
Machine learning for high performance organic solar cells: current scenario and future prospects
Asif
Mahmood
 and
Jin-Liang
Wang
*
and
Jin-Liang
Wang
*
Key Laboratory of Cluster Science of Ministry of Education, Beijing Key Laboratory of Photoelectronic/Electrophotonic Conversion Materials, School of Chemistry and Chemical Engineering, Beijing Institute of Technology, Beijing, 100081, China. E-mail: jinlwang@bit.edu.cn
First published on 26th November 2020
Abstract
Machine learning (ML) is a field of computer science that uses algorithms and techniques for automating solutions to complex problems that are hard to program using conventional programming methods. Owing to the chemical versatility of organic building blocks, a large number of organic semi-conductors have been used for organic solar cells. Selecting a suitable organic semi-conductor is like searching for a needle in a haystack. Data-driven science, the fourth paradigm of science, has the potential to guide experimentalists to discover and develop new high-performance materials. The last decade has seen impressive progress in materials informatics and data science; however, data-driven molecular design of organic solar cell materials is still challenging. The data-analysis capability of machine learning methods is well known. This review is written about the use of machine learning methods for organic solar cell research. In this review, we have outlined the basics of machine learning and common procedures for applying machine learning. A brief introduction on different classes of machine learning algorithms as well as related software and tools is provided. Then, the current research status of machine learning in organic solar cells is reviewed. We have discussed the challenges in anticipating the data driven material design, such as the complexity metric of organic solar cells, diversity of chemical structures and necessary programming ability. We have also proposed some suggestions that can enhance the usefulness of machine learning for organic solar cell research enterprises.
 Asif Mahmood | Asif Mahmood, received his PhD degree from the National Center for Nanoscience and Technology (NCNST), China. Currently, he is a Post-Doctoral at School of Chemistry and Chemical Engineering, Beijing Institute of Technology under the supervision of Prof. Jin-Liang Wang. His research interests include the design and synthesis of organic semi-conductor materials for organic solar cells and computational analysis of organic solar cells. |
 Jin-Liang Wang | Jin-Liang Wang, received his PhD degree in College of Chemistry and Molecular Engineering, Peking University under the supervision of Prof. Jian Pei, from 2003-2008. From 2008 to 2012, he was a Postdoctoral research fellow in The University of Akron and The University of North Carolina at Chapel Hill. In 2013, he was awarded the junior thousand talent award and joined Beijing Institute of Technology as a full professor. His major is organic optoelectronic materials chemistry and his interests focus on synthesis of functional organic molecular materials for optoelectronic molecular devices and investigation of the relationship of chemical structure, computational analysis, optimization of film morphology and device performances. |
Broader contextTo provide an environment friendly solution to the energy crisis, extensive research in various fields of solar cells is going on. Organic solar cells are the most prestigious candidate. After tremendous success of machine learning in language translation and image recognition, it has gained significant attention from material scientists. Finding efficient materials for organic solar cells is a prime task for further improvement. An innovative approach is required to replace the tedious and expensive trial and error method because a large number of organic semi-conductors are possible due to the chemical versatility of organic building blocks. In this review, we have discussed the current state of machine learning assisted material design for organic solar cells. There are serious challenges for the scientific community to accelerate the data-driven research paradigm. However, this field has the potential to overcome obstacles. We have proposed suggestions to overcome these obstacles. Machine learning based design can be a cost-effective way to foster innovation in organic solar cell research. |
1. Introduction
Organic solar cells (OSCs) have gained huge attention due to their unique qualities such as lightweight, transparency, flexibility and low-cost.1–5 In recent years, the power conversion efficiency (PCE) has significantly increased.6–8 The great success is the result of device architecture design, design of active layer materials and processing.9–13 New design ideas are based on knowledge obtained from expensive and time-consuming synthesis, characterization and device optimization. The results are further used for redesigning.14 This approach leads to an extended iterative cycle that not always leads to high performance in the end. A large number of donors and acceptors have been tried for organic solar cell devices. Still, an infinite number of candidate materials are possible due to intrinsic degrees of freedom in organic molecules. Selection of materials is carried out through a trial and error method that is time consuming and laborious. From chemical structure, it is hard to guess whether a material will work well or not because the working of organic solar cells is complex. It is based on light absorption, formation, migration, and dissociation of excitons and transportation of generated charges to respective electrodes. Complicated multiple mechanisms and donor–acceptor morphology make it more difficult to design a rule.15–17 Human experts are required to select suitable candidates, synthesize and fabricate devices, and then determine the performance. In this scenario, computer assisted prediction of performance of organic solar cells is an ultimate need.Scharber's model was developed to predict photovoltaic efficiency of organic solar cells.18 It uses few electronic parameters of active layer materials (donor and acceptor) for prediction. Its scope is limited due to assumptions based on it. There are so many obstacles in its extension i.e. it is hard to include other descriptors such structural, topological and thermodynamic. The performance of this model is quite poor.19–21
Data driven research has started a new paradigm and shown promising results within materials science. It can provide an understanding of fundamental factors that govern the performance of materials for specific applications. Material discovery through a data-driven paradigm is efficient and effective to utilize relevant information.22,23 The systematic way of doing this is known as machine learning, which learns from past data and helps to screen the candidates for laboratory work. The discovery of excellent candidate materials for organic solar cells can be made fast and cheaper through multidimensional design by combining machine learning (ML), DFT calculations and available experimental data survey (Fig. 1).
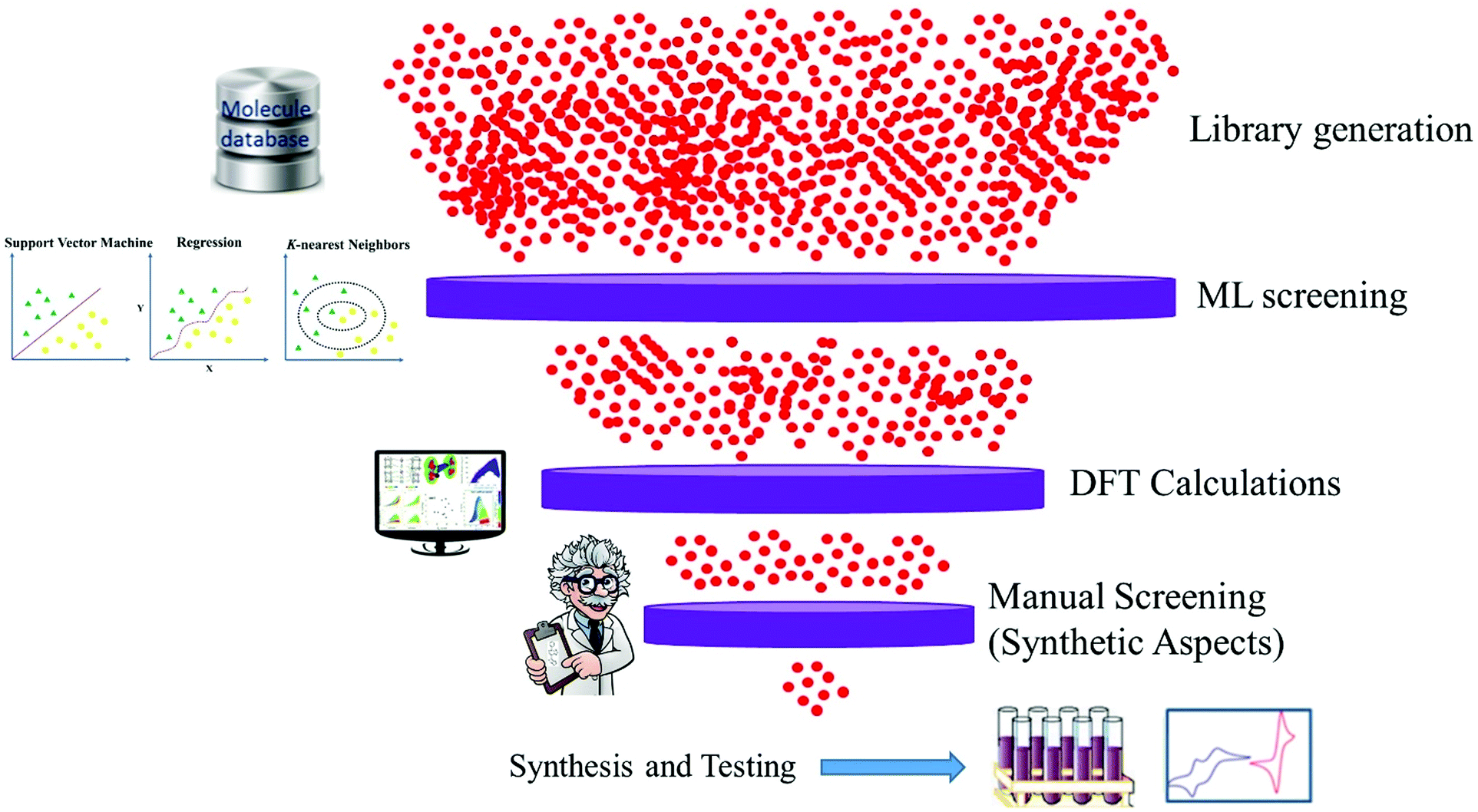 | ||
| Fig. 1 Computer assisted design and screening of materials for organic solar cells. | ||
The use of machine learning (ML) should be encouraged because it can provide a chance to get hidden information and trends. It uses a digitized form of chemical structures, and theoretically calculated and experimentally measured properties.24–26 Further development in the ML model's explainability and interpretability will increase their usefulness. As compared to other fields of materials science, the use of ML for organic photovoltaic is limited. However, in the last year, the number of published papers has increased significantly as compared to the past. To the best of our knowledge, this is the first review article related to ML use in organic solar cell research.
This review article is written to provide a comprehensive view of the machine learning methods used for organic photovoltaic materials. In Section 2, a brief description of machine learning and its classes is provided. In Section 3, basic steps to train ML models are given. In Section 4, the research status with respect to the applications of machine learning in organic solar cells is discussed. Section 5 discusses the limitations and drawbacks of machine learning and possible solutions.
2. Machine-learning
Machine learning is a branch of artificial intelligence. Machine learning is based on the collection of sufficient amounts of reliable materials data (from experiment or computation) that best explain behavior or properties of materials or their applications to develop a model that is used to discover new materials without performing the same experiment or computation. Machine learning allows us to find hidden insights from data. Machine learning methods learn the rules from a given dataset and build a model to make predictions. A brief description of types of machine learning is given in Fig. 2. Generally speaking, machine learning techniques can be divided into three groups, namely, supervised learning, unsupervised learning, and reinforcement learning.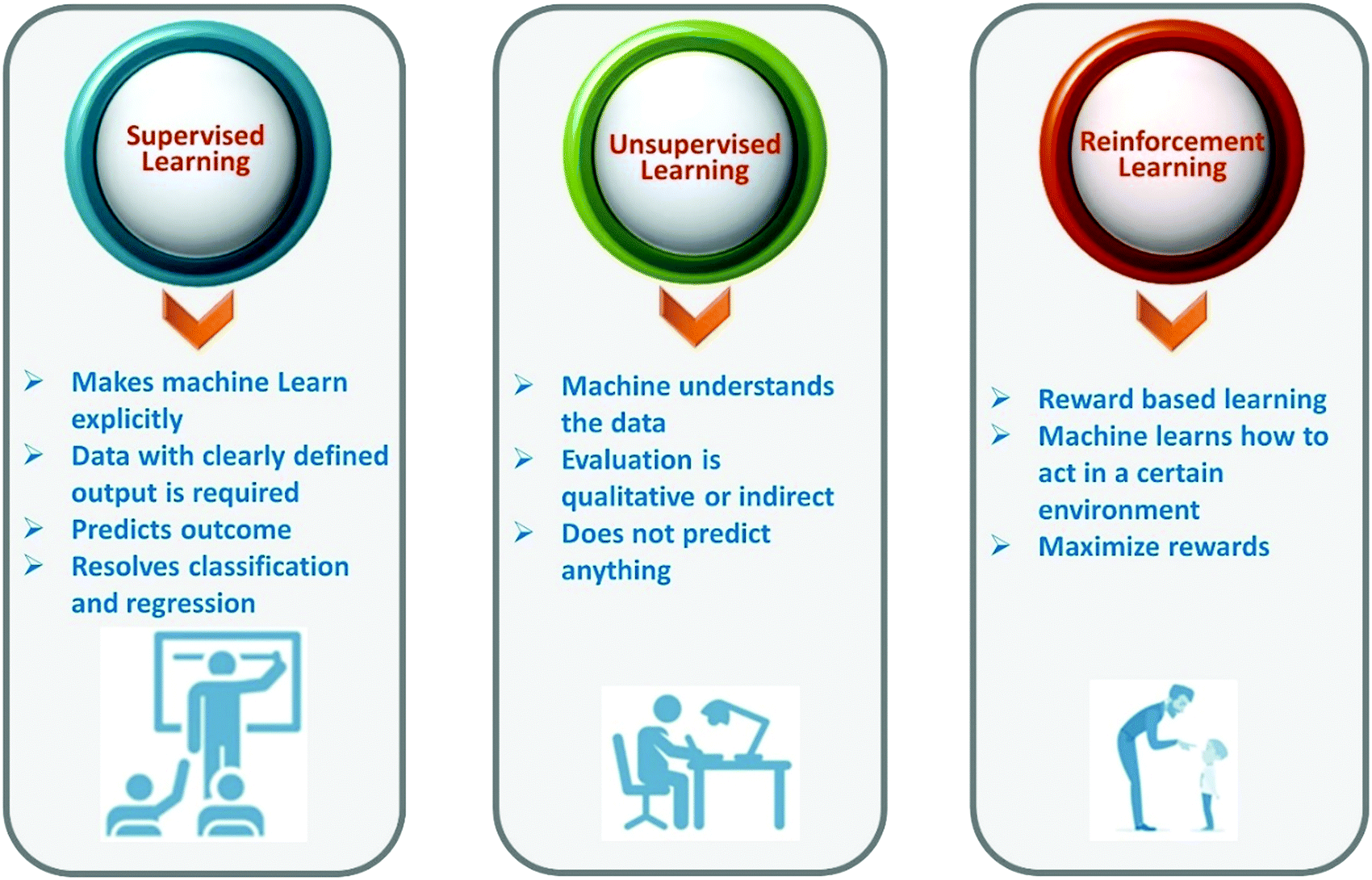 | ||
| Fig. 2 Different types of machine learning. | ||
The selection of an appropriate ML algorithm plays a key role since it significantly affects the prediction performance. There is no single best method for all cases. There are so many machine learning algorithms (Fig. 3). To achieve a highly effective model, it is crucial to select an appropriate algorithm that is mostly done on the basis of a trial and error process. Detailed literature search can help in this aspect. The learner should have enough knowledge of algorithms. It can help to select the best algorithms. A range of learning algorithms can be applied (Fig. 3), depending on the type of data and the question posed. The scope of this review article does not allow discussion of all the algorithms. Readers interested in a more comprehensive understanding of different algorithms can look at the following review articles.27–30 Information about popular machine learning frameworks and libraries is summarized in Table 1.
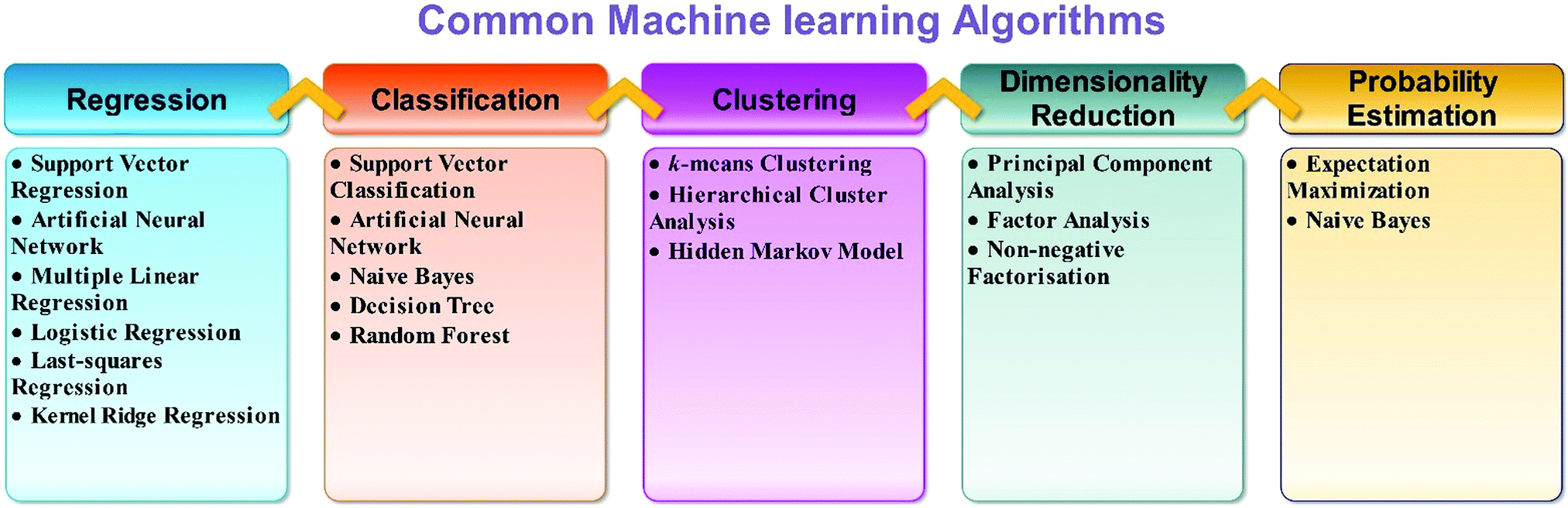 | ||
| Fig. 3 Different types of machine learning algorithms. | ||
| Name | Description | Website |
|---|---|---|
| Scikit-learn | Free python-based library of machine learning algorithms | https://scikit-learn.org/stable/index.html |
| TensorFlow | Open-source software library for dataflow and programming | https://www.tensorflow.org |
| CRAN-machine learning | The Comprehensive R Archive Network (CRAN) is based on R programming language and contains verity of machine learning algorithms | https://cran.rstudio.com/web/views/MachineLearning.html |
| Keras | Open-source python library, supports data mining using deep learning algorithms | https://keras.io |
| Weka | Waikato Environment for Knowledge Analysis (Weka) offers verity of ML algorithms with graphical user interface | https://www.cs.waikato.ac.nz/ml/weka |
| Amazon Web Services machine learning | Offers fully managed solution for machine learning through various libraries such keras, TensorFlow, and Apache Spark ML etc. | https://aws.amazon.com/machine-learning |
| IBM Watson machine learning | Efficient and fast machine learning facilities based on Apache Spark libraries. | https://www.ibm.com/cloud/machine-learning |
| Google artificial intelligence | Cloud-based artificial intelligence machine learning framework. | https://ai.google/ |
3. Basic steps of machine learning
Machine learning analysis consists of four steps.3.1. Sample collection
The first step is to collect data. Data can be from computational simulations and experimental measurements. Sometimes data cleaning or data transformation should be performed to avoid noise and inconsistency. Data can be split in various ratios; split sizes can also differ. Depending on the situation, it could be 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40, 70:30 and 80:20, or can even be 90:10. The data can be split into training and testing sets in a number of ways. The simplest way is to select non-overlapping subsets of data while preserving the order of the data records, i.e. first 70% as the training set and the remaining 30% as the test set. However, this method will be problematic when the response is not evenly distributed. Random sampling is another way, selecting data randomly for the training set and test set. The range of the response values of both subsets should be covered from the lowest to the highest value. The data split strongly influences the performance of a model.31 High performance of a model can be due to chance variations in the training and testing sets rather than a robust association between the features and the target variable.32
:40, 70:30 and 80:20, or can even be 90:10. The data can be split into training and testing sets in a number of ways. The simplest way is to select non-overlapping subsets of data while preserving the order of the data records, i.e. first 70% as the training set and the remaining 30% as the test set. However, this method will be problematic when the response is not evenly distributed. Random sampling is another way, selecting data randomly for the training set and test set. The range of the response values of both subsets should be covered from the lowest to the highest value. The data split strongly influences the performance of a model.31 High performance of a model can be due to chance variations in the training and testing sets rather than a robust association between the features and the target variable.32
The required quantity of data varies model to model but a general rule of thumb is that at least 50 data points are necessary for a reasonable ML model. However, some models such as neural networks require much larger quantities. The data quantity and quality are perhaps the main challenges in the application of ML in materials science. A large fraction of data is available in journal publications.
3.2. Data preparation and processing
As the machine learning model learns the hidden patterns of given data and predicts the pattern in new data. To improve the quality of modelling, data are cleaned to find missing data and outliers. If the data is better, the ML model will perform well.Values of different descriptors fall in different scales. It is better to normalize the values, as this will make it easy to find the difference between observations and use within a single algorithm. If the number of features (descriptors) is higher than the number of observations or features highly correlate with each other, then dimensionality reduction tools are used. Principal component analysis (PCA), discriminant analysis (LDA) and independent component analysis (ICA) are the most famous ones. They help to reduce the dimension of the feature space and identify the most relevant features. These tools also visualize the data.
3.3. Model building
In organic solar cells, a complex relationship exists between parameters and performance. The accuracy and generalization ability of a machine learning model strongly depends on the machine learning algorithm. Every algorithm has its own advantages and disadvantages. Classification, clustering, regression and probability estimation are the most widely used types of ML algorithms for materials science. Classification, clustering and regression are used for material property prediction. Probability estimation is used for new materials discovery.3.4. Model evaluation
A model will be considered good, if it performs well on both the training and test data. Statistical analysis is used to check the performance of machine-learning models. For this purpose, root mean squared error (RMSE) and coefficient of determination (R2) are used.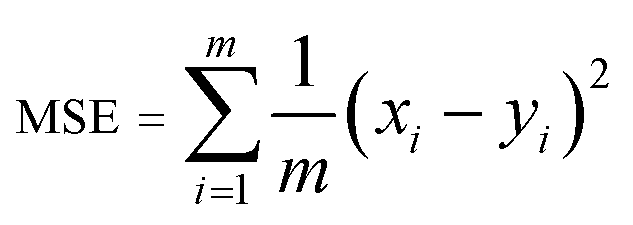 | (1) |
 | (2) |
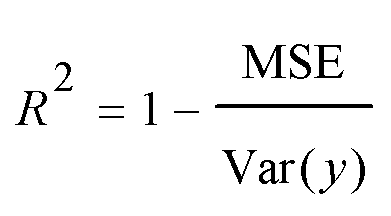 | (3) |
An alternative approach for estimating the model's performance is splitting the available data into training/test sets several times, each time with a different group of observations in each set. This is a way to check performance several times, then sum up for each train/test split. This process also provides us with a measure of variability and stability of the model performance. This method can be executed using the cross validation (CV) and bootstrapping procedures.
4. Machine learning analysis of organic solar cells
For the successful screening of potential candidates for organic solar cells, it is a prerequisite to understand the relationships between molecular properties and PCE. However, it is also important to study the relationship between molecular properties and specific device performance parameter to fulfil the requirements of a device for a particular application such as solar-to-fuel energy conversion (high open-circuit voltage (VOC)) or solar window applications (high short circuit current (JSC)). In this regard, ML has wide scope in organic solar cells research. The prediction ability of a ML model strongly depends on the type of used descriptors. The descriptors are the languages that researchers use to speak with the database. They play a significant role in making trustful predictions. If the target property is not clear then selection of candidate descriptors will become difficult.33Generally, a material property strongly depends on specific factors. Selection of suitable descriptors for specific properties is a crucial step before applying the ML process, especially microscopic descriptors that are experimentally and computationally expensive to determine. A good material descriptor should at least meet the following three criteria: It should be (i) a unique characterization of the material, (ii) sensitive to the target property, and (iii) easy to calculate.
We have grouped the reported studies on the basis of input data. However, this grouping is not highly accurate due to the use of multiple types of inputs in some studies.
4.1. Molecular descriptors
Molecular descriptors are driven from a compound's molecular structure, and represent physical and chemical properties of a molecule. They range from simple features such as count of specific atoms to more complex features such as charge distribution. There are thousands of molecular descriptors of various categories, zero-dimensional (0D) to three-dimensional (3D).35 0D descriptors describe molecular information without topological or atom connectivity inferences such as atom number, atom types, and molecular weight. 1D descriptors describe chemical fragment types and counts. In contrast, 2D descriptors define topological and topo-chemical molecular features. Finally, 3D descriptors capture geometrical data and include conformation data such as molecular volume and partial surface charges. An ideal expression must provide most of the features of the molecule and be free from redundant information. Various expressions for the same molecule comprise vastly different chemical information, or this information is presented in different abstract levels. There are many expressions, some are shown in Fig. 4. Molecular descriptors are easy and fast to calculate, which allows the rapid screening of a huge number of materials. There are many free and commercial tools to calculate molecular descriptors. A list of software and website resources is presented in Table 2.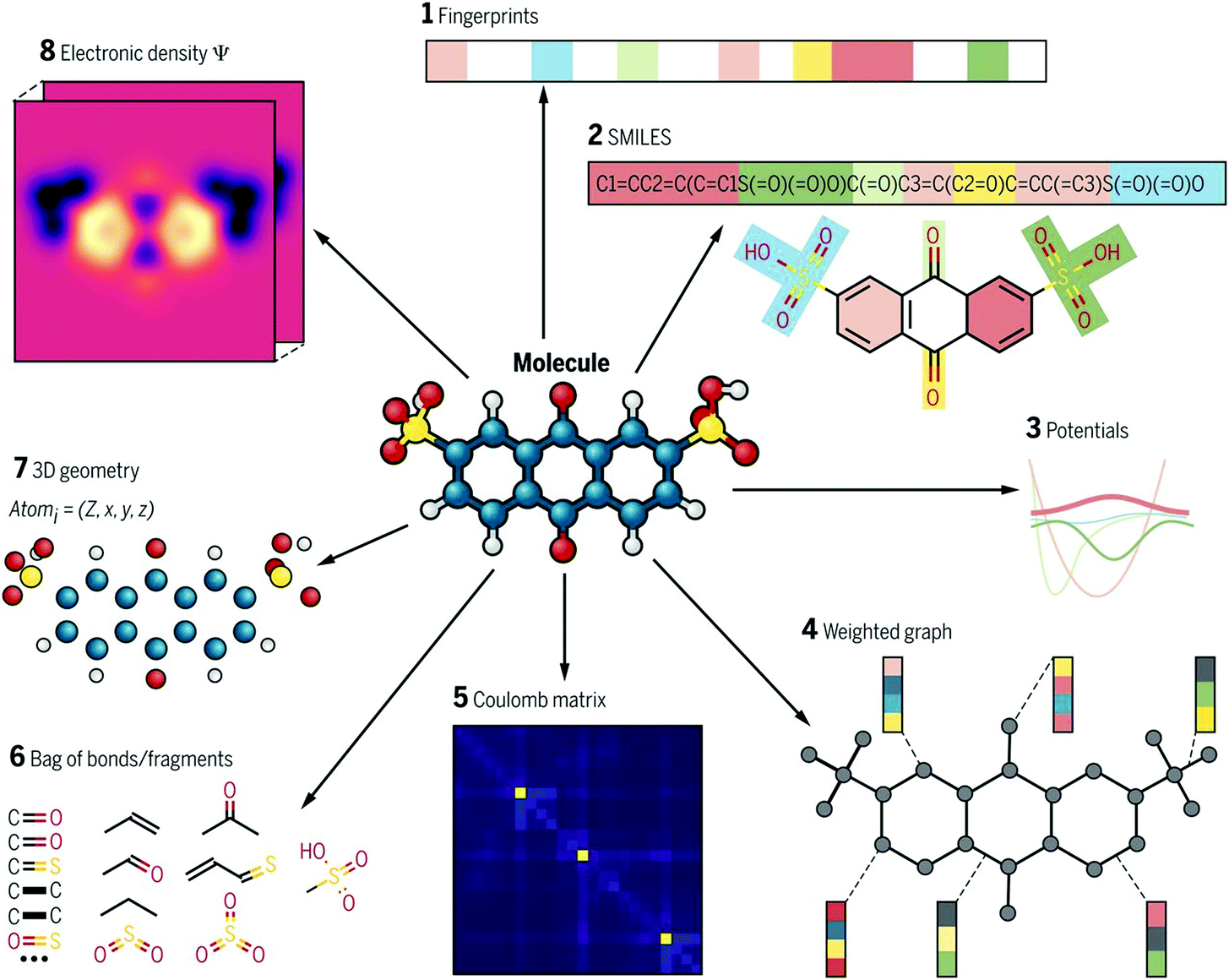 | ||
| Fig. 4 Different types of molecular representations applied to one molecule. Adopted with permission from ref. 34 Copyright 2018, AAAS. | ||
| Name | Description | Website | Ref. |
|---|---|---|---|
| DRAGON | Software, it can calculate 5270 descriptors | http://www.talete.mi.it | 36 |
| E-DRAGON | Web version of DRAGON can calculate over 3000 descriptors for molecules with atoms 150 atoms | http://www.vcclab.org/lab/edragon/ | 37 |
| Mold2 | Software to calculate 779 descriptors | https://www.fda.gov/science-research/ | 38 |
| Mordred | Software, it can calculate over 1800 2D and 3D descriptors | https://github.com/mordred-descriptor/ | 39 |
| PaDEL-Descriptor | Tool, can calculate 1444 1D and 2D descriptors, 431 3D descriptors, and 12 different types of fingerprint | http://www.yapcwsoft.com/dd/ | 40 |
| MOE | tool to calculate over 300 topological, physical and structural descriptors | http://www.chemcomp.com/ | |
| MOLGEN QSPR | software to calculate 708 arithmetical, topological, and geometrical descriptors | http://molgen.de/ | 41 |
| ChemoPY | Python package to calculate 1135 2D and 3D descriptors | http://code.google.com/p/pychem/ | 42 |
| BlueDesc | software to calculate 174 descriptors | http://www.ra.cs.uni-tuebingen.de/software/bluedesc/ | 43 |
| PowerMV | PC software for calculation of 1000 descriptors | https://www.niss.org/research/software/ | 44 |
In 2011, Aspuru Guzik's group used ML for the discovery of promising OPV donor materials.45 Current–voltage properties of 2.6 million molecular motifs were modelled using linear regression. On the basis of feature predictions, they identified benzothiadiazole, pyridinethiadazole and thienopyrrole as the top candidates.
Zhang et al. established a data set of 111000 molecules and trained an ML model using random forest (RF).46 Through this model, they predicted the LUMO and HOMO with below 0.16 eV error without any DFT calculations. This can speed up high-throughput screening of organic semi-conductors for solar cells.
Su et al. designed a series of novel acceptors based on multi-conformational bistricyclic aromatic ene (BAE) derivative.47 They have predicted their PCE using an ML model build using experimental data through cascaded support vector machine (CasSVM). The CasSVM model is a novel two-level network (Fig. 5), which consists of three subset SVM models taking JSC, VOC, and FF, respectively, as outputs in the first level. Then, the second level was used to establish the relationship between the first level outputs and the ultimate endpoint PCE. The best established CasSVM model has predicted the PCE value of OPVs with mean absolute error (MAE) of 0.35 (%), which is approximately 10% (3.89%) of the mean PCE. The value of R2 was 0.96. This approach can be very useful for experimental chemists to screen the potential candidates before synthesis.
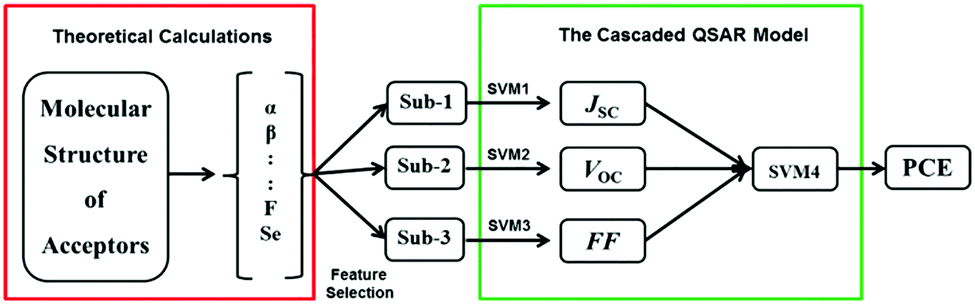 | ||
| Fig. 5 The structure of the cascaded SVM QSAR model. Sub-1–3 are the input descriptors, respectively, for JSC, VOC, and FF. SVM1∼4 are the subset SVM models used for the prediction of JSC, VOC, FF and PCE, respectively. Reprinted with permission from ref. 47 Copyright 2018, Wiley-VCH Verlag GmbH & Co. KGaA. | ||
4.2. Molecular fingerprints
Molecular fingerprints are a digitized form of chemical structures. They do not include structural data such as coordinates. They are used to search the database and find the similarity between two molecules. There are several methods to transfer a molecular structure into digitized form. Keys-based fingerprints, circular fingerprints and topological or path-based fingerprints are famous types of fingerprints. These types have further subtypes. We are suggesting a few articles for further reading.48–50In recent years, non-fullerene acceptors have been extensively used in organic photovoltaics (OPVs).3–5,51–55 In 2017, Aspuru-Guzik et al. collected a data set of over 51000 non-fullerene acceptors based on benzothiadiazole (BT), diketopyrrolopyrroles (DPPs), perylene diimides (PDIs), tetraazabenzodifluoranthenes (BFIs) and fluoranthene-fused imides from Harvard Clean Energy Project (HCEP).20 A data set of 94 experimentally reported molecules was used to calibrate the DFT methods for the calculation of HOMO and LUMO values of new non-fullerene acceptors. Gaussian process regression was used instead of the widely used linear regression because of the absence of a linear trend. They used the Scharber model to calculate the PCE for organic solar cells based on designed non-fullerene acceptors and poly[N-90-heptadecanyl-2,7-carbazole-alt-5,5-(40,70-di-2-thienyl-20,10,30-benzothiadiazole)] (PCDTBT), a standard electron-donor material. DFT calculated HOMO and LUMO values of acceptors and experimentally reported HOMO and LUMO values of PCDTBT were used as input for the Scharber model. To check the PCE prediction ability of the Scharber model, they utilized a set of 49 reported experimental values and predicted their Scharber model PCE values. A weak correlation was found (r = 0.43 and R2 = 0.11).
Prediction of PCE and specific device properties are equally important. To improve a specific property, it is important to find the relationship between the specific property and molecular descriptors.56 For example, most high performing OSC devices showed lower open-circuit voltage (VOC). In BHJ OSCs, charge separation is typically associated with large voltage losses because of the extra energy required to split excitons into free carriers. This voltage loss in OSCs with high performance is usually around 0.6 V, which is 0.2–0.3 V higher than that for c-Si and GaAs-based solar cells.57 Non-fullerene acceptors with extended thin-film absorption and suitable energy levels can help to achieve a balanced trade-off between VOC and JSC.58 Their structural versatility allows highly tunable absorption and molecular energy levels. Machine learning can speed up the screening of suitable materials. Predication of specific parameters will be helpful to further enhance the PCE. Aspuru Guzik et al. calibrated the VOC and JSC values, which were calculated from the Scharber model with available experimental data using structural similarity.21 Information on the molecular graph is extracted with extended connectivity fingerprints, and exploited using a Gaussian process. This calibration method reduced the functional dependence of the calculated properties, and it will help to ease the high-throughput virtual screening.
In 2019, Sun et al. collected the dataset of 1719 donor materials.59 They tested different inputs including seven types of molecular fingerprints, two types of descriptors, ASCII strings and images. Classification of donor materials was carried out in two categories “low” and “high” PCE. Models developed using fingerprints showed the best performance in predicting the PCE class (86.76% accuracy). They have verified the ML results by synthesizing 10 donor materials. The model classified eight molecules into the correct category. Experimental results were in good agreement with predicted results. Classification in just two categories (0–2.9%, 3–14.6%) is very easy as compared to predicating the PCE of individual semi-conductors. This study does not have much practical value i.e. the second category is too wide.
In the same year, Saeki et al. extracted 2.3 million molecules from the Harvard Clean Energy Project database.60 1000 molecules were selected on the basis of calculated PCE. They used molecular access system (MACCS) fingerprints and the extended connectivity fingerprint (ECFP6) key to train the ML model. 149 molecules were selected using random forest (RF) screening (Fig. 6). The RF method for PCE prediction showed an accuracy of 48%. They selected one polymer on the basis of synthetic feasibility. A solar cell device based on the new polymer showed a PCE of 0.53% that is much lower than the RF prediction (5.0–5.8%). There are two reasons behind this failure. Firstly, the PCE calculated from the Scharber model is used to train the RF model. The performance of the Scharber model is very poor. Secondly, the structures of polymer donors reported in the literature are more complex than that of semi-conductors in the HCEP database. Even if we ignore these factors, the PCE prediction accuracy of the RF model is very low. Therefore, the ML model should be more accurate and multiple materials should be synthesized for experimental validation.
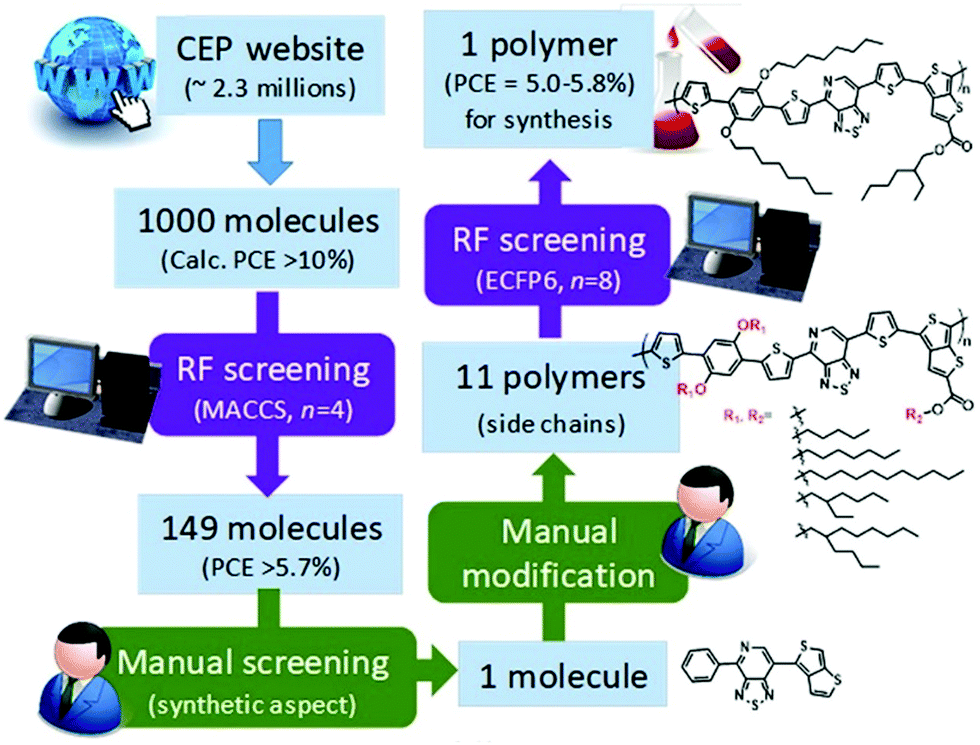 | ||
| Fig. 6 Scheme of polymer design by combining RF screening and manual screening/modification. Adopted with permission from ref. 60 Copyright 2018, American Chemical Society. | ||
Schmidt and co-workers collected a dataset of 3,989 monomers and trained a model using a grammar variational autoencoder (GVA).61 The trained model can predict the lowest optical transition energy and lowest unoccupied molecular orbital (LUMO) without the knowledge of the atomic positions. Moreover, this model can generate new molecular structures with the desired LUMO and optical gap energies. The prediction accuracy of the deep neural network (DNN) was higher than that of GVA. However, it is necessary to perform DFT calculations to find the atomic position that is necessary to predict the LUMO. So, in the case of the DNN model, it impossible to skip the DFT calculations.
Paul et al. used extremely randomized tree to predict the HOMO values for donor compounds.62 Their proposed models showed better results than neural networks trained on molecular fingerprints, SMILES, Chemception and Molecular Graph.
Peng and Zhao used convolutional neural network (CNN) to construct generative and prediction models for the design and analysis of non-fullerene acceptors.63 Different molecular descriptors have been used such as extended-connectivity fingerprints, coulomb matrix, molecular graph, bagof-bonds, and SMILES string. The depth of convolutional layers influences the diversity of the generated NFAs. Quantum chemistry calculations were performed to verify the predicted molecules. In the prediction model, the dilated convolution layers are adopted for feature extraction, and the attention mechanism is used as an interpretable module. The authors concluded that graph representation is better than a string representation.
In most of the experimental studies, donor and acceptor materials are optimized separately. Just optimization of one of the two components of the cell results in a limited exploration of the space of combinations. Troisi used ML to find the answer to the question: can components be optimized separately or should the optimization occur simultaneously.64 They used molecular fingerprints as input. Combinations of 262 donors (D) and 76 acceptors (A) were collected from the literature. They have predicted the PCE of BHJ solar cells based on combination of donor and acceptor materials. A high accuracy (r = 0.78) was obtained even though the data set was small. The best combination is proposed for experimental investigation. Min et al. reported an excellent work. They trained five ML models using linear regression (LR), multiple linear regression (MLR), boosted regression trees (BRT), RF, and ANN algorithms. 565 D/A combinations collected from the literature were used as training data sets.65 For polymer-NFA OSC devices, correlation between D/A pairs and the PCE predication is validated. BRT and RF models showed higher prediction ability, with an r value of 0.71 and 0.70, respectively. These two models were used to predict PCE of >32 million D/A combinations. Six D/A pairs were selected and incorporated into OSC devices. Experimental PCEs were close to the predicted ones. All the synthesized non-fullerene acceptors belong to high performing Y6 series. The workflow of the whole study is summarized in Fig. 7.
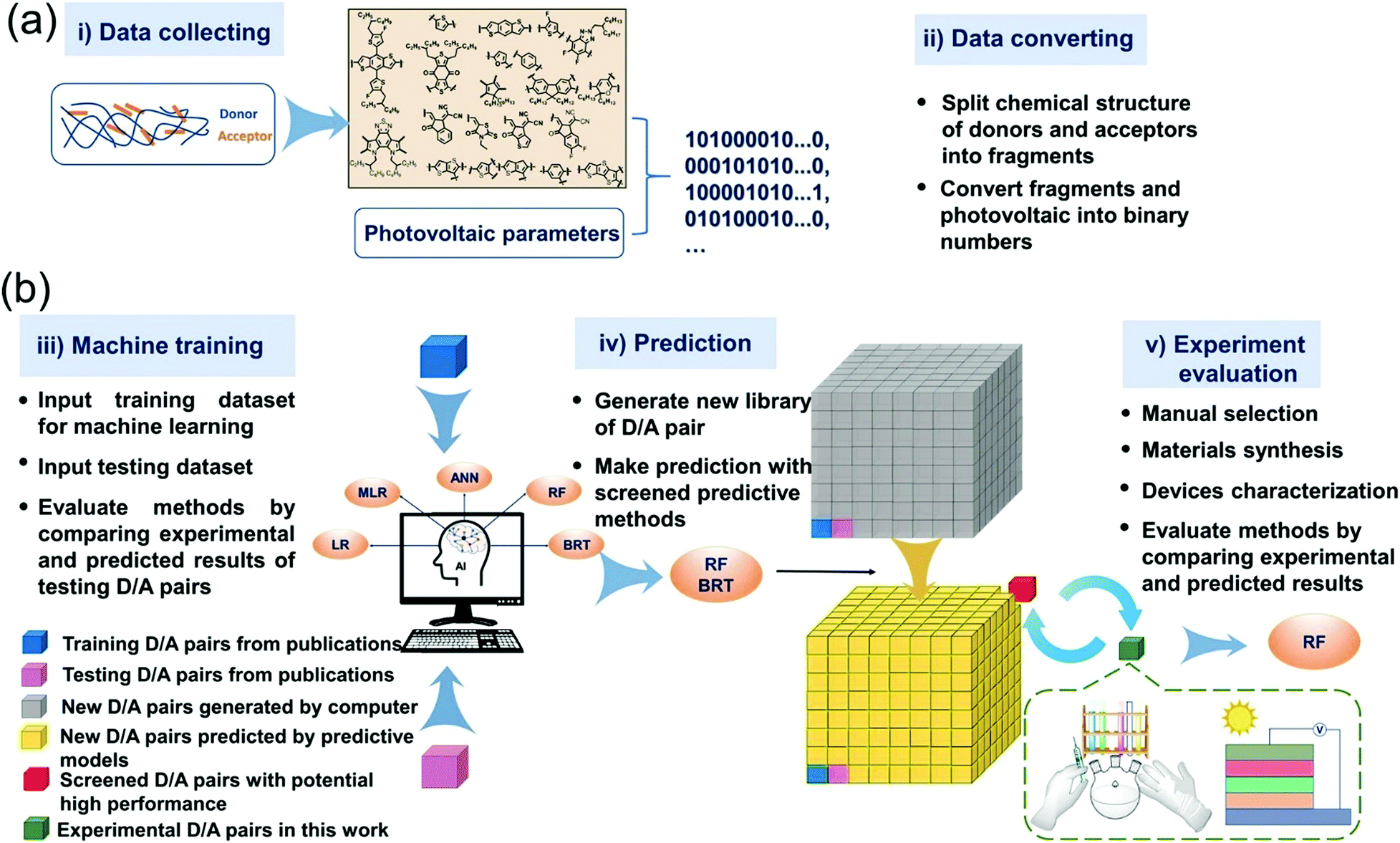 | ||
| Fig. 7 Workflow of building, application, and evaluations of machine learning methods. (a) Scheme of collecting experimental data and converting chemical structure to digitized data. (b) Scheme of machine training, predicting, and method evaluation. Adopted from ref. 65 Creative Commons Attribution 4.0 International License. | ||
4.3. Images
Machine learning has achieved tremendous success in image recognition. It identifies features out of a complicated background and associates them with the same output. Sun et al. used a deep neural network that can recognize the chemical structures and automatically classify them to predict the PCE.66 Therefore, pictures of chemical structures were used without any transformation. This model was fast with low computational cost, and possible to run on a personal computer. It showed 91.02% accuracy in the prediction of PCE of donor materials. The working pattern of this study is given in Fig. 8. There are several drawbacks in this study; firstly, data from the Harvard Clean Energy Project (HCEP) is used to train the ML model for the screening of OSC materials. However, molecules reported in the literature are usually much more complicated than those in the HCEP. Secondly, DFT calculated energy levels are used as input for the Scharber model to estimate PCE. Both are not accurate. The working of organic solar cells is very complex, and the active layer materials, solubility, solvent additives, crystallinity and molecular orientation affect the performance of solar cells. In this study just, pictures of chemical structures of donor materials without any transformation were used as input. Such kind of approach cannot provide realistic results. Molecular descriptors are a better option as compared with just a picture of the structure. So far use of images as input for ML analysis of OSCs is rare (Table 3). | ||
| Fig. 8 Structure of the convolutional neural network (CNN). Reprinted with permission from ref. 66 Copyright 2018, Wiley-VCH Verlag GmbH & Co. KGaA. | ||
| Data set | Source | Input | Method | ML models | Performance* | Experimental validation | Ref. |
|---|---|---|---|---|---|---|---|
| *PCE unless mention. ** mean absolute errors. *** Between ML predicted and Scharber's model calculated PCE. AOC = Accuracy of classification. | |||||||
| 2.3 million | HCEP | Descriptors | regression | Linear regression | 0.84 R2 | No | 45 |
| 111000 |
Literature + database | Descriptors | Regression | RF | HOMO(0.85), LUMO (0.94) R2 | No | 46 |
| 161 | Literature | Descriptors | Regression | CasSVM | 0.96R2 | No | 47 |
| 51000 |
HCEP | Fingerprints | Regression | GPR | 0.43(r) | No | 20 |
| 2.3 million | HCEP | Fingerprints | Regression | GPR | 0.65(r) | No | 21 |
| 1719 | Literature | Fingerprints | Classification (two groups) | RF | 86.67% AOC | Yes (successful) | 59 |
| 1000 | HCEP | Fingerprints | Classification (four groups) | RF | 48% AOC | Yes (failed) | 60 |
| 3989 | Quantum-Machine.org | Fingerprints | Regression | deep tensor network | HOMO(45) LUMO(31)** | No | 61 |
| 350 | HOPV15 | Fingerprints | Regression | decision trees | HOMO (0.74) R2 | No | 62 |
| 51 000 | HCEP | Fingerprints | Regression | CNN | 0.91 (r)*** | No | 63 |
| 320 | Literature | Fingerprints | Regression | KRR | 0.78 (r) | No | 64 |
| 565 | Literature | Fingerprints | Regression | BRT | 0.71 | Yes (successful) | 65 |
| 5000 | HCEP | Image | Classification (two groups) | CNN | 91.02% AOC *** | No | 66 |
| 270 | Literature | Microscopic properties | Regression | GB | 0.79(r) | No | 67 |
| 300 | Literature | Microscopic properties | Regression | GBRT | 0.78(r) | No | 68 |
| 566 | Literature | Microscopic properties | Regression | k-NN | 0.72(r) | No | 69 |
| 290 | Literature | Microscopic properties | Regression | GBRT | 0.80 (r) | No | 70 |
| 249 | Literature | Microscopic properties | Regression | kRR | 0.68 (r) | No | 19 |
| 2.3 million | HCEP | Energy level | Scharber's model | No | 71 | ||
| 380 | Auto-generation | Energy level | Scharber's model | No | 72 | ||
| 135 | Literature | Energy level | regression | RF | 0.80R2 | No | 73 |
| 124 | Literature | Energy level | regression | RF | 0.77R2 | No | 74 |
| 121 | Literature | Energy level | regression | RF | 0.77(VOC)R2 | No | 75 |
| 70 | Literature | Energy level | regression | RF | 0.69R2 | No | 76 |
| 1800 | Simulation | Simulated properties | No | 77 | |||
| 65000 |
Simulation | Simulated properties | Classification | CNN | 95.80%(JSC) AOC | No | 78 |
| 20000 |
Simulation | Simulated properties | ANN | Yes | 79 | ||
4.4. Microscopic properties
The efficiency of OSCs depends on many microscopic properties of the organic materials such as optical gap, charge-carrier mobility, ionization potential of the donor, electron affinity of the acceptor, hole–electron binding energy etc. These microscopic descriptors are more realistic for solar cell applications as compared with simple topological descriptors. However, they are expensive to compute or experimentally determined. Ma et al. used 13 microscopic properties as descriptors to train a model for the prediction of PCE. They used the dataset of 270 small molecules.67 Different techniques such as random forest, gradient boosting and artificial neural network were used. The gradient boosting model showed an impressive performance (Pearson's coefficient (r) = 0.79). However, the used descriptors are computationally expensive such as polarizability and excited state, which is an obstacle for large-scale high-throughput virtual screening of potential molecules.Ma et al. used RF and Gradient boosting regression tree (GBRT) algorithms to predict device characteristics (VOC, JSC, and FF) through microscopic properties.68JSC (r = 0.78) and FF (r = 0.73) showed strong correlation with PCE, however, VOC (r = 0.15) showed very weak correlation with PCE, these results are consistent with recently reported results.60 The JSC and FF are found to be poorly correlated (r = 0.33), with almost no correlation between VOC and JSC (r = −0.18) as well as VOC and FF (r = −0.09).
To analyze the effect of descriptor types on the prediction ability of ML models, Trois et al. trained k-Nearest Neighbors (k-NN), Kernel Ridge Regression (KRR) and Support Vector Regression (SVR) models on data of 566 donor/acceptor pairs collected from the literature.69 Both structural (topology properties) and physical descriptors (energy-levels, molecular size, light absorption and mixing properties) are used. The structural descriptors are the ones that contributed most to the ML models. Some physical properties showed high correlation with PCE but didn’t improve the model prediction ability because information they carry was already encoded in the structural descriptors.
To achieve push–pull conjugated systems, different types of building blocks such as electron-deficient, electron-rich and π-spacer are used to design organic semi-conductors. Ma et al. performed ML modelling to screen 10000 molecules designed from 32 building blocks.70 The purpose of this study was to understand the effect of the nature and arrangement of building blocks. Descriptors were calculated from ground and excited states of candidate molecules. 126 potential candidates with predicted efficiencies ≥8% were selected on the basis of GBRT and ANN models. This approach is efficient to screen the potential candidates for organic solar cells.
Troisi et al. used electronic and geometrical properties to train ML models to predict device parameters, and it performed much better than the Scharber model (Pearson's coefficient (r) = 0.68).19
In organic solar cells, the thermodynamics of mixing of active layer materials controls the evolution of film morphology, and consequently, changes the charge-transport and light-harvesting, so controls the overall performance and stability of the final device.80,81 It is important to study the relationship between molecular interaction parameters and phase behaviour of thin films. For this purpose, Perea et al. used the ANN model and the Flory–Huggins solution theory to study the phase evolution of polymers and fullerenes.82 The ANN model was used to predict solubility parameters by surface charge distribution. Combined with solubility parameters, a figure of merit was established to describe the stability of polymer–fullerene blends (Fig. 9).
 | ||
| Fig. 9 Computational flowchart describing the routine for determining the relative stability capable of describing the microstructure of polymer:fullerene blends. (i) Creation of the σ-profile from the conductor-like screening model (COSMO); (ii) σ-moments as extracted from COSMO are fed into an artificial neural network (ANN) to determine Hansen solubility parameters (HSPs); (iii) HSPs are used to calculate the qualitative Flory–Huggings interaction parameters (χ1,2); (iv) implementation of moiety-monomer-structure properties (reduced molar volumes/weights); (v) spinodal demixing diagrams resulting from polymer blend theory; and (vi) figure of merit (FoM) defined as the ratio of the Flory–Huggins intermolecular parameter and the spinodal diagram forms the basis of a relative stability metric. Adopted with permission from ref. 82 Copyright 2017, American Chemical Society. | ||
4.5. Energy levels
Energy levels of donor and acceptor materials are among the factors that affect the performance of organic solar cells. Energy level mismatch between the donor and acceptor reduces the PCE of OSCs due to large energy loss resulting from radiative recombination loss.83In 2017, Aspuru Guzik's group investigated millions of molecular motifs using 150 million DFT calculations.71 PCE was predicted using Scharber's model,18 and the calculated energy level was used as input. Candidates with a PCE of more than 10% were identified. In 2017, Imamura et al. reported the automatic generation of thiophene-based polymers from donor and acceptor units, estimation of the orbital levels by Hückel-based models and an evaluation of photovoltaic characteristics.72 PCE was calculated using Scharber's model, but its performance is very poor.19–21 Molecular descriptors and microscopic properties of semi-conductors were totally ignored.
Min-Hsuan Lee performed Random Forest (RF) modelling on a database of >100 bulk heterojunction solar cells and achieved high prediction accuracy (R2 of 0.85 and 0.80 for the training set and testing set, respectively).73 In this study, the number of descriptors was small (HOMO, LUMO and band gap). Inclusion of parameters such as solubility parameters, interaction parameters and surface energies as input for the ML model can further enhance the usefulness of similar studies.
Various examples of ML of binary solar cells are discussed above. Generally, ternary OSCs show higher performance than binary OSCs. Binary OSCs are facing the problem of insufficient light harvesting due to narrow absorption range of organic semi-conductors. In ternary OSCs, the third component, can be a donor or an acceptor, and not only works as an additional absorber to enhance photon harvesting but also helps to achieve favourable morphology.84 As the working of ternary solar cells is complex as compared with binary solar cells,85,86 it is a challenging task to find ideal third components for ternary solar cells. Min-Hsuan Lee has trained an ML model for ternary solar cells using Random Forest, Gradient Boosting, k-Nearest Neighbors (k-NN), Linear-Regression and Support Vector Regression. The value of LUMO for the donor (D1) showed a noticeable linear correlation with PCE (r = −0.55), and the correlations of other indicators with PCE were weak.74 The value of VOC is highly correlated with the HOMO of the donor (r = −0.54) and the LUMO of the donor (r = −0.54), respectively, suggesting that the energy levels of the donor need further consideration to find the origin of VOC in ternary OSCs. The Random Forest model showed higher R2 (0.77 on test set) value among all ML methods. In another study, he trained the ML model to predict the VOC of fullerene derivatives based ternary organic solar cells. The descriptors were the same as in a previous study.75 The Random Forest model showed an R2 value of 0.77. In both studies, just energy levels of organic semi-conductors were used as descriptors and other molecular descriptors and the thin film morphology effect were ignored. Therefore, it is required to develop a hybrid modelling framework that should include thin-film characteristics (e.g., the appropriate ratio of three components) and fabrication conditions (e.g., annealing temperature and solvent additive). Optimization of all the factors can enhance charge generation and reduce voltage loss, and resultantly can improve device efficiency.87 Theoretical analysis of the morphology of the three components is much more complex than that of two components.
Tandem organic solar cells have shown superior power conversion efficiency (PCE). A tandem organic solar cell consists of two sub-cells. The major purposes of this device architecture are to widen the photon response range, and suppress the transmission loss and thermalization loss.88 It is more challenging to develop a relationship between efficiency and physical properties of active layer materials due to the great diversity of organic materials that leads to more candidate materials. To solve this problem, Min-Hsuan Lee used ML algorithms to predict the efficiency of tandem OSCs and identify sufficient bandgap combinations for tandem OSCs.76 Random Forest regression was used to perform machine learning using energy levels as input to predict the efficiency. The results indicate that energy offset in the LUMO level between the donor and the acceptor material should be optimized to improve the process of electron transfer and device performance.
4.6 Simulated properties
Film morphology controls the efficiency of organic solar cells. For further improvement of PCE, a clear understanding of the film morphology is essential. Along with the experimental work, mathematical simulation is another way to explore the film morphology and find the effect of various parameters on it. These kinds of simulations have two distinct stages: the representation stage and the mapping stage. In the first stage, a mathematical framework is developed to generate microstructures. In the second stage, the generated microstructures are related to a target property. The use of graph theory for microstructure analysis of OSCs is increasing.89–91 For example, Ganapathysubramanian et al. studied morphology descriptors related to various processes in OSCs such as photon absorption, exciton diffusion, charge separation, and charge transport through a graph-based approach.77 High correlation between the graph-based approach and computationally intensive method was achieved. In another study, they used CNN to relate the film morphology with short-circuit current (JSC).78 They solved the thermodynamically consistent CahnHilliard equation92 for binary phase separation using an in-house finite element library.93 A total of ∼65000 morphologies were generated. JSC, was evaluated for each morphology using the excitonic drift-diffusion equation.94 CNN using morphologies as input and JSC as output showed a classification accuracy of 95.80% (Fig. 10).
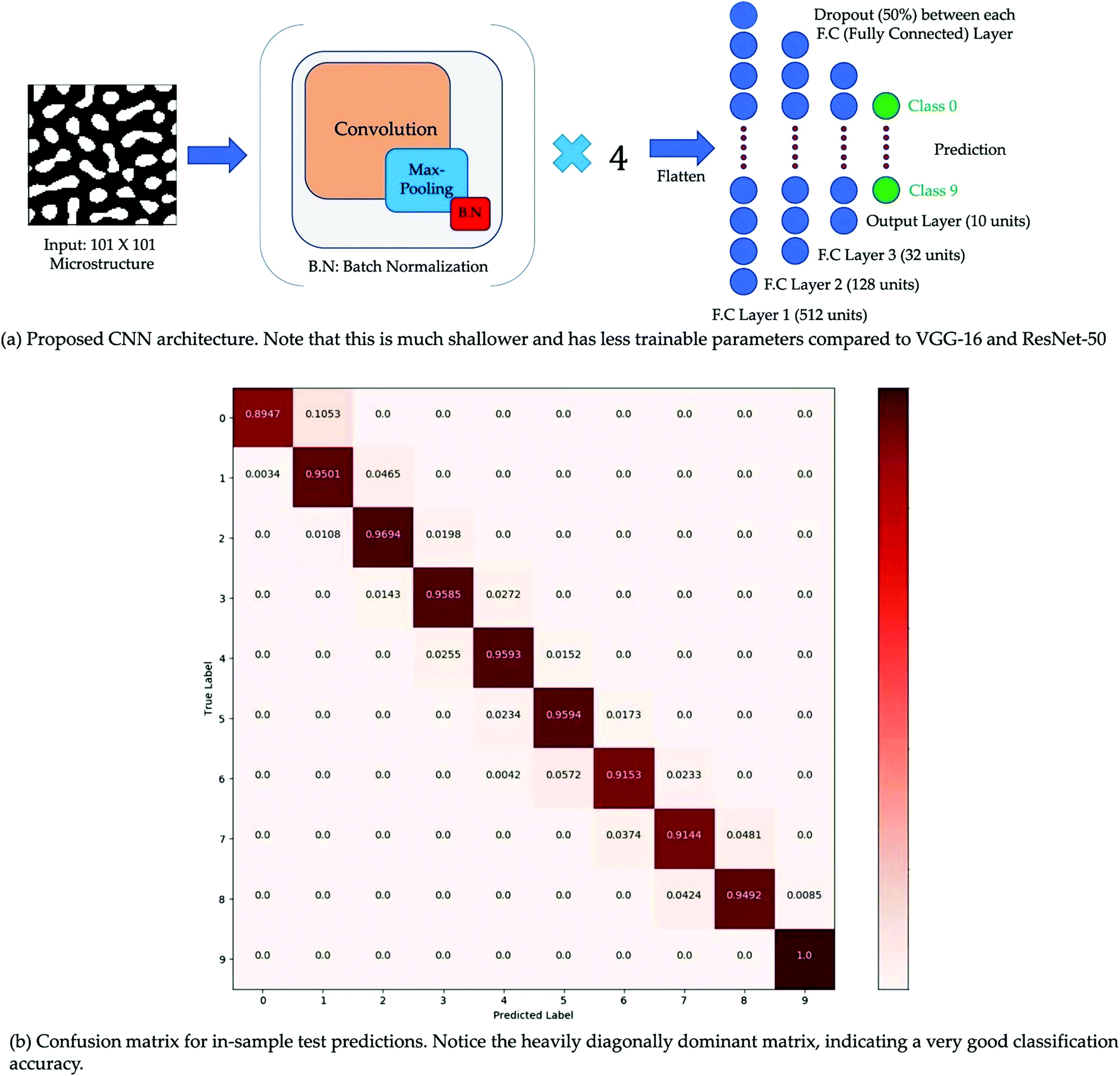 | ||
| Fig. 10 (a) Simple sketch of CNN architecture and (b) Confusion matrix. Adopted from ref. 78 Creative Commons Attribution 4.0 International License. | ||
MacKenzie et al. used the Shockley–Read–Hall based drift-diffusion model to simulate current/voltage (JV) curves.79 They generated a set of 20000 devices and calculated electrical parameters such as carrier trapping rates, energetic disorder, trap densities, recombination time constants, and parasitic resistances. Simulated data were used to train the neural network. Then the trained model was used to study the effect of surfactant choice and annealing temperature on charge carrier dynamics of some well-known OSC devices.
The solubility of the active layer materials in a specific solvent controls the film morphology and resultantly affects the performance of the device. Risko et al. calculated the free energy of mixing using molecular dynamics (MD) simulation.95 They also used Bayesian statistics to calculate the free energy of mixing. This approach is an effective and fast way to study the large number of solvent and solvent additives.
5. Problems and future prospects
In spite of the increasing number of publications on machine learning, its use for organic solar cells is still not very efficient due to the complex nature of organic solar cells. The performance of OSCs depends on the processing solvent, solvent additive, crystallinity and molecular orientation of the active layer materials. Charge-separation at the donor–acceptor interface strongly depends on morphological properties. Further work is required for efficient use of ML for photovoltaic materials.5.1. Data infrastructure
In many studies data from Harvard Clean Energy Project (HCEP) is used to contract the ML model for the screening of OPV materials. However, molecules reported in the literature are usually much more complicated than those in the HCEP. The differences in structures may lead to inaccurate ML predictions. Some groups have already collected data from the published literature.For effective training of an ML model, a large amount of data is required. Availability of data is not a problem for fields such as image recognition, i.e. millions of input datasets are available. However, in organic solar cells data are in hundreds or thousands. It is reported that accuracy is increased with increase of the data points (number of molecules).31,66,76 In the case of ML models trained using descriptors related to power conversion processes, it is hard to include huge data due to time consuming DFT calculations. For limited data sets, meta-learning is a promising solution, whereby knowledge is learned within and across problems. A Bayesian framework is also a good option for limited data. A dual strategy is required to maintained balance between the availability of the data and the predictive capability of models. Instead of affecting the model precision directly, the effect of data size can be mediated by the degree of freedom (DoF) of the model, resulting in the phenomenon of association between precision and DoF. This concept is originated from the statistical bias-variance trade-off. In this regard, Zhang and Ling proposed a strategy to apply machine learning to small datasets.96
5.2. Descriptor selection
Selection of molecular descriptors is a very important task. Molecular descriptors and fingerprints are fast and easy to calculate but not completely suitable for organic solar cell modelling. The microscopic properties that contain information related to photovoltaic phenomena require high accuracy quantum calculations. Their high computational cost makes it impossible to use them for large-scale fast virtual screening. A suitable trade-off between speed and accuracy is required. In this situation, accurate and easily accessible fingerprints and as well as a new generation of descriptors specifically designed for organic semiconductors are urgently needed.5.3. Multidimensional design
Most of the models correlate the chemical structures with PCE but ignore many factors such miscibility and film morphology. The use of Flory and Huggins theory may enhance the usefulness of ML methods. Inclusion of transmission electron microscopy (TEM), atomic force microscopy (AFM), grazing-incidence small-angle X-ray scattering (GISAXS) and grazing incidence wide angle x-ray scattering (GIWAXS) data may improve the prediction accuracy.97 Although machine learning is so successful on routine life picture analysis, images from the above techniques are very different. Compared to routine life images, images from the above techniques have featured characters, therefore, ML analysis of these images is very difficult. For example, microscopy images are associated with high-level noise and distortions.98 In addition, information provided by images from experimental techniques strongly associates with physiochemical properties of materials, their mixing and experimental environments. All these factors make the situation much more sophisticated. Additionally, images from different techniques have different physical meanings. Therefore, different strategies will be required for different techniques. Considering this situation, machine learning analysis of experimental images should be carried out with enough knowledge of machine learning and physicochemical properties and underlying phenomena.In this route, the first step is collection of images, which will be manual due to the presence of various images for one compound under different experimental conditions. Automatic image extraction and sorting will be very difficult. Therefore, human assistance is essential. The second step will be task specification and analysis, for example decision of the data label or target properties. The morphology of the active layer strongly influences the FF values.99 Therefore, it will be more realistic to select FF as the target rather than PCE. Then FF along with the other factors can correlate with the PCE. Another thing to decide is the use of classification or regression. In the case of small data sets, classification can be preferred and vice versa. The third step will be training of the model and extraction of the pattern from the data to make a prediction. The last step will be experimental validation. It feels a hard journey to connect images to performance, however, it will be fruitful.
Although no study is reported where experimental morphologies were used as input for machine learning analysis of organic solar cells, there are several studies that used simulated images and properties to train an ML model. Some studies are discussed in Section 4.6 and some are cited. These studies will pave the way toward the utilization of experimental morphologies for machine learning models.100
5.4. Experimental validation
The review of the above mention literature has showed increasing use of machine learning in organic solar cell research. Further advances in high-throughput screening are expected. Generally, heuristic rules are used to screen the materials, and there is no guaranty that they are synthesizable (their synthesis procedures are not necessarily known). Therefore, collaboration with experimental experts is necessary. Collaboration can improve the success rate of machine prediction. Potential candidates selected from machine learning should be further manually screened on the basis of synthetic aspects. In addition, machine learning predictions should be followed by experimental validation. However, relatively few examples are present where the final candidates are validated using experiments. Sun et al. synthesized 10 donor materials to verify the ML results. The model has classified eight molecules into the correct category.59 Saeki et al. synthesized one donor and fabricated an OSC device, and it showed a PCE of 0.53% that is much lower than the RF prediction (5.0–5.8%).60 Min et al. fabricated six D/A pairs, and most of the devices showed a PCE close to the predicted values.655.5. Development of better software
Currently, some degree of programming ability is required to use the majority of machine learning tools. Therefore, ML is only accessible to those who have deep experience in computer programming and data science. However, they do not have enough knowledge of underlying processes. Sometime, it leads to the wrong interpretation of results. There are so many experimental scientists working in the field of organic solar cells. However, they do not have background of machine learning. To solve this problem, software with user-friendly graphical interface should be developed for material experts. In this way, material experts will able to explore all the powers of data driven science without the problems of unfamiliar syntax and arcane tuning parameters.6. Summary
Different types of input such as molecular descriptors, molecular fingerprints, images, microscopic properties, energy levels and simulated properties are using to train machine learning models. The types of input and machine learning model (algorithm) strongly affect the performance of the machine learning model. In spite of pitfalls, machine learning is still essential to speed-up the discovery of efficient organic semi-conductors for organic solar cells. The number of publications based on machine learning is growing at an extraordinary rate. Despite of success stories, still there are many obstacles for the use of machine learning in organic solar cell research. We have mentioned some obstacles and proposed possible solutions. Further improvement will increase the use of machine learning in organic solar cell research. It will replace the experimental trial and error methods. Therefore, the use of machine learning in organic solar cells research should be encouraged. The availability of open-source tools and data sharing have enhanced the potential of ML to revolutionize materials discovery. There is a large gap between what machine learning can deliver and what currently has been achieved. Soon, machine learning will be a necessary method for organic solar cells as the existing experimental and computational techniques are.Abbreviations
| ML | Machine learning |
| BHJ | Bulk-heterojunction |
| GBRT | Bradient boosting regression tree |
| RF | Random forest |
| SVM | Support vector machine |
| ANN | Artificial neural network |
| DNN | Deep neural network |
| CNN | Convolutional neural network |
| KRR | Kernel ridge regression |
| SVR | Support vector regression |
| GPR | Gaussian processes regression |
| SMILES | Simplified molecular-input line-entry system |
| PCE | Power conversion efficiency |
| V OC | Open-circuit voltage |
| J SC | Short-circuit current density |
| FF | Fill factor |
| HOMO | Highest occupied molecular orbitals |
| LUMO | Lowest occupied molecular orbitals |
| DFT | Density functional theory |
| r | Correlation coefficient |
| R 2 | Coefficient of determination |
| HCEP | Harvard Clean Energy Project |
| HOPV15 | The Harvard Organic Photovoltaic Dataset |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the support from the National Natural Science Foundation of China (No. 21672023, 21971014, and 21950410533) and the National Key Research and Development Program of China (2018YFA0901800). Jin-Liang Wang was supported by the Thousand Youth Talents Plan of China and BIT Teli Young Fellow Recruitment Program. The authors thank the Analysis & Testing Center, Beijing Institute of Technology.References
- S. Günes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324–1338 CrossRef.
- H. Imahori, T. Umeyama and S. Ito, Acc. Chem. Res., 2009, 42, 1809–1818 CrossRef CAS.
- A. Mahmood, J.-Y. Hu, B. Xiao, A. Tang, X. Wang and E. Zhou, J. Mater. Chem. A, 2018, 6, 16769–16797 RSC.
- A. Mahmood, J. Hu, A. Tang, F. Chen, X. Wang and E. Zhou, Dyes Pigm., 2018, 149, 470–474 CrossRef CAS.
- C. Zhang, X. Song, K.-K. Liu, M. Zhang, J. Qu, C. Yang, G.-Z. Yuan, A. Mahmood, F. Liu, F. He, D. Baran and J.-L. Wang, Small, 2020, 16, 1907681 CrossRef CAS.
- Q. Liu, Y. Jiang, K. Jin, J. Qin, J. Xu, W. Li, J. Xiong, J. Liu, Z. Xiao, K. Sun, S. Yang, X. Zhang and L. Ding, Sci. Bulletin, 2020, 65, 272–275 CrossRef CAS.
- C. Zhu, J. Yuan, F. Cai, L. Meng, H. Zhang, H. Chen, J. Li, B. Qiu, H. Peng, S. Chen, Y. Hu, C. Yang, F. Gao, Y. Zou and Y. Li, Energy Environ. Sci., 2020, 13, 2459–2466 RSC.
- L. Zhan, S. Li, T.-K. Lau, Y. Cui, X. Lu, M. Shi, C.-Z. Li, H. Li, J. Hou and H. Chen, Energy Environ. Sci., 2020, 13, 635–645 RSC.
- J.-L. Wang, K.-K. Liu, J. Yan, Z. Wu, F. Liu, F. Xiao, Z.-F. Chang, H.-B. Wu, Y. Cao and T. P. Russell, J. Am. Chem. Soc., 2016, 138, 7687–7697 CrossRef CAS.
- S.-S. Wan, X. Xu, Z. Jiang, J. Yuan, A. Mahmood, G.-Z. Yuan, K.-K. Liu, W. Ma, Q. Peng and J.-L. Wang, J. Mater. Chem. A, 2020, 8, 4856–4867 RSC.
- Y. Wang, Y. Wang, L. Zhu, H. Liu, J. Fang, X. Guo, F. Liu, Z. Tang, M. Zhang and Y. Li, Energy Environ. Sci., 2020, 13, 1309–1317 RSC.
- D. Hu, Q. Yang, H. Chen, F. Wobben, V. M. Le Corre, R. Singh, T. Liu, R. Ma, H. Tang, L. J. A. Koster, T. Duan, H. Yan, Z. Kan, Z. Xiao and S. Lu, Energy Environ. Sci., 2020, 13, 2134–2141 RSC.
- C. Yang, S. Zhang, J. Ren, M. Gao, P. Bi, L. Ye and J. Hou, Energy Environ. Sci., 2020, 13, 2864–2869 RSC.
- X. Wan, C. Li, M. Zhang and Y. Chen, Chem. Soc. Rev., 2020, 49, 2828–2842 RSC.
- G. Han, T. Hu and Y. Yi, Adv. Mater., 2020, 32, 2000975 CrossRef CAS.
- J.-L. Brédas, J. E. Norton, J. Cornil and V. Coropceanu, Acc. Chem. Res., 2009, 42, 1691–1699 CrossRef.
- A. Wadsworth, M. Moser, A. Marks, M. S. Little, N. Gasparini, C. J. Brabec, D. Baran and I. McCulloch, Chem. Soc. Rev., 2019, 48, 1596–1625 RSC.
- M. C. Scharber, D. Mühlbacher, M. Koppe, P. Denk, C. Waldauf, A. J. Heeger and C. J. Brabec, Adv. Mater., 2006, 18, 789–794 CrossRef CAS.
- D. Padula, J. D. Simpson and A. Troisi, Mater. Horiz., 2019, 6, 343–349 RSC.
- S. A. Lopez, B. Sanchez-Lengeling, J. de Goes Soares and A. Aspuru-Guzik, Joule, 2017, 1, 857–870 CrossRef CAS.
- E. O. Pyzer-Knapp, G. N. Simm and A. Aspuru Guzik, Mater. Horiz., 2016, 3, 226–233 RSC.
- G. H. Gu, J. Noh, I. Kim and Y. Jung, J. Mater. Chem. A, 2019, 7, 17096–17117 RSC.
- C. Chen, Y. Zuo, W. Ye, X. Li, Z. Deng and S. P. Ong, Adv. Energy Mater., 2020, 10, 1903242 CrossRef CAS.
- M.-H. Lee, Adv. Electron. Mater., 2019, 5, 1900573 CrossRef CAS.
- T. Wang, C. Zhang, H. Snoussi and G. Zhang, Adv. Funct. Mater., 2020, 30, 1906041 CrossRef CAS.
- R. Hu, J. Song, Y. Liu, W. Xi, Y. Zhao, X. Yu, Q. Cheng, G. Tao and X. Luo, Nano Energy, 2020, 72, 104687 CrossRef CAS.
- A. O. Oliynyk and J. M. Buriak, Chem. Mater., 2019, 31, 8243–8247 CrossRef CAS.
- T. Cova and A. Pais, Front. Chem., 2019, 7, 809 CrossRef CAS.
- G. R. Schleder, A. C. M. Padilha, C. M. Acosta, M. Costa and A. Fazzio, J. Phys. Mater., 2019, 2, 032001 CrossRef CAS.
- T. Zhou, Z. Song and K. Sundmacher, Engineering, 2019, 5, 1017–1026 CrossRef CAS.
- E. O. Pyzer-Knapp, K. Li and A. Aspuru-Guzik, Adv. Funct. Mater., 2015, 25, 6495–6502 CrossRef CAS.
- S. Vieira, W. H. Lopez Pinaya and A. Mechelli, in Machine Learning, ed. A. Mechelli and S. Vieira, Academic Press, 2020, pp. 21–44, DOI:10.1016/B978-0-12-815739-8.00002-X.
- Y. Iwasaki, I. Takeuchi, V. Stanev, A. G. Kusne, M. Ishida, A. Kirihara, K. Ihara, R. Sawada, K. Terashima, H. Someya, K.-i. Uchida, E. Saitoh and S. Yorozu, Sci. Rep., 2019, 9, 2751 CrossRef.
- B. Sanchez-Lengeling and A. Aspuru-Guzik, Science, 2018, 361, 360–365 CrossRef CAS.
- A. H. Vo, T. R. Van Vleet, R. R. Gupta, M. J. Liguori and M. S. Rao, Chem. Res. Toxicol., 2020, 33, 20–37 Search PubMed.
- A. Mauri, V. Consonni, M. Pavan and R. Todeschini, MATCH-Commun. Math. Co., 2006, 56, 237–248 Search PubMed.
- I. V. Tetko, J. Gasteiger, R. Todeschini, A. Mauri, D. Livingstone, P. Ertl, V. A. Palyulin, E. V. Radchenko, N. S. Zefirov, A. S. Makarenko, V. Y. Tanchuk and V. V. Prokopenko, J. Comput. Aided Mol. Des., 2005, 19, 453–463 CrossRef CAS.
- H. Hong, Q. Xie, W. Ge, F. Qian, H. Fang, L. Shi, Z. Su, R. Perkins and W. Tong, J. Chem. Inf. Model., 2008, 48, 1337–1344 CrossRef CAS.
- H. Moriwaki, Y.-S. Tian, N. Kawashita and T. Takagi, J. Cheminform., 2018, 10, 4 CrossRef.
- C. W. Yap, J. Comput. Chem., 2011, 32, 1466–1474 CrossRef CAS.
- A. Kerber, R. Laue, M. Meringer and C. Ucker, MATCH-Commun. Math. Co., 2004, 51, 187–204 CAS.
- D.-S. Cao, Q.-S. Xu, Q.-N. Hu and Y.-Z. Liang, Bioinformatics, 2013, 29, 1092–1094 CrossRef CAS.
- BlueDesc, http://www.ra.cs.uni-tuebingen.de/software/bluedesc/, accessed July 12, 2020.
- K. Liu, J. Feng and S. S. Young, J. Chem. Inf. Model., 2005, 45, 515–522 CrossRef CAS.
- R. Olivares-Amaya, C. Amador-Bedolla, J. Hachmann, S. Atahan-Evrenk, R. S. Sánchez-Carrera, L. Vogt and A. Aspuru-Guzik, Energy Environ. Sci., 2011, 4, 4849–4861 RSC.
- F. Pereira, K. Xiao, D. A. R. S. Latino, C. Wu, Q. Zhang and J. Aires-de-Sousa, J. Chem. Inf. Model., 2017, 57, 11–21 CrossRef CAS.
- M.-Y. Sui, Z.-R. Yang, Y. Geng, G.-Y. Sun, L. Hu and Z.-M. Su, Sol. RRL, 2019, 3, 1900258 CrossRef CAS.
- A. Cereto-Massagué, M. J. Ojeda, C. Valls, M. Mulero, S. Garcia-Vallvé and G. Pujadas, Methods, 2015, 71, 58–63 CrossRef.
- L. Pattanaik and C. W. Coley, Chem, 2020, 6, 1204–1207 CAS.
- I. Muegge and P. Mukherjee, Expert Opin. Drug Discovery, 2016, 11, 137–148 CrossRef CAS.
- A. Mahmood, A. Tang, X. Wang and E. Zhou, Phys. Chem. Chem. Phys., 2019, 21, 2128–2139 RSC.
- A. Mahmood, J. Yang, J. Hu, X. Wang, A. Tang, Y. Geng, Q. Zeng and E. Zhou, J. Phys. Chem. C, 2018, 122, 29122–29128 CrossRef CAS.
- G.-Z. Yuan, H. Fan, S.-S. Wan, Z. Jiang, Y.-Q. Liu, K.-K. Liu, H.-R. Bai, X. Zhu and J.-L. Wang, J. Mater. Chem. A, 2019, 7, 20274–20284 RSC.
- J.-L. Wang, K.-K. Liu, L. Hong, G.-Y. Ge, C. Zhang and J. Hou, ACS Energy Lett., 2018, 3, 2967–2976 CrossRef CAS.
- K.-K. Liu, X. Xu, J.-L. Wang, C. Zhang, G.-Y. Ge, F.-D. Zhuang, H.-J. Zhang, C. Yang, Q. Peng and J. Pei, J. Mater. Chem. A, 2019, 7, 24389–24399 RSC.
- Y. Xie, W. Wang, W. Huang, F. Lin, T. Li, S. Liu, X. Zhan, Y. Liang, C. Gao, H. Wu and Y. Cao, Energy Environ. Sci., 2019, 12, 3556–3566 RSC.
- T. Linderl, T. Zechel, M. Brendel, D. Moseguí González, P. Müller-Buschbaum, J. Pflaum and W. Brütting, Adv. Energy Mater., 2017, 7, 1700237 CrossRef.
- J. Zhang, W. Liu, M. Zhang, Y. Liu, G. Zhou, S. Xu, F. Zhang, H. Zhu, F. Liu and X. Zhu, iScience, 2019, 19, 883–893 CrossRef CAS.
- W. Sun, Y. Zheng, K. Yang, Q. Zhang, A. A. Shah, Z. Wu, Y. Sun, L. Feng, D. Chen, Z. Xiao, S. Lu, Y. Li and K. Sun, Sci. Adv., 2019, 5, eaay4275 CrossRef CAS.
- S. Nagasawa, E. Al-Naamani and A. Saeki, J. Phys. Chem. Lett., 2018, 9, 2639–2646 CrossRef CAS.
- P. B. Jørgensen, M. Mesta, S. Shil, J. M. G. Lastra, K. W. Jacobsen, K. S. Thygesen and M. N. Schmidt, J. Chem. Phys., 2018, 148, 241735 CrossRef.
- A. Paul, A. Furmanchuk, W.-k. Liao, A. Choudhary and A. Agrawal, Mol. Inform., 2019, 38, 1900038 CrossRef CAS.
- S.-P. Peng and Y. Zhao, J. Chem. Inf. Model., 2019, 59, 4993–5001 CrossRef CAS.
- D. Padula and A. Troisi, Adv. Energy Mater., 2019, 9, 1902463 CrossRef CAS.
- Y. Wu, J. Guo, R. Sun and J. Min, NPJ Comput. Mater., 2020, 6, 120 CrossRef CAS.
- W. Sun, M. Li, Y. Li, Z. Wu, Y. Sun, S. Lu, Z. Xiao, B. Zhao and K. Sun, Adv. Theory Simul., 2019, 2, 1800116 CrossRef.
- H. Sahu, W. Rao, A. Troisi and H. Ma, Adv. Energy Mater., 2018, 8, 1801032 CrossRef.
- H. Sahu and H. Ma, J. Phys. Chem. Lett., 2019, 10, 7277–7284 CrossRef CAS.
- Z.-W. Zhao, M. del Cueto, Y. Geng and A. Troisi, Chem. Mater., 2020, 32, 7777–7787 CrossRef CAS.
- H. Sahu, F. Yang, X. Ye, J. Ma, W. Fang and H. Ma, J. Mater. Chem. A, 2019, 7, 17480–17488 RSC.
- J. Hachmann, R. Olivares-Amaya, A. Jinich, A. L. Appleton, M. A. Blood-Forsythe, L. R. Seress, C. Román-Salgado, K. Trepte, S. Atahan-Evrenk, S. Er, S. Shrestha, R. Mondal, A. Sokolov, Z. Bao and A. Aspuru-Guzik, Energy Environ. Sci., 2014, 7, 698–704 RSC.
- Y. Imamura, M. Tashiro, M. Katouda and M. Hada, J. Phys. Chem. C, 2017, 121, 28275–28286 CrossRef CAS.
- M.-H. Lee, Organ. Electron., 2020, 76, 105465 CrossRef CAS.
- M.-H. Lee, Adv. Energy Mater., 2019, 9, 1900891 CrossRef.
- M.-H. Lee, Adv. Intelligent Syst., 2020, 2, 1900108 CrossRef.
- M.-H. Lee, Energy Technol., 2020, 8, 1900974 CrossRef CAS.
- O. Wodo, S. Tirthapura, S. Chaudhary and B. Ganapathysubramanian, Organ. Electron., 2012, 13, 1105–1113 CrossRef CAS.
- B. S. S. Pokuri, S. Ghosal, A. Kokate, S. Sarkar and B. Ganapathysubramanian, NPJ Comput. Mater., 2019, 5, 95 CrossRef.
- N. Majeed, M. Saladina, M. Krompiec, S. Greedy, C. Deibel and R. C. I. MacKenzie, Adv. Funct. Mater., 2020, 30, 1907259 CrossRef CAS.
- L. Ye, W. Zhao, S. Li, S. Mukherjee, J. H. Carpenter, O. Awartani, X. Jiao, J. Hou and H. Ade, Adv. Energy Mater., 2017, 7, 1602000 CrossRef.
- D. T. Duong, B. Walker, J. Lin, C. Kim, J. Love, B. Purushothaman, J. E. Anthony and T.-Q. Nguyen, J. Polym. Sci. B Polym. Phys., 2012, 50, 1405–1413 CrossRef CAS.
- J. D. Perea, S. Langner, M. Salvador, B. Sanchez-Lengeling, N. Li, C. Zhang, G. Jarvas, J. Kontos, A. Dallos, A. Aspuru-Guzik and C. J. Brabec, J. Phys. Chem. C, 2017, 121, 18153–18161 CrossRef CAS.
- H. Z. Jun Yuan, R. Zhang, Y. Wang, J. Hou, M. Leclerc, X. Zhan, F. Huang, F. Gao, Y. Zou and Y. Li, Chem, 2020, 6, 2147–2161 Search PubMed.
- Q. Yue, W. Liu and X. Zhu, J. Am. Chem. Soc., 2020, 142, 11613–11628 CrossRef CAS.
- J. Gao, W. Gao, X. Ma, Z. Hu, C. Xu, X. Wang, Q. An, C. Yang, X. Zhang and F. Zhang, Energy Environ. Sci., 2020, 13, 958–967 RSC.
- T. Liu, R. Ma, Z. Luo, Y. Guo, G. Zhang, Y. Xiao, T. Yang, Y. Chen, G. Li, Y. Yi, X. Lu, H. Yan and B. Tang, Energy Environ. Sci., 2020, 13, 2115–2123 RSC.
- Z. Zhou, S. Xu, J. Song, Y. Jin, Q. Yue, Y. Qian, F. Liu, F. Zhang and X. Zhu, Nat. Energy, 2018, 3, 952–959 CrossRef CAS.
- G. Liu, J. Jia, K. Zhang, X. E. Jia, Q. Yin, W. Zhong, L. Li, F. Huang and Y. Cao, Adv. Energy Mater., 2019, 9, 1803657 CrossRef.
- P. Du, A. Zebrowski, J. Zola, B. Ganapathysubramanian and O. Wodo, NPJ Comput. Mater., 2018, 4, 50 CrossRef.
- S. Pfeifer, B. S. S. Pokuri, P. Du and B. Ganapathysubramanian, Mater. Disc, 2018, 11, 6–13 Search PubMed.
- R. Noruzi, S. Ghadai, O. R. Bingol, A. Krishnamurthy and B. Ganapathysubramanian, Comput. Aided Des., 2020, 118, 102771 CrossRef.
- J. W. Cahn, Acta Metall., 1961, 9, 795–801 CrossRef CAS.
- O. Wodo and B. Ganapathysubramanian, Comput. Mater. Sci., 2012, 55, 113–126 CrossRef CAS.
- H. K. Kodali and B. Ganapathysubramanian, Model. Simul. Mater. Sci. Eng., 2012, 20, 035015 CrossRef.
- S. Li, B. S. S. Pokuri, S. M. Ryno, A. Nkansah, C. De’Vine, B. Ganapathysubramanian and C. Risko, J. Chem. Inf. Model., 2020, 60, 1424–1431 CrossRef CAS.
- Y. Zhang and C. Ling, NPJ Comput. Mater., 2018, 4, 25 CrossRef.
- A. Mahmood and J.-L. Wang, Sol. RRL, 2020, 4, 2000337 CrossRef CAS.
- L. Jones and P. D. Nellist, Microsc. Microanal., 2013, 19, 1050–1060 CrossRef CAS.
- M. Zawodzki, R. Resel, M. Sferrazza, O. Kettner and B. Friedel, ACS Appl. Mater. Interfaces, 2015, 7, 16161–16168 CrossRef CAS.
- B. S. S. Pokuri, J. Stimes, K. O’Hara, M. L. Chabinyc and B. Ganapathysubramanian, Comput. Mater. Sci., 2019, 163, 1–10 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |