
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLow-oxidation state cobalt–magnesium complexes: ion-pairing and reactivity†
John A.
Kelly
a,
Johannes
Gramüller
b,
Ruth M.
Gschwind
 b and
Robert
Wolf
*a
b and
Robert
Wolf
*a
aUniversity of Regensburg, Institute of Inorganic Chemistry, 93040 Regensburg, Germany. E-mail: robert.wolf@ur.de
bUniversity of Regensburg, Institute of Organic Chemistry, 93040 Regensburg, Germany
First published on 20th September 2021
Abstract
Magnesium cobaltates (Arnacnac)MgCo(COD)2 (1–3) were synthesised by reacting (Arnacnac)MgI(OEt2) with K[Co(η4-COD)2] (COD = 1,5-cyclooctadiene) [Arnacnac = CH(ArNCMe)2; Ar = 2,4,6-Me3-C6H2 (Mes), 2,6-Et2-C6H3 (Dep), 2,6-iPr2-C6H3Mes (Dipp)]. Compounds 1–3 form contact ion-pairs in toluene, while solvent separated ion-pairs are formed in THF. The effect of ion-pairing on the reactivity is illustrated by reaction of 2 with tert-butylphosphaalkyne, which affords distinct 1,3-diphosphacyclobutadiene complexes. The heteroleptic sandwich complex [(Depnacnac)MgCo(P2C2tBu2)]2 (4) is selectively formed in toluene, while the homoleptic bis(1,3-diphosphacyclobutadiene) complex [(Depnacnac)Mg(THF)3][Co(P2C2tBu2)2] (5) is obtained in THF. Complex 4 is a precursor to further unusual phosphaorganometallic compounds. Substitution of the labile COD ligand in 4 by white phosphorus (P4) enabled the synthesis of the phosphorus-rich sandwich compound [(Depnacnac)MgCoP4(P2C2tBu2)]2 (6). The heterobimetallic complex (Cp*NiP2C2tBu2)Co(COD) (7) was isolated after treatment of 4 with Cp*Ni(acac) (Cp* = C5Me5, acac = acetylacetonate).
Introduction
Unsaturated hydrocarbons are able to stabilize transition metal atoms in negative oxidation states by back-donation of electron density from the metal atom into low-lying ligand π* orbitals.1 Due to these efficient π acceptor properties, highly reactive alkene and polyarene metalates are accessible with a wide range of d-block elements.2,3 As a result of the labile coordination of the hydrocarbon ligands, metalates may be used as synthetic equivalents of the parent, ‘naked’ transition metal anions. Early and late transition metalates have found diverse applications in the synthesis of unusual organometallic molecules such as highly reduced carbonyl complexes and anionic sandwich compounds.4 Moreover, various applications in homogeneous catalysis, e.g. transition-metal-catalyzed hydrogenations and cross couplings, have emerged recently.5Surprisingly, the possible influence of countercations on the stability and reactivity of such metalate complexes has largely been neglected. Arene and alkene metalates are typically synthesised via the reduction of transition metal halides or metallocenes with alkali metals (typically lithium, sodium and potassium). Interactions between the alkali metal cations are frequently observed in the solid-state molecular structures, but the effect of such anion–cation interactions (if present in solution) on reactivity is unknown.
To our knowledge, no examples of alkene or arene metalates incorporating alkaline earth metals have yet been reported. This is despite a steady and long-standing concurrent interest in compounds bearing unsupported magnesium-transition-metal bonds (Mg-TM), the first example of which, Cp(DPPE)2FeMgBr(THF)2 (Cp = cyclopentadienyl; DPPE = 1,2-(Ph2P)2C2H4), was synthesised almost fifty years ago.6 Notably, the vast majority of transition metal fragments in such Mg-TM compounds are based upon the carbonyl complexes e.g. [CpFe(CO)2]−, [Co(CO)4]− or [Co(CO)3(PR3)]− (see Fig. 1a for examples with cobalt), although there are singular examples with other moieties.7 The use of carbonyl ligands in these compounds is beneficial for isolation, as they stabilise the negatively charged metal atom via strong d–π* back donation. However, this added stability significantly limits any further reactivity and can lead to weakening of the metal–metal interaction due to bridging of the CO ligands.8 As a consequence, the further chemistry of these Mg-TM compounds has scarcely been studied, and there is a major outstanding need for the preparation and investigation of new and structurally-distinct examples of this compound class.
 | ||
| Fig. 1 (a) Previously reported compounds bearing magnesium–cobalt bonds A–C and alkali metal cobaltates D–F, (b) newly synthesised magnesium cobaltates and how their ionic form alters their reactivity (Dep = 2,6-Et2-C6H3). | ||
Recent work from our group has shown that alkali metal salts of metalate anions are effective in a wide range of applications, including as hydrogenation catalysts, scaffolds for white phosphorus (P4) functionalisation and in the synthesis of mixed metal clusters.9 Notably, the use of labile ligands, such as 1,5-cyclooctadiene (COD) in complexes D and E shown in Fig. 1a has allowed for significant further reactivity at the TM centre. Because of these studies we were interested in investigating how different countercations may affect the structure and reactivity of the resulting metalate complex. To that end, we report the synthesis and reactivity of new and highly reactive magnesium cobaltate compounds. The influence of the Mg2+ cation is strongly apparent, on both the molecular structure and the reactivity of the cobalt(-I) complexes, allowing for the isolation of a range of hitherto inaccessible (phospha)organometallic compounds.
Results
Magnesium cobaltates, (Mesnacnac)MgCo(COD)2 (1), (Depnacnac)MgCo(COD)2 (2) and (Dippnacnac)MgCo(COD)2 (3) were synthesised according to Scheme 1, via the simple salt metathesis reaction of (Arnacnac)MgI(OEt2) with potassium cobaltate, K[Co(COD)2] (E) in toluene/diethyl ether (Arnacnac = CH(ArNCMe)2, Mes = 2,4,6-Me3-C6H2, Dep = 2,6-Et2-C6H3, Dipp = 2,6-iPr2-C6H3). Yields are moderate for 1 (59%), good for 2 (88%) and low for 3 (30%). The physical properties of 1–3 are all similar to each other and all are significantly more soluble in common, unchlorinated organic solvents compared to the potassium analogue E. The magnesium cobaltates are also appreciably easier to prepare and handle under anaerobic conditions compared to E, which is very sensitive to air and moisture. | ||
| Scheme 1 Synthesis of magnesium cobaltates 1–3. | ||
X-ray quality crystals of 1–3 were obtained from concentrated toluene solutions stored at 5 °C in the form of yellow blocks. When crystallised in this manner, the compounds form contact ion-pairs with a direct magnesium–cobalt interaction, which is comparable to other magnesium-transition metal compounds.10 The cobalt centre is in the –I oxidation state making each complex diamagnetic.
Fig. 2 shows the molecular structures of 1 and 2; the structure of 3, which is analogous to that of 2, is shown in the ESI.† In all three compounds, the magnesium moiety is bonded to the cobalt centre perpendicularly to the COD ligands. The biggest disparity when comparing 1–3 is the cobalt–magnesium bond length, which is longer for 1 than for 2 and 3 (2.6314(7) Å for 1vs. 2.5459(5) and 2.5584(7) Å for 2 and 3, respectively). Surprisingly, 1 bearing the smaller Mesnacnac ligand possesses the longest Mg–Co bond. This can be explained by the highly distorted Mg(nacnac) moiety in 2 and 3, which pins back the aryl rings allowing for a closer magnesium–cobalt interaction (Mg–Cent(N1,N2)–C2 = 146.40° and 137.91°, respectively, where Cent(N1,N1) refers to the midpoint of the N1–N2 vector). In contrast, the Mg(nacnac) moiety is completely planar in 1 (Mg–Cent(N1,N2)–C2 = 180°). The Mg–Co bond length in 1–3 is within a similar range compared to the few other structurally characterized compounds containing a cobalt–magnesium bond (A–C, Co–Mg 2.480(4)–2.6163(6) Å, see Fig. 1a).11
 | ||
| Fig. 2 Thermal ellipsoid plots (30% probability surface) of the molecular structures of 1 (top) and 2 (bottom). Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°) for 1: Co1–Mg1 2.6314(7), Mg1–N1 2.0749(13), N1–Mg1–N1′ 91.69(7), N1–Mg1–Co1 134.15(4); for 2: Co1–Mg1 2.5459(5), N1–Mg1 2.0483(12), N2–Mg1 2.0937(12), N1–Mg1–N2 92.58(5), N1–Mg1–Co1 134.19(4), N2–Mg1–Co1 132.52(4). | ||
To investigate the nature of the magnesium–cobalt interaction we performed DFT calculations (TPSS D3BJ/def2-TZVP) on (Depnacnac)MgCo(COD)2 (2).12 An analysis of the intrinsic bond orbitals (IBOs) indicates there are no bond-like localised orbitals involving magnesium and cobalt (see the ESI† for details).13 The only shared orbital between Mg and Co is a delocalised orbital between cobalt, magnesium and a vinyl group from the COD ligand, but this has a very low contribution from magnesium (Co 19.6%, Mg 7.9%, C 14.9%, C 49.9%, see Fig. S45 ESI†). NBO analysis shows no Lewis or non-Lewis shared orbitals between the magnesium and cobalt centres. The second order perturbation theory analysis furthermore indicates that the donor–acceptor interaction between a lone pair of electrons on the cobalt centre towards a Rydberg orbital on the magnesium centre is negligibly small (0.51 kcal mol−1). Note that the NBO, IBO and AIM analyses provide little evidence for an interaction between the COD vinyl carbons and the magnesium cation (see the ESI† for details). The Wiberg bond index for the Mg–Co bond is 0.027, and the AIM (atoms in molecules) analysis reveals no bond critical point between the Mg and Co centres (Fig. 3).
 | ||
| Fig. 3 A Laplacian contour plot of the DFT-calculated electron density of 2; bond critical points are shown in orange. | ||
Altogether, this data clearly indicates that the nature of the magnesium–cobalt interaction is highly electrostatic, which was also the case for the previously reported carbonyl iron–magnesium compound, (Dippnacnac)MgFeCp(CO)2.2c The ionic character of the Mg–Co bond is also in line with Mulliken, Löwdin and intrinsic atomic orbital (IAO) population analyses, which consistently show that the cobalt atom is more negatively charged than the magnesium atom (see the ESI† for details).
We were interested to see how these highly ionic, heterobimetallic complexes would behave in solution, especially comparing the effect of non-donor/donor solvents. The 1H NMR spectra of 1–3 in C6D6 share similar motifs, the main one being the abundance of broad multiplets within the range δ = 1–4 ppm attributed to the COD ligands (in some cases overlapping with the signals for the nacnac moiety) on the Co centre (see ESI†). However, when 1–3 were dissolved in d8-THF only two multiplets are observed for the COD ligands (e.g. at δ = 1.84 and 2.14 ppm for compound 2). It should be noted that these signals are very similar to that of potassium cobaltate, E (δ = 1.86 and 2.18 ppm). The appearance of the 1H NMR spectrum in a non-donor solvent (C6D6) seems to indicate that the solid-state structure featuring a contact ion-pair is maintained in solution. This is due to the COD protons being inequivalent due to the proximity of the (nacnac)Mg moiety. It then appears that the Mg–Co interaction is broken when dissolved in a donor solvent (d8-THF).
This was confirmed via X-ray crystallography and DOSY NMR spectroscopy. It was possible to isolate [(Depnacnac)Mg(THF)3][Co(COD)2] (2·THF), from a solution of 2 in 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 THF:n-hexane stored at −30 °C (Fig. 4). The molecular structure of 2·THF clearly shows the cationic [(Depnacnac)Mg]+ moiety is separated from the [Co(COD)2]− unit due to the coordination of three molecules of THF to the former. The DOSY NMR experiment gave further insight into the solution behavior of 2 (see ESI† for full analysis). When conducted in C6D6, all suitable signals of both ligands show similar diffusion coefficients (≈6.6 × 10−10 m2 s−1) in the 1H NMR spectrum. In d8-THF however, the region that coincides with the signals for the COD ligands (δ = 1.9–2.3 ppm) has a higher diffusion coefficient (D ≈ 7.6 × 10−10 m2 s−1) compared to those for the nacnac moiety (D ≈ 6.7 × 10−10 m2 s−1), validating that they are separate entities in solution (see ESI† for further details). When a solution of 2·THF was dried in vacuo and the residue dissolved in C6D6, the 1H NMR spectrum showed the reformation of the contact ion-pair 2. To our knowledge, this is the first clear example of reversible ion-pairing in a molecular transition metal-magnesium complex.14 When a stronger ligand is added, such as DMAP, the solvent separated species (2·DMAP) is observed even in non-donor solvents, indicating that metal–metal bond formation does not occur in this case (see the ESI† for further details, DMAP = dimethylaminopyridine). It should be noted that the ionic form has little effect on the UV-Vis spectra of 2/2·THF, both having similar absorption maxima at 351 and 347 nm, respectively (see ESI†).
:20 THF:n-hexane stored at −30 °C (Fig. 4). The molecular structure of 2·THF clearly shows the cationic [(Depnacnac)Mg]+ moiety is separated from the [Co(COD)2]− unit due to the coordination of three molecules of THF to the former. The DOSY NMR experiment gave further insight into the solution behavior of 2 (see ESI† for full analysis). When conducted in C6D6, all suitable signals of both ligands show similar diffusion coefficients (≈6.6 × 10−10 m2 s−1) in the 1H NMR spectrum. In d8-THF however, the region that coincides with the signals for the COD ligands (δ = 1.9–2.3 ppm) has a higher diffusion coefficient (D ≈ 7.6 × 10−10 m2 s−1) compared to those for the nacnac moiety (D ≈ 6.7 × 10−10 m2 s−1), validating that they are separate entities in solution (see ESI† for further details). When a solution of 2·THF was dried in vacuo and the residue dissolved in C6D6, the 1H NMR spectrum showed the reformation of the contact ion-pair 2. To our knowledge, this is the first clear example of reversible ion-pairing in a molecular transition metal-magnesium complex.14 When a stronger ligand is added, such as DMAP, the solvent separated species (2·DMAP) is observed even in non-donor solvents, indicating that metal–metal bond formation does not occur in this case (see the ESI† for further details, DMAP = dimethylaminopyridine). It should be noted that the ionic form has little effect on the UV-Vis spectra of 2/2·THF, both having similar absorption maxima at 351 and 347 nm, respectively (see ESI†).
 | ||
| Fig. 4 Thermal ellipsoid plot (30% probability surface) of the molecular structure of 2·THF. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Mg1–N1 2.1140(15), Mg–N2 2.0707(15), Mg1–O1 2.0790(13), Mg1–O2 2.1461(13), Mg1–O3 2.0806(14), N1–Mg1–N2 90.01(6). | ||
In order to investigate the potential ramifications of the different ionic forms (contact ion-pair vs. solvent separated ion-pair) reactivity studies were performed with 2, which is the most readily accessible compound among 1–3. We initially studied the reactivity of 2 with phosphaalkynes. It has been previously shown that when potassium cobaltate E was treated with phosphaalkynes, the orange homoleptic sandwich complex, K[Co(P2C2R2)2] (G) (R = tBu, adamantyl) was formed exclusively.15 It was not possible to form the related heteroleptic sandwich complex, K[Co(COD)(P2C2R2)] with G being the only observed product formed (alongside unreacted E) when sub-stoichiometric amounts of phosphaalkyne were used. In contrast, treatment of 2 in toluene with the tert-butyl-phosphaalkyne, tBuCP, affords a yellow-green solution (Scheme 2). Subsequent addition of n-hexane after 1–2 days causes the precipitation of the heteroleptic sandwich complex (Depnacnac)MgCo(P2C2tBu2)(COD) (4) as a green powder. Single crystals of 4 were grown from a benzene solution stored at ambient temperatures. X-ray analysis elucidated the structure of 4 as a contact ion-pair with one COD ligand replaced with a 1,3-diphosphacyclobutadiene moiety (Fig. 5). To fully illustrate the effect of the cation on reactivity we also attempted to synthesise a heteroleptic complex with lithium cobaltate Li[Co(COD)2] (D). The reaction of D with 2 equivalents of tBuCP was conducted in C6D6 and monitored via1H and 31P NMR spectroscopy, both of which indicated the formation of homoleptic sandwich complex Li[Co(P2C2tBu2)] as the only product (see Fig. S28 and S29, ESI†). This observation clearly highlights the distinct reactivity offered by the Mg salt, 2.
 | ||
| Scheme 2 Synthesis of 4, 5, 6 and 7 (Cp* = C5Me5, acac = acetylacetonate). | ||
 | ||
| Fig. 5 Thermal ellipsoid plot (30% probability surface) of the molecular structure of 4 (hydrogens omitted, selected substituents are displayed in wireframe for clarity). Selected bond lengths (Å) and angles (°): Mg1–Co1 2.6762(14), Mg1–P1 2.6272(16), Mg–N1 2.064(2), Co1–P1 2.3069(11), Co1–P2 2.2951(11), P1–C14 1.807(3), P2–C14 1.788(3), N1–Mg1–N1′ 92.89(14), P1–Co1–P2 72.07(4). | ||
Compound 4 shares a similar structural motif to 1–3, in that the (nacnac)Mg moiety is bound perpendicularly to the cobalt sandwich anion. The Mg–Co bond distance in 4 is elongated compared to 2 (2.6762(14) vs. 2.5459(5) Å, respectively) and the previously reported Mg–Co complexes (vide supra) due to the larger size of the (P2C2tBu2) group compared to COD and additional coordination of the P atom from the 1,3-diphosphocyclobutadiene ring to the Mg centre. The Mg1–P1 bond distance (2.6272(16) Å) is similar to other cationic Mg-PR3 compounds, such as [(Dippnacnac)Mg(PPh3)][Al{OC(CF3)3}4] (2.59 Å) and [(Dippnacnac)Mg(PCy3)][B(C6F5)4] (2.65 Å, Cy = cyclohexyl).16 The 31P{1H} NMR spectra (in C6D6) show two singlets in 1:1 ratio at δ = 76.5 ppm and −100.6 ppm. This observation evidences that the contact ion-pair is retained in non-coordinating solvents.‡
DFT calculations (TPSS D3BJ/def2-TZVP level) were employed to investigate the effect the 1,3-diphosphacyclobutadiene ligand on the magnesium–cobalt interaction in 4. Much like with 2, IBO and NBO analysis shows there are no shared orbitals or donor–acceptor interaction between the Mg and Co centres. The AIM analysis also shows no bond critical point between the metal centres, indicating the interaction remains highly electrostatic (see ESI†).
We next attempted the same reaction with tBuCP in a donor solvent to see whether the reactivity may differ with the solvent separated ion pair, and indeed, the difference was immediately apparent with an instant colour change to a deep orange-red when adding tBuCP to a THF solution of 2·THF. This contrasts the yellow-green colour observed when the reaction is conducted in toluene. The 1H and 31P{1H} NMR spectra of the reaction mixture show the clean formation of [(Depnacnac)Mg(THF)3][Co(P2C2tBu2)2]. The 31P{1H} NMR spectrum shows a singlet at δ = 3.2 ppm (see ESI†), which is reminiscent of the potassium analogue (2.4 ppm).14 It was not possible to crystallographically characterise the solvent separated ion-pair, as it precipitates out of solution as a powder due to its low solubility in THF. Attempts to recrystallise it from toluene instead formed the coordination compound (Depnacnac)Mg(THF)Co(P2C2tBu2)2 (5, see ESI†). It should be noted that if there is THF present in the reaction of tBuCP and 2, the formation of 5 alongside 4 is observed. The solid-state structure consists of a bis(diphosphacyclobutadiene) cobalt unit with a (Depnacnac)Mg moiety coordinated to one of the P atoms. In contrast to the heteroleptic species 4, there is no appreciable interaction between the magnesium and cobalt centres. Although the coordination of a heterometal to the diphosphacyclobutadiene ring in metal sandwich complexes is a common motif, complexes 4 and 5 are the first instances of such heterometallic compounds involving a group 2 metal.17
As complex 4 contains a labile COD ligand, we envisioned this would allow for further functionalisation, forming novel heteroleptic metallocenes. Generally, heteroleptic sandwich complexes found in literature are composed of a Cp ligand and either a polycyclic phosphorus or diphosphacyclobutadiene unit.18,19 This limits the variety of heteroleptic metallocene-like species available. We were then interested in substituting the COD ligand in 4 with another cyclic ligand, in particular cyclo-P4. To this end, we treated 4 with P4 in toluene (Scheme 2). Upon heating to 75 °C for 18 hours the heteroleptic sandwich dimer [(Depnacnac)MgCoP4(P2C2tBu2)]2 (6) was isolated as a red solid.
Compound 6 is a heteroleptic sandwich dimer, coordinating via the cyclo-P4 ring to the (DepnacnacMg) moiety (Fig. 6). Like 5, there is no magnesium-cobalt interaction, with the magnesium cation being coordinatively saturated by binding to two P atoms from two distinct P4 rings. The formation of 6 from 4 goes via a simple ligand substitution reaction with the release of the COD ligand, which was observed in the 1H NMR spectrum of the reaction mixture. The Mg–P interactions in 6 (2.6747(6)/2.6847(6) Å) are elongated compared to 4 (2.6272(16) Å) and the other Mg-PR3 compounds mentioned earlier (vide supra). The P–P bond lengths of the cyclo-P4 unit are slightly inequivalent with the P1–P2/P1–P4 distances (2.2083(5)/2.2184(5) Å, respectively) elongated compared to P3–P2/P3–P4 (2.1641(5)/2.1740(5) Å, respectively). The cyclo-P4 moiety has been previously reported for cobalt centres, but they are generally stabilised with bulky cyclopentadienyl or multidentate ligands.19 Aside from cobalt, low valent iron complexes are the most commonly able to activate P4 to give the cyclo-P4 moiety, as well as sparse examples with niobium, vanadium, tantalum and molybdenum.20 Compound 6 marks the first example of an anionic cobaltate sandwich compound bearing a cyclo-P4 ligand. In addition, it is noteworthy that it is a rare example of a highly phosphorus-rich sandwich complex, having six P atoms in its ligand scaffold.4a,21
 | ||
| Fig. 6 Thermal ellipsoid plots (30% probability surface) of the molecular structure or 6. Hydrogen atoms are omitted and the tBu and Dep groups are displayed in wireframe for clarity. Selected bond lengths (Å) and angles (°) for 6: (°): Co–P1 2.2460(4), Co1–P2 2.3322(4), Co1–P3 2.2520(4), Co1–P4 2.2988(4), Co1–P5 2.2514(4), Co1–P6 2.2494(4), Mg1–P1 2.6747(6), Mg1′–P3 2.6847(6), P1–P2 2.2083(5), P2–P3 2.1641(5), P3–P4 2.1740(5), P4–P1 2.2184(5), P1–P2–P3 88.553(19), P2–P3–P4 92.834(19), P3–P4–P1 88.047(19), P4–P1–P2 90.451(19). | ||
Due to the poor solubility of 6 in most organic solvents it was not possible to obtain high resolution NMR spectra (see ESI† for further details). Investigations to further characterize 6 by solid-state NMR spectroscopy are currently in progress.
We anticipated that 4 could be utilised for the synthesis of novel heterobimetallics stabilised by the 1,3-diphosphacyclobutadiene ligand. To that end, 4 was treated with Cp*Ni(acac) at ambient temperature (Scheme 2). A deep red colour was observed, and monitoring via NMR spectroscopy indicated 4 was completely consumed (Cp* = C5Me5, acac = acetylacetonate). After workup and recrystallization from n-hexane, dark red blocks of the heterobimetallic compound, (Cp*NiP2C2tBu2)Co(COD) (7) were isolated. The XRD analysis (Fig. 7) shows that the Cp*Ni fragment has inserted into the diphosphacyclobutadiene ring forming a 1,4-diphospha-2-nickelacyclopentadiene, which is coordinating to the Co(COD) moiety. This is the second example of such an insertion, the first being (Cp*NiP2C2tBu2)Co(P2C2tBu2) (H) synthesised from the reaction of [K(THF)2{Co(P2C2tBu2)2}] with [Ni2Cp3]BF4.22
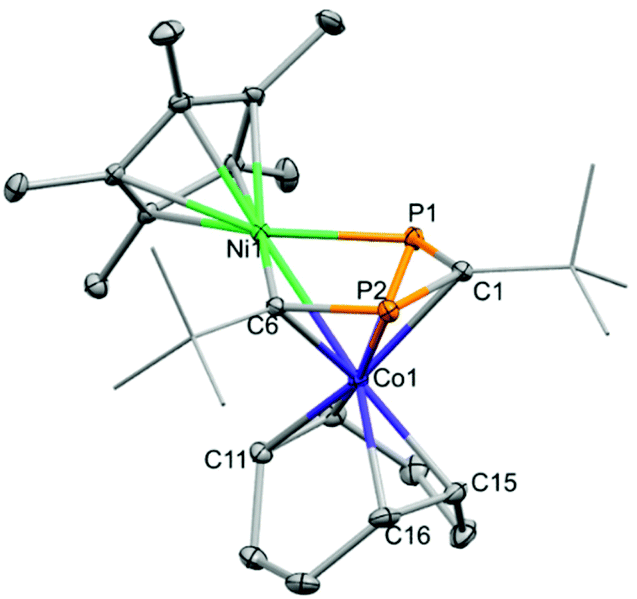 | ||
| Fig. 7 Thermal ellipsoid plots (30% probability surface) of the molecular structure of 7. Hydrogen atoms are omitted and selected tBu groups are displayed in wireframe for clarity. Selected bond lengths (Å) and angles (°) for 7: Co1–Ni1 2.5065(3), Ni1–P1 2.2051(4), Ni1–C6 1.9269(14), P1–C1 1.7388(15), C1–P2 1.7663(14), P2–C6 1.7625(14), C6–Ni1–P1 92.66(4), C6–P2–C1 101.10(7), P1–C1–P2 120.41(8). | ||
The Ni–C and Ni–P bonds lengths are within the range expected for a single bond (1.9269(14) and 2.2051(4) Å, respectively) and the similar P1–C1, C1–P2 and P2–C6 bond lengths indicates delocalisation (1.7388(15), 1.7663(14) and 1.7625(14) Å, respectively). The Ni–Co bond length is slightly shorter when compared to that found in H (2.5065(3) vs. 2.536(1) Å, respectively) assumedly due to the less bulky COD ligand present in 7. The NMR spectra are in agreement with the asymmetrical molecular structure observed in the solid state. The 1H NMR spectrum shows two singlets at δ = 1.53 and 2.07 ppm for the tBu groups, the former is overlapping with the signal for the Cp* moiety. There are various (partially overlapping) multiplet signals for the COD protons at δ = 1.32, 2.32, 3.16, 3.56 and 4.37 ppm. The 31P NMR spectrum shows two doublet signals at δ = 197.1 and 460.3 ppm with a 2JPP coupling constant of 28 Hz; the strongly downfield shifted signal is attributed to the P atom bonded to the Ni centre. Similar shifts were observed for H (δ = 191.4 and 492.2 ppm).
Conclusions
The synthesis of magnesium cobaltates has enabled a first investigation of the previously unrecognized influence of the countercation on the reactivity of low oxidation state transition metalates. Detailed structural and NMR spectroscopic studies on magnesium cobaltates 1–3 have revealed that these species reversibly adopt either a contact or solvent separated ion-pair motif depending on the solvent used. Ion-pairing has a direct effect on the reactivity and selectivity, exemplified by the reaction of 2 with tBuCP in toluene vs. THF. When using 2 it is possible to isolate the heteroleptic sandwich complex 4, whereas only the homoleptic complex can be formed when using the alkali metal cobaltates D and E. This divergent reactivity is attributed to the formation of an intimate ion-pair between [(Depnacnac)Mg]+ cation and the [Co(COD)2]− anion for 2. The presence of a substitutionally labile COD ligand in the structure of 4 has permitted the synthesis of the phosphorus-rich heteroleptic sandwich complex 6, which was previously inaccessible by other routes. Lastly, the potential of using magnesium cobaltates to synthesise unusual heterobimetallic compounds was demonstrated with the synthesis of 7. Only through utilising 4 can the heteroleptic compound 7 be isolated. It is also noteworthy that the presence of the labile COD ligand on the Co centre of 7 should stimulate significant further chemistry and catalysis.This initial investigation calls attention to how varying the cation has a profound impact on the structural features and reactivity of transition metalates. The facile and reversible formation of highly ionic Mg–Co bonds in 1–3 should open them up to applications in ion-pairing catalysis. On-going investigations are focusing on utilising magnesium cobaltates, 1–4 and 7 in hydrogenation catalysis and in the formation of a range of new heterobimetallic compounds. The results of these on-going studies will be reported on in due course.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Ilya G. Shenderovich for NMR measurements. Funding by the Deutsche Forschungsgemeinschaft (DFG RTG 2620), the European Research Council (ERC CoG 772299) and Fonds der Chemischen Industrie (fellowship to J. G.) is gratefully acknowledged.Notes and references
- Reviews:
(a) J. E. Ellis, Inorg. Chem., 2006, 45, 3167–3186 CrossRef CAS PubMed
; (b) J. E. Ellis, Dalton Trans., 2019, 48, 9538–9563 RSC
- Selected examples:
(a) K. Jonas, Angew. Chem., Int. Ed. Engl., 1975, 14, 752 CrossRef
-
(a) H. C. Clark, D. G. Ibbott, N. C. Payne and A. Shaver, J. Am. Chem. Soc., 1975, 12, 3556 Search PubMed
-
(a) E. Urnéžius, W. W. Brennessel, C. J. Cramer, J. E. Ellis and P. von Ragué Schleyer, Science, 2002, 295, 832 CrossRef PubMed
-
(a) A. Fürstner, H. Krause and C. W. Lehmann, Angew. Chem., Int. Ed., 2006, 45, 440 CrossRef PubMed
-
(a) H. Felkin, P. J. Knowles and B. Meunier, J. Chem. Soc., Chem. Commun., 1974, 44 RSC
-
(a) J. Hicks, C. E. Hoyer, B. Moubaraki, G. L. Manni, E. Carter, D. M. Murphy, K. S. Murray, L. Gagliardi and C. Jones, J. Am. Chem. Soc., 2014, 136, 5283 CrossRef CAS PubMed
-
(a) M. P. Blake, N. Kaltsoyannis and P. Mountford, J. Am. Chem. Soc., 2011, 133, 15358 CrossRef CAS PubMed
-
(a) P. Büschelburger, D. Gärtner, E. Reyes-Rodriguez, F. Kreyenschmidt, K. Koszinowski, A. Jacobi v. Wangelin and R. Wolf, Chem. – Eur. J., 2017, 23, 3139 CrossRef PubMed
-
(a) M. P. Blake, N. Kaltsoyannis and P. Mountford, Chem. Commun., 2013, 49, 3315 RSC
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171 CrossRef CAS PubMed
-
(a) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC
- G. Knizia, J. Chem. Theory Comput., 2013, 9, 4834 CrossRef CAS PubMed
- Reversible metal–metal bond formation has been observed for a few 4d and 5d metal complexes:
(a) J. Bauer, H. Braunschweig and R. Dewhurst, Chem. Rev., 2012, 112, 4329 CrossRef CAS PubMed
-
(a) R. Wolf, A. W. Ehlers, C. J. Slootweg, M. Lutz, D. Gudat, M. Hunger, A. L. Spek and K. Lammertsma, Angew. Chem., Int. Ed., 2008, 47, 4584 CrossRef CAS PubMed
-
(a) L. Garcia, M. D. Anker, M. F. Mahon, L. Maron and M. S. Hill, Dalton Trans., 2018, 47, 12684 RSC
-
(a) J. Malberg, T. Wiegand, H. Eckert, M. Bodensteiner and R. Wolf, Eur. J. Inorg. Chem., 2014, 1638 CrossRef CAS
-
(a) P. Binger, R. Milczarek, R. Mynott, M. Regitz and W. Rösch, Angew. Chem., Int. Ed. Engl., 1986, 25, 644 CrossRef
-
(a) M. Scheer and U. Becker, J. Organomet. Chem., 1997, 545, 451–460 CrossRef
-
(a) O. J. Scherer, J. Vondung and G. Wolmershäuser, Angew. Chem., Int. Ed. Engl., 1989, 28, 1355 CrossRef
-
(a) J. Müller, S. Heinl, C. Schwarzmaier, G. Balázs, M. Keilwerth, K. Meyer and M. Scheer, Angew. Chem., Int. Ed., 2017, 56, 7312 CrossRef PubMed
- C. Rödl and R. Wolf, Chem. – Eur. J., 2019, 25, 8332 CrossRef PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2038297, 2054748–2054754 and 2081284. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt02621f |
| ‡ Decomposition to an intractable mixture was observed when 4 was dissolved in THF. |
| This journal is © The Royal Society of Chemistry 2021 |