
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImidosulfonate scorpionate ligands in lanthanide single-molecule magnet design: slow magnetic relaxation and butterfly hysteresis in [ClDy{Ph2PCH2S(NtBu)3}2]†
Jochen
Jung
,
Christina M.
Legendre
 ,
Serhiy
Demeshko
,
Regine
Herbst-Irmer
and
Dietmar
Stalke
*
,
Serhiy
Demeshko
,
Regine
Herbst-Irmer
and
Dietmar
Stalke
*
Georg-August Universität Göttingen, Institut für Anorganische Chemie, Tammannstraße 4, 37077 Göttingen, Germany. E-mail: dstalke@chemie.uni-goettingen.de
First published on 25th October 2021
Abstract
Single-molecule magnets (SMMs) harbour vast opportunities for potential pioneering applications upon optimization like big data storage and quantum computing. Lanthanides were found to be highly suitable candidates in the design of such molecules, as they intrinsically hold a large unquenched orbital momentum and a strong spin–orbit coupling, warranting a high magnetic anisotropy. An indispensable element in successfully tailoring SMMs is the ligand design. Polyimido sulfur ligands offer a promising choice because the polar S+–N−-bond facilitates both electronic and geometric adaptability to various f-metals. In particular, the acute N–Ln–N bite angle generates advantageous magnetic properties. The [Ph2PCH2S(NtBu)3]− anion, introduced from [(thf)3K{Ph2PCH2S(NtBu)3}] (2) to a series of complexes [ClLn{Ph2PCH2S(NtBu)3}2] with Ln = Tb (3a), Dy (3b), Er (3c), Ho (3d), and Lu (3e), provides tripodal shielding of the metal's hemisphere as well as a side-arm donation of a soft phosphorus atom. For the Tb and Er complexes 3a and 3d, slow magnetic relaxation (Ueff = 235 and 34.5 cm−1, respectively) was only observed under an applied dc field. The dysprosium congener 3b, however, is a true SMM with relaxation at zero field (Ueff = 66 cm−1) and showing a butterfly hysteresis close to 3.5 K. Upon magnetic dilution with the diamagnetic and isostructural lutetium complex 3e or application of a magnetic field, the energy barrier to spin reversal is increased to 74 cm−1.
Introduction
The realm of single-molecule magnets (SMMs) has continuously expanded since their discovery in 1993.1 These molecules can be magnetized and remain so upon the removal of the external magnetic field, however only at helium-level temperatures for the vast majority. Their remarkable magnetic properties, potentially transferable to industry for applications such as high-density data storage and spintronics,2 are mainly due to the magnetic anisotropy created by the paramagnetic metal centre in a particular crystal field. Lanthanides were found to be highly promising candidates in the design of such molecules as they intrinsically possess a large unquenched orbital momentum and a strong spin–orbit coupling.3 This warrants a high magnetic anisotropy, even with only one trivalent lanthanide ion as the sole paramagnetic centre in the SMM. These discoveries spurred the studies of mononuclear, lanthanide containing complexes in recent years.4,5–7 The design of dysprosocenium sandwich cations with bulky, non-coordinating counter-ions allowed to reach operating temperatures above the liquid nitrogen threshold, narrowing the gap between fundamental research and industrial applications. After these major advances in organometallic chemistry, the development of mononuclear SMMs currently suffers stalling and the scientific community is turning to main group chemistry to look for new ligands enhancing SMM properties. Some studies already indicate the underlying potential in the marriage of molecular magnetism with main group chemistry.8,9–11 More recently, soft elements were even exploited to strongly couple paramagnetic transition metals with lanthanide ions to build promising heterometallic single-molecule magnets.12Known for several years but not sufficiently promoted in the field of molecular magnetism are the polyimido sulfur ligand species S(NR)nm− (n = 2, 3, and 4 and m = 0 and 2) that have been studied extensively within our group for several years.13–28 We published many examples of sulfur-imido ligated s,13–15 p16 and d-block13,17–19 metal complexes and now exploit that ligand versatility in f-element20 chemistry to give molecular magnets. The S–N ligand class25 is particularly beneficial in SMM chemistry because it features a sulfur–imido bond with both ionic and covalent character.15,21 This renders the ligands highly adaptable and responsive to the various requirements of different metals (size, different electronegativity and redox potential, etc.), both geometrically and electronically.
The first results with this ligand class18,19,22,23 indicated the next steps for synthetic and structural improvements in lanthanide complexes,20 so far scarcely investigated.20,24 From the plethora of our SN ligands, we identified the [Ph2PCH2S(NtBu)3]− scorpionate anion28 as the most suitable candidate for the following reasons. In addition to three responsive Sδ+–Nδ− bonds arranged in a potential cap shaped tripodal fashion, it holds a soft P-donating side-arm. For transition metal SMMs, various examples demonstrate how the introduction of heavier and softer p-block donor atoms improved the magnetic properties.23 Phosphorus donating ligands in trigonal planar Fe(II)19,26,27 and Co(II)19,27 complexes raise the effective energy barriers Ueff to undergo a significant spin reversal.19,23 The enhancement of the zero-field splitting, thanks to the introduction of donor atoms with more diffuse orbitals, was also observed in a series of tetrahedral Co(II) complexes with EPh ligands (E = O, S and Se) that exhibit increasing zero-field splitting descending the group.9,29,30 Lanthanide-based SMMs with heavier donor atoms are rare and to the best of our knowledge the only example with P-donation improving the magnetic properties is the phospholyl Er(III) complex, in which the Er–PCp* interaction, weaker than Er–CCp*, results in a stronger, more favorable equatorial field.31 The performance of the bis(phospholyl) Dy(III) cation11 is not quite on par with either the [Cp*DyCp5-iPr]+5 or the [(Cp3-tBu)2Dy]+ cation.6,7 Approaches involving the stacking of magnetically well-performing building blocks32 or the design of asymmetric complexes modulating the relaxation pathways33 have proven to be more successful for Er(III) based SMMs. For Dy(III), however, introducing softer atoms is generally considered a good strategy. For example, a series of group 15 bridged dysprosocenes, with the soft donor ligands in the equatorial positions and a strong axial crystal field generated by the cyclopentadienyl ligands, revealed increasing anisotropy barriers from phosphorus to antimony.10,34,35 These results not only clearly show the necessity to reduce the equatorial crystal field for the development of high-temperature SMMs but also demonstrate the option to improve the molecular magnetism on the basis of softer donor atoms.
For this purpose, we herein propose the [Ph2PCH2S(NtBu)3]− anion, generated from [(thf)3K{Ph2PCH2S(NtBu)3}] (2), as a potentially suitable candidate for both requests: theoretically an N,N,N- or N,N,P-tripodal ligand, it can shield one hemisphere of the f-metal ion alike Cp-derivatives, and it holds a P-donor side-arm. Thus, we synthesized a series of ligated complexes [ClLn{Ph2PCH2S(NtBu)3}2] with Ln = Tb (3a), Dy (3b), Er (3c), Ho (3d), and Lu (3e) and analysed their magnetic properties.
Results and discussion
Synthesis and crystallography
Deprotonation of 1 with K(HMDS) in thf gives the potassium complex [(thf)3K{Ph2PCH2S(NtBu)3}] (2) (Scheme 1) that is employed as the ligand source in the subsequent lanthanide transmetallation reactions. Crystallization of 2 in a mixture of n-pentane/thf at −34 °C yielded colourless crystals suitable for X-ray structure analysis. [K(thf)3{Ph2PCH2S(NtBu)3}] (2) loses the coordinating thf molecules when stored at room temperature over time and all thf molecules can be removed under reduced pressure (see Fig. S7†). 2 crystallizes in the monoclinic space group P21/n with one molecule in the asymmetric unit (Fig. 1). The potassium ion is five-coordinated by only one nitrogen and the phosphorus atom, both from the triimidosulfonate ligand and the additional three thf solvent molecules. In contrast to the other structural motifs of this ligand, it does not even function as an N,N′ chelating ligand. The three S1–N bond distances in 2 are similar (av. 1.5578 Å) but due to potassium coordination, the S1–N1 bond (1.5685(14) Å) is the longest. Nevertheless, it is still short compared to the S–N(H) bond distance of 1.6498(13) Å in 1.19,28 The coordination of the hard proton has therefore a much more pronounced impact than that of the soft potassium metal. The K1–P1 bond distance of 3.3301(7) Å is in the normal range for a potassium phosphorus bond. In comparison, the P–Li distance of the lithium species [(tmeda)Li{Ph2PCH2S(NtBu)3}]13 is 3.718 Å, too long to be considered a bond (Fig. 2). The K1–N1 bond distance of 2.7434(14) Å falls as well in the normal range covered by a five-fold coordinated potassium ion. The coordination sphere of the potassium atom is completed by three thf solvent molecules with K–O bond lengths from 2.648(5) to 2.7511(14) Å. Through reduced pressure, they can be removed to give a donor-free powder, insoluble in nonpolar hydrocarbons. The addition of small amounts of thf-d8 enables NMR analysis in solution. In 1H NMR, the three NtBu groups are equivalent and resonate at 1.65 ppm. The two methylene protons show a doublet centred at 3.94 ppm (2JHP = 5.0 Hz) and the phenyl signals can be found in the normal aromatic region. The potassium complex 2 was then used as a precursor for the synthesis of a series of lanthanide complexes [ClLn{Ph2PCH2S(NtBu)3}2] 3a–e. First attempts with neat thf as the reaction solvent were unsuccessful and the target products could not be isolated. Thus, to trigger KCl precipitation, we selected the more nonpolar solvent toluene. In the synthesis of 3a–e, thf-free 2 was first suspended in toluene and approximately 2% by volume of thf was added to form the soluble ligand precursor [(thf)3K{Ph2PCH2S(NtBu)3}] (2). After the addition of the appropriate lanthanide(III) chloride salt, the reaction mixture was stirred for one day to allow the formation of the soluble complexes [ClLn{Ph2PCH2S(NtBu)3}2] 3a–e (a: Ln = Tb, b: Dy, c: Ho, d: Er, and e: Lu). Subsequently, the precipitated KCl was filtered off and all volatiles were removed under reduced pressure. The raw product was then dissolved in thf and layered with n-pentane. After a few days, crystals suitable for X-ray structure analysis were grown at room temperature. 3a–e are isomorphous and crystallize in the monoclinic space group C2/c with one molecule in the asymmetric unit. Disordered lattice thf and n-pentane are also present (Fig. S2–6†). The central lanthanide(III) ion is six-fold coordinated. One [Ph2PCH2S(NtBu)3]− anion coordinates N,N,P-tripodal, while the second is only N,N′-chelating with a pendant P-donor side-arm. The remaining coordination site is occupied by a chlorine atom. As anticipated from the decreasing radii, the Ln–Cl1 bond distances decrease from 2.6174(7) Å in 3a to 2.5467(5) Å in 3e. All nitrogen atoms are crystallographically independent but the Ln–N distances in all the complexes span over a relatively narrow range of 2.3259(17) Å (Ln–N5) to 2.4085(18) Å (Ln–N2) in 3a (largest distances) and from 2.2577(11) Å (Ln–N5) to 2.3393(11) Å (Ln–N2) in 3e (shortest distances in the 3a–e series). The four metal-coordinated S–N bonds are almost identical, even among the different complexes 3a–e and average at 1.604 Å. The pendant S1–N3 and S2–N6 bonds, however, are significantly shorter with an average value of 1.515 Å. Although the Ln–P1 distances, decreasing from 3.2291(7) Å in the terbium complex 3a to 3.1900(5) Å in the lutetium complex 3e, are at the end of the range covered by other lanthanide–phosphorus bond distances, this has to be judged as an attractive interaction because the second pendant P-donor is idle, similar to [Dy{PPh2S(NtBu)2}2(μ-Cl2)Li(THF)2].20b The average N–Ln–N angle of 60.7° is very acute and ranges from 59.7° in 3a to 62.2° in 3e. For superior magnetic properties, the linear arrangement of the ligands seems to be advantageous. A key feature in this respect is the S⋯Ln⋯S angle. In the series of [Dy{PPh2S(NtBu)2}2(μ-Cl2)Li(THF)2], [Dy{PhS(NtBu)2}2(μ-Cl2)Li-(THF)2] and [Dy{MeS(NtBu)3}2(μ-Cl2)Li (THF)2],20b it ranges from 108 to 136°, improving among others Ueff at 1000 Oe from 56.8 to 72.4 cm−1 in the latter complex, which shows slow relaxation even at zero field (Ueff = 59.3 cm−1). Here, the average S⋯Ln⋯S angle for 3a–3e is 142°, even closer to linearity. Attempts to characterize the paramagnetic lanthanide complexes 3a–d by NMR spectroscopy were not successful, while it confirmed the formation of diamagnetic 3e. The 1H NMR spectra in C6D6 compared with the protonated ligand [Ph2PCH2S(NtBu)2NHtBu] (1) and with the thf free potassium precursor [(thf)3K{Ph2PCH2S(NtBu)3}] (2) indeed shows the conversion to [LuCl{Ph2PCH2S(NtBu)3}2] (3e) (Fig. S12†). The singlet in 2 at 1.65 ppm splits into two singlets. The smaller one at δ = 1.50 ppm corresponds to the pendant NtBu groups. The larger signal at 1.65 ppm is attributed to the coordinating NtBu groups. The methylene protons in 2 (doublet, 3.94 ppm) are shifted as a multiplet to δ = 4.77–5.06. The 31P NMR spectrum shows only one singlet at −24.59 ppm at room temperature in C6D6, indicating a fast on/off coordination exchange among both phosphorus sites. At −20 °C in d8-toluene, the two sites, starting from −24.64 ppm, could be deconvoluted to give two signals at −23.84 and 27.74 ppm, respectively, at −60 °C (Fig. S23–25†). The obtained LIFDI mass spectra and elemental analyses for 3a–e further substantiate the successful syntheses and isolation of bulk material, whose magnetic properties were further investigated. | ||
| Scheme 1 Synthesis of the potassium starting material [(thf)3K{Ph2PCH2S(NtBu)3}] (2) and the lanthanide complexes [ClLn{Ph2PCH2S(NtBu)3}2] 3a–e (a: Ln = Tb, b: Dy, c: Ho, d: Er, and e: Lu). | ||
 | ||
| Fig. 1 Crystal structure of [(thf)3K{Ph2PCH2S(NtBu)3}] (2). Anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms and minor positions of the disordered thf molecules are omitted for clarity. Selected bond lengths [Å] and angles [°]: K1–N1 2.7434(14), K1–P1 3.3301(6), N1–S1–N2 109.56(7), N1–S1–N3 110.32(7), N2–S1–N3 122.66(7), N1–S1–C13 108.68(7), N2–S1–C13 108.07(7), N3–S1–C13 96.13(7), N1–K1–P1 69.44(3). | ||
 | ||
| Fig. 2 Crystal structure of 3a–e. Anisotropic displacement parameters are depicted at the 50% probability level for the Tb complex 3a. Hydrogen atoms and disordered thf/n-pentane lattice solvent are omitted for clarity. For full bond lengths [Å] and angles [°] of all complexes see the ESI.† | ||
Magnetic susceptibility measurements
We initially measured the temperature dependency of the product χMT for the complexes 3a–d (Fig. 3). The obtained high-temperature χMT values for 3a–d 11.81, 14.09, 13.41 and 10.86 cm3 mol−1 K are very close to the expected values for every single ion (expected 11.82, 14.17, 14.04 and 11.48 cm3 mol−1 K, respectively). The minute differences are attributed to the splitting of the ground state. Upon cooling, for complexes 3a–d, the χMT value first slowly decreases, indicating the thermal depopulation of the Stark sublevels, and then drastically decreases to reach χMT = 7.46, 8.43, 8.29 and 5.39 cm3 mol−1 K, respectively. | ||
| Fig. 3 Temperature dependency of χMT for 3a–d under a dc field of 0.5 T. | ||
We probed the presence of slow magnetic relaxation with a measurement of the temperature dependence of in-phase (χ′M) and out-of-phase (χ′′M) ac magnetic susceptibility at 0 Oe and 1000 Oe (see Fig. S26, 30, 39 and 41†). For the Tb (3a) and Er (3d) complexes, slow magnetic relaxation was only observed under an applied dc field and up to 25 K and 5 K, respectively (see the ESI Fig. S26† for 3a and S41† for 3d). The optimal dc field was probed with an additional measurement of the field dependence of the ac magnetic susceptibility at 25 and 5 K, respectively (see Fig. S26 and S41†). The maximal ac magnetic susceptibility values were already reached by 1000 Oe for both Tb and Er complexes. The magnetic relaxation times τ were extracted from the fitting plots of χ′′Mvs. χ′M using the CCfit program36 (Fig. S29 and S43†) and were then used to obtain the temperature dependence plot of the relaxation time (Fig. 4).
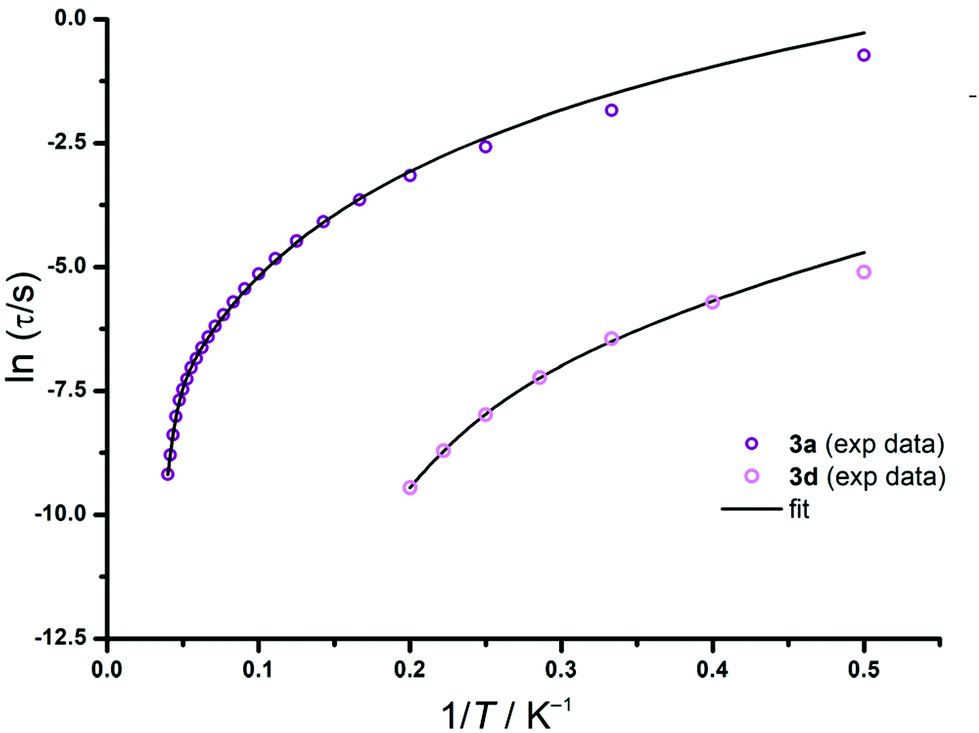 | ||
| Fig. 4 Temperature dependency plots of the relaxation time plots for complexes 3a and 3d under an applied field of 1000 Oe and their corresponding fits (solid black line) according to eqn (1). | ||
These plots indicated the nature of the involved relaxation pathways and the height of the energy barrier associated with the Orbach-type relaxation. The relaxation time τ data measured under 1000 Oe for 3a and 3d were fitted according to the following equation taking Orbach and Raman processes into account:
τ−1 = τ0−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) e(Ueff/kBT) + τQTM−1 + CTn e(Ueff/kBT) + τQTM−1 + CTn | (1) |
| Complex | 3a | 3b | 3d |
|---|---|---|---|
| H dc (Oe) | 1000 | 1000 | 1000 |
| U eff (cm−1) | 235 | 73.9 | 34.5 |
| τ 0 (s) | 1.9 × 10−9 | 2.7 × 10−8 | 7.3 × 10−9 |
| C (s−1 K−n) | 0.17 | 0.012 | 5.2 |
| n | 3.0 | 4.2 | 4.4 |
| Complex | 3b | 3b@Lu | 3b@Lu |
| H dc (Oe) | 0 | 0 | 1000 |
| U eff (cm−1) | 66.5 | 71.0 | 74.3 |
| τ 0 (s) | 1.2 × 10−7 | 2.9 × 10−8 | 2.3 × 10−8 |
| C (s−1K−n) | 6.5 × 10−2 | 1.6 × 10−2 | 6.6 × 10−3 |
| n | 4.0 | 4.4 | 4.5 |
| τ QTM (s) | 1.1 × 10−3 | — | — |
The holmium analog 3c does not show slow magnetic relaxation even under an applied dc field as Ho3+ ions are non-Kramers ions with a high mJ state mixing rate in the absence of perfectly symmetric ligand fields.3b Since dysprosium-containing molecules are more prone to show SMM behaviour, we thus expected even better magnetic properties for the Dy complex 3b. Remarkably, peaks in the out-of-phase ac dynamic susceptibility were indeed observed for 3b up to 12 K without any applied dc field, indicative of true SMM behaviour (Fig. S30–32†). At low temperatures, the peaks decrease in intensity without showing any frequency dependence, which strongly suggests the presence of quantum tunneling processes until 5.6 K. The temperature dependence plot of the relaxation time obtained from the fitting of the χ′′Mvs. χ′M Cole–Cole plot reflects the presence of multiple relaxation pathways. A fit according to eqn (1) yields an energy barrier Ueff = 66.5 cm−1 (Table 1, eqn (1) and Fig. S32†). Along with the Orbach relaxation, the slow relaxation of the magnetization mainly happens through a Raman process (n ≈ 4.0) at higher temperatures and through QTM at lower temperatures. The presence of quantum tunneling typically prevents magnetic blocking. Consequently, 3b (Dy) shows a butterfly hysteresis even at 2 K. The hysteresis completely closes at 3.5 K (Fig. 5, top).
 | ||
| Fig. 5 Magnetic hysteresis for 3b (top) and 3b@Lu (bottom) with a sweep rate of 2.7 mT s−1. | ||
In order to suppress the quantum tunneling effects, we conducted magnetic dilution with the diamagnetic and isostructural lutetium analogue 3e in a 1:10 (Dy:Lu) ratio. Despite the magnetic dilution, the ac magnetic susceptibility measurements of 3b@Lu still show a temperature independent process until 4.4 K is reached (Fig. S33–35†). However, the out-of-phase signals are shifted to the left and a translation of the peaks to the higher frequencies is noticeable while the temperature is increased (Fig. S33†). Subsequent fitting of the high temperature data gives a thermal energy barrier to spin reversal of Ueff = 71.0 cm−1 with a relaxation time of τ0 = 2.9 10−8 s (Fig. S35†).
Quite surprisingly, applying an external field on 3b or on 3b@Lu shows a similar effect than the magnetic dilution: the maxima for ac dynamic susceptibility were reached at 1000 Oe and data analysis yielded energy barriers of Ueff = 73.9 cm−1 and 74.3 cm−1, respectively (Table 1, Fig. S38† and Fig. 6). Since the energy barrier values were almost the same for the diluted sample 3b@Lu and for the pure sample of 3b at the 1000 Oe dc field, we concluded that further dilution attempts would not improve the energy barrier any further.
 | ||
| Fig. 6 In- and out-of-phase signals of the ac susceptibility at 1000 Oe (top), Cole–Cole plot and Arrhenius plot fitted according to eqn (1) (bottom) for 3b. | ||
Noteworthy, upon magnetic dilution, the shape of the hysteresis obtained for 3b@Lu slightly improved in comparison with pure 3b: the wings of the butterfly hysteresis are wider and close at a higher absolute coercive field value (+30 Oe) (Fig. 5, bottom). The wings of the hysteresis, however, completely close at 3.5 K for both the pure and magnetically diluted samples.
Furthermore, we calculated the orientation of the main magnetic axis of the ground state in 3b (Fig. 7, see section S3.4† for details) based on an electrostatic model.37 The main magnetic axis is found to be located close to the coordinating nitrogen atoms with the chlorine atom Cl1 perpendicular to it. This suggests that Cl1 contributes to the transverse anisotropy and that the magnetic properties of 3b could further benefit from its removal. This would probably promote the second phosphorus side arm to coordinate the metal as well as the linear arrangement of the ligands. Thus, the benefits of the soft donor could be fully assessed and guide further research.
 | ||
| Fig. 7 Orientation of the main magnetic axis of the ground state for the entire molecule of 3b (brown line). Hydrogens are omitted for better clarity. Orange, green, yellow, pink, dark blue, and grey represent dysprosium, chlorine, sulfur, phosphorus, nitrogen, and carbon atoms, respectively. | ||
Conclusions
The N,N,P-tripodal scorpionate anion [Ph2PCH2S(NtBu)3]− has emerged to be an ideal ligand in SMMs containing f-metal atoms as paramagnetic centres. The responsive Sδ+–Nδ− bonds easily adapt to the electronic and geometric requirements of various metals like terbium to lutetium. Due to their cap-shaped character, they provide a similar steric demand as the omnipresent cyclopentadienide ligands and arrange at an angle of 142°, closer to linearity. This and the acute N–Ln–N angle of 60.7° causes slow magnetic relaxation (Ueff = 235 and 34.5 cm−1, respectively) under an applied dc field for the Tb and Er complexes 3a and 3d. The dysprosium congener 3b, however, is an SMM at zero field (Ueff = 66 cm−1), showing in addition a butterfly hysteresis closing at 3.5 K. The dynamic magnetic properties are slightly improved upon dilution in the diamagnetic lutetium matrix (3e) or application of a dc field (Ueff increased to 73.9 cm−11). This study shows that the potassium starting material is more promising than its lithium analogue as it prevents alkaline metal halide co-complexation. The next area of action focuses on the removal of the halide coordinated orthogonally to the main magnetic axis, spoiling the magnetic properties of 3b and preventing the second Ln–P coordination, as well as the increase of the S⋯Ln⋯S angle to approach linearity.Experimental section
General considerations
All experiments were performed under inert conditions in N2 or Ar, using Schlenk techniques or in an Ar glovebox. Solvents were dried over sodium or potassium, distilled prior to use and stored over molecular sieves (3 Å). Starting materials were purchased commercially and used without further purification. (Ph2PCH2)S(HNtBu)(NtBu)2 was synthesized according to the literature known procedure.13,19,28 NMR spectra were recorded using a Bruker Avance III HD 500 and referenced to a deuterated solvent signal. LIFDI-MS spectra were recorded using a Jeol AccuTOF spectrometer. Elemental analysis (C, H, N, and S) was performed at the Analytische Labor, Institut für Anorganische Chemie, University of Göttingen.Single crystals were selected under cooling using an X-Temp2 device.38 The datasets were collected using an Incoatec Ag Microsource39 with mirror optics and an APEX II detector with a D8 goniometer. The data were integrated with SAINT.40 A multi-scan absorption correction was applied using SADABS.41 The structures were solved by SHELXT42 and refined on F2 using SHELXL43 in the graphical user interface ShelXle.44
Magnetic data were recorded with a Quantum-Design MPMS-XL-5 SQUID magnetometer equipped with a 5 T magnet. The crystalline solid sample was crushed, charged into a gel capsule and covered with a few drops of a low viscosity inert oil (perfluoropolyether Fomblin YL VAC 25/6; this oil is liquid above 215 K); the use of oil prevents magnetic torqueing and the loss of solvent molecules. The capsule was placed in a non-magnetic sample holder, which was then inserted in the SQUID magnetometer for measurement. The data were analyzed thanks to the CC-fit36 and Origin programs (see the ESI† for more details).
Synthesis of [(thf)3K{Ph2PCH2S(NtBu)3}] (2)
A solution of K{N(SiMe)3}2 (1.119 g, 5.610 mmol) in thf (15 mL) was added to a solution of Ph2PCH2S(NtBu)2NHtBu (1) (2.500 g, 5.610 mmol) in thf (10 mL) at ambient temperature. After stirring for 1 d, the solvent was removed under reduced pressure. The residue was dissolved in a mixture of n-pentane/thf (15 mL/3 mL) and filtered. Crystallization started within minutes after storing at −34 °C yielding colourless crystals suitable for X-ray analysis. The product was isolated, washed with n-pentane (2 × 2 mL) and dried under reduced pressure. Yield: 2.384 g, 4.928 mmol, (88%); 1H NMR (500.13 MHz, 298 K, C6D6): δ [ppm] = 1.65 (s, 27H, 3 NC(CH3)3), 3.94 (d, 2JHP = 5.0 Hz, 2H, PCH2), 7.07–7.09 (m, 2H, 2 p-Ph-H), 7.15–7.18 (m, 4H, 2 m-Ph-H), 7.84–7.87 (s, 2H, o-Ph-H); 13C{1H} NMR (125.76 MHz, 298 K, C6D6): δ [ppm] = 34.00 (s, NC(CH3)3), 52.73 (s, NC(CH3)3), 66.11 (d, 1JCP = 21.1 Hz, PCH2), 128.22 (s, p-Ph-C), 128.37 (d, 3JCP = 6.3 Hz, m-Ph-C), 133.74 (d, 2JCP = 17.4 Hz, o-Ph-C), 142.28 (d, 1JCP = 9.4 Hz, ipso-Ph-C); 15N NMR (50.70 MHz, 298 K, C6D6): δ [ppm] = −253.33 (s); 31P{1H} NMR (202.46 MHz, 298 K, C6D6): δ [ppm] = −18.14 (s); elemental analysis for C25H39KN3PS (found (calc.) [%]): C 61.73 (62.07), H 7.84 (8.13), N 8.85 (8.69), S 7.44 (6.63).General synthesis for [ClLn{Ph2PCH2S(NtBu)3}2] (3a–e)
The donor solvent free potassium precursor [K{Ph2PCH2S(NtBu)3}] (500.0 mg, 1.034 mmol) and LnCl3 (0.517 mmol) were suspended in toluene (40 mL) and thf (0.8 mL) was added. Subsequently, the mixture was stirred for 1 d at ambient temperature. Precipitated KCl was filtered off and the solvent was removed under reduced pressure. The residue was extracted with thf, filtered and the solvent was removed in vacuo again. To receive the target compound as crystalline material, the raw product was dissolved in thf (2.5 mL) and layered with n-pentane (12.5 mL). Crystallization started within hours up to several days at ambient temperature yielding crystals that were isolated which were then washed with n-pentane (2 × 1 mL).3a: yield: 240.4 mg, 0.2147 mmol (42%); LIFDI-MS: m/z: 1047.3 [M − Cl]+; elemental analysis for C50H78ClN6P2S2Tb(C2.12H4.47O0.38) (found (calc.) [%]): C 55.78 (55.91), H 7.37 (7.42), N 7.50 (7.51), S 5.82 (5.73). Colour: pale yellow.
3b: yield: 285.3 mg, 0.2540 mmol (49%); LIFDI-MS: m/z: 1052.4 [M − Cl]+; elemental analysis for C50H78ClN6P2S2Dy(C2.25H4.99O0.25) (found (calc.) [%]): C 55.80 (55.87), H 7.21 (7.45), N 7.57 (7.48), S 5.92 (5.71). Colour: pale yellow.
3c: yield: 298.6 mg, 0.2653 mmol (51%); LIFDI-MS: m/z: 1053.3 [M − Cl]+; elemental analysis for C50H78ClN6P2S2Ho(C2.14H4.56O0.36) (found (calc.) [%]): C 54.75 (55.63), H 7.30 (7.39), N 7.36 (7.47), S 5.91 (5.70). Colour: pale orange.
3d: yield: 259.8 mg, 0.2303 mmol (45%); LIFDI-MS: m/z: 1056.3 [M − Cl]+; elemental analysis for C50H78ClN6P2S2Er(C2.09H4.38O0.41) (found (calc.) [%]): C 54.53 (55.46), H 7.20 (7.36), N 7.33 (7.45), S 5.70 (5.68). Colour: pink.
3e: yield: 175.3 mg, 0.1543 mmol (30%); 1H NMR (500.13 MHz, 298 K, C6D6): δ [ppm] = 1.50 (s, 18H, 2 NC(CH3)3), 1.65 (s, 36H, 4 Lu-NC(CH3)3), 4.77–5.06 (m, 4H, 2 PCH2), 6.96–7.13 (m, 12H, m,p-Ph-H), 7.63 (s, 4H, o-Ph-H), 7.99 (s,4H, o-Ph-H); 13C{1H} NMR (125.76 MHz, 298 K, C6D6): δ [ppm] = 33.42 (s, NC(CH3)3), 34.24 (s, Lu-NC(CH3)3), 54.24 (s, Lu-NC(CH3)3), 55.18 (s, NC(CH3)3), 64.79 (s br, PCH2), 128.69–128.77 (m, m,m,p-Ph-C), 129.53 (s, p-Ph-C), 133.03 (d, 2JCP = 16.6 Hz, o-Ph-C), 135.27 (d, 2JCP = 18.6 Hz, o-Ph-C), 138.77 (s, ipso-Ph-C); 15N NMR (50.70 MHz, 298 K, C6D6): δ [ppm] = −216.5 (s, Lu-NC(CH3)3), −218.7 (s, Lu-NC(CH3)3) −255.2 (s, (NC(CH3)3)); 31P{1H} NMR (202.46 MHz, 298 K, C6D6): δ [ppm] = −24.59 (s); 31P{1H} NMR (161.98 MHz, 298 K, toluene-d8): δ [ppm] = −24.64 (s); 31P{1H} NMR (161.98 MHz, 213 K, toluene-d8): δ [ppm] = −23.84 (s), −27.74 (s); LIFDI-MS: m/z: 1063.6 [M − Cl]+; elemental analysis for C50H78ClN6P2S2Lu(C2.27H5.08O0.23) (found (calc.) [%]): C 55.03 (55.28), H 7.12 (7.37), N 7.48 (7.40), S 6.12 (5.65). Colour: colourless.
Author contributions
D. St. conceived the project idea and supervised the investigation; J. J. synthesized all compounds and generated and analysed the spectroscopic, diffraction and other experimental data; C. M. L. and S. D. conducted the magnetic measurements and analysed the data; and R. H.-I. supervised the diffraction data refinement. All authors wrote and edited the manuscript with input from everybody in the author list.Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. St. thanks the DFG (STA 334/28-1) for funding. C. M. L. thanks the Fonds der Chemischen Industrie for a PhD grant.References
- R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141 CrossRef CAS.
- K. V. Raman, A. M. Kamerbeek, A. Mukherjee, N. Atodiresei, T. K. Sen, P. Lazić, V. Caciuc, R. Michel, D. Stalke, S. K. Mandal, S. Blügel, M. Münzenberg and J. S. Moodera, Nature, 2013, 493, 509 CrossRef CAS PubMed.
- (a) N. Ishikawa, M. Sugita, T. Ishikawa, S.-Y. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694 CrossRef CAS PubMed; (b) J. D. Rinehart and J. R. Long, Chem. Sci., 2011, 2, 2078 RSC.
- (a) K. R. McClain, C. A. Gould, K. Chakarawet, S. J. Teat, T. J. Groshens, J. R. Long and B. G. Harvey, Chem. Sci., 2018, 9, 8492 RSC; (b) C. A. Gould, K. R. McClain, J. M. Yu, T. J. Groshens, F. Furche, B. G. Harvey and J. R. Long, J. Am. Chem. Soc., 2019, 141, 12967 CrossRef CAS PubMed.
- F.-S. Guo, B. M. Day, Y.-C. Chen, M.-L. Tong, A. Mansikkamäki and R. A. Layfield, Science, 2018, 362, 1400 CrossRef CAS PubMed.
- C. A. P. Goodwin, F. Ortu, D. Reta, N. F. Chilton and D. P. Mills, Nature, 2017, 548, 439 CrossRef CAS PubMed.
- F.-S. Guo, B. M. Day, Y.-C. Chen, M.-L. Tong, A. Mansikkamäki and R. A. Layfield, Angew. Chem., Int. Ed., 2017, 56, 11445 CrossRef CAS PubMed.
- (a) F.-S. Guo, A. K. Bar and R. A. Layfield, Chem. Rev., 2019, 119, 8479 CrossRef CAS PubMed; (b) J. P. Durrant, J. Tang, A. Mansikkamäki and R. A. Layfield, Chem. Commun., 2020, 56, 4708 RSC; (c) L. R. Thomas-Hargreaves, M. J. Giansiracusa, M. Gregson, E. Zanda, F. O'Donnell, A. J. Wooles, N. F. Chilton and S. T. Liddle, Chem. Sci., 2021, 12, 3911 RSC.
- J. M. Zadrozny and J. R. Long, J. Am. Chem. Soc., 2011, 133, 20732 CrossRef CAS PubMed.
- T. Pugh, V. Vieru, L. F. Chibotaru and R. A. Layfield, Chem. Sci., 2016, 7, 2128 RSC.
- P. Evans, D. Reta, G. F. S. Whitehead, N. F. Chilton and D. P. Mills, J. Am. Chem. Soc., 2019, 141, 19935 CrossRef CAS PubMed.
- L. E. Darago, M. D. Boshart, B. D. Nguyen, E. Perlt, J. W. Ziller, W. W. Lukens, F. Furche, W. J. Evans and J. R. Long, J. Am. Chem. Soc., 2021, 143, 8465 CrossRef CAS PubMed.
- E. Carl and D. Stalke, Chem. – Eur. J., 2014, 20, 15849 CrossRef CAS PubMed.
- (a) R. Fleischer, S. Freitag, F. Pauer and D. Stalke, Angew. Chem., Int. Ed. Engl., 1996, 35, 204–206 CrossRef CAS; (b) R. Fleischer, S. Freitag, F. Pauer and D. Stalke, Angew. Chem., Int. Ed. Engl., 1996, 35, 204 CrossRef CAS.
- R. Fleischer, B. Walfort, A. Gbureck, P. Scholz, W. Kiefer and D. Stalke, Chem. – Eur. J., 1998, 4, 2266 CrossRef CAS.
- (a) E. Carl and D. Stalke, Eur. J. Inorg. Chem., 2015, 2052 CrossRef CAS; (b) B. Walfort, A. P. Leedham, C. R. Russell and D. Stalke, Inorg. Chem., 2001, 40, 5668 CrossRef CAS PubMed.
- J. Matussek, R. Herbst-Irmer, I. Objartel and D. Stalke, Dalton Trans., 2014, 43, 15944 RSC.
- E. Carl, S. Demeshko, F. Meyer and D. Stalke, Chem. – Eur. J., 2015, 21, 10109 CrossRef CAS PubMed.
- J. Jung, C. M. Legendre, S. Demeshko, R. Herbst-Irmer and D. Stalke, Inorg. Chem., 2021, 60, 9580 CrossRef CAS PubMed.
- (a) J. Jung, F. Benner, R. Herbst-Irmer, S. Demir and D. Stalke, Chem. – Eur. J., 2021, 27, 12310 CrossRef CAS PubMed; (b) C. M. Legendre, R. Herbst-Irmer and D. Stalke, Inorg. Chem., 2021, 60, 13982–13989 CrossRef CAS PubMed.
- (a) D. Leusser, B. Walfort and D. Stalke, Angew. Chem., 2002, 114, 2183 ( Angew. Chem., Int. Ed. , 2002 , 41 , 2079 ) CrossRef; (b) D. Leusser, B. Walfort and D. Stalke, Angew. Chem., Int. Ed., 2002, 41, 2079 CrossRef CAS; (c) D. Leusser, J. Henn, N. Kocher, B. Engels and D. Stalke, J. Am. Chem. Soc., 2004, 126, 1781 CrossRef CAS PubMed; (d) J. Henn, D. Ilge, D. Leusser, D. Stalke and B. Engels, J. Phys. Chem. A, 2004, 108, 9442 CrossRef CAS.
- J. Jung, C. M. Legendre, R. Herbst-Irmer and D. Stalke, Inorg. Chem., 2021, 60, 967 CrossRef CAS PubMed.
- C. M. Legendre, E. Damgaard-Møller, J. Overgaard and D. Stalke, Eur. J. Inorg. Chem., 2021, 3108–3114 CrossRef CAS.
- S. V. Klementyeva, N. P. Gritsan, M. M. Khusniyarov, A. Witt, A. A. Dmitriev, E. A. Suturina, N. D. D. Hill, T. L. Roemmele, M. T. Gamer, R. T. Boeré, P. W. Roesky, A. V. Zibarev and S. N. Konchenko, Chem. – Eur. J., 2017, 23, 1278 CrossRef CAS PubMed.
- T. Chivers and R. S. Laitinen, Chalcogen-nitrogen chemistry, from fundamentals to applications in biological, physical and material sciences, World Scientific, 2021 Search PubMed.
- P.-H. Lin, N. C. Smythe, S. I. Gorelsky, S. Maguire, N. J. Henson, I. Korobkov, B. L. Scott, J. C. Gordon, R. T. Baker and M. Murugesu, J. Am. Chem. Soc., 2011, 133, 15806 CrossRef CAS PubMed.
- A. Eichhöfer, Y. Lan, V. Mereacre, T. Bodenstein and F. Weigend, Inorg. Chem., 2014, 53, 1962 CrossRef PubMed.
- E. Carl, Metal Complexes of a New Polyimido Sulfur Phosphanyl Ligand, PhD Thesis, Göttingen University, 2014; http://hdl.handle.net/11858/00-1735-0000-0022-5DB0-9 Search PubMed.
- J. M. Zadrozny, J. Telser and J. R. Long, Polyhedron, 2013, 64, 209 CrossRef CAS.
- J. M. Zadrozny and J. R. Long, J. Am. Chem. Soc., 2011, 133(51), 20732–20734 CrossRef CAS PubMed.
- S.-M. Chen, J. Xiong, Y.-Q. Zhang, Q. Yuan, B.-W. Wang and S. Gao, Chem. Sci., 2018, 9, 7540 RSC.
- K. L. M. Harriman and M. Murugesu, Acc. Chem. Res., 2016, 49(6), 1158–1167 CrossRef CAS PubMed.
- M. He, X. Chen, T. Bodenstein, A. Nyvang, S. F. M. Schmidt, Y. Peng, E. Moreno-Pineda, M. Ruben, K. Fink, M. T. Garner, A. K. Powell and P. W. Roesky, Organometallics, 2018, 37(21), 3708–3717 CrossRef CAS.
- T. Pugh, F. Tuna, L. Ungur, D. Collison, E. J. L. McInnes, L. F. Chibotaru and R. A. Layfield, Nat. Commun., 2015, 6, 7492 CrossRef PubMed.
- T. Pugh, N. F. Chilton and R. A. Layfield, Chem. Sci., 2017, 8, 2073 RSC.
- D. Reta and N. F. Chilton, Phys. Chem. Chem. Phys., 2019, 21, 23567 RSC.
- N. F. Chilton, D. Collison, E. J. L. McInnes, R. E. P. Winpenny and A. Soncini, Nat. Commun., 2013, 4, 2551 CrossRef PubMed.
- (a) T. Kottke and D. Stalke, J. Appl. Crystallogr., 1993, 26, 615 CrossRef; (b) D. Stalke, Chem. Soc. Rev., 1998, 27, 171 RSC.
- T. Schulz, K. Meindl, D. Leusser, D. Stern, J. Graf, C. Michaelsen, M. Ruf, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2009, 42, 885 CrossRef CAS.
- Bruker AXS Inc., SAINT, Madison, 2016 Search PubMed.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic, spectroscopic and magnetic details. CCDC 2104539 (2), 2104540 (3a), 2104541 (3b), 2104542 (3c), 2104543 (3d) and 2104544 (3e). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt03555j |
| This journal is © The Royal Society of Chemistry 2021 |