
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Titanium compounds containing naturally occurring dye molecules†
Wei-Hui
Fang
 *ab,
Rosa
Müller
b,
Rajesh B.
Jethwa
b,
Victor
Riesgo-González
b,
Ning
Li
bc,
Sebastian D.
Pike
b,
Andrew D.
Bond
b,
He-Kuan
Luo
d,
Cheng
Zhang
e and
Dominic S.
Wright
*b
*ab,
Rosa
Müller
b,
Rajesh B.
Jethwa
b,
Victor
Riesgo-González
b,
Ning
Li
bc,
Sebastian D.
Pike
b,
Andrew D.
Bond
b,
He-Kuan
Luo
d,
Cheng
Zhang
e and
Dominic S.
Wright
*b
aState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian 350002, China
bHusuf Hamied Department of Chemistry, University of Cambridge, CB2 1EW, UK. E-mail: dsw1000@cam.ac.uk
cInstitute of Bioengineering and Bioimaging, 31 Biopolis Way, The Nanos, #07-01, Singapore 138669
dInstitute of Materials Research and Engineering, Agency for Science, Technology and Research, 2 Fusionopolis Way, #08-03, Innovis, Singapore 138634
eCollege of Chemical Engineering, Zhejiang University of Technology, Hangzhou, 310014, P. R. China
First published on 9th November 2021
Abstract
A range of titanium compounds containing the naturally occurring dyes quinizarin (QH2) and alizarin (AH2) was synthesized and structurally characterized in the solid state. Among these is the first examples of a discrete metallocyclic arrangement formed exclusively using quinizarin ligands and the first examples of lanthanide containing titanium compounds of the alizarin family of ligands.
Introduction
Natural dyes can be derived from plants, invertebrates, or minerals. Plant-based dyes such as woad, indigo, saffron, and madder are available commercially and have been important trade goods in Asia and Europe for millennia.1 The madder plant (Rubia) has been used since antiquity as a dye material, principally for textile fabrics (e.g., Turkey Red), and as a principal component of paint pigments (e.g., Rose Madder). In modern times madder has also been used as a staining agent in biological research, and colour indicator for calcium.The active dye materials in madder are a series of related di- and tri-hydroxyl-anthraquinones, two of which are alizarin and quinizarin (Fig. 1).2–4 Recently, both of these materials have received much attention as low-cost photosensitizing dyes in semiconductor solar cells, photographic processes, and in the photo-degradation of pollutants.5 Many state-of-the-art photochemical devices are based on the high band-gap semiconductor titania (TiO2).6,7 However, a major problem faced using TiO2 (Eg = 3.2 eV) is that the majority of ambient solar light (ca. 95%) is of too low energy for photoexcitation from the valence to the conduction band.8,9 One option to circumvent this is to dope TiO2 with other metals or non-metals, which has the result of shifting the absorption edge into the visible region.10–13 A further way by which this can be achieved is the incorporation of a visible-light absorbing dye sensitizer on the semiconductor surface.14–19 Expensive inorganic compounds such as ruthenium(II) poly-pyridyl complexes have been commonly used as molecular sensitizers in solar cells and water-splitting devices.20–22 However, the use of sustainable natural dyes derived from plants to increase the rate and efficiency of light-capturing is clearly an attractive prospect in regard to mass production of photochemical devices.5 For example, it is reported that the photoexcitation of alizarin coupled to the surface of TiO2 films leads to ultrafast electron transfer from the dye to the TiO2 conduction band on the sub-100 fs timescale.23
 | ||
| Fig. 1 Structures of alizarin (AH2) and quinizarin (QH2). | ||
In the past decade there has been considerable interest in the development of titanium-oxo- and metal-doped titanium oxo-cages,24 which have proved to be particularly effective in the low-temperature, solution deposition of photocatalytic films of un-doped and metal-doped TiO2.25 A more recent innovation is the incorporation of dye molecules as supporting ligands in these species, which can potentially be used to deposit dye-sensitized metal-doped titania films in a single step.26 The potential for the use of naturally occurring dye molecules was recently demonstrated in a study of the use of titanium compounds containing alizarin ligands in the deposition of dye-sensitized TiO2.27 In the current work, we have synthesized a series of titanium compounds containing alizarin and quinizarin ligands. Apart from their structural novelty, the incorporation of lanthanides into this type of arrangement provides potential precursors for dye sensitized functional materials.28 As a prelude to future exploration of their applications, we also demonstrate the hydrolytic deposition of these materials as dye-containing titania and lanthanide-doped titania thin films on a range of substrates.
Results and discussion
The new homometallic TiIV metallocycle [Ti4(Q)4(iPrO)8·2iPrOH] (1) (QH2 = quinizarin) and the series of isostructural heterometallic lanthanide/TiIV compounds [Ti2LnCl2(A)2(iPrOH)2(OiPr)6]·xiPrOH [AH2 = alizarin, Ln = Eu (2), Gd (3), Tb (4), Dy (5), Ho (6), Er (7), Yb (8), x = 0 for compounds 2–5, 8; x = 1 for complexes 6 and 7] can be obtained via the room-temperature reactions of Ti(OiPr)4 with QH2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 equivalents) in iPrOH (for 1) and the reaction of excess Ti(OiPr)4 with AH2 and LnCl3·xH2O in iPrOH (for 2–8) under inert N2 atmosphere. Dark red crystals of 1 (53%) and deep-purple crystals of 2–8 (ca. 40%) are obtained by storage of the reaction solutions at room temperature. All of the compounds were characterised using a combination of 1H NMR, IR and solution UV-visible spectroscopy, chemical analysis and single-crystal X-ray diffraction studies. In addition, the experimental powder X-ray diffraction (PXRD) patterns for 1 and 2–8 are all in agreement with the patterns derived from their single-crystal X-ray structures, further confirming the purity of the bulk materials (ESI, Fig. S6–S8†). The paramagnetic nature of 2–8 made NMR investigation of little value. However, the presence of [Q]2− ligands in 1 and [A]2− ligands in 2–8 is shown by a comparison of the UV-vis spectra of the complexes with those of the authentic [Q]2− and [A]2− anions, which were generated by double-deprotonation of QH2 and AH2 (see Fig. 6, later).
:1 equivalents) in iPrOH (for 1) and the reaction of excess Ti(OiPr)4 with AH2 and LnCl3·xH2O in iPrOH (for 2–8) under inert N2 atmosphere. Dark red crystals of 1 (53%) and deep-purple crystals of 2–8 (ca. 40%) are obtained by storage of the reaction solutions at room temperature. All of the compounds were characterised using a combination of 1H NMR, IR and solution UV-visible spectroscopy, chemical analysis and single-crystal X-ray diffraction studies. In addition, the experimental powder X-ray diffraction (PXRD) patterns for 1 and 2–8 are all in agreement with the patterns derived from their single-crystal X-ray structures, further confirming the purity of the bulk materials (ESI, Fig. S6–S8†). The paramagnetic nature of 2–8 made NMR investigation of little value. However, the presence of [Q]2− ligands in 1 and [A]2− ligands in 2–8 is shown by a comparison of the UV-vis spectra of the complexes with those of the authentic [Q]2− and [A]2− anions, which were generated by double-deprotonation of QH2 and AH2 (see Fig. 6, later).
A prominent feature of the solid-state structure of compound 1 (which crystallises as the solvate 1·(iPrOH)2) is the presence of an almost square-planar Ti4 core which is held together by κ2/κ2-bridging [Q]2− dianions, with two terminal iPrO groups bonded to each metal centre (Fig. 2a). The [Q]2− dianions are tilted significantly from the perpendicular to the metallocyclic Ti4-plane and form a cavity measuring ca. 8.2–8.5 Å along its edges by 7.3 Å deep (Fig. 2b). The cavity is occupied by an iPrOH molecule, and additional iPrOH molecules are present in the crystal lattice. Isolation under vacuum results in the complete removal of these solvent molecules in solid samples of 1, as revealed by elemental analysis. While the same κ2/κ2-metal bridging mode for the [Q]2− dianion has been observed in a number of discrete metallocyclic and polymeric arrangements, such as the polymer [FeQ]∞ and tetranuclear [{Re(CO)3}4(Q)2(bipy)2],291 is the first example in which the assembly of a discrete metallocyclic arrangement is achieved exclusively using the [Q]2− dianion.30 A related, square Ti4 metallocycle has been observed for the structure of Ti4O4(C2O4)8·4C2N2H10·4H2O, but here using a mixed ligand set to accomplish cyclisation.31
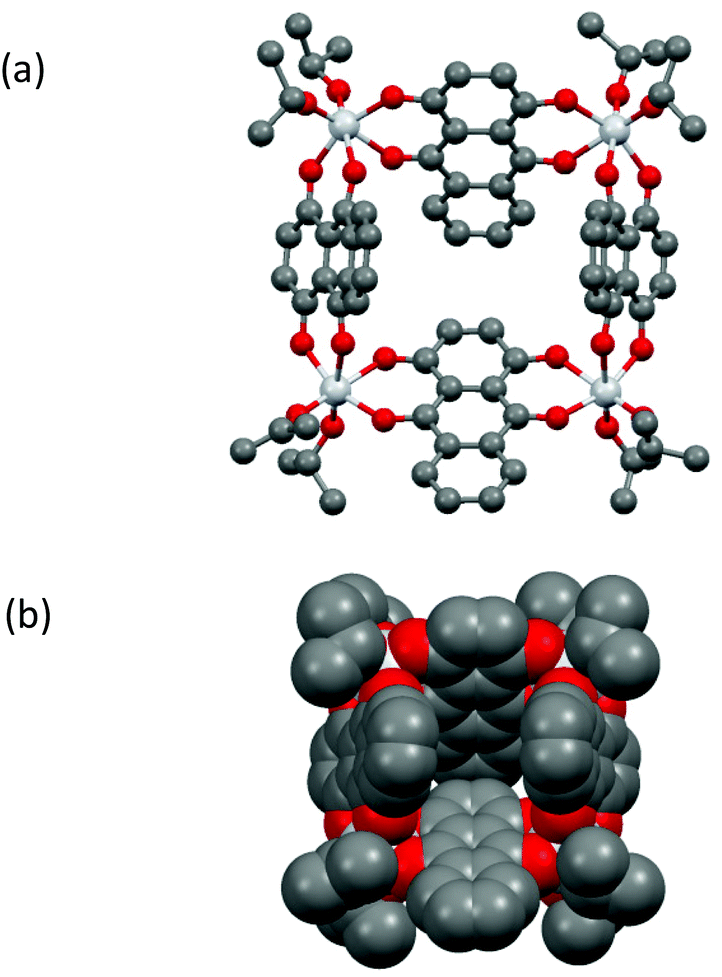 | ||
| Fig. 2 (a) Molecular structure of metallocycle 1 in the solid state, (b) space-filling representation in the same orientation as in (a). The iPrOH molecule in the void is not shown. For bond lengths and angles in 1 see Fig. S11.† | ||
The structure of 1 is maintained in toluene solution at room-temperature, as indicated by the presence of four (1:1:1:1) iPr-resonances in the room-temperature 1H NMR spectrum in d8-toluene. This results from the CS-mirror symmetry of the molecule which leads to the inequivalence of iPr-groups above and below the Ti4-mean plane (Fig. 3). The spectrum does not change significantly on raising the temperature to 60 °C.
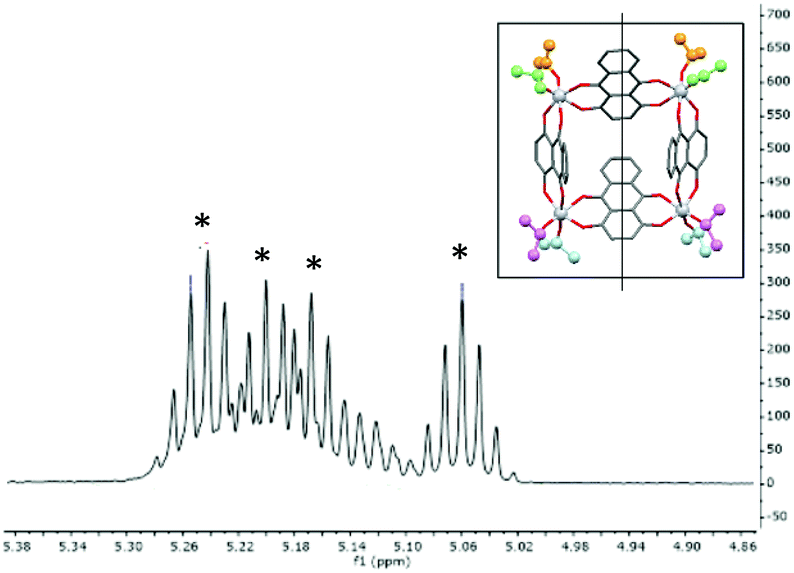 | ||
| Fig. 3 1H NMR spectrum of 1 in the iPr (CHMe2) region, showing the four environments resulting from molecular Cs symmetry in the solid state and solution. The inset shows the molecular mirror plane, with the four environments highlighted (these could not be assigned unambiguously in the spectrum). | ||
Cyclic voltammetry (CV) of 1 in DCM shows a complicated series of overlapping redox processes at negative potentials (Fig. 4, ESI, Fig. S28 and S29†) in which the quinizarin ligand is likely to be involved. The electrochemistry was found to contain electrochemically irreversible redox processes, and to change slightly with time over the course of ca. 100 cycles with a blue solid being deposited on the electrode surface and the walls of the electrochemical cell. This is distinctly different from the behaviour of QH2 in DCM (Fig. 4, ESI, Fig. S31†), which shows two reversible one-electron processes in a similar region to the redox waves in 1, or to QH2 in basic aqueous conditions (ESI, Fig. S32†), which shows a single reversible two-electron process. Overall, this suggests that the electronic character of the ligand is changed significantly by deprotonation and metal coordination within 1.
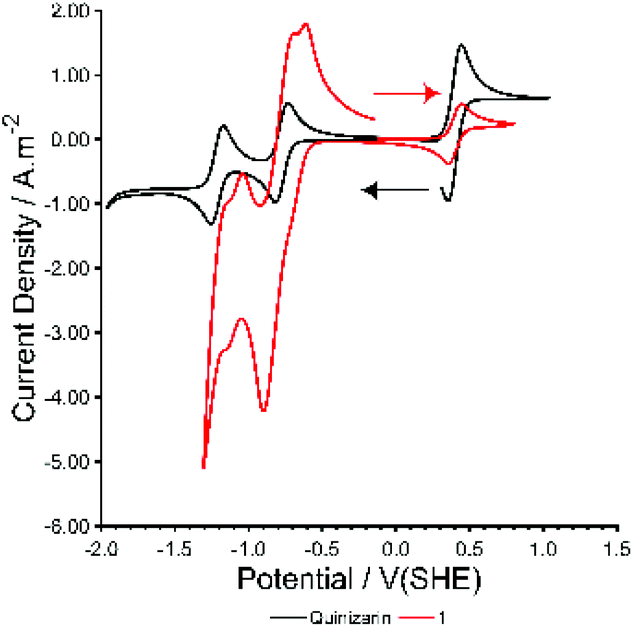 | ||
| Fig. 4 CV of Species 1 and quinizarin at 50 mV s−1 in dry dichloromethane with 0.1 M TBAPF6 and 0.3 mM Ferrocene (at 0.4 V(SHE)). | ||
Compounds 2–8 all have formula [Ti2LnCl2(A)2(iPrOH)2(OiPr)5], and differ only in the number of iPrOH molecules in the crystal lattice. The crystal structures comprise of two isomorphous sets (2–5, 8, with no lattice iPrOH; 6–7 with one lattice iPrOH per formula unit) The structure of the Eu compound 2 is presented in Fig. 5. Despite the historical importance of metal-alizarin complexes in ancient dyes, there are surprisingly few structurally characterized metal complexes containing alizarin and, to the best of our knowledge, no examples of lanthanide complexes.32–36 The closest analogue to 2–8 is the heterometallic TiIV/Ca2+ complex [Ca2Ti4(A)4(μ3-O)2], containing doubly-deprotonated AH2.32
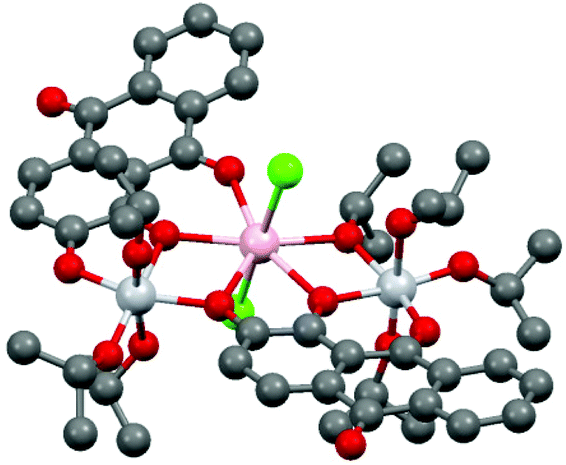 | ||
| Fig. 5 Structure of 2. H-atoms have been omitted for clarity. Colour codes for atoms: Ti-light grey; Eu-pink; O-red; C-dark grey; Cl-green. For bond lengths and angles in 2–8 see Table S1 (ESI).† | ||
Compounds 2–8 have the same trinuclear arrangement in which the Ln3+ cations are seven coordinate (pentagonal bipyramidal), being bonded to four O atoms of two [A]2− dianions as well as one iPrO O atom and two Cl ions (Fig. 5). The presence of two neutral iPrOH groups (which are necessary to balance the charge with the complexes) is indicated by the elongation of two of the terminal Ti–O(iPr) bonds in 2–8 by ca. 0.4 Å. The OH groups of these molecules form clear hydrogen bonds to the Cl ions on the Ln3+ cations. The overall molecular arrangement is distinctly asymmetrical. Although the two [A]2− ions adopt the same (unprecedented) κ3-μ1:μ2:μ2 bonding mode, the two [A]2− anions use different combinations of their C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and 1- and 2-O atoms to bridge the TiIV and Ln3+ centres, i.e., μ1 (2-O):μ2 (1-O):μ2 (CO) for one and μ1 (CO):μ2 (2-O):μ2 (1-O) for the other. This results in chemically-distinct environments for the two TiIV centres in 2–8, both being six coordinate, but with one bonded to three [A]2− O-atoms and three iPrO O-atoms and the other bonded to two [A]2− O-atoms and four iPrO O-atoms. Looking across the lanthanide series, there is a general reduction observed in the framework Ln–O and Ln–Cl bond lengths (by ca. 0.1 Å for both), in line with the expected lanthanide contraction. Bond valence sum calculations (ESI, Table 2†) are consistent with the oxidation states of the metal ions concluded from X-ray and spectroscopic analysis (i.e., TiIV and LnIII).37
O and 1- and 2-O atoms to bridge the TiIV and Ln3+ centres, i.e., μ1 (2-O):μ2 (1-O):μ2 (CO) for one and μ1 (CO):μ2 (2-O):μ2 (1-O) for the other. This results in chemically-distinct environments for the two TiIV centres in 2–8, both being six coordinate, but with one bonded to three [A]2− O-atoms and three iPrO O-atoms and the other bonded to two [A]2− O-atoms and four iPrO O-atoms. Looking across the lanthanide series, there is a general reduction observed in the framework Ln–O and Ln–Cl bond lengths (by ca. 0.1 Å for both), in line with the expected lanthanide contraction. Bond valence sum calculations (ESI, Table 2†) are consistent with the oxidation states of the metal ions concluded from X-ray and spectroscopic analysis (i.e., TiIV and LnIII).37
The UV visible spectra of 1 and 2–8 are shown in Fig. 6. All of the compounds show significant absorption in the visible region, arising directly from the presence of the [Q]2− and [A]2− anion transitions. The similarity of the UV-vis spectrum of the [Q]2− dianion (generated by deprotonation of QH2 with 2 equivalents of nBuLi) and the spectrum of 1 (Fig. 6a) provides further confirmation for the presence of the [Q]2− dianion in 1. The [Q]2− dianion in 1 (λmax = 610 nm) is significantly red-shifted with respect to [QH2] (λmax = 490 nm) and slightly red-shifted with respect to the [Q]2− dianion (λmax = 595 nm). This is probably due to the presence of charge transfer [ligand π-Ti(d)] transitions in 1, cf. π–π* transitions in QH2 and [Q]2−. The observed red shift is also consistent with the colour change from QH2 (yellow) to [Q]2− and 1 (both violet). The isostructural compounds 2–8 exhibit very similar spectra in the visible region, with no obvious correlation between the Zeff of LnIII ions and λmax (being ca. 510 nm for all) (Fig. 6b). Not surprisingly, the [A]2− absorptions in 2–8 are red-shifted with respect to [AH2] (consistent with the colour change from yellow to red) but are very similar to the spectrum of the [A]2− anion itself (generated by reaction of AH2 with 2 equivalent of nBuLi). One interesting feature of the optical behaviour of 2–8 is that the compounds exhibit markedly different molar extinction coefficients, depending on the Ln3+ ion present, with most absorbing more strongly than [A]2−. This is likely to be due to the magnetic anisotropy of the respective Ln3+ ions, which influences the electronic transitions upon light irradiation.38
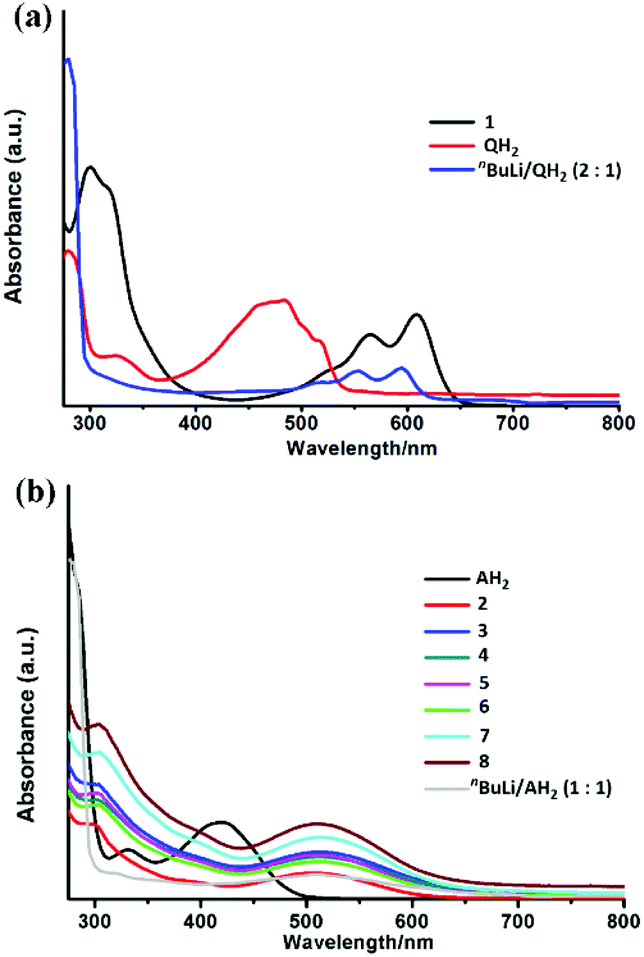 | ||
| Fig. 6 (a) UV-visible absorbance spectra of quinizarin, compound 1 (0.075 g L−1) and [Q]2− (all 0.075 g L−1 in dichloromethane) and (b) UV-visible absorbance spectra of alizarin, compounds 2–8 and [AH]− (all solutions ca. 6.1–6.2 × 10–5 mol L−1). | ||
PXRD studies of 1 and 2–8 reveal that crystalline samples are stable in ambient air for at least one hour. The decomposition of 1 and 2 (as an example of the lanthanide series) in the presence of atmospheric or deliberately added water were followed in detail by IR (ESI, Fig. S15 and S16†) and UV-visible spectroscopy (Fig. 7), PXRD (ESI, Fig. S9 and S10†), luminescence spectroscopy (ESI, Fig. S25–27†), scanning electron microscopy (SEM) (ESI, Fig. 4†) and EDX (ESI, Fig. S17–24†) studies.
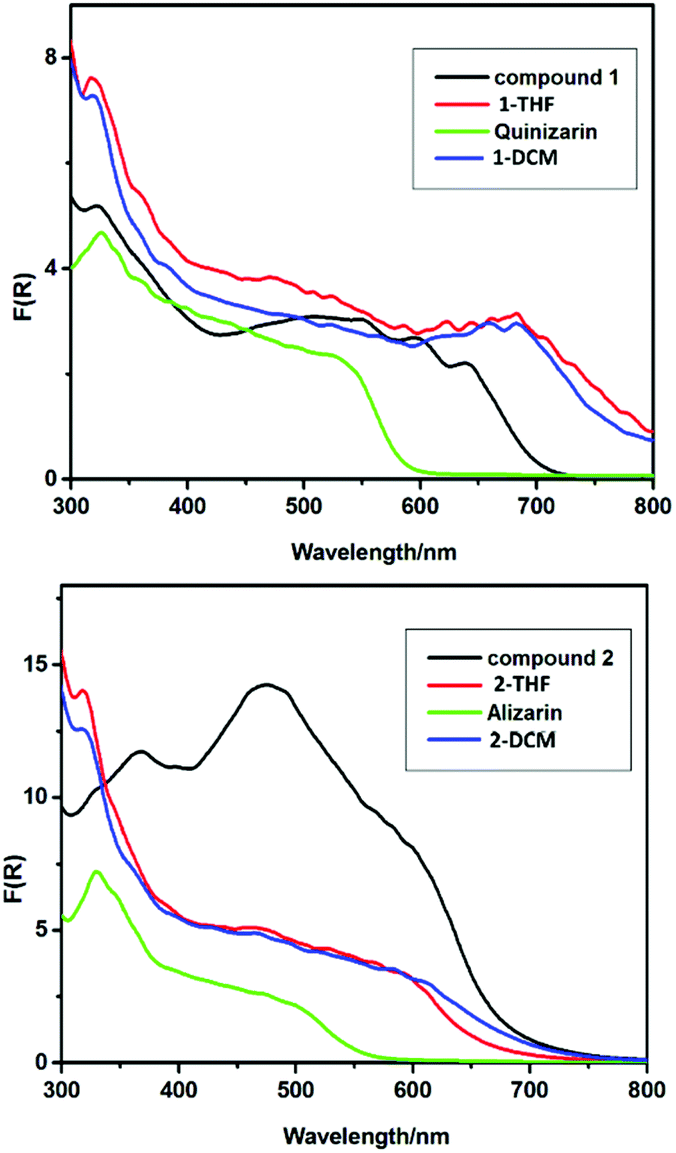 | ||
| Fig. 7 Solid-state UV-vis spectra of (a) drop-cast films of (hydrolysed) 1 from THF (red trace) and DCM (blue trace), compared to solid powder samples of 1 and QH2, (b) drop-cast films of (hydrolysed) 2 from THF (red trace) and DCM (blue trace), compared to solid powder samples of 2 and AH2. | ||
The PXRD patterns of the solid decomposition products of 1 and 2 after stirring in wet CH2Cl2 for 16 h indicate that both are amorphous. In the case of 1, however, low-intensity peaks for QH2 are visible. Although no residual ligand peaks are found in the PXRD of 2, the solid-state IR spectra of the solid residues are consistent with the formation of QH2 (for 1) and AH2 (for 2). Further support for the conclusion that hydrolysis results in the protonation of the dianionic ligands comes from luminescence measurements of 1, which show that the emission spectrum of the hydrolysed compound is identical to that of QH2 (ESI, Fig. S27†) (but distinctly different to that of 1 itself, ESI Fig. S26†). Not surprisingly, after annealing at 800 °C no traces of the ligands (QH2 and AH2) are observed, with PXRD patterns indicating that the solids are composed of rutile (in the case of 1) and rutile/E2O3 (in the case of 2) (ESI Fig. S9 and S10†). While SEM-EDX analysis of the annealed product of 1 is uninformative (showing only Ti and O), element mapping of the product of 2 shows Eu, Ti and O, with EDX area analysis indicating inhomogeneous distribution of these elements (Eu range 20.5–42.3 at%, Ti 4.1–13.2 at% and O 37.2–54.2 at%) (ESI Fig. S17–21†). Overall, the data is consistent with the formation of TiO2/QH2 for 1 and Eu-doped TiO2/Eu2O3/AH2 for 2 before annealing.
In further studies it was shown that the organically soluble precursors 1 and 2 can be successfully drop-cast into films from solution at room temperature (ESI, Fig. S24†). The morphologies of the resulting films were found to depend on the solvent used (THF or CH2Cl2), the substrate (FTO or brass) and the pH (see ESI, Fig. S24†). The solid-state UV-vis spectra of hydrolytically decomposed films of 1 and 2 from THF or CH2Cl2 without annealing (Fig. 7) both show much lower-energy absorption onsets than the TiO2 or pristine organic dyes themselves, extending well above 800 nm for 1 (cf. ca. 600 nm for QH2) and ca. 700 nm for 2 (cf. 550 nm for AH2). This appears to indicate that these TiO2/QH2 and Eu-doped TiO2/Eu2O3 composite materials exhibit lower band gaps compared to the separate TiO2 and dye components. This supports the view that there is some integration of the dyes into the surface or bulk structure of TiO2.
Disappointingly, films prepared by spin-coating 1 and 2 in CH2Cl2 onto BiVO4 coated ITO glass followed by air drying at room temperature exhibited no photocurrent response. This contrasts with the previous report of the photochemical behaviour of alizarin dye-sensitized TiO2 films generated using [Ti2(OiPr)5(A)(4-X-C6H4CO2)] (X = NO2, F, Br), which assumed that the A2− ligands survive hydrolysis and showed that the films generated exhibited a significantly increased photocurrent compared to TiO2 or a TiO2/AH2 mixture.27 There could be several reasons for the different behaviour of our systems. In particular, the absence of the benzoate ligand component in precursors may be a significant factor.
In summary, we have prepared a series of new titanium compounds containing the dianions of the naturally occurring dye molecules alizarin and quinizarin. Apart from the various new structural and chemical features observed, we have shown these can be used as single-source precursors to dye-containing titania composites. The presence of natural dyes results in significant visible light absorption and lower bandgap in the deposited materials. However, our studies indicate that the dye components (which are present as the neutral dye molecules themselves) may not have direct bearing on the photocurrent of titania films.
Author contributions
W.-H. F., R. M., V. R.-G. and N. L. did the synthetic work, A. D. B. collected and refined all the X-ray data, R. B. J. did the electrochemical measurements, and H.-K. L., C. Z. and D. S. W. supervised various aspects of the project and wrote the paper with W.-H. F.Conflicts of interest
There are no conflicts of interest.Acknowledgements
This work was supported by National Natural Science Foundation of China (92061104 and 21771181), Youth Innovation Promotion Association CAS (2017345), Shanghai Key Laboratory of Rare Earth Functional Materials. We also acknowledge support from China Scholarship Council (W.-H. Fang), the A*STAR Graduate Academy for a Ph.D. Scholarship (N. L.), and the Herchel Smith Fund (S.D.P.). Financial support from SERC-A*STAR (1526004162) and IMRE-A*STAR (IMRE/15-2C0252) is also acknowledged. We thank the EPRSC and Shell for the I-Case studentship EP/R511870/1. (R. B. J.) We also thank Lambda Energy Ltd. (U.K.) for further financial support.References
- K. Baghalian, M. Maghsodi and M. R. Naghavi, Ind. Crops Prod., 2010, 31, 557–562 CrossRef CAS.
- A. Shotipruk, J. Kiatsongserm, P. Pavasant, M. Goto and M. Sasaki, Biotechnol. Prog., 2004, 20, 1872–1875 CrossRef CAS PubMed.
- D. Cheuk, M. Svard, C. Seaton, P. McArdle and A. C. Rasmuson, CrystEngComm, 2015, 17, 3985–3997 RSC.
- M. K. Cyrański, M. H. Jamróz, A. Rygula, J. C. Dobrowolski, Ł. Dobrzycki and M. Baranska, CrystEngComm, 2012, 14, 3667–3676 RSC.
- S. Gholamrezaei and M. Salavati-Niasari, J. Mater. Sci.: Mater. Electron., 2016, 27, 2467–2472 CrossRef CAS.
- H. Gaminian and M. Montazer, J. Photochem. Photobiol., A, 2017, 332, 158–166 CrossRef CAS.
- R. Huber, S. Sporlein, J. E. Moser, M. Gratzel and J. Wachtveitl, J Phys. Chem. B, 2000, 104, 8995–9003 CrossRef CAS.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- J. Yuan, X. Huang, M. Chen, J. Shi and W. Shangguan, Catal. Today, 2013, 201, 182–188 CrossRef CAS.
- Y. Matsumoto, M. Murakami, T. Shono, T. Hasegawa, T. Fukumura, M. Kawasaki, P. Ahmet, T. Chikyow, S. Koshihara and H. Koinuma, Science, 2001, 291, 854–856 CrossRef CAS PubMed.
- S. In, A. Orlov, R. Berg, F. Garcia, S. Pedrosa-Jimenez, M. S. Tikhov, D. S. Wright and R. M. Lambert, J. Am. Chem. Soc., 2007, 129, 13790–13791 CrossRef CAS PubMed.
- Y. Wang, R. Zhang, J. Li, L. Li and S. Lin, Nanoscale Res. Lett., 2014, 9, 46–46 CrossRef PubMed.
- G. J. Meyer, Inorg. Chem., 2005, 44, 6852–6864 CrossRef CAS PubMed.
- M. Gratzel, Nature, 2001, 414, 338–344 CrossRef CAS.
- A. Hagfeldt and M. Gratzel, Chem. Rev., 1995, 95, 49–68 CrossRef CAS.
- Y.-Y. Wu, X.-W. Lu, M. Qi, H.-C. Su, X.-W. Zhao, Q.-Y. Zhu and J. Dai, Inorg. Chem., 2014, 53, 7233–7240 CrossRef CAS PubMed.
- C. F. A. Negra, K. J. Young, M. Belén Oviedo, L. J. Allan, C. G. Sánchez, K. N. Jarembska, J. B. Benedict, R. H. Crabtree, P. Coppens, G. W. Brudvig and V. S. Batista, J. Am. Chem. Soc., 2014, 136, 16420 CrossRef PubMed.
- For a recent review of this area, see: Q.-Y. Zhu and J. Dai, Coord. Chem. Rev., 2021, 430, 213664 CrossRef CAS.
- J. J. Concepcion, J. W. Jurss, M. K. Brennaman, P. G. Hoertz, A. O. T. Patrocinio, N. Y. Murakami Iha, J. L. Templeton and T. J. Meyer, Acc. Chem. Res., 2009, 42, 1954–1965 CrossRef CAS PubMed.
- B. D. Sherman, Y. Xie, M. V. Sheridan, D. Wang, D. W. Shaffer, T. J. Meyer and J. J. Concepcion, ACS Energy Lett., 2017, 2, 124–128 CrossRef CAS.
- A. El-Shafei, M. Hussain, A. Islam and L. Han, Prog. Photovoltaics, 2014, 22, 958–969 CAS.
- L. Dworak, V. V. Matylitsky and J. Wachtveitl, ChemPhysChem, 2009, 10, 384–391 CrossRef CAS PubMed.
- S. Eslava, M. McPartlin, R. I. Thomson, J. M. Rawson and D. S. Wright, Inorg. Chem., 2010, 49, 11532–11540 CrossRef CAS PubMed.
- P. D. Matthews, T. C. King and D. S. Wright, Chem. Commun., 2014, 50, 12815–12823 RSC.
- N. Li, P. D. Matthews, H. K. Luo and D. S. Wright, Chem. Commun., 2016, 52, 11180–11190 RSC; H.-T. Lv, Y. Cui, Y.-M. Zhang, H.-M. Li, G.-D. Zou, R.-H. Duan, J.-T. Cao, Q.-S. Jing and Y. Fan, Dalton Trans., 2017, 46, 12313–12319 RSC; J. B. Benedict and P. Coppen, J. Am. Chem. Soc., 2010, 132, 2938–2944 CrossRef CAS; R. C. Snoeberger III, K. J. Young, J. Tang, A. J. Allen, R. h. Crabtree, J. W. Brudvig, P. Coppens, V. S. Batista and J. B. Benedict, J. Am. Chem. Soc., 2012, 134, 8911–8917 CrossRef PubMed; J. Hou, Q. Zhang, Y. Wu, Y. Liu, L. Du, C.-H. Tung and Y. Wang, Inorg. Chim. Acta, 2016, 443, 279–283 CrossRef.
- Y. Guo, J.-L. Hou, W. Luo, Z.-Q. Li, D.-H. Zou, Q.-Y. Zhu and J. Dai, J. Mater. Chem. A, 2017, 5, 18270 RSC.
- B. M. van der Ende, L. Aarts and A. Mejerink, Phys. Chem. Chem. Phys., 2009, 11, 11081 RSC.
- (a) D. Bhattacharya, M. Sathiyendiran, T. T. Luo, C. H. Chang, Y. H. Cheng, C. Y. Lin, G. H. Lee, S. M. Peng and K. L. Lu, Inorg. Chem., 2009, 48, 3731–3742 CrossRef CAS PubMed; (b) S. Agrawal, S. M. Clarke, I. J. Vitorica-Yrezabal, C. Liu, W. Fang, P. T. Wood and D. S. Wright, Mol. Phys., 2019, 117, 3424 CrossRef CAS.
- M. J. Maroney, R. O. Day, T. Psyris, L. M. Fleury and J. P. Whitehead, Inorg. Chem., 1989, 28, 173–175 CrossRef CAS.
- Y. L. Fu, Y. L. Liu, Z. Shi, B. Z. Li and W. Q. Pang, J. Solid State Chem., 2002, 163, 427–435 CrossRef CAS.
- C.-H. Wunderlich and G. Bergerhoff, Z. Kristallogr., 1993, 207, 185–188 CAS.
- C.-H. Wunderlich and G. Bergerhoff, Chem. Ber., 1994, 127, 1185–1190 CrossRef CAS.
- M. R. Churchill, K. M. Keil, F. V. Bright, S. Pandey, G. A. Baker and J. B. Keister, Inorg. Chem., 2000, 39, 5807–5816 CrossRef CAS PubMed.
- B. Zhang, S. Z. Du, X. M. Lu, G. Wang and J. Fen, Inorg. Chem., 2013, 52, 9470–9478 CrossRef CAS PubMed.
- S. Du, J. Feng, X. Lu and G. Wang, Dalton Trans., 2013, 42, 9699–9705 RSC.
- I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 244 CrossRef.
- N. Li, P. D. Matthews, J. Xiao, T. E. Rosser, E. Reisner, H.-K. Luo and D. S. Wright, Dalton Trans., 2017, 46, 4287 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, synthesis and physical measurements; crystallographic data. CCDC 1547945-1547952 for compounds 1–8. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt03377h |
| This journal is © The Royal Society of Chemistry 2021 |