
Photocatalytic and electrocatalytic transformations of C1 molecules involving C–C coupling
Shunji
Xie
 a,
Wenchao
Ma
a,
Xuejiao
Wu
a,
Haikun
Zhang
a,
Qinghong
Zhang
*a,
Yangdong
Wang
*b and
Ye
Wang
*a
a,
Wenchao
Ma
a,
Xuejiao
Wu
a,
Haikun
Zhang
a,
Qinghong
Zhang
*a,
Yangdong
Wang
*b and
Ye
Wang
*a
aState Key Laboratory of Physical Chemistry of Solid Surfaces, Collaborative Innovation Center of Chemistry for Energy Materials, National Engineering Laboratory for Green Chemical Productions of Alcohols, Ethers and Esters, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China. E-mail: zhangqh@xmu.edu.cn; wangye@xmu.edu.cn
bState Key Laboratory of Green Chemical Engineering and Industrial Catalysis, Shanghai Research Institute of Petrochemical Technology, SINOPEC Corp., Shanghai 201208, China. E-mail: wangyd.sshy@sinopec.com
First published on 31st October 2020
Abstract
Selective transformation of one-carbon (C1) molecules, which are abundant or easily available and inexpensive carbon feedstocks, into value-added multi-carbon (C2+) compounds is a very attractive but highly challenging research target. Photocatalysis and electrocatalysis have offered great opportunities for the activation and controllable C–C coupling of C1 molecules under mild and environmentally benign conditions. This article provides a critical review on recent advances in photocatalytic and electrocatalytic conversions of major C1 molecules, including CO, CO2, CH4, CH3OH and HCHO, into C2+ compounds, such as C2H4, C3H6, ethanol and ethylene glycol, which play essential roles in the current chemical or energy industry. Besides the photocatalysts and electrocatalysts reported for these conversions, the structure–performance relationships and the key factors that control the activity and product selectivity are analysed to provide insights into the rational design of more efficient catalysts for the synthesis of C2+ compounds from C1 feedstocks. The active species, reaction intermediates and reaction or catalyst-functioning mechanism are discussed to deepen the understanding of the chemistry for the activation and selective C–C coupling of C1 molecules in the presence of solar energy or electrical energy.
 Shunji Xie | Shunji Xie received his BS and MSc degrees from Hunan University of China in 2008 and 2011, and obtained his PhD degree from Xiamen University in 2014. He then carried out postdoctoral research at the Collaborative Innovation Center of Chemistry for Energy Materials (iChEM). He is currently a senior engineer in State Key Laboratory of Physical Chemistry of Solid Surfaces of Xiamen University. His research interest focuses on photocatalysis and electrocatalysis for C1 and sustainable chemistry, including CO2 reduction, CH4 oxidation, biomass conversion and ethylene glycol synthesis. |
 Wenchao Ma | Wenchao Ma received his BSc degree from Xiamen University in June 2016. He is currently a PhD candidate in Professor Ye Wang's group in the Collaborative Innovation Center of Chemistry for Energy Materials (iChEM) at Xiamen University. His research interests focus on photo- and/or electro-catalytic upgrading of C1 molecules. |
 Xuejiao Wu | Xuejiao Wu received her BSc degree from Xiamen University in June 2015. She is currently a PhD candidate in Professor Ye Wang's group in the Collaborative Innovation Center of Chemistry for Energy Materials (iChEM) at Xiamen University. Her research interest focuses on the valorisation of lignocellulose, in particular by photocatalytic approaches. |
 Qinghong Zhang | Qinghong Zhang received her BSc and MSc degrees from Nanjing University in 1989 and 1992, and obtained her PhD degree from Hiroshima University of Japan in 2002. She joined Xiamen University in October 2002 and was promoted to a full professor in 2010. Her research interests include the synthesis and characterization of novel materials with advanced catalytic properties. |
 Yangdong Wang | Yangdong Wang received his PhD degree in the Department of Physical Chemistry from Nanjing University in 2000. After that he joined the faculty at SINOPEC Shanghai Research Institute of Petrochemical Technology, where he worked as a research director of Chemistry. His research fields include the heterogeneous catalysis of porous materials and their application in petrochemical processes such as methanol conversion, and the production and transformation of olefins. |
 Ye Wang | Ye Wang received his BS degree from Nanjing University and obtained his PhD degree in 1996 from Tokyo Institute of Technology. He then worked at Tokyo Institute of Technology, Tohoku University and Hiroshima University, and was promoted to associate professor at Hiroshima University in 2001. He became a full professor of Xiamen University in the August of 2001. He serves as an associate editor of ACS Catalysis and is a council member of International Association of Catalysis Societies. The research interest of Prof. Ye Wang's group is catalysis for C1 and sustainable chemistry, including C–H activation and C–C coupling of C1 molecules and C–O/C–C cleavage chemistry for cellulose/lignin valorization. |
Broader contextUnder the current background of looking for alternative carbon resources to replace crude oil for the production of chemicals and liquid fuels, the transformation of C1 molecules, which are typically either abundant in nature or can be readily produced from non-petroleum carbon resources, into important C2+ compounds such as ethylene, propylene, ethanol and ethylene glycol, has attracted much recent attention. The activation of stable C1 molecules (e.g., CH4 and CO2) and controllable C–C coupling are two of the most challenging research goals in chemistry. As compared to thermocatalysis, which is usually performed under harsh conditions and has difficulty in controlling product selectivity, photocatalysis and electrocatalysis hold great potential to activate and selectively convert C1 molecules to C2+ compounds under mild conditions with solar or electrical energy. This article highlights the state-of-the-art advances in photocatalytic/electrocatalytic transformations of C1 molecules into important C2+ compounds. The challenges and opportunities in this field will be analysed. The present article may stimulate further research effort to develop efficient photocatalytic/electrocatalytic systems for C1 chemistry and to deepen the understanding of mechanisms for activation and selective C–C coupling of C1 molecules in the presence of solar or electrical energy. |
1. Introduction
C1 chemistry, i.e., the chemistry for the transformation of C1 molecules into value-added products, is experiencing a renaissance because of the rapidly growing demand for the harvesting of carbon-based energy and chemicals from alternative resources other than petroleum, such as the emerging shale gas in the US, coal in China, renewable biomass and CO2.1–4 Traditional homogeneous and heterogeneous catalysis has played crucial roles in C1 chemistry by transforming C1 molecules such as CO, CO2, CH4 and CH3OH into liquid fuels (e.g., gasoline, diesel and jet fuel) or building-block chemicals (e.g., C2H4, C3H6, aromatics and ethanol).5–26 Several C1-chemistry-based commercial processes such as the carbonylation of methanol to acetic acid,4 the conversion of natural gas to liquid fuels (GTL) or the conversion of coal to liquid fuels (CTL) via synthesis gas (CO/H2) by Fischer–Tropsch synthesis,7 and the conversion of methanol to gasoline (MTG) or the conversion of methanol to lower olefins (MTO)23–26 have contributed largely to the chemical and energy industries. Most of these reactions are performed under harsh conditions such as high temperatures and/or high pressures, and the selectivity of the products, in particular, a specific C2+ compound, is difficult to control. Some developments based on the design of homogeneous and heterogeneous catalysts have been achieved for selective conversions of C1 molecules into C2+ compounds. For example, the design of bi(multi)functional catalysts or catalytic systems that work in tandem have shown great potential for the synthesis of C2–C4 olefins, aromatics and C2+ oxygenates by hydrogenation of CO and CO2.8,13–15,27–38Nevertheless, the activation of stable C1 molecules (e.g., CO2 and CH4) under mild conditions and the precise C–C coupling of C1 molecules remain two of the most challenging research targets in chemistry.
Photocatalysis and electrocatalysis have been emerging as promising tools for the activation and selective conversion of C1 molecules into various types of products under mild conditions.39–42 The transformation of C1 molecules by photocatalysis/electrocatalysis has become a booming research field, and the number of academic publications in the field has increased enormously over the past decade (Fig. 1). At the same time, many review articles have been published on either photocatalytic or electrocatalytic conversions of a single C1 molecule, in particular CO2.43–56 However, to the best of our knowledge, so far few critical reviews have been contributed to photocatalytic/electrocatalytic conversions of multiple C1 molecules with a focus on C–C coupling.
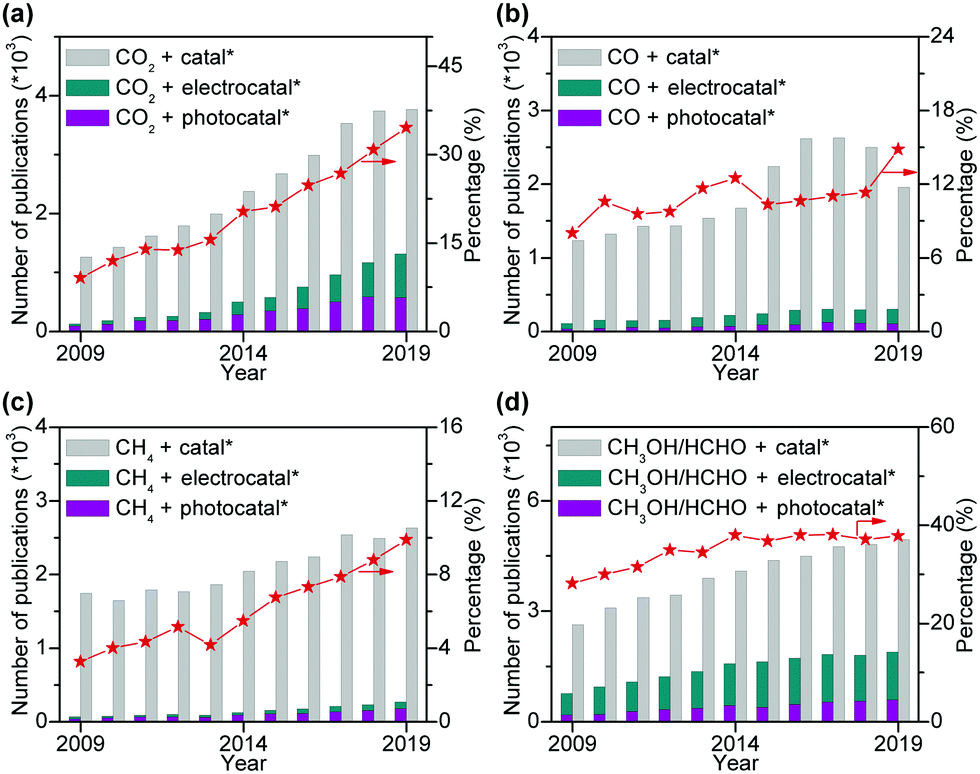 | ||
| Fig. 1 Numbers of annual publications and percentages of electrocatalysis and photocatalysis for different C1 molecules based on Web of Science with keywords of C1 molecules plus “catal*”, “electrocatal*” and “photocatal*”. (a) CO2. (b) CO. (c) CH4. (d) CH3OH and HCHO. | ||
The present article is devoted to reviewing recent advances in the photocatalytic/electrocatalytic conversions of typical C1 molecules including CO2, CO, CH4, CH3OH and HCHO into C2+ compounds involving C–C coupling (Fig. 2). The products include both C2+ hydrocarbons (e.g., C2H4, C3H6 and C2+ alkanes) and C2+ oxygenates (e.g., ethanol, acetate, n-propanol and ethylene glycol), which are important building blocks/synthetic intermediates in the current chemical industry or fuel additives/alternative fuels. The photocatalytic/electrocatalytic systems that are capable of catalysing the formation of C2+ compounds from C1 molecules and the active catalyst structures are summarized and analysed to provide insights into the design of efficient catalysts. The mechanistic insights such as reactive species, reaction intermediates and reaction pathways are discussed to offer in-depth understanding of the chemistry for the activation of C1 molecules and selective C–C coupling with solar or electrical energy. The challenges, opportunities and future trends in photocatalytic/electrocatalytic transformations of typical C1 molecules are further described to advance the emerging research area of solar energy- or electrical energy-driven C–C coupling of abundant C1 feedstocks to form high-value C2+ compounds.
 | ||
| Fig. 2 Major topics of the present article. | ||
2. Fundamental aspects of photocatalysis and electrocatalysis
The essence of chemical reactions is the rearrangement of chemical bonds, i.e., the breaking of some chemical bonds in reactants and the formation of new bonds in the products. This process usually needs to overcome an activation barrier. Thus, a sufficiently high temperature is required to overcome the activation energy without supply of external energy. For traditional thermocatalysis, the activation energy can be decreased by addition of a catalyst, which can chemically interact with the reactants, and thus can activate the reactants and change the reaction path. On the other hand, for photocatalysis and electrocatalysis, the supplied photo or electrical energy can be applied to the reactants via the photocatalyst/electrocatalyst and the reactants are then excited, and thus the reaction can proceed without the need to overcome a high activation energy. Furthermore, photocatalysis and electrocatalysis can also drive the thermodynamically unfavourable reactions that are impossible for thermocatalysis. Therefore, photocatalysis and electrocatalysis hold great potential to bring about breakthroughs toward new reactions or more efficient catalytic processes.The matching of the energy-band position in semiconductor photocatalysis or the electrode potential in electrocatalysis with the redox potentials of reactants is the key to accomplishing photocatalytic or electrocatalytic reactions due to the thermodynamic requirement. For a semiconductor-based photocatalytic reaction, the conduction-band edge should be higher (more negative) than the reduction potential of the reactant for the reduction half reaction, while the valence-band edge should be lower (more positive) than the oxidation potential of the reactant for the oxidation-half reaction (Fig. 3a). Similarly, for an electrocatalytic reaction, the cathode potential should be higher than the reduction potential and the anode potential should be lower than the oxidation potential of the reactants for the reduction- and oxidation-half reactions, respectively (Fig. 3b). The energy-band position of the semiconductor or the electrode potential can be tuned by choosing a suitable semiconductor or changing the applied potential. The redox potentials of some possible reactions related to the conversions of typical C1 molecules to some C1 and C2 compounds are displayed in Fig. 3c. They are all located in the range of −0.2 to 0.6 V versus normal hydrogen electrode (NHE) at 298 K and can be easily matched by controlling the energy-band structure of the semiconductor catalyst or the electrode potential. However, selectivity control would be difficult from a thermodynamic consideration, because the redox potentials for different products are very close. Therefore, it can be expected that kinetic control by designing proper photocatalysts/electrocatalysts is crucial to accomplish highly selective synthesis of specific C2+ compounds.
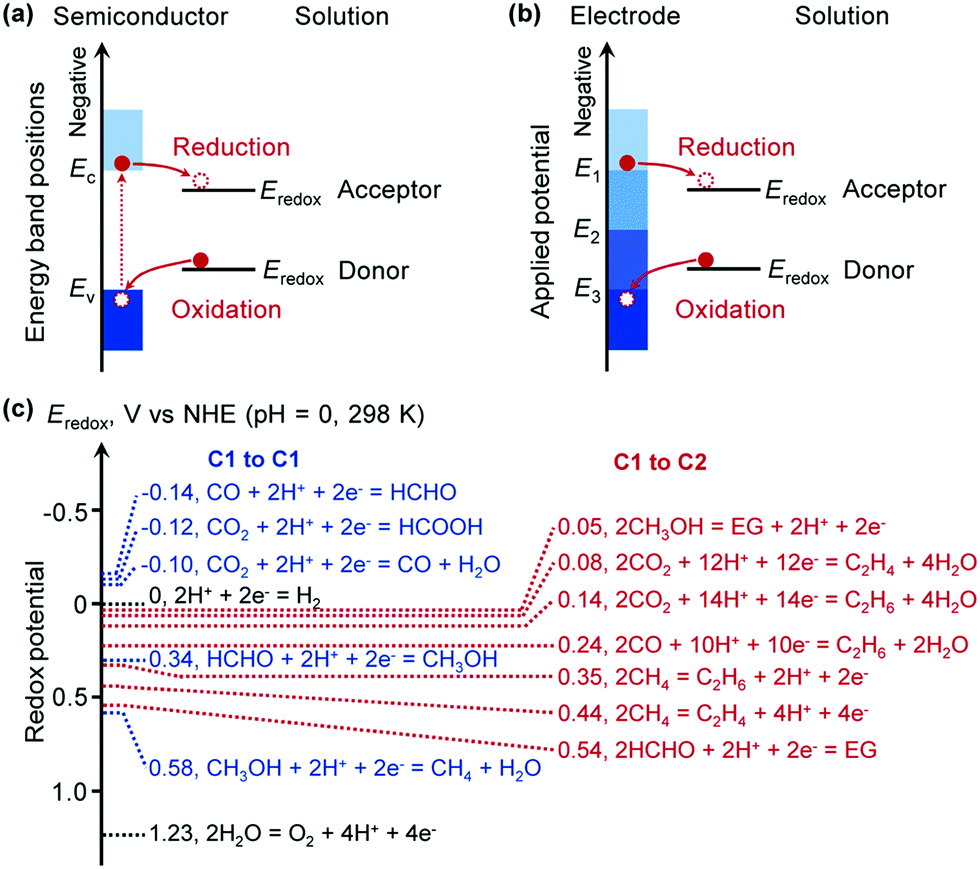 | ||
| Fig. 3 (a) Positions of semiconductor energy-band edges (Ec, Ev) and substrate redox potentials (Eredox), and the thermodynamics of electron transfer between a semiconductor and substrate. (b) Positions of electrode applied potentials (E1, E2, E3) and substrate redox potentials (Eredox), and the thermodynamics of electron transfer between an electrode and a substrate. (c) The redox potentials of some reactions related to the conversion of C1 molecules to other C1 or C2 products. The redox potentials are obtained from the thermochemical software “HSC Chemistry”. | ||
3. Photocatalytic conversions of CO2 and CO
3.1. Photocatalytic conversion of CO2
CO2 is one of the most stable molecules and also the most probable terminal state of carbon after full utilization of carbon resources. The natural photosynthesis using solar energy to convert CO2 and H2O to biomass is the principal step for the carbon cycle in nature.44 Many approaches have been proposed and developed for establishing anthropogenic chemical recycling of CO2 to make the carbon cycle more controllable.44 However, the chemical transformation of CO2 is very challenging due to the thermodynamic stability and kinetic inertness of CO2. Photocatalytic reduction of CO2 with H2O to organic compounds, also known as artificial photosynthesis, is an ideal route for the transformation of CO2 and the storage of solar energy in chemical bonds.45–50Since the pioneering work reported by Fujishima, Honda and their co-workers in 1979,57 a lot of semiconductors, such as TiO2, g-C3N4, BiVO4, Bi2WO6, CdS, SrNb2O6, NaTaO3, Zn2GeO4 and Zn2GaO4, have been reported for photocatalytic reduction of CO2 with H2O.45–50 The products in these systems are often C1 compounds, such as CO, HCOOH, CH3OH and CH4. The direct photocatalytic reduction of CO2 to longer carbon-chain products involving C–C coupling is more challenging not only due to the multi-electron/multi-proton transfer but also because of the requirement of C–C coupling active sites.58–61 Nevertheless, some advances have been achieved for the synthesis of C2+ compounds such as C2H4, C2H6 and C2H5OH by photocatalytic reduction of CO2. Some semiconductors, such as TiO2,62 g-C3N4,63 BiVO4,64 Bi2WO6,65 InTaO4,66 and CdS,67 have been reported to be capable of catalysing the formation of C2+ compounds, and some factors are known to play key roles in determining the catalytic performances. Here, we will analyse the effects of doping and co-catalysts, hybridisation of different semiconductors, hybridisation of semiconductors and carbon materials and the presence of plasmonic components on photocatalytic reduction of CO2 with H2O to C2+ compounds. Some typical photocatalysts and performances are summarized in Table 1.62–64,66–87
| Photocatalyst | Formation rate (μmol g−1 h−1) | Light source, reaction mode | Ref. |
|---|---|---|---|
| Effects of semiconductor types and doping | |||
| TiO2 (effect of electrolytes) | NaOH: CH4, 0.23; CH3OH, 2.5; HCOOH, 0.23; C2H5OH, 11; CH3CHO, 29 | Xe lamp, solid–liquid | 62 |
| H2O: CH4, 0.32; CH3OH, 0.04; HCOOH, 1.7; C2H4, 0.06 | |||
| Mesoporous flake-like g-C3N4 (u-g-C3N4) | u-g-C3N4: CH3OH, 6.3; C2H5OH, 4.5; O2, 21 | Xe lamp (>420 nm), solid–liquid | 63 |
| Non-porous flaky g-C3N4 (m-g-C3N4) | m-g-C3N4: C2H5OH, 3.6; O2, 10 | ||
| BiVO4 | Monoclinic: C2H5OH, 101 | Xe lamp (>400 nm), solid–liquid | 64 |
| Tetragonal: C2H5OH, 5.6 | |||
| 1 wt% Co-doped TiO2 | H2, 63; CH3OH, 26; C2H5OH, 18; CH3CHO, 19 | Halogen lamp (>380 nm), solid–vapour | 68 |
| Cu2+-Doped TiO2 | CH3OH, 24; C2H5OH, 47 | 365 nm LED lamp, solid–vapour | 69 |
| TiO2 | CH3OH, 6.7; C2H5OH, 6.3 | ||
| Hybridisation of semiconductors and a semiconductor with a nanocarbon material | |||
| 4 nm PbS QD–Cu/TiO2 | CH4, 0.58; CO, 0.82; C2H6, 0.31 | Xe lamp (>420 nm), solid–vapour | 70 |
| AgBr–TiO2 | CH4, 26; CH3OH, 16; CO, 6; C2H5OH, 2.7 | Xe lamp (>420 nm), solid–liquid | 71 |
| AgBr–NG–g-C3N4 | CH3OH, 21; C2H5OH, 51 | Xe lamp (>420 nm), solid–liquid | 72 |
| CNT–TiO2 | CH4, 12; HCOOH, 19; C2H5OH, 30 | UV lamp (365 nm), solid–vapour | 73 |
| GO–TiO2 | CH3OH, 12; C2H5OH, 145 | Hg lamp, solid–liquid | 74 |
| 2% G–TiO2 | CH4, 8.0; C2H6, 16.8 | Xe lamp, solid–vapour | 75 |
| 0.5G–TiO2−x | CH4, 7.4; C2H6, 1.2 | Solar simulator (AM 1.5), solid–vapour | 76 |
| 1% Pt–0.5G–TiO2−x | CH4, 37; C2H6, 11 | ||
| Carbon-coated Cu2O nanorods | CH4, 0.014; C2H4, 0.019; O2, 0.15 μmol h−1 | Xe lamp (>420 nm), solid–liquid | 77 |
| Effect of co-catalyst | |||
| 1% Pd/TiO2 | CH4, 0.37; CO, 0.033; C2H6, 0.067 | Hg lamp, solid–liquid | 78 |
| Nafion-modified Pd/TiO2 | CH4, 2.2; C2H6, 1.0 μmol h−1 | UV lamp (>300 nm), solid–liquid | 79 |
| Pd/TiO2 | CH4, 2.1; C2H6, 0.47 μmol h−1 | ||
| Rh nanowires/TiO2 | H2, 11; CH4, 4.5; CO, 14; C2H5OH, 12 | Xe lamp (<400 nm), solid–vapour | 80 |
| 2.6% NiO/InTaO4 | CH3CHO, 0.3 | Xe lamp (AM 1.5), solid–vapour | 66 |
| NiO/Na1−xLaxTaO3+x | H2, 0.2; CH4, 0.02; CH3OH, 60; C2H6, 0.01; C2H4, 0.14; C2H5OH, 19; CH3CHO, 2.2; C3H6, 0.4 | Hg lamp (300–700 nm), solid–liquid | 81 |
| CdS/(Cu–Na1.5H0.5Ti3O7) | CH4, 28; C2H6, 17; C2H4, 0.1; C3H8, 9.7; C3H6, 0.8 μL g−1 h−1 | Xe lamp (>420 nm), solid–liquid | 67 |
| 5% Cu/TiO2 | CH4, 0.29; C2H6, 0.02; C2H4, 0.54 μL g−1 h−1 | Xe lamp, solid–liquid | 82 |
| Cu–Pt/nitrogen-doped TiO2 nanotube array | H2, 160; CH4, 75; other alkanes, 25; olefin, 8.5; branched paraffin, 2.5 ppm cm−2 h−1 | Outdoor sunlight (AM 1.5), solid–vapour | 83 |
| Cu0.33Pt0.67/PMTiNT | CH4, 2.6; C2H6, 0.47; C2H4, 0.24 mL g−1 h−1 | Solar simulator (AM 1.5), solid–vapour | 84 |
| Cu(0.5 wt%)–Fe(0.5 wt%)/TiO2 | Glass plate: CH4, 0.06; C2H4, 0.05 | Hg lamp (320–580 nm), solid–vapour | 85 |
| Optical fiber: CH4, 0.91; C2H4, 0.58 | |||
| Plasmonic effect | |||
| Au/TiO2 | CH4, 231; CH3OH, 87; HCHO, 135; C2H6, 162 μmol m−2 | Hg lamp (254 nm), solid–vapour | 86 |
| Au nanoparticle in 5% EMIM-BF4 aqueous solution | TOF: CH4, 4.5; C2H6, 0.99; C2H4, 1.1; C3H8, 0.56; C3H6, 0.93 NP−1 h−1 | 532 nm laser, solid–liquid | 87 |
Visible-light responsive semiconductors have also been investigated for photocatalytic reduction of CO2 to C2+ compounds.63–67 For example, g-C3N4 was reported to catalyse the formation of C2H5OH together with CH3OH during the reduction of CO2 in 1.0 M NaOH solution under visible-light (λ ≥ 420 nm) irradiation.63 The yield of C2H5OH increased proportionally to the irradiation time together with those of CH3OH and O2 (Fig. 4). The morphology of g-C3N4 was found to influence the catalytic performance; a mesoporous flake-like g-C3N4 (denoted as u-g-C3N4) showed higher activity than a non-porous flaky g-C3N4 (denoted as m-g-C3N4). After 12 h of reaction, the yields of C2H5OH, CH3OH and O2 with u-g-C3N4 were 10.8, 15.1 and 51.2 μmol, respectively, whereas the yields of C2H5OH and O2 over m-g-C3N4 were 8.7 and 24.6 μmol, respectively. The mesoporous structure and large surface area of u-g-C3N4 might lead to higher charge separation ability and photocatalytic activity than m-g-C3N4. It is surprising that CH3OH and C2H5OH were both formed on u-g-C3N4, whereas only C2H5OH was observed on m-g-C3N4. It is speculated that the H+-deficient feature of the system, in particular m-g-C3N4, might favour the CO2 reduction and dimerization to form C2H5OH as mentioned in an early work.88 Considering that the formations of one mole of CH3OH and C2H5OH require 6 and 12 moles of electrons, respectively, and the formation of 1 mole of O2 from H2O requires 4 moles of holes, the ratio of the consumed electrons to holes in the photoredox reactions is nearly 1. This evaluation implies that the results may be reliable. Nevertheless, the confirmation of the source of products by using for example 13CO2 as the reactant is necessary for the carbon-containing photocatalysts or photocatalytic systems.
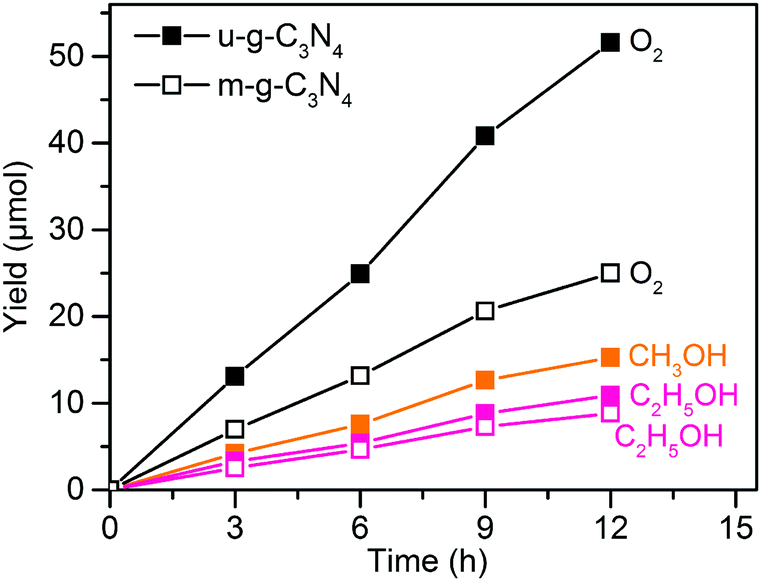 | ||
| Fig. 4 Photocatalytic conversion of CO2 with H2O to CH3OH, C2H5OH and O2 over u-g-C3N4 and m-g-C3N4 under visible-light irradiation.63 | ||
The doping of a metal or non-metal element is a useful strategy to tune the energy-band structure of semiconductors for CO2 photoreduction.48,49 Generally, the metal doping can create intra-band states below the conduction band, whereas the non-metal doping is capable of forming intra-band states above the valence band. Both may narrow the bandgap and extend the light absorption range. Moreover, the metal or non-metal doping may also generate surface vacancies or new active sites, thus affecting the CO2 activation and reduction. In this case, the metal dopant may also function like a co-catalyst.
Some studies have investigated the effect of metal or metal cation dopants, such as Cu, Fe, V, Cr, Co and Rh, in TiO2 on the photocatalytic reduction of CO2 to C2 compounds.68,69 As compared to the UV-light responsive TiO2, the cationic V-, Cr- and Co-doped TiO2 catalysts extended the absorption spectrum into the visible-light region, thus enabling the photocatalytic reduction of CO2 under visible-light irradiation.68 It was found that the Co2+-doped TiO2 showed higher photocatalytic activity than the other metal cation-doped catalysts, but the product selectivities for the V-, Cr- and Co-doped TiO2 were similar, and the major reduction products were H2, CH3OH, C2H5OH and CH3CHO.68 The performance was also strongly dependent on the doping amount of Co2+, and the formation rates of H2, CH3OH, C2H5OH and CH3CHO over the 1 wt% Co-doped TiO2 reached 63, 26, 18 and 19 μmol g−1 h−1, respectively.68 A Cu2+-doped TiO2 nanorod thin film catalyst was also claimed to be efficient for the gas-phase reduction of CO2 with H2O vapour to CH3OH and C2H5OH under UV-light irradiation.69 The reaction was carried out in a flow-type optofluidic planar microreactor, and the formation rates of CH3OH and C2H5OH with Cu2+-doped TiO2 reached 24 and 47 μmol g−1 h−1, respectively at a CO2 flow rate of 2 mL min−1 and 60 °C, whereas they were only 6.7 and 6.3 μmol g−1 h−1, respectively, with TiO2 alone. The increase in temperature to 80 °C could further increase the formation rates of CH3OH and C2H5OH to 36 and 79 μmol g−1 h−1, respectively.69 Besides the decreased band-gap energy and the extended light absorption, the doped Cu2+ ions were supposed to serve as active sites of electron traps and suppress the electron–hole recombination. Thus, the doped metal cation centre may also play a role as a co-catalyst, which will be discussed later.
 | ||
| Fig. 5 Schematic illustrations of hybridisation of two different semiconductors for photocatalytic CO2 reduction to C2+ compounds. (a) p–n junction. (b) Z-scheme. | ||
Some hybrid photocatalysts were reported for the photocatalytic reduction of CO2 to products including C2+ products such as C2H5OH and C2H6 together with C1 compounds.70,71,89 For example, the combination of PdS quantum dots (QDs) with TiO2 could increase the rate of CO2 photocatalytic reduction by a factor of 5 because of the sensitisation effect of PbS QDs, which made the utilisation of visible and near-infrared light possible.70 CO, CH4 and C2H6 were the major products over the PbS–TiO2 nanocomposite. Similarly, AgBr–TiO2 could catalyse the reduction of CO2 to CH4, CH3OH, CO and C2H5OH under visible-light irradiation because of the sensitisation effect of AgBr.71 The light-harvesting complex II (LHCII), which was extracted from spinach and functioned as a sensitizer, was combined with Rh-doped TiO2 for photocatalytic reduction of CO2 in aqueous solution under visible-light irradiation, offering acetaldehyde, CO and methyl formate as major products.89 The formation rates of CO, acetaldehyde and methyl formate were 0.57, 3.2 and 0.26 μmol g−1 h−1. Han, Niu and their co-workers loaded AgBr nanoparticles onto g-C3N4-decorated N-doped graphene (NG) and found that this nano-composite showed excellent performance for the photocatalytic reduction of CO2 to C2H5OH under visible-light irradiation.72 The reaction was proposed to proceed by the scheme of Fig. 5a and the reduction of CO2 took place on AgBr nanoparticles. The rate of C2H5OH formation reached 51 μmol g−1 h−1, 2.4 times that of CH3OH over the ternary composite.72
Nanocarbon materials, such as graphene (G), graphene oxide (GO) and carbon nanotubes (CNTs), have been widely used to fabricate hybrid photocatalysts, owing to their high conductivity, good electron mobility, favourable Fermi levels and large surface areas.17 The nanocarbon can serve as an electron collector or transporter to enhance the charge separation, an active surface to facilitate the adsorption of reactants or a protector to inhibit semiconductor photo-corrosion. Some nanocarbon–semiconductor hybrid photocatalysts, such as CNT–TiO2, GO–TiO2, G–TiO2, G–TiO2−x and carbon–Cu2O, could offer C2+ compounds during photocatalytic reduction of CO2.73–77
A CNT–TiO2 nanocomposite was reported to provide CH4, HCOOH and C2H5OH as the major products during the reduction of CO2 with H2O vapour under UV-light (λ = 365 nm) irradiation, and the formation rate of C2H5OH reached ∼30 μmol g−1 h−1, higher than those of CH4 (12 μmol g−1 h−1) and HCOOH (19 μmol g−1 h−1).73 A GO–TiO2 hybrid suspended in an aqueous medium was also claimed to offer C2H5OH together with CH3OH during the reduction of CO2 under UV-vis-light irradiation.74 It was found that the pH value of the aqueous solution influenced the products; CH3OH was the major product at a lower pH value, whereas C2H5OH became the major product at a higher pH value. The formation rate of C2H5OH reached ∼145 μmol g−1 h−1 at a pH of 11.74 It was speculated that the pH value of the aqueous solution might affect the point of zero charge of the catalyst surface and thus the interaction between carbonate species and the surface.74 It is, however, surprising that C2H5OH was also reported to be the major product with a formation rate of ∼110 μmol g−1 h−1 over TiO2 (P25) alone. Such a high C2H5OH formation rate for TiO2 alone has not been reported by other groups.
On the other hand, Zou and co-workers showed that a G–TiO2 hybrid photocatalyst with 2D sandwich-like nanosheet morphology could function for the reduction of CO2 with H2O vapour to C2H6 along with CH4 under UV-light irradiation.75 The G–TiO2 nanocomposite was fabricated by a simultaneous reduction-hydrolysis strategy in ethylenediamine/H2O solvent, by which the reduction of GO to graphene by ethylenediamine and the hydrolysis of Ti4+ to TiO2 took place simultaneously. The pure TiO2 synthesized by the same procedure showed a better performance for CO2 reduction than P25 probably owing to the abundant Ti3+ sites on TiO2, and the formation rates of CH4 and C2H6 were 10 and 7.2 μmol g−1 h−1, respectively. Upon increasing the content of graphene in G–TiO2 from 0 to 2 wt%, the formation rate of CH4 somewhat decreased but that of C2H6 increased significantly (Fig. 6a).75 Although a further increase in the content of graphene decreased the formation rate of the total products, the molar ratio of C2H6 to CH4 increased monotonically with the content of graphene to 5 wt%, suggesting that the introduction of graphene favoured the formation of C2H6. The 2% G–TiO2 offered the highest total product formation rate, and the formation rates of CH4 and C2H6 reached 8.0 and 16.8 μmol g−1 h−1, respectively. Both Ti3+ sites on the TiO2 surfaces and the presence of graphene are proposed to play key roles in the photosynthesis of C2H6 from CO2. It is speculated that the abundant surface Ti3+ sites can work for trapping photogenerated electrons and the electrons may be further accumulated on graphene, both favouring the multi-electron reduction of CO2 to CH3˙. The electron-rich graphene may also be beneficial for stabilising CH3˙, the reaction intermediate, and thus limit the combination of CH3˙ with H+ and e− into CH4. At the same time, the accumulation of CH3˙ on graphene may contribute to increasing the probability of coupling of CH3˙ to form C2H6.75
 | ||
| Fig. 6 (a) Photocatalytic reduction of CO2 to CH4 and C2H6 over a graphene–TiO2 hybrid under UV-light irradiation.75 (b) Schematic illustration of photocatalytic reduction of CO2 over a graphene–TiO2 hybrid.76 | ||
In and co-workers fabricated graphene-wrapped reduced blue titania (G–TiO2−x) loaded with a small amount of Pt nanoparticles and found that this nanocomposite showed a high formation rate of C2H6 and CH4 in the photocatalytic reduction of CO2 in a continuous flow-through (CO2, H2O) photo-reactor under one sun AM1.5G illumination.76 P25 was inactive under such circumstances and TiO2−x catalysed the formation of CH4 with a rate of 1.0 μmol g−1 h−1. The presence of graphene not only enhanced the formation of CH4 but also induced the formation of C2H6, and the loading to a small amount of Pt could further accelerate the formations of both CH4 and C2H6. The formation rates of CH4 and C2H6 could reach 37 and 11 μmol g−1 h−1, corresponding to CH4 and C2H6 selectivities of 77% and 23%, respectively.76 This work provided solid evidence for the formation of CH4 and C2H6 from CO2 by 13CO2 isotopic control experiments. A CH3˙ radical was proposed to be the intermediate for the formations of both CH4 and C2H6. In other words, CH4 is formed by the reaction of CH3˙ with H+ and e−, whereas the coupling of two CH3˙ radicals gives C2H6. However, different from Zou and co-workers,77 In and co-workers proposed through transient absorption spectroscopy studies that photogenerated holes rather electrons migrate to graphene, where the oxidation of H2O occurs, whereas the reduction of CO2 takes place on TiO2−x surfaces, where the electrons accumulate.76 Nevertheless, In and co-workers also speculated that graphene might stabilise CH3˙ radicals, enabling the increased probability for C2H6 formation (Fig. 6b).
Yu et al. reported that the coating of a carbon layer onto mesoporous Cu2O nanorods deposited on Cu foils could also enhance the photocatalytic reduction of CO2, but they observed the formation of CH4 and C2H4.77 The photocatalytic reaction was carried out in KHCO3 aqueous solution under visible-light irradiation. Cu2O nanorods on Cu foils alone could offer CH4 and C2H4 with almost the same amount, but the amounts of CH4 and C2H4 did not increase with time after ∼6 h of reaction because of the photo-corrosion of Cu2O. The coating of a carbon layer enhanced the formation of C2H4, the amount of which became ∼1.3 times that of CH4, and the amounts of C2H4 and CH4 kept increasing with time at least in 12 h over the carbon-coated Cu2O nanorods. The total amounts of C2H4 and CH4 were 0.037 μmol after 12 h of reaction. The carbon layer was proposed to work as a protective layer to quench the photo-corrosion of Cu2O and to extract photogenerated electrons, thus accelerating the separation of electron–hole pairs and the transfer of a multitude of electrons for the formation of C2H4. Although the formation rates of C2H4 and CH4 reported in this work were quite low, it is noteworthy to point out that the 13C-labelling isotopic experiment using 13CO2 and the measurement of the evolved O2 provided solid evidence that C2H4 and CH4 were produced by the reduction of CO2 in such a carbon-containing catalyst system.77
Noble and coinage metals (such as Pt, Pd, Rh, Au, Ag and Cu) have been used as co-catalysts for TiO2-based photocatalytic reduction of CO2 with H2O.49,92 Among the noble and coinage metal co-catalysts, Pt is known to be the most efficient in extracting electrons from a semiconductor and in promoting CO2 reduction, but the reduction of H2O to H2 is also enhanced at the same time because of the fast recombination of H atoms on Pt. A co-catalyst that can efficiently extract conduction-band electrons but has lower ability of H atom recombination to H2 is beneficial for CO2 reduction.
Pd is a good candidate of such a co-catalyst because of the retarded recombination of H atoms on Pd surfaces. Ishitani and co-workers found that the photocatalytic reduction of CO2 in aqueous solution with TiO2 suspended provided CO as the major product and the loading of Pd onto TiO2 remarkably enhanced the formation of CH4 (Fig. 7a).78 It is noteworthy that the formation of C2H6 was observed over the Pd/TiO2, although the rate of C2H6 formation was about 6 times lower than that of CH4 (Fig. 7a).78 The acid/base property of the support, which was used to load the semiconductor, was found to influence the selectivity of photocatalytic reduction of CO2 to C1 or C2+ products.93 This result implied that the introduction of an acidic support favoured the formation of C1 products, whereas a basic support such as MgO or basic promoter (e.g., Li+) was beneficial for the formation of C2+ products such as C2H6, which were supposed to be formed by dimerization of radical intermediates (e.g., CH3˙).93 On the other hand, Choi and co-workers demonstrated that the introduction of a thin layer of acidic Nafion (perfluorinated polymer with sulfonated groups) onto Pd/TiO2 could significantly enhance the photocatalytic reduction of CO2 in aqueous solution with different pH values not only to CH4 but also to C2H6 (Fig. 7b).79 In particular, the effect of Nafion was still remarkable for the formation of C2H6 even at a pH value of 1, whereas the enhancement in CH4 formation due to the Nafion layer was insignificant at such a low pH value. A small amount of C3H8 was detected over the Nafion-modified Pd/TiO2 but not over the Pd/TiO2. The formation of C2H6 and C3H8 requires more protons and electrons than that of CH4. Choi and co-workers strengthened the crucial role of proton-coupled multi-electron transfer in the formation of C2H6 and C3H8. It is speculated that at a lower pH value (pH = 1), protons may be sufficient at the surface active site for the formation of CH4 without the need of Nafion.79 The presence of a Nafion layer accelerated the formation of C2H6 and C3H8 even at an acidic environment (pH = 1) because the Nafion layer might enhance the local proton activity to facilitate the proton-coupled multi-electron transfer processes. The Nafion layer might also stabilise the intermediates (probably radicals) involved in the formation of C2H6 and C3H8.
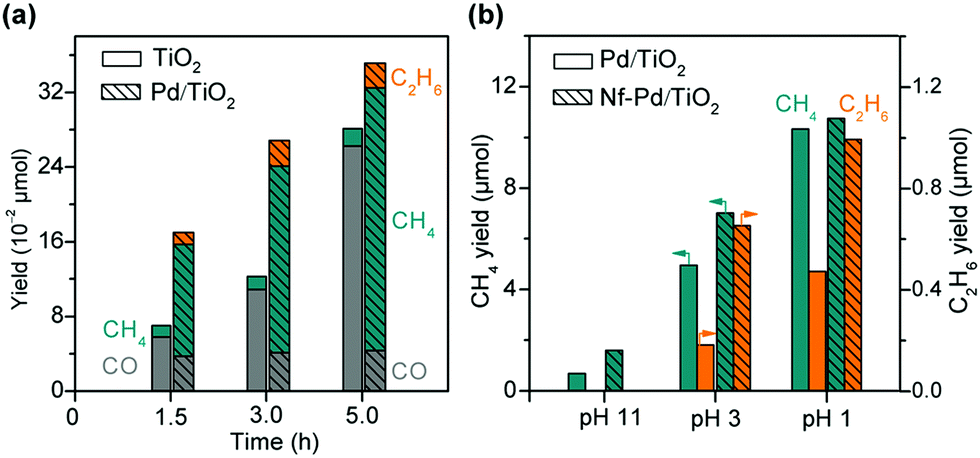 | ||
| Fig. 7 (a) Photocatalytic reduction of CO2 over TiO2 and 1% Pd/TiO2.78 (b) Effect of Nafion (Nf) layer deposited on Pd/TiO2 on photocatalytic reduction of CO2.79 | ||
The morphology of a co-catalyst may affect the interface between the co-catalyst and semiconductor, thus influencing the photocatalytic reduction of CO2. Rh and Pd nanowires loaded onto TiO2 nanosheets were found to be more efficient co-catalysts than the corresponding nanoparticles for photocatalytic reduction of CO2 with H2O under UV-light irradiation.80 The use of nanowire co-catalysts reduced the H2 evolution and enhanced the reduction of CO2. The larger density of grain boundaries between the co-catalyst and TiO2 in the nanowire-based photocatalyst was proposed to be the key. CO, CH4 and C2H5OH were the major products, and the rates of CO, CH4 and C2H5OH formation reached 4.5, 14 and 12 μmol g−1 h−1 over the Rh-nanowire-loaded TiO2 nanosheet catalyst.80 The molar ratios of CO, CH4 and C2H5OH were comparable between Rh nanowire- and Rh nanoparticle-based catalysts.
NiO was reported to be an efficient co-catalyst for photocatalytic reduction of CO2 in a few studies.66,81 NiO/InTaO4 photocatalysts with different NiO contents were investigated for photocatalytic reduction of CO2 with H2O vapour in a monolith reactor with optical fibres inserted inside, i.e., an internally illuminated monolith reactor.66 It is of interest that acetaldehyde was formed with a considerable amount under UV-light or simulated-sunlight irradiation. The formation rate of acetaldehyde increased with the content of NiO and reached 0.21 μmol g−1 h−1 over the 2.6 wt% NiO/InTaO4 catalyst at 30 °C under AM1.5G irradiation.66 The increase in temperature to 70 °C slightly increased the formation rate of acetaldehyde to 0.30 μmol g−1 h−1. A NiO/Na1−xLaxTaO3+x catalyst was reported to offer CH3OH and C2H5OH during photocatalytic reduction of CO2 in 0.2 M NaOH aqueous solution under UV-light irradiation.81 The rates of CH3OH and C2H5OH reached 59.6 and 18.8 μmol g−1 h−1, respectively over the NiO/Na1−xLaxTaO3+x catalyst, whereas they were 35.0 and 6.4 μmol g−1 h−1, respectively in the absence of NiO. Thus, NiO might play a crucial role in accelerating the formation of C2 oxygenates by increasing the charge separation and C2 oxygenate selectivity.
Among noble or coinage metals, Cu is a very attractive co-catalyst that can accelerate the formation of C2+ hydrocarbons.58,67,94 An early study showed that Cu-loaded TiO2 powders suspended in aqueous solution, which was pressurised CO2 (2.8 MPa), could catalyse the reduction of CO2 into CH4, C2H4 and C2H6 with yields of 21.8, 26.2 and 2.7 μL g−1 under optimised conditions.82 A Cu2+-doped TiO2 nanorod thin film catalyst was recently reported to offer CH3OH and C2H5OH as the major products during the gas-phase reduction of CO2 with H2O vapour under UV-light irradiation in a flow-type optofluidic planar microreactor.69 The formation rates of CH3OH and C2H5OH reached 24 and 47 μmol g−1 h−1, respectively. For a nitrogen-doped TiO2 nanotube array, the presence of Cu as a co-catalyst enhanced the formation of hydrocarbons including CH4 and C2–C6 paraffins as well as minor olefins during the photocatalytic reduction of CO2 with H2O vapour under outdoor sunlight.83 On the other hand, the employment of a Pt co-catalyst mainly enhanced the formation of H2 by accelerating the reduction of H2O under the same reaction circumstance.
Hoffmann and co-workers reported an interesting CdS/Cu–TNTs photocatalyst fabricated by coating CdS QDs on Cu-deposited sodium trititanate nanotubes (TNTs, NaxH2−xTi3O7) and found that this ternary photocatalyst was capable of working for photocatalytic reduction of CO2 in aqueous solution to C1–C3 hydrocarbons under visible-light (λ ≥ 420 nm) irradiation.67 Over the ternary catalyst, photogenerated electrons can transfer from the CdS QDs to the Cu particle surface through the titanate nanotube framework and the reduction of CO2 to C1–C3 hydrocarbons may mainly proceed on the Cu surfaces. The formation rates of CH4, C2H6 and C3H8 were 28, 17 and 9.7 μL g−1 h−1, and minor amounts of C2H4 (0.1 μL g−1 h−1) and C3H6 (0.8 μL g−1 h−1) were also formed. The isotopic labelling experiments using 13CO2 confirmed that most of the hydrocarbons were formed by the reduction of CO2, and a part of CH4 may be formed by organic compound contamination.67 Cu was found to be crucial in the formation of C2 and C3 hydrocarbons. Hoffmann and co-workers performed a mechanistic study using the CdS/Cu–TNTs catalyst. The formation of CH3˙ was confirmed through the EPR characterization of the formed radicals using DMPO as a spin trapping reagent. It is speculated that CO2 reduction is initiated most likely by a one-electron reduction to CO2˙−, which then reacts with H˙ to produce hydrocarbons through CH3˙ radicals (Fig. 8).67 The Fischer–Tropsch route for the formation of hydrocarbons may not occur in the present system because no evolution of H2 and CO was observed at the low temperature. Lower-intensity of signals ascribed to CH3˙ was observed in the absence of Cu, suggesting that Cu played a key role in accelerating the formation of CH3˙.
 | ||
| Fig. 8 Proposed reaction pathways for photocatalytic reduction of CO2 to hydrocarbons over the CdS/Cu–TNTs catalyst.67 | ||
As compared to a single metal co-catalyst, a bimetallic or alloy co-catalyst may have potential to further improve the catalytic activity and/or selectivity of a semiconductor photocatalyst. Grimes and co-workers co-loaded Cu and Pt nanoparticles on nitrogen-doped titania nanotube arrays (NT), and conducted photocatalytic reduction of CO2 under outdoor sunlight irradiation.58,83 The formation rates of H2 and hydrocarbons were 61 and 104 ppm cm−2 h−1 over the Cu/NT, respectively, whereas they were 190 and 82 ppm cm−2 h−1 over the Pt/NT.83 The formation rate of hydrocarbons over the Cu–Pt/NT was 111 ppm cm−2 h−1, which was close to that over Cu/NT, while the formation rate of H2 was 160 ppm cm−2 h−1 over the bimetallic catalyst, close to that of Pt/NT. The co-loading of Cu and Pt nanoparticles did not show a significant synergistic effect on tuning the selectivity of C2+ hydrocarbons. The fabrication of a core–shell structured Pt@Cu co-catalyst with Pt as the core and Cu as the shell to cover Pt could promote both the extraction of photogenerated electrons by Pt and the reduction of CO2 to CH4.95 Shankar and co-workers synthesized CuPt alloy co-catalysts with different Cu/Pt molar rations on a periodically modulated double-walled TiO2 nanotube (PMTiNT) array and found that the Cu0.33Pt0.67/PMTiNT showed the highest performance for photocatalytic reduction of CO2 to CH4, C2H4, and C2H6 under simulated one-sun AM1.5 illumination.84 It is of interest that the present system could work for the reduction of not only concentrated CO2 (99.9%) but also diluted CO2 (0.998%). The formation rates of CH4, C2H4 and C2H6 over Cu0.33Pt0.67/PMTiNT reached 2.60, 0.24 and 0.47 mL g−1 h−1, respectively.84 In contrast, the formation rates of hydrocarbons over the Cu/PMTiNT and Pt/PMTiNT were both quite a bit lower (0.61 and 0.20 mL g−1 h−1, respectively), further suggesting the role of the CuPt alloy co-catalyst in photocatalytic reduction of CO2. A recent work by In and co-workers further adopted the bimetallic Cu–Pt nanoparticles as co-catalysts of blue titania (BT) for photocatalytic reduction of CO2 with H2O vapour in a flow-through reactor under AM1.5 illumination.96 It was found that the configuration of bimetallic nanoparticles had a significant effect on catalytic performance; the deposition of Cu atop Pt particles directly attached on blue titania was optimal. Photogenerated electrons were believed to be extracted from blue titania to Pt, due to the interfacial charge transfer phenomenon, which was confirmed by photoluminescence spectroscopy. All extracted photogenerated electrons were then transferred to Cu, resulting in an enriched electron density within Cu, and CO2 was activated and reduced on Cu surfaces. Over a 6 h period, the Cu1.00%–Pt0.35%–BT catalyst offered CH4 and C2H6 with yields of 3.0 and 0.15 mmol g−1, respectively, and the sunlight to fuel (CH4 and C2H6) photoconversion efficiency reached 1% with an apparent quantum yield of 86%.96 This sunlight to fuel (CH4 and C2H6) photoconversion efficiency is significantly higher than those reported before (∼0.03%).58
In addition, a Cu and Fe co-doped TiO2 photocatalyst was studied for photocatalytic reduction of CO2 with H2O vapour under UV-light irradiation.85 The photocatalyst was intriguingly coated on optical fibres, which were assembled into a flow reactor. This configuration of reactor showed significant advantages in improving the CO2 reduction activity. The presence of Fe together with Cu was found to suppress the formation of CH4 but enhanced that of C2H4, and the Cu(0.5 wt%)–Fe(0.5 wt%)/TiO2 catalyst offered a C2H4 formation rate of 0.58 μmol g−1 h−1.
 | ||
| Fig. 9 (a) Photocatalytic reduction of CO2 over TiO2, Au/TiO2 and Au under light irradiation with different wavelengths.86 (b) Hydrocarbon selectivity and total hydrocarbon turnover frequency in photocatalytic reduction of CO2 over plasmonic Au nanoparticle photocatalysts in aqueous solutions with different concentrations of EMIM-BF4.87 | ||
The alloying effect such as the loading of Au–Cu or Au–Pd alloy onto a semiconductor could further accelerate the photocatalytic reduction of CO2.98,99 For example, Kuang and co-workers found that the Au nanoparticles loaded on {101} facets of the well-defined truncated bipyramidal TiO2 nanocrystals offered mainly CO during the photocatalytic reduction of CO2 with H2O vapour.99 The use of an Au–Pd alloy instead of Au significantly decreased the selectivity of CO and increased that of hydrocarbons. Over the Au6Pd1/TiO2 {101} catalyst, the selectivities of CH4 and C2 hydrocarbons (C2H6 and C2H4) were 71% and 14%, respectively. It was argued that selectivity depended on the synergistic functions of several parameters including: (i) the LSPR effect of Au, (ii) the hydrogenation performance of Pd and (iii) the adsorption–desorption behaviour of the reaction intermediate (COads) on the Au and Pd sites.99 These parameters enable the optimisation of activity and selectivity by tuning the elemental composition of the alloy co-catalyst.
Jain and co-workers reported the use of semiconductor- or support-free colloidal Au nanoparticles with diameters of ∼12 nm and a characteristic LSPR band centred at 520 nm for photocatalytic reduction of CO2 under visible-light irradiation in aqueous solution.100 In the presence of isopropanol, which acted as a sacrificial reagent to scavenge holes, the reduction of CO2 by the photo-excited electrons generated on the oxide- or support-free Au nanoparticles could offer CH4 and C2H6 as major products. The formation rates of CH4 and C2H6 were 0.68 and 0.56 NP(nanoparticle)−1 h−1. Both the photon energy and the light intensity play crucial roles in C–C coupling. No C2H6 was observed using 532 nm-light irradiation, and under 488 nm-light irradiation, there existed an onset of C2H6 generation at a light intensity of 300 mW cm−2, above which the formation rate of C2H6 increased with an increase in light intensity.100 CO2˙− or its hydrogenated form is proposed to be the intermediate, which undergoes a series of hot-electron- and proton-transfer steps to form products. The C–C coupling was proposed to occur between the two CO2˙− pair. The occurrence of C–C coupling requires simultaneous harvesting of two electrons, and thus higher rate of hot electron transfer and higher light intensity.
Considering that an ionic liquid, i.e., 1-ethyl-3-methylimidazolium tetra-fluoroborate (EMIM-BF4) could stabilise the high-energy CO2˙− intermediate, Yu and Jain further adopted aqueous solution for photocatalytic reduction of CO2 using the plasmonic Au nanoparticle under green light (532 nm) irradiation.87 As shown in Fig. 9b, the activity and selectivity of CO2 reduction depended on the concentration of EMIM-BF4.87 The activity was very low in pure water or EMIM-BF4. The presence of EMIM-BF4 in water significantly accelerated the photocatalytic reduction of CO2 and the highest activity was found in 5 mol% EMIM-BF4 solution. Besides CH4, C2 (C2H4 and C2H2) and C3 (C3H8 and C3H6) were also formed with considerable amounts. The EMIM-BF4 concentration affected the selectivity of C–C coupling products. The selectivity of C2+ hydrocarbons was ∼50% in 1–10 mol% EMIM-BF4 solutions, and it decreased to ∼20% in 25 mol% EMIM-BF4 solution. The 13C isotope labelling experiment and the fluorogenic test confirmed that the hydrocarbons were generated from CO2 and that the photogenerated holes oxidized H2O to H2O2 instead of O2. The studies further showed that EMIM-BF4 enhanced the activation of CO2 and promoted the electron transfer from Au nanoparticles to CO2, thus improving the performance for CO2 reduction. Moreover, the CO2˙− intermediate formed at the Au nanoparticle/solution interface may be stabilized due to the complexation by EMIM+ ([EMIM*–CO2]) and the higher concentration of the activated [EMIM*–CO2] complex would lead to higher probability of C–C coupling. The drops in activity and selectivity at higher EMIM-BF4 concentrations might arise from the fact that the adsorption of BF4− on Au nanoparticles inhibited the adsorption of CO2 and the transfer of electrons to CO2.
 | ||
| Fig. 10 (a) Possible adsorption structures of CO2 on TiO2 surfaces.106 (b) Proposed mechanisms for photocatalytic CO2 reduction involving C–C coupling. | ||
After CO2 activation, a series of elementary steps may take place including the transfer of electrons and protons, formation of intermediates, the cleavage of the C–O bond, and the creation of C–H and C–C bonds, resulting in C2+ products. Many studies have been devoted to studying the mechanism for CO2 photocatalytic reduction, but efforts focusing on C–C coupling to C2+ compounds are limited. Most studies reported to date have proposed that the adsorbed CO2 first accepts an electron to form surface CO2˙−. Several possible routes have been proposed for the subsequent conversion of CO2˙− and C–C coupling. These routes include the oxalic acid pathway, glyoxal pathway and methyl radical pathway (Fig. 10b). In the oxalic acid pathway, the C–C coupling is proposed to occur between the CO2˙− pair to oxalate (C2O42−), and oxalic acid is a primary C2 product, which can further be reduced to other C2+ compounds.59,100,107,108 Eggins and co-workers found the formation of C2O42− as the major product together with glyoxylate, glycolate and tartrate during the photocatalytic reduction of CO2 on ZnS in the presence of tetramethylammonium chloride, providing evidence for the oxalic acid pathway.107,108 In the glyoxal pathway, CO2˙− is converted to formyl radicals (˙CHO), which can not only be reduced to C1 derivatives but also be dimerized into glyoxal, followed by further reduction to other C2+ compounds. Shkrob and co-workers investigated the intermediates on TiO2 for CO2 photocatalytic reduction by ESR spectroscopy and the result suggested that ˙CHO is the intermediate of glyoxal.109 Glyoxal can be reduced to glycolaldehyde and acetaldehyde, which may undergo oxidation and decarbonylation to CH3˙, the intermediate of CH4 or C2H6. CH3˙ radicals may also be generated through other paths,45,104 and the formation of CH3˙ has been confirmed through the ESR characterization.67 CH3˙ has been proposed as the key intermediate for C–C coupling to form C2H6 in many studies, although it may also react with H˙ or ˙OH to yield CH4 or CH3OH (Fig. 10b).
3.2. Photocatalytic conversion of CO
CO can be transformed into C2+ chemicals and fuels by thermocatalysis via hydrogenation.6–8 Fischer–Tropsch synthesis is the most intensively studied heterogeneous reaction for hydrogenation of CO to C2+ hydrocarbons, but the reaction should be performed at high temperatures and pressures, and suffers from the limited selectivity of a specific C2+ hydrocarbon.7 The most accepted scenario for C2+ formation on Fischer–Tropsch catalyst surfaces is schematically illustrated in Fig. 11. In brief, the adsorption and dissociation of CO and H2 on catalyst surfaces lead to the generation of CHx (x = 1–3) species, which undergo C–C coupling to form surface CnHm intermediates (n ≥ 2), followed by the dehydrogenation or hydrogenation to olefins or paraffins, respectively.7 The uncontrollable C–C coupling of CHx species on Fischer–Tropsch catalysts causes a wide distribution of hydrocarbon products.7 Developing novel methods or new catalytic systems with enhanced activity and selectivity for the transformation of syngas to C2+ products has drawn great research interests.6–8 | ||
| Fig. 11 Simplified reaction mechanism for photo-driven CO hydrogenation. | ||
Solar energy-driven hydrogenation of CO or Fischer–Tropsch synthesis has recently attracted research attention.42 The solar light can either trigger the photo-thermal effect or induce free charge carriers on a catalyst, which may have potential to enhance the adsorption/activation of CO and H2 to increase the activity or to regulate the growth of C–C bonds and the termination of reactions to control the selectivity (Fig. 11). Co, Fe and Ru, which are typical Fischer–Tropsch catalysts owing to their balanced capacities for CO/H2 activation and C–C coupling,7 have mainly been studied for the hydrogenation of CO under light irradiation (Table 2).110–115
| Catalyst | External heating (°C) | Light wavelength (nm) | CO conv. (%) | CO2 selectivity (%) | Selectivity without CO2 (%) | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|
| CH4 | C=2–4 | C02–4 | C5+ | ||||||
| Co–Co3O4/ZnO–Al2O3 | No | 200–800 | 15.4 | 47.6 | 47.6 | 36.0 | 5.9 | 10.1 | 110 |
| Fe–Fe3O4/ZnO–Al2O3 | No | 200–800 | 20.9 | 11.4 | 28.6 | 42.4 | 9.0 | 8.6 | 111 |
| Fe5C2 | No | 200–1100 | 49.5 | 18.9 | 33.1 | 55.5 | 5.1 | 6.3 | 112 |
| Ru/graphene | 150 | 400–800 | 1.44 | — | 2.6 | — | 15.7 | 83.9 | 113 |
| 20% Co/TNT | 220 | UV | 63.9 | 17.3 | 34.6 | 0.3 | 22.4 | 42.7 | 114 |
| Ni-Based catalyst | No | 200–800 | 27.7 | 2.7 | 38.4 | — | 21.7 | 39.9 | 115 |
| Ni-Based catalyst | No | 400–800 | 20.9 | 2.0 | 31.5 | — | 23.3 | 45.2 | 115 |
Zhang and co-workers reported that under UV-vis-light irradiation without external heating, a Co-based catalyst, Co–Co3O4/ZnO–Al2O3, which was fabricated by H2 reduction of ZnCoAl-layered double-hydroxide (LDH) nanosheets at 450 °C, showed a CO conversion of 15.4% and C2–C4 olefin selectivity of 36.0% (ratio of olefins to paraffins, 6.1).110 This Co-based catalyst was found to exhibit strong light adsorption ability across the UV-vis region, and the temperature of the catalyst bed could increase rapidly from 25 to 195 °C by irradiating the catalyst under UV-vis light irradiation without external heating. It is proposed that the solar-driven CO hydrogenation with the present Co catalyst involves a photo-thermal rather than a photocatalytic pathway.110 Control experiments for thermocatalytic hydrogenation of CO at 195 °C without light irradiation showed a similar conversion of CO and distribution of hydrocarbon products, confirming that the photo-thermal effect contributed to the CO hydrogenation. The catalytic performance depended on the temperature adopted for H2 reduction, and it was clarified that the co-existence of Co3O4 and Co played a crucial role in hydrogenation of CO to lower olefins. The oxide-decorated metallic Co nanoparticle heterostructure was proposed to weaken the hydrogenation capability of metallic Co, leading to the formation of lower olefins over Co-based catalysts.
Using a similar strategy, Zhang and co-workers further fabricated a Fe-based catalyst, denoted as Fe–Fe3O4/ZnO–Al2O3, by H2 reduction of ZnFeAl-LDH nanosheets at 500 °C for photo-thermal catalytic hydrogenation of CO.111 This catalyst, which was composed of Fe0 and FeOx nanoparticles, showed a CO conversion of 21% and C2–C4 olefin selectivity of 42% at a syngas pressure of 0.18 MPa under UV-vis irradiation without external heating. The reaction could also be performed under visible-light irradiation, offering a CO conversion of 11% and C2–C4 olefin selectivity of 41%. It is of interest that the selectivity towards CO2 using this system is quite low (11% under UV-vis light irradiation) as compared with the traditional thermocatalysis. The heterostructure of Fe0 and FeOx may also play a crucial role in the hydrogenation. The temperature of the catalyst bed for Fe–Fe3O4/ZnO–Al2O3 increased to 230 °C under UV-vis light irradiation. The control experiments with external heating and without light irradiation showed a lower CO conversion (9.9%) compared to the light-driven hydrogenation of CO (21%).111 Furthermore, the selectivity of C2–C4 olefins was 37.2% under a thermocatalytic reaction, lower than that obtained in a solar-driven reaction, while the selectivity of CO2 was 60.8%, much higher than that under light-driven CO hydrogenation (Fig. 12a). DFT calculations revealed that the catalyst was in an excited state under light irradiation and had different ability for CO2 formation and adsorption/hydrogenation of olefin compared to the ground state under thermocatalytic conditions (Fig. 12b). Thus, the Fe-based system may involve a photocatalytic mechanism.
 | ||
| Fig. 12 (a) Product distributions in photo-driven CO hydrogenation over Fe–Fe3O4/ZnO–Al2O3 and Fe5C2. The filled bars are for the reaction performed under light irradiation, while the bars with slashes are for the reaction without light irradiation but with external heating.111,112 (b) The potential energy profile for CO2 production and olefin hydrogenation over ground-state and excited-state Fe–Fe3O4/ZnO–Al2O3.111 (c) Distributions of hydrocarbon products over Ru/graphene with or without light irradiation at different temperatures. The filled bars and bars with slashes are for the reactions performed under light irradiation and without light irradiation, respectively.113 (d) Proposed roles of photogenerated electrons in tuning the selectivity of photo-driven CO hydrogenation over Ru/graphene and Co/TNT.113,114 | ||
Ma and co-workers further employed Fe5C2 for photo-thermal hydrogenation of CO at atmospheric pressure under UV-vis light irradiation in the absence of external heating.112 The conversion of CO and the selectivity of C2–C4 olefins were 49.5% and 55.5%, respectively. This photo-thermal catalytic system was particularly favourable for the production of olefins and the ratio of C2–C4 olefins to paraffins reached 10.9. At the same time, the selectivity of CO2 was 18.9%, relatively lower than that reported in the conventional FTO process. This performance is quite surprising because usually heavy modification of Fe5C2 is required to gain considerable selectivity of lower olefins in the conventional thermocatalytic reaction operated under a high pressure. Fe5C2 was found to exhibit a remarkable photo-thermal effect, which might result from excitation of the surface plasmon band and the consequential fast thermal relaxation of the excitation, and the catalyst bed could reach 490 °C under UV-vis-light irradiation. The control experiments for thermocatalytic hydrogenation of CO over Fe5C2 at 300–500 °C showed that the CO conversion was low at a low temperature, and upon increasing the temperature gradually to 500 °C, CO conversion reached 80.5%, but CH4 was the predominant product with a CO2 selectivity of 36% and no C2–C4 olefins were formed (Fig. 12a).112 It was clarified that the Fe5C2 surface was oxidized to oxides at the high temperature under traditional thermal catalytic reaction conditions. On the other hand, the characterisation indicated that the surface of the Fe5C2 catalyst was partially decorated by O atoms under light irradiation. The DFT calculations further revealed that the O-decorated Fe5C2 surface could accelerate the desorption of olefin molecules, thus increasing the selectivity of C2–C4 olefins.
Photo-irradiation of a Ru/graphene catalyst was found to accelerate the hydrogenation of CO to C5+ hydrocarbons.113 The nanostructured worm-like Ru dispersed on graphene sheets could catalyse the hydrogenation of CO under visible-light irradiation under relatively mild reaction conditions (150 °C, 2.0 MPa CO and 1.0 MPa H2), and the catalytic activity reached 14.4 molCO molRu−1 h−1, significantly higher than that in the absence of photo-irradiation (7.8 molCO molRu−1 h−1).113 The reaction rate was found to be dependent on the light intensity or the light wavelength, confirming that light irradiation played a key role in the enhancement of CO conversion activity. Furthermore, at the same temperature, the selectivity of the C5+ hydrocarbons was higher under irradiation than that without irradiation (Fig. 12c). It was claimed that the enhancement in CO conversion to C5+ hydrocarbons mainly resulted from the light-excited hot electrons in Ru nanoparticles. The injecting of photo-excited hot electrons into the 2π* orbital of CO molecules can accelerate the activation of CO molecules, thus increasing the rate of CO dissociation. Furthermore, it is speculated that the excited electrons under light irradiation may increase the unsaturation of the d-band of Ru metal, which results in the donation of electrons from surface CHx species to the metal centre and decreases the Pauli repulsion between the CHx species and CnHm intermediates (Fig. 12d).113 The decrease in the Pauli repulsion may increase the chance of chain growth, resulting in high selectivity of the C5+ product.
TiO2 nanotube (TNT)-supported Co catalysts were examined for the hydrogenation of CO under UV-light illumination at 220 °C and syngas pressure of 2.0 MPa.114 Under such conditions, the thermocatalytic conversion of syngas could also occur, but the UV-light illumination was found to enhance the CO conversion. For example, the CO conversion was increased from 9.2% to 64%, while the C5+ selectivity was kept at 42–43%, when 20 wt% Co/TNT was illuminated under UV light.114 The selectivity of CH4 was enhanced from 25% to 35% and the ratio of C2–C4 olefins to paraffins decreased from 11 to 1.4 at the same time. It was speculated that the transfer of photogenerated electrons from TNTs to Co sites might occur, thus promoting the adsorption and activation of CO molecules to enhance activity and induce the hydrogenation of olefin to paraffins or hydrogenolysis of heavier hydrocarbons to shorter-chain paraffins (Fig. 12d).114
Ni-Based catalysts showed an unexpected activity in the hydrogenation of CO to C2+ hydrocarbons under photo-irradiation without external heating. The traditional thermocatalytic conversion of syngas usually leads to the formation of CH4. Zhang, Ma and their co-workers found that the irradiation of a Ni-based catalyst, which was synthesized by H2 reduction of NiAl-LDH nanosheets at 525 °C, under UV-vis illumination could convert syngas (0.8 MPa, H2/CO = 3/1) into C2–C7 hydrocarbons with a selectivity of 60% at a CO conversion of 27.7%.115 The temperature of the catalyst bed increased to 150 °C under UV-vis illumination. The control experiment carried out at 150 °C without photo-irradiation only afforded a CO conversion of 6% with CO2 as a predominant product. The catalyst could also work for the formation of C2–C7 hydrocarbons under visible-light (λ ≥ 400 nm) irradiation; the selectivity of C2–C7 hydrocarbons was 67% at a CO conversion of 21%. Under visible-light illumination, the temperature of the catalyst bed only increased to around 81 °C. It is proposed that the CO hydrogenation to C2+ hydrocarbons is photo-activated rather than a photo-thermal process. The characterisation suggested that the catalyst was composed of Ni and NiOx. The DFT calculations indicated that the modification of metallic Ni by surface oxides changed the reaction path of the surface CHx species by increasing the barrier for CH4 formation and decreasing that for C–C coupling.115 This is similar to the phenomena observed for the Co- and Fe-based catalysts,110–112 forming a concept that the oxide-decorated surfaces modulate the product selectivity in these photo-enhanced CO hydrogenation systems.
Photo-driven Fischer–Tropsch reaction enables the production of C2+ hydrocarbons through hydrogenation of CO in the absence of external heat source. Photo-thermal effect induces temperature increment at the catalyst bed, thus providing energy required for CO/H2 activation and C–C coupling. Photo-excited charge carriers may modify the oxidation state and electronic structure of the active site on catalyst surfaces, thus regulating the product selectivity and making the product distribution different from that in thermocatalysis. High yields of C2–C4 olefins have been obtained over oxide-decorated Co- and Fe-based catalysts under light irradiation.110–112 Decoration with the corresponding oxides can change the hydrogenation ability of the metal centre110,111 or accelerate the desorption of olefins from the surface,112 thus contributing to the high selectivity of lower olefins. C5+ hydrocarbons could be formed as major products using Ru/graphene113 or Co/TNT.114 Photo-generated electrons have been proposed to play a key role in the activation of CO/H2 and the favourable production of these C5+ paraffins. A Ni-based catalyst, which usually catalyses the formation of CH4 in thermocatalysis, has shown an unexpectedly high selectivity of C2–C7 hydrocarbons in the photo-driven hydrogenation of CO.115 The modification of Ni by NiOx is proposed to contribute to the selectivity switching by changing the energy barriers for CH4 formation and C–C coupling.
4. Electrocatalytic conversions of CO2 and CO
Electrocatalytic conversions of CO2 and CO to C2+ products such as olefins and alcohols have become one of the most promising routes to utilize the abundant CO2 and CO feedstocks under ambient conditions, owing to recent progress in generating electricity from renewable energy sources, such as solar, wind and hydropower.40,116–118 An analysis comparing the electrocatalytic, biocatalytic and fossil fuel-derived chemical-production routes has shown that the electrocatalytic route has great potential to yield the greatest reduction in CO2 emission, provided that a steady supply of clean electricity is available.40 Different from photocatalytic reactions, in which the reduction and oxidation reactions usually take place in the same cell, the electrocatalytic reaction typically separates reduction and oxidation half reactions into two cells, and thus is capable of avoiding the re-oxidation of reduction products. The activity and selectivity of electrocatalytic reactions can be modulated and optimised by regulating the applied potential. Furthermore, the use of a gas diffusion electrode in a flow cell has technically pushed the current densities to industrially relevant levels (>200 mA cm−2).119–121 Thus, electrocatalytic reductions of CO2 and CO into high-value C2+ compounds have become a very promising and appealing research area in chemistry.Metal catalysts have been typically employed in an electrocatalytic CO2 reduction reaction (CO2RR) with high activity and selectivity. It is generally accepted that the metal–CO binding strength, expressed by ΔHCO, plays an important role in determining the selectivity of a metal catalyst for electrocatalytic CO2RR (Fig. 13).122,123 This is known as the Sabatier principle in electrocatalytic CO2RR.123 In brief, metals with too weak CO binding energy, such as In, Sn, Pb and Hg, tend to be beneficial for the formation of formate,116,123–125 while those with relatively weak CO binding energy, such as Zn, Au, and Ag, offer CO as the main product.116,122,126 The metals with too strong CO adsorption (such as Fe, Ni, Co, and Pt) can cause the poisoning of electrocatalysts, and thus usually favour H2 evolution.126 In the single-metal electrocatalysts, only Cu with moderate CO binding energy is able to catalyse the formation of C2+ products with considerable efficiency.53,117,118,127
 | ||
| Fig. 13 General relationship between the primary product formed and ΔHCO over different metals for electrocatalytic CO2RR.122 Reproduced from ref. 122, with permission of Wiley-VCH Verlag GmbH&Co. KGaA, copyright 2017. | ||
For the synthesis of C2+ products, it is generally believed that CO is the key reaction intermediate in electrocatalytic CO2RR.53,118,127,128 The study of an electrocatalytic CO reduction reaction (CORR) would be beneficial for developing more efficient electrocatalysts for CO2RR to C2+ products and understanding the C–C coupling mechanism.127,128 Furthermore, because better selectivity to C2+ products and improved stability may be expected for electrocatalytic CORR than for direct CO2RR, electrocatalytic CORR would be useful for implementing a tandem strategy to synthesize C2+ products from CO2via CO.43,129,130 An analysis demonstrated that a two-step tandem electrocatalytic reactor, in which the first reactor converts CO2 to CO or formate and the second reactor converts CO or formate to C2H5OH or C2H4 may offer higher optimal solar-to-fuel conversion efficiencies.131
Here, we highlight recent advances in developing Cu-based catalysts for the synthesis of four C2+ products, i.e., ethylene, ethanol, acetate and n-propanol, by electrocatalytic CO2RR and CORR. It is noteworthy that the synthesis of a single product is still very difficult. Therefore, we mainly review the studies that can offer one of these C2+ compounds as the major product. The electrocatalysts other than Cu that are capable of forming C2+ products are also briefly touched upon. The possible reaction mechanism will be discussed with an emphasis on C–C coupling.
4.1. Electrocatalytic CO2RR and CORR into ethylene over Cu-based catalysts
Ethylene is one of the most important building blocks in the current chemical industry for the production of a wide range of chemicals, in particular plastics.7,8 It is primarily produced by thermal cracking of crude oil-derived naphtha. C2H4 can be produced by hydrogenation of CO via direct Fischer–Tropsch synthesis, an indirect route via methanol synthesis followed by methanol-to-olefins (MTO) or a direct route via a methanol intermediate using a bifunctional catalyst.7,8 However, the selectivity of C2H4 by these thermocatalysis routes is generally not high (<50%) even though the selectivity of C2–C4 olefins has recently made a significant step-forward by using the bifunctional catalyst.27,28,132,133 The thermocatalytic hydrogenation of CO2 also suffers from lower selectivity of C2H4.8–15 CO2RR or CORR with H2O by electrocatalysis provides a promising avenue for the synthesis of C2H4 as a major product under mild conditions. This section contributes to summarizing recent advances in Cu-catalysed CO2RR and CORR to C2H4 with the aim to offer insights into the key factors controlling the CO2 or CO conversion activity and faradaic efficiency (FE) of C2+ compounds, in particular C2H4. The effects of different influencing factors such as the exposed facet, chemical state of surface Cu and dopant-modification of Cu catalysts are discussed. Some engineering factors such as cell configuration, electrolyte engineering and reaction conditions will also be analysed. Some typical results for electrocatalytic CO2 and CORR are displayed in Tables 3 and 4.134–175| Electrocatalyst | Cell | E (V) | j (mA cm−2) | FE (%) | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|
| Ethylene | Acetate | Ethanol | Propanol | C2+ | |||||
| a The potential refers to RHE. b The potential represents the full-cell potential (V). | |||||||||
| Cu(100) nanocubes | H-cell | −0.96 | −68 | 32 | 0 | 13 | 15.5 | 60.5 | 134 |
| 44 nm Cu cubes | H-cell | −1.1 | −5.6 | 41.1 | 0.2 | 3.7 | 2.7 | 46.4 | 135 |
| Plasma-activated Cu | H-cell | −0.9 | −23 | 60 | 0 | 0 | 0 | 60 | 136 |
| Cu2O derived Cu | H-cell | −0.99 | −25 | 40 | 0 | 8.7 | 0 | 48.7 | 137 |
| Nano-defective Cu nanosheets | H-cell | −1.18 | −60 | 83.2 | 0 | 0 | 0 | 83.2 | 138 |
| B-Doped Cu | H-cell | −1.1 | −70 | 52 | 0 | 27 | 0 | 79 | 139 |
| N-Doped Cu | H-cell | −0.95 | −22 | 39.3 | 0 | 18.4 | 6.0 | 63.7 | 140 |
| Cu_I | H-cell | −0.90 | −39 | 45 | 0.7 | 27 | 7.3 | 80 | 141 |
| F–Cu | Alkaline GDE | −0.89 | −1600 | 65 | 2 | 12 | 1 | 80 | 142 |
| Thermally deposited Cu | Alkaline GDE | −0.67 | −750 | 65 | 4 | 11 | 0 | 80 | 143 |
| Cu–Al | Alkaline GDE | −1.8 | −600 | 80 | ∼2 | ∼13 | ∼2 | ∼97 | 144 |
| Catalyst:ionomer bulk heterojunction | Alkaline GDE | −0.91 | ∼−1750 | ∼75 | — | — | — | ∼75 | 145 |
| Arypyridiniums-Cu | Neutral GDE | −0.83 | −320 | 71.5 | 1.5 | 10.5 | 2.1 | 85.6 | 146 |
| MEA | −3.65b | −120 | 60 | — | — | — | 60 | ||
| Mesoporous Cu | H-cell | −1.3 | −14.3 | 2 | 46 (ethane) | — | — | 48 | 147 |
| Iodine-derived Cu | H-cell | −1.0 | −20.5 | 0.8 | 4.0 (ethane) | 1.9 | — | 6.7 | 148 |
| Cu4Zn | H-cell | −1.05 | −8.2 | 10.7 | 0.96 | 29.1 | 4.4 | 45.16 | 149 |
| Phase-blended Ag–Cu2O | H-cell | −1.2 | −3 | 9.5 | 5.6 | 34.15 | 0 | 49.25 | 150 |
| Nanoporous CuAg-wire | Alkaline GDE | −0.7 | −300 | 60 | 0 | 25 | 0 | 85 | 151 |
| Ag0.14/Cu0.86 alloy | Alkaline GDE | −0.67 | −250 | 35 | 5 | 41 | 0 | 81 | 152 |
| Cu2S@Cu vacancy | Alkaline GDE | −0.92 | −400 | 21.2 | 3.0 | 24.7 | 6.9 | 55.8 | 153 |
| FeTPP[Cl]/Cu | Neutral GDE | −0.82 | −300 | 39 | 2 | 41 | 3 | 85 | 154 |
| Cu/N-doped carbon nanospike | H-cell | −1.2 | −2 | 0 | 0 | 63 | 0 | 63 | 155 |
| 34% N–C/Cu | Alkaline GDE | −0.68 | −300 | 37.5 | 2.3 | 52.3 | 1.4 | 93.5 | 156 |
| N–C/Cu nanorods | Alkaline GDE | −0.9 | −281 | 24 | 4 | 45 | 7.4 | 80.4 | 157 |
| Cu single atom/C | H-cell | −0.7 | −1.23 | 0 | 0 | 91 | 0 | 91 | 158 |
| N-Doped nanodiamond | H-cell | −1.0 | −7 | 0 | 91 | 0 | 0 | 91 | 159 |
| B- and N-co-doped nanodiamond | H-cell | −1.0 | −0.5 | 0 | 0 | 93.2 | 0 | 93.2 | 160 |
| Mesoporous N-doped carbon | H-cell | −0.56 | −2 | 0 | 0 | 78 | 0 | 78 | 161 |
| N-Doped graphene quantum dots | Alkaline GDE | −0.75 | −28 | 31 | 6 | 14 | 4 | 55 | 162 |
| Catalyst | Cell | E (V) | j (mA cm−2) | FE (%) | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|
| Ethylene | Acetate | Ethanol | Propanol | C2+ | |||||
| a The potential refers to RHE. b The potential represents the full-cell potential (V). | |||||||||
| Oxide-derived Cu | Alkaline GDE | −0.67 | −700 | 41.8 | 13.9 | 26.7 | 8.3 | 90.7 | 163 |
| Oxide-derived Cu (5% CO/N2) | Alkaline GDE | −0.52 | −120 | 72 | — | — | — | 72 | 164 |
| Oxide-derived Cu | H-cell | −0.3 | −0.285 | 0.6 | 13.6 | 42.9 | 0 | 57.1 | 165 |
| Cu/carbon nanotubes | H-cell | −0.3 | −0.37 | 0 | 36 | 36 | 0 | 72 | 166 |
| Cu nanowires | H-cell | −0.30 | −0.22 | 0.66 | 14 | 50 | 0 | 65 | 167 |
| Pd0.07Cu | Alkaline GDE | −0.62 | −700 | 37 | 14.9 | 33.2 | 6.3 | 91.4 | 168 |
| Cu nanoflower | H-cell | −0.23 | −0.21 | 0.39 | 23.76 | 15.38 | 59.32 (aldehyde) | 99 | 169 |
| Cu–Ag nanoflower | H-cell | −0.335 | −0.61 | 1.2 | 1.8 | 2.6 | 70 (aldehyde) | 75.6 | 170 |
| Cu nanoparticles | MEA | −2.4b | −144 | 35.2 | 30.4 | 3.6 | 1.8 | 71 | 171 |
| Cu nanosheet | Alkaline GDE | −0.736 | −200 | 16.3 | 48 | 2.4 | 2 | 68.7 | 172 |
| Cu nanocavity | Alkaline GDE | −0.56 | −37 | 21 | 7.8 | 12.5 | 21 | 62.3 | 173 |
| Fragmented Cu | Alkaline GDE | −0.45 | −50 | 20 | 7 | 13 | 20 | 60 | 174 |
| Cu adparticle | Alkaline GDE | −0.44 | −48 | 29.6 | 4.7 | 14.6 | 23 | 71.9 | 175 |
The control of the exposed facet of a Cu catalyst may modulate its electrocatalytic performance, because different crystalline facets have different surface atomic arrangements and electronic structures, and may have different abilities to adsorb and activate the reactants and intermediates. Model studies using Cu single crystals showed that the product selectivity in electrocatalytic reduction of CO2 or CO depended strongly on the facet.176–178 Two early studies by Hori et al. showed that the electrocatalytic reduction of CO2 in 0.1 M KHCO3 aqueous solution using Cu(111) and Cu(100) yielded CH4 and C2H4 as the major products, respectively.176,177 The introduction of (111) or (110) step atoms to the (100) basal plane could significantly enhance the formation of C2H4. The Cu(110) facet favoured the formation of C2+ oxygenates such as CH3COOH, CH3CHO and C2H5OH.177 Koper and co-workers found a similar trend in the electrocatalytic reduction of CO in aqueous solution over Cu single crystals with different facets; CH4 was favoured on Cu(111), while C2H4 was favoured on Cu(100).178 The reduction of CO on Cu(110) exhibited a similar potential dependence to that on Cu(111), but more C2H5OH was observed.178 A study on electrocatalytic CO2RR over Cu thin films on single crystals by epitaxial growth with the physical vapour deposition technique demonstrated that the Cu(100) film and the Cu(751) film that has a heterogeneous kinked surface with a (110) terrace were more active and selective for C–C coupling than Cu(111) film, but the Cu(751) film was more selective toward C2+ oxygenate formation.179
Yeo and co-workers performed experimental and computational studies for electrocatalytic CO2RR on Cu(100), Cu(111) and Cu(110) single-crystal surfaces.180 It was found that the onset potentials for C2H4 and CO were very closely related and Cu(100) showed the earliest onset potentials for CO and C2H4 formation, whereas Cu(111) exhibited the latest onset potentials for CO and C2H4 formation. The DFT calculations suggested that Cu(100) had the lowest energy barrier for CO dimerization and the energy barrier could be further lowered by increasing the *CO coverage.180 These results indicate the importance of Cu(100) surfaces and *CO coverage in CO2RR to C2H4. Recently, Wang, Chan and their co-workers performed detailed DFT calculations on the energetics of the initial C–C coupling steps on different Cu facets in CO2 reduction.134 The result suggested that relative to Cu(111) facet, the Cu(100) and stepped (211) facets were more favourable for CO dimerization, and thus were beneficial for the formation of C2+ products. They synthesized Cu nanocubes with preferentially exposed (100) facets through a metal ion battery cycling method and found that the 100-cycled Cu nanocubes exhibited a six-fold increase in the ratio of C2+ (mainly C2H4) to C1 products as compared to the pristine polycrystalline Cu foil primarily with (111) facets (Fig. 14a).134 The FEs of C2+ products and C2H4 could reach 60% and 32%, respectively, over the 100-cycled Cu nanotubes, which were much higher than those over the pristine polycrystalline Cu foil.
 | ||
| Fig. 14 (a) Comparisons of C2+ FEs and ratios of C2+/C1 over different catalysts for electrocatalytic CO2RR. 100-Cycle Cu mainly with (100) facets exhibits a six-fold improvement as compared to the polished Cu foil primarily with (111) facets.134 (b) C2H4 and CH4 FEs for plasma-treated Cu foils. From left to right, the insets show SEM images of the low surface area H2 plasma-treated metallic Cu foil, the O2 20 W 2 min plasma-treated Cu foil with optimised C2H4 selectivity, and the high surface area nanoneedles on the O2 100 W 10 min oxidized sample after the reaction. Scale bars: 500 nm.136 Reproduced from ref. 136 with the permissions of Springer-Nature, copyright 2016. (c) Comparison of C2+ FEs on Cu(H), Cu(C) and Cu(B).139 (d) Comparisons of C2+ FEs and ratios of C2+/CH4 on Cu, Cu-on-Cu2O and Cu-on-Cu3N.140 | ||
A few studies reported the fabrication of Cu nanocubes by other techniques for electrocatalytic reduction of CO2 to C2H4.135,181 For example, Buonsanti and co-workers fabricated cube-shaped Cu nanocrystals with three different sizes (edge lengths) of 24, 44 and 63 nm by a colloidal chemistry-based method and found that the cubes with 44 nm edge length showed the best performance for C2H4 formation.135 The FE of C2H4 over the Cu cubes with 44 nm edge length was 41%, significantly higher than those over the smaller or bigger Cu cubes as well as Cu foil. The H2 evolution reaction was very serious over the smaller Cu cubes.
Many groups found that as compared to the pure metallic Cu catalyst, the oxide-derived Cu catalysts showed enhanced C2+ selectivity.136–138,182–188 This may result from the differences in surface structures and local environments such as the density of defects, roughness factors, chemical states and oxygen modifiers. Cuenya and co-workers fabricated an oxide-derived Cu catalyst by treating Cu foil with O2 plasma for electrocatalytic CO2RR.136 After O2-plasma treatment of Cu foil, the formation of C2H4 was significantly enhanced, while the formation of CH4 was suppressed. The FE of C2H4 reached 60% at −0.9 V versus reversible hydrogen electrode (RHE) for the Cu catalyst with 20 W-2 min O2 plasma-treatment (Fig. 14b). On the other hand, the changes in the FEs of CH4 and C2H4 were limited with H2-plasma treatment (Fig. 14b). The O2-plasma treatment could increase both the roughness factor and the surface Cu+ sites. It was found that when the O2 plasma-activated Cu foil was further subjected to H2-plasma treatment, the FE of C2H4 decreased significantly despite similar roughness. The H2-plasma treatment could deplete the Cu+ sites. Thus, it was argued that Cu+ species played a key role in enhancing the formation of C2H4 on the oxide-derived Cu catalysts.
Through theoretical calculations, Goddard III and co-workers proposed a unique electrocatalyst composed of Cu metal embedded in oxidised matrix for CO2RR to C2+ products.182 It was shown that the fully oxidised catalyst model could not explain the enhanced electrocatalytic performance and such a catalyst was also unstable under CO2RR conditions. Both Cu+ and Cu0 were proposed to contribute to CO2RR to C2+ products and they worked synergistically to improve the kinetics and thermodynamics for both CO2 activation and CO dimerization.182 It was proposed that in the initial step, the Cu+ site could bind H2O molecules at the edge of a Cu0 region and this H2O on surface Cu+ may form strong hydrogen bonds to CO2, stabilising both the transition state and the final state of CO2 activation on nearby Cu0, facilitating the formation of CO (Fig. 15a). In the next step, the carbon atom of CO on the Cu+ site may be positively charged (Mulliken charge, +0.11), whereas that on the Cu0 site may be negatively charged (Mulliken charge, −0.31) because of the back donation (Fig. 15b). The electrostatic attraction between the two carbon atoms would assist C–C coupling to form C2+ products (Fig. 15c). The theoretical calculation in combination with ambient-pressure X-ray photoelectron spectroscopy (XPS) studies by Goddard III and co-workers further pointed out the importance of the presence of a thin layer of suboxide below the metallic Cu surface in CO2 sorption and activation.183
 | ||
| Fig. 15 Schematic illustration of the synergistic effects of Cu+ and Cu0.182 (a) CO2 activation. (b) Chemical state of CO. (c) C–C coupling. Reproduced from ref. 182 with the permissions of the National Academy of Sciences, copyright 2017. | ||
The nature of active sites leading to high C2+ FE over oxide-derived Cu catalysts is still under debate. In situ ambient-pressure XPS and quasi in situ electron energy loss spectroscopy (EELS) studies showed no residual copper oxide over the oxide-derived Cu catalysts in electrocatalytic CO2RR, but subsurface oxygen species may exist after oxidation–reduction cycles.184 The subsurface oxygen species may increase the CO binding energy and contribute to enhanced C–C coupling owing to the increased CO coverage on the catalyst surfaces. It is noteworthy that the reduced oxide-derived Cu catalysts could be re-oxidised rapidly in air or H2O environments, making the ex situ characterisation to determine the true chemical state of Cu under CO2RR conditions highly challenging. Lum and Ager prepared an 18O-enriched oxide-derived Cu catalyst by oxidation/reduction cycling in H218O and the residual 18O in the Cu catalyst after the electrocatalytic CO2RR was measured by secondary-ion mass spectrometry (SIMS).185 The result showed that only a small fraction (<1%) of the original 18O was sustained after the CO2RR, whereas the catalyst maintains a high FE (∼60%) towards C2 and C3 products.185 Ager and co-workers further studied the possible active sites of a series of Cu2O samples with different morphologies for electrocatalytic CO2RR by in situ Raman in combination with real-time selected-ion flow tube mass spectrometry.186 The result showed that the reduction of Cu2O is kinetically and energetically more favourable than the CO2RR, thus excluding the possibility of Cu2O as active sites for the electrocatalytic CO2RR.186 Although further operando spectroscopy studies are needed, it is generally believed that the Cuδ+ sites on the catalyst surfaces play pivotal roles in the formation of C2+ products.
Xu, Jiao and co-workers recently demonstrated that oxygen-containing surface species are unlikely involved in facilitating the formation of C2+ oxygenates in CORR.189 By using in situ surface-enhanced Raman spectroscopy, they showed that oxygen-containing species do exist under the CORR conditions in an alkaline electrolyte for four commonly used Cu surfaces, i.e., Cu foil, Cu micro/nanoparticles, electrochemically deposited Cu film and oxide-derived Cu. The relative abundance of surface CuOx on Cu foils is higher than that on other catalysts, while the opposite is true for the surface CuOx/(OH)y species. The surface speciation on different catalysts has been correlated with initial degree of oxidation of the Cu surfaces prior to the exposure to negative potentials. However, the CORR experiments showed different trends in product selectivity. Thus, they concluded that the oxygen-containing surface species might not be the active species in the formation of C2+ oxygenates.189
The doping of a heteroatom can modify the structure, and the electronic and chemical properties of Cu catalysts, and thus plays an important role in governing the activity and selectivity for electrocatalytic CO2RR by influencing the adsorption and activation of reactants and intermediates. Boron- and nitrogen-doped Cu catalysts have been found to show significantly improved performance for electrocatalytic CO2RR to C2H4.139,140,190 On the other hand, the doping of chalcogen (such as sulfur, selenium and tellurium) has been reported to improve the formation of formate.191–193 For example, Sargent and co-workers synthesized a boron-doped Cu (Cu(B)) catalyst by reducing CuCl2 with NaBH4.139 Two reference samples, Cu(C), which consisted of oxidized nano-copper, and Cu(H) that were synthesized by using hydrazine hydrate as the reducing reagent were also prepared. The FE of C2H4 over Cu(B) reached 52% at −1.1 V versus RHE, which was 1.6 and 2.4 times higher than those of Cu(C) and Cu(H), respectively (Fig. 14c). The FE of total C2 products (mainly C2H4 and C2H5OH) reached 79% with a current density of 70 mA cm−2 under the same conditions over the Cu(B) catalyst, which was also much better than those over the Cu(H) and Cu(C) catalysts. The DFT calculations and characterisation results revealed that boron was located at the subsurface of the catalyst. In situ X-ray absorption near-edge spectroscopy (XANES) studies indicated that the average oxidation state of Cu was between 0 and +1, and the slightly positive oxidation state of Cu was stable during the electrocatalytic CO2RR. The electrocatalytic CO2RR performance of the Cu(B) catalyst kept stable in 40 h of continuous operation, which was much better than other oxide-derived Cu catalysts.139 Han and co-workers also reported the superior performance of a B-doped oxide-derived Cu catalyst, which was prepared by reduction of B2O3–Cu(OH)2 film, for electrocatalytic CO2RR.190 An FE toward total C2+ products (C2H4, C2H6 and C2H5OH) of 48% was achieved at a current density of 33.4 mA cm−2 at −1.05 V vs. RHE over the B-doped catalyst, better than that over the oxide-derived Cu catalyst without B (FE of C2 products was 30.5% with a slightly lower current density at −1.05 V).190 The XPS studies indicated that the doping of boron could stabilise a larger fraction of Cu+ on the Cu surfaces.
The group of Sargent further synthesized Cu deposited on Cu3N (Cu-on-Cu3N) catalyst, which achieved a 6.3-fold enhancement in the ratio of C2+ to CH4 as compared to Cu deposited on Cu2O (Cu-on-Cu2O) catalyst and a 40-fold enhancement relative to a pure Cu catalyst (Fig. 14d).140 The highest FE towards total C2+ products over the Cu-on-Cu3N catalyst reached 64% at −0.95 V vs. RHE, with the FEs of C2H4, C2H5OH and C3H7OH being 39%, 19% and 6%, respectively. The Cu-on-Cu3N catalyst also showed a better stability than the Cu-on-Cu2O and Cu catalysts. The characterisation by XPS and in situ XANES suggested that Cu3N could stabilise Cu+ to a larger extent as compared to Cu2O during CO2RR. DFT calculations further indicated that the modulated partial oxidation state by nitride enabled Cu-on-Cu3N to achieve the lowest CO dimerization barrier energy. Therefore, both the boron and nitride modification contribute to stabilising Cu+ sites during electrocatalytic CO2RR, which likely play crucial roles in CO adsorption and dimerization to form C2+ products.139,140,190
A few groups reported that the modification of Cu by sulphur and other chalcogen modifiers enhanced the formation of formate during electrocatalytic CO2RR.191–193 For example, Pérez-Ramírez, López and co-workers found that the FE of formate increased markedly from 26% to 78% after doping of sulfur on Cu surfaces at −0.8 V versus RHE.193 It was proposed that the chalcogen modifier on Cu surfaces actively participated in the CO2RR either by tethering CO2 or transferring a hydride, thus suppressed the formation of CO and C2+ products. The different behaviours of heteroatoms doped on Cu catalysts reveal diversified functioning mechanisms of heteroatoms and offer us opportunities to explore more heteroatom dopants to narrow product distribution for electrocatalytic CO2RR.
Cuenya and co-workers reported that the addition of a halide ion into KHCO3 electrolyte could enhance the electrocatalytic CO2RR into C2+ products over a plasma-oxidised polycrystalline Cu foil catalyst.194 The CO2 reduction rate was found to increase in the order of Cl− < Br− < I−, while the high FE of C2+ products kept almost unchanged. The enhancement in CO2 reduction activity was proposed to arise from the adsorption of halide ions on Cu surfaces, which may facilitate the formation of a carboxyl intermediate (*COOH), which was a precursor of CO, by partial charge donation from halide to CO2. The effect of electrolytes on electrocatalytic CO2RR over Cu(100) and Cu(111) surfaces was investigated by Yeo and co-workers.195 They found that the FEs of C2H4 and C2H5OH increased in the sequence of KClO4 < KCl < KBr < KI over both Cu surfaces. The FE of total C2+ products reached 74% with those of C2H4 and C2H5OH being 50% and 16%, respectively, over the Cu(100) surface in the case of KI. KI roughened the Cu surface but the increase in the roughness was not the major reason for the enhancement in performance. It was proposed that the increase in the population of adsorbed CO on Cu surfaces in the presence of KI might contribute to enhancing the formation of C2+ products. The effect of alkali metal cations in an electrolyte was also examined for electrocatalytic CO2RR in MHCO3 (M = Li, Na, K, Cs) over an O2-plasma-activated Cu catalyst.196 The increase in the size of alkali-metal cations increased the formation of C2+ products (mainly C2H4), and the co-existence of Cs+ and I− further enhanced the partial current of C2+ compounds (Fig. 16). The highest FE of C2+ products reached ∼69% with a C2+ partial current density of −45.5 mA cm−2.
 | ||
| Fig. 16 Effects of cations and anions added in the electrolyte on CO2RR.196 Reproduced from ref. 196 with the permissions of American Chemical Society, copyright 2018. | ||
Recently, the modification of Cu by halides has shown promising performances in CO2RR to C2+ products.141,142 Cuenya and co-workers prepared halide-modified Cu (denoted as Cu_X, X = Cl, Br or I) catalysts by cycling electropolished Cu foils in 0.1 M KX solution and found that the Cu_I catalyst showed the best performance in electrocatalytic CO2RR to C2+ products (mainly C2H4).141 The highest FE of a C2+ product was ∼80% with a partial current density of ∼31.2 mA cm−2 at −0.90 V vs. RHE. The nanostructure of Cu changed significantly for the Cu_I catalyst, and it was proposed that the enhancement in the formation of C2+ products arose from the reconstructed surface structures and adsorbed I− species, which probably led to unique electronic and chemical effects to stabilise the subsurface oxygen and Cu+ species.
A recent work by our group demonstrated that a fluorine-modified Cu catalyst, which was fabricated by electroreduction of a Cu(OH)F precursor synthesized by a solvothermal method, showed excellent performance for electrocatalytic CO2RR to C2+ products in a flow cell with a gas diffusion electrode (GDE).142 The FE of C2+ products (mainly C2H4 and C2H5OH) reached ∼80% with an ultrahigh current density of 1.6 A cm−2 at −0.89 V vs. RHE. The FE and formation rate of C2H4 reached 65% and ∼3.2 mmol h−1 cm−2, much better than those reported to date. The carbon-based selectivity and single-pass yield of C2–4 products (mainly C2H4 and C2H5OH) reached 85.8% and 16.5%, respectively, outperforming those achieved in high-temperature and high-pressure thermocatalysis.8 It was proposed that besides stabilising the Cu+ sites to enhance CO adsorption, the fluoride species on Cu surfaces facilitated the activation of H2O and the hydrogenation of CO to CHO intermediate, which could undergo C–C coupling more facilely.142
Most studies for electrocatalytic CO2RR and CORR have typically been conducted in an H-cell. The limited CO2 or CO solubility in water (33 mM for CO2 and 1 mM for CO at 25 °C and 1 atm) usually leads to low reaction rates (<100 mA cm−2 for CO2RR and <10 mA cm−2 for CORR), which are not viable for commercialisation, although the hydrophobic treatment can further enhance the CORR reaction rate in an H-cell.197,198 As already mentioned, the flow cell with GDE configuration enables the direct feeding of gaseous reactants to the electrode–electrolyte interface to circumvent the mass-transport limitation of CO2 or CO, resulting in high reaction rates.119–121 Flow cells with different configurations (Fig. 17) have shown great potential in electrocatalytic CO2RR to C2+ products.142–146,199,200
 | ||
| Fig. 17 Configurations of three types of flow cells, stability of the cell voltage and gas products for 120 min in different flow cells with an applied current density of 150 mA cm−2.200 (a) Alkaline flow cell. (b) Neutral flow cell. (c) MEA flow cell. Reproduced from ref. 200, with permission of Elsevier, copyright 2019. | ||
One significant merit of the flow cell is that it can be operated in a strong alkaline electrolyte (e.g., KOH) for CO2RR (Fig. 17a),200 whereas the use of a strong alkaline electrolyte is restricted in the H-cell configuration because of the acidic nature of the CO2 molecule. The current consensus is that a high local pH can significantly promote the C–C coupling and suppress the formation of H2 and methane.143,163,201,202 Thus, the alkaline flow cell particularly favours the formation of C2+ products, offering high FEs of C2+ compounds at impressively high current densities.142–146,202 For example, Sargent and co-workers reported that upon increasing the concentration of KOH from 1 to 10 M, the onset potential of C2H4 for CO2RR decreased to only −0.165 V versus RHE, close to the CO onset potential, whereas the onset potential of H2 remained almost unchanged (Fig. 18a).143 As such, a Cu catalyst with a thickness of 25 nm deposited on a carbon-based gas diffusion layer could achieve an C2H4 FE of 66% with a current density of 275 mA cm−2 at a potential of −0.54 V versus RHE in a 10 M KOH electrolyte. By further optimising the electrolyte to (3.5 M KOH + 5.0 M KI), the current density could increase to 750 mA cm−2 with C2H4 FE of 65% at −0.67 V versus RHE.
 | ||
| Fig. 18 (a) C2H4 and CO FEs versus applied potential during CO2RR at different KOH concentrations.143 (b) Partial current densities of C2+ products in CORR in KOH with different concentrations.163 (c) Partial current densities of C2+ products in CORR in electrolytes with different Na+ and OH− concentrations.203 Reproduced from ref. 143, 163 and 203, with the permissions of American Association for the Advancement of Science, Springer-Nature and Wiley-VCH Verlag GmbH&Co. KGaA, copyright 2018 and 2020. | ||
Very recently, a Cu–Al catalyst, which demonstrated further improved C2H4 selectivity in electrocatalytic CO2RR, was developed through machine learning-accelerated high-throughput DFT studies.144 The de-alloyed nanoporous Cu–Al catalyst, which was fabricated by both thermal evaporation and co-sputtering followed by chemical etching, on a carbon-based gas diffusion layer showed an FE of C2H4 as high as 80% at a current density of 600 mA cm−2 in a 1 M KOH flow cell.
Similarly, by using the alkaline flow cell, Jiao and co-workers achieved excellent performances for electrocatalytic CORR to C2+ compounds.163 An oxide-derived Cu catalyst exhibited a C2+ FE of 91% and a C2+ partial current density of 635 mA cm−2 at a potential of −0.67 V versus RHE (Fig. 18b).163 It is noteworthy that the current density is very low (∼10 mA cm−2) in electrocatalytic CORR with H-cell configuration due to the extremely low solubility of CO in aqueous electrolyte solution. Thus, the work by Jiao and co-workers is a significant step-forward in electrocatalytic CORR to C2+ compounds.
A recent work revealed that a high concentration of the cation (i.e., Na+) rather than OH− might be the determining factor in promoting the formation of C2+ products during electrocatalytic CORR.203 By systematically varying the concentration of cations (Na+) and OH− at identical absolute electrode potential, that is on the standard hydrogen electrode (SHE) scale, Lu, Xu and their co-workers found that the partial currents of C2+ products, including C2H4, CH3COO−, C2H5OH and n-C3H7OH, all increased with an increase in the concentration of Na+ but not OH− (Fig. 18c).203 The promotional effect of OH− at the same potential on the RHE scale was attributed to the larger overpotential at higher electrolyte pH. The chelation of Na+ with a crown ether to form a bulky organic cation while maintaining the concentration of OH− significantly decreased the current density and the FE of C2+ products, providing further evidence that Na+ played a pivotal role in the formation of C2+ products from CO.
To further increase the efficiencies for CO2RR in an alkaline flow cell, Sargent, Sinton and their co-workers recently designed a catalyst/ionomer bulk heterojunction (CIBH) architecture, which could decouple gas, ion and electron transport.145 The ionomer layer with hydrophobic and hydrophilic functionalities was assembled into an architecture with differentiated domains that favoured gas and ion transport routes on the metal catalyst surfaces. Gas transport was promoted through a side chain of hydrophobic domains, leading to extended gas diffusion, while water uptake and ion transport took place through hydrophilic domains. The CIBH configuration with a Cu catalyst in a 7 M KOH electrolyte could offer an FE of C2H4 in the range of 65–75% and a peak partial current density of ∼1.3 A cm−2 at a cathodic energy efficiency of 45%.145 This C2H4 partial current density is the highest one reported to date.
The alkaline GDE design successfully circumvents mass-transport limitations associated with the low solubility of CO2/CO in aqueous solutions and enables superior performances to be achieved at high reaction rates, resulting in a significant step toward future practical applications. However, challenges remain in sustaining the high reaction rate for CO2RR in the alkaline flow cell over a long period of time. The flooding and the salt accumulation in the triple-phase boundary are two major reasons that can cause decreases in activity and selectivity in electrocatalytic CO2RR using the alkaline flow cell. To solve the problem of the formation of carbonate salt due to the undesirable consumption of CO2 by KOH in the alkaline GDE, neutral electrolytes, such as KHCO3 and K2SO4, were employed (Fig. 17b).146,200 However, the activity in neutral GDE is significantly lower than that of alkaline GDE because of the high Ohmic resistance and overpotential. Moreover, to maintain the stability of catholyte in neutral electrolytes still remains an issue due to the reaction-driven pH increase. A recent work demonstrated that the electrodeposition of N-aryl-substituted-tetrahydro-bipyridine films on a Cu catalyst could achieve 72% FE of C2H4 with a current density of 320 mA cm−2 at a potential of −0.83 V versus RHE during electrocatalytic CO2RR in a liquid-electrolyte flow cell using 1.0 M KHCO3 as the electrolyte.146 It was found that, although the large current densities in the flow cell drove up the local pH, the tetrahydro-bipyridine layer did not create a further pH gradient near the active Cu surface, and the layer was chemically robust to the locally alkaline environment. When the substituents in the organic modifier were changed to tune the electronic structure of organic films, the configuration of CO adsorption changed. A volcano-shaped relationship was observed between the FE of C2H4 and the ratio of atop-bound CO to bridge-bound CO. Thus, there is an optimum ratio of atop-bound CO to bridge-bound CO for the formation of C2H4. The DFT calculations indicated that the coupling of CO molecules located on atop–bridge pair sites was more facile than that of CO molecules on the bridge–bridge pair sites or top–top sites. The adhered N-aryl-substituted-tetrahydro-bipyridine molecules may help to optimise the pairs of atop- and bridge-bound CO.
The membrane electrode assembly (MEA) cell, which has been widely applied to the H2–O2 fuel cell, is emerging as an alternative for CO2RR to solve the stability issue.200 In the MEA cell, the cathodic GDE contacts directly with an ion-exchange membrane instead of a catholyte (Fig. 17c). The stability issues associated with GDE flooding, electrolyte consumption with CO2 and electrolyte ohmic loss may be solved. Sargent and co-workers integrated the Cu catalyst sputtered onto a porous polytetrafluoroethylene gas diffusion layer and modified with N-aryl-substituted-tetrahydro-bipyridine films into an MEA device.146 They found that the system operated at a full-cell voltage of 3.65 V was stable at least for 190 h, offering a stable C2H4 FE of 64% and a stable current density of ∼120 mA cm−2.146 This is probably the best stability achieved for electrocatalytic CO2RR with a current density of >100 mA cm−2. In general, the reaction rate in MEA is still lower than that in alkaline flow cell. Future studies should be focused on improving the ion conductivity of the membrane and the assembly craft.
As already mentioned, excellent performances could also be achieved for electrocatalytic CORR by using an alkaline flow cell.163 The electrocatalytic CORR in an alkaline flow cell has advantages over the CO2RR because of no salt accumulation problem and potentially higher efficiencies towards C2+ products.43,163 Moreover, no formation of carbonates in an alkaline electrolyte can increase the process economics, since the regeneration of KOH and CO2 from the formed carbonates would be costly in the case of CO2RR in an alkaline flow cell.
It is noteworthy that the C2+ products in electrocatalytic CORR in an alkaline flow cell contained a large fraction of C2+ oxygenates (such as C2H5OH, CH3CHO, CH3COO−, n-C3H7OH) and the FE of C2H4 was not very high (∼40%).43,163 A recent work suggested that the coverage of CO on Cu surfaces determined the path for C2H4 formation or for C2H5OH formation.164 Conventionally, it is generally believed that a higher CO coverage on Cu surfaces is beneficial for the formation of C2+ products.180 Through analysing the possible paths for C2H4 and C2H5OH formation, Sinton and co-workers proposed that the higher CO coverage may favour the formation of C2 oxygenates, while the lower CO coverage may favour the formation of C2H4.164 They found that the concentration of CO significantly influenced the product selectivity in electrocatalytic CORR in an alkaline flow cell using KOH as the electrolyte. At a fixed potential of −0.44 V vs. RHE, the CORR in 1 M KOH offered C2+ products containing C2H4, C2H5OH, CH3COO− and C3H7OH, and the FE of C2H4 was ∼30%. The decrease in CO concentration to 10% or 5% increases the FE of C2H4 to ∼50% at the expense of mainly C3H7OH, but a further decrease in CO concentration led to a significant increase in H2 formation. By tuning the conditions to constrain local CO availability, C2H4 FEs of 65–72% could be achieved with high cathodic energy efficiencies (35–44%) and current densities (120–1250 mA cm−2).164 The DFT calculations indicated that a lower CO coverage favoured the dehydroxylation of *CHCOH, which was probably a common intermediate for C2H4 and C2 oxygenates, to *CCH, leading to C2H4. On the other hand, the higher CO coverage would be beneficial for the hydrogenation of *CHCOH to *CHCHOH, resulting in oxygenates.
Recently, Strasser and co-workers found that the co-feeding of CO and CO2 could enhance the formation of C2H4 during electrocatalysis over an oxide-derived Cu catalyst.204 There existed an optimum ratio of CO/CO2 in the gas mixture for C2H4 formation. Using an operando differential electrochemical mass spectrometry capillary flow cell with millisecond time resolution, it was demonstrated that the enhanced C2H4 formation mainly originated from a cross-coupling between two *CO species derived from CO2 and CO (the so-called CO2–CO pathway). It was speculated that the co-fed CO did not compete with CO2 for adsorption sites, implying non-competent and reactant-specific sites for CO and CO2 on Cu surfaces. These insights enabled the design of a tandem system of oxide-derived Cu in combination with Ni–N-functionalised carbon (NiNC), which involved NiNC for CO formation from CO2 and Cu for CO–CO coupling, for electrocatalytic CO2RR to C2H4 with doubled C2H4 yields.204
Besides C2H4, C2H6 could also be obtained as a C2 hydrocarbon product in electrocatalytic CO2RR, although the selectivity of C2H6 was typically low in most cases. Nam and co-workers found that the selectivities of C2H4 and C2H6 could be tuned by using mesoporous Cu with different morphologies.147 The mesoporous Cu catalysts with different pore widths and depths were fabricated by a thermal deposition method on anodized aluminium oxide. As the pore width decreased and the pore depth increased, the FE of C1 products gradually decreased and that of C2 products increased. The FE of C2H4 reached 38% at the pore width and depth of 30 and 40 nm, respectively. As the pore depth increased to 70 nm, the major C2 product changed to C2H6 and the FE of C2H6 was 46% (Fig. 19a). Through computational simulations, they proposed that the mesopore can change the local pH and prolong the retention time of key intermediates, thus favouring deep hydrogenation of intermediates and promoting C2H6 formation.147
 | ||
| Fig. 19 (a) FEs of different CO2RR products at −1.3 V versus RHE over polycrystalline Cu and mesoporous Cu catalysts with different widths and depths (inset: SEM image of mesoporous Cu).147 (b) Reaction pathway toward the formations of ethane and ethanol via an ethoxy intermediate.148 Reproduced from ref. 147 and 148, with the permissions of Wiley-VCH Verlag GmbH&Co. KGaA, copyright 2017 and 2020. | ||
Recently, Qiao and co-workers reported that an iodide-derived Cu was more selective for electrocatalytic CO2RR to C2H6 than an oxide-derived Cu.148 The iodide-derived Cu offered a normalized C2H6 selectivity of 72% (normalized to the total C2 + C3 products) at −1.0 V versus RHE, whereas that of oxide-derived Cu was only 27%. Through in situ X-ray absorption fine-structure (XAFS) and Raman spectroscopy studies, an O-bound ethoxy (*OCH2CH3) intermediate was detected during C2H6 formation and *OCH2CH3 was also responsible for C2H5OH formation (Fig. 19b).148 The optimized oxidation state of Cu on iodide-derived Cu may stabilize the *OCH2CH3 intermediate and steer the reaction into C2H6 over C2H5OH.
4.2. Electrocatalytic CO2RR and CORR into ethanol over Cu-based catalysts
Ethanol is a key chemical in both chemical and energy industries as well as in our daily life. It has been widely used as a fuel additive, alternative fuel, solvent or disinfectant, and can also serve as a versatile feedstock for the production of various chemicals and polymers.205 Traditionally, C2H5OH is primarily produced through the fermentation of sugars, and recently, the catalytic conversions of syngas derived from fossil resources (in particular natural or shale gas and coal) and cellulose have attracted much attention.38,206–212As described above, single Cu catalysts (in particular oxide-derived Cu catalysts) and some non-metallic dopant (e.g., boron and halogen)-modified Cu catalysts usually offer C2H4 as the major products during electrocatalytic CO2RR. C2H5OH is typically formed as the second largest product, and the ratio of FE of C2H5OH to that of C2H4 is typically lower than 0.5. DFT calculations suggested that the formation of C2H4 is more favourable than the formation of C2H5OH from a CH2CHO* intermediate over a Cu(100) surface.213 It is generally believed that the incorporation of a different atom in the Cu lattice may alter its geometric and electronic structures, thus changing its selectivity and activity during CO2RR. The design of bimetallic catalysts containing Cu and another guest metal such as Zn, Au or Ag has shown potential to improve the selectivity of C2+ oxygenates especially C2H5OH in electrocatalytic CO2RR.149–152,214,215
Oxide film-derived CuxZn bimetallic catalysts were reported to exhibit enhanced selectivity towards C2H5OH.149 Cu alone showed twice higher FE of C2H4 as compared to that of C2H5OH at most potentials studied, whereas CO was the major product along with a small fraction of HCOO− on Zn alone. By increasing the Zn content in the bimetallic catalysts, the maximum FEs of C2H5OH increased from 11.3% (on Cu) to 29.1% (on Cu4Zn) and then decreased to 18.1% (on Cu2Zn). Meanwhile, the maximum FEs of C2H4 decreased monotonically from 26.5% (on Cu) to 4.1% (on Cu2Zn). The selectivities of C2H5OH versus C2H4 production, defined as the ratio of their FEs, could be modulated from 0.48 (on Cu) to 6 (on Cu2Zn). The formation of C2H5OH was maximized on a Cu4Zn catalyst at −1.05 V versus RHE with a FE of 29.1% (Fig. 20a).149 Although CO was a major product on Zn in the whole potential ranges investigated, the formation of CO was suppressed at potentials more negative than −1.0 V, where C2H5OH formation was significantly accelerated, over the Cu4Zn and Cu2Zn catalysts. Furthermore, the formation of HCOO− was also inhibited over the bimetallic catalysts. These results suggest that Zn works synergistically with Cu to enhance the formation of C2H5OH. A dual-site mechanism has been proposed for the enhanced formation of C2H5OH. In brief, both Cu and Zn sites catalyse the reduction of CO2 to CO, but Cu sites can work further for the formation of CHO or CHx (x = 1–3) intermediates via CO. Zn sites provide CO, which might undergo spill-over to a nearby Cu site and then insert itself into the bond between Cu and *CHO or *CHx, eventually forming C2H5OH.149
 | ||
| Fig. 20 (a) Maximum FEs of C2H4 and C2H5OH as well as the average FEethanol/FEethylene ratio (calculated on the basis of the ratios measured at different potentials) in CO2RR on CuxZn.149 (b) Comparison of C2H5OH FEs on Ag–Cu2OPS, Ag–Cu2OPB and Cu2O.150 | ||
The deposition of Au nanoparticles on Cu foils without forming an alloy was found to enhance C2H5OH and C3H7OH formation at low overpotentials.214 The Au/Cu catalyst had an earlier onset potential for alcohol production than either Cu or Au alone and it showed higher selectivity for alcohols as compared with hydrocarbons at lower overpotentials. Mechanistic studies suggest that Au nanoparticles work for the reduction of CO2 to CO, driving a high CO coverage on the nearby Cu surface, enhancing the dimerization of CO and further reduction to C2+ compounds.
Several groups have studied Cu–Ag bimetallic catalysts for electrocatalytic CO2RR to C2H5OH.150–152,215 For example, Lee and co-workers reported another interesting example to enhance the formation of C2H5OH by incorporating Ag, a CO formation electrocatalyst, into Cu2O.150 Two types of Ag–Cu2O, phase-separated (Ag–Cu2OPS) and phase-blended (Ag–Cu2OPB) were fabricated by electrochemical co-deposition. The Ag–Cu2OPB catalyst had more intimate contact between Ag and Cu atoms than the Ag–Cu2OPS. The maximum FEs of C2H5OH on Ag–Cu2OPS and Ag–Cu2OPB reached 20.1% and 34.2% at −1.2 V versus RHE, being twice and three times higher than that on pure Cu2O, respectively (Fig. 20b).150 This result also indicated the importance of intimate contact between Ag and Cu for synergistic formation of C2H5OH. Lee also pointed out the CO insertion mechanism for the enhanced C2H5OH. In other words, CO formed on Ag sites could migrate to Cu sites to couple with the CHx intermediates, contributing to the formation of C2H5OH. The closer distance between Ag and Cu was vital for facilitating efficient CO transfer to Cu sites, thus affecting the ethanol selectivity. Nanoporous Cu–Ag alloy nanowires, which were fabricated by co-electrodeposition in the presence of 3,5-diamino-1,2,4-triazole (DAT), an additive to inhibit nucleation, and contained Cu and Ag atoms mixed homogeneously, demonstrated excellent performances for electrocatalytic CO2RR to C2H4 and C2H5OH in an alkaline flow cell.151 The FEs of C2H4 and C2H5OH reached ∼60% and 25% and a current density of −300 mA cm−2 at a cathode potential of only −0.7 V vs. RHE over the Cu–Ag alloy nanowires containing 6% Ag.151 The studies suggest that the presence of Ag may stabilise the Cu2O overlayer and keep optimal availability of the CO intermediate, contributing to the enhanced formation of C2+ compounds. On the other hand, Bell and co-workers proposed a compressive-strain mechanism to explain the role of Ag in enhancing C2+ oxygenate formation over bimetallic Cu–Ag catalysts with surface alloy.215 Briefly speaking, the formation of Cu–Ag surface alloy induced compressed strain in the Cu lattice and thus modified the electronic structure of Cu, leading to decreases in the binding energies of H and O relative to CO. This results in the enhancement of the formation of C2+ oxygenates at the expense of C2H4 due to the reduced coverage of adsorbed H and the reduced oxophilicity of the compressively strained Cu.
Recently, Sargent and co-workers proposed through DFT calculations that after the introduction of Ag on Cu surfaces, the intermediates for the formation of C2H5OH would become more favourable than the formation of C2H4.152 They fabricated Ag/Cu catalysts with different Ag/Cu ratios by a co-sputtering technique. The morphology and in situ XAS characterization indicated that Cu and Ag were distributed homogeneously in the bimetallic catalysts, forming Cu–Ag alloy phase. The best CO2RR performance was achieved over the Ag0.14/Cu0.86 catalyst, which exhibited a maximum FE of C2H5OH of 41% with a current density of 250 mA cm−2 at −0.67 V vs. RHE in an alkaline flow cell using a 1 M KOH electrolyte.152 The cathodic energy efficiency reached 25%. Further in situ Raman spectroscopy studies suggest that the introduction of Ag into Cu could lead to multiple different binding configurations. The C2H4 formation path would be suppressed due to the unsaturated nature of C2H4 formation intermediates.
DFT calculations also suggest that the presence of vacancies on Cu surfaces, in particular on a Cu shell with a Cu2S core, may increase the activation energy barrier for C2H4 formation via a *CH2CHO intermediate, which is believed as a common intermediate for both C2H4 and C2H5OH formations, while leaving that for C2H5OH formation mostly unaffected.153 The Cu2S@Cu-V catalyst with a Cu2S core and surface vacancy-enriched Cu shell, which is known as a core–shell-vacancy engineering (CSVE) catalyst, showed enhanced formation of C2H5OH and C3H7OH. In an alkaline flow cell with 1 M KOH as an electrolyte, the Cu2S@Cu-V catalyst exhibited maximum FEs of C2H5OH and C3H7OH of 25 and 7% with a total current density of 400 mA cm−2, respectively.153 Cuenya and co-workers recently investigated the roles of surface defects and Cu(I) sites on Cu(100) surfaces in the formation of C2+ compounds.216 They controlled the generation of surface defects and Cu(I) species on Cu(100) by designing a pulsed potential sequence applied, i.e., a brief pulse at an anodic potential to reconstruct the Cu(100) surface and generate Cu(I), followed by a pulse at a cathodic potential (typically −1.0 V) for CO2RR. The result indicated that the continuous regeneration of defects and Cu(I) species synergistically enhanced the C–C coupling especially to C2H5OH.216
A tandem CO2 electrocatalysis composed of two working electrodes (Ag and Cu) in a flow cell with 0.1 M CsHCO3 electrolyte showed high FEs of C2+ oxygenates (Fig. 21).217 Ag in the upstream worked for electrocatalytic CO2RR to CO, which was then transported by convective flow to Cu to be converted to C2+ products. The ratio of oxygenates to C2H4 reached a maximum by operating Ag and Cu electrodes at −1 and −0.8 V, respectively, and the maximum FE of C2+ oxygenates was ∼30%.217 This tandem-catalysis concept has also been exploited to design and fabricate electrocatalysts such as Cu–Zn, Cu–Au and Cu–Ag bimetallic systems for CO2RR to enhance the selectivity towards the formation of C2H5OH.149–152,214 Furthermore, porphyrin-based metallic complex-immobilised Cu catalysts were recently reported to enhance the formation of C2H5OH during CO2RR via the tandem mechanism.154 A FeTPP[Cl] (5,10,15,20-tetraphenyl-21H,23H-porphine iron(III) chloride)-modified Cu catalyst showed a maximum FE of C2H5OH of 41% with a partial current density of >100 mA cm−2 at −0.82 V in a flow cell with a 1 M KHCO3 electrolyte. It was confirmed using 13CO2 that the carbon in C2H5OH originated from CO2. DFT calculations and control experiments suggested that the molecular complex mainly functioned for the reduction of CO2 to CO, generating a rich *CO environment at molecule-metal interface, increasing the coverage of *CO on Cu surfaces.154 This lowers the energy barrier for C–C coupling and favours the formation of C2H5OH.
 | ||
| Fig. 21 Schematic illustration of the tandem-catalysis strategy for CO2RR to C2+ products.217 Reproduced from ref. 217 with the permissions of American Chemical Society, copyright 2019. | ||
Carbon materials have also shown potential to modulate the products on Cu/carbon-catalysed CO2RR.155–158,218 An early study reported that Cu nanoparticles loaded on a highly textured N-doped carbon nanospike film (CNS) could achieve high ethanol selectivity in electrocatalytic CO2RR, whereas there was no ethanol formation over Cu/glassy carbon or bare CNS.155 It was claimed that the Cu/CNS offered 63% FE of C2H5OH at −1.2 V versus RHE, whereas CO and CH4 were the major CO2RR products on CNS or Cu/glassy carbon catalysts (Fig. 22a). A synergistic effect between Cu and CNS was speculated to accelerate the formation of C2H5OH. Very recently, Sargent and co-workers developed an electrocatalyst composed of Cu nanoparticles surrounded by N-doped carbon layers (denoted as N–C/Cu), which displayed high FEs of C2H5OH at high reaction rates.156 The CO2RR in a flow cell with 1 M KOH electrolyte using the 34% N–C/Cu (the percentage of N in the N–C layer = 34%) catalyst offered a total C2+ FE of 93% and a C2H5OH FE of 52% at a current density of 300 mA cm−2 at −0.68 V vs. RHE.156 This C2H5OH FE was significantly higher than that on Cu alone and the use of a carbon-only layer to cover Cu did not improve the FE of C2H5OH as compared to Cu alone (Fig. 22b).156 It is noteworthy that no C2H5OH was formed on the bare 34% N–C layer. These observations indicate the crucial role of the N–C layer in Cu surfaces in the formation of C2H5OH. The indispensability of nitrogen might imply that the electron-donating ability of the capping layer is a key. DFT calculations suggest that the confinement effect of Cu by an N–C layer may stabilise the C–O bond of *CH–CHOH, the intermediate for the formation of either C2H5OH or C2H4, thus facilitating the path toward C2H5OH and suppressing that toward C2H4.
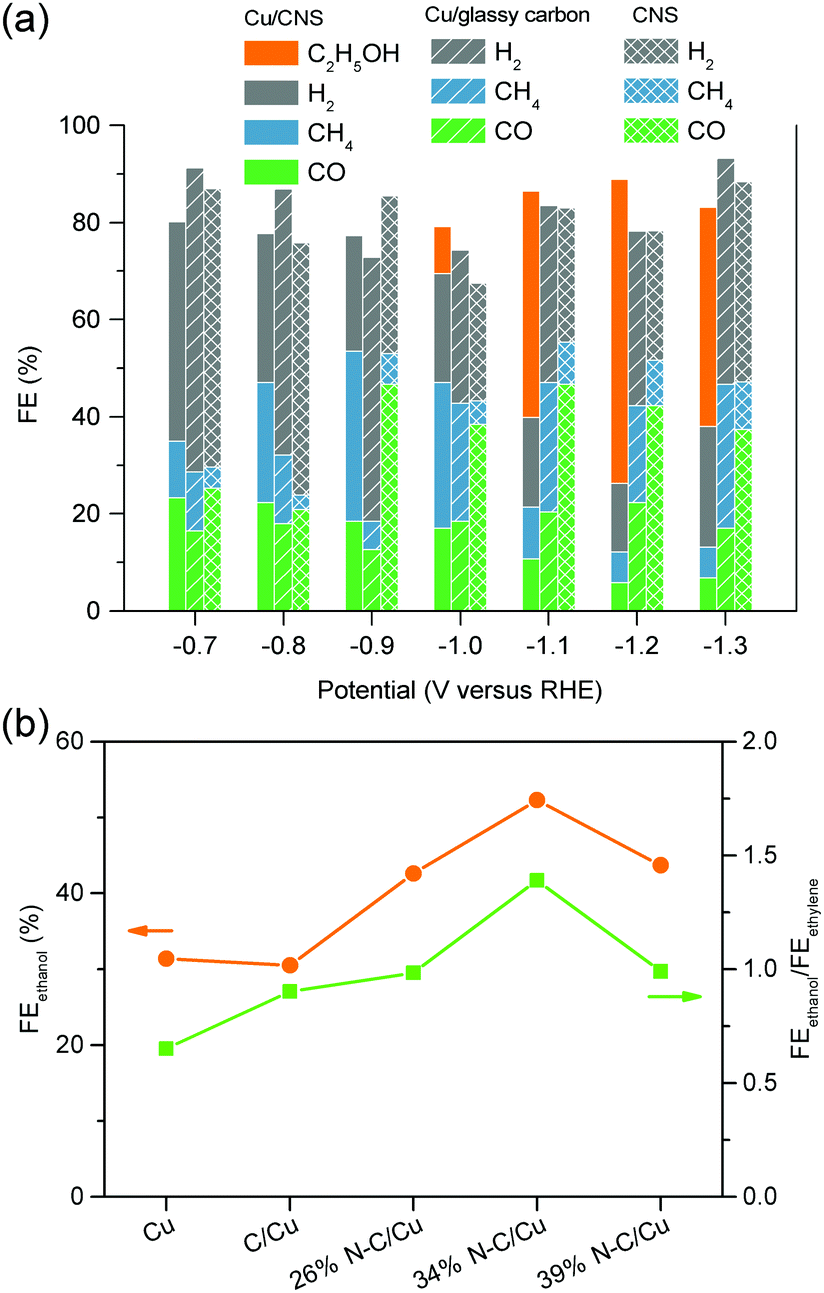 | ||
| Fig. 22 (a) FEs of CO2RR products at different potentials on Cu/CNS, Cu/glassy carbon and CNS catalysts.155 (b) FEs of C2H5OH and the ratios of FEethanol to FEethylene at 300 mA cm−2 on different catalysts.156 | ||
It is noteworthy that g-C3N4 cannot only be applied to photocatalytic CO2 reduction,63 but also holds the potential to tune the selectivity of C2+ products on a Cu catalyst during the electrocatalytic CO2RR. From DFT calculations, Qiao and co-workers revealed that g-C3N4 could modify the electronic structure of Cu in a Cu–C3N4 composite by shifting its d-orbital position toward the Fermi level, thus leading to a stronger adsorption of intermediates.218 In addition, the Cu–C3N4 composite shows an intramolecular synergistic effect with dual active centres (Cu and C) on electrocatalytic CO2RR, different from the widely studied bifunctional catalysts composed of a metallic site and a support. Actually, the fabricated Cu–C3N4 catalyst was able to produce a wider variety of C2 products (C2H4, C2H6 and C2H5OH) in electrocatalytic CO2RR, whereas a Cu/N-doped graphene catalyst only provided C1 products.218
A comparison of CORR and CO2RR on polycrystalline Cu has demonstrated that there is a large decrease in the overpotential for C–C coupling products during CORR.202 Some studies have demonstrated that the electrocatalytic CORR on single Cu catalysts operated at a low overpotential can afford C2H5OH as the major product.163–167,219 whereas C2H4 is usually formed as the major product in CO2RR on single Cu catalysts operated at higher overpotentials. For example, Kanan and co-workers found that an oxide-derived nanocrystalline Cu catalyst, which was fabricated by annealing polycrystalline Cu foil at 500 °C in air followed by electroreduction, offered C2+ oxygenates (C2H5OH, CH3COO− and C3H7OH) with an FE of 57% (C2H5OH FE: 43%) at −0.3 V versus RHE for electrocatalytic CORR.165 On the other hand, a commercial Cu-nanoparticle catalyst provided H2 as the major product under the same conditions. Characterisation of the oxide-derived Cu catalyst revealed particles with sizes of 30–100 nm and interconnected nanocrystalline networks with distinct grain boundaries between nanocrystallites. The oxide-derived Cu catalyst reduced by H2 at 130 °C instead of electroreduction also showed nearly 50% FE towards C2+ oxygenates (C2H5OH and CH3COO−) in electrocatalytic CORR.165 However, the annealing of the oxide-derived Cu catalyst in N2 at 350 °C resulted in significant decreases in surface-area corrected current density and FE of CO reduction (<5%) as well as a remarkable increase in grain sizes. It is thus proposed that the excellent C2+ oxygenates (mainly C2H5OH) selectivity on oxide-derived Cu is not a consequence of nanocrystalline size or morphology. Instead, the grain boundaries play a pivotal role in electrocatalytic CORR to C2+ oxygenates.165,219
Kanan and co-workers further fabricated Cu/carbon nanotube (Cu/CNT) catalysts with different average grain-boundary densities by depositing Cu onto a film of superaligned CNTs using e-beam evaporation.166 TEM characterisation showed that CNTs were decorated with Cu NPs, most of which consisted of multiple Cu crystallites connected by grain boundaries. The density of grain boundaries, defined as the sum of surface grain boundary lengths divided by the sum of NP surface areas, was evaluated by TEM, and the fabricated Cu/CNT had a grain boundary of 40.6 ± 4.3 μm−1. The density of grain boundaries decreased monotonically by increasing the annealing temperature from 200 to 500 °C in N2. The Cu/CNT catalyst showed an FE of 72% towards C2H5OH and CH3COO− with a current density of 0.37 mA cm−2 at −0.3 V during the electrocatalytic CORR.166 The catalyst after annealing showed increased FE of H2 evolution at the expense of CO-reduction products. A strong relationship was found between the density of surface grain boundaries and the CO electrocatalytic activity.
Cu nanowires, which were fabricated by reducing CuO nanowires by H2 at 150 °C, also showed higher selectivity of C2+ oxygenates during CORR.167 The FE of C2H5OH reached 50% with 65% FE towards total CO reduction products at a current density of −0.36 mA cm−2 at −0.22 V vs. RHE.167 The H2 reduction of CuO nanowires at higher temperatures led to significantly lower FE of CO reduction. Characterisation and DFT calculations suggested that the coordinately unsaturated (110) surface sites on Cu nanowires might be responsible for CORR to ethanol.167 Jaramillo, Hahn and co-workers recently demonstrated that the roughness factor of a Cu electrode was a key to determining the product selectivity of CO reduction.169 By comparing Cu catalysts with different morphologies, such as nanoflowers, nanodendrites, nanowires and nanorods, they found that the activity of CORR and selectivity of C2+ oxygenates increased almost linearly with an increase in the roughness factor. It is of interest that although the current density was quite low, the nanoflower Cu catalysts with the highest roughness factor could offer a nearly 100% selectivity to C2 oxygenates (C2H5OH, CH3CHO and CH3COO−) without detectable H2 evolution at an applied potential of only −0.23 V versus RHE. Acetaldehyde was the major product with an FE of ∼60% and the current density was ∼0.2 mA cm−2 at this applied potential. The change in the applied potential to −0.33 V increased the current density to ∼0.6 mA cm−2, and meanwhile C2H5OH became the major product with an FE of ∼60%. It was proposed that the porous mesostructure might contribute to suppressing H2 evolution and enabling highly selective formation of C2 oxygenates at a quite low overpotential.169
Very recently, Jaramillo, Hahn and co-workers demonstrated that a Cu–Ag bimetallic electrocatalyst could achieve a high FE of CH3CHO during electrocatalytic CORR in a 0.1 M KOH electrolyte.170 The FE of CH3CHO reached 50% and the carbon-based selectivity of CH3CHO reached 90% at a potential of −0.536 V vs. RHE. The FE of CH3CHO could be further enhanced to 70% by using a porous bimetallic Cu–Ag nanoflower electrocatalyst with an increased roughness factor. DFT calculations suggested that the Ag-adatoms on Cu weakened the binding energy of the reduced acetaldehyde intermediate, thus suppressing its further reduction to C2H5OH.
4.3. Electrocatalytic CORR into acetate over Cu-based catalysts
Acetate is a versatile chemical in the current chemical industry, which can be used for the production of vinyl acetate monomer, cellulose acetate, acetate esters and polyhydroxyalkanoate (PHA) or used as a solvent in many processes such as the production of polyethylene terephthalate (PET). Recent studies demonstrated that the electrocatalytic synthesis of acetic acid would also be a very promising route for the utilization of CO2via CO.43As described above, although electrocatalytic CORR could offer high FEs of C2 oxygenates at relatively low overpotentials, the current density in the conventional H-cell was typically very low (<10 mA cm−2) mainly due to the low solubility of CO.165–167,169,170 Recently, Kanan and co-workers designed a flow cell with GDE to circumvent the problem of low CO solubility.171 It is reported that the electrocatalytic CORR in the flow cell with a Cu catalyst and NaOH electrolyte could achieve not only high current density (>100 mA cm−2) and high C2+ FE but also high CO single-pass conversion. At a full cell potential of 2.4 V, the CO single-pass conversion reached 43% and the acetate concentration reached 1.1 M over 24 h. Meanwhile, the FE of acetate and current density were 30% and 144 mA cm−2, respectively. It is noteworthy that in the work reported by Kanan and co-workers, very limited CO flow rate (1.0 mL min−1) and high CO pressure (4 bar) were used.171
At the same time, Jiao and co-workers discovered that after switching the reactant from CO2 to CO over an oxide-derived Cu catalyst in an alkaline flow cell, the selectivity of C2+ oxygenates especially for acetate increased significantly and the FE of acetate could reach ∼20% (Fig. 23a).163 The C18O isotopic-labelling studies revealed that one oxygen of acetate originated from CO and the other oxygen originated from the electrolyte, probably from an OH−. The pH-gradient simulations for CO2/CO reduction under various current densities clarified that the pH at electrode surfaces during CO reduction was much higher than that during CO2 reduction, because of the carbonate formation through a fast chemical reaction between CO2 and KOH. They thus attributed the high acetate selectivity in CORR to higher local pH at the electrode–electrolyte interface.
 | ||
| Fig. 23 (a) CO/CO2 reduction on OD-Cu at 300 mA cm−2 in 1 M KOH over 2 h.163 (b) Acetate molar fraction excluding hydrogen for CO reduction over (111) Cu nanosheet, 25 nm Cu particle and 1 μm Cu nanoparticle catalysts.172 Reproduced from ref. 163 and 172, with the permission of Springer-Nature, copyright 2018 and 2019. | ||
The same group further reported a freestanding triangle-shaped two-dimensional Cu-nanosheet catalyst, which exhibited an acetate FE of 48% with a current density of 200 mA cm−2 at −0.736 V vs. RHE for electrocatalytic CORR in a flow cell with a 2 M KOH electrolyte.172 This value of FE is the highest one reported to date for electrocatalytic synthesis of acetate. The Cu nanosheet catalyst, which was fabricated by chemical reduction of Cu(II) nitrate with ascorbic acid in the presence of hexadecyltrimethylammonium bromide (CTAB) and hexamethylenetetramine (HMTA), selectively exposed the (111) surface. As compared to Cu nanoparticles with polycrystalline surfaces, the Cu-nanosheet catalyst exhibited a similar intrinsic activity towards acetate formation but much suppressed intrinsic activities towards the formations of ethylene and ethanol, thus leading to a higher selectivity of acetate (Fig. 23b). Previous studies indicated that (100) and (110) facets were responsible for the formations of C2H4 and C2H5OH, respectively.178 Thus, the preferentially exposed (111) surface of the Cu-nanosheet catalyst was proposed to contribute to the high acetate selectivity.172 DFT calculations suggested that the formation of acetate may proceed through a ketene intermediate (CH2CO) with the oxygen from a CO molecule, and the incorporation of another oxygen from H2O (or OH−) resulted in the formation of acetic acid or acetate.
4.4. Electrocatalytic CORR into n-propanol over Cu-based catalysts
Higher alcohols, such as n-propanol, which have impressive volumetric energy densities (27 MJ L−1 for n-C3H7OH) and excellent octane numbers (118 for n-C3H7OH), can be used as engine fuels. Typically, n-C3H7OH can be observed accompanied by C2H5OH and/or C2H4 in electrocatalytic CO2RR or CORR on Cu catalysts under suitable conditions, but its selectivity is usually lower. For example, n-C3H7OH was formed with an FE of ∼5% in CO2RR on a Cu catalyst composed of monodisperse Cu nanoparticles deposited on a carbon paper support.220 The Cu2S@Cu-V catalyst with a Cu2S core and surface vacancy-enriched Cu shell offered a n-C3H7OH FE of ∼7%.153 A high FE (∼13%) of n-C3H7OH was once reported during electrocatalytic CO2RR on a Cu mesh-supported oxide-derived Cu catalyst at a potential of −0.9 V vs. RHE.221 In electrocatalytic CORR, the oxide-derived Cu catalyst provided an FE of n-C3H7OH of ∼10%.165 It is expected that the key step in n-propanol formation may be the coupling between surface-adsorbed C1 and C2 intermediates. However, the inadequate stabilization of C2 intermediates on pristine Cu surfaces would result in desorption rather than further intermolecular coupling with C1 intermediates to form C3 products. A few recent studies have contributed to developing strategies to design selective catalysts for the formation of n-C3H7OH.173–175The first strategy is the confinement effect to enhance the concentration of C2 intermediates. Sargent, Sinton and co-workers fabricated open Cu-nanocavity catalysts with tuneable geometry by acidic etching of Cu2O nanoparticles and found that suitable opening could significantly promote the formation of n-propanol during electrocatalytic CORR.173 The FE of n-propanol reached 21% with a partial current density of 7.8 mA cm−2 at −0.56 V versus RHE in an alkaline GDE, much better than solid and fragment Cu catalysts (Fig. 24a). Through finite element method (FEM) simulations, they found that the nanocavity geometry could concentrate C2 species via steric confinement and reduce the desorption of C2 species, thus promoting the coupling to form a C3 product.173 DFT calculations suggest that the coupling between C2 species and CO is the most likely route for the formation of C3 species. A lower C2 surface coverage would reduce the likelihood that CO and C2 species meet to form C3 species, and thus the enhancement in the concentration of C2 species is vital to increasing the C3 selectivity.
 | ||
Fig. 24 (a) FEs of C2+ products in CORR at −0.56 V versus RHE on Cu-based catalysts with morphologies of solid, cavity I, cavity II and fragment as well as the corresponding SEM images. Scale bars, 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) nm.173 (b) FEs of C2+ products in CORR at −0.45 V versus RHE on high-fragment Cu (HF-Cu), medium-fragment Cu (MF-Cu) and low-fragment Cu (L-Cu) catalysts.174 (c) FEs of C2+ products in CORR at different potentials on Cu adparticle catalysts.175 (d) Maximum FEs of n-propanol and ratios of n-propanol FE to C2+ FE on Cu nanoparticle (NP), Cu nanobump (NB) and Cu adparticle (AD).175 Reproduced from ref. 173–175, with the permission of Springer-Nature, copyright 2018 and 2019. nm.173 (b) FEs of C2+ products in CORR at −0.45 V versus RHE on high-fragment Cu (HF-Cu), medium-fragment Cu (MF-Cu) and low-fragment Cu (L-Cu) catalysts.174 (c) FEs of C2+ products in CORR at different potentials on Cu adparticle catalysts.175 (d) Maximum FEs of n-propanol and ratios of n-propanol FE to C2+ FE on Cu nanoparticle (NP), Cu nanobump (NB) and Cu adparticle (AD).175 Reproduced from ref. 173–175, with the permission of Springer-Nature, copyright 2018 and 2019. | ||
The second strategy is to create interfaces of (100) and (111) facets, which are selective towards C2 and C1 species, respectively.174 For this purpose, a highly fragmented (HF) Cu catalyst was fabricated by a slow hydrolysis and oxidation of CuI in aqueous solution to Cu2O with a variety of crystalline phases, followed by in situ reduction on a GDE during CORR. TEM confirmed that the HF-Cu catalyst possessed Cu(100) and Cu(111) facets that were adjacent and comprised mostly fragments below 200 nm2 (all fragments <700 nm2). The electrocatalytic CORR in a flow cell with GDE and 1 M KOH electrolyte showed that the HF-Cu catalyst exhibited a maximum FE of n-C3H7OH of 20.3% at −0.45 V vs. RHE (an overpotential of 0.55 V, iR corrected) (Fig. 24b).174 The partial current density for n-C3H7OH formation was 8.5 mA cm−2 at this electrode potential and the full cell electric power to chemical energy conversion efficiency was 10.8%. The control experiments with medium-fragmented (MF) and low-fragmented (LF) Cu catalysts further demonstrated that the interface between Cu(100) and Cu(111) played a critical role in the formation of n-C3H7OH (Fig. 24b).
The third strategy is to load Cu adparticles on Cu surfaces to enhance the adsorption of CO and binding energy of C2 intermediates.175 DFT calculations indicated that the presence of adatoms on pristine Cu surfaces, i.e., Cu(100), Cu(111) and Cu(211), could increase the CO adsorption. The adsorption of C2 intermediates (*CCH2 and *OCCOH) was stabilised on the Cu(111) surface in the presence of Cu adatoms. The reaction energies for *CO dimerization and the coupling between *CO and *CCH2 or *OCCOH were lowered in the presence of Cu adatoms on the pristine Cu surfaces. Adparticle-covered Cu nanoparticle catalysts were fabricated by in situ electroreduction of a nanoparticulate copper oxide precursor deposited on a GDE under rich CO conditions, which enabled simultaneous rapid reduction of oxide and growth of adparticles. Electrocatalytic CORR in a flow cell with a 1 M KOH electrolyte showed a maximum FE of n-C3H7OH of 23% at −0.44 to −0.47 V vs. RHE (Fig. 24c). The partial current of n-C3H7OH reached 11 mA cm−2 at −0.47 V vs. RHE. The control experiments using adparticle-free Cu nanoparticles and the catalyst by reduction of the nanoparticulate copper oxide under N2 instead of CO, which contained nanobumps (NBs) on the surfaces, showed lower maximum FEs of n-C3H7OH (Fig. 24d). The partial pressure of CO played a key role in the formation of n-C3H7OH and it was found that the FE of n-C3H7OH increased at the expense of that of C2H4 upon increasing the CO partial pressure.175 This further supports the hypothesis that n-C3H7OH is formed by coupling between CO and the C2 intermediate, which may also be converted to C2H4.
4.5. Electrocatalytic CO2RR and CORR into C2+ over catalysts other than Cu
High FEs towards C2+ compounds have made Cu a star catalyst for electrocatalytic CO2RR and CORR. One of the major reasons for the high C2+ FEs of Cu-based catalysts is believed to be the appropriate CO-binding ability. If a metal binds CO too weakly, CO would desorb rapidly as a final product, whereas a too strong CO adsorption of a metal would increase the difficulty for the further conversion of surface CO species. The CO2RR to C2+ products on Cu usually require potentials more negative than or close to −1.0 V to achieve high current densities. The search for other electrocatalytic materials other than Cu might enable the CO2RR to C2+ products to proceed at lower overpotential, thus resulting in alternative lower-energy pathways.Several bimetallic catalysts that do not contain Cu have shown potential to offer C2+ products during CO2RR. Kortlever et al. demonstrated that a Pd–Au bimetallic catalyst fabricated by electrodeposition of Pd, which could bind CO strongly, on Au without strong binding to CO afforded C1–C5 hydrocarbons and C2+ oxygenates including ethanol and acetate besides HCOOH and CH3OH.222 The onset potential for the formation of hydrocarbons was −0.8 V vs. RHE. Electrocatalytic CORR to C1–C5 hydrocarbons could also take place on the Pd–Au catalyst with a slightly lower onset potential of −0.6 V, suggesting that CO was a key intermediate. The active sites were proposed to be Pd-rich Au–Pd alloy thin layer on the catalyst surface. The FE of C2+ products was <5%. Several Ni-based bimetallic catalysts have also been investigated for electrocatalytic CO2RR to C2+ compounds.223–225 The Ni–Ga intermetallic compound is an interesting catalyst, since it has similar oxygen adsorption energy to Cu and has shown comparable or even better performance as compared to the well-known Cu–Zn–Al catalyst in the hydrogenation of CO2 to CH3OH.223 In electrocatalytic CO2RR, the Ni–Ga intermetallic catalyst could work for the formation of C2+ hydrocarbons, mainly C2H4 and C2H6, in addition to CH4.224 The onset potential for the formation of C2H4 and C2H6 on the Ni–Ga was −0.48 V vs. RHE, even more positive than that reported for Cu catalysts. The Ni3Al intermetallic compound was reported to show the formation of C3 oxygenates, including n-C3H7OH and acetone, during the electrocatalytic CO2RR.225 The FEs of C2+ products using these bimetallic electrocatalysts that do not contain Cu are still quite low (<5%).
Nitrogen-doped carbon-based materials have been reported to be candidates for metal-free electrocatalytic CO2RR to C2+ products, in particular C2 oxygenates.159–162,226–228 Quan and co-workers reported that a nitrogen-doped nanodiamond (NDD) catalyst deposited on a Si rod array could work for the electrocatalytic CO2RR to CH3COO− and HCOO− in a NaHCO3 electrolyte.159 The total FE toward CO2 reduction was 91.8% with a current density of ∼7 mA cm−2 at −1.0 V vs. RHE. The FE of CH3COO− reached ∼78% at the same time. The high overpotential for H2 evolution on the NDD catalyst was proposed to favour the FE of CO2 reduction. In situ FT-IR studies suggested the appearance of OOC–COO species, which might be a reaction intermediate. Further studies showed that the presence of nitrogen with suitable content (≥∼2 at%) and its chemical state played crucial roles. The N–sp3C was found to be more active than the N–sp2C. It was speculated that the defect sites and the polarised carbon atoms adjacent to nitrogen might stabilise the CO2˙− through electronic interactions. The coupling between CO2˙− was proposed to account for the formation of CH3COO−.159
A boron- and nitrogen-co-doped nanodiamond (BND) catalyst deposited on a Si substrate was found to be very selective for the formation of C2H5OH during the electrocatalytic CO2RR in H-cell with a 0.1 M NaHCO3 electrolyte.160 A surprisingly high C2H5OH FE of 93.2% was claimed on a BND catalyst with 2.5 at% boron and 4.9 at% nitrogen at −1.0 V versus RHE in spite of the low current density (∼−1 mA cm−2). The increase in nitrogen content in BND catalysts from 3.1 at% to 3.6 at% and further to 4.9 at% (from BND1 to BND2 and further to BND3) while keeping the boron content at 2.4–2.5 at% increased the FE of C2H5OH (Fig. 25a). It is of interest that the nitrogen-doped nanodiamond (NDD) mainly provided CH3COO− and HCOO−, whereas HCHO and HCOO− were the major products on the boron-doped nanodiamond (BDD) catalyst (Fig. 25a). Thus, the doping of nitrogen may be the key to C–C coupling. DFT calculations on the (111) facet of BND suggested that the CO2RR might proceed via the following intermediates: CO2 → *COOH → *CO → *COCO → *COCOH → *COCHOH → *COCH2OH → *CHOCH2OH → *CH2OCH2OH → CH3CH2OH.160 The doped nitrogen and boron would work synergistically for the formation of C2H5OH.
 | ||
| Fig. 25 (a) FEs for CO2 reduction on NDD, BDD, BND1, BND2 and BND3 at −1.0 V versus RHE.160 (b) FEs for CO2 reduction on cylindrical mesoporous carbon (c-NC) and inverse mesoporous carbon (i-NC) catalysts at various applied potentials.226 (c) FEs for CO2 reduction at various applied potentials for NGQDs.162 Reproduced from ref. 160, 226 and 162, with the permission of Wiley-VCH Verlag GmbH&Co. KGaA and Springer-Nature, copyright 2017 and 2016. | ||
Chen, Sun and co-workers demonstrated that a N-doped ordered cylindrical mesoporous carbon (denoted as c-NC) catalyst showed high selectivity toward C2H5OH during CO2RR in a 0.1 M KHCO3 electrolyte.226 The FE of C2H5OH reached 77% with a current density of ∼0.2 mA cm−2 at −0.56 V vs. RHE (Fig. 25b). The presence of nitrogen was also confirmed to play key roles in CO2 reduction and C–C coupling, since H2 evolution was the major reaction on the ordered cylindrical mesoporous carbon without nitrogen. The cylindrical mesoporous structure was also important, and the reference N-doped inverse mesoporous carbon only showed a much lower FE of C2H5OH (∼44%) at −0.50 V vs. RHE and CO was formed as a major product. DFT calculations suggested that the pure carbon or graphitic N sites were highly unfavourable for the formation of *CO, which was the key intermediate for C2 formation, whereas both pyridinic and pyrrolic N sites could work for the *CO formation.226 The formation of *CO proceeded preferentially on the pyridinic N site. The ordered channel surface with high electron density might stabilise *CO, thus favouring the coupling between *CO, and might also facilitate the subsequent multiple electron and proton transfer to form C2H5OH. A recent work from the same group demonstrated that the pore-structure engineering by constructing hierarchical micro-/mesoporous N-doped carbon materials could further enhance the formation of C2H5OH.161 The creation of medium micropores (∼0.52 nm) in the channel walls of the N-doped ordered mesoporous carbon resulted in an FE of C2H5OH of 78% with a current density of ∼2 mA cm−2 at −0.56 V vs. RHE during CO2RR in H-cell with a 0.1 M KHCO3 electrolyte. The formation rate of C2H5OH reached 2.3 mmol g−1 h−1 at −0.8 V vs. RHE, which was one order of magnitude higher than that on the corresponding catalyst without micropores. Further experiments and DFT calculations suggested that the microporous structure with the active pyridinic and pyrrolic N sites might lead to fast kinetics of charge transfer and high electric driving potentials to enhance the formation of ethanol.161
N-Doped graphene quantum dots (NGQDs) were also found to be efficient for the electrocatalytic CO2RR into C2+ products.162 C2H4, C2H5OH, CH3COO− and n-C3H7OH were all formed in CO2RR in a flow cell with DGE and 1 M KOH electrolytes. The maximum FE of C2H4 was 31% at −0.75 V versus RHE, and the total FE of C2+ products was 55% at the same time (Fig. 25c).162 The maximum FE of C2+ oxygenates was 26% at −0.78 V versus RHE with 16% FE of C2H5OH. The flow cell with DGE also resulted in high current density, and the partial current densities of CO, C2H4 and C2H5OH were 23, 46 and 21 mA cm−2 at −0.86 V vs. RHE. On the other hand, CO and HCOO− were the major products on the graphene quantum dots without nitrogen or the nitrogen-doped reduced graphene at similar applied potentials. Thus, it was confirmed again that the nitrogen sites played crucial roles in the formation of C2+ products. Further, the result indicated that the pyridinic N at edge sites were more efficient for C–C coupling than the pyridinic N at basal planes.
Recently, some studies have demonstrated that the addition of a catalyst component that can accelerate the CO2RR to CO and the N-doped carbon can enhance the formation of C2 oxygenates (Fig. 26).229–231 For example, the immobilisation of a molecular catalyst, i.e., Ru(II) polypyridyl carbine complex (RuPC), which was efficient for electrocatalytic CO2RR to CO, onto N-doped porous carbon (NPC) was found to show synergistic effects in the formation of C2+ products.229 The NPC alone could catalyse the electrocatalytic CO2RR in H-cell with a 0.5 M KHCO3 electrolyte, forming C2H5OH, CH3COO−, methanol, HCOO− and CO. The immobilisation of RuPC onto NPC significantly enhanced the formation of C2H5OH. At −0.97 V vs. NHE, the FEs of C2H5OH and CH3COO− were 27.5% and ∼10%, respectively, on the RuPC/NPS, whereas they were ∼15% and 10% on NPC. It was speculated that the RuPC enhanced the electrocatalytic reduction of CO2 to CO and the increased coverage of *CO species on NPC or at the interface between RuPC and NPC would facilitate the C–C coupling to form C2H5OH. The loading of Ag nanoparticles, which are known to catalyse the CO2RR to CO, onto a 3D graphene-wrapped nitrogen doped carbon foam (G–NCF) has also been found to enhance C2H5OH formation during electrocatalytic CO2RR in a H-cell with 0.1 M KHCO3 electrolyte.230 The FE of C2H5OH reached 85.2% at −0.6 V vs. RHE despite the low current density (<0.5 mA cm−2). The anchoring of CoO onto N-doped carbon materials composed of mesoporous carbon and carbon nanotube resulted in an efficient catalyst (Co/MC–CNT) for electrocatalytic CO2RR to ethanol.231 The maximum FE of C2H5OH on this catalyst was 60.1% at −0.32 V vs. RHE during the electrocatalytic CO2RR in H-cell with a 0.5 M KHCO3 electrolyte. CH3CHO, probably the precursor of C2H5OH, and H2 were observed as the only by-products under such conditions. The FE of CH3CHO was 10% and the partial current of (C2H5OH + CH3CHO) was 5.1 mA cm−2 at −0.32 V vs. RHE. Further studies indicated that CoO facilitated the formation of *CO intermediate that might undergo spill-over to MC–CNT, where the C–C coupling took place.231 The pyridinic N and pyrrolic N on MC–CNT as well as the mesoporous structure may stabilise *CO species and promote C–C coupling.
 | ||
| Fig. 26 Simplified mechanism on bifunctional catalyst for CO2RR to ethanol by integrating CO synthesis and C–C coupling. | ||
4.6. Mechanism of C–C coupling for electrocatalytic CO2RR and CORR to C2+ compounds
The most important step for the synthesis of C2+ hydrocarbons or oxygenates from CO2 or CO is the C–C coupling. To understand how the C–C coupling occurs and how to control the C–C coupling as well as the formation of different products would be very helpful for rational design of highly selective catalysts for the formation of C2+ compounds.The well-known catalytic system involving the coupling of C1 molecules is Fischer–Tropsch synthesis, i.e., the hydrogenation of CO to C2+ hydrocarbons, which typically proceeds on Fe-, Co- or Ru-based heterogeneous catalysts at high temperatures and pressures.7 The hydrogenation of CO2 to C2+ hydrocarbons on related heterogeneous catalysts usually proceeds via CO intermediate, i.e., the reverse water-gas shift reaction (H2 + CO2 → CO + H2O) followed Fischer–Tropsch synthesis. The mechanism for Fischer–Tropsch synthesis is relatively mature. Take the formation of C2H4 as an example, it is accepted that the reaction proceeds through the following key elementary steps on catalyst surfaces: (1) adsorption and dissociation of CO and H2; (2) formation of surface CHx species; (3) C–C coupling of CHx species; (4) hydrogenation or dehydrogenation of C2Hx into C2H4 (Fig. 27a).7 However, the selective formation of C2H4 on the Fischer–Tropsch catalyst is very difficult because the C–C coupling is generally uncontrollable. Actually, the polymerisation of CHx species to form CnHm with different carbon numbers, also known as chain growth, can occur on the Fischer–Tropsch catalyst surfaces. Thus, the selectivity of products generally follows the Anderson–Schulz–Flory distribution, which is determined by the rate of chain growth and that of chain termination.7 According to the ideal ASF model, the maximum selectivity of C2–C4 (including olefins and paraffins) is only 58%. For the formation of ethanol by hydrogenation of CO over a heterogeneous catalyst, it is generally believed that both dissociated and non-dissociated CO species should exist on the catalyst surface.209 The formation of C2H5OH requires the coupling of CHx species and *CO species, and the formed CHxCO intermediate undergoes further hydrogenation to ethanol (Fig. 27b).
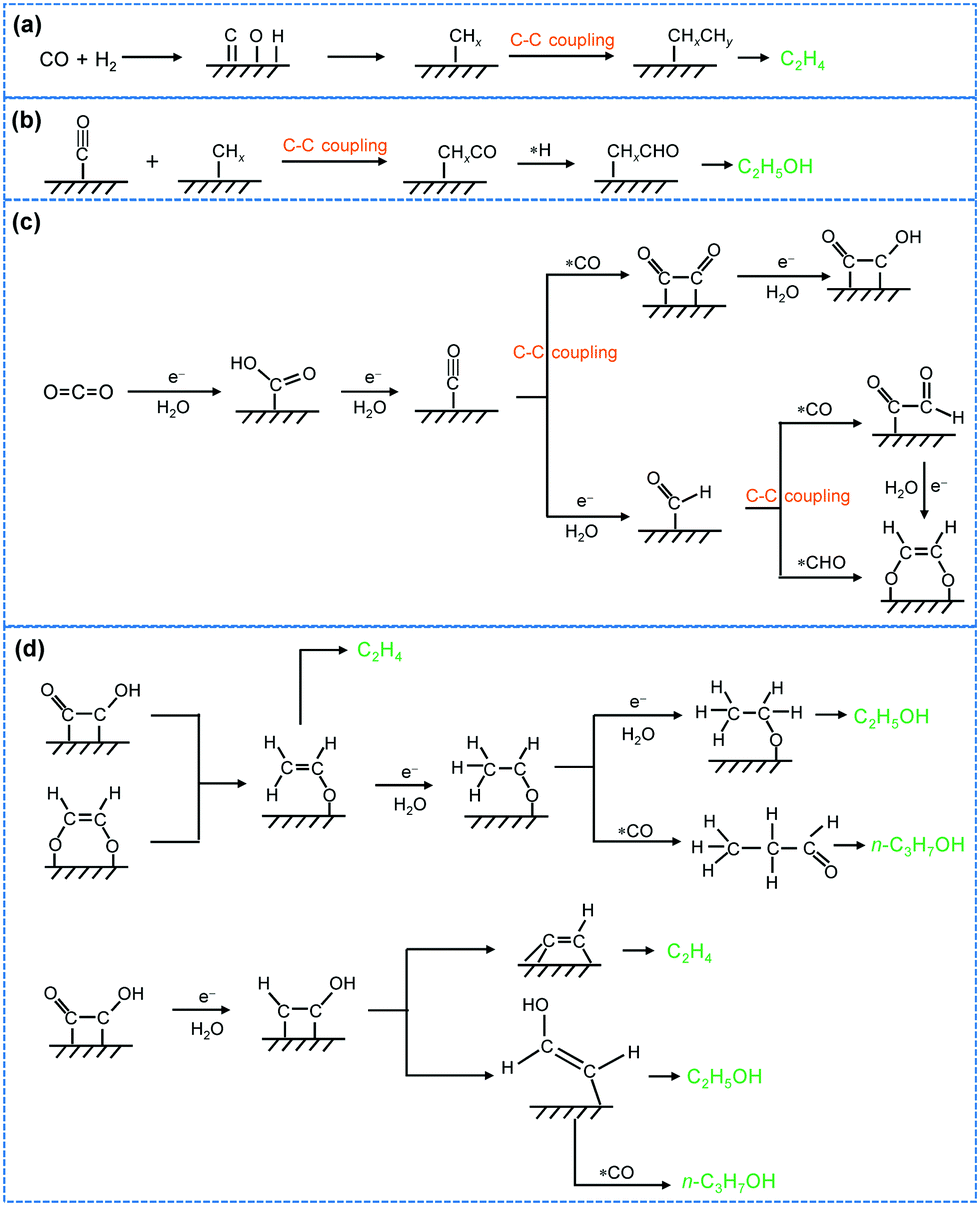 | ||
| Fig. 27 (a) Mechanism for the conversion of syngas to C2H4 in Fischer–Tropsch synthesis. (b) Mechanism for the conversion of syngas to C2H5OH on thermocatalysts. (c) Three possible C–C coupling mechanisms in electrocatalytic CO2RR. (d) Two possible mechanisms for the formation of C2H4, C2H5OH and n-C3H7OH. | ||
A few studies have claimed that similar to the thermocatalysis, the C–C coupling may take place between surface *CH2 species to form C2H4 on Cu nanoparticles during electrocatalytic CO2RR, and meanwhile the coupling between *CH2 and *CO or the insertion of CO contributes to the formation of C2H5OH or CH3CHO.199,201 However, most studies have pointed out that the electrocatalytic CO2RR or CORR under mild conditions (typically ambient temperature and pressure) would proceed in different mechanisms.232–238 The current consensus is that CO is the key intermediate in electroreduction CO2RR. The direct dimerization of CO2 or activated CO2 is very rare.159 Thus, the electrocatalytic CO2RR to C2+ products proceeds via the same pathway as that for CORR after the reduction of CO2 to CO.
Several possibilities have been proposed for C–C coupling of CO or CO-derived intermediates. The direct coupling of *CO to form *OCCO species followed by hydrogenation to C2H4 and C2H5OH has been proposed in most of the studies reported to date (Fig. 27c).232–234 The pathway of direct *CO dimerization has mainly been supported by the DFT calculations. Recently, Calle-Vallejo, Koper and co-workers observed the appearance of adsorbed species with vibrational bands at 1191 and 1584 cm−1 in in situ FT-IR spectroscopic studies during CO reduction on the Cu(100) surface in a LiOH electrolyte, which were assignable to the C–O–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations of *OCCOH species based on DFT calculations.235 The *OCCOH species was a hydrogenated CO dimer intermediate, and thus this provided evidence for the direct dimerization of *CO species. It is of interest that the *OCCOH species could not be observed on the Cu(111) surface, confirming that the coupling of CO is a structure-sensitive reaction. This result also agrees well with the fact that the Cu(100) with square symmetry is more selective for the formation of C2+ products.178,180
O stretching vibrations of *OCCOH species based on DFT calculations.235 The *OCCOH species was a hydrogenated CO dimer intermediate, and thus this provided evidence for the direct dimerization of *CO species. It is of interest that the *OCCOH species could not be observed on the Cu(111) surface, confirming that the coupling of CO is a structure-sensitive reaction. This result also agrees well with the fact that the Cu(100) with square symmetry is more selective for the formation of C2+ products.178,180
On the other hand, some studies have demonstrated the importance and participation of *CHO species in the C–C coupling step (Fig. 27c).142,236,237,239 Nørskov and co-workers studied the kinetic barriers to the formation of a C–C bond between adsorbates on the Cu(211) surface derived from CO using DFT calculations.236 The result demonstrated that the kinetic barriers for C–C coupling decreased significantly with the degree of hydrogenation of reacting adsorbates. It was confirmed that this trend was not affected by the electric field present during the solid–electrolyte interface. The DFT calculations offered energy barriers of 1.619, 0.675, 0.564, 0.364 and 0.203 eV for the coupling steps of (*CO + *CO), (*CO + *CHO), (*CHO + *CHO), (*CHO + *CH2O) and (*CH2O + *CH2O), respectively.236 Generally, the coupling reaction with a kinetic barrier of <0.7 eV can proceed facilely at ambient temperature. Thus, these coupling steps may occur except for the direct dimerization of *CO. It is believed that the unique feature of the electrochemical environment lies in that the chemical potential of hydrogen (electrons and protons) can be tuned by the applied potential. The computational work by Goddard III and co-workers tended to suggest that the *CHO species can be an intermediate for the formation of both CH4 and C2H4.237 Bell, Head-Gorden and co-workers performed DFT calculations using an implicit electrolyte model, which enables a better simulation of the effect of an applied potential by varying on the surface to match the Fermi level to the target potential of electrode electrons, for CO2RR on Cu(100) and Cu(111) surfaces.239 These results strongly suggest that the coupling between *CO and *CHO plays a pivotal role in the formation of C2 compounds and *COCHO is the common intermediate for the formation of C2H4 and C2H5OH.239 Our recent work demonstrated that the coupling of (*CHO + *CHO) could also play a key role in the formation of C2 compounds during CO2RR on a fluoride-modified Cu catalyst, which exhibited excellent FEs and formation rates of C2+ compounds (C2H4 and C2H5OH).142In situ electrochemical attenuated total reflection Fourier-transform infrared spectroscopy (ATR-FTIRS) measurements provided evidence for the generation of *CHO species on the fluoride-modified Cu surface, whereas the *CHO could not be detected on the Cu surface without fluoride modification probably due to the very low concentration.142
Two possible mechanisms have been proposed for the formation of C2+ compounds including C2H4, C2H5OH and C3H7OH (Fig. 27d).127,237,239–241 Koper, Bell and co-workers proposed that after a series of protonation and electron transfer steps, the *OCCOH or *OCHCHO* species is converted to *CH2CHO, which serves as the selectivity-determining intermediate to C2+ products.213,239–241 The hydrogenolysis of *CH2CHO intermediate directs a pathway to C2H4, while the hydrogenation of *CH2CHO results in *CH3CHO and consequently C2H5OH. DFT calculations revealed that the barrier for C2H5OH formation via *CH3CHO was ∼0.2 eV higher than that for C2H4 formation via *CH2CHO.213 This may explain why higher C2H4 selectivity over C2H5OH is usually observed on Cu-based catalysts. Further, n-C3H7OH can be formed through a (*CH3CHO + *CO) coupling step. Alternatively, Goddard III and co-workers suggested that the selectivity-determining intermediate may be formed earlier for the formation of different C2+ products.237 They proposed that the hydrogenation of *OCCOH gave *CHCOH intermediate, which was the selectivity-determining intermediate. After the formation of *CHCOH, the dehydroxylation of *CHCOH to *CCH leads to C2H4, whereas the hydrogenation of *CHCOH to *CHCHOH leads to C2H5OH.
5. Photocatalytic and electrocatalytic conversions of CH4
Methane is the major component of natural gas, shale gas, biogas and methane hydrate, and has abundant reserves and vast availability. Nowadays, CH4 has become a major source for energy and fuels owing to its cleaner feature as compared with other fossil resources, but the chemical use of CH4 is still insufficient.20,242,243 The activation and selective conversion of CH4 is one of the most challenging goals and is regarded a “holy grail” in chemistry because of the much higher stability of CH4 as compared to a potential target product (such as CH3OH and C2H4).244 In the current chemical industry, the transformation of CH4 is performed indirectly via syngas (CO/H2), i.e., the steam reforming of CH4 to syngas followed by conversion of syngas to CH3OH or C2+ hydrocarbons by CH3OH synthesis or Fischer–Tropsch synthesis. The steam reforming of CH4 is a high-cost and energy-intensive process. Although a lot of efforts have been devoted to transforming CH4 directly into chemicals or liquid fuels via thermocatalysis, the selectivity of the target product is generally low at a reasonably high CH4 conversion, because the reactive target product can undergo consecutive dehydrogenation or oxidation to carbon or CO2 under severe conditions. Photocatalysis and electrocatalysis may provide new opportunities for the transformation of CH4 to chemicals and fuels under mild conditions by introducing photo or electrical energy. Several review articles have been published for photocatalytic and electrocatalytic conversions of CH4,55,56,245–248 but those are devoted to the photocatalytic or electrocatalytic conversion of CH4 to C2+ compounds are still quite limited. Here, we focus on recent advances in the transformation of CH4 to C2+ compounds via photocatalysis or electrocatalysis.5.1. Photocatalytic conversion of CH4 to C2+ compounds
Photocatalytic conversion of CH4 alone by a non-oxidative dehydrogenation process mainly offered C2+ hydrocarbons. In the presence of H2O, not only C2+ hydrocarbons but also C2+ oxygenates could be formed in CH4 photocatalytic conversions. Some typical systems for photocatalytic coupling of CH4 in the absence and presence of H2O are summarized in Table 5.249–265| Photocatalyst | Formation rate (μmol g−1 h−1) | Quantum efficiency (QE) | Light source | Ref. |
|---|---|---|---|---|
| Photocatalytic coupling of CH4 alone | ||||
| SiO2–Al2O3 | C2H4, 0.01; C2H6, 0.10; C3H6, 0.005; C3H8, 0.02; C4H8, 0.003; C4H10, 0.002 | — | Xe lamp | 249 |
| SiO2–Al2O3–TiO2 | C2H6, 0.69; C3H8, 0.056 | — | Xe lamp (220–300 nm) | 250 |
| FSM-16 | C2H6, 0.09; C3H8, 0.003 | — | Xe lamp (220–300 nm) | 251 |
| MgO/SiO2 | C2H6, 0.046; C3H8, 0.0008 | — | Xe lamp (220–300 nm) | 252 |
| GaxO/SiO2 | C2H4, 0.005; C2H6, 0.11; C3H6, 0.002; C3H8, 0.002 | 0.01% (220–270 nm) | Xe lamp (220–300 nm) | 253 |
| CexO/Al2O3 | C2H4, 0.053; C2H6, 0.24; C3H6, 0.038; C3H8, 0.0049 | — | Xe lamp (220–300 nm) | 254 |
| Zn2+-ZSM-5 | C2H6, 3.0 | 0.55% (300–400 nm) | High-pressure Hg lamp | 255 |
| Ga3+-ETS-10 | C2H4, 0.61; C2H6, 10.89; C3, 1.18; C4, 0.74 | — | High-pressure Hg lamp | 256 |
| Au/ZnO | C2H6, 11.5 | 0.08% (320–2500 nm) | Xe lamp (320–2500 nm) | 257 |
| Si-Doped GaN | Benzene, 55.6; toluene, 0.5; ethane, 3.3; ethylene, 0.5 | 0.72% (290–380 nm) | Xe lamp (290–380 nm) | 258 |
| Ag–HPW–TiO2 | C2H6, 23; C3H8, 1.2 | 3.5% (362 nm) | Hg–Xe lamp (280–400 nm) | 259 |
| Photocatalytic coupling of CH4 in the presence of H2O | ||||
| BiVO4 | CH3OH, 21; C2H6, 2 | — | Medium-pressure mercury lamp (>185 nm) | 260 |
| Beta zeolite | CH3OH, 242; HCHO, 117; HCOOH, 122 | — | Deep UV irradiation (<200 nm) | 261 |
| BiVO4/V2O5/beta zeolite | CH3OH, 10.7; C2H6, 2.46 | — | Medium-pressure Hg lamp | 262 |
| Pt–TiO2 | C2H6, 50.6 | 3.3% (254 nm) | UV lamp (254 nm) | 263 |
| Pd–TiO2 | C2H6, 54.7 | 2.76% (254 nm) | UV lamp (254 nm) | 264 |
| Cu-0.5/PCN | CH3OH, 24.5; C2H5OH, 106 | — | Xe lamp | 265 |
| 2CH4 → C2H6 + H2, ΔG(298K) = 68.6 kJ mol−1 | (1) |
| 6CH4 → C6H6 + 9H2, ΔG(298K) = 434 kJ mol−1 | (2) |
A series of studies by Yoshida and co-workers showed that porous SiO2 or metal oxide-loaded SiO2 or Al2O3 (e.g., MgO/SiO2, GaOx/SiO2, and CeOx/Al2O3) could catalyse the dehydrogenative coupling of CH4 to C2H6 and H2 under UV light irradiation at mild temperatures.247–254 A SiO2–Al2O3 composite evacuated at 800 °C was first reported to be active for non-oxidative coupling of CH4 to C2+ hydrocarbons (including C2H6, C2H4, C3H8, C3H6, C4H10 and minor C5–6) with C2H6 as the major product under UV-vis light irradiation using a 250 W Xe lamp at 37 °C.249 A later study found that the ternary composite of SiO2–Al2O3–TiO2 (Al 10 mol%, Ti 0.5 mol%) after 800 °C evacuation showed higher performance, offering a C2H6 yield of ∼2% on a molar carbon basis during the conversion of 200 μmol CH4 with a 1.0 g catalyst in a closed system for 3 h under UV-vis light irradiation using a 250 W Xe lamp at 37 °C.250 The pair site composed of dispersed AlO4 and TiO4 in the silica matrix with Ti4+–O2−–Al3+ might function as the active site. Nevertheless, pure SiO2, in particular mesoporous silica such as MCM-41 or FSM-16 could also work for this reaction in spite of lower activity.251 The photocatalytic sites on pure SiO2 were proposed to be surface defect sites such as non-bridging oxygen hole sites (![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Si–O˙), which were generated during pretreatment at a high temperature (e.g., 800 °C) to remove surface hydroxyl groups. The photoexcitation of this site was believed to be the first step, followed by CH4 activation to CH3˙ and H˙ and the coupling of radical intermediates into C2H6 and H2.251 The mesoporous silica with a thinner silica wall such as FSM-16 showed better performance than that with a thicker wall (MCM-41) and amorphous SiO2. The excitation occurred at 258 nm and thus only a part of UV light could be harnessed for this photochemical process. The highest yield of C2+ hydrocarbons (∼91% C2H6 with 8.8% C3H8 and minor C2H4) on FSM-16 (0.20 g) after 3 h of the reaction for 200 μmol CH4 under UV-vis irradiation was ∼0.1%.251 The loading of MgO improved the performance of amorphous silica.252 Ga2O3 alone exhibited much higher yield of C2H6 during the photocatalytic conversion of CH4, but the formation of H2 was very low possibly due to the occurrence of other side-reactions such as coke deposition.253 The dispersion of Ga3+ on SiO2 (0.1 mol% Ga/SiO2) could smoothly catalyse the dehydrogenation of CH4 to C2H6 and H2 with enhanced product formation. The yield of C2H6 reached 0.14% after 3 h irradiation under UV-vis light, which was ∼35 times higher than the pure amorphous silica.253 The bulk Ga2O3 could work as a semiconductor photocatalyst and thus showed high activity, but side-reactions would also occur. On the other hand, the highly dispersed tetrahedral Ga3+ sites on SiO2 surfaces may function as another type of photoactive sites and the photoexcitation may take place on localised isolated sites, thus leading to the selective dehydrogenation of CH4 to C2H6 and H2.253 Similarly, the highly dispersed Ce3+ sites on SiO2 or Al2O3 surfaces could also accelerate the non-oxidative dehydrogenation coupling of CH4 to C2H6 and H2.254
Si–O˙), which were generated during pretreatment at a high temperature (e.g., 800 °C) to remove surface hydroxyl groups. The photoexcitation of this site was believed to be the first step, followed by CH4 activation to CH3˙ and H˙ and the coupling of radical intermediates into C2H6 and H2.251 The mesoporous silica with a thinner silica wall such as FSM-16 showed better performance than that with a thicker wall (MCM-41) and amorphous SiO2. The excitation occurred at 258 nm and thus only a part of UV light could be harnessed for this photochemical process. The highest yield of C2+ hydrocarbons (∼91% C2H6 with 8.8% C3H8 and minor C2H4) on FSM-16 (0.20 g) after 3 h of the reaction for 200 μmol CH4 under UV-vis irradiation was ∼0.1%.251 The loading of MgO improved the performance of amorphous silica.252 Ga2O3 alone exhibited much higher yield of C2H6 during the photocatalytic conversion of CH4, but the formation of H2 was very low possibly due to the occurrence of other side-reactions such as coke deposition.253 The dispersion of Ga3+ on SiO2 (0.1 mol% Ga/SiO2) could smoothly catalyse the dehydrogenation of CH4 to C2H6 and H2 with enhanced product formation. The yield of C2H6 reached 0.14% after 3 h irradiation under UV-vis light, which was ∼35 times higher than the pure amorphous silica.253 The bulk Ga2O3 could work as a semiconductor photocatalyst and thus showed high activity, but side-reactions would also occur. On the other hand, the highly dispersed tetrahedral Ga3+ sites on SiO2 surfaces may function as another type of photoactive sites and the photoexcitation may take place on localised isolated sites, thus leading to the selective dehydrogenation of CH4 to C2H6 and H2.253 Similarly, the highly dispersed Ce3+ sites on SiO2 or Al2O3 surfaces could also accelerate the non-oxidative dehydrogenation coupling of CH4 to C2H6 and H2.254
A Zn+-modified ZSM-5 catalyst was found to show significantly higher activity for non-oxidative coupling of CH4 to C2H6 and H2 than SiO2- or Al2O3-based catalysts.255 The Zn2+-ZSM-5− sample, which was fabricated by a solid-vapour reaction between dehydrated H-ZSM-5 and metallic Zn vapour, contained delocalised electrons on the zeolite framework. The irradiation of Zn2+-ZSM-5− under UV light from a 150 W high-pressure Hg lamp led to the generation of Zn+ as confirmed by ESR. The obtained (Zn+,Zn2+)-ZSM-5− catalyst could work for the non-oxidative coupling of CH4 to C2H6 and H2 under either UV or sunlight irradiation. The formation rate of C2H6 was 9.8 μmol h−1 g−1 under UV-light irradiation from a high-pressure Hg lamp. Almost an equimolar amount of H2 was formed and the selectivity of C2H6 was close to 100%. The conversion of 200 μmol CH4 with a 1.0 g (Zn+,Zn2+)-ZSM-5− catalyst offered a 17.5% C2H6 yield in 24 h UV-light irradiation, and the quantum efficiency was around 0.55%.255 The highest CH4 conversion reached 23.8% with 99% C2H6 selectivity under the optimised conditions. Further studies suggested a two-stage mechanism (Fig. 28a): (1) the generation of (Zn+,Zn2+)-ZSM-5− in the first stage that required irradiation by UV light of 278–390 nm; (2) the dehydrogenative coupling of CH4 by the (Zn+,Zn2+)-ZSM-5− catalyst that could work under visible light of <700 nm.255
 | ||
| Fig. 28 (a) Schematic energy diagram for the processes of the photocatalytic conversion of CH4 to C2H6 over Zn2+-ZSM-5− (inset: optimized geometry of the adsorbed CH4 on Zn+ active site; red: O, blue: Si, pink: Al, gray: C, white: H, and green: the 4s electron of Zn+).255 (b) Photocatalytic performance for the conversion of CH4 over different cation-modified ETS-10 catalysts.256 (c) Proposed reaction mechanism for the photocatalytic conversion of CH4 over Ga3+-ETS-10.256 Reproduced from ref. 255 and 256, with permission of Wiley-VCH Verlag GmbH&Co. KGaA, copyright 2011 and 2012. | ||
A latter study from the same group found that the Ga3+-modified zeolite ETS-10, which was a microporous titanosilicate with a framework containing one-dimensional O–Ti–O–Ti–O semiconducting nanowires (diameter, 0.67 nm) insulated from each other by the surrounding SiO2 matrix, could work for photocatalytic dehydrogenative coupling of CH4, offering a CH4 conversion rate of 29.8 μmol g−1 h−1, which was about 3 times higher than that of (Zn+,Zn2+)-ZSM-5−.256 The ETS-10 samples containing exchanged cations of Ga3+, Al3+, Zn2+ and Fe3+ were all active, and the Ga3+-ETS-10 catalyst was the most efficient for photocatalytic conversion of CH4 (Fig. 28b). During the conversion of 200 μmol CH4 with a 0.2 g catalyst in a closed system, CH4 conversion reached 14.9% with C2H6 selectivity of 70% after 5 h irradiation under UV light from a high-pressure Hg lamp.256 Other products included C2H4, C3H8, C3H6, C4H10 and C4H8. The catalyst was also efficient for photocatalytic conversion of C2H6, offering C4H10 with a selectivity of 57% at C2H6 conversion of 11% after 5 h of reaction. Ga3+ was found to be a key to the reaction, but the Ga3+-ZSM-5 and Ga3+-Y catalyst only showed very poor performances for photocatalytic conversion of CH4. Therefore, the Ga3+ ions and TiO2-wire units in the ETS-10 framework both played crucial roles in photocatalytic conversion of CH4. The in-depth studies by Chen and co-workers indicated a dual active site mechanism (Fig. 28c).256 In other words, the extraframework metal cation (Ga3+) and the Ti4+–OH group on the titanate wire interacted synergistically under UV-light irradiation, leading to the cleavage of a C–H bond in CH4. Probably, CH4 was adsorbed and polarized through the interaction between Ga3+ and a methyl group to weaken the C–H bond, and then the hydrogen abstraction from CH4 took place by the ˙OH radical in the vicinity generated by photoexcitation of the surface Ti–OH species. C2H6 is formed through the C–C coupling of two CH3 surface species or radicals.
Semiconductor-based photocatalysts have also been examined for non-oxidative photocatalytic conversion of CH4 to C2+ compounds.257,258,266 Long and co-workers fabricated a plasmonic photocatalyst consisting of Au nanoparticles and ZnO nanosheets with exposed (001) facets.257 C2H6 and H2 with formation rates of about 11 and 10 μmol g−1 h−1 could be achieved on the optimized Au/ZnO catalyst under simulated solar-light irradiation, and the solar-to-C2H6 energy conversion efficiency was 0.08%. It was proposed that the strong polarisation of the (001) facet of ZnO nanosheets would promote the polarization and activation of the C–H bond of CH4. Meanwhile, the SPR effect of Au nanoparticles offered photogenerated holes and electrons for the reduction of H+ to H atoms to give H2 and the oxidation of CH3− to CH3˙, which coupled to produce C2H6 (Fig. 29).
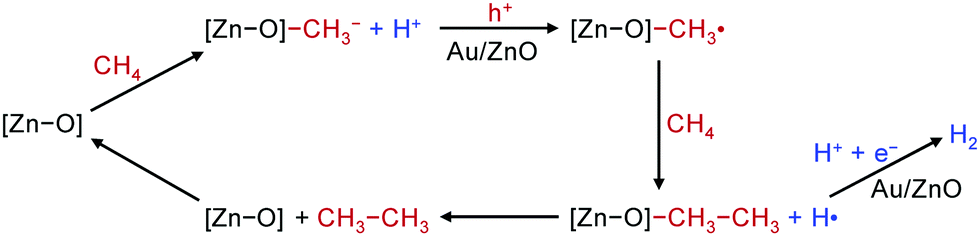 | ||
| Fig. 29 Proposed reaction mechanism for photocatalytic conversion of CH4 over a plasmonic Au/ZnO catalyst.257 | ||
Li and co-workers found that GaN nanowires with a 3% top c-plane and 97% lateral m-plane showed high selectivity of C6H6 during non-oxidative photocatalytic conversion of CH4 under UV light irradiation at room temperature.258 The selectivity of C6H6 in hydrocarbon products was about 97% and H2 was also formed with a ratio of benzene to H2 of 1:9, in agreement with the stoichiometry (eqn (2)). Further studies with GaN samples with different morphologies and different fractions of exposed facets revealed that the reaction was structure sensitive (Fig. 30a). The analysis of results indicated that the lateral m-plane, which was composed of Ga3+ and N3− tetrahedrally coordinated with each other, contributed to CH4 conversions, whereas the c-place made up of only Ga or N atoms had no contribution to the photocatalytic conversion of CH4 (Fig. 30b). Mechanistic studies suggested that the Ga3+ and N3− on the m-plane could interact with CH4, leading to polarisation of Hδ+–CH3δ− and heterolytic dissociation of CH4. The photogenerated electrons and holes would function for the reduction and oxidation of H+ and CH3− to H˙ and CH3˙, respectively. The coupling of radical intermediates could give H2 and C2H6, and benzene was formed by the dehydrogenation of C2H6 to C2H4, followed by cyclization and dehydrogenation processes (Fig. 30b).258
 | ||
| Fig. 30 (a) Photocatalytic conversion of CH4 over GaN catalysts with different morphologies and different exposed surface areas of the m-plane.258 (b) Schematic diagram for CH4 C–H bond polarization on the m-plane of GaN.258 Reproduced from ref. 258, with permission of the American Chemical Society, copyright 2014. | ||
Very recently, Khodakov and co-workers reported an interesting photochemical looping strategy for CH4 conversion to C2H6 over a silver–phosphotungstic acid–titania nanocomposite (Ag–HPW/TiO2) photocatalyst.259 When a batch photo-reactor was used, the selectivity of C2H6 could reach 90% with small amounts of C3H8 and CO2, and the C2H6 yield was lower than 0.1%. The yield of C2H6 could reach 9% after 5 h irradiation in a capillary photo-reactor with a C2H6 selectivity of ∼55%. It was proposed that the photochemical looping involved two important steps, i.e., a CH4 coupling step and silver regeneration step (Fig. 31).259 During the CH4 coupling step, C2H6 was formed through oxidative coupling by photogenerated holes, while the photogenerated electrons were responsible for the reduction of cationic Ag+ to metallic Ag0. Ag0 was then re-oxidized to Ag+ under light irradiation in the presence of air in the silver regeneration step. The cationic Ag+ dispersed in the HPW layer over Ag–HPW/TiO2 was essential for the coupling of CH4 to C2H6.
 | ||
| Fig. 31 Schematic illustration of the photochemical looping process for coupling of CH4 to C2H6.259 | ||
Si–OH was activated by deep UV light irradiation to Si–O˙ and H˙, and the Si–O˙ functioned for the activation of CH4 to CH3˙, followed by the transformation of CH3˙ to CH3OH. Actually, the formation of C2 products, mainly C2H6, which can be formed via the coupling of CH3˙, have also been observed in some of these systems using semiconductor- or zeolite-based catalysts.260,261 The formation rate of C2H6 could reach 10 μmol g−1 h−1 on a beta zeolite under light irradiation by a medium-pressure Hg lamp, and the loading of V2O5 or BiVO4/V2O5 heterojunction could shift the product to CH3OH.262
Pt- or Pd-loaded semiconductor TiO2 was reported to catalyse the conversion of CH4 to C2H6 in an aqueous medium under UV-light irradiation.263,264 H2 and CO2 were also formed in considerable amounts. The studies using electron or hole scavengers suggested that H2 was formed by photogenerated electrons, while holes participated in the formation of C2H6 and CO2. The selectivity of C2H6 was quite low on TiO2 alone because of the formation of a large fraction of CO2 from CH4. The loading of Pt or Pd not only enhanced H2 formation but also improved the formation rate and the selectivity of C2H6 (Fig. 32a).263,264 It is of interest that although Pd is not as efficient as Pt in promoting H2 formation, it is more suitable for the formation of C2H6. The C2H6 selectivity and CH4 conversion quantum efficiency could reach 73% and 2.8% over the Pd/TiO2 catalyst, respectively.264 Electron spin resonance (ESR) spectroscopy studies showed the generation of ˙OH and CH3˙ radicals. Thus, the ˙OH species generated by the oxidation of H2O by photoexcited holes was proposed to be responsible for abstracting a hydrogen atom from CH4 to generate CH3˙, the coupling of which afforded C2H6. Although it was assumed that Pd nanoparticles on TiO2 surfaces might participate in the activation of the C–H bond of CH4,264 there was no evidence to support this hypothesis.
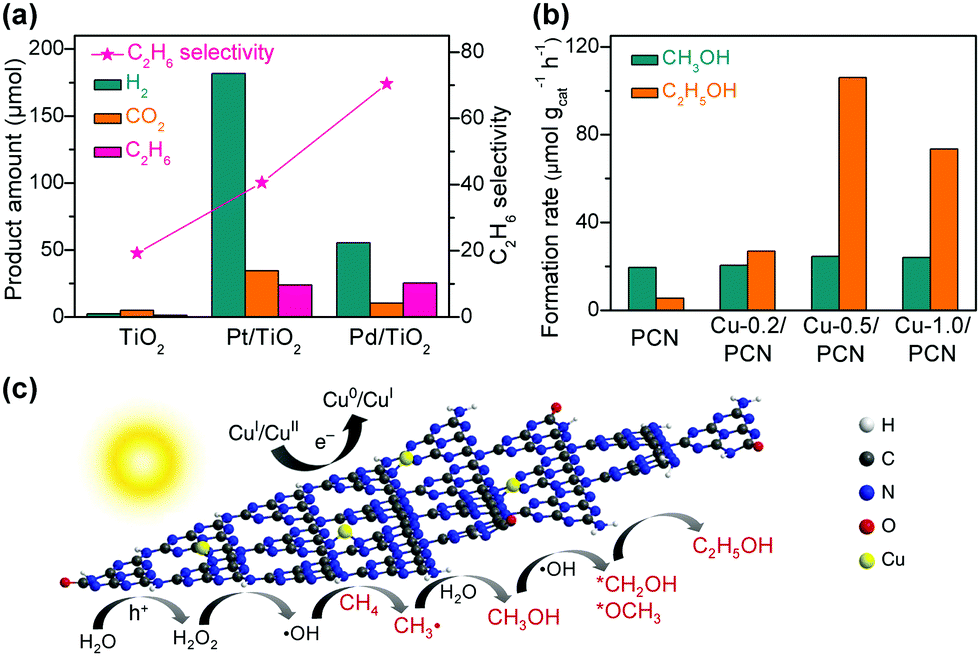 | ||
| Fig. 32 (a) Photocatalytic conversion of CH4 over TiO2, Pt/TiO2 and Pd/TiO2.263,264 (b) Photocatalytic conversion of CH4 over Cu-X/PCN photocatalysts with different amounts of Cu.265 (c) Proposed mechanism for photocatalytic conversion of CH4 with H2O over Cu-0.5/PCN.265 Reproduced from ref. 265, with the permission of Springer-Nature, copyright 2019. | ||
Designing a photocatalytic system that can generate ˙OH in a controllable manner could be beneficial for the conversion of CH4. Wang and co-workers recently reported that a Cu-modified polymeric carbon nitride (PCN) catalyst could work for photocatalytic conversion of CH4 and H2O to C2H5OH under UV-vis light irradiation.265 PCN with a bandgap of 2.97 eV and valence band position of 1.82 eV can work for the oxidation of H2O to H2O2 but not to ˙OH. The control experiment for photocatalytic conversion of H2O on PCN confirmed the formation of H2O2. The photocatalytic conversion of CH4 on PCN led to the formation of CH3OH as a major product together with a small amount of C2H5OH. The loading of Cu onto PCN from 0 to 0.5% significantly increased the formation rate of C2H5OH from 5.5 to 106 μmol g−1 h−1, whereas the formation rate of CH3OH remained almost unchanged (20–25 μmol g−1 h−1) (Fig. 32b).265 The Cu sites could accelerate the decomposition of H2O2 to ˙OH radicals and the oxidised Cu+ or Cu2+ could be reduced by photogenerated electrons. The Cu sites might also function for the adsorption and activation of CH4 to help the formation of CH3˙ by the ˙OH species. The synergistic effect between the Cu sites and the adjacent C atom in PCN has been proposed to play key roles in the formation of C2 product. Understanding the mechanism for C–C coupling would be very helpful for the design of more efficient photocatalytic systems for the conversion of CH4 to C2 compounds. Wang and co-workers considered that C2H5OH was formed via a CH3OH intermediate on the Cu/PCN catalyst and the C–C coupling might proceed through hydroxymethyl and methoxy species formed by interaction of CH3OH with ˙OH radicals (Fig. 32c).265 However, this mechanism is too speculative and there exist many other possibilities. Considering that Cu-based catalysts show interesting performances also in C–C coupling of C1 intermediates from CO2/CO during photocatalysis or electrocatalysis, further studies on the Cu-based CH4 photocatalytic conversion are definitely needed in the future.
5.2. Electrocatalytic conversion of CH4 to C2+ compounds
The electrocatalytic partial oxidation of CH4 could produce C1 (such as CH3OH, HCHO) or C2+ (such as C2H6, C2H4) compounds.54,55,246,270,271 Previous studies tend to suggest that the major useful products obtained from low-temperature electrocatalytic reaction systems were C1 derivatives, whereas useful C2+ compounds could be formed at high temperatures (>600 °C).270,271 Some typical electrocatalysts and performances are summarized in Table 6.272–283| Catalyst | Reaction condition | E (V) | j (mA cm−2) | CH4 conv. (%) | C2+ selectivity (%) | C2+ yield (%) | Ref. |
|---|---|---|---|---|---|---|---|
| a The potential represents the full-cell potential (V). b The potential refers to RHE; RT: room temperature. | |||||||
| Bi2O3–Ag | SOE, CH4|O2, 700 °C | 1.3a | — | — | C2H4 + C2H6: 47 | — | 272 |
| Porous Ag | SOE, CH4|O2, 800 °C | — | 5 | 97 | C2H4 + C2H6: 91 | 88 | 273 |
| Mn–Ce–Na2WO4/SiO2 | SOE, CH4|O2, 800 °C | — | 470 | 61 | C2H4: 36; C2H6: 6.2 | 23 | 274 |
| Ag | SOE, CH4|H2O, 820 °C | — | 400 | 26 | C2H4: 26; C2H6: 4.3 | 8 | 275 |
| Ag + Ce–Na2WO4/SiO2 | SOE, CH4|H2O, 800 °C | — | 150 | 23 | C2H4: 44; C2H6: 17 | 13 | 276 |
| Ag + SrZr0.95Y0.05O3 | SOE, CH4|H2O, 720 °C | — | 250 | 18 | C2H4: 21; C2H6: 21 | 7.6 | 277 |
| Ag + Mn–Ce–Na2WO4/SiO2 | SOE, CH4|H2O, 800 °C | — | 70 | 34 | C2H4: 36; C2H6: 7.4 | 15 | 278 |
| 0.075Fe–Sr2Fe1.5Mo0.5O6−δ | SOE, CH4|O2, 850 °C | 1.6a | 1000 | 41 | C2H4 + C2H6: 82 | 33 | 279 |
| 0.075Fe–Sr2Fe1.5Mo0.5O6−δ | SOE, CH4|CO2, 850 °C | 1.6a | 750 | 14 | C2H4 + C2H6: 76 | 10 | 279 |
| Co3O4/ZrO2 | Na2CO3 as the electrolyte, CH4|H2O, RT | 2.0a | 8.7 | 40 | 1-Propanol: 31; 2-propanol: 30 | 24 | 280 |
| ZrO2:NiCo2O4 | Na2CO3 as the electrolyte, CH4|H2O, RT | 2.0a | 2.3 | 48 | Propionic acid: 65 | 31 | 281 |
| NiO@NiHF | NaOH as the electrolyte, CH4|H2O, RT | 1.5b | 0.04 | C2H5OH: 85 (FE) | 282 | ||
| NiO/Ni | Proton exchange membrane, CH4|H2O, RT | 1.4b | 3.0 mA gNiO−1 | C2H5OH: 89 (FE), 25 μmol gNiO−1 h−1 (formation rate) | 283 | ||
The oxidative coupling of methane (OCM) to C2 compounds (mainly C2H6 and C2H4) is a well-known thermocatalytic reaction for CH4 transformation at high temperatures.20,242,243 The selectivity of C2H6 and C2H4 usually decreases with an increase in the conversion of CH4, thus limiting the C2 yield. It is generally accepted that the molecular oxygen in the gas phase would lead to the formation of CO and CO2, whereas the oxygen species on the catalyst surfaces are responsible for the OCM reaction.20,242,243 To suppress the gas-phase non-selective oxidation of CH4, the O2 concentration in the gas phase should be minimized. The solid oxide electrolyte electrochemical reactor, which uses oxygen ion conductors as the electrolyte, has been harnessed for the electrocatalytic oxidation of CH4 with the aim to improve the selectivity of C2 products. This electrochemical reactor provides an oxidant through transferring oxygen ions from the cathode side to the anode side, and thus can avoid the co-feeding of CH4 and O2. An electrocatalytic reactor with solid oxide electrolyte (SOE) mainly consists of two porous electrodes (i.e., anode and cathode) and a dense SOE to separate the cathode and anode compartments (Fig. 33). The anode compartment is usually fed with CH4, while the cathode compartment can be fed either with O2 (Fig. 33a) or another reactant (e.g., H2O or CO2) that is capable of providing oxygen ions (Fig. 33b). It is noteworthy that the feeding of H2O or CO2 can simultaneously produce H2 or CO.
 | ||
| Fig. 33 (a) Schematic illustration of the solid oxide electrolyte reactor for O2/CH4 conversion. (b) Schematic illustration of a solid oxide electrolyte reactor for H2O/CH4 or CO2/CH4 conversion. | ||
Otsuka and co-workers reported a pioneering work for CH4 coupling in an SOE electrocatalytic reactor in 1985.272 A Y2O3-stabilized ZrO2 (YSZ) with high oxygen ion conductivity was used as a solid oxide electrolyte, and Ag and Bi2O3–Ag were used as anode catalysts for electrocatalytic conversion of CH4 at 700 °C. CH4 and O2 were fed into the anode and cathode compartments separately (Fig. 33a). The oxygen transfer flux through the YSZ could be controlled by the externally applied potential. The CH4 conversion rate increased with an increase in oxygen flux over Ag and Bi2O3–Ag catalysts, but the selectivity of C2 compounds (C2H4 and C2H6) decreased upon increasing the oxygen flux. The Bi2O3–Ag catalyst reached a maximum C2 formation rate of ∼1.85 μmol min−1 (selectivity, 47%) at an oxygen flux of 12 μmol min−1, whereas the C2 formation rate was only 0.42 μmol min−1 (selectivity, 30%) at the same oxygen flux over the Ag catalyst (Fig. 34a).
 | ||
| Fig. 34 (a) Electrocatalytic conversion of CH4 over Ag and Bi2O3–Ag catalysts at the same oxygen flux of 12 μmol min−1.272 (b) Catalytic performances of porous Ag catalyst without and with C2 hydrocarbons trapping and gas recycling.273 | ||
After that, many catalysts and reaction systems based on the SOE electrocatalytic reactor have been developed.273,274,284,285 Vayenas and co-workers developed a reaction system with product separation and CH4 recycling abilities for electrocatalytic conversion of CH4 to C2 hydrocarbons.273 The C2 hydrocarbons could be separated by molecular sieve trap, thus significantly inhibiting the over-oxidation of C2 hydrocarbons and improving the total yield of C2 hydrocarbons (Fig. 34b). Over a porous Ag catalyst, at an applied current density of 5 mA cm−2 at 800 °C, CH4 was oxidatively coupled to C2 hydrocarbons with a C2 hydrocarbon yield of 88% and a C2H4 yield of 85%. Some typical OCM catalysts in thermocatalysis, such as Mn/Na2WO4/SiO2 and La-doped SrTiO3, were also examined for the electrocatalytic conversion of CH4 to C2 hydrocarbons.274,284,285 Tang and co-workers integrated a Mn–Ce–Na2WO4/SiO2 catalyst with SOE tubular membrane reactor for electrocatalytic coupling of CH4.274 The reaction could be operated at a high applied current density of 470 mA cm−2 at 800 °C with a CH4 single-pass conversion of 61% and C2+ selectivity of 42%. The C2+ selectivity still needs to be improved.
The oxygen for electrocatalytic oxidation of CH4 could be provided by H2O instead of O2, enabling the simultaneous formation of H2 and C2 hydrocarbons (C2H4 and C2H6).275–278,286,287 A single-chamber reactor configuration with Ag/YSZ/Pt (anode/solid electrolyte/cathode) co-fed with CH4 and H2O mixture was reported to show a maximum C2 yield of ∼8% under the optimised conditions at 820 °C.275 The addition of 5 wt% Ce–5 wt% Na2WO4/SiO2 to the single-chamber Ag/YSZ/Pt reactor could enable thermocatalytic oxidation of CH4 by the O2 released to the gas phase, enhancing the C2 yield to 13% at 800 °C.276 The double-chamber reactor with CH4 and H2O vapours fed separately to the anode and cathode chambers has the potential to obtain higher C2 selectivity. In the double-chamber reactor with Ag/YSZ/Pt configuration (Fig. 35a), Stoukides and co-workers obtained a C2 yield of 5.7% at 840 °C at CH4 conversion of 16.2% and C2 selectivity of 35.1%.277 Although O2 was not co-fed with CH4, some excess oxygen ions electrochemically pumped from the cathode to the anode may form O2 and evolve to the CH4 stream, leading to the gas-phase non-selective oxidation of CH4. To enhance the selective oxidation of CH4 on anode surfaces, a layer of OCM catalyst was deposited on the top of an anodic electrode (Fig. 35b). When a perovskite OCM catalyst (SrZr0.95Y0.05O3−a) was deposited, the C2 yield was improved to 7.6% with a CH4 conversion of 18% and C2 selectivity of 42.2%, and the reaction temperature could decrease to 720 °C.277 In a Ag/YSZ/Ag double-channel reactor, the addition of the Mn–Ce–Na2WO4/SiO2 catalyst could promote the C2 yield to 15% at 800 °C.278
 | ||
| Fig. 35 Schematic diagram of an O2−-conducting solid oxide electrolyte reactor. (a) Without OCM catalyst. (b) With OCM catalyst. | ||
CO2 could also be used as the source of oxygen for the electrocatalytic oxidation of CH4. Xie and co-workers reported simultaneous electrocatalytic reduction of CO2 to CO on the cathode and the oxidation of CH4 on the anode (Fig. 36a).279,288 The utilisation of a redox-reversible layered perovskite, i.e., Sr2Fe1.5Mo0.5O6−δ (SFMO), as the anode and the in situ construction of metal/oxide interfaces at nanoscale using Sr2Fe1.5+x–Mo0.5O6−δ (x = 0–0.1) to grow iron nanoparticles on the SFMO scaffold (xFe–SFMO) were demonstrated to be an efficient approach for the electrocatalytic oxidation of CH4 to C2 compounds by oxygen from CO2.279 A La0.9Sr0.1Ga0.8Mg0.2O3−δ (LSGM) was used as the solid electrolyte and Ce0.8Sm0.2O2−δ (SDC) was mixed with xFe–SFMO to fabricate a single cell composed of xFe–SFMO–SDC/LSGM/SFMO–SDC (anode/solid electrolyte/cathode). DFT calculations suggested that the Fe–SFMO metal–oxide interface favoured the cleavage of the C–H bond of CH4 with a lower energy barrier than the SFMO alone (Fig. 36b). Such a metal/oxide interface structure also improved the coking resistance and the thermal stability. The 0.075Fe–SFMO showed the best catalytic performances at an applied voltage of 1.6 V and a temperature of 850 °C (Fig. 36c and d).279 When O2 was fed to the cathode, the conversion of CH4, selectivity and yield of C2 products (C2H4 and C2H6) could reach 41%, 81.2% and 33%, respectively (Fig. 36c). The conversion of CH4, selectivity and yield of C2 products were 13.7%, 75.6% and 10.3%, respectively, as CO2 was fed to the cathode (Fig. 36d). The decreased performance in the case of electrocatalytic conversion of CH4–CO2 as compared to that of CH4–O2 may probably arise from the higher thermodynamic and kinetic barriers for CO2 electrolysis than O2 splitting to generate oxygen ions on the cathode. The simultaneous electrocatalytic conversion of CH4 and CO2 to C2H4 and CO is an interesting and challenging direction and is worth further study.
 | ||
| Fig. 36 (a) Schematic diagram of electrocatalytic conversion of CH4 on the anode and reduction of CO2 on the cathode. (b) Energy diagrams for adsorption and C–H bond cleavage of CH4 on SFMO(001) and Fe/SFMO(001) surfaces including their initial, transition and final states. Inset: Electronic charge density difference for the transition state of Fe/SFMO(001). (c) Anodic product concentrations of xFe–SFMO catalysts in the electrocatalytic conversion of CH4 on the anode and O2 feeding on the cathode. (d) Anodic product concentrations of xFe–SFMO catalysts in the electrocatalytic conversion of CH4 on the anode and reduction of CO2 on the cathode.279 Reproduced from ref. 279, with the permission of Springer-Nature, copyright 2019. | ||
The studies on electrocatalytic conversion of CH4 to C2+ compounds under mild conditions are rare. Park and co-workers claimed that CH4 could be electrocatalytically oxidized to C2+ alcohols, acids and ketones at ambient temperature.280,281 It is very surprising that 2-propanol and 1-propanol were obtained as major products when a Co3O4/ZrO2 nanocomposite loaded on carbon paper (Alfar) was employed as the anode for CH4 oxidation with Na2CO3 as the electrolyte and Pt as the cathode for H2O reduction (Fig. 37a).280 Acetaldehyde was formed at shorter times and was proposed to be the precursor for the formation of 2-propanol and 1-propanol. When a ZrO2:NiCo2O4 quasi-solid solution catalyst loaded on graphite foil was used as the anode for the oxidation of CH4, propionic acid was obtained as the major product along with 2-propanol, 1-propanol, acetone and acetic acid. The conversion of CH4 could reach 47.5% after 20 h of reaction and the formation rate of propionic acid was 1173 μmol gcat−1 h−1 (selectivity ∼65%) at ambient temperature operated in a two-electrode system at 2.0 V versus a Pt counter electrode.281 The reaction-time-course studies suggested that 1-propanol, acetaldehyde and 2-propanol were the major products in the first 5 h of reaction, and these products were further oxidized to propionic acid, acetic acid and acetone in 20 h of reaction, respectively (Fig. 37b). Although the phenomenon that the electrocatalytic conversion of CH4 could offer C2+ alcohols, acids and ketones (with C3 alcohols or acids as the major products) at room temperature is very interesting, some solid evidence, such as 13CH4-labelling isotopic experimental results, is expected to further confirm these very surprising results obtained in the oxidation of CH4 with such kinds of carbon-containing catalyst systems.
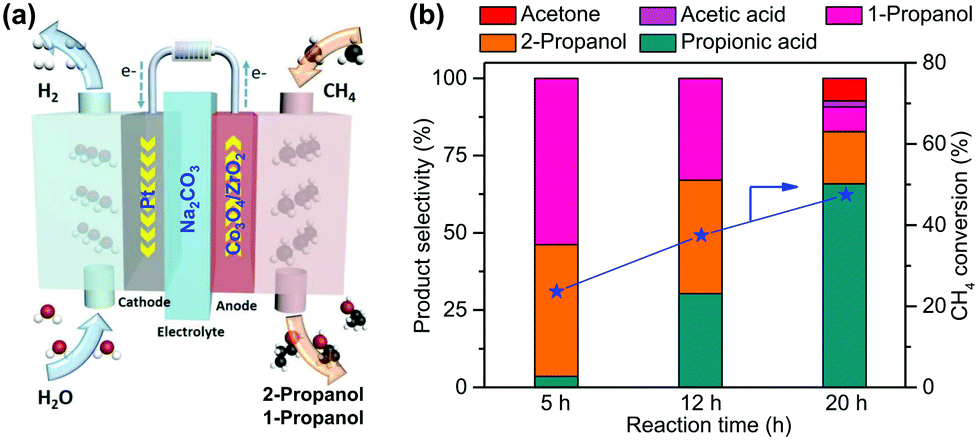 | ||
| Fig. 37 (a) Schematic reaction processes for electrocatalytic conversion of CH4.280 (b) Catalytic performances of a ZrO2:NiCo2O4 quasi-solid solution catalyst for electrocatalytic conversion of CH4 to C2+ compounds.281 Reproduced from ref. 280, with permission of Wiley-VCH Verlag GmbH&Co. KGaA, copyright 2017. | ||
Recently, Chen, Sun and co-workers reported that CH4 could be converted to C2H5OH with high selectivity by electrocatalytic oxidation over a NiO/Ni catalyst.282,283 They constructed catalysts with a series of NiO/Ni interfaces (Fig. 38a) by controlling the calcination temperature of Ni foam.283 The 3.0NiO/Ni catalyst calcined at 500 °C with a NiO content of 3.0 wt% showed the best performance. At an applied potential of 1.4 V vs. RHE, the FE and formation rate of C2H5OH reached 89% and 25 μmol gNiO−1 h−1, respectively.283 The optimised NiO/Ni interface with appropriate NiO content showed balanced charge transfer ability and the highest electrochemically active surface area, thus enabling efficient C–H activation and C–C coupling. DFT calculations further demonstrated a thermodynamically favourable route on the NiO(200)/Ni(111) interface for C2H5OH formation with the elementary steps of CH4* → CH3* + H*, CH3* → CH2* + H*, CH2* + OH* → CH2OH* and CH3* + CH2OH* → C2H5OH* in sequence (Fig. 38b).283 The NiO(200)/Ni(111) interface favours the dissociation of CH4* to CH3* with a relatively low activation barrier (0.30 eV). Although the steps of CH3* to CH2* and CH3OH* had similar activation barriers, the exothermic reaction energy of CH3* to CH2* was significantly lower than that of CH3* to CH3OH*. This may be the main reason for the low CH3OH selectivity observed over the NiO/Ni catalyst. The subsequent hydroxylation of CH2* and the C–C coupling of CH3* and CH2OH* to C2H5OH* are both more energy favourable steps.
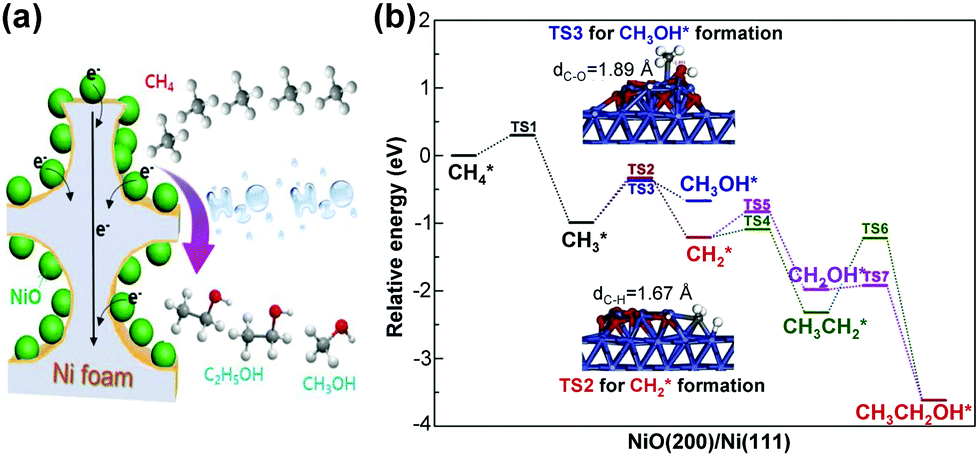 | ||
| Fig. 38 (a) Schematic illustration of electrocatalytic conversion of CH4 to C2H5OH on the NiO/Ni interface. (b) Reaction energy profiles for electrocatalytic conversion of CH4 to CH3OH and C2H5OH at the NiO(200)/Ni(111) interface from DFT calculations.283 Reproduced from ref. 283, with permission of Elsevier, copyright 2020. | ||
5.3. Mechanism of C–C coupling for photocatalytic and electrocatalytic conversions of CH4 to C2+ compounds
Many studies have been devoted to the transformation of CH4 by thermocatalysis.56 For the oxidative coupling of methane (OCM) at high temperatures (typically 873–1073 K), a gas-phase methyl radical (CH3˙) has long been proposed as the key intermediate for the C–C coupling into C2H6, which undergoes dehydrogenation to C2H4. The generation of CH3˙ during OCM was confirmed by synchrotron VUV photoionization mass spectroscopy.289 CH3˙ has also been proposed as the intermediate for the C–C coupling of CH4 to C2H6 in photocatalysis,251,256–258,264 and the formation of CH3˙ has been detected by in situ electron spin resonance (ESR) spectroscopy.264 The selectivity of C2H4 is lower by photocatalysis. This is probably because the dehydrogenation of C2H6 becomes difficult at low temperatures used for photocatalysis. C2H5OH could also be formed by photocatalytic conversion of CH4 on a Cu/PCN catalyst, and CH3OH was proposed to be the intermediate.265 However, this mechanism still lacks solid evidence. On the other hand, in the electrocatalytic CH4 transformations, C2H4 and C2H6 could be obtained at high temperatures,272,273,279 whereas 1-propanol and C2H5OH may be obtained at low temperatures.280–283 The studies on the mechanism of electrocatalytic C–C coupling of CH4 are very scarce. In the future, the advanced characterization techniques used for thermocatalytic CH4 studies such as synchrotron VUV photoionization mass spectroscopy should be exploited for mechanistic studies for photocatalytic and electrocatalytic CH4 conversions to offer deeper understanding of the C–C coupling and other elementary steps.6. Photocatalytic and electrocatalytic conversions of CH3OH and HCHO
Methanol is an abundant and key C1 feedstock, which can be produced from either fossil resources (in particular coal and natural/shale gas via syngas) or renewable carbon resources (biomass via syngas or CO2 hydrogenation).290 Various types of C2+ chemicals, including both hydrocarbons and oxygenates, can be produced from methanol. The selective oxidation of methanol to formaldehyde, carbonylation of methanol into acetic acid and the conversion of methanol to olefins (MTO) and gasoline (MTG) are well-established commercial processes. The direct conversion of methanol into a high-value C2+ product with high selectivity is still the most attractive and challenging target in methanol chemistry. Here, we highlight recent studies on the conversion of methanol or formaldehyde to C2+ compounds under mild conditions using photocatalysis and electrocatalysis (Table 7).291–306| Catalyst | Target product | Reaction conditions | Formation rate (mmol g−1 h−1) | Selectivity (%) | Yield (%) | Ref. |
|---|---|---|---|---|---|---|
| a The potential refers to RHE. b The potential represents the full-cell potential (V). | ||||||
| Photocatalytic conversion of CH3OH | ||||||
| ZnS | EG | Hg arc lamp | 1.0 | 75 | 0.3 (60 h) | 291 |
| MoS2/CdS | EG | Xe lamp (420–780 nm) | 11 | 90 | 16 (100 h) | 292 |
| CoP/Zn2In2S5 | EG | Xe lamp (AM 1.5) | 18.9 | 90 | 4.5 (12 h) | 293 |
| GaN | C2H5OH | Xe lamp (290–380 nm) | ∼4.0 | 100 | ∼5.4 (12 h) | 294 |
| Ni2P/CdS | 1,1-Dimethoxymethane | Xe lamp (>400 nm) | 188 | 83 | 1.7 (3 h) | 295 |
| Pt/TiO2 | EG | UV light (320–400 nm) | ∼2 | 80 | ∼0.4 (10 h) | 296 |
| Pt/TiO2 | Aliphatic alcohols | Xe lamp (200–400 nm) | 0.013–0.094 | >90 | 0.94–6.6 (15 h) | 297 |
| CdS | Aliphatic alcohols | Xe lamp (>420 nm) | 0.0035–0.0070 | >70 | 0.35–0.70 (20 h) | 298 |
| Electrocatalytic conversion of CH3OH (including dielectric barrier discharge, DBD) | ||||||
| Co3ZnC/NC | C2H5OH | j: 10 mA cm−2, E: 1.54 Va | 24.4 mg cm−2 h−1 | 95 (FE: 12) | 3.2 (4.5 h) | 299 |
| — | EG | DBD, AC 16.8 kV,b 300 °C | ∼1.7 mmol h−1 | 72 | 11.3 | 300 |
| — | EG | DBD, AC 16.8 kV,b 300 °C | ∼3.3 mmol h−1 | 75 | 22.5 | 301 |
| — | n-C4H10 | DBD, AC 30 kV,b 140 °C | — | 37.5 | 15 | 302 |
| Photocatalytic conversion of HCHO | ||||||
| BiVO4 | EG, CH3CHO, HOCH2CHO | Xe lamp (320–780 nm) | 11 | 74 | 8 (12 h) | 303 |
| MnOx–Pt@MoOx/BiVO4 | EG, CH3CHO, HOCH2CHO | Xe lamp (320–780 nm) | 54 | 74 | 21 (12 h) | 304 |
| Electrocatalytic conversion of HCHO | ||||||
| Mercury | EG | j: 250 mA cm−2, E: ∼−1.0 Va | — | FE: 30 | — | 305 |
| Graphite | EG | j: 400 mA cm−2, E: −1.0 Va | — | FE: 97 | ∼10.4 | 306 |
6.1. Photocatalytic and electrocatalytic conversions of CH3OH
The dehydrogenative coupling of methanol to ethylene glycol (EG) (eqn (3), denoted as MTEG) is a very attractive but highly challenging reaction. The product of this reaction, i.e., EG, is an important chemical with many important applications, in particular as a monomer for the production of poly(ethylene terephthalate) (PET). Currently, EG is primarily produced from ethylene by hydration. The development of a methanol-based EG production route would provide an alternative non-petroleum route.
| 2CH3OH → HOCH2CH2OH + H2, ΔG(298K) = 9.3 kJ mol−1 | (3) |
In an early communication, Yanagida and co-workers reported that the oxidation of methanol over ZnS (Eg = 3.6 eV, corresponding to λ = 345 nm) under UV-light irradiation formed HCHO and EG.291 The rate and selectivity of EG were ∼1 mmol g−1 h−1 and ∼75%, respectively.
Recently, our group demonstrated that the dehydrogenative coupling of methanol into ethylene glycol could proceed over CdS (Eg = 2.4 eV, corresponding to λ = 518 nm) under visible-light irradiation.292 ZnS and CdS were found to be unique photocatalysts for the formation of EG and other types of semiconductors such as TiO2, ZnO, C3N4 and CuS provided HCHO, HCOOH, CO and CO2. Nanorod-shaped CdS showed better performances than CdS nanoparticles. Different types of co-catalysts were examined to enhance the performance of CdS nanorods and MoS2 was found to be the best co-catalyst. Upon loading 5 wt% three-dimensional MoS2 nanofoams to CdS nanorods (Fig. 39a), the formation rate of EG increased from 0.46 to 11 mmol g−1 h−1 and the selectivity of EG on a molar carbon basis increased from 71% to 90%. The yield of EG on the molar carbon basis was calculated to be ∼2.5% after 12 h of the reaction under visible-light (λ = 420–780 nm) irradiation. The increase in reaction time to obtain higher yield of EG in the conventional batch-type reactor led to significant decreases in EG selectivity due to the consecutive oxidation. The design of a process-intensified reactor with simultaneous reaction and CH3OH/EG separation ability could keep the EG selectivity at 90% during 100 h of reaction, affording an EG yield of 16% (Fig. 39b).292 The mechanistic studies revealed that EG was formed by oxidative coupling of CH3OH by photogenerated holes, while photogenerated electrons contributed to H2 formation. CdS was found to be quite unique in the preferential activation of the C–H bond in CH3OH by the holes without affecting the O–H group, generating a ˙CH2OH intermediate, which coupled to give EG. DFT calculations suggested that the weak absorption of CH3OH on CdS surfaces offered a possibility of preferential activation of the C–H bond instead of the O–H bond (Fig. 39c). The cleavage of the C–H bond in CH3OH on CdS surfaces to produce ˙CH2OH radicals occurred through a concerted proton–electron transfer (CPET) mechanism with a low activation barrier. The weakly adsorbed ˙CH2OH could readily desorb from CdS surfaces for subsequent C–C coupling. On the other hand, the stronger adsorption of CH3OH on TiO2 led to a very easy proton transfer in the first step, forming CH3O− species on TiO2 surfaces, which consequently resulted in the formation of HCHO (Fig. 39c).
 | ||
| Fig. 39 (a) Schematic illustration of MoS2 foam/CdS for photocatalytic synthesis of EG and H2 from CH3OH. (b) Catalytic performance of MoS2 foam/CdS with process intensification. (c) Reaction energy profiles via ˙CH2OH and CH3O˙ on CdS(100) and rutile TiO2(110).292 Reproduced from ref. 292, with the permission of Springer-Nature, copyright 2018. | ||
Very recently, instead of CdS, an environmentally friendly sheet-like Zn2In2S5 semiconductor was further found to be efficient for selective conversion of CH3OH to EG under visible-light irradiation.293 The loading of 0.25 wt% CoP onto Zn2In2S5 enhanced the EG selectivity to 90%. Under the simulated sunlight irradiation (AM1.5), the formation rate of EG on the 0.25 wt% CoP/Zn2In2S5 catalyst reached 18.9 mmol g−1 h−1 and the EG yield on the molar carbon basis was 4.5% after 12 h of reaction.293 In addition to the conversion of CH3OH to EG, the CoP/Zn2In2S5 catalyst could also work for the photocatalytic dehydrogenative coupling of ethanol to 2,3-butanediol. Under visible-light irradiation, the selectivity and formation rate of 2,3-butanediol were 53% and 3.2 mmol g−1 h−1, respectively.293 ESR measurements confirmed that the reaction proceeded via the ˙CH(OH)CH3 intermediate. The photocatalytic dehydrogenative coupling of ethanol to 2,3-butanediol was also reported previously using a TiO2 catalyst by Zhu and co-workers.308 The pre-treatment of TiO2 at high temperatures to remove the surface hydroxyl groups was found to be key to obtaining high selectivity of 2,3-butanediol from ethanol, and this might indicate that avoiding the interaction of the –OH of ethanol with the surface was vital.
Photocatalytic conversion of CH3OH could also afford C2H5OH as the major product. Li and co-workers showed that GaN nanowires could catalyse the one-step conversion of CH3OH to C2H5OH under UV-vis-light irradiation at ambient temperature.294 The cylinder-shaped GaN-nanowire catalyst with average diameters of 100 nm and lengths of 800 nm was fabricated and the catalyst exposed two types of surfaces, i.e., the polar surface (c-plane) located at the end of the rod and the non-polar surface (m-place) located at the side of the rod. The conversion of 25 μL CH3OH over 0.35 mg GaN nanowires doped with a trace amount of Mg2+ (p-GaN) under UV-vis-light irradiation offered 17 μmol C2H5OH in 12 h, showing a formation rate of C2H5OH of ∼4.0 mmol g−1 h−1.294 Further studies indicated that the polar plane (c-plane) was mainly responsible for the formation of C2H5OH. Mechanistic studies suggested that a methyl carbene (:CH2) intermediate might be generated on the polar c-plane of GaN surfaces for subsequent insertion into the C–H bond of CH3OH to form C2H5OH (Fig. 40). Moreover, CH3OH could be converted to n-C3H7OH over GaN nanowires under UV-vis-light irradiation by lowering the reaction temperature from 15 to 0 °C, and this indicated that this reaction system may have potential for the synthesis of a wide range of lower to higher alcohols.
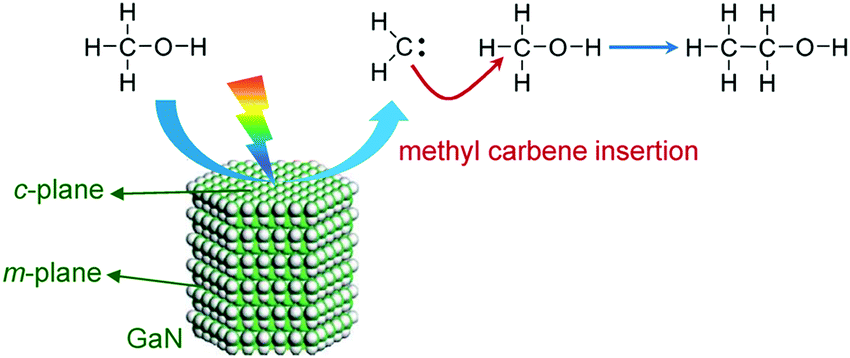 | ||
| Fig. 40 Proposed mechanism for the conversion of CH3OH to C2H5OH on GaN surface.294 | ||
Besides EG and C2H5OH, 1,1-dimethoxymethane (DMM) could also be synthesized by photocatalytic conversion of CH3OH. DMM is a product without a C–C bond but is of importance as the precursor of polyoxymethylene dimethyl ether (POMM), which is an environmentally friendly embalming agent to replace HCHO. When Ni2P/CdS was used as a photocatalyst for the conversion of CH3OH under visible-light irradiation, the major product was found to shift from EG to DMM by adding a small amount of sulphuric acid.295 The selectivity and formation rate of DMM reached 83% and 188 mmol g−1 h−1 at a H2SO4 concentration of 40 mM. It was speculated that the presence of H+ induced the reaction between the formed HCHO and CH3OH to produce DMM.
Photocatalytic coupling of CH3OH with other molecules has been reported by Zhong, Sun and co-workers.296–298 For example, the photocatalytic coupling of CH3OH and HCHO could proceed over a Pt/TiO2 catalyst under UV-light irradiation, offering EG with a selectivity of 80%.296 It was proposed that CH3OH was first oxidised by photogenerated holes to form ˙CH2OH and then the ˙CH2OH intermediate reacted with HCHO to give HOCH2CH2O˙, which was finally transformed to EG by photogenerated electrons and protons. The coupling of ˙CH2OH could also afford EG. The photocatalytic coupling of CH3OH with terminal olefins could proceed efficiently over Pt/TiO2 or CdS to form higher aliphatic alcohols.297,298 In the reaction, CH3OH worked as a hydroxymethylation agent and a wide range of terminal olefins were successfully functionalized to the corresponding longer aliphatic alcohols with high anti-Markovnikov selectivity. Mechanistic studies indicated that CH3OH was oxidised by photogenerated holes to a ˙CH2OH radical, which then attacked the terminal olefins to generate a new C–C bond and another radical intermediate for subsequent reduction by photogenerated electrons to aliphatic alcohols.
| Cathode: 4CH3OH + 2e− → 2OH− + H2 + 2C2H5OH | (4) |
| 2H2O + 2e− → 2OH− + H2 | (5) |
| Anode: 2H2O → O2 + 4e− + 4H+ | (6) |
Dielectric barrier discharge (DBD) is the electrical discharge between two electrodes separated by an insulating dielectric barrier, which can readily generate free radicals in the gas phase for radical reactions. The DBD has been used for methanol conversion to C2+ compounds such as EG and n-C4H10.300–302,310 Guo and co-workers have systematically studied the direct synthesis of EG by co-feeding of CH3OH and H2 in a double dielectric barrier discharge reactor.300,301 Under the optimised reaction conditions, under a H2/CH3OH molar ratio of ∼7.2 and 300 °C, the CH3OH conversion and EG selectivity reached 30% and 75%, respectively. The co-feeding of H2 could improve the CH3OH conversion and EG selectivity, and played a catalytic role by releasing ˙H radicals for the abstraction of hydrogen from CH3OH to form ˙CH2OH intermediates for subsequent C–C coupling to EG (Fig. 41a).301 Tu and co-workers demonstrated that CH3OH could be converted to n-C4H10 and H2 in a DBD reactor.302 The optimised selectivities of n-C4H10, H2 and CO were 37.5%, 28.9% and 14%, respectively, with a CH3OH conversion of 40% at a CH3OH inlet concentration of 18 mol% and a reaction temperature of 140 °C using N2 as a carrier gas. It was proposed that CH3˙ radicals were generated by electron impact dissociation and then the CH3˙ radicals dimerized to C2H6, which could further be converted to C2H5˙ radicals through hydrogen abstraction and then undergo C–C coupling to n-C4H10 (Fig. 41b). The presence of several competitive pathways for the formation of CO, CH4, and C2H5OH reduced the selectivity of n-C4H10.

6.2. Photocatalytic and electrocatalytic conversions of HCHO
Photocatalysis and electrocatalysis also offer opportunities for the direct conversion of formaldehyde to C2+ compounds, in particular EG.303–306 Our group first developed a photocatalytic route for coupling of HCHO to C2 oxygenates.303 BiVO4 and Bi2WO6 were found to catalyse the conversion of HCHO, providing EG, glycolaldehyde, and acetaldehyde as the major products with a total C2 selectivity of 74%. BiVO4 nanocrystals with truncated tetragonal bipyramidal morphology and controllable {010} and {110} facets were fabricated. The BiVO4 nanocrystal catalyst with an equal fraction of {010}/{110} showed a C2 yield of 12% during the conversion of HCHO aqueous solution under UV-vis-light irradiation for 12 h.304 This catalyst had the highest ability to separate the photogenerated electrons and holes probably because photogenerated electrons and holes migrated separately to {010} and {110} facets, respectively. The effects of co-catalysts were further investigated. The photo-deposition of co-catalysts showed that Pt@MoOx with a Pt core and MoOx shell was selectively deposited on the {010} facets, whereas MnOx was deposited selectively on the {110} facets (Fig. 42a).304 This provided further evidence for the migration of photogenerated electrons and holes towards different facets for reduction and oxidation. The single Pt or MoOx did not significantly promote the catalytic activity of BiVO4, whereas the Pt@MoOx co-catalyst accelerates the formation of C2 oxygenates, in particular EG (Fig. 42b). The presence of MnOx as an oxidation co-catalyst further enhanced EG formation. The yields of C2 compounds and EG over the 3% MnOx–0.5% Pt@3% MoOx/BiVO4 catalyst reached 21% and 11%, respectively, under UV-vis light irradiation for 12 h. Mechanistic studies revealed that Pt worked for the extraction of photogenerated electrons and accelerates the electron–hole separation, while MoOx provided active sites for the activation of HCHO. In brief, Mo6+ was proposed to be reduced to Mo5+ by accepting a photogenerated electron passed from the Pt core, and the Mo5+ site could adsorb HCHO and transfer the electron (ET) to the adsorbed HCHO (Fig. 42c). Then, the proton transfer (PT) led to the formation of ˙CH2OH, which coupled to give EG after desorption. | ||
| Fig. 42 (a) Model of the MnOx–Pt@MoOx/BiVO4 catalyst. (b) Effect of co-catalysts on the performance of BiVO4 for photocatalytic conversion of HCHO. (c) Proposed mechanism for the conversion of HCHO to EG over MnOx–Pt@MoOx/BiVO4.304 | ||
The electrocatalytic reductive coupling of HCHO is also an important route for the synthesis of EG under mild conditions (eqn (7) and (8)).305,306 The materials with high H2 evolution overpotential, such as Cd, Hg, Pb, Ti, and graphite, could be used as a cathode to suppress the competitive H2 evolution reaction (eqn (9)). Mazur and co-workers found that graphite showed the best performance for the formation of EG.306 A large fraction of HCHO was in the hemiacetal form in the presence of methanol inhibitor, and a high pH could cause the disproportionation of HCHO to CH3OH and HCOO− (eqn (10)), whereas a lower pH would lead to the polymerization of HCHO by acid catalysis (eqn (11)). Thus, the control of the reaction conditions to obtain a high concentration of free HCHO for C–C coupling and to suppress side reactions is essential to EG formation. The optimum reaction conditions required a high concentration of aqueous HCHO solution, near-neutral pH of 5 to 8, the addition of quaternary ammonium salt and a reaction temperature of about 80 °C. Under the optimized conditions, with an applied current density of 300 mA cm−2 and HCHO concentration of 52%, the FE of EG was higher than 80% and the EG concentration reached 23%.306 However, with an increase in HCHO conversion, the FE of EG decreased significantly. The separation of EG from the electrolyte solution is also a challenging issue that needs to be solved for commercialisation.
| Cathode: 2HCHO + 2H+ + 2e− → EG | (7) |
| Anode: 2H2O → O2 + 4e− + 4H+ | (8) |
| Side reaction: 2H+ + 2e− → H2 | (9) |
| 2HCHO + OH− → CH3OH + HCO2− | (10) |
| nHCHO → polymer | (11) |
6.3. Mechanism of C–C coupling for photocatalytic and electrocatalytic conversions of CH3OH and HCHO to C2+ compounds
The thermocatalytic activation of CH3OH usually leads to the cleavage of its C–O or O–H bond, and the resulting CHx species may undergo C–C bond formation. The use of zeolite catalysts like in the MTO process can partially control the product selectivity due to their shape-selective effect.22 Interestingly, the preferential activation of the C–H bond of CH3OH has been successfully achieved by photocatalysis using CdS-based catalysts, and EG could be obtained with a selectivity of 90% via the subsequent coupling of the ˙CH2OH intermediate.292,293 To the best of our knowledge, this cannot be accomplished through thermocatalysis. The formation of ˙CH2OH in photocatalytic conversion of CH3OH has been confirmed by in situ ESR.292,293 In a DBD reactor, CH3OH could also be converted to ˙CH2OH with electrical energy, which undergoes coupling to EG.300,301 Furthermore, EG can be produced by photocatalytic or electrocatalytic reduction of HCHO. Similarly, ˙CH2OH is the key intermediate during photocatalytic/electrocatalytic conversion of HCHO to EG.303–306 Besides EG, C2H5OH was reported to be obtained through photocatalytic or electrocatalytic conversion of CH3OH.294,299 It is proposed that C2H5OH is formed by insertion of methyl carbine (:CH2) into the C–H bond of CH3OH on the polar plane of GaN in photocatalysis.294 On the other hand, CH3˙ and ˙CH2OH intermediates were confirmed by in situ ESR on Co3ZnC/NC in electrocatalysis, and thus C2H5OH might be the coupling product between these two radicals.299 In the DBD reactor, CH3OH may also be transformed into a CH3˙ intermediate, which then undergoes C–C coupling to C2H6. C2H6 could be further converted to n-C4H10via C2H5˙.302 Thus, the nature of the intermediate generated from CH3OH or HCHO in photocatalysis/electrocatalysis has a significant effect on the final C–C coupling product.7. Conclusions and outlook
The selective transformation of C1 molecules to value-added C2+ compounds through controllable C–C coupling is a highly attractive but challenging research goal in chemistry. The control of C–C bond formation in the conversion of C1 molecules by thermocatalysis under harsh reaction conditions is generally difficult. Photocatalysis and electrocatalysis have offered promising routes for activation and selective transformation of C1 molecules to C2+ compounds under mild conditions by harnessing photo or electrical energy. The present article has highlighted key advances in photocatalytic and electrocatalytic conversions of typical C1 molecules, including CO2, CO, CH4, CH3OH, and HCHO, to C2+ compounds, in particular C2H4, C2H6, C2H5OH, ethylene glycol and n-propanol.Photocatalytic reduction of CO2 to C2+ compounds requires efficient transfer of multi-electrons/multi-protons and suitable active sites to catalyse not only CO2 activation but also C–C coupling. Many semiconductors, e.g., TiO2,62 g-C3N4,63 BiVO4,64 Bi2WO665 and InTaO4,66 have been reported to be capable of working for the reduction of CO2 to C2+ compounds, but the efficiencies were low. Several strategies have been reported to increase the activity and the selectivity of C2+ compounds. To increase the light harvesting by semiconductor doping, combination with sensitizer and loading of SPR metals could enhance the activity for CO2 photocatalytic reduction in particular under visible-light irradiation. The enhancement in the separation of photogenerated electrons and holes by the hybridisation of different semiconductors or a semiconductor with a carbon nanomaterial and loading of co-catalysts may contribute not only to increasing the activity but also improving the selectivity to C2+ products by promoting the multi-electron transfer. Several studies demonstrated that the hybridisation of TiO2 with CNT or graphene could enhance the formation of C2H5OH or C2H6.73–76 The nanostructure of a photocatalyst may also affect the C–C coupling. For example, the construction of mesoporous g-C3N4 was found to be beneficial to the formation of C2H5OH. The surface structure is the key to the surface reaction, and thus the modification of the catalyst surfaces may modify the selectivity of C2+ products. For example, the coating of a thin Nafion layer on the catalyst surface may increase the local proton concentration, thus favouring the multi-step proton-coupled electron transfer (PCET) reactions to facilitate the formation of C2+ products.79 The co-catalyst can not only enhance the electron–hole separation but also provide active sites for CO2 activation and C–C coupling. Several co-catalysts such as Pd,78,80 Rh,80 NiO,66,81 Cu,67,83,94 and bimetallic Cu–Pt83,84,96 have been reported to hold potential to promote the formation of C2+ compounds. Among these co-catalysts, Cu has received particular interest in the formation of C2+ hydrocarbons and the enhancing effect of Cu or Cu-based co-catalysts has been confirmed in many studies.67,69,82,83,85,94 The loading of co-catalysts on semiconductors has been demonstrated to be one of the most effective strategies for improving the photocatalytic activity and tuning the C–C coupling selectivity in CO2 reduction. The findings suggest that the co-catalysts are necessary for developing highly efficient photocatalysts for photocatalytic reduction of CO2 to C2+ compounds. In addition, the reaction conditions and reaction medium could also exert influences on the selectivity of C2+ compounds. For example, the increase in CO2 pressure and the use of a basic medium were reported to be beneficial for the formation of C2+ products.62 The addition of ionic liquid (EMIM-BF4) into aqueous solution with a proper concentration could promote the selectivity of C2 and C3 hydrocarbons during photocatalytic reduction of CO2 on plasmonic Au nanoparticles under green-light irradiation because of the enhanced CO2 activation and accelerated electron transfer from Au nanoparticles to CO2.87 Despite the progress achieved, the formation rates of C2+ compounds are still quite low, being <100 μmol g−1 h−1 in most cases (Table 1). It is noteworthy that the results with such low rates should be treated carefully, because the carbon sources contained in the reaction system including CO2 may all have possibilities to be reduced to C2+ products. Therefore, to carry out isotopic 13C-labelling experiments to confirm that the products are derived from CO2 as well as the quantification of O2 formation is crucial to providing solid results. This is particularly necessary when carbon-containing photocatalysts are studied.
Three pathways, i.e., the oxalic acid pathway, glyoxal pathway and methyl radical pathway, have been proposed for C–C coupling in photocatalytic reduction of CO2 (Fig. 10b).45,59,104 A deep understanding of how the surface structure and other influencing factors determine the CO2 activation and the C–C coupling is still lacking. The mechanism for the formation of C2+ compounds needs further studies.
Photo-driven Fischer–Tropsch synthesis is the major research area in photocatalytic conversion of CO to C2+ compounds. It is demonstrated that the conversion of syngas to C2+ hydrocarbons could proceed over several supported metal catalysts under light irradiation without external heating.110–115 The typical Fischer–Tropsch metal (i.e., Co, Fe and Ru)-based and Ni-based catalysts all showed activities in light-driven syngas conversions. The photo-thermal effect by metal catalysts and the photogenerated charge carriers on catalyst surfaces may both contribute to the light-driven CO hydrogenation to C2+ hydrocarbons. The photo-thermal effect could provide sufficient temperature at the catalyst bed for CO hydrogenation. The photogenerated charges on the catalyst surfaces may create a unique oxidation state and electronic structure of the catalyst, thus regulating the C–C coupling and hydrogenation ability to obtain unique product selectivity. For example, lower olefins were formed with higher selectivity in the Co- and Fe (or Fe5C2)-catalysed photo-driven syngas conversions.110–112 Ni-Based catalysts could also work for the formation of C2+ hydrocarbons under light irradiation, whereas CH4 was the only hydrocarbon product over Ni catalysts under thermocatalytic conditions.115 Despite uniqueness, the distribution of hydrocarbon products is still quite broad in the photo-driven CO hydrogenation, indicating that the C–C coupling is still uncontrollable.
Electrocatalytic reduction has recently been proven to be very promising for the synthesis of C2+ compounds from CO2 or CO. Although Cu is still the only selective catalyst for electrocatalytic reduction of CO2 to C2+ compounds owing to its moderate CO binding energy, significant advances have been achieved in developing active and selective Cu-based electrocatalysts for the formation of various C2+ compounds, including C2H4, C2H5OH, CH3COOH and n-C3H7OH. Model studies using Cu single crystals reveal that the electrocatalytic reduction of CO2 is structure sensitive; the Cu(111) surfaces favour the formation of CH4, whereas Cu(100) and Cu(110) surfaces can offer C2H4 and C2+ oxygenates, respectively.176–178 As compared to pure metallic Cu, the oxide-derived Cu catalyst showed enhanced C2+ selectivity. Although the nature of the active sites leading to high C2+ FE over oxide-derived Cu catalysts is still under debate, the current consensus is that the presence of surface Cuδ+ sites plays a pivotal role in the C–C coupling and the formation of C2+ products. Many studies have been devoted to improving the C2+ formation activity on Cu-based catalysts and high C2+ FEs have been reported. For example, the doping of a heteroatom such as boron, nitrogen or halogen on the surfaces of Cu nanoparticles could offer high FEs (∼80%) towards C2+ compounds with C2H4 as the major product.139–142 A nano-defective Cu nanosheet catalyst derived from CuO nanosheets showed an FE of C2H4 as high as 83.2%.138 Electrolyte engineering could also improve C2+ FEs, and larger sizes of anions or cations (e.g., I− or Cs+) added into the electrolyte showed significant promoting effects.194–196 Recent studies using the flow cell with GDE instead of H-cell to circumvent the mass-transport limitation have achieved high C2+ FEs at high current densities. Typically, the alkaline GDE design has successfully enabled superior performances to be achieved at commercially-relevant levels (≥200 mA cm−2).
Many studies have attempted to improve the FE toward C2+ oxygenate formation. The fabrication of bimetallic catalysts containing Cu and another guest metal such as Zn, Au or Ag has shown great promising for the formation of C2+ oxygenates, in particular C2H5OH, in electrocatalytic CO2RR. The FE of C2H5OH could be enhanced to 41% on an Ag0.14/Cu0.86 alloy catalyst at a current density of 400 mA cm−2 in the alkaline flow cell.152 The design of carbon materials also holds potential to modulate the selectivity of C2H5OH in Cu/carbon-catalysed CO2RR. For example, Cu nanoparticles surrounded by N-doped carbon layers displayed a C2H5OH FE of 52% with a total C2+ FE of 93% at a current density of 300 mA cm−2 in an alkaline flow cell.156
As compared to electrocatalytic CO2RR, Cu-catalysed electrocatalytic CORR has typically demonstrated lower overpotentials for C–C coupling and higher selectivity towards C2+ oxygenate formation. For example, a nanoflower Cu catalyst with a high roughness factor offered a nearly 100% selectivity to C2 oxygenates at a potential of only −0.23 V vs. RHE despite the low current density.169 Not only C2H5OH but also CH3CHO, CH3COO− and n-C3H7OH can be produced with high selectivity during electrocatalytic CORR. CH3COO− formation was typically enhanced after switching CO2 reactant to CO, which was attributed to higher local pH at the electrode–electrolyte interface in CORR. A 48% FE of CH3COO− was achieved at a current density of 200 mA cm−2 during electrocatalytic CORR over a Cu nanosheet catalyst.172 It is expected that the key step in n-C3H7OH formation is the coupling between C1 and C2 intermediates, however, the inadequate stabilization of C2 intermediates on Cu surfaces usually results in desorption rather than further coupling with C1. Several strategies have been developed to stabilize C2 species and promote the C1–C2 coupling to n-C3H7OH, including the confinement effect, creating interfaces of Cu(100) and Cu(111) facets and loading Cu adparticles on Cu surfaces. The maximum FE of n-C3H7OH achieved to date is 20–25%.173–175
The moderate CO binding strength is believed to be one major reason for the excellent performance of Cu catalysts in electrocatalytic CO2RR and CORR to C2+ products. The electrocatalytic CO2RR to C2+ products on Cu usually requires potentials more negative than or close to −1.0 V to achieve industrial-relevant current densities. The search for electrocatalytic materials other than Cu may enable the CO2RR to C2+ products at lower overpotentials. The fabrication of Cu-free bimetallic catalysts (e.g., Pd–Au, Ni–Ga and Ni–Al), which combine one metal that binds CO strongly with the other that binds CO weakly, could offer C2+ products during CO2RR. However, the C2+ FEs of these bimetallic catalysts are still quite low (<5%). Nitrogen-doped carbon-based materials have been exploited for metal-free electrocatalytic CO2RR to C2+ products. A surprisingly high FE of C2H5OH (>90%) has been claimed on a boron- and nitrogen-co-doped nanodiamond catalyst despite of the low current density (∼1 mA cm−2).160 In addition, some studies have demonstrated that the fabrication of a bifunctional catalyst composed of a catalyst component that can accelerate the CO2RR to CO and the N-doped carbon can enhance the formation of C2 oxygenates, in particular C2H5OH.229–231
CO is generally believed to be the key intermediate in the electrocatalytic CO2RR to C2+ compounds. Several mechanisms have been proposed for C–C coupling of CO or CO-derived intermediates on Cu-based catalysts. The direct coupling of *CO to form a *OCCO intermediate followed by hydrogenation to C2H4 and C2H5OH is believed to be the major C–C coupling route in most studies reported to date. Some studies have demonstrated the importance of *CHO species in the C–C coupling step, and the (*CO + *CHO) coupling or (*CHO + *CHO) coupling may participate in the formation of C2 compounds. Two major mechanisms have been proposed for the formation of C2H4 and C2 oxygenates. One assumes that *CH2CHO serves as the selectivity-determining intermediate, while the other considers that *CHCOH is the key intermediate. In both cases, it is accepted that the hydrogenolysis of the selectivity-determining intermediate leads to C2H4 formation, while the hydrogenation results in C2+ oxygenate.
As compared to photocatalytic CO2RR or CORR, electrocatalytic CO2RR and CORR have made a significant step-forward in controlling the product selectivity. Excellent selectivity of C2H4 or C2 oxygenates such as C2H5OH or CH3COO− has been achieved at industrially relevant current densities. Nevertheless, challenges still remain for electrocatalytic CO2 and CO conversions to C2+ products. First, the most efficient alkaline GDE suffers from stability issues. More effort should be put into improving the resistance of GDE flooding. Electrocatalytic CORR can avoid the salt accumulation under alkaline conditions, and thus improved stability may be expected for electrocatalytic CORR than for direct CO2RR. Thus, to implement a tandem strategy to synthesize C2+ products from CO2via CO would be useful. Second, most electrocatalytic CO2RR and CORR studies have only touched on the cathodic half-reaction, and the oxygen evolution reaction in the anode has seldom been considered. Combining an efficient oxygen-evolution catalyst with CO2RR or CORR to improve the full-cell energy conversion efficiency would be important for future industrial applications. Third, it still remains challenging to achieve a sufficient CO2/CO single-pass conversion or single-pass target product yield that is comparable to the commercial Fischer–Tropsch synthesis process.
As compared to the traditional thermocatalytic OCM and CH4 dehydrogenative coupling processes, the photocatalytic conversion of CH4 to C2+ compounds can be performed under very mild conditions, although the efficiency is quite limited. Metal cations or oxides highly dispersed on SiO2, Al2O3 and zeolites (e.g., Zn+-ZSM-5255 and Ga-EST-10256) and some semiconductors (Au/ZnO,257 GaN258 and Ag+–HPW/TiO2259) have been reported for the non-oxidative dehydrogenation of CH4 to C2H6 or C6H6 under UV-light irradiation. The active site, such as Zn2+ and Ga3+ ions, could promote the adsorption of CH4 and the polarization of C–H bonds to form CH3 surface species or radicals for C–C coupling to C2H6. The formation of C6H6 is more difficult, because it needs multi-step dehydrogenation and cyclization processes. The non-oxidative reaction condition may facilitate the C–C bond formation to C2+ hydrocarbons. In the presence of H2O, in addition to hydrocarbons, CH3OH and C2H5OH are also formed. Hydroxyl radicals (˙OH) formed through the oxidation of H2O by photogenerated holes have been detected and proposed to be the reactive oxygen species for abstracting a hydrogen atom from CH4 to generate CH3˙. The ˙OH radical can be easily generated through the decomposition of H2O2. Thus, the design of photocatalysts with high H2O2 generation activity may be promising for CH4 conversion.
Electrocatalytic conversion of CH4 to C2 hydrocarbons could be performed in solid oxide-electrolyte reactors at high temperatures similar to that used for OCM reactions. However, the gas-phase non-selective oxidation of CH4 may be inhibited, because CH4 and O2 are fed separately in the anode and cathode compartments. Many modified OCM catalysts have been employed as the electrocatalysts for CH4 conversion. By using a reaction system with product separation and CH4 recycling abilities, the yields of C2 hydrocarbons and C2H4 could reach 88% and 85%, respectively, over a porous Ag catalyst.273 Over a Fe-doped layered perovskite anode (Sr2Fe1.575–Mo0.5O1−δ) for CH4 oxidation, the single-pass conversion of CH4 could reach 41% and the yield of C2 hydrocarbons (C2H4 and C2H6) reached 33%, when O2 was fed to the cathode.279 O2 on the cathode could be replaced by H2O or CO2 and H2 or CO would be formed simultaneously with CH4 oxidative coupling products. Because of the higher thermodynamic and kinetic barriers for H2O and CO2 electrolysis to generate oxygen ions on the cathode, the performance of electrocatalytic conversion of CH4 to C2 hydrocarbons using H2O or CO2 is still not high. However, the full use of the anode and cathode half reaction to produce useful chemicals is important, and much effort should be directed in the future to developing high efficiency electrocatalysts for both cathode and anode reactions. Studies on electrocatalytic conversion of CH4 to C2+ compounds at moderate temperatures have also emerged in recent years, and this direction is definitely worthy of further study.
The photocatalytic and electrocatalytic conversions of CH3OH and HCHO to C2+ compounds are important targets that have not been widely studied. Some novel reactions, such as the visible-light-driven dehydrogenative coupling of CH3OH to EG,292,293 light-driven one-step conversion of CH3OH to C2H5OH,294 photocatalytic coupling of HCHO to EG,303,304 and electrocatalytic conversion of CH3OH to C2H5OH,299 have been developed in recent years. A high selectivity (90%) and yield (16%) of EG have been achieved through photocatalytic coupling of CH3OH to EG by using a MoS2 foam-modified CdS nanorod catalyst under visible-light irradiation.292 It has been demonstrated that the C–H bond of CH3OH is activated by the photogenerated hole to ˙CH2OH, which couples to give EG.292 Thus, these studies also offer an opportunity for the preferential activation of the C–H bond of functionalised molecules. It is of interest that ˙CH2OH is a common intermediate for the photocatalytic and electrocatalytic coupling of CH3OH or HCHO to EG. Thus, CH3OH or HCHO could be utilised as a platform for hydroxymethylation reaction to construct new C–C couplings to produce diols or higher aliphatic alcohols in synthetic chemistry.
Some catalysts have shown promising performances for photocatalytic and electrocatalytic conversions of C1 molecules into C2+ compounds. For photocatalytic reduction of CO2, a Pt–0.5G–TiO2−x catalyst achieved formation rates of CH4 and C2H6 of 37 and 11 μmol g−1 h−1, respectively, under one sun AM 1.5G illumination with solid evidence for the formation of CH4 and C2H6 from CO2 by 13CO2 isotopic control experiments.76 The apparent quantum yield of (CH4 + C2H6) reached 7.9% and the catalyst was stable during 42 h of reaction. A Cu-modified polymeric carbon nitride (PCN) could work for photocatalytic conversion of CH4 with H2O to C2H5OH under UV-vis light irradiation, and the formation rate of C2H5OH reached 106 μmol g−1 h−1.265 Despite the progress, the state-of-the-art performances for photocatalytic conversions of CO2 and CH4 are far from commercial consideration. On the other hand, the performances for photocatalytic conversions of CO, CH3OH and HCHO are usually significantly better than those for photocatalytic conversions of CO2 and CH4. For example, without external heating, the Fe5C2 for photo-thermal hydrogenation of CO at atmospheric pressure under UV-vis light irradiation provided a CO conversion of 49.5% (conversion rate, 17 mmol g−1 h−1) with C2–C4 olefin selectivity of 55.5% (CO2 selectivity, 18.9%).112 For photocatalytic conversion of methanol, the EG formation rate could reach 11 mmol g−1 h−1 over a MoS2/CdS catalyst under visible-light irradiation, with EG selectivity, yield and quantum yield of 90%, 16% and 5%, respectively.292 HCHO is more reactive and the formation rate of C2 compounds (EG, CH3CHO, HOCH2CHO) could reach 54 mmol g−1 h−1 from HCHO over a MnOx–Pt@MoOx/BiVO4 catalyst under UV-vis light irradiation.304 Furthermore, as compared to photocatalysis, electrocatalysis has achieved much better performances for the transformations of inert CO2 and CH4. For electrocatalytic CO2RR, the FE and partial current density of C2H4 over Cu-based catalysts could reach 83% and 1.3 A cm−2, respectively.138,145 A fluorine-modified Cu catalyst offered C2–4 products (mainly C2H4 and C2H5OH) with selectivity of 85.8% and single-pass yield of 16.5%, and the formation rate of C2H4 reached 3.2 mmol h−1 cm−2 (12.8 mol g−1 h−1).142 This performance outperforms those achieved in high-temperature and high-pressure thermocatalysis.8 A Fe–Sr2Fe1.5Mo0.5O6−δ catalyst showed a current density of 1.0 A cm−2 in the electrocatalytic oxidation of CH4 at an applied voltage of 1.6 V and a temperature of 850 °C.279 The conversion of CH4, and the selectivity and yield of C2 products (C2H4 and C2H6) reached 41%, 81.2% and 33%, respectively.279 It is noteworthy that recent studies on electrocatalytic conversion of CH3OH or HCHO to C2+ compounds are very limited.
With the flourishing development of photocatalytic and electrocatalytic systems for the transformation of C1 molecules into C2+ products, the catalyst stability has become an important issue for future applications. Better stability would reduce the associated maintenance and consumable costs, and is one of the keys for commercial applications of photocatalytic and electrocatalytic conversions of C1 molecules into C2+ compounds. Many photocatalysts and electrocatalysts reported for the conversion of C1 molecules showed good stability of tens or hundreds of hours. For a potential photocatalytic process such as H2 evolution by water splitting, a stability of 1000 h with a solar to chemical efficiency of 10% is required for commercial applications.311 Considering the comparability, the catalyst stability for photocatalytic conversions of C1 molecules to C2+ compounds should exceed 1000 h at a sufficiently high quantum efficiency (at least >10%). As described above, the FE (>80%) and current density (>1 A cm−2) reported for the electrocatalytic CO2RR to C2+ products (mainly C2H4, C2H5OH) could meet the industrial requirement, but the long-term catalyst stability is still a challenge. The best stability achieved to date for electrocatalytic CO2RR to C2+ compounds is 190 h at a current density of ∼120 mA cm−2.146 It was once considered that a catalyst stability of about 5000 h is required as the first cornerstone for an electrocatalytic process.312 There is large room for the future studies to improve the catalyst stability in photocatalytic and electrocatalytic transformations of C1 molecules to C2 compounds.
An understanding of the reaction mechanism would not only deepen our knowledge of C1 chemistry but also help to develop more efficient photocatalysts and electrocatalysts for valorisation of C1 molecules. Generally, the photocatalytic and electrocatalytic conversions of C1 molecules to C2+ compounds include two major steps: the activation of C1 molecules to C1 intermediates and C–C coupling of C1 intermediates to C2+ compounds. The C1 intermediates may also be easily converted into other C1 molecules, and thus controlling C–C coupling of C1 intermediates is the key to obtaining high selectivity of C2+ products. Mechanistic studies have pointed out that the typical C1 molecules, including CO2, CO, CH4, CH3OH and HCHO, are transformed into specific C1 surface species or radicals, which then undergo C–C coupling to C2+ compounds through multi-electron/multi-proton transfer processes (Fig. 43). For example, CO2˙−, ˙CHO and CH3˙ have been proposed as the C1 intermediates for C–C coupling to form oxalic acid, glyoxal and C2H6, respectively, in photocatalytic CO2 reduction.67,107–109 In electrocatalytic CO2RR and CORR, *CO species has been proposed to be the major intermediate for C–C coupling in most studies,232–234 but recent studies have demonstrated that the coupling between *CHO species is a more facile pathway.142,236,237,239 The ˙CH2OH radical is the intermediate for C–C coupling in photocatalytic and electrocatalytic conversions of CH3OH and HCHO to EG.292,301,304,306 Meanwhile, the CH3˙ radical is the key intermediate for the C–C coupling in either photocatalytic or electrocatalytic conversion of CH4 to C2H6.251,256–258,264,279 It is noteworthy that the insights into the detailed mechanisms for C1-molecule activation, reaction intermediate formation, C–C coupling and product formation are still very limited. The nature of active sites and the synergy between the catalytically active sites and electrons/holes/protons are not well documented. The state-of-the-art knowledge cannot enable rational design of efficient photocatalysts or electrocatalysts with controlled product selectivity.
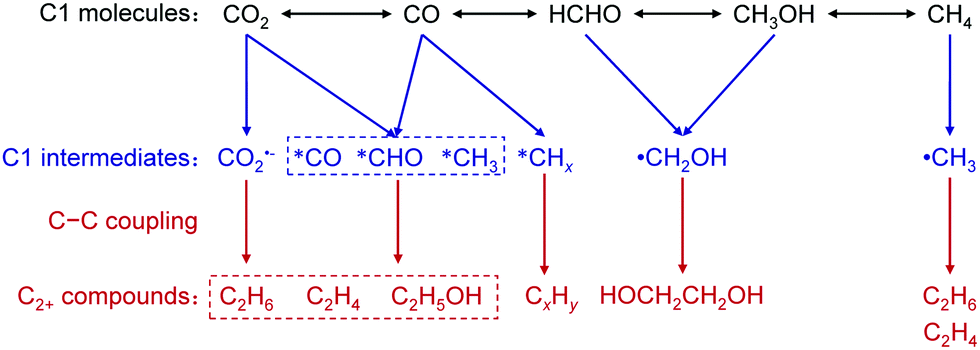 | ||
| Fig. 43 Proposed C1 intermediates in photocatalytic and electrocatalytic conversions of C1 molecules to C2+ compounds. “*” in the figure represents surface or radical species. | ||
Advanced characterization techniques have also promoted the progress in photocatalytic and electrocatalytic transformations of C1 molecules, in particular for the elucidation of active sites and reaction intermediates. Take electrocatalytic CO2RR and CORR as examples. To understand the nature of the active sites leading to high C2+ FE over oxide-derived Cu catalysts, multiple in situ characterization techniques have been employed to investigate the oxidation state of Cu under reaction conditions, including secondary-ion mass spectrometry,185 surface-enhanced Raman spectroscopy189 and time-resolved X-ray absorption spectroscopy.313In situ spectroscopic studies have also been used to detect the reaction intermediate. Through in situ FT-IR spectroscopic studies, our group observed *CHO species during electrocatalytic CO2RR,142 while *OCCOH species were detected during electrocatalytic CORR by Koper and co-workers.235 These insights provide deeper understanding of the intermediate formation and C–C coupling process. However, it is noteworthy that most of the in situ characterization techniques are performed under conditions with low reaction rates, which are different from those adopted for achieving high reaction rates. In the future, characterization techniques should be developed to improve the capability of working under real reaction conditions.
Theoretical calculations have largely contributed to the understanding of the active sites and reaction mechanisms for the conversion of C1 molecules to C2+ products as well as the insights that guide the selectivity control. Elementary steps including the activation of C1 molecules, proton/electron transfer, C1 intermediate formation, C–C coupling of C1 intermediates and product formation for different systems have been studied on molecular or atomic levels by using DFT calculations. However, most of the theoretical calculations are based on gas-phase reaction models, whereas the effects of coverage, solution, electrolyte and electric field have seldom been considered, although these effects are known to be very important for photocatalytic and electrocatalytic reactions. Theoretical studies have recently also been exploited to accelerate the catalyst discovery. For example, Sargent and co-workers developed a machine-learning-accelerated, high-throughput DFT framework to screen electrocatalysts.144 After studying 144 different Cu-containing intermetallic materials, they discovered that a Cu–Al alloy had multiple sites and surface orientations with near-optimal CO binding. The experimentally fabricated de-alloyed nanoporous Cu–Al catalyst showed an FE of C2H4 as high as 80%.144 More efforts should be put into this area in the future.
Photocatalysis and electrocatalysis have been enriching the area of C1 chemistry. More sustainable routes for the utilisation of C1 molecules including CO, CO2, CH4, CH3OH and HCHO can be developed to produce C2+ compounds such as C2H4, C2H5OH and EG, which play important roles in the current chemical or energy industry. Not only could some existing C1-chemistry reactions, such as the hydrogenation of CO to C2+ hydrocarbons, be performed under very mild conditions, offering better selectivity toward target products, but photocatalysis and electrocatalysis also enable new transformations of C1 molecules involving C–C coupling, in particular the thermodynamically limited reactions. With the assistance of photo or electrical energy, the inert CO2 and CH4 molecules can be efficiently activated to reactive intermediates for subsequent C–C coupling, selectively offering C2+ compounds under mild conditions. It is noteworthy that the electrocatalytic conversion of CO2 to C2H4 and C2H5OH has achieved significant progress in recent years. Furthermore, the inert C–H bond in CH3OH could be preferentially activated by photocatalysis with the OH group keeping intact, generating ˙CH2OH radicals for C–C coupling to EG. Photocatalysis and electrocatalysis have brought many opportunities and also challenges in the area of C1 chemistry. We are expecting further breakthroughs in this emerging field.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the National Key Research and Development Program of Ministry of Science and Technology (2017YFB0602201) and the National Natural Science Foundation of China (No. 91945301, 21972115 and 21503176) is greatly acknowledged.Notes and references
- C. Mesters, Annu. Rev. Chem. Biomol. Eng., 2016, 7, 223–238 CrossRef.
- C. B. Roberts and N. O. Elbashir, Fuel Process. Technol., 2003, 83, 1–9 CrossRef CAS.
- Energy Information Administration (EIA), Annual Energy Outlook 2018, (Washington, DC, July 2018) (https://www.eia.gov/pressroom/presentations/capuano_07242018.pdf).
- W. Keim, Pure Appl. Chem., 1986, 58, 825–832 CAS.
- W. Cui, G. Zhang, T. Hu and X. Bu, Coord. Chem. Rev., 2019, 387, 79–120 CrossRef CAS.
- J. Bao, G. Yang, Y. Yoneyama and N. Tsubaki, ACS Catal., 2019, 9, 3026–3053 CrossRef CAS.
- K. Cheng, J. Kang, D. L. King, V. Subramanian, C. Zhou, Q. Zhang and Y. Wang, Adv. Catal., 2017, 60, 125–208 CAS.
- W. Zhou, K. Cheng, J. Kang, C. Zhou, V. Subramanian, Q. Zhang and Y. Wang, Chem. Soc. Rev., 2019, 48, 3193–3228 RSC.
- T. Sakakura, J. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS.
- R. W. Dorner, D. R. Hardy, F. W. Williams and H. D. Willauer, Energy Environ. Sci., 2010, 3, 884–890 RSC.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- M. D. Porosoff, B. Yan and J. G. Chen, Energy Environ. Sci., 2016, 9, 62–73 RSC.
- H. Yang, C. Zhang, P. Gao, H. Wang, X. Li, L. Zhong, W. Wei and Y. Sun, Catal. Sci. Technol., 2017, 7, 4580–4598 RSC.
- L. Guo, J. Sun, Q. Ge and N. Tsubaki, J. Mater. Chem. A, 2018, 6, 23244–23262 RSC.
- A. Dokania, A. Ramirez, A. Bavykina and J. Gascon, ACS Energy Lett., 2019, 4, 167–176 CrossRef CAS.
- J. Zhong, X. Yang, Z. Wu, B. Liang, Y. Huang and T. Zhang, Chem. Soc. Rev., 2020, 49, 1385–1413 RSC.
- X. Jiang, X. Nie, X. Guo, C. Song and J. G. Chen, Chem. Rev., 2020, 120, 7984–8034 CrossRef CAS.
- M. Ravi, M. Ranocchiari and J. A. van Bokhoven, Angew. Chem., Int. Ed., 2017, 56, 16464–16483 CrossRef CAS.
- A. I. Olivos-Suarez, À. Szécsényi, E. J. M. Hensen, J. Ruiz-Martinez, E. A. Pidko and J. Gascon, ACS Catal., 2016, 6, 2965–2981 CrossRef CAS.
- P. Schwach, X. Pan and X. Bao, Chem. Rev., 2017, 117, 8497–8520 CrossRef CAS.
- N. J. Gunsalus, A. Koppaka, S. H. Park, S. M. Bischof, B. G. Hashiguchi and R. A. Periana, Chem. Rev., 2017, 117, 8521–8573 CrossRef CAS.
- U. Olsbye, S. Svelle, M. Bjørgen, P. Beato, T. V. W. Janssens, F. Joensen, S. Bordiga and K. P. Lillerud, Angew. Chem., Int. Ed., 2012, 51, 5810–5831 CrossRef CAS.
- P. Tian, Y. Wei, M. Ye and Z. Liu, ACS Catal., 2015, 5, 1922–1938 CrossRef CAS.
- S. Xu, Y. Zhi, J. Han, W. Zhang, X. Wu, T. Sun, Y. Wei and Z. Liu, Adv. Catal., 2017, 61, 37–122 CAS.
- I. Yarulina, A. D. Chowdhury, F. Meirer, B. M. Weckhuysen and J. Gascon, Nat. Catal., 2018, 1, 398–411 CrossRef CAS.
- M. Yang, D. Fan, Y. Wei, P. Tian and Z. Liu, Adv. Mater., 2019, 31, 1902181 CrossRef CAS.
- F. Jiao, J. Li, X. Pan, J. Xiao, H. Li, H. Ma, M. Wei, Y. Pan, Z. Zhou, M. Li, S. Miao, J. Li, Y. Zhu, D. Xiao, T. He, J. Yang, F. Qi, Q. Fu and X. Bao, Science, 2016, 351, 1065–1068 CrossRef CAS.
- K. Cheng, B. Gu, X. Liu, J. Kang, Q. Zhang and Y. Wang, Angew. Chem., Int. Ed., 2016, 55, 4725–4728 CrossRef CAS.
- K. Cheng, W. Zhou, J. Kang, S. He, S. Shi, Q. Zhang, Y. Pan, W. Wen and Y. Wang, Chem, 2017, 3, 334–347 CAS.
- S. Kasipandi and J. W. Bae, Adv. Mater., 2019, 31, 1803390 CrossRef.
- Z. Li, J. Wang, Y. Qu, H. Liu, C. Tang, S. Miao, Z. Feng, H. An and C. Li, ACS Catal., 2017, 7, 8544–8548 CrossRef CAS.
- X. Liu, M. Wang, C. Zhou, W. Zhou, K. Cheng, J. Kang, Q. Zhang, W. Deng and Y. Wang, Chem. Commun., 2018, 54, 140–143 RSC.
- P. Gao, S. Dang, S. Li, X. Bu, Z. Liu, M. Qiu, C. Yang, H. Wang, L. Zhong, Y. Han, Q. Liu, W. Wei and Y. Sun, ACS Catal., 2018, 8, 571–578 CrossRef CAS.
- Y. Ni, Z. Chen, Y. Fu, Y. Liu, W. Zhu and Z. Liu, Nat. Commun., 2018, 9, 3457 CrossRef.
- Z. Li, Y. Qu, J. Wang, H. Liu, M. Li, S. Miao and C. Li, Joule, 2019, 3, 570–583 CrossRef CAS.
- C. Zhou, J. Shi, W. Zhou, K. Cheng, Q. Zhang, J. Kang and Y. Wang, ACS Catal., 2020, 10, 302–310 CrossRef CAS.
- W. Zhou, J. Kang, K. Cheng, S. He, J. Shi, C. Zhou, Q. Zhang, J. Chen, L. Peng, M. Chen and Y. Wang, Angew. Chem., Int. Ed., 2018, 57, 12012–12016 CrossRef CAS.
- J. Kang, S. He, W. Zhou, Z. Shen, Y. Li, M. Chen, Q. Zhang and Y. Wang, Nat. Commun., 2020, 11, 827 CrossRef CAS.
- G. Chen, G. I. N. Waterhouse, R. Shi, J. Zhao, Z. Li, L. Wu, C. Tung and T. Zhang, Angew. Chem., Int. Ed., 2019, 58, 17528–17551 CrossRef CAS.
- P. D. Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef.
- Y. Jiang, R. Long and Y. Xiong, Chem. Sci., 2019, 10, 7310–7326 RSC.
- Y. Zhao, G. I. N. Waterhouse, G. Chen, X. Xiong, L. Wu, C. Tung and T. Zhang, Chem. Soc. Rev., 2019, 48, 1972–2010 RSC.
- M. Jouny, G. S. Hutchings and F. Jiao, Nat. Catal., 2019, 2, 1062–1070 CrossRef CAS.
- G. A. Olah, G. K. Prakash and A. Goeppert, J. Am. Chem. Soc., 2011, 133, 12881–12898 CrossRef CAS.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS.
- S. Xie, Q. Zhang, G. Liu and Y. Wang, Chem. Commun., 2016, 52, 35–59 RSC.
- X. Chang, T. Wang and J. Gong, Energy Environ. Sci., 2016, 9, 2177–2196 RSC.
- K. Li, B. Peng and T. Peng, ACS Catal., 2016, 6, 7485–7527 CrossRef CAS.
- X. Li, J. Yu, M. Jaroniec and X. Chen, Chem. Rev., 2019, 119, 3962–4179 CrossRef CAS.
- C. F. Shih, T. Zhang, J. Li and C. Bai, Joule, 2018, 2, 1925–1949 CrossRef CAS.
- C. Chen, J. F. K. Kotyk and S. W. Sheehan, Chem, 2018, 4, 2571–2586 CAS.
- L. Zhang, Z. Zhao, T. Wang and J. Gong, Chem. Soc. Rev., 2018, 47, 5423–5443 RSC.
- D. Gao, R. M. Arán-Ais, H. S. Jeon and B. R. Cuenya, Nat. Catal., 2019, 2, 198–210 CrossRef CAS.
- K. Elouarzaki, V. Kannan, V. Jose, H. S. Sabharwal and J. M. Lee, Adv. Energy Mater., 2019, 9, 1900090 CrossRef.
- S. Xie, S. Lin, Q. Zhang, Z. Tian and Y. Wang, J. Energy Chem., 2018, 27, 1629–1636 CrossRef.
- X. Meng, X. Cui, N. P. Rajan, L. Yu, D. Deng and X. Bao, Chem, 2019, 5, 2296–2325 CAS.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637–638 CrossRef CAS.
- S. C. Roy, O. K. Varghese, M. Paulose and C. A. Grimes, ACS Nano, 2010, 4, 1259–1278 CrossRef CAS.
- M. R. Hoffmann, J. A. Moss and M. M. Baum, Dalton Trans., 2011, 40, 5151–5158 RSC.
- L. Wei, C. Yu, Q. Zhang, H. Liu and Y. Wang, J. Mater. Chem. A, 2018, 6, 22411–22436 RSC.
- F. M. Mota and D. H. Kim, Chem. Soc. Rev., 2019, 48, 205–259 RSC.
- T. Mizuno, K. Adachi, K. Ohta and A. Saji, J. Photochem. Photobiol., A, 1996, 98, 87–90 CrossRef CAS.
- J. Mao, T. Peng, X. Zhang, K. Li, L. Ye and L. Zan, Catal. Sci. Technol., 2013, 3, 1253–1260 RSC.
- Y. Liu, B. Huang, Y. Dai, X. Zhang, X. Qin, M. Jiang and M.-H. Whangbo, Catal. Commun., 2009, 11, 210–213 CrossRef CAS.
- W. Dai, J. Yu, Y. Deng, X. Hu, T. Wang and X. Luo, Appl. Surf. Sci., 2017, 403, 230–239 CrossRef CAS.
- P. Liou, S. Chen, J. Wu, D. Liu, S. Mackintosh, M. Maroto-Valer and R. Linforth, Energy Environ. Sci., 2011, 4, 1487–1494 RSC.
- H. Park, H. Ou, A. Colussi and M. R. Hoffmann, J. Phys. Chem. A, 2015, 119, 4658–4666 CrossRef CAS.
- O. Ola and M. M. Maroto-Valer, Appl. Catal., A, 2015, 502, 114–121 CrossRef CAS.
- M. Cheng, S. Yang, R. Chen, X. Zhu, Q. Liao and Y. Huang, Int. J. Hydrogen Energy, 2017, 42, 9722–9732 CrossRef CAS.
- C. Wang, R. L. Thompson, P. Ohodnicki, J. Baltrus and C. Matranga, J. Mater. Chem., 2011, 21, 13452–13457 RSC.
- M. A. Asi, C. He, M. Su, D. Xia, L. Lin, H. Deng, Y. Xiong, R. Qiu and X. Li, Catal. Today, 2011, 175, 256–263 CrossRef.
- H. Li, S. Gan, H. Wang, D. Han and L. Niu, Adv. Mater., 2015, 27, 6906–6913 CrossRef CAS.
- X. Xia, Z. Jia, Y. Yu, Y. Liang, Z. Wang and L. Ma, Carbon, 2007, 45, 717–721 CrossRef CAS.
- L. M. Pastrana-Martínez, A. M. T. Silva, N. N. C. Fonseca, J. R. Vaz, J. L. Figueiredo and J. L. Faria, Top. Catal., 2016, 59, 1279–1291 CrossRef.
- W. Tu, Y. Zhou, Q. Liu, S. Yan, S. Bao, X. Wang, M. Xiao and Z. Zou, Adv. Funct. Mater., 2013, 23, 1743–1749 CrossRef CAS.
- S. Sorcar, J. Thompson, Y. Hwang, Y. H. Park, T. Majima, C. A. Grimes, J. R. Durrant and S. In, Energy Environ. Sci., 2018, 11, 3183–3193 RSC.
- L. Yu, G. Li, X. Zhang, X. Ba, G. Shi, Y. Li, P. Wong, J. C. Yu and Y. Yu, ACS Catal., 2016, 6, 6444–6454 CrossRef CAS.
- T. Yui, A. Kan, C. Saitoh, K. Koike, T. Ibusuki and O. Ishitani, ACS Appl. Mater. Interfaces, 2011, 3, 2594–2600 CrossRef CAS.
- W. Kim, T. Seok and W. Choi, Energy Environ. Sci., 2012, 5, 6066–6070 RSC.
- Y. Zhu, Z. Xu, Q. Lang, W. Jiang, Q. Yin, S. Zhong and S. Bai, Appl. Catal., B, 2017, 206, 282–292 CrossRef CAS.
- V. Jeyalakshmi, R. Mahalakshmy, K. R. Krishnamurthy and B. Viswanathan, Catal. Today, 2016, 266, 160–167 CrossRef CAS.
- K. Adachi, K. Ohta and T. Mizuno, Sol. Energy, 1994, 53, 187–190 CrossRef CAS.
- O. K. Varghese, M. Paulose, T. J. LaTempa and C. A. Grimes, Nano Lett., 2009, 9, 731–737 CrossRef CAS.
- X. Zhang, F. Han, B. Shi, S. Farsinezhad, G. P. Dechaine and K. Shankar, Angew. Chem., Int. Ed., 2012, 51, 12732–12735 CrossRef CAS.
- T. Nguyen and J. C. S. Wu, Appl. Catal., A, 2008, 335, 112–120 CrossRef CAS.
- W. Hou, W. Hung, P. Pavaskar, A. Goeppert, M. Aykol and S. B. Cronin, ACS Catal., 2011, 1, 929–936 CrossRef CAS.
- S. Yu and P. K. Jain, Nat. Commun., 2019, 10, 2022 CrossRef.
- J. Paul and F. M. Hoffmann, Catal. Lett., 1988, 1, 445–456 CrossRef CAS.
- C. Lee, R. Antoniou Kourounioti, J. C. S. Wu, E. Murchie, M. Maroto-Valer, O. E. Jensen, C. Huang and A. Ruban, J. CO2 Util., 2014, 5, 33–40 CrossRef CAS.
- J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900–1909 CrossRef CAS.
- Y. Zhang, B. Xia, J. Ran, K. Davey and S.-Z. Qiao, Adv. Energy Mater., 2020, 10, 1903879 CrossRef CAS.
- S. Xie, Y. Wang, Q. Zhang, W. Deng and Y. Wang, ACS Catal., 2014, 4, 3644–3653 CrossRef CAS.
- M. Subrahmanyam, S. Kaneco and N. Alonso-Vante, Appl. Catal., B, 1999, 23, 169–174 CrossRef CAS.
- M. B. Gawande, A. Goswami, F.-X. Felpin, T. Asefa, X. Huang, R. Silva, X. Zou, R. Zboril and R. S. Varma, Chem. Rev., 2016, 116, 3722–3811 CrossRef CAS.
- Q. Zhai, S. Xie, W. Fan, Q. Zhang, Y. Wang, W. Deng and Y. Wang, Angew. Chem., Int. Ed., 2013, 52, 5776–5779 CrossRef CAS.
- S. Sorcar, Y. Hwang, J. Lee, H. Kim, K. M. Grimes, C. A. Grimes, J. Jung, C. Cho, T. Majima, M. R. Hoffmann and S. In, Energy Environ. Sci., 2019, 12, 2685–2696 RSC.
- M. Tahir, Appl. Catal., B, 2017, 219, 329–343 CrossRef CAS.
- Q. Kang, T. Wang, P. Li, L. Liu, K. Chang, M. Li and J. Ye, Angew. Chem., Int. Ed., 2015, 54, 841–845 CrossRef CAS.
- Q. Chen, X. Chen, M. Fang, J. Chen, Y. Li, Z. Xie, Q. Kuang and L. Zheng, J. Mater. Chem. A, 2019, 7, 1334–1340 RSC.
- S. Yu, A. J. Wilson, J. Heo and P. K. Jain, Nano Lett., 2018, 18, 2189–2194 CrossRef CAS.
- M. A. Asi, L. Zhu, C. He, V. K. Sharma, D. Shu, S. Li, J. Yang and Y. Xiong, Catal. Today, 2013, 216, 268–275 CrossRef.
- W. H. Koppenol and J. D. Rush, J. Phys. Chem., 1987, 91, 4429–4430 CrossRef CAS.
- H.-J. Freund and M. W. Roberts, Surf. Sci. Rep., 1996, 25, 225–273 CrossRef.
- N.-N. Vu, S. Kaliaguine and T.-O. Do, Adv. Funct. Mater., 2019, 29, 1901825 CrossRef.
- L. Mino, G. Spoto and A. M. Ferrari, J. Phys. Chem. C, 2014, 118, 25016–25026 CrossRef CAS.
- H. He, P. Zapol and L. A. Curtiss, Energy Environ. Sci., 2012, 5, 6196–6205 RSC.
- B. R. Eggins, P. K. J. Robertson, J. H. Stewart and E. Woods, J. Chem. Soc., Chem. Commun., 1993, 349–350 RSC.
- B. R. Eggins, P. K. J. Robertson, E. P. Murphy, E. Woods and J. T. S. Irvine, J. Photochem. Photobiol., A, 1998, 118, 31–40 CrossRef CAS.
- I. A. Shkrob, T. W. Marin, H. He and P. Zapol, J. Phys. Chem. C, 2012, 116, 9450–9460 CrossRef CAS.
- Z. Li, J. Liu, Y. Zhao, G. I. N. Waterhouse, G. Chen, R. Shi, X. Zhang, X. Liu, Y. Wei, X.-D. Wen, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Mater., 2018, 30, 1800527 CrossRef.
- Y. Zhao, Z. Li, M. Li, J. Liu, X. Liu, G. I. N. Waterhouse, Y. Wang, J. Zhao, W. Gao, Z. Zhang, R. Long, Q. Zhang, L. Gu, X. Liu, X. Wen, D. Ma, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Mater., 2018, 30, 1803127 CrossRef.
- W. Gao, R. Gao, Y. Zhao, M. Peng, C. Song, M. Li, S. Li, J. Liu, W. Li, Y. Deng, M. Zhang, J. Xie, G. Hu, Z. Zhang, R. Long, X.-D. Wen and D. Ma, Chem, 2018, 4, 2917–2928 CAS.
- X. Guo, Z. Jiao, G. Jin and X. Guo, ACS Catal., 2015, 5, 3836–3840 CrossRef CAS.
- L. Wang, Y. Zhang, X. Gu, Y. Zhang and H. Su, Catal. Sci. Technol., 2018, 8, 601–610 RSC.
- Y. Zhao, B. Zhao, J. Liu, G. Chen, R. Gao, S. Yao, M. Li, Q. Zhang, L. Gu, J. Xie, X. Wen, L.-Z. Wu, C.-H. Tung, D. Ma and T. Zhang, Angew. Chem., Int. Ed., 2016, 55, 4215–4219 CrossRef CAS.
- L. Zhang, Z.-J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2017, 56, 11326–11353 CrossRef CAS.
- L. Fan, C. Xia, F. Yang, J. Wang, H. Wang and Y. Lu, Sci. Adv., 2020, 6, eaay3111 CrossRef CAS.
- D. U. Nielsen, X.-M. Hu, K. Daasbjerg and T. Skrydstrup, Nat. Catal., 2018, 1, 244–254 CrossRef CAS.
- D. M. Weekes, D. A. Salvatore, A. Reyes, A. Huang and C. P. Berlinguette, Acc. Chem. Res., 2018, 51, 910–918 CrossRef CAS.
- D. Higgins, C. Hahn, C. Xiang, T. F. Jaramillo and A. Z. Weber, ACS Energy Lett., 2019, 4, 317–324 CrossRef CAS.
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 RSC.
- J. He, K. E. Dettelbach, D. A. Salvatore, T. Li and C. P. Berlinguette, Angew. Chem., Int. Ed., 2017, 56, 6068–6072 CrossRef CAS.
- W. Ma, S. Xie, X.-G. Zhang, F. Sun, J. Kang, Z. Jiang, Q. Zhang, D.-Y. Wu and Y. Wang, Nat. Commun., 2019, 10, 892 CrossRef.
- W. Ma, M. Xie, S. Xie, L. Wei, Y. Cai, Q. Zhang and Y. Wang, J. Energy Chem., 2021, 54, 422–428 CrossRef.
- A. Vasileff, X. Zhi, C. Xu, L. Ge, Y. Jiao, Y. Zheng and S.-Z. Qiao, ACS Catal., 2019, 9, 9411–9417 CrossRef CAS.
- R. P. Jansonius, L. M. Reid, C. N. Virca and C. P. Berlinguette, ACS Energy Lett., 2019, 4, 980–986 CrossRef CAS.
- Y. Zheng, A. Vasileff, X. Zhou, Y. Jiao, M. Jaroniec and S.-Z. Qiao, J. Am. Chem. Soc., 2019, 141, 7646–7659 CrossRef CAS.
- X. Liu, J. Xiao, H. Peng, X. Hong, K. Chan and J. K. Nørskov, Nat. Commun., 2017, 8, 15438 CrossRef CAS.
- H. Zhang, J. Li, M. Cheng and Q. Lu, ACS Catal., 2019, 9, 49–65 CrossRef CAS.
- N. S. Romero Cuellar, K. Wiesner-Fleischer, M. Fleischer, A. Rucki and O. Hinrichsen, Electrochim. Acta, 2019, 307, 164–175 CrossRef CAS.
- X. Zhou and C. Xiang, ACS Energy Lett., 2018, 3, 1892–1897 CrossRef CAS.
- X. Liu, M. Wang, H. Yin, J. Hu, K. Cheng, J. Kang, Q. Zhang and Y. Wang, ACS Catal., 2020, 10, 8303–8314 CrossRef CAS.
- X. Liu, W. Zhou, Y. Yang, K. Cheng, J. Kang, L. Zhang, G. Zhang, X. Min, Q. Zhang and Y. Wang, Chem. Sci., 2018, 9, 4708–4718 RSC.
- K. Jiang, R. B. Sandberg, A. J. Akey, X. Liu, D. C. Bell, J. K. Nørskov, K. Chan and H. Wang, Nat. Catal., 2018, 1, 111–119 CrossRef CAS.
- A. Loiudice, P. Lobaccaro, E. A. Kamali, T. Thao, B. H. Huang, J. W. Ager and R. Buonsanti, Angew. Chem., Int. Ed., 2016, 55, 5789–5792 CrossRef CAS.
- H. Mistry, A. S. Varela, C. S. Bonifacio, I. Zegkinoglou, I. Sinev, Y.-W. Choi, K. Kisslinger, E. A. Stach, J. C. Yang, P. Strasser and B. R. Cuenya, Nat. Commun., 2016, 7, 12123 CrossRef.
- D. Ren, Y. Deng, A. D. Handoko, C. S. Chen, S. Malkhandi and B. S. Yeo, ACS Catal., 2015, 5, 2814–2821 CrossRef CAS.
- B. Zhang, J. Zhang, M. Hua, Q. Wan, Z. Su, X. Tan, L. Liu, F. Zhang, G. Chen, D. Tan, X. Cheng, B. Han, L. Zheng and G. Mo, J. Am. Chem. Soc., 2020, 142, 13606–13613 CrossRef CAS.
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. De Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero-Bermudez, T.-K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974–980 CrossRef CAS.
- Z.-Q. Liang, T.-T. Zhuang, A. Seifitokaldani, J. Li, C.-W. Huang, C.-S. Tan, Y. Li, P. De Luna, C. T. Dinh, Y. Hu, Q. Xiao, P.-L. Hsieh, Y. Wang, F. Li, R. Quintero-Bermudez, Y. Zhou, P. Chen, Y. Pang, S.-C. Lo, L.-J. Chen, H. Tan, Z. Xu, S. Zhao, D. Sinton and E. H. Sargent, Nat. Commun., 2018, 9, 3828 CrossRef.
- D. Gao, I. Sinev, F. Scholten, R. M. Arán-Ais, N. J. Divins, K. Kvashnina, J. Timoshenko and B. R. Cuenya, Angew. Chem., Int. Ed., 2019, 58, 17047–17053 CrossRef CAS.
- W. Ma, S. Xie, T. Liu, Q. Fan, J. Ye, F. Sun, Z. Jiang, Q. Zhang, J. Cheng and Y. Wang, Nat. Catal., 2020, 3, 478–487 CrossRef CAS.
- C.-T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. García de Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS.
- M. Zhong, K. Tran, Y. Min, C. Wang, Z. Wang, C. T. Dinh, P. De Luna, Z. Yu, A. S. Rasouli, P. Brodersen, S. Sun, O. Voznyy, C. S. Tan, M. Askerka, F. Che, M. Liu, A. Seifitokaldani, Y. Pang, S. C. Lo, A. Ip, Z. Ulissi and E. H. Sargent, Nature, 2020, 581, 178–183 CrossRef CAS.
- F. P. G. de Arquer, C. T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D. H. Nam, C. Gabardo, A. Seifitokaldani, X. Wang, Y. C. Li, F. Li, J. Edwards, L. J. Richter, S. J. Thorpe, D. Sinton and E. H. Sargent, Science, 2020, 367, 661–666 CrossRef.
- F. Li, A. Thevenon, A. Rosas-Hernández, Z. Wang, Y. Li, C. M. Gabardo, A. Ozden, C. T. Dinh, J. Li, Y. Wang, J. P. Edwards, Y. Xu, C. McCallum, L. Tao, Z.-Q. Liang, M. Luo, X. Wang, H. Li, C. P. O’Brien, C.-S. Tan, D.-H. Nam, R. Quintero-Bermudez, T.-T. Zhuang, Y. C. Li, Z. Han, R. D. Britt, D. Sinton, T. Agapie, J. C. Peters and E. H. Sargent, Nature, 2020, 577, 509–513 CrossRef CAS.
- K. D. Yang, W. R. Ko, J. H. Lee, S. J. Kim, H. Lee, M. H. Lee and K. T. Nam, Angew. Chem., Int. Ed., 2017, 56, 796–800 CrossRef CAS.
- A. Vasileff, Y. Zhu, X. Zhi, Y. Zhao, L. Ge, H. M. Chen, Y. Zheng and S.-Z. Qiao, Angew. Chem., Int. Ed., 2020, 59, 19649–19653 CrossRef CAS.
- D. Ren, B. S.-H. Ang and B. S. Yeo, ACS Catal., 2016, 6, 8239–8247 CrossRef CAS.
- S. Lee, G. Park and J. Lee, ACS Catal., 2017, 7, 8594–8604 CrossRef CAS.
- T. T. H. Hoang, S. Verma, S. Ma, T. T. Fister, J. Timoshenko, A. I. Frenkel, P. J. A. Kenis and A. A. Gewirth, J. Am. Chem. Soc., 2018, 140, 5791–5797 CrossRef CAS.
- Y. C. Li, Z. Wang, T. Yuan, D.-H. Nam, M. Luo, J. Wicks, B. Chen, J. Li, F. Li, F. P. G. de Arquer, Y. Wang, C.-T. Dinh, O. Voznyy, D. Sinton and E. H. Sargent, J. Am. Chem. Soc., 2019, 141, 8584–8591 CrossRef CAS.
- T. T. Zhuang, Z. Q. Liang, A. Seifitokaldani, Y. Li, P. De Luna, T. Burdyny, F. Che, F. Min, R. Quintero-Bermudez, C. T. Dinh, Y. Pang, M. Zhong, B. Zhang, J. Li, P. N. Chen, X. L. Zheng, H. Liang, W. N. Ge, B. L. J. Ye, D. Sinton, S. H. Yu and E. H. Sargent, Nat. Catal., 2018, 1, 421–428 CrossRef CAS.
- F. Li, Y. C. Li, Z. Wang, J. Li, D.-H. Nam, Y. Lum, M. Luo, X. Wang, A. Ozden, S.-F. Hung, B. Chen, Y. Wang, J. Wicks, Y. Xu, Y. Li, C. M. Gabardo, C.-T. Dinh, Y. Wang, T.-T. Zhuang, D. Sinton and E. H. Sargent, Nat. Catal., 2020, 3, 75–82 CrossRef CAS.
- Y. Song, R. Peng, D. K. Hensley, P. V. Bonnesen, L. Liang, Z. Wu, H. M. Meyer, M. Chi, C. Ma, B. G. Sumpter and A. J. Rondinone, ChemistrySelect, 2016, 1, 6055–6061 CrossRef CAS.
- X. Wang, Z. Wang, F. P. G. de Arquer, C. T. Dinh, A. Ozden, Y. C. Li, D. H. Nam, J. Li, Y. S. Liu, J. Wicks, Z. Chen, M. Chi, B. Chen, Y. Wang, J. Tam, J. Y. Howe, A. Proppe, P. Todorović, F. Li, T. T. Zhuang, C. M. Gabardo, A. R. Kirmani, C. McCallum, S. F. Hung, Y. Lum, M. Luo, Y. Min, A. Xu, C. P. O’Brien, B. Stephen, B. Sun, A. H. Ip, L. J. Richter, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Energy, 2020, 5, 478–486 CrossRef CAS.
- C. Chen, X. Yan, S. Liu, Y. Wu, Q. Wan, X. Sun, Q. Zhu, H. Liu, J. Ma, L. Zheng, H. Wu and B. Han, Angew. Chem., Int. Ed., 2020, 59, 16459–16464 CrossRef CAS.
- H. Xu, D. Rebollar, H. He, L. Chong, Y. Liu, C. Liu, C.-J. Sun, T. Li, J. V. Muntean, R. E. Winans, D.-J. Liu and T. Xu, Nat. Energy, 2020, 5, 623–632 CrossRef CAS.
- Y. Liu, S. Chen, X. Quan and H. Yu, J. Am. Chem. Soc., 2015, 137, 11631–11636 CrossRef CAS.
- Y. Liu, Y. Zhang, K. Cheng, X. Quan, X. Fan, Y. Su, S. Chen, H. Zhao, Y. Zhang, H. Yu and M. R. Hoffmann, Angew. Chem., Int. Ed., 2017, 56, 15607–15611 CrossRef CAS.
- Y. Song, S. Wang, W. Chen, S. Li, G. Feng, W. Wei and Y. Sun, ChemSusChem, 2020, 13, 293–297 CrossRef CAS.
- J. Wu, S. Ma, J. Sun, J. I. Gold, C. Tiwary, B. Kim, L. Zhu, N. Chopra, I. N. Odeh, R. Vajtai, A. Z. Yu, R. Luo, J. Lou, G. Ding, P. J. A. Kenis and P. M. Ajayan, Nat. Commun., 2016, 7, 13869 CrossRef CAS.
- M. Jouny, W. Luc and F. Jiao, Nat. Catal., 2018, 1, 748–755 CrossRef CAS.
- J. Li, Z. Wang, C. McCallum, Y. Xu, F. Li, Y. Wang, C. M. Gabardo, C.-T. Dinh, T.-T. Zhuang, L. Wang, J. Y. Howe, Y. Ren, E. H. Sargent and D. Sinton, Nat. Catal., 2019, 2, 1124–1131 CrossRef CAS.
- C. W. Li, J. Ciston and M. W. Kanan, Nature, 2014, 508, 504–507 CrossRef CAS.
- X. Feng, K. Jiang, S. Fan and M. W. Kanan, ACS Cent. Sci., 2016, 2, 169–174 CrossRef CAS.
- D. Raciti, L. Cao, K. J. T. Livi, P. F. Rottmann, X. Tang, C. Li, Z. Hicks, K. H. Bowen, K. J. Hemker, T. Mueller and C. Wang, ACS Catal., 2017, 7, 4467–4472 CrossRef CAS.
- J. Li, A. Xu, F. Li, Z. Wang, C. Zou, C. M. Gabardo, Y. Wang, A. Ozden, Y. Xu, D.-H. Nam, Y. Lum, J. Wicks, B. Chen, Z. Wang, J. Chen, Y. Wen, T. Zhuang, M. Luo, X. Du, T.-K. Sham, B. Zhang, E. H. Sargent and D. Sinton, Nat. Commun., 2020, 11, 3685 CrossRef CAS.
- L. Wang, S. Nitopi, A. B. Wong, J. L. Snider, A. C. Nielander, C. G. Morales-Guio, M. Orazov, D. C. Higgins, C. Hahn and T. F. Jaramillo, Nat. Catal., 2019, 2, 702–708 CrossRef CAS.
- L. Wang, D. C. Higgins, Y. Ji, C. G. Morales-Guio, K. Chan, C. Hahan and T. F. Jaramillo, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 12572–12575 CrossRef CAS.
- D. S. Ripatti, T. R. Veltman and M. W. Kanan, Joule, 2019, 3, 240–256 CrossRef CAS.
- W. Luc, X. Fu, J. Shi, J.-J. Lv, M. Jouny, B. H. Ko, Y. Xu, Q. Tu, X. Hu, J. Wu, Q. Yue, Y. Liu, F. Jiao and Y. Kang, Nat. Catal., 2019, 2, 423–430 CrossRef CAS.
- T. T. Zhuang, Y. Pang, Z.-Q. Liang, Z. Wang, Y. Li, C.-S. Tan, J. Li, C. T. Dinh, P. De Luna, P.-L. Hsieh, T. Burdyny, H.-H. Li, M. Liu, Y. Wang, F. Li, A. Proppe, A. Johnston, D.-H. Nam, Z.-Y. Wu, Y.-R. Zheng, A. H. Ip, H. Tan, L.-J. Chen, S.-H. Yu, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Catal., 2018, 1, 946–951 CrossRef CAS.
- Y. Pang, J. Li, Z. Wang, C.-S. Tan, P.-L. Hsieh, T.-T. Zhuang, Z.-Q. Liang, C. Zou, X. Wang, P. De Luna, J. P. Edwards, Y. Xu, F. Li, C.-T. Dinh, M. Zhong, Y. Lou, D. Wu, L.-J. Chen, E. H. Sargent and D. Sinton, Nat. Catal., 2019, 2, 251–258 CrossRef CAS.
- J. Li, F. Che, Y. Pang, C. Zou, J. Y. Howe, T. Burdyny, J. P. Edwards, Y. Wang, F. Li, Z. Wang, P. De Luna, C.-T. Dinh, T.-T. Zhuang, M. I. Saidaminov, S. Cheng, T. Wu, Y. Z. Finfrock, L. Ma, S.-H. Hsieh, Y.-S. Liu, G. A. Botton, W.-F. Pong, X. Du, J. Guo, T.-K. Sham, E. H. Sargent and D. Sinton, Nat. Commun., 2018, 9, 4614 CrossRef.
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, J. Phys. Chem. B, 2002, 106, 15–17 CrossRef CAS.
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, J. Mol. Catal. A: Chem., 2003, 199, 39–47 CrossRef CAS.
- K. J. P. Schouten, E. Pérez Gallent and M. T. M. Koper, ACS Catal., 2013, 3, 1292–1295 CrossRef CAS.
- C. Hahn, T. Hatsukade, Y.-G. Kim, A. Vailionis, J. H. Baricuatro, D. C. Higgins, S. A. Nitopi, M. P. Soriaga and T. F. Jaramillo, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5918–5923 CrossRef CAS.
- Y. Huang, A. D. Handoko, P. Hirunsit and B. S. Yeo, ACS Catal., 2017, 7, 1749–1756 CrossRef CAS.
- F. S. Roberts, K. P. Kuhl and A. Nilsson, Angew. Chem., Int. Ed., 2015, 54, 5179–5182 CrossRef CAS.
- H. Xiao, W. A. Goddard III, T. Cheng and Y. Liu, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6685–6688 CrossRef CAS.
- M. Favaro, H. Xiao, T. Cheng, W. A. Goddard III, J. Yano and E. J. Crumlin, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6706–6711 CAS.
- A. Eilert, F. Cavalca, F. S. Roberts, J. Osterwalder, C. Liu, M. Favaro, E. J. Crumlin, H. Ogasawara, D. Friebel, L. G. M. Pettersson and A. Nilsson, J. Phys. Chem. Lett., 2017, 8, 285–290 CrossRef CAS.
- Y. Lum and J. W. Ager, Angew. Chem., Int. Ed., 2018, 57, 551–554 CrossRef CAS.
- L. Mandal, K. R. Yang, M. R. Motapothula, D. Ren, P. Lobaccaro, A. Patra, M. Sherburne, V. S. Batista, B. S. Yeo, J. W. Ager, J. Martin and T. Venkatesan, ACS Appl. Mater. Interfaces, 2018, 10, 8574–8584 CrossRef CAS.
- C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 7231–7234 CrossRef CAS.
- J.-J. Velasco-Vélez, T. Jones, D. Gao, E. Carbonio, R. Arrigo, C.-J. Hsu, Y.-C. Huang, C.-L. Dong, J.-M. Chen, J.-F. Lee, P. Strasser, B. Roldan Cuenya, R. Schlögl, A. Knop-Gericke and C.-H. Chuang, ACS Sustainable Chem. Eng., 2019, 7, 1485–1492 CrossRef.
- Y. Zhao, X. Chang, A. S. Malkani, X. Yang, L. Thompson, F. Jiao and B. Xu, J. Am. Chem. Soc., 2020, 142, 9735–9743 CAS.
- C. Chen, X. Sun, L. Lu, D. Yang, J. Ma, Q. Zhu, Q. Qian and B. Han, Green Chem., 2018, 20, 4579–4583 RSC.
- T. Shinagawa, G. O. Larrazábal, A. J. Martín, F. Krumeich and J. Pérez-Ramírez, ACS Catal., 2018, 8, 837–844 CrossRef CAS.
- Y. Huang, Y. Deng, A. D. Handoko, G. K. L. Goh and B. S. Yeo, ChemSusChem, 2018, 10, 1–8 Search PubMed.
- R. García-Muelas, F. Dattila, T. Shinagawa, A. J. Martín, J. Pérez-Ramírez and N. López, J. Phys. Chem. Lett., 2018, 9, 7153–7159 CrossRef.
- D. Gao, F. Scholten and B. R. Cuenya, ACS Catal., 2017, 8, 5112–5120 CrossRef.
- Y. Huang, C. W. Ong and B. S. Yeo, ChemSusChem, 2018, 11, 3299–3306 CrossRef CAS.
- D. Gao, I. T. McCrum, S. Deo, Y. W. Choi, F. Scholten, W. Wan, J. G. Chen, M. J. Janik and B. R. Cuenya, ACS Catal., 2018, 8, 10012–10020 CrossRef CAS.
- J. Li, K. Chang, H. Zhang, M. He, W. A. Goddard, J. G. Chen, M.-J. Cheng and Q. Lu, ACS Catal., 2019, 9, 4709–4718 CrossRef CAS.
- R. Chen, H.-Y. Su, D. Liu, R. Huang, X. Meng, X. Cui, Z.-Q. Tian, D. H. Zhang and D. Deng, Angew. Chem., Int. Ed., 2020, 59, 154–160 CrossRef CAS.
- S. Ma, M. Sadakiyo, R. Luo, M. Heima, M. Yamauchi and P. J. A. Kenis, J. Power Sources, 2016, 301, 219–228 CrossRef CAS.
- C. M. Gabardo, C. P. O’Brien, J. P. Edwards, C. McCallum, Y. Xu, C.-T. Dinh, J. Li, E. H. Sargent and D. Sinton, Joule, 2019, 3, 2777–2791 CrossRef CAS.
- M. Ma, K. Djanashvili and W. A. Smith, Angew. Chem., Int. Ed., 2016, 55, 6680–6684 CrossRef CAS.
- L. Wang, S. A. Nitopi, E. Bertheussen, M. Orazov, C. G. Morales-Guio, X. Liu, D. C. Higgins, K. Chan, J. K. Nørskov, C. Hahn and T. F. Jaramillo, ACS Catal., 2018, 8, 7445–7454 CrossRef CAS.
- J. Li, D. Wu, A. S. Malkani, X. Chang, M. J. Cheng, B. Xu and Q. Lu, Angew. Chem., Int. Ed., 2020, 59, 4464–4469 CrossRef CAS.
- X. Wang, J. F. de Araújo, W. Ju, A. Bagger, H. Schmies, S. Kühl, J. Rossmeisl and P. Strasser, Nat. Nanotechnol., 2019, 14, 1063–1070 CrossRef CAS.
- V. Subramani and S. K. Gangwal, Energy Fuels, 2008, 22, 814–839 CrossRef CAS.
- J. Pang, M. Zheng and T. Zhang, Adv. Catal., 2019, 64, 89–191 CAS.
- A. Hira, Energy Policy, 2011, 39, 6925–6935 CrossRef CAS.
- Q. Liu, H. Wang, H. Xin, C. Wang, L. Yan, Y. Wang, Q. Zhang, X. Zhang, Y. Xu, G. W. Huber and L. Ma, ChemSusChem, 2019, 12, 3977–3987 CrossRef CAS.
- H. T. Luk, C. Mondelli, D. C. Ferré, J. A. Stewart and J. Pérez-Ramírez, Chem. Soc. Rev., 2017, 46, 1358–1426 RSC.
- H. Song, P. Wang, S. Li, W. Deng, Y. Li, Q. Zhang and Y. Wang, Chem. Commun., 2019, 55, 4303–4306 RSC.
- M. Yang, H. Qi, F. Liu, Y. Ren, X. Pan, L. Zhang, X. Liu, H. Wang, J. Pang, M. Zheng, A. Wang and T. Zhang, Joule, 2019, 3, 1937–1948 CrossRef CAS.
- C. Li, G. Xu, C. Wang, L. Ma, Y. Qiao, Y. Zhang and Y. Fu, Green Chem., 2019, 21, 2234–2239 RSC.
- F. Calle-Vallejo and M. T. M. Koper, Angew. Chem., Int. Ed., 2013, 52, 7282–7285 CrossRef CAS.
- C. G. Morales-Guio, E. R. Cave, S. A. Nitopi, J. T. Feaster, L. Wang, K. P. Kuhl, A. Jackson, N. C. Johnson, D. N. Abram, T. Hatsukade, C. Hahn and T. F. Jaramillo, Nat. Catal., 2018, 1, 764–771 CrossRef CAS.
- E. L. Clark, C. Hahn, T. F. Jaramillo and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 15848–15857 CrossRef CAS.
- R. M. Arán-Ais, F. Scholtenm, S. Kunze, R. Rizo and B. R. Cuenya, Nat. Energy, 2020, 5, 317–325 CrossRef.
- Gurudayal, D. Perone, S. Malani, Y. Lum, S. Haussener and J. W. Ager, ACS Appl. Energy Mater., 2019, 2, 4551–4559 CrossRef CAS.
- Y. Jiao, Y. Zheng, P. Chen, M. Jaroniec and S.-Z. Qiao, J. Am. Chem. Soc., 2017, 139, 18093–18100 CrossRef CAS.
- A. Verdaguer-Casadevall, C. W. Li, T. P. Johansson, S. B. Scott, J. T. McKeown, M. Kumar, I. E. L. Stephen, M. W. Kanan and I. Chorkendorff, J. Am. Chem. Soc., 2015, 137, 9808–9811 CrossRef CAS.
- D. Kim, C. S. Kley and P. Yang, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 10560–10565 CrossRef CAS.
- M. Rahaman, A. Dutta, A. Zanetti and P. Broekmann, ACS Catal., 2017, 7, 7946–7956 CrossRef CAS.
- R. Kortlever, I. Peters, C. Balemans, R. Kas, Y. Kwon, G. Mul and M. T. M. Koper, Chem. Commun., 2016, 52, 10229–10232 RSC.
- F. Studt, I. Sharafutdinov, F. Abild-Pedersen, C. F. Elkjær, J. S. Hummelshøj, S. Dahl, I. Chorkendorff and J. K. Nøskov, Nat. Chem., 2014, 6, 320–324 CrossRef CAS.
- D. A. Torelli, S. A. Francis, J. C. Crompton, A. Javier, J. R. Thompson, B. S. Brunschwig, M. P. Soriaga and N. S. Lewis, ACS Catal., 2016, 6, 2100–2104 CrossRef CAS.
- A. R. Paris and A. B. Bocarsly, ACS Catal., 2017, 7, 6815–6820 CrossRef CAS.
- Y. Song, W. Chen, C. Zhao, S. Li, W. Wei and Y. Sun, Angew. Chem., Int. Ed., 2017, 56, 10840–10844 CrossRef CAS.
- X. Zhi, Y. Jiao, Y. Zheng and S.-Z. Qiao, Small, 2019, 15, 1804224 CrossRef.
- J. Liu, C. Guo, A. Vasileff and S.-Z. Qiao, Small Methods, 2017, 1, 1600006 CrossRef.
- Y. Liu, X. Fan, A. Nayak, Y. Wang, B. Shan, X. Quan and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 26353–26358 CrossRef CAS.
- K. Lv, Y. Fan, Y. Zhu, Y. Yuan, J. Wang, Y. Zhu and Q. Zhang, J. Mater. Chem. A, 2018, 6, 5025–5031 RSC.
- J. Du, S. Li, S. Liu, Y. Xin, B. Chen, H. Liu and B. Han, Chem. Sci., 2020, 11, 5098–5104 RSC.
- K. J. P. Schouten, Y. Kwon, C. J. M. van der Ham, Z. Qin and M. T. M. Koper, Chem. Sci., 2011, 2, 1902–1909 RSC.
- J. H. Montoya, C. Shi, K. Chan and J. K. Nørskov, J. Phys. Chem. Lett., 2015, 6, 2032–2037 CrossRef CAS.
- X. Liu, P. Schlexer, J. Xiao, Y. Ji, L. Wang, R. B. Sandberg, M. Tang, K. S. Brown, H. Peng, S. Ringe, C. Hahn, T. F. Jaramillo, J. K. Nørskov and K. Chan, Nat. Commun., 2019, 10, 32 CrossRef CAS.
- E. Pérez-Gallent, M. C. Figueiredo, F. Calle-Vallejo and M. T. M. Koper, Angew. Chem., Int. Ed., 2017, 56, 3621–3624 CrossRef.
- J. H. Montoya, A. A. Peterson and J. K. Nørskov, ChemCatChem, 2013, 5, 737–742 CrossRef CAS.
- T. Cheng, H. Xiao and W. A. Goddard III, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1795–1800 CrossRef CAS.
- X. Zhi, Y. Jiao, Y. Zheng, A. Vasileff and S.-Z. Qiao, Nano Energy, 2020, 71, 104601 CrossRef CAS.
- A. J. Darza, A. T. Bell and M. Head-Gordon, ACS Catal., 2018, 8, 1490–1499 CrossRef.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- I. Ledezma-Yanez, E. P. Gallent, M. T. M. Koper and F. Calle-Vallejo, Catal. Today, 2016, 262, 90–94 CrossRef CAS.
- Z. Guo, B. Liu, Q. Zhang, W. Deng, Y. Wang and Y. Yang, Chem. Soc. Rev., 2014, 43, 3480–3524 RSC.
- P. Tang, Q. Zhu, Z. Wu and D. Ma, Energy Environ. Sci., 2014, 7, 2580–2591 RSC.
- B. A. Arndtsen, R. G. Bergman, T. A. Mobley and T. H. Peterson, Acc. Chem. Res., 1995, 28, 154–162 CrossRef CAS.
- H. Song, X. Meng, Z. Wang, H. Liu and J. Ye, Joule, 2019, 3, 1606–1636 CrossRef CAS.
- J. Baltrusaitis, I. Jansen and J. D. Schuttlefield Christus, Catal. Sci. Technol., 2014, 4, 2397–2411 RSC.
- L. Yuliati and H. Yoshida, Chem. Soc. Rev., 2008, 37, 1592–1602 RSC.
- K. Shimura and H. Yoshida, Catal. Surv. Asia, 2014, 18, 24–33 CrossRef CAS.
- Y. Kato, H. Yoshida and T. Hattori, Chem. Commun., 1998, 2389–2390 RSC.
- H. Yoshida, N. Matsushita, Y. Kato and T. Hattori, J. Phys. Chem. B, 2003, 107, 8355–8362 CrossRef CAS.
- L. Yuliati, M. Tsubota, A. Satsuma, H. Itoh and H. Yoshida, J. Catal., 2006, 238, 214–220 CrossRef CAS.
- L. Yuliati, T. Hattori and H. Yoshida, Phys. Chem. Chem. Phys., 2005, 7, 195–201 RSC.
- L. Yuliati, T. Hattori, H. Itoh and H. Yoshida, J. Catal., 2008, 257, 396–402 CrossRef CAS.
- L. Yuliati, T. Hamajima, T. Hattori and H. Yoshida, J. Phys. Chem. C, 2008, 112, 7223–7232 CrossRef CAS.
- L. Li, G. Li, C. Yan, X. Mu, X. Pan, X. Zou, K. Wang and J. Chen, Angew. Chem., Int. Ed., 2011, 50, 8299–8303 CrossRef CAS.
- L. Li, Y. Cai, G. Li, X. Mu, K. Wang and J. Chen, Angew. Chem., Int. Ed., 2012, 51, 4702–4706 CrossRef CAS.
- L. Meng, Z. Chen, Z. Ma, S. He, Y. Hou, H. Li, R. Yuan, X. Huang, X. Wang, X. Wang and J. Long, Energy Environ. Sci., 2018, 11, 294–298 RSC.
- L. Li, S. Fan, X. Mu, Z. Mi and C. Li, J. Am. Chem. Soc., 2014, 136, 7793–7796 CrossRef CAS.
- X. Yu, V. L. Zholobenko, S. Moldovan, D. Hu, D. Wu, V. V. Ordomsky and A. Y. Khodakov, Nat. Energy, 2020, 5, 511–519 CrossRef CAS.
- S. Murcia-López, K. Villa, T. Andreu and J. R. Morante, ACS Catal., 2014, 4, 3013–3019 CrossRef.
- F. Sastre, V. Fornés, A. Corma and H. García, J. Am. Chem. Soc., 2011, 133, 17257–17261 CrossRef CAS.
- S. Murcia-López, M. C. Bacariza, K. Villa, J. M. Lopes, C. Henriques, J. R. Morante and T. Andreu, ACS Catal., 2017, 7, 2878–2885 CrossRef.
- L. Yu, Y. Shao and D. Li, Appl. Catal., B, 2017, 204, 216–223 CrossRef CAS.
- L. Yu and D. Li, Catal. Sci. Technol., 2017, 7, 635–640 RSC.
- Y. Zhou, L. Zhang and W. Wang, Nat. Commun., 2019, 10, 506 CrossRef CAS.
- F. Amano, C. Akamoto, M. Ishimaru, S. Inagaki and H. Yoshida, Chem. Commun., 2020, 56, 6348–6351 RSC.
- C. E. Taylor and R. P. Noceti, Catal. Today, 2000, 55, 259–267 CrossRef CAS.
- C. E. Taylor, Catal. Today, 2003, 84, 9–15 CrossRef CAS.
- M. A. Gondal, A. Hameed and A. Suwaiyan, Appl. Catal., A, 2003, 243, 165–174 CrossRef CAS.
- M. Stoukides, J. Appl. Electrochem., 1995, 25, 899–912 CrossRef CAS.
- M. Stoukides, Res. Chem. Intermed., 2006, 32, 187–204 CrossRef CAS.
- K. Otsuka, S. Yokoyama and A. Morikawa, Chem. Lett., 1985, 319–322 CrossRef CAS.
- Y. Jiang, I. V. Yentekakis and C. G. Vayenas, Science, 1994, 264, 1563–1566 CrossRef CAS.
- K. Liu, J. Zhao, D. Zhu, F. Meng, F. Kong and Y. Tang, Catal. Commun., 2017, 96, 23–27 CrossRef CAS.
- A. Caravaca, A. de Lucas-Consuegra, J. González-Cobos, J. L. Valverde and F. Dorado, Appl. Catal., B, 2012, 113–114, 192–200 CrossRef CAS.
- A. Caravaca, V. J. Ferreira, A. de Lucas-Consuegra, J. L. Figueiredo, J. L. Faria, J. L. Valverde and F. Dorado, Int. J. Hydrogen Energy, 2013, 38, 3111–3122 CrossRef CAS.
- V. Kyriakou, I. Garagounis and M. Stoukides, Int. J. Hydrogen Energy, 2014, 39, 675–683 CrossRef CAS.
- A. Caravaca, A. de Lucas-Consuegra, V. J. Ferreira, J. L. Figueiredo, J. L. Faria, J. L. Valverde and F. Dorado, Appl. Catal., B, 2013, 142–143, 298–306 CrossRef CAS.
- C. Zhu, S. Hou, X. Hu, J. Lu, F. Chen and K. Xie, Nat. Commun., 2019, 10, 1173 CrossRef.
- M. Ma, B. Jin, P. Li, M. Jung, J. Kim, Y. Cho, S. Kim, J. Moon and J. Park, Adv. Sci., 2017, 4, 1700379 CrossRef.
- M. Ma, C. Oh, J. Kim, J. Moon and J. Park, Appl. Catal., B, 2019, 259, 118095 CrossRef.
- Z. Guo, W. Chen, Y. Song, X. Dong, G. Li, W. Wei and Y. Sun, Chin. J. Catal., 2020, 41, 1067–1072 CrossRef CAS.
- Y. Song, Y. Zhao, G. Nan, W. Chen, Z. Guo, S. Li, Z. Tang, W. Wei and Y. Sun, Appl. Catal., B, 2020, 270, 118888 CrossRef CAS.
- N. Lapeña-Rey and P. H. Middleton, Appl. Catal., A, 2003, 240, 207–222 CrossRef.
- S. Kodama, R. Kikuchi, N. Fujiwara, S. Tada, Y. Kobayashi and S. T. Oyama, ECS Trans., 2019, 91, 2697–2705 CrossRef CAS.
- V. Kyriakou, C. Athanasiou, I. Garagounis, A. Skodra and M. Stoukides, Int. J. Hydrogen Energy, 2012, 37, 16636–16641 CrossRef CAS.
- A. de Lucas-Consuegra, N. Gutiérrez-Guerra, A. Caravaca, J. C. Serrano-Ruiz and J. L. Valverde, Appl. Catal., A, 2014, 483, 25–30 CrossRef CAS.
- J. Lu, C. Zhu, C. Pan, W. Lin, J. P. Lemmon, F. Chen, C. Li and K. Xie, Sci. Adv., 2018, 4, eaar5100 CrossRef.
- L. Luo, X. Tang, W. Wang, Y. Wang, S. Sun, F. Qi and W. Huang, Sci. Rep., 2013, 3, 1625 CrossRef.
- G. A. Olah, Angew. Chem., Int. Ed., 2013, 52, 104–107 CrossRef CAS.
- S. Yanagida, T. Azuma, H. Kawakami, H. Kizumoto and H. Sakurai, J. Chem. Soc., Chem. Commun., 1984, 1, 21–22 RSC.
- S. Xie, Z. Shen, J. Deng, P. Guo, Q. Zhang, H. Zhang, C. Ma, Z. Jiang, J. Cheng, D. Deng and Y. Wang, Nat. Commun., 2018, 9, 1181 CrossRef.
- H. Zhang, S. Xie, J. Hu, X. Wu, Q. Zhang, J. Cheng and Y. Wang, Chem. Commun., 2020, 56, 1776–1779 RSC.
- M. Liu, Y. Wang, X. Kong, R. T. Rashid, S. Chu, C. Li, Z. Hearne, H. Guo, Z. Mi and C. Li, Chem, 2019, 5, 858–867 CAS.
- Y. Chao, J. Lai, Y. Yang, P. Zhou, Y. Zhang, Z. Mu, S. Li, J. Zheng, Z. Zhu and Y. Tan, Catal. Sci. Technol., 2018, 8, 3372–3378 RSC.
- Y. Fan, J. Bao, L. Shi, S. Li, Y. Lu, H. Liu, H. Wang, L. Zhong and Y. Sun, Catal. Lett., 2018, 148, 2274–2282 CrossRef CAS.
- Y. Fan, S. Li, J. Bao, L. Shi, Y. Yang, F. Yu, P. Gao, H. Wang, L. Zhong and Y. Sun, Green Chem., 2018, 20, 3450–3456 RSC.
- J. Bao, Y. Fan, S. Zhang, L. Zhong, M. Wu and Y. Sun, Catal. Lett., 2019, 149, 1651–1659 CrossRef CAS.
- Y. Li, J. Lu, X. Wang, H. Zhang, X. Wu, K. Zhang, J. Ye and D. Zhan, ChemCatChem, 2019, 11, 2277–2282 CrossRef CAS.
- J. Zhang, Q. Yuan, J. Zhang, T. Li and H. Guo, Chem. Commun., 2013, 49, 10106–10108 RSC.
- J. Zhang, T. Li, D. Wang, J. Zhang and H. Guo, Chin. J. Catal., 2015, 36, 274–282 CrossRef CAS.
- L. Wang, S. Liu, C. Xu and X. Tu, Green Chem., 2016, 18, 5658–5666 RSC.
- Z. Shen, S. Xie, W. Fan, Q. Zhang, Z. Xie, W. Yang, Y. Wang, J. Lin, X. Wu, H. Wan and Y. Wang, Catal. Sci. Technol., 2016, 6, 6485–6489 RSC.
- S. Xie, Z. Shen, H. Zhang, J. Cheng, Q. Zhang and Y. Wang, Catal. Sci. Technol., 2017, 7, 923–933 RSC.
- M. I. Montenegro, D. Pletcher, E. A. Liolios, D. J. Mazur and C. Zawodzinski, J. Appl. Electrochem., 1990, 20, 54–59 CrossRef CAS.
- N. L. Weinberg and D. J. Mazur, J. Appl. Electrochem., 1991, 21, 895–901 CrossRef CAS.
- Y. Ma, X. Wang, Y. Jia, X. Chen, H. Han and C. Li, Chem. Rev., 2014, 114, 9987–10043 CrossRef CAS.
- H. Lu, J. Zhao, L. Li, L. Gong, J. Zheng, L. Zhang, Z. Wang, J. Zhang and Z. Zhu, Energy Environ. Sci., 2011, 4, 3384–3388 RSC.
- H. Liu, C. Song, L. Zhang, J. Zhang, H. Wang and D. P. Wilkinson, J. Power Sources, 2006, 155, 95–110 CrossRef CAS.
- J. T. Franclemont, X. Fan, R. Li, R. K. Singh, T. M. Holsen and S. Mededovic Thagard, Plasma Processes Polym., 2018, 15, e1180019 Search PubMed.
- A. Nawaz, A. Kuila, A. Rani, N. S. Mishra, L. C. Sim, K. H. Leong and P. Saravanan, in Industrial Applications of Nanomaterials, ed. S. Thomas, Y. Grohens and Y. B. Pottathara, Elsevier, 2019 Search PubMed.
- A. J. Martin, G. O. Larrazabal and J. Perez-Ramirez, Green Chem., 2015, 17, 5114–5130 RSC.
- S.-C. Lin, C.-C. Chang, S.-Y. Chiu, H.-T. Pai, T.-Y. Liao, C.-S. Hsu, W.-H. Chiang, M.-K. Tsai and H. M. Chen, Nat. Commun., 2020, 11, 3525 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |