migration and has been experimentally shown to correlate with the capacity loss.11–15 However, the ordered occupation of TM ions in the Li layer and Li ions in the TM layer does not inhibit Li migration, but may actually facilitate it by creating a 3D network of Li pathways since Li ions can jump into the adjacent TM layer. Using Li MAS NMR and XRD, it was shown that Li ions could be extracted from both Li and TM layers at the first charge from Li[Ni0.5Mn0.5]O2, wherein the preponderance of cation ordering (including Li in the TM layers) was observed.24,25 In addition, the ordered structure can stabilize the delithiated structure.26 Removing an entire row of Li ions in the deeply charged state can cause a localized collapse of the TM slabs and will highly strain the lattice around the collapsed region. In the ordered structure, the TM ions in the Li layer act as pillars to suppress the massive collapse of the TM slabs in the highly delithiated state, as shown in Fig. 3h. In generating the cation-ordered structure, F− ions (only 1 mol%) alone are not sufficient to create the ordered structure throughout the particles in a substantial fraction. It is believed that F− ions merely trigger and promote the migration of TM ions by stabilizing the delithiated structure. F− initially creates a small concentration of Ni2+ ions which migrate to the Li layer and partially stabilize the delithiated structure by preventing the collective collapse of the Li layer. In the partially stabilized structure, the transition metal (TM) ions further migrate into the Li layer to form the observed structure.
, resulting from the Li ions occupying alternate TM sites to form a supercell with 2ahex × 2ahex × chex in Fig. 3g. Cation mixing, i.e., random presence of TM ions in the Li layer, in a layered structure is believed to impede Li migration and has been experimentally shown to correlate with the capacity loss.11–15 However, the ordered occupation of TM ions in the Li layer and Li ions in the TM layer does not inhibit Li migration, but may actually facilitate it by creating a 3D network of Li pathways since Li ions can jump into the adjacent TM layer. Using Li MAS NMR and XRD, it was shown that Li ions could be extracted from both Li and TM layers at the first charge from Li[Ni0.5Mn0.5]O2, wherein the preponderance of cation ordering (including Li in the TM layers) was observed.24,25 In addition, the ordered structure can stabilize the delithiated structure.26 Removing an entire row of Li ions in the deeply charged state can cause a localized collapse of the TM slabs and will highly strain the lattice around the collapsed region. In the ordered structure, the TM ions in the Li layer act as pillars to suppress the massive collapse of the TM slabs in the highly delithiated state, as shown in Fig. 3h. In generating the cation-ordered structure, F− ions (only 1 mol%) alone are not sufficient to create the ordered structure throughout the particles in a substantial fraction. It is believed that F− ions merely trigger and promote the migration of TM ions by stabilizing the delithiated structure. F− initially creates a small concentration of Ni2+ ions which migrate to the Li layer and partially stabilize the delithiated structure by preventing the collective collapse of the Li layer. In the partially stabilized structure, the transition metal (TM) ions further migrate into the Li layer to form the observed structure.
The ordered structure is essential for the ultra-long cycling stability of the F1-GC80 cathode as the cation-ordered structure persists after 5000 cycles (Fig. S6, ESI†). As further evidence of the cooperative rather than random migration of Ni ions into Li slabs is required to form such an ordered structure, Fig. 3i shows a [100] zone HRTEM image of a region whose structure is based on a different superlattice from the F1-GC80 that was charged to 4.3 V. The FT images of the two regions (marked as I and II) clearly reveal the extra superlattice reflections at every one-third position along the c-direction. The extra superlattice reflections are an indication of a new stacking sequence for TM slabs with a periodicity of 3 × d003, which can be clearly observed in the accompanying magnified image of the fault region, which has a three-times larger repeating periodicity than that of the regular layered lattice. The superlattice is probably created from the concerted migration of Ni ions into the tetrahedral sites of Li slabs, which are not visible as the interatomic spacing is beyond the microscope resolution. The Ni migration occurs on every two other rows to form the superlattice. Apparently, this kind of superlattice was frequently observed in the first charged and cycled cathodes, often in the vicinity of the cation-ordered region. Since the concerted migration of multiple Ni ion hopping into the adjacent Li slabs is a prerequisite to create the proposed 2ahex × 2ahex × chex superlattice, the persistent appearance of such structural faults supports that the cation ordering is maintained, thereby helping to extend the de facto battery life.
4. Theoretical calculations
To substantiate the beneficial effect of the cation-ordered structure, density functional theory is used to study its relative phase stability. To reduce the computational complexity, LiNiO2 is used as a model layered cathode, whereas to determine the relative stability and quantify the effects of F doping, the enthalpy of formation for pristine and cation-ordered LiNiO2 and half delithiated Li0.5NiO2 was calculated along with fully delithiated NiO2 and NiO (formed by ½O2 evolution) for both pristine and F-doped structures (4% F). The calculated enthalpies are plotted with different voltage references to assess the electrochemical phase stabilities as shown in Fig. 4a. For both the pristine and F-doped LiNiO2 cathodes, cation ordering (dashed line) is energetically more favorable than maintaining the layered structure at the half and fully delithiated phases, confirming that cation ordering energetically stabilizes the layered structure.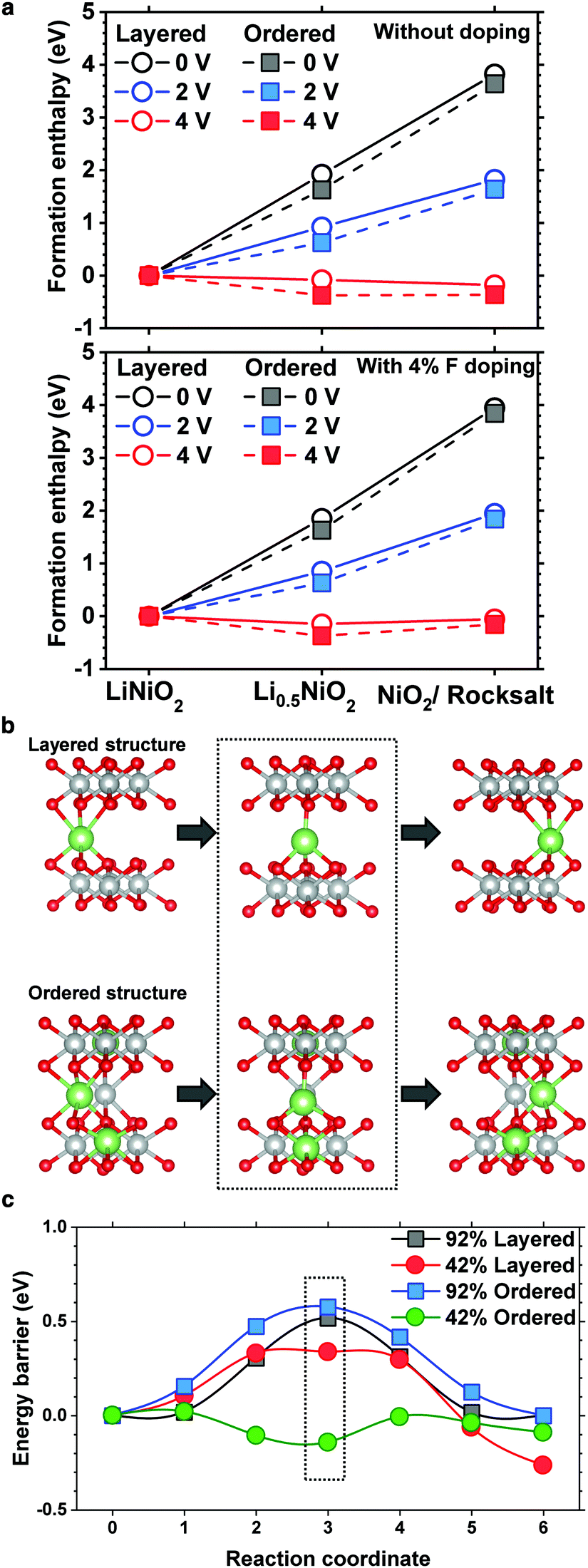 | ||
| Fig. 4 Theoretical calculation for the structural stability and Li migration barrier for the cation-ordered structure. (a) Formation energies of cathode phases during delithiation are shown for both F-doped and pristine materials. Initial layered (solid line) phases and ordered (dashed line) phases formed by cation mixing are shown at 0, 2, and 4 V references to compare the relative electrochemical stability during the charging cycles. (b) Diffusion path (octahedral–tetrahedral–octahedral) in the ordered structure, with a tetrahedral hop between adjacent octahedral sites. This is the preferred path in both the layered and ordered polymorph, but the local coordination environment of the ordered structure is unique as a result of cation mixing. (c) Li-ion diffusion between adjacent octahedral sites (reaction coordinates 0 and 6) in the layered and ordered polymorphs of LixNiO2. The intermediate position (reaction coordinate 3) is the tetrahedral site representing the energetic barrier in all, except the delithiated ordered composition. | ||
The cell with a 92% occupancy depicts the vacancy diffusion in the initial fully charged state, whereas the 42% cell is targeted at the diffusion in the intermediate charged state. In all the structures, two adjacent octahedral sites were used as endpoints for the NEB calculation. In the normal layered structure, all the Li octahedral sites are equivalent, whereas, in the ordered structure, the sites were carefully chosen to reflect the local coordination resulting from the cation mixing. The diffusion pathway in the ordered structure is shown in Fig. 4b. The results of the diffusion calculations are summarized in Fig. 4c. The 0.33 eV barrier of the 42% layered structure is consistent with that obtained previously.27 The cell with 92% occupancy exhibits higher barriers because every site surrounding the diffusing vacancy is occupied, prohibiting the same equilibrium tetrahedral configuration for the intermediate stage. Moreover, the barrier in the Li 92% ordered phase is uniformly greater than that in the layered phase by approximately 0.1 eV. This is due to the presence of the Ni ion in the Li layer adjacent to the diffusion path leading to an increased coulombic repulsion. However, in the 42% occupancy cell, Ni apparently does not have a significantly adverse effect because the diffusing ion is free to move laterally. The diffusion barrier in the half delithiated ordered phase is very low, approximately 0.15 eV. Notably, the energetic summit of the diffusion path is no longer located at the central tetrahedral site. A detailed analysis shows that this is caused by the presence of the Li+ ion located in the TM layer directly underneath the diffusing Li ion. The effect of the cation ordering on the diffusion kinetics of Li was also investigated using nudged elastic band (NEB) calculations. As mentioned previously, the pathway is the same in the layered and ordered phases, but in the former, the diffusing ion is exclusively coordinated above and below Ni. Substituting Li into those sites changes the coordination and corresponding electronic structure of the nearby oxygen atoms, facilitating the Li diffusion through the interlayer where the substitutions exist. The detailed density of state plots are provided in Fig. S7 and S8 (ESI†). The Li diffusion calculations show that cation ordering lowers the energy barrier and expedites Li migration or at least does not impede Li migration. Li migration is hampered by the presence of TM ions in Li slabs only when TM ions randomly occupy Li sites in a rocksalt layer, which is often observed on the surface of cycled NCM and NCA cathodes.8,18,28–30
Direct evidence on the structural stability engendered by F doping is obtained from in situ XRD measurements during cathode charging (Fig. 5). The unit cell for the cathode is contracted in both the a- and c-directions at the end of charging. While the dimensional contraction was monotonic in the a-direction, the lattice initially expanded owing to the coulombic repulsion between the anion layers and contracted rather abruptly at the onset of the H2 → H3 phase transition in the highly charged state of above 4.2 V Li+/Li. The magnitude of the contraction is much larger in the c-direction than that in the a-direction, making the overall volume contraction highly anisotropic and detrimental to the mechanical stability of the cathodes. Comparing the extent of the anisotropic volume contraction for the GC80 and F1-GC80 cathodes, both cathodes incurred a similar maximum contraction of −6.9% during the first charging (Fig. 5a). However, the F1-GC80 cathode reclaimed after 2000 cycles exhibited substantially less contraction in the charged state than the pristine GC80 cathode after 2000 cycles (Fig. 5b). The maximum unit cell volume contraction for the F1-GC80 cathode at full charging was −5.9%, whereas that for the GC80 cathode was −6.3%, confirming the structural stabilization resulting from F doping and subsequent cation ordering. In addition, it was recently shown that the presence of TM ions in Li slabs (Li-deficient Li layer) on the surface of the Li-rich cathode eliminated oxygen release and helped to maintain the crystal structure during cycling.31 We believe that the cation ordering observed in the F1-GC80 cathode should also improve the chemical stability of the cathode against electrolyte attack.
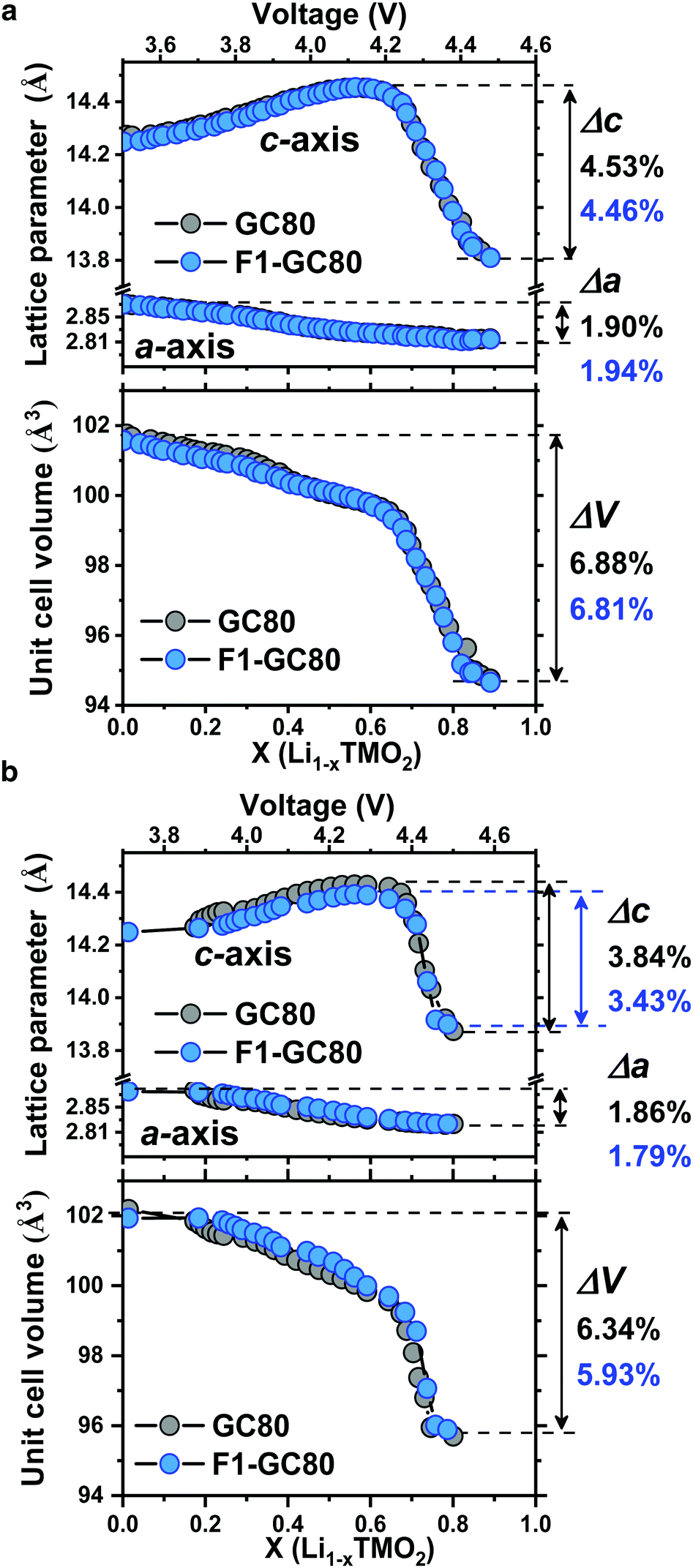 | ||
| Fig. 5 Unit cell contraction of GC80 and F1-GC80 cathodes during delithiation. The lattice parameter and unit cell volume change for GC80 and F-GC80 cathodes by in situ XRD analysis; (a) first cycle and (b) after 2000 cycles. | ||
5. Post-mortem analysis
To assess the surface damage accumulated on both the external and internal surfaces of the cathodes during extended cycling (GC-80 after 2000 cycles and F1-GC80 after 5000 cycles), the spatial distribution of Ni oxidation states was mapped using scanning transmission X-ray microscopy (STXM) in conjunction with spatially resolved X-ray absorption spectroscopy (XAS). STXM mapping in Fig. 6a, b and Fig. S9 (ESI†) shows that interparticle microcracks and surfaces of both cycled cathodes have enhanced Ni2+ features, which indicate the formation of a NiO-like impurity phase near the cathode/electrolyte interface. The GC80 cathode after 2000 cycles has clearly visible wide cracks, whose vicinity contains a high density of Ni2+ resulting from direct contact with the infiltrated electrolyte through the open microcrack. In comparison, in the F1-GC80 cathode after 5000 cycles, although its microcracks reached the surface, electrolyte attack and the structural deterioration of the particle interior were minimized.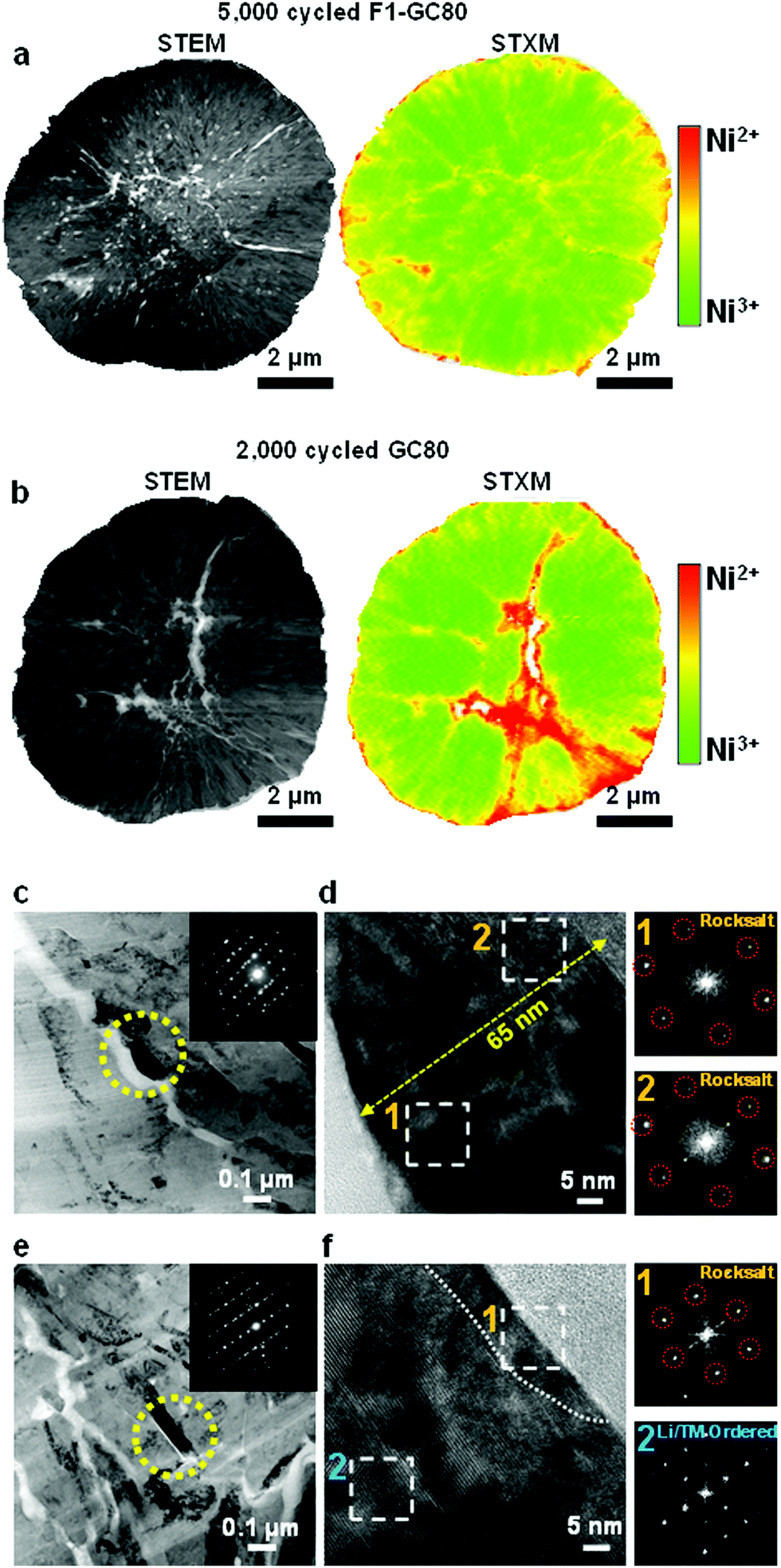 | ||
| Fig. 6 Post-mortem analysis of the cycled cathode at the discharge state using TEM and STXM. (a and b) Mapping of Ni oxidation states for the cycled F1-GC80 (5000 cycles) and GC80 (2000 cycles) cathodes. The presence of pure Ni3+ and Ni2+ are assigned colors green and red, respectively. (c) Low-magnification TEM images of the cycled CG80 particle core with the corresponding electron diffraction pattern (inset), (d) high-magnification TEM image of the region marked by the yellow circle in (c), and Fourier transforms of the regions marked by Arabic numerals in (d). (e) Low-magnification TEM images of the cycled F1-CG80 particle core with the corresponding electron diffraction pattern (inset), (f) high-magnification TEM images of the region marked by the yellow circle in (e), and Fourier transforms of the regions marked by Arabic numerals in (f). | ||
In agreement with the STXM data, the surface damage caused by the infiltrated electrolyte on the crack faces is apparent in the TEM image of the CG80 cathode after 2000 cycles, as shown in Fig. 6c. The surface of the primary particle along a microcrack were covered by a 65 nm thick NiO-like impurity layer, as revealed by the FT image of the regions near the microcrack in Fig. 6d. The FT images of the damaged regions are missing the characteristic diffraction spots corresponding to the layered structure (see the SAED pattern that covers the entire marked primary particles in the inset of the bright-field image); rather, they conform to a rocksalt structure. In contrast, the TEM image in Fig. 6e and f from the F1-GC80 cathode after 5000 cycles shows that despite its long cycling, the particle surface was covered by an impurity layer that was less than 10 nm thick. Moreover, the cation-ordered structure was also preserved, as verified by the FT image and HR-TEM of the selected region in Fig. 6f. It is speculated that the cation-ordered region probably retarded the inward progress of the electrolyte attack because the rocksalt structure pattern requires random occupation of Li and TM ions, which is impeded by the already-stabilized structure through cation ordering. The STXM and TEM analyses of the cycled cathodes confirm their structural and chemical stability against electrolyte attack that has been rendered by cation ordering.
The chemical stability of the F1-GC80 cathode in the presence of the electrolyte was assessed using differential scanning calorimetry of the charged cathodes (4.3 V) immersed in the electrolyte solution (Fig. S10, ESI†). In the as-prepared state prior to cycling, the charged GC80 and F1-GC80 cathodes underwent an exothermic reaction at approximately 240 °C, whereas the onset temperature and heat released from the F1-GC80 cathode were slightly lower than those of the pristine GC80 cathode. This difference considerably widened when the experiment was repeated using the reclaimed cathodes after 20 cycles. The exothermic peak for the GC80 cathode was observed at a much lower temperature with substantially higher heat released than that for the F1-GC80 cathode whose exothermic peak remained at approximately 240 °C even after 20 cycles. The thermal (chemical) stability of the GC80 cathode, which is substantially improved by F doping after some cycles, indicates that the cation ordering induced by F doping requires activation cycles to allow for the migration of TM ions before its stabilizing effect becomes apparent.
To further demonstrate the enhanced chemical stability of the ordered structure produced by F doping, time-of-flight secondary ion mass spectrometry (TOF-SIMS) was employed for surface analysis using depth profiling and 3D analysis, as shown in Fig. S11 (ESI†). We analyzed the GC80 cathode after 2000 cycles and the F1-GC80 cathode after 2000 and 5000 cycles, including their respective graphite anodes. The depth profiles and 3D mapping images, as shown in Fig. S11 (ESI†), show that a broad range of fragments of interest is detected on the cycled anodes and cathodes. Ni-Containing secondary ion fragments (Ni− and NiO−) represent cathode dissolution, whereas C- and P-containing species (C3− and PO−) originate from the graphite anode and LiPF6–EC based electrolyte, respectively. After 2000 cycles, a 30 nm thick Ni fragment layer (Ni−, NiO−) formed on the graphite anode of the GC80 cell. These Ni fragments likely came from the dissolution of the cathode to form an SEI layer on the anode surface. In comparison, a relatively thin layer was observed on the anode from the F1-GC80 cell, confirming its enhanced chemical stability. On the GC80 cathode surface, PO-fragments were found, indicating that the electrolyte penetrated the cathode bulk probably through microcracks. In contrast, even after 2000 cycles, the PO-fragments detected on the F1-GC80 cathode surface were negligible, confirming the remarkable mechanical stability of the cathode.
Experimental
Synthesis of cathode materials
Synthesis of GC80 (Li[Ni0.80Co0.05Mn0.15]O2): first, NiSO4·6H2O and MnSO4·H2O aqueous solutions (Samchun Chemical, Korea) comprising Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mn = 98:2 molar ratio were reacted for synthesizing a spherical NM [Ni0.98Mn0.02](OH)2 precursor as a starting material for co-precipitation synthesis. Then, the Ni-less metal sulfate hydrate solution (molar ratio of Ni:Co:Mn = 76:6:18) in tank 2 was slowly pumped into tank 1. The homogeneously mixed solution in tank 1 was gradually fed into a batch reactor (17 L), including a solution of deionized (DI) water, NH4OH (aq), and NaOH under N2 atmosphere. Concurrently, 4 M NaOH (aq, Junsei, Japan; molar ratio of NaOH to TM = 2.0) and NH4OH (aq, Samchun, Korea; molar ratio of NH4OH to TM = 1.2) were pumped separately into the reactor for maintaining the synthetic conditions. At the end of the co-precipitation reaction, the final synthesis product was washed using distilled water to eliminate residual chemicals. Finally, a GC-type precursor was obtained through filtering and drying in a vacuum oven at 110 °C for 12 h. For cathode lithiation, the precursor, [Ni0.80Co0.05Mn0.15](OH)2, was mixed with LiOH·H2O (Li:(Ni + Co + Mn) = 1.01:1 molar ratio) and calcined at 770 °C for 10 h under an oxygen atmosphere.
:1) for 2 h using a zirconia ball. The mechanically mixed powder was heated at 400 °C for 5 h with oxygen gas to form an F-coating layer on the surface of the primary particles.
:Mn = 98:2 molar ratio were reacted for synthesizing a spherical NM [Ni0.98Mn0.02](OH)2 precursor as a starting material for co-precipitation synthesis. Then, the Ni-less metal sulfate hydrate solution (molar ratio of Ni:Co:Mn = 76:6:18) in tank 2 was slowly pumped into tank 1. The homogeneously mixed solution in tank 1 was gradually fed into a batch reactor (17 L), including a solution of deionized (DI) water, NH4OH (aq), and NaOH under N2 atmosphere. Concurrently, 4 M NaOH (aq, Junsei, Japan; molar ratio of NaOH to TM = 2.0) and NH4OH (aq, Samchun, Korea; molar ratio of NH4OH to TM = 1.2) were pumped separately into the reactor for maintaining the synthetic conditions. At the end of the co-precipitation reaction, the final synthesis product was washed using distilled water to eliminate residual chemicals. Finally, a GC-type precursor was obtained through filtering and drying in a vacuum oven at 110 °C for 12 h. For cathode lithiation, the precursor, [Ni0.80Co0.05Mn0.15](OH)2, was mixed with LiOH·H2O (Li:(Ni + Co + Mn) = 1.01:1 molar ratio) and calcined at 770 °C for 10 h under an oxygen atmosphere.
:1) for 2 h using a zirconia ball. The mechanically mixed powder was heated at 400 °C for 5 h with oxygen gas to form an F-coating layer on the surface of the primary particles.
Analytical techniques
The chemical compositions of the prepared powders were determined by inductively coupled plasma (ICP, OPIMA 8300, PerkinElmer). The morphologies and structures of the prepared particles were observed by FE-SEM (Verios G4 UC, FEI). To observe the cross section of the charged cathode electrode (particles) by SEM, half-cells were charged in CC/CV mode until each upper cutoff voltage of the cells was reached (4.3 and 4.5 V) and subsequently disassembled in an Ar-filled glove box. The obtained cathode was rinsed immediately using dimethyl carbonate (DMC) (Deajung Chemical & Metal Co., Korea) and completely dried under vacuum. The electrode was cut using a cross-sectional polisher (JEOL IB-19520 CCP). Powder and in situ XRD (Empyrean, Panalytical) using Cu-Kα radiation was employed to identify the crystalline phases of the cathodes. XRD data were obtained between 2θ = 10° and 110° with a step size of 0.02°, and the collected XRD data were analyzed by the Rietveld refinement program, FullProf. The in situ XRD experiment of the cathodes was implemented using a laminated pouch-type cell with a graphite anode. The cell was charged with 0.05C constant current and the XRD pattern at each SOC was recorded in transmission mode every 40 min. The TEM samples were prepared by focused ion beam (FEI, FIB, SCIOS). Structural information and elemental distribution of F are investigated by transmission electron microscopy (TEM, JEM 2100F, JEOL). The compositional distribution of the transition metal atoms (Ni, Co, Mn) in the cathode material was determined by an electron probe micro analyzer (EPMA, JXA-8100, JEOL). Time of flight secondary ion mass spectroscopy (TOF.SIMS-5, ION-TOF) analyses were performed with a TOF-SIMS instrument equipped with a 30 keV Bi3+ primary ion gun and a 0.5 keV Cs+ sputter gun. For depth profiling, the sputtering area was 200 × 200 μm2.Chemical phase mapping by soft X-ray transmission X-ray microscopy (STXM)
Chemical composition maps (e.g. Ni oxidation states) were visualized by spectral-sensitive imaging endstation (Nanosurvery2) combined with the sensitivity of X-ray absorption spectroscopy (XAS).32 STXM image-sequences across the Ni L-edges were recorded with the finest energy steps of 0.25 eV. After registration, the STXM image-sequences were converted to the optical densities by following Beer–Lambert law. The X-ray absorption spectra on a pixel-by-pixel basis were quantified as a function of chemical composition by linear combination fits of standard Ni2+ and Ni3+ spectra which were collected from LiMn1.5Ni0.5O4 spinel and LiNi0.8Co0.15Al0.05O2 layered oxide, respectively (Fig. S9, ESI†). The justification of the reference spectra is confirmed by non-negative matrix analysis.33 The quality of fit for each pixel was confirmed by the R-factor defined as R = ∑(data-fit)2/∑(data)2. Pixels showing poor signal-to-noise ratios were filtered out by a lower bound of the R-factor (0.15).DFT calculations
Computational studies were performed using density functional theory as implemented in the Vienna ab initio Simulation Package.34 All the calculations used the generalized gradient approximation along with a Dudarev-type Hubbard U-Correction of 6.2 eV for Ni, accounting for the strong 3d electron–electron correlations.35 Projector augmented-wave pseudopotentials were used with an energy cutoff of 520 eV and a force convergence criterion of 0.001 eV nm−1.34 The van der Waals correction method reported by Grimme et al.36 was incorporated to ensure accurate interlayer spacing and unit cell parameters. The k-space sampling of the Brillouin zone was done using a gamma centered grid with a mesh density of at least 0.003 nm−1 for all cells. The spin-polarized magnetic configurations were optimized to produce the most accurate energies, as well as monitor changes in local magnetic moments.Electrochemical testing
To fabricate the cathodes, the synthesized powders were mixed with carbon black and poly-(vinylidene fluoride) (90:5.5:4.5) in N-methyl-2-pyrrolidone. The slurry was coated onto an Al foil, vacuum dried, and roll pressed. Half-cell tests for understanding the basic electrochemical properties were performed using 2032 coin-type cells with lithium metal as the anode. The cells were charged (discharged) with a constant current density of 90 mA g−1 (0.5C rate) in a constant temperature chamber retained at 30 °C. Capacity retention during long-term cycling was tested using a laminated pouch-type full cell (50 mA h) with artificial graphite as an anode. N/P ratio calculated from the areal capacity of each electrode was in the range of 1.15–1.20. The cells were charged and discharged between 3.0 and 4.2 V with a constant 1C current (200 mAg−1) at 25 °C. Every electrochemical test in this research was performed using 1.2 M LiPF6 in ethylene carbonate–ethyl methyl carbonate (EC:EMC = 3:7 by vol%) with 2 wt% vinylene carbonate additive as an electrolyte.
At every 10% SOC, a hybrid pulse power characterization was conducted to measure the direct current resistance of the pouch-type full cell. Each cell was charged to 4.2 V (SOC 100%) by CC/CV mode with a 0.1C current (20 mA g−1) and underwent a pulse test with 1C current (200 mA g−1). One sequence of pulse test was composed of 30 s discharge, 40 s rest, 10 s charges, and 60 s rest. After testing a sequence of pulses at each SOC, the cells were discharged to another SOC using 0.1C current (20 mA g−1) and rested for 1 h for electrochemical equilibrium. Every direct current resistance of each SOC was analyzed from voltage responses and calculated in accordance with ref. 22 and 23.
Thermal stability testing
For thermal stability analyses, 2032 coin-type half-cells were charged to 4.3 V and disassembled to recover the cathodes in an Ar-filled glovebox. After the remaining electrolyte had been carefully washed with dimethyl carbonate (DMC), the cathode materials were recovered from the current collector. A stainless-steel sealed pan with Au-plated Cu seals was used to accommodate 7–8 mg samples with 100 μl of fresh electrolyte (1.2 M LiPF6 in EC:EMC = 3:7 by vol% with 2 wt% vinylene carbonate). The measurements were carried out by differential scanning calorimetry (DSC, DSC 214 Polyma, Netzsch) at a temperature scan rate of 5 °C min−1.
Conclusions
Herein, the ability of an NCM cathode to maintain its capacity upon extended cycling was improved to an unprecedented level (>8000 cycles) by introducing F ions into a compositionally graded cathode. The result stands out from previous long-term cycling literature data for NCM cathodes because the cathode was cycled at 100% DOD, thereby fully using its available capacity, in addition to its exceedingly long cycling life. We believe that the main reason for the observed ultra-long battery life demonstrated by the F1-GC80 cathode stems from the cation ordering created by the formation of a 2ahex × 2ahex × chex superlattice through alternate occupations of Li ions in the TM slab and TM ions in the Li slab. The ultra-long battery life of the F1-GC80 cathode has the potential to significantly increase the energy density for EV application because of the 100% DOD at which the cathode operates, thereby presenting opportunities for used EV batteries to be recycled in ESS applications, thus doubly relieving the environmental pressure by reducing CO2 emissions and the cost of dead battery disposal.Author contributions
U.-H. K. and Y.-K. S. conceived and designed the research. U.-H. K. and G.-T. P. performed the experiments and characterization of materials. U.-H. K., C. S. Y. and Y.-K. S. analyzed the data. Y.-S. Y. and D. S. performed the chemical phase mapping by soft X-ray transmission X-ray microscopy. P. C., N. A., and K. C. performed the theoretical calculations. U.-H. K., C. S. Y., F. M., S.-J. K., P. L., and Y.-K. S. contributed to the discussion of the results. C. S. Y. and Y.-K. S. wrote the manuscript. All the authors commented on and revised the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korea Government Ministry of Education and Science Technology (MEST) (no. NRF-2018R1A2B3008794). Additionally, this work was also supported by the Human Resources Development Program (no. 20184010201720) of the Korea Institute of Energy Technology Evaluation and Planning funded by the Ministry of Trade, Industry and Energy of the Korean Government.Notes and references
- R. Schmuch, R. Wagner, G. Horpel, T. Placke and M. Winter, Nat. Energy, 2018, 3, 267–278 CrossRef CAS.
- D. Andre, S.-J. Kim, P. Lamp, S. F. Lux, F. Maglia, O. Paschos and B. Stiaszny, J. Mater. Chem. A, 2015, 3, 6709–6732 RSC.
- H.-J. Noh, S. Youn, C. S. Yoon and Y.-K. Sun, J. Power Sources, 2013, 233, 121–130 CrossRef CAS.
- G. W. Nam, N.-Y. Park, K.-J. Park, J. Yang, J. Liu, C. S. Yoon and Y.-K. Sun, ACS Energy Lett., 2019, 4, 2995–3001 CrossRef CAS.
- X. Wang, T.-L. Ding and Z. Chen, Adv. Energy Mater., 2020, 10, 1903864 CrossRef CAS.
- R. B. Wright, J. P. Christophersen, C. G. Motloch, J. R. Belt, C. D. Ho, V. S. Battaglia, J. A. Barnes, T. Q. Duong and R. A. Sutula, J. Power Sources, 2003, 119–121, 865–869 CrossRef CAS.
- S. Watanabe, M. Kinoshita, T. Hosokawa, K. Morigaki and K. Nakura, J. Power Sources, 2014, 260, 50–56 CrossRef CAS.
- K.-J. Park, J.-Y. Hwang, H.-H. Ryu, F. Maglia, S.-J. Kim, P. Lamp, C. S. Yoon and Y.-K. Sun, ACS Energy Lett., 2019, 4, 1394–1400 CrossRef CAS.
- Y.-K. Sun, S.-T. Myung, B.-C. Park, J. Prakash, I. Belharouak and K. Amine, Nat. Mater., 2009, 8, 320–324 CrossRef CAS.
- U.-H. Kim, H.-H. Ryu, J.-H. Kim, R. Mücke, P. Kaghazchi, C. S. Yoon and Y.-K. Sun, Adv. Energy Mater., 2019, 9, 1803902 CrossRef.
- J. Morales, C. Pérez-Vicente and J. L. Tirado, Mater. Res. Bull., 1990, 25, 623–630 CrossRef CAS.
- S. B. Schougaard, J. Bréger, M. Jiang, C. P. Grey and J. B. Goodenough, Adv. Mater., 2006, 18, 905–909 CrossRef CAS.
- A. Rougier, P. Gravereau and C. Delmas, J. Electrochem. Soc., 1996, 143, 1168–1175 CrossRef CAS.
- Y. Makimura, T. Sasaki, T. Nonaka, Y. F. Nishimura, T. Uyama, C. Okuda, Y. Itou and Y. Takeuchi, J. Mater. Chem. A, 2016, 4, 8350–8358 RSC.
- K. Kang, Y. S. Meng, J. Bréger, C. P. Grey and G. Ceder, Science, 2006, 311, 977–980 CrossRef CAS.
- S.-U. Woo, B.-C. Park, C. S. Yoon, S.-T. Myung, J. Prakash and Y.-K. Sun, J. Electrochem. Soc., 2007, 154, A649–A655 CrossRef CAS.
- R. D. Shannon, Acta Cryst., 1976, A32, 751–767 CrossRef CAS.
- H.-H. Ryu, K.-J. Park, C. S. Yoon and Y.-K. Sun, Chem. Mater., 2018, 30, 1155–1163 CrossRef CAS.
- A. O. Kondrakov, A. Schmidt, J. Xu, H. Geßwein, R. Mönig, P. Hartmann, H. Sommer, T. Brezesinski and J. Jankek, J. Phys. Chem. C, 2017, 121, 3286–3294 CrossRef CAS.
- J.-M. Lim, T. S. Hwang, D. H. Kim, M.-S. Park, K. J. Cho and M. H. Cho, Sci. Rep., 2017, 7, 39669 CrossRef CAS.
- H. H. Sun, H.-H. Ryu, U.-H. Kim, J. A. Weeks, A. Heller, Y.-K. Sun and C. B. Mullins, ACS Energy Lett., 2020, 5, 1136–1146 CrossRef CAS.
- G. Hunt, PNGV battery test manual, presented in part at Tech. Rep. DOE/ID-10597, National Institute of Electricity and Clean Energy, 2001.
- J. P. Christopherson, Battery test manual for electric Vehicles, presented in part at INL/EXT-15-34184, The Idaho National Laboratory, 2015.
- W.-S. Yoon, Y. Paik, X.-Q. Yang, M. Balasubramanian, J. McBreen and C. P. Grey, Electrochem. Solid-State Lett., 2002, 5, A263–A266 CrossRef CAS.
- Y. S. Meng, G. Ceder, C. P. Grey, W.-S. Yoon, M. Jiang, J. Bréger and Y. Shao-Horn, Chem. Mater., 2005, 17, 2386–2394 CrossRef CAS.
- U.-H. Kim, G.-T. Park, B.-K. Son, G. W. Nam, J. Liu, L.-Y. Kuo, P. Kaghazchi, C. S. Yoon and Y.-K. Sun, Nat. Energy, 2020, 5, 860–869 CrossRef CAS.
- F. Kong, C. Liang, R. C. Longo, D.-H. Yeon, Y. Zheng, J.-H. Park, S.-G. Doo and K. Cho, Chem. Mater., 2016, 28, 6942–6952 CrossRef CAS.
- C. S. Yoon, H.-H. Ryu, G.-T. Park, J.-H. Kim, K.-H. Kim and Y.-K. Sun, J. Mater. Chem. A, 2018, 6, 4126–4132 RSC.
- H.-H. Sun and A. Manthiram, Chem. Mater., 2017, 29, 8486–8493 CrossRef CAS.
- F. Lin, I. M. Markus, D. Nordlud, T.-C. Weng, M. D. Asta, H. L. Xin and M. M. Doeff, Nat. Commun., 2014, 5, 3529 CrossRef.
- Z. Zhu, D. Yu, Y. Yang, C. Su, Y. Huang, Y. Dong, I. Waluyo, B. Wang, A. Hunt, X. Yao, J. Lee, W. Xue and J. Li, Nat. Energy, 2019, 4, 1049–1058 CrossRef CAS.
- D. A. Shapiro, Y.-S. Yu, T. Tyliszczak, J. Cabana, R. Celestre, W. Chao, K. Kaznatcheev, A. L. D. Kilcoyne, F. Maia, S. Marchesini, Y. S. Meng, T. Warwick, L. L. Yang and H. A. Padmore, Nat. Photonics, 2014, 8, 765–769 CrossRef CAS.
- D. C. Langreth and M. J. Mehl, Phys. Rev. B, 1984, 29, 2310 CrossRef.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B, 1998, 57, 1505–1509 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and S. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ee03774e |
| This journal is © The Royal Society of Chemistry 2021 |