
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCoupling of CO2 and epoxides catalysed by novel N-fused mesoionic carbene complexes of nickel(II)†
Fabian A.
Watt‡
a,
Benedikt
Sieland‡
a,
Nicole
Dickmann‡
a,
Roland
Schoch
a,
Regine
Herbst-Irmer
 b,
Holger
Ott
c,
Jan
Paradies
a,
Dirk
Kuckling
a and
Stephan
Hohloch
*d
b,
Holger
Ott
c,
Jan
Paradies
a,
Dirk
Kuckling
a and
Stephan
Hohloch
*d
aPaderborn University, Faculty of Science, Department of Chemistry, Warburger Straße 100, 33098 Paderborn, Germany
bUniversity of Göttingen, Institute of Inorganic Chemistry, Tammannstraße 4, 37077 Göttingen, Germany
cBruker AXS GmbH, Östliche Rheinbrückenstraße 49, 76187 Karlsruhe, Germany
dUniversity of Innsbruck, Faculty of Chemistry and Pharmacy, Institute of General, Inorganic and Theoretical Chemistry, Innrain 80-82, 6020 Innsbruck, Austria. E-mail: Stephan.Hohloch@uibk.ac.at
First published on 27th October 2021
Abstract
We report the syntheses of two rigid mesoionic carbene (MIC) ligands with a carbazole backbone via an intramolecular Finkelstein–cyclisation cascade and investigate their coordination behavior towards nickel(II) acetate. Despite the nickel(II) carbene complexes 4a,b showing only minor differences in their chemical composition, they display curious differences in their chemical properties, e.g. solubility. Furthermore, the potential of these novel MIC complexes in the coupling of carbon dioxide and epoxides as well as the differences in reactivity compared to classical NHC-derived complexes are evaluated.
Introduction
Pincer-type ligands have opened many new avenues in modern-day chemistry, giving rise to stable and robust catalysts as well as allowing for the isolation of highly reactive metal complexes.1,2 Therefore, a large variety of neutral and monoanionic pincer ligands have been synthesised in the past decades (Fig. 1).2,3 Due to their unique profiles and modular chemical designs, especially regarding the nature of the coordinating donor atoms (ranging from PNP,4 PCP,5 POP,6 OCO,7 SCS,8 CNC,9 or CCC10 just to mention a few), numerous breakthroughs have been achieved. One moiety of ever-increasing focus in this context is the monoanionic carbazole fragment. Its planar and rigid geometry in combination with its persistent fluorescence and its unique ability to partake in redox-reactions led to a plethora of applications in catalysis and small molecule activation.11 However, until now most of the carbazole systems have been derived from the classical PNP substitution pattern, while CNC coordination motifs, e.g. by using N-heterocyclic carbenes (NHCs), have found less attention. Nevertheless, seminal work by Kunz and co-workers12 has shown that carbazole derived CNC coordinating ligands are valuable precursors for the design of effective catalysts13 and can stabilize highly reactive group 10 metal complexes (Fig. 2, top left).14 This is supported by a recent report of Lee and co-workers who showed that nickel(II) complexes (Fig. 2, top right) are potent catalysts in the copolymerisation of CO2 and cyclohexene oxide.15 However, given the relatively high temperatures required for catalysis, especially in comparison to systems with early transition metals,16 we felt that the improvement of these catalysts should be a valuable goal.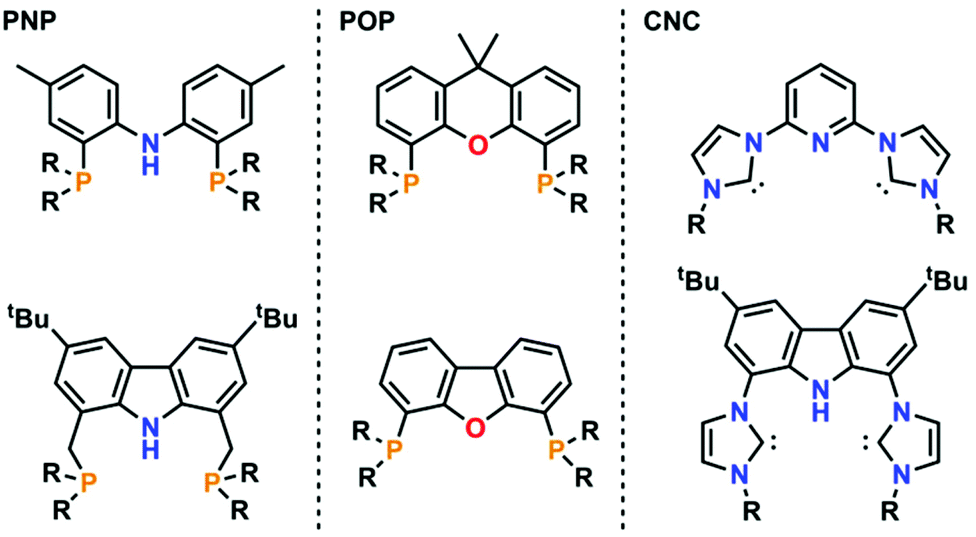 | ||
| Fig. 1 Selected examples of the most commonly used pincer-type ligands scaffolds with PNP (left), POP (middle) and CNC (right) coordination modes. | ||
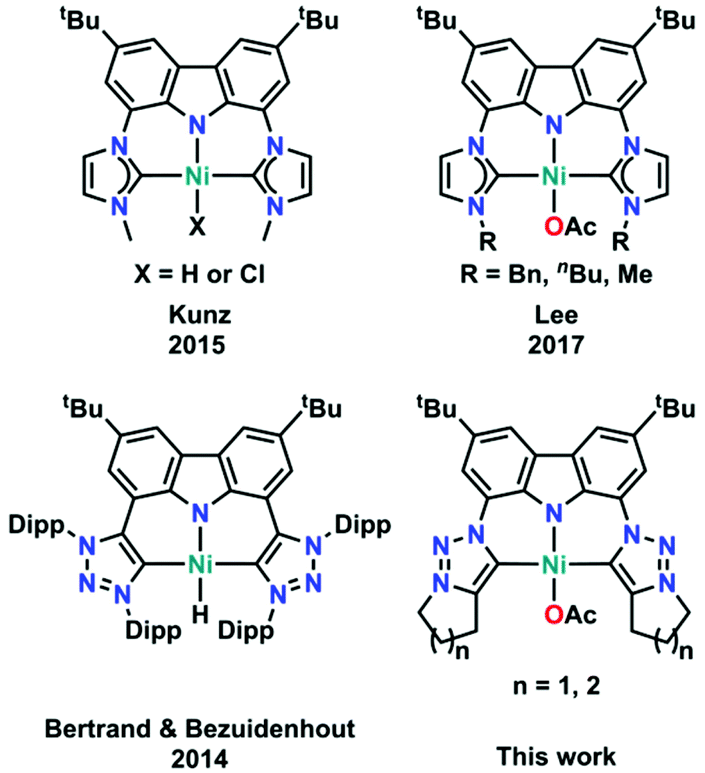 | ||
| Fig. 2 Overview of all literature known carbene decorated nickel(II) carbazolide complexes, including the two new examples reported in this work. | ||
One strategy for the improvement of the catalytic potential of NHC-derived catalysts in recent years was the exchange of the NHC donors for mesoionic carbene (MIC) donors.17j,18 Pioneering work by Sarkar and co-workers has shown that MIC-derived catalysts often exceed the catalytic potential of their NHC congeners.18,19,20–26 Transferring this concept to carbazolide-derived mesoionic carbenes, Bertrand and Bezuidenhout reported the first CNC pincer-type MIC ligands in 2014 (Fig. 2, bottom left).27 While this ligand was capable of stabilising a rare rhodium(I)–dioxygen adduct28 as well as a rare T-shaped gold(I) complex,29 its synthesis is tedious and requires the use of harmful and explosive reagents such as tert-butylhypochlorite and in situ generated 3-chlorotriazenes. Additionally, this synthetic route is limited by the availability of stable triazene derivatives, which means that it only serves to synthesise sterically demanding systems.
Aiming for a less hazardous synthetic protocol and reducing the steric bulk of carbazole-derived triazolium salts, we recently reported a tandem Finkelstein–cyclisation strategy which gave access to highly luminescent bis-N-fused triazolylidene carbazolide complexes of lithium and magnesium.30 In this contribution we investigate the coordination behaviour of these novel carbazole functionalized triazolylidene ligands towards nickel(II) and their application in the copolymerisation of CO2 with epoxides.
Results and discussion
To expand the versatility of our recently reported synthetic strategy to prepare bis(triazolylidene)-carbazolide ligands,30 in addition to the known compound 3b with a six-membered piperidine-based ring, we also synthesised ligand precursor 3a with a five-membered pyrrolidine-based annulated ring, which has a slightly lower steric bulk (Scheme 1). Compound 3a was synthesised using standard CuAAC (copper catalysed alkyne azide cycloaddition) conditions with 5-chloro-1-pentyne and 1,8-diazido-3,6-di-tert-butyl-carbazole,31 resulting in the clean formation of the desired chloro-propyl 1,2,3-triazole 2a in 70% yield (Scheme 1, middle). Indicative for the successful formation of the desired triazole is the disappearance of the characteristic azide stretching band in the IR spectrum at 2099 cm−1 (compare Fig. S53 and S54†). Additionally, successful formation of 2a was evidenced by 1H NMR spectroscopy, showing a characteristic triazole-5H low-field resonance at δ = 8.02 ppm. Addition of an excess of potassium iodide (20 equiv.) and heating the mixture to 90 °C for 48 h led to the formation of the desired N-fused triazolium salt 3a in quantitative yields (Scheme 1, right). Successful cyclisation was evident by various features in the NMR spectra: (i) the typical low-field shift of the triazolium-5H resonance in the 1H NMR spectrum from δ = 8.02 ppm for 2a to δ = 8.67 ppm for 3a, (ii) the low-field shift of the resonance of the (former) –CH2Cl protons from δ = 3.69 ppm for 2a to δ = 4.90 ppm for 3a, and (iii) the coupling of this methylene group to the triazole nitrogen atoms (δ(15N) = 266.6 ppm) as seen in the 1H–15N HMBC NMR spectrum (compare Fig. S10†). Finally, X-ray quality crystals32 of 3a were obtained by layering a concentrated dichloromethane solution with n-hexane in a NMR tube and storage at room temperature for several days. Compound 3a crystallises in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with five dichloromethane solvent molecules in the crystal lattice and two independent molecules in the asymmetric unit. Despite the relatively small change from an N-fused piperidine to a pyrrolidine ring, the structures of 3a,b reveal notable differences. While 3a shows hydrogen bonding of the carbazole-NH proton H10 to one of the iodide counterions (Fig. 3, left), in 3b such an interaction is absent.30 Apart from the hydrogen bonding, the structural parameters in 3a reflect the ones in 3b30 and previously reported 1,2,3-triazolium salts.20,22 A detailed discussion of the structural parameters is therefore omitted. For more details on the structural metrics please see Tables S1 and S2 in the ESI† (Fig. 3).
with five dichloromethane solvent molecules in the crystal lattice and two independent molecules in the asymmetric unit. Despite the relatively small change from an N-fused piperidine to a pyrrolidine ring, the structures of 3a,b reveal notable differences. While 3a shows hydrogen bonding of the carbazole-NH proton H10 to one of the iodide counterions (Fig. 3, left), in 3b such an interaction is absent.30 Apart from the hydrogen bonding, the structural parameters in 3a reflect the ones in 3b30 and previously reported 1,2,3-triazolium salts.20,22 A detailed discussion of the structural parameters is therefore omitted. For more details on the structural metrics please see Tables S1 and S2 in the ESI† (Fig. 3).
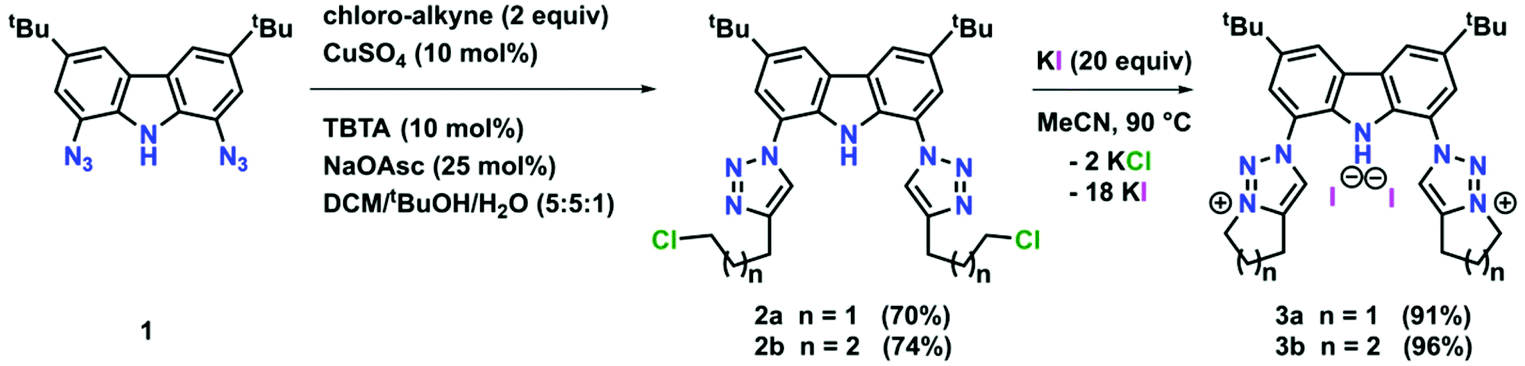 | ||
| Scheme 1 Synthesis of N-fused triazolium salts 3a and 3b from the corresponding chloroalkyl-triazoles 2a and 2b, which can be obtained following standard CuAAC reaction conditions between 1,8-diazido-3,6-di-tert-butylcarbazole 1 and the corresponding chloro-alkynes 5-chloro-1-pentyne or 6-chloro-1-hexyne. | ||
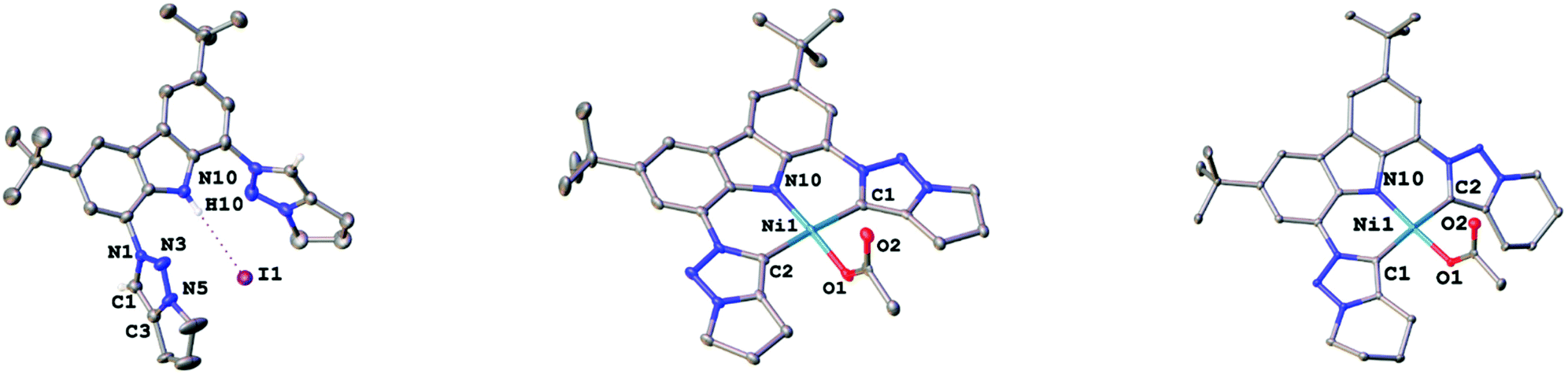 | ||
| Fig. 3 Thermal ellipsoid plots of 3a, 4a and 4b (left to right). Second iodide counteranion (3a), solvent atoms and hydrogen atoms have been omitted for clarity. Thermal ellipsoids are shown at a probability level of 50%. | ||
Although mesoionic carbene complexes of nickel(II) are still scarce and often rely on the use of precursors which are unstable under ambient (air and moisture) conditions, such as nickelocene,33,34 we found that complexation could be achieved using Ni(OAc)2·4H2O in the presence of excess triethylamine at 80 °C (see Scheme 2). Despite the fact that 4a and 4b differ only by one methylene group, their solubility differs drastically. While complex 4b is soluble in a range of coordinating, aromatic and halogenated solvents, 4a does only dissolve to a minimal extent in halogenated or coordinating solvents, but is insoluble in aromatic solvents. Successful formation of the new triazolylidene nickel(II) complexes was supported by several features in the 1H and 13C{1H} NMR spectra of the complexes: (i) the disappearance of the carbazole-NH and triazolium-5H resonances in the 1H NMR spectra of the complexes (Fig. S11 and S16†) as well as the NH absorption band in the IR spectrum (Fig. S56 and S57†), (ii) a high-field shift of the resonance of the methylene protons adjacent to the triazole nitrogen atoms from δ = 4.90 ppm and δ = 4.79 ppm for 3a and 3b to δ = 4.48 ppm and δ = 4.49 ppm for 4a and 4b (compare Fig. S11 and S16†), and (iii) the presence of the characteristic triazolylidene-5C resonance at δ = 152.0 ppm and δ = 146.9 ppm in the 13C{1H} NMR spectra of 4a and 4b (compare Fig. S12 and S17†), which are in a comparable range of previously reported nickel(II) triazolylidene complexes.33,34 Additionally, a signal at δ = 177.7 ppm and δ = 177.1 ppm in the 13C{1H} NMR spectra of 4a and 4b was detected, corresponding to the carbonyl carbon atom of the acetate anion. Ultimate prove for the successful formation of the desired triazolylidene complexes 4a and 4b was given by X-ray diffraction analysis. Single crystals suitable for structure analysis were obtained by layering a concentrated dichloromethane solution of the respective complexes with n-hexane in an NMR tube at room temperature (Fig. 3, middle and right). Both compounds crystallise in the triclinic space group P as either a water solvate (4a) or a dichloromethane solvate (4b). In both complexes the nickel(II) ion is in a slightly distorted square planar coordination environment, with N10–Ni1–O1 and C1–Ni1–C2 angles of 175.44(7)°/176.30(9)° (4a) and 176.11(7)°/173.39(7)° (4b), as well as τ′4 values of 0.06 and 0.07 in 4a and 4b, respectively. The Ni1–N10 distances are 1.854(2) Å (4a) and 1.861(2) Å (4b) and lie in the range of previously reported carbazolide and pyrrolide coordinated nickel(II) ions.14,27 The nickel–carbene carbon atom distances were found to be 1.945(2) Å and 1.947(2) Å in 4a and 1.943(2) Å and 1.931(2) Å in 4b for Ni1–C1 and Ni1–C2, respectively. These distances also compare well to those of previously reported nickel(II) triazolylidene complexes.33,34 The intra ligand distances within the triazolylidene ring are in the expected region and comparable to previously reported triazolylidene complexes.21–26,33,34 Further information regarding the crystal structures and selected bond lengths and angles can be found in the ESI, Tables S1 and S2.†
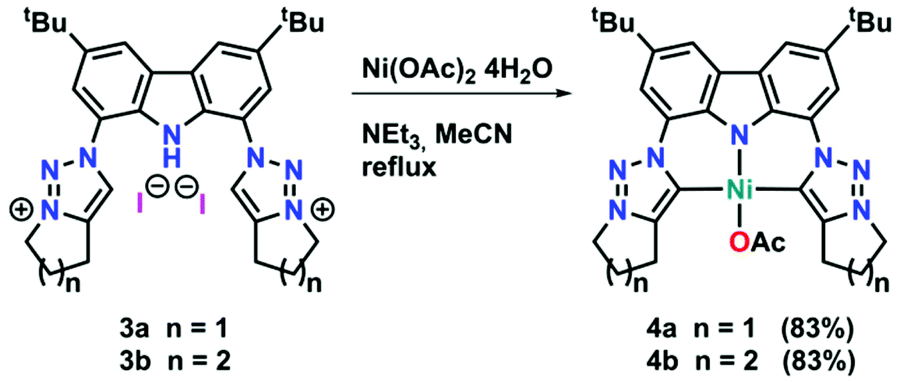 | ||
| Scheme 2 Synthesis of the nickel(II) triazolylidene complexes 4a and 4b. | ||
Having the new complexes in hand, and given the fact, that MIC-derived complexes are known to often surpass their NHC congeners’ activities in catalytic reactions,18 we next turned our focus towards their catalytic potential in the copolymerisation of carbon dioxide and cyclohexene oxide. Lee et al.15 have previously shown that corresponding NHC-derived catalysts (Fig. 1, top right) are moderately active catalysts for the polymerisations of carbon dioxide and cyclohexene oxide to give copolymers. Thus, we applied similar conditions to our catalysts and used cyclohexene oxide (CHO, Fig. 4) as monomer as well. The results for the catalysed coupling reactions of epoxide and carbon dioxide (CO2) are shown in Table 1. It is noteworthy that compared to the report by Lee and co-workers, who were able to apply a carbon dioxide pressure of 500 psi (34.5 bar), our synthetic setup was limited to a pressure of 290 psi (20 bar). Although it has been accepted as a common rule, that higher pressures favour the formation of polycarbonates,35,36 recent literature reports show that nickel(II)-based catalysts can efficiently couple carbon dioxide and epoxides to the desired polycarbonates even at 20 bar.37 In an initial check, wether 4a and 4b could be active (pro)-catalysts in the copolymerisation of CHO and CO2, we applied different catalyst loadings 0.5, 0.25 and 0.1 mol% of 4a or 4b (entries 1–5 and 7), similar to the experiments performed by Lee and co-workers.15 Crude 1H NMR spectra of the reaction mixtures indicated the possible formation of polymers showing broad peaks (Fig. S21†). However, after work-up cyclic carbonate (CC) 5a was isolated as the sole product from these reactions. Notably, when the reaction was performed either without catalysts or without carbon dioxide, small amounts of polyether (PE) could be isolated (vide infra, entries 15–17).
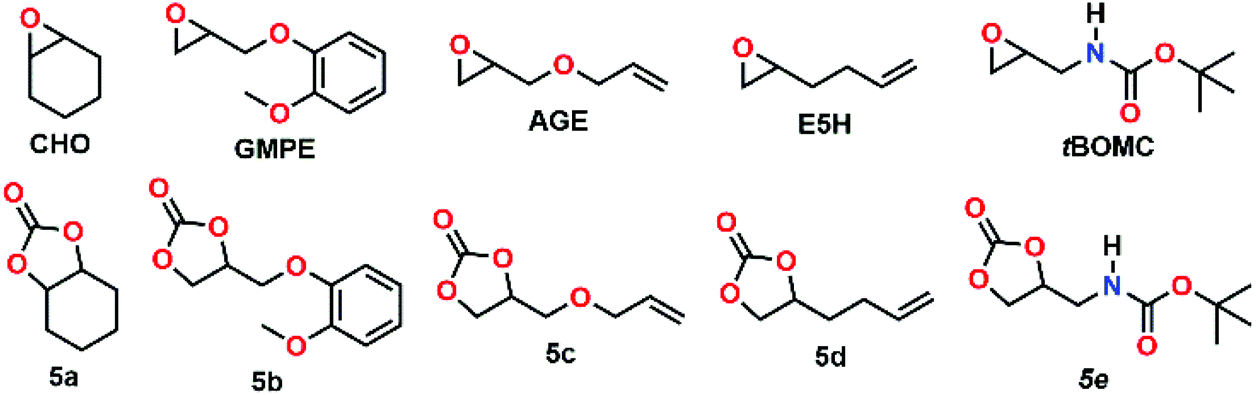 | ||
| Fig. 4 Epoxides used for the coupling reaction with CO2. The corresponding cyclic carbonates 5a–5e are listed underneath. | ||
| Entry | Epoxide | Cat. (mol%) | Co-catalyst | Time [h] | T [°C] | pCO2 [bar] | Epoxide conv.a [%] | Ratio CC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PEa :PEa |
TONb,d | TOFc,d |
|---|---|---|---|---|---|---|---|---|---|---|
| a Determined by 1H NMR. b TON = moles of CC formed by mole of catalyst. c TOF = TON per hour. d Both, TON and TOF were calculated on the basis of the epoxide conversion determined by 1H NMR. e Reaction performed in a 3.06 mmol scale. f Performed in DCM. | ||||||||||
| 1 | CHO | 4a (0.5) | — | 18 | 130 | 20 | 53 | 64:36 |
67 | 4 |
| 2 | CHO | 4b (0.5) | — | 18 | 130 | 20 | 19 | 63:37 |
24 | 1 |
| 3 | CHO | 4a (0.25) | — | 18 | 130 | 20 | 50 | 33:67 |
67 | 4 |
| 4 | CHO | 4b (0.25) | — | 18 | 130 | 20 | 55 | 24:76 |
53 | 3 |
| 5e | CHO | 4a (0.1) | — | 18 | 130 | 20 | 38 | 15:85 |
58 | 3 |
| 6e | CHO | 4a (0.1) | [PPN]Cl | 18 | 130 | 20 | 46 | 43:57 |
195 | 11 |
| 7 | CHO | 4b (0.1) | — | 18 | 130 | 20 | 69 | 8:92 |
55 | 3 |
| 8 | CHO | 4b (0.1) | [PPN]Cl | 18 | 130 | 20 | 70 | 32:68 |
220 | 12 |
| 9 | CHO | 4a (0.1) | — | 120 | 130 | 20 | 73 | 53:47 |
392 | 3 |
| 10 | CHO | 4a (0.1) | [PPN]Cl | 120 | 130 | 20 | >99 | 100:0 |
>999 | 8 |
| 11 | CHO | 4b (0.1) | — | 120 | 130 | 20 | 95 | 78:22 |
739 | 6 |
| 12 | CHO | 4b (0.1) | [PPN]Cl | 120 | 130 | 20 | >99 | 93:7 |
930 | 8 |
| 13 | CHO | 4a (0.1) | [PPN]Cl | 18 | 130 | 2 | 4 | 37:63 |
16 | 1 |
| 14 | CHO | 4b (0.1) | [PPN]Cl | 18 | 130 | 2 | 7 | 27:73 |
19 | 1 |
| 15 | CHO | — | — | 18 | 130 | 20 | 85 | 0:100 |
0 | 0 |
| 16 | CHO | — | [PPN]Cl | 18 | 130 | 20 | 56 | 53:47 |
301 | 17 |
| 17 | CHO | — | — | 18 | 130 | — | 99 | 0:100 |
0 | 0 |
| 18f | GMPE | 4a (0.1) | — | 18 | RT | 20 | 0 | — | 0 | 0 |
| 19f | GMPE | 4a (0.1) | [PPN]Cl | 18 | RT | 20 | 0 | — | 0 | 0 |
| 20f | GMPE | 4a (0.1) | — | 18 | 80 | 20 | 0 | — | 0 | 0 |
| 21f | GMPE | 4a (0.1) | [PPN]Cl | 18 | 80 | 20 | 15 | 100:0 |
153 | 8 |
| 22 | GMPE | 4a (0.1) | — | 18 | 130 | 20 | 41 | 100:0 |
411 | 23 |
| 23 | GMPE | 4a (0.1) | [PPN]Cl | 18 | 130 | 20 | >99 | 100:0 |
>999 | 55 |
| 24 | GMPE | 4b (0.1) | — | 18 | 130 | 20 | >99 | 100:0 |
>999 | 55 |
| 25 | GMPE | 4b (0.1) | [PPN]Cl | 18 | 130 | 20 | >99 | 100:0 |
>999 | 55 |
| 26 | GMPE | — | — | 18 | 130 | 20 | 0 | — | 0 | 0 |
| 27 | GMPE | — | [PPN]Cl | 18 | 130 | 20 | 11 | 100:0 |
110 | 6 |
| 28 | AGE | 4a (0.1) | — | 18 | 130 | 20 | 73 | 100:0 |
733 | 41 |
| 29 | AGE | 4a (0.1) | [PPN]Cl | 18 | 130 | 20 | 98 | 100:0 |
981 | 54 |
| 30 | AGE | 4b (0.1) | — | 18 | 130 | 20 | 96 | 100:0 |
962 | 53 |
| 31 | AGE | 4b (0.1) | [PPN]Cl | 18 | 130 | 20 | 98 | 100:0 |
975 | 54 |
| 32 | E5H | 4a (0.1) | — | 18 | 130 | 20 | 52 | 100:0 |
519 | 29 |
| 33 | E5H | 4a (0.1) | [PPN]Cl | 18 | 130 | 20 | 77 | 100:0 |
770 | 43 |
| 34 | E5H | 4b (0.1) | — | 18 | 130 | 20 | 33 | 100:0 |
331 | 18 |
| 35 | E5H | 4b (0.1) | [PPN]Cl | 18 | 130 | 20 | 84 | 100:0 |
839 | 47 |
| 36 | tBOMC | 4a (0.1) | — | 18 | 130 | 20 | >99 | 48:52 |
482 | 27 |
| 37 | tBOMC | 4a (0.1) | [PPN]Cl | 18 | 130 | 20 | >99 | 82:18 |
820 | 46 |
| 38 | tBOMC | 4b (0.1) | — | 18 | 130 | 20 | >99 | 64:36 |
638 | 35 |
| 39 | tBOMC | 4b (0.1) | [PPN]Cl | 18 | 130 | 20 | >99 | 81:19 |
808 | 45 |
While the TONs to CC seem to be barely affected by the catalyst loading for 4a (entries 1, 3 and 5), the formation of PE by-products increased with lower catalyst loadings. Contrary, for catalyst 4b (entries 2, 4 and 7), TONs as well as the formation of PE by-product increased concurrently with lower catalyst loadings. Applying the established cocatalyst bis(triphenyl-phosphine) iminium chloride ([PPN]Cl) (entries 6 and 8) resulted in higher conversion of the monomer and TONs/TOFs up to 220/12 h−1. Since no significant polycarbonate formation could be detected for all of these conditions, we proceeded with the lowest catalyst loading (0.1 mol%) to improve the conditions leading to cyclic carbonates. Increasing the reaction time from 18 h to 120 h (entries 9–12) led to increasing amounts of cyclic carbonate 5a, while polyether formation was suppressed. Similarly, the addition of [PPN]Cl shifted the selectivity of the reaction towards the formation of cyclic carbonates. Furthermore, complex 4a seems to have a lower activity at this catalyst loading compared to 4b, which is most likely related to the lower solubility of 4a. Reducing the carbon dioxide pressure to 2 bars (entries 13 and 14) resulted in a drastic drop of the conversions and a preferred formation of polyether. Notably, Mayilmurugan et al. have recently reported a nickel(II) complex, which is capable of performing efficient CC coupling at 1 bar and 100 °C.38 No catalyst (entry 15) for CHO gave solely PE, only co-catalyst resulted in lower TOFs and PE by-product of 47% (entry 16). Heating of the monomer led to pure PE (entry 17).
Next we turned our interest towards the reactivity of other, functionalised epoxides, especially glycidyl ethers (see Fig. 4),39 in order to probe if the trends for cyclohexene oxide can be reproduced for other epoxides. Treating 3-(o-methoxyphenoxy)-1,2-epoxypropane (GMPE) (entries 18–27) under the same conditions as applied for our initial cyclohexene oxide studies (130 °C, 20 bar of CO2 and 18 h), the overall conversions for GMPE were found to be higher (entries 22–25). The TOF for complex 4b without co-catalyst was >55 h−1 and the TOF for 4a only 23 h−1. Therefore, a higher reactivity of 4b for the reaction of GMPE to 5b can be assumed, which might as well be related to the higher solubility of 4b. With increasing temperature, the formation of CC is entropically favoured over PC formation. Therefore, experiments with GMPE at RT (entries 18 and 19) and at 80 °C (entries 20 and 21) were performed. While at RT no reaction was observed, at 80 °C the use of complex 4b gave CC with a moderate TOF of 8 h−1. In all cases, the addition of [PPN]Cl resulted in a drastic enhancement of the catalytic activity of the systems. Using no catalyst and only co-catalyst for GMPE resulted in no conversion. Using no catalyst and only [PPN]Cl gave low TOFs of 6 h−1 (entries 26 and 27). The same trends were also observed for other glycidyl ethers such as allyl glycidyl ether (AGE, entries 28–31), and 1,2-epoxyhex-5-ene (E5H, entries 32–35).
Finally, we examined the influence of a protic substrate, for which purpose we synthesized tert-butyl(oxiran-2-ylmethyl)carbamate (tBOMC). Similar to the glycidyl ethers, full conversion of the starting material was achieved for both catalysts 4a and 4b. However, for tBOMC the carbonate selectivity of the complexes dropped and the PE by-product was formed in significant amounts. Nevertheless, PE formation can be decreased by addition of [PPN]Cl as co-catalyst (entries 36–39).
Conclusion
We extended the route to 1,8-bis(1,2,3-triazolylidene)-carbazolide ligands by applying a straight-forward intramolecular cyclisation reaction, avoiding hazardous and explosive tert-butylhypochlorite. Furthermore, following a simple deprotonation protocol using only triethylamine as a base, we have been able to synthesise new nickel(II) complexes 4a,b.In contrast to their imidazolylidene Ni(II) congeners, which previously have been found to be good polymerisation catalysts,15 the 1,2,3-triazolylidene complexes 4a,b presented in this work turned out to be moderate catalysts for the cyclisation of carbon dioxide and epoxides to cyclic carbonates. However, it should be pointed out again, that the selectivity of cyclisation vs. polymerisation is strongly balanced by electronic and steric effects of the catalysts, as well as the applied pressure of CO2, and even subtle changes in catalyst design can have a large impact.36 Nevertheless, these results represent a rare case, in which the replacement of normal by mesoionic carbenes in a catalyst has not led to an increase of its catalytic potential, but to an inversion of selectivity. We are currently investigating the cause of this selectivity switch and further applications of the new carbazole-derived MIC ligands in early transition metal coordination chemistry and photochemistry.
Experimental section
General remarks
If not otherwise mentioned, all transformations involving nickel precursors were carried out under inert conditions using the Schlenk technique or an argon-filled glovebox. Organic syntheses were carried out under ambient conditions without taking precautions to exclude moisture or air. Solvents were dried by a MBraun SPS system and stored over activated molecular sieves (3 Å) for at least 24 h. The deuterated solvents CDCl3 and CD2Cl2 were used as received without any prior drying. IR spectra were recorded at room temperature under inert conditions using a Bruker Vertex 70 with ATR equipment. NMR spectra were collected at 298 K on a Bruker AV-500 or an Ascent 700 spectrometer using regular NMR tubes. All chemical shifts (δ) are reported in ppm. 1H and 13C chemical shifts were calibrated to residual solvent peaks. 15N chemical shifts were calibrated to liquid ammonia (NH3). Elemental analyses were performed using an Elementar vario microcube instrument at the University of Paderborn. 1,8-Diazido-3,6-di-tert-butylcarbazole 131 and triazolium salt 3b30 were prepared following literature known procedures. All reagents were used as received without further purification.Synthetic procedures
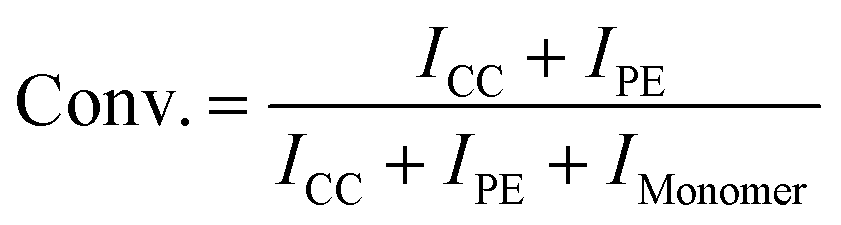
In order to isolate the cyclic carbonate, the solvent was removed under reduced pressure and purified by column chromatography (silica, CH/EE).
:1, Rf = 0.58) to obtain a colorless solid with 85% yield. (44.7 mmol, 7.03 g) 1H NMR (CDCl3, 303 K, 500 MHz, in ppm): 5.84 (m, 1H, Allyl-CH), 5.18 (m, 1H, Allyl-CH2), 5.11 (m, 1H, Allyl-CH2), 4.75 (br., 1H, NH), 3.74 (s, 2H, NCH2), 1.45 (s, 9H, C(CH3)3); 13C{1H} NMR (CDCl3, 303 K, 175 MHz, in ppm): 155.9 (Cq), 135.1 (Allyl-CH), 115.8 (Allyl-CH2), 79.5 (C(CH3)3), 43.2 (NCH2), 28.5 (C(CH3)3); Hi-Res mass (ESI+) calcd. for [C8H15O2N + Na+] 180.1000 found: 180.1000.
:1, Rf = 0.33) to yield in 81% of a slightly yellow oil. (51.5 mmol, 8.92 g) 1H NMR (CDCl3, 303 K, 500 MHz, in ppm): 4.81 (br., 1H, NH), 3.47 (br, 1H, N-CH2), 3.18 (m, 1H, Epoxide-CH), 3.05 (br., 1H, NCH2), 2.75 (dd, 2JHH = 4.7 Hz, 3JHH,trans = 4.2 Hz, 1H, Epoxide-CH2), 2.57 (dd, 2JHH = 4.8 Hz, 3JHH,cis = 2.7 Hz, 1H, Allyl-CH2), 1.42 (s, 9H, C(CH3)3). 13C{1H} NMR (CDCl3, 303 K, 175 MHz, in ppm): 155.9 (Cq), 79.6 (C(CH3)3), 50.9 (Epoxide-CH), 45.1 (NCH2), 41.8 (Epoxide-CH2), 28.5 (C(CH3)3); Hi-Res mass (ESI+) calcd. for [C8H15O2N + Na+] 196.0944 found: 196.0950.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Daimler and Benz Foundation, to the Fonds der Chemischen Industrie, to the Young Academy of the North Rhine-Westphalian Academy of Sciences, Humanities, and the Arts, the University of Paderborn and the University of Innsbruck for funding of this work. Prof. Dr Matthias Bauer and Prof. Dr Thomas Kühne are acknowledged for helpful discussions.Notes and references
- E. Peris and R. H. Crabtree, Chem. Soc. Rev., 2018, 47, 1959–1968 RSC.
- M. Fritz and S. Schneider, in Catalytic, materials, biological and medical applications, ed. A. Bhagi-Damodaran, Springer International Publishing, Cham, 2019, vol. 182, pp. 1–36 Search PubMed.
- (a) S. Schneider, J. Meiners and B. Askevold, Eur. J. Inorg. Chem., 2012, 2012, 412–429 CrossRef CAS; (b) L. Alig, M. Fritz and S. Schneider, Chem. Rev., 2019, 119, 2681–2751 CrossRef CAS PubMed; (c) M. R. Mills, C. L. Barnes and W. H. Bernskoetter, Inorg. Chem., 2018, 57, 1590–1597 CrossRef CAS PubMed.
- (a) D. Delony, M. Kinauer, M. Diefenbach, S. Demeshko, C. Würtele, M. C. Holthausen and S. Schneider, Angew. Chem., Int. Ed., 2019, 58, 10971–10974 CrossRef CAS PubMed; (b) J. Abbenseth and S. Schneider, Z. Anorg. Allg. Chem., 2020, 55, 1690 Search PubMed; (c) M. Kinauer, M. Diefenbach, H. Bamberger, S. Demeshko, E. J. Reijerse, C. Volkmann, C. Würtele, J. van Slageren, B. de Bruin, M. C. Holthausen and S. Schneider, Chem. Sci., 2018, 9, 4325–4332 RSC; (d) F. Schendzielorz, M. Finger, J. Abbenseth, C. Würtele, V. Krewald and S. Schneider, Angew. Chem., Int. Ed., 2019, 58, 830–834 CrossRef CAS PubMed; (e) F. Schneck, M. Finger, M. Tromp and S. Schneider, Chem. – Eur. J., 2017, 23, 33–37 CrossRef CAS PubMed; (f) F. Schneck, F. Schendzielorz, N. Hatami, M. Finger, C. Würtele and S. Schneider, Angew. Chem., Int. Ed., 2018, 57, 14482–14487 CrossRef CAS PubMed; (g) G. A. Silantyev, M. Förster, B. Schluschaß, J. Abbenseth, C. Würtele, C. Volkmann, M. C. Holthausen and S. Schneider, Angew. Chem., Int. Ed., 2017, 56, 5872–5876 CrossRef CAS PubMed; (h) R. S. van Alten, F. Wätjen, S. Demeshko, A. J. M. Miller, C. Würtele, I. Siewert and S. Schneider, Eur. J. Inorg. Chem., 2020, 1, 490 Search PubMed; (i) T. Kurogi, B. Pinter and D. J. Mindiola, Organometallics, 2018, 37, 3385–3388 CrossRef CAS; (j) U. J. Kilgore, X. Yang, J. Tomaszewski, J. C. Huffman and D. J. Mindiola, Inorg. Chem., 2006, 45, 10712–10721 CrossRef CAS PubMed; (k) U. J. Kilgore, J. Tomaszewski, H. Fan, J. C. Huffman and D. J. Mindiola, Organometallics, 2007, 26, 6132–6138 CrossRef CAS; (l) U. J. Kilgore, C. A. Sengelaub, H. Fan, J. Tomaszewski, J. A. Karty, M.-H. Baik and D. J. Mindiola, Organometallics, 2009, 28, 843–852 CrossRef CAS; (m) B. C. Bailey, J. C. Huffman, D. J. Mindiola, W. Weng and O. V. Ozerov, Organometallics, 2005, 24, 1390–1393 CrossRef CAS; (n) D. Adhikari, S. Mossin, F. Basuli, B. R. Dible, M. Chipara, H. Fan, J. C. Huffman, K. Meyer and D. J. Mindiola, Inorg. Chem., 2008, 47, 10479–10490 CrossRef CAS PubMed; (o) T. Kurogi, M. E. Miehlich, D. Halter and D. J. Mindiola, Organometallics, 2018, 37, 165–167 CrossRef CAS; (p) T. Kurogi, M. Kamitani, B. C. Manor, P. J. Carroll and D. J. Mindiola, Organometallics, 2017, 36, 74–79 CrossRef CAS; (q) T. Kurogi, P. J. Carroll and D. J. Mindiola, J. Am. Chem. Soc., 2016, 138, 4306–4309 CrossRef CAS PubMed; (r) M. Kamitani, K. Searles, C.-H. Chen, P. J. Carroll and D. J. Mindiola, Organometallics, 2015, 34, 2558–2566 CrossRef CAS; (s) A. R. Fout, B. C. Bailey, D. M. Buck, H. Fan, J. C. Huffman, M.-H. Baik and D. J. Mindiola, Organometallics, 2010, 29, 5409–5422 CrossRef CAS; (t) S. Mossin, B. L. Tran, D. Adhikari, M. Pink, F. W. Heinemann, J. Sutter, R. K. Szilagyi, K. Meyer and D. J. Mindiola, J. Am. Chem. Soc., 2012, 134, 13651–13661 CrossRef CAS PubMed; (u) D. Adhikari, M. Pink and D. J. Mindiola, Organometallics, 2009, 28, 2072–2077 CrossRef CAS; (v) S.-Y. Baek, T. Kurogi, D. Kang, M. Kamitani, S. Kwon, D. P. Solowey, C.-H. Chen, M. Pink, P. J. Carroll, D. J. Mindiola and M.-H. Baik, J. Am. Chem. Soc., 2017, 139, 12804–12814 CrossRef CAS PubMed; (w) M. Schlagbauer, F. Kallmeier, T. Irrgang and R. Kempe, Angew. Chem., Int. Ed., 2020, 59, 1485–1490 CrossRef CAS PubMed.
- (a) D. Benito-Garagorri and K. Kirchner, Acc. Chem. Res., 2008, 41, 201–213 CrossRef CAS PubMed; (b) W. Eder, B. Stöger and K. Kirchner, Chem. Mon., 2019, 150, 1235–1240 CrossRef CAS; (c) J. He, N. W. Waggoner, S. G. Dunning, A. Steiner, V. M. Lynch and S. M. Humphrey, Angew. Chem., Int. Ed., 2016, 55, 12351–12355 CrossRef CAS PubMed; (d) D. Himmelbauer, M. Mastalir, B. Stöger, L. F. Veiros, M. Pignitter, V. Somoza and K. Kirchner, Inorg. Chem., 2018, 57, 7925–7931 CrossRef CAS PubMed; (e) G. van Koten, in Organometallic Pincer Chemistry, ed. G. van Koten and D. Milstein, Springer, Berlin, Heidelberg, 2013, vol. 40, pp. 1–20 Search PubMed; (f) Organometallic Pincer Chemistry, ed. G. van Koten and D. Milstein, Springer, Berlin, Heidelberg, 2013, vol. 40 Search PubMed; (g) S. K. Gibbons, Z. Xu, R. P. Hughes, D. S. Glueck and A. L. Rheingold, Organometallics, 2018, 37, 2159–2166 CrossRef CAS.
- G. M. Adams and A. S. Weller, Coord. Chem. Rev., 2018, 355, 150–172 CrossRef CAS.
- (a) C. Romain, D. Specklin, K. Miqueu, J.-M. Sotiropoulos, C. Fliedel, S. Bellemin-Laponnaz and S. Dagorne, Organometallics, 2015, 34, 4854–4863 CrossRef CAS; (b) C. Romain, B. Heinrich, S. B. Laponnaz and S. Dagorne, Chem. Commun., 2012, 48, 2213–2215 RSC; (c) C. Romain, S. Choua, J.-P. Collin, M. Heinrich, C. Bailly, L. Karmazin-Brelot, S. Bellemin-Laponnaz and S. Dagorne, Inorg. Chem., 2014, 53, 7371–7376 CrossRef CAS PubMed; (d) U. J. Kilgore, X. Yang, J. Tomaszewski, J. C. Huffman and D. J. Mindiola, Inorg. Chem., 2006, 45, 10712–10721 CrossRef CAS PubMed; (e) W. Kai, H. Qian, D. Liu and Z. Ye, J. Saudi Chem. Soc., 2019, 23, 274–283 CrossRef CAS; (f) C. F. Harris, C. S. Kuehner, J. Bacsa and J. D. Soper, Angew. Chem., Int. Ed., 2018, 57, 1311–1315 CrossRef CAS PubMed; (g) C. F. Harris, M. B. Bayless, N. P. van Leest, Q. J. Bruch, B. N. Livesay, J. Bacsa, K. I. Hardcastle, M. P. Shores, B. de Bruin and J. D. Soper, Inorg. Chem., 2017, 56, 12421–12435 CrossRef CAS PubMed; (h) L. Gravogl, F. W. Heinemann, D. Munz and K. Meyer, Inorg. Chem., 2020, 59(8), 5632–5645 CrossRef CAS PubMed; (i) C. Gandara, C. Philouze, O. Jarjayes and F. Thomas, Inorg. Chim. Acta, 2018, 482, 561–566 CrossRef CAS; (j) E. Despagnet-Ayoub, M. K. Takase, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 2015, 137, 10500–10503 CrossRef CAS PubMed; (k) J. D. Carter and Y. Schrodi, Organometallics, 2020, 39, 378–382 CrossRef CAS; (l) E. Borré, G. Dahm, A. Aliprandi, M. Mauro, S. Dagorne and S. Bellemin-Laponnaz, Organometallics, 2014, 33, 4374–4384 CrossRef; (m) M. Baltrun, F. A. Watt, R. Schoch, C. Wölper, A. G. Neuba and S. Hohloch, Dalton Trans., 2019, 48, 14611–14625 RSC; (n) M. Baltrun, F. A. Watt, R. Schoch and S. Hohloch, Organometallics, 2019, 38, 3719–3729 CrossRef CAS; (o) S. Liu, J. I. Amaro-Estrada, M. Baltrun, I. Douair, R. Schoch, L. Maron and S. Hohloch, Organometallics, 2021, 40, 107–118 CrossRef CAS; (p) F. R. Neururer, S. Liu, D. Leitner, M. Baltrun, K. R. Fisher, H. Kopacka, K. Wurst, L. J. Daumann, D. Munz and S. Hohloch, Inorg. Chem., 2021, 60, 15421–15434 CrossRef CAS PubMed.
- A. L. Speelman, K. L. Skubi, B. Q. Mercado and P. L. Holland, Inorg. Chem., 2021, 60, 1965–1974 CrossRef CAS PubMed.
- (a) J. M. Darmon, R. P. Yu, S. P. Semproni, Z. R. Turner, S. C. E. Stieber, S. DeBeer and P. J. Chirik, Organometallics, 2014, 33, 5423–5433 CrossRef CAS PubMed; (b) L. A. Fredin, M. Pápai, E. Rozsályi, G. Vankó, K. Wärnmark, V. Sundström and P. Persson, J. Phys. Chem. Lett., 2014, 5, 2066–2071 CrossRef CAS PubMed; (c) Y. Liu, T. Harlang, S. E. Canton, P. Chábera, K. Suárez-Alcántara, A. Fleckhaus, D. A. Vithanage, E. Göransson, A. Corani, R. Lomoth, V. Sundström and K. Wärnmark, Chem. Commun., 2013, 49, 6412–6414 RSC; (d) K. Magra, E. Domenichini, A. Francés-Monerris, C. Cebrián, M. Beley, M. Darari, M. Pastore, A. Monari, X. Assfeld, S. Haacke and P. C. Gros, Inorg. Chem., 2019, 58, 5069–5081 CrossRef CAS PubMed; (e) L. Maron and D. Bourissou, Organometallics, 2009, 28, 3686–3690 CrossRef CAS; (f) T. H. T. Myren, A. M. Lilio, C. G. Huntzinger, J. W. Horstman, T. A. Stinson, T. B. Donadt, C. Moore, B. Lama, H. H. Funke and O. R. Luca, Organometallics, 2019, 38, 1248–1253 CrossRef CAS; (g) D. Pugh, J. A. Wright, S. Freeman and A. A. Danopoulos, Dalton Trans., 2006, 775–782 RSC; (h) S. M. Rummelt, J. M. Darmon, R. P. Yu, P. Viereck, T. P. Pabst, Z. R. Turner, G. W. Margulieux, S. Gu and P. J. Chirik, Organometallics, 2019, 38, 3159–3168 CrossRef CAS PubMed; (i) M. Sheng, N. Jiang, S. Gustafson, B. You, D. H. Ess and Y. Sun, Dalton Trans., 2015, 44, 16247–16250 RSC; (j) B. Yang and D. G. Truhlar, Organometallics, 2018, 37, 3917–3927 CrossRef CAS; (k) R. P. Yu, J. M. Darmon, J. M. Hoyt, G. W. Margulieux, Z. R. Turner and P. J. Chirik, ACS Catal., 2012, 2, 1760–1764 CrossRef CAS PubMed; (l) R. P. Yu, J. M. Darmon, C. Milsmann, G. W. Margulieux, S. C. E. Stieber, S. DeBeer and P. J. Chirik, J. Am. Chem. Soc., 2013, 135, 13168–13184 CrossRef CAS PubMed; (m) P. Zimmer, L. Burkhardt, A. Friedrich, J. Steube, A. Neuba, R. Schepper, P. Müller, U. Flörke, M. Huber, S. Lochbrunner and M. Bauer, Inorg. Chem., 2018, 57, 360–373 CrossRef CAS PubMed; (n) P. Zimmer, L. Burkhardt, R. Schepper, K. Zheng, D. Gosztola, A. Neuba, U. Flörke, C. Wölper, R. Schoch, W. Gawelda, S. E. Canton and M. Bauer, Eur. J. Inorg. Chem., 2018, 2018, 5203–5214 CrossRef CAS; (o) L. C. Tolley, I. Strydom, W. J. Louw, M. A. Fernandes, D. I. Bezuidenhout and G. Guisado-Barrios, ACS Omega, 2019, 4, 6360–6374 CrossRef CAS; (p) P. Dierks, A. Kruse, O. S. Bokareva, M. J. Al-Marri, J. Kalmbach, M. Baltrun, A. Neuba, R. Schoch, S. Hohloch, K. Heinze, M. Seitz, O. Kühn, S. Lochbrunner and M. Bauer, Chem. Commun., 2021, 57, 6640–6643 RSC.
- (a) M. Bauer, J. Steube, A. Päpcke, O. Bokareva, T. Reuter, S. Demeshko, R. Schoch, S. Hohloch, F. Meyer, K. Heinze, O. Kühn and S. Lochbrunner, Res. Square, ver. 1., 2020 DOI:10.21203/rs.3.rs-64316/v1; (b) A. Sattler and G. Parkin, J. Am. Chem. Soc., 2012, 134, 2355–2366 CrossRef CAS PubMed.
- (a) N. Grüger, L.-I. Rodríguez, H. Wadepohl and L. H. Gade, Inorg. Chem., 2013, 52, 2050–2059 CrossRef PubMed; (b) L. Gade, H. Wadepohl and J. Ott, Angew. Chem., Int. Ed., 2020, 59, 9448–9452 CrossRef PubMed; (c) K. R. D. Johnson and P. G. Hayes, Organometallics, 2009, 28, 6352–6361 CrossRef CAS; (d) J. Higuchi, S. Kuriyama, A. Eizawa, K. Arashiba, K. Nakajima and Y. Nishibayashi, Dalton Trans., 2018, 47, 1117–1121 RSC; (e) L. S. Merz, C. K. Blasius, H. Wadepohl and L. H. Gade, Inorg. Chem., 2019, 58, 6102–6113 CrossRef CAS PubMed; (f) J. C. Ott, C. K. Blasius, H. Wadepohl and L. H. Gade, Inorg. Chem., 2018, 57, 3183–3191 CrossRef CAS PubMed; (g) J. C. Ott, H. Wadepohl, M. Enders and L. H. Gade, J. Am. Chem. Soc., 2018, 140, 17413–17417 CrossRef CAS PubMed.
- (a) B. Wucher, M. Moser, S. A. Schumacher, F. Rominger and D. Kunz, Angew. Chem., 2009, 121, 4481–4485 CrossRef; (b) M. Moser, B. Wucher, D. Kunz and F. Rominger, Organometallics, 2007, 26, 1024–1030 CrossRef CAS; (c) E. Jürgens, K. N. Buys, A.-T. Schmidt, S. K. Furfari, M. L. Cole, M. Moser, F. Rominger and D. Kunz, New J. Chem., 2016, 40, 9160–9169 RSC; (d) T. Maulbetsch and D. Kunz, Angew. Chem., Int. Ed., 2021, 60, 2007–2012 CrossRef CAS PubMed.
- (a) E. Jürgens, O. Back, J. J. Mayer, K. Heinze and D. Kunz, Z. Naturforsch., B: J. Chem. Sci., 2016, 71, 1011–1018 CrossRef; (b) E. Jürgens, B. Wucher, F. Rominger, K. W. Törnroos and D. Kunz, Chem. Commun., 2015, 51, 1897–1900 RSC; (c) A. Seyboldt, B. Wucher, M. Alles, F. Rominger, C. Maichle-Mössmer and D. Kunz, J. Organomet. Chem., 2015, 775, 202–208 CrossRef CAS; (d) Y. Tian, E. Jürgens and D. Kunz, Chem. Commun., 2018, 54, 11340–11343 RSC; (e) Y. Tian, E. Jürgens, K. Mill, R. Jordan, T. Maulbetsch and D. Kunz, ChemCatChem, 2019, 11, 4028–4035 CrossRef CAS; (f) Y. Tian, T. Maulbetsch, R. Jordan, K. W. Törnroos and D. Kunz, Organometallics, 2020, 39, 1221–1229 CrossRef CAS.
- A. Seyboldt, B. Wucher, S. Hohnstein, K. Eichele, F. Rominger, K. W. Törnroos and D. Kunz, Organometallics, 2015, 34, 2717–2725 CrossRef CAS.
- T.-Y. Lee, Y.-J. Lin, Y.-Z. Chang, L.-S. Huang, B.-T. Ko and J.-H. Huang, Organometallics, 2017, 36, 291–297 CrossRef CAS.
- (a) J. A. Garden, A. J. P. White and C. K. Williams, Dalton Trans., 2017, 46, 2532–2541 RSC; (b) J. Hessevik, R. Lalrempuia, H. Nsiri, K. W. Törnroos, V. R. Jensen and E. Le Roux, Dalton Trans., 2016, 45, 14734–14744 RSC; (c) K. Nakano, K. Kobayashi and K. Nozaki, J. Am. Chem. Soc., 2011, 133, 10720–10723 CrossRef CAS PubMed; (d) T. Ohkawara, K. Suzuki, K. Nakano, S. Mori and K. Nozaki, J. Am. Chem. Soc., 2014, 136, 10728–10735 CrossRef CAS PubMed; (e) C. C. Quadri, R. Lalrempuia, J. Hessevik, K. W. Törnroos and E. Le Roux, Organometallics, 2017, 36, 4477–4489 CrossRef CAS; (f) C. C. Quadri and E. Le Roux, Dalton Trans., 2014, 43, 4242–4246 RSC.
- (a) J. M. Aizpurua, M. Sagartzazu-Aizpurua and Z. Monasterio, in Chemistry of 1,2,3-triazoles, ed. W. Dehaen and B. Abarca, Springer, Cham, 2015, vol. 40, pp. 211–267 Search PubMed; (b) Chemistry of 1,2,3-triazoles, ed. W. Dehaen and B. Abarca, Springer, Cham, 2015, vol. 40 Search PubMed; (c) S. C. Sau, P. K. Hota, S. K. Mandal, M. Soleilhavoup and G. Bertrand, Chem. Soc. Rev., 2020, 49, 1233–1252 RSC; (d) D. Schweinfurth, L. Hettmanczyk, L. Suntrup and B. Sarkar, Z. Anorg. Allg. Chem., 2017, 643, 554–584 CrossRef CAS; (e) G. Guisado-Barrios, M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2018, 51, 3236–3244 CrossRef CAS PubMed; (f) A. J. Mantanona, D. R. Tolentino, K. S. Cay, M. Gembicky, R. Jazzar, G. Bertrand and J. D. Rinehart, Dalton Trans., 2020, 49, 2426–2430 RSC; (g) Á. Vivancos, C. Segarra and M. Albrecht, Chem. Rev., 2018, 118, 9493–9586 CrossRef PubMed; (h) S. Hohloch, D. Scheiffele and B. Sarkar, Eur. J. Inorg. Chem., 2013, 2013, 3956–3965 CrossRef CAS; (i) S. Hohloch, C.-Y. Su and B. Sarkar, Eur. J. Inorg. Chem., 2011, 2011, 3067–3075 CrossRef CAS; (j) D. Munz, Organometallics, 2018, 37, 275–289 CrossRef CAS.
- S. Hohloch, B. Sarkar, L. Nauton, F. Cisnetti and A. Gautier, Tetrahedron Lett., 2013, 54, 1808–1812 CrossRef CAS.
- (a) L. Suntrup, S. Klenk, J. Klein, S. Sobottka and B. Sarkar, Inorg. Chem., 2017, 56, 5771–5783 CrossRef CAS PubMed; (b) S. Klenk, L. Suntrup and B. Sarkar, Nachr. Chem., 2018, 66, 717–721 CrossRef CAS; (c) S. Klenk, S. Rupf, L. Suntrup, M. van der Meer and B. Sarkar, Organometallics, 2017, 36, 2026–2035 CrossRef CAS; (d) S. Hohloch, L. Suntrup and B. Sarkar, Inorg. Chem. Front., 2016, 3, 67–77 RSC; (e) S. Hohloch, L. Hettmanczyk and B. Sarkar, Eur. J. Inorg. Chem., 2014, 2014, 3164–3171 CrossRef CAS; (f) L. Hettmanczyk, S. J. P. Spall, S. Klenk, M. van der Meer, S. Hohloch, J. A. Weinstein and B. Sarkar, Eur. J. Inorg. Chem., 2017, 2017, 2112–2121 CrossRef CAS; (g) L. Hettmanczyk, D. Schulze, L. Suntrup and B. Sarkar, Organometallics, 2016, 35, 3828–3836 CrossRef CAS; (h) L. Hettmanczyk, S. Manck, C. Hoyer, S. Hohloch and B. Sarkar, Chem. Commun., 2015, 51, 10949–10952 RSC; (i) L. Suntrup, F. Stein, G. Hermann, M. Kleoff, M. Kuss-Petermann, J. Klein, O. S. Wenger, J. C. Tremblay and B. Sarkar, Inorg. Chem., 2018, 57, 13973–13984 CrossRef CAS PubMed; (j) M. van der Meer, E. Glais, I. Siewert and B. Sarkar, Angew. Chem., Int. Ed., 2015, 54, 13792–13795 CrossRef CAS PubMed; (k) M. Rigo, L. Hettmanczyk, F. J. L. Heutz, S. Hohloch, M. Lutz, B. Sarkar and C. Müller, Dalton Trans., 2016, 46, 86–95 RSC.
- L. Suntrup, S. Hohloch and B. Sarkar, Chem. – Eur. J., 2016, 22, 18009–18018 CrossRef CAS PubMed.
- S. Hohloch, L. Suntrup and B. Sarkar, Organometallics, 2013, 32, 7376–7385 CrossRef CAS.
- S. Hohloch, S. Kaiser, F. L. Duecker, A. Bolje, R. Maity, J. Košmrlj and B. Sarkar, Dalton Trans., 2015, 44, 686–693 RSC.
- L. Hettmanczyk, L. Suntrup, S. Klenk, C. Hoyer and B. Sarkar, Chem. – Eur. J., 2017, 23, 576–585 CrossRef CAS PubMed.
- A. Bolje, S. Hohloch, D. Urankar, A. Pevec, M. Gazvoda, B. Sarkar and J. Košmrlj, Organometallics, 2014, 33, 2588–2598 CrossRef CAS.
- A. Bolje, S. Hohloch, J. Košmrlj and B. Sarkar, Dalton Trans., 2016, 45, 15983–15993 RSC.
- J. Beerhues, K. Fauché, F. Cisnetti, B. Sarkar and A. Gautier, Dalton Trans., 2019, 48, 8931–8936 RSC.
- D. I. Bezuidenhout, G. Kleinhans, G. Guisado-Barrios, D. C. Liles, G. Ung and G. Bertrand, Chem. Commun., 2014, 50, 2431–2433 RSC.
- G. Kleinhans, G. Guisado-Barrios, D. C. Liles, G. Bertrand and D. I. Bezuidenhout, Chem. Commun., 2016, 52, 3504–3507 RSC.
- G. Kleinhans, M. M. Hansmann, G. Guisado-Barrios, D. C. Liles, G. Bertrand and D. I. Bezuidenhout, J. Am. Chem. Soc., 2016, 138, 15873–15876 CrossRef CAS PubMed.
- P. Pinter, C. M. Schüßlbauer, F. A. Watt, N. Dickmann, R. Herbst-Irmer, B. Morgenstern, A. Grünwald, T. Ullrich, M. Zimmer, S. Hohloch, D. M. Guldi and D. Munz, Chem. Sci., 2021, 12, 7401–7410 RSC.
- I. Pryjomska-Ray, D. Zornik, M. Pätzel, K. B. Krause, L. Grubert, B. Braun-Cula, S. Hecht and C. Limberg, Chem. – Eur. J., 2018, 24, 5341–5349 CrossRef CAS PubMed.
- (a) L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed; (c) O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS; (d) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed; (e) A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed; (f) C. M. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
- Y. Wei, S.-X. Liu, H. Mueller-Bunz and M. Albrecht, ACS Catal., 2016, 6, 8192–8200 CrossRef CAS.
- Y. Wei, A. Petronilho, H. Mueller-Bunz and M. Albrecht, Organometallics, 2014, 33, 5834–5844 CrossRef CAS.
- G. W. Coates and D. R. Moore, Angew. Chem., Int. Ed., 2004, 43, 6618–6639 CrossRef CAS PubMed.
- A. J. Kamphuis, F. Picchioni and P. P. Pescarmona, Green Chem., 2019, 21, 406–448 RSC.
- Y.-C. Su and B.-T. Ko, Inorg. Chem., 2021, 60, 852–865 CrossRef CAS PubMed.
- (a) S. Muthuramalingam, M. Velusamy and R. Mayilmurugan, Dalton Trans., 2021, 50, 7984–7994 RSC; (b) S. Muthuramalingam, M. Sankaralingam, M. Velusamy and R. Mayilmurugan, Inorg. Chem., 2019, 58, 12975–12985 CrossRef CAS PubMed.
- (a) C.-S. Tan and T.-W. Kuo, J. Appl. Polym. Sci., 2005, 98, 750–757 CrossRef CAS; (b) C.-S. Tan, C.-C. Juan and T.-W. Kuo, Polymer, 2004, 45, 1805–1814 CrossRef CAS; (c) J. Hilf, M. Scharfenberg, J. Poon, C. Moers and H. Frey, Macromol. Rapid Commun., 2015, 36, 174–179 CrossRef CAS PubMed; (d) H. Zhang and M. W. Grinstaff, J. Am. Chem. Soc., 2013, 135, 6806–6809 CrossRef CAS PubMed; (e) M. Scharfenberg, J. Hilf and H. Frey, Adv. Funct. Mater., 2018, 28, 1704302 CrossRef.
- W. J. Kruper and D. D. Dellar, J. Org. Chem., 1995, 60, 725–727 CrossRef CAS.
- A. A. Bredikhin, Z. A. Bredikhina, D. V. Zakharychev and A. V. Pashagin, Tetrahedron: Asymmetry, 2007, 18, 1239–1244 CrossRef CAS.
- D.-W. Park, N.-Y. Mun, K.-H. Kim, I. Kim and S.-W. Park, Catal. Today, 2006, 115, 130–133 CrossRef CAS , https://www.sciencedirect.com/science/article/pii/S0920586106001209.
- C. J. Whiteoak, A. Nova, F. Maseras and A. W. Kleij, ChemSusChem, 2012, 5, 2032–2038 CrossRef CAS PubMed.
- C. J. Whiteoak, N. Kielland, V. Laserna, E. C. Escudero-Adán, E. Martin and A. W. Kleij, J. Am. Chem. Soc., 2013, 135, 1228–1231 CrossRef CAS PubMed.
- R. Inkum, A. Teerawutgulrag, P. Puangsombar and N. Rakariyatham, Maejo Int. J. Sci. Technol., 2012, 6, 372–378 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1996487, 1996486 and 1996485. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt03311e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |