
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBi- and trinuclear coinage metal complexes of a PNNP ligand featuring metallophilic interactions and an unusual charge separation†
Milena
Dahlen
a,
Max
Kehry
 b,
Sergei
Lebedkin
c,
Manfred M.
Kappes
bc,
Wim
Klopper
bc and
Peter W.
Roesky
*a
b,
Sergei
Lebedkin
c,
Manfred M.
Kappes
bc,
Wim
Klopper
bc and
Peter W.
Roesky
*a
aInstitute of Inorganic Chemistry, Karlsruhe Institute of Technology (KIT), Engesserstr. 15, 76131 Karlsruhe, Germany. E-mail: roesky@kit.edu
bInstitute of Physical Chemistry, Karlsruhe Institute of Technology (KIT), Fritz-Haber-Weg 2, 76131 Karlsruhe, Germany
cInstitute of Nanotechnology, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
First published on 3rd September 2021
Abstract
A selective synthesis of bi- and trinuclear complexes featuring a tetradentate monoanionic PNNP ligand is presented. The binuclear coinage metal complexes show a typical fourfold coordination for Cu and Ag, which changes to a bifold coordination for Au. The latter is accompanied by an unusual charge separation. Optical properties are investigated using photoluminescence spectroscopy and complemented by time-dependent density-functional-theory calculations. All compounds demonstrate clearly distinguished features dependent on the metals chosen and differences in the complex scaffold.
Introduction
First predominantly known for gold compounds (“aurophilicity”), the concept of “metallophilicity” has meanwhile been expanded to numerous other systems, such as d10–d10, d10–d8, d8–d8 or d10–d10s2 electron configurations.1–22 The term “metallophilic” interactions was first introduced by Pyykkö in 1994![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 3 and has since then been established for the description of short distances between closed shell metal centres with distances often below the sum of their van der Waals radii.23–35 While aurophilic and argentophilic attractions fall in the range of strong hydrogen bonds (30–50 kJ mol−1), cuprophilic contacts are considered to be approximately three times weaker (up to 15 kJ mol−1).36–39 Despite intensive research, aspects of these attractions are still under debate.37,40,41 Metallophilic interactions are often induced through a supporting ligand system. Fig. 1 illustrates the general types of ligand assistance, namely “fully supported” (A) and “semi supported” (B), as well as the “unsupported” configuration (C) with only intermolecular metal–metal contacts.32
3 and has since then been established for the description of short distances between closed shell metal centres with distances often below the sum of their van der Waals radii.23–35 While aurophilic and argentophilic attractions fall in the range of strong hydrogen bonds (30–50 kJ mol−1), cuprophilic contacts are considered to be approximately three times weaker (up to 15 kJ mol−1).36–39 Despite intensive research, aspects of these attractions are still under debate.37,40,41 Metallophilic interactions are often induced through a supporting ligand system. Fig. 1 illustrates the general types of ligand assistance, namely “fully supported” (A) and “semi supported” (B), as well as the “unsupported” configuration (C) with only intermolecular metal–metal contacts.32
 | ||
| Fig. 1 Different types of ligand assistance for metallophilic interactions. L = neutral ligand; X = anionic ligand.32 | ||
Compounds which exhibit metallophilic contacts often feature interesting and rich photophysical properties due to the influence of such contacts on the electronic structure.42 For example, when compared to a mononuclear metal complex, a ligand to metal charge transfer (LMCT) process may alter to a LMMCT in a binuclear complex featuring metallophilic interactions.37,38 However, establishment of systematic correlations between PL properties and structural parameters of complexes with metallophilic contacts remains challenging.43–45 There is consequently a demand for systematic investigations of the photophysical properties of defined ligand systems with varying metal loadings.46,47
We therefore aimed to study a series of coinage metal complexes with an increasing number of metal centres while keeping the ligand scaffold virtually unchanged. Specifically, we have used a ligand which comprises different “hard/soft” coordination compartments. Accordingly, for gold as the “softest” cation in this row, a preference towards the “softer” coordination site would be expected. N,N′-Bis[(2-diphenylphosphino)phenyl]-formamidinate (dpfam−; Scheme 1), a monoanionic PNNP ligand, introduced in 2002 by Tsukada et al.,48 has already been shown to incorporate several different metal centres and was therefore chosen for this work.48–52 Herein, we present the respective series of homometallic bi- and trinuclear complexes of copper, silver and gold. Photophysical properties were investigated from 20 K to room temperature and complemented by quantum chemical calculations using time-dependent density-functional theory.
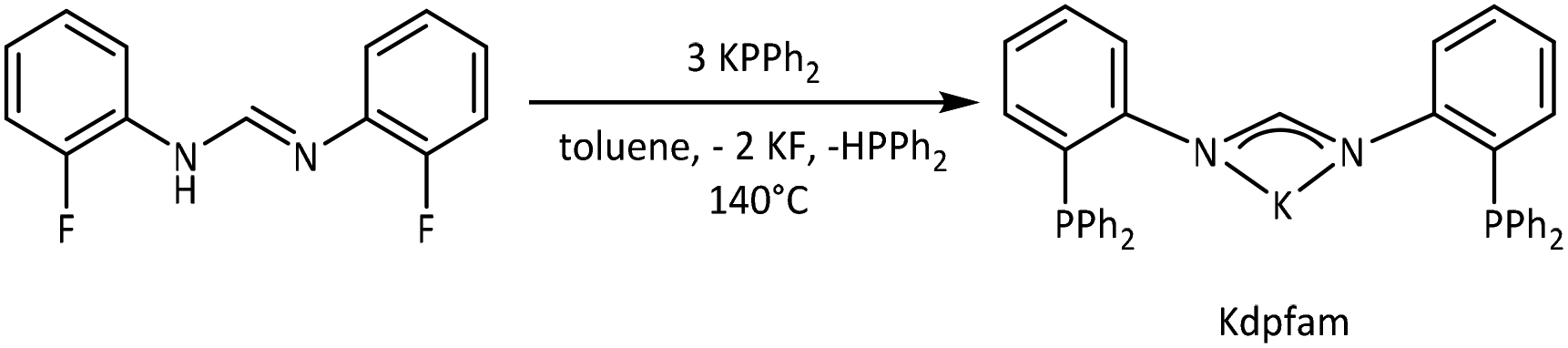 | ||
| Scheme 1 Synthesis of Kdpfam. | ||
Results and discussion
The known dpfam− ligand48 was obtained with a new synthetic protocol adapted from related PNNP ligand synthesis.53–55 It directly leads to the potassium salt Kdpfam which can be readily isolated and applied as a versatile precursor. Specifically, N,N′-bis(2-fluorophenyl)formamidine was reacted with potassium diphenyl phosphide in toluene to yield the desired product in excellent yield (see ESI;†Scheme 1). It provides four coordination sites with two being rather “soft” – the phosphine moieties and two being rather “hard” – the negatively charged amidinate nitrogen atoms.Binuclear complexes
To access a series of complexes with successively higher metal loading, we first synthesized the neutral binuclear complexes. For this purpose, Kdpfam was reacted with soluble salts of the coinage metals (Scheme 2). | ||
| Scheme 2 Synthesis of binuclear complexes 1–3. | ||
The dicopper complex [dpfam2Cu2] (1) was obtained by reaction of Kdpfam with [Cu(MeCN)4][PF6] and subsequent crystallization through vapor diffusion. It features a yellow luminescence when irradiated with UV light. The copper atoms are each coordinated by two nitrogen atoms of the amidinate moieties and two phosphines in a distorted tetrahedral environment (Fig. 2). The molecular structure in the solid state reveals nearly identical N–C bond lengths (N1–C1 1.307(6) and N2–C1 1.319(6) Å, N3–C38 1.321(6) and N4–C38 1.322(6) Å), indicating a delocalization of the negative charge. The Cu–N bonds (2.042(4)–2.120(4) Å) are slightly shorter than the respective Cu–P bonds (2.2266(14)–2.2648(14) Å). No cuprophilic interaction is observed (Cu1–Cu2 3.63 Å), which is in contrast to similar copper bis(amidinate) complexes with short Cu–Cu contacts and metallophilic interactions.56,57 For 1, the additional phosphine coordination most likely stabilizes the favoured tetrahedral coordination and thus suppresses a cuprophilic interaction.58 However, the amidinate angles (N–C–N 120.8(4) and 120.6(4)°) in 1 are consistent with the values reported in literature.48,56,57 The 31P{1H} NMR spectrum reveals a singlet resonance at δ = −18.4 ppm. In the 1H NMR spectrum, the resonance of the isolated proton in the amidinate backbone is shifted by approximately 1 ppm to δ = 9.51 ppm as compared to Kdpfam (8.59 ppm) and by almost 2 ppm as compared to the protonated ligand (7.57 ppm), respectively.48
 | ||
| Fig. 2 Molecular structures of the binuclear complexes 1 (left) and 2 (right) in the solid state. Hydrogen atoms and co-crystallized solvent molecules are omitted for clarity. Selected bond lengths and angles are given in the ESI, section 6.† | ||
The silver complex [dpfam2Ag2] (2) was obtained by applying the same reaction protocol as for the synthesis of 1 but by using AgBF4 as metal precursor (Scheme 2). The product was obtained as pale yellow crystals demonstrating blueish luminescence upon UV irradiation. The overall molecular structure of 2 in the solid state is similar to that of 1. However, additionally an argentophilic contact is observed, resulting in a distorted trigonal bipyramidal coordination sphere (Fig. 2). The intermetallic distance of 3.44 Å indicates a metallophilic interaction and is in good agreement with literature values.38 The Ag–N bond lengths vary between 2.302(2) and 2.453(2) Å, while the Ag–P distances are in the range of 2.4263(5)–2.4793(5) Å. Literature values for Ag–P and Ag–N bond lengths in comparable coordination geometries agree with those found in 2.59,60
The 31P{1H} spectrum features a broad pseudo triplet of triplets at −16.6 ppm, most likely through coupling of the 31P nucleus with 107Ag and 109Ag.61–63 The observed proton NMR resonances could be assigned by COSY-NMR spectroscopy and 1H{31P} NMR experiments. The resonance of the NCHN proton in the 1H{31P} NMR spectrum is detected as a pseudo triplet at δ = 9.73 ppm showing 1H–107/109Ag coupling.
The corresponding digold complex [dpfam2Au2] (3) was obtained from a reaction of Kdpfam with one equivalent of [AuCl(tht)] (tht = tetrahydrothiophene) in THF (Scheme 2). Compound 3 was isolated as yellow crystals featuring a yellow luminescence at room temperature when excited with a UV lamp. The molecular structure in the solid state reveals a metal coordination mode differing significantly from that in 1 and 2. Instead of being coordinated by both amidinate and phosphine moieties, a slightly bent bis(phosphine) gold coordination mode is realized (P2–Au1–P1 163.60(6), P3–Au2–P4B 168.6(3), P4A–Au2–P3 163.5(4) (Fig. 3). The Au–P distances vary from 2.277(2) Å to 2.333(2) Å and are in the expected range for this structural motif.64,65 The observed molecular structure in combination with our DFT calculations suggests an unusual charge separation. Apparently, the negative charges are delocalized over the pairs of nitrogen atoms (N1 and N2, N3 and N4, respectively), as all C–N bonds have approximately the same length.66 The corresponding positive charges are situated on the gold cations and are therefore more “isolated” from the negative charges than in 1 and 2. The amidinate angles are as much as 9° wider than for 1 and 2 (130.5(7)° and 129.6(6)°) most likely due to the different coordination mode. NMR experiments were carried out in THF-d8 (other deuterated solvents did not allow an interpretation of the spectra due to either insolubility or solvent effects). Variable temperature 1H NMR experiments at temperatures down to 213 K allow to follow the dynamic behaviour of 3 in solution (ESI, Fig. S5.3-3†).
 | ||
| Fig. 3 Molecular structure of the binuclear complex 3 in the solid state. Hydrogen atoms and co-crystallized solvent molecules are omitted for clarity. Both disordered parts are displayed. Selected bond lengths and angles are given in the ESI, section 6.† | ||
At 213 K, the NCHN proton is detected at δ = 7.65 ppm which is a smaller value than for 1 and 2 and which can be rationalized by reduced metal coordination of the nitrogen atoms. In the 31P{1H} NMR spectrum, two doublet resonances (δ = 35.9 ppm (2JP,P = 339.3 Hz) and 27.6 ppm (2JP,P = 338.8 Hz)) are observed. The latter implies strong coupling of the phosphorus nuclei as well as a slightly different chemical environment in solution. An additional roof effect indicates additional higher order coupling.
Trinuclear complexes
A selective metal chain extension was accomplished by a simple variation of the stochiometric ratio. The trinuclear copper complex [dpfam2Cu3(MeCN)][PF6] (4) was obtained by reacting [Cu(MeCN)4][PF6] and Kdpfam in a 3:2 ratio (Scheme 3) and subsequently crystallized from THF/MeCN/n-pentane as yellow crystals which exhibit pale yellow luminescence at room temperature upon UV excitation.
 | ||
| Scheme 3 Synthesis of the trinuclear complexes 4, 4a, and 5. | ||
Although a linear arrangement of copper atoms is often reported in the literature,67 the molecular structure of 4 in the solid state revealed a tilted Cu3 chain with an angle of 117.84(1)° while the two ligand molecules are shifted against each other (Fig. 4). Not taking the connecting cuprophilic contacts into account, Cu1 is set in an almost trigonal planar coordination environment by two phosphine moieties and one nitrogen atom, whilst Cu2 is almost linearly coordinated by two nitrogen atoms (172.21(8)°). Cu3 is set in the same coordination sphere as Cu1 but is additionally coordinated by one molecule of acetonitrile, resulting in a distorted tetragonal coordination sphere. Such a configuration has been known in the literature and the obtained Cu–MeCN distance aligns well with published values.68–72 Intermetallic distances were found to be 2.5984(4) Å (Cu1–Cu2) and 2.7792(4) Å (Cu2–Cu3), which are in the range of cuprophilic interactions.39 The Cu–N bond distances are found between 1.872(2) Å and 2.098(2) Å, concurring with literature values.56,57 P–Cu coordination bond lengths vary from 2.2126(6) Å to 2.3158(6) Å and agree with literature values reported for similar systems.68 In the 1H NMR spectrum the NCHN proton is detected at δ = 9.06 ppm, which is shifted upfield by 0.5 ppm as compared to the dicopper complex 1.
 | ||
| Fig. 4 Molecular structures of trinuclear complexes 4 (left), 4a (middle) and 5 (right) in the solid state. Hydrogen atoms and co-crystallized non-coordinating solvent molecules are omitted for clarity. Note that PF6− for 4 is half occupied. For 4a only main part (A) is displayed, see Fig. S6-6† for more details. Selected bond lengths and angles are given in the ESI, section 6.† | ||
Assignment of the 1H NMR resonances (two sets) was confirmed by COSY-NMR experiments.
The respective 31P{1H} NMR spectrum resembles that of complex 3, featuring two doublets (δ = −17.0 and −19.5 ppm). This coupling again indicates slightly different chemical environments for the phosphorus nuclei and a strong coupling between the phosphorus atoms through P–Cu–P or the P–Cu–Cu–Cu–P chain. The positively charged ion [4-MeCN]+ was identified by HRMS.
By maintaining the same reaction conditions as for 4 and only varying the crystallization conditions (no MeCN), [dpfam2Cu3][PF6] (4a) was obtained (Scheme 3). The disordered solid-state structure (ratio 84:16) reveals a very similar scaffold to that of 4 with a bent Cu3 chain (angle 122.07(3)°), but without any coordinated solvent molecules. As no acetonitrile molecule is attached, both Cu–Cu-distances are roughly of the same length (2.5736(7) Å and 2.5577(8) Å) and even shorter than the Cu1–Cu2 distance in 4. Disregarding the cuprophilic contacts, the outer copper atoms are set in a distorted trigonal planar coordination sphere by two phosphorus atoms and one nitrogen atom, whilst the central Cu2 is linearly coordinated by two nitrogen atoms. 4a also exhibits yellow luminescence at room temperature. Yet, a different emission curve shape than for 4 is observed (see photoluminescence section). However, no significant differences between 4 and 4a can be observed by NMR spectroscopy, indicating a similar behaviour in solution and a loss of the MeCN-coordination of 4 in solution.
Similarly, the Ag3 complex [dpfam2Ag3(thf)2][BF4] (5) was obtained by reacting Kdpfam with 1.5 equivalents of AgBF4 to yield compound 5 as pale-yellow crystals (Scheme 3). The latter emit a barely visible orange luminescence upon irradiation with UV light. The structural and coordination motif in the solid state resembles 4 (Fig. 4). However, in 5, there is one THF molecule close to both outer silver atoms resulting in a more symmetrical arrangement than in 4. Comparing the oxygen-silver distances (Ag1–O2 2.674(2) Å and Ag3–O1 2.552(2) Å) with literature values classifies them as rather weakly bound (ranking between dative bonding and van der Waals interaction) and within typical Ag–THF distances.73–77 The silver atoms feature a bent chain with an angle of 133.861(10)°. The intermetallic distances of 2.8987(3) (Ag1–Ag2) and 2.8893(3) Å (Ag2–Ag3) are in the range of argentophilic interactions.38 The coordination of Ag2 by N1 and N4 is approximately linear (173.66(9)°). Ag–N and Ag–P bond distances are consistent with literature values, except for Ag1–P2, which is slightly elongated.59,78,79
In the 31P{1H} NMR spectrum, a broad misshaped doublet at δ = −12.0 ppm (nJ = 385.8 Hz) is observed, indicating higher order coupling with the silver nuclei and/or through the P–Ag–P or P–Ag3–P chain.61,62,80,81 In the 1H NMR spectrum, the NCHN resonance was detected as a multiplet at δ = 7.61–7.52 ppm. This might be attributed to a coupling to the silver atoms. The elemental composition of 5 could be verified by HRMS with a signal at m/z = 1447.079, which corresponds to [5-THF]+ (cal. [C37H58M4P4Ag3]+m/z = 1447.076).
The synthesis of an analogous trinuclear gold complex was attempted under various conditions but it could not be isolated as a crystalline product. However, it was detected in ESI HRMS as the positively charged [dpfam2Au3]+ fragment at m/z = 1717.266 (cal. [C74H58N4P4Au3]+ 1717.260).
Photoluminescence properties
The presented copper and gold complexes (1, 3, 4 and 4a) appear yellow or pale orange in the solid (polycrystalline) state, whereas the silver complexes (2 and 5) are nearly colourless. All of them show an efficient visible photoluminescence (PL) upon UV excitation at temperatures below 100 K. The PL excitation (PLE) and emission spectra over a temperature interval of 20–295 K are collected in Fig. 5 and 6 for di- and, trinuclear complexes, respectively. All major emission bands correspond to phosphorescence (except complex 2 at T > 100 K), as indicated by long PL decay times (τ) under pulsed UV laser excitation (ESI, Fig. S2.2-1†).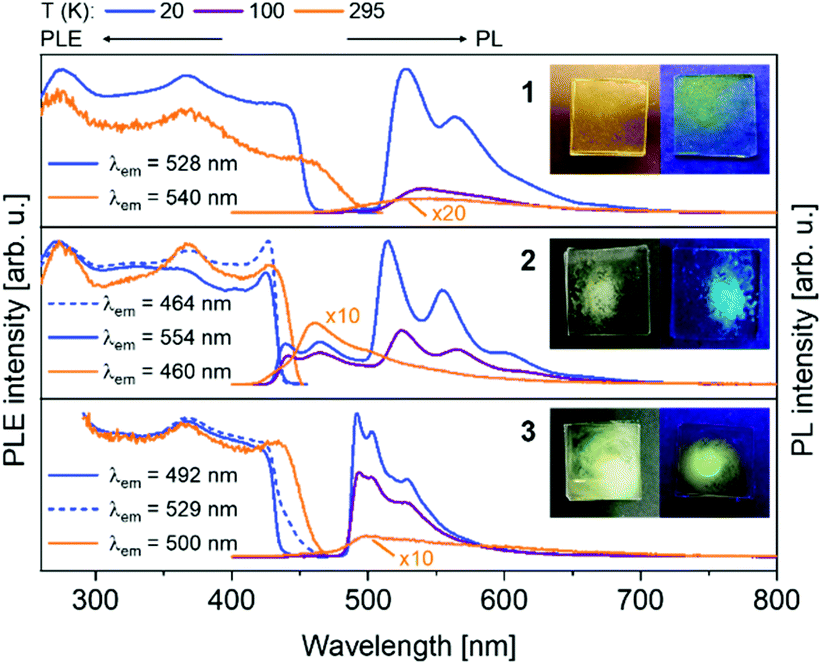 | ||
| Fig. 5 Photoluminescence emission (PL) and excitation (PLE) spectra of solid (polycrystalline) binuclear complexes 1, 2 and 3 at different temperatures. PL was excited at λexc = 350 nm and PLE spectra were recorded at the depicted wavelengths (λem). Photographs of the PL samples: left: daylight; right: UV lamp (λexc = 365 nm). | ||
 | ||
| Fig. 6 Photoluminescence emission (PL) and excitation (PLE) spectra of polycrystalline solid 4, 4a and 5 at different temperatures. PL was excited at λexc = 350 nm (330 nm for 5) and PLE spectra were recorded at the depicted wavelengths (λem). PL of 5 is not displayed at 295 K due to a very weak signal. Photographs of the PL samples: left: daylight; right: UV lamp (λexc = 365 nm). | ||
Specifically, the binuclear complexes 1–3 show similar PLE spectra with the onset at ca. 450 nm and phosphorescence bands centred at ca. 520 (1, 2) and 500 nm (3). The vibronic emission patterns observed at low temperatures are similar for 1 and 2 and distinct for 3, in line with the structural differences between these complexes (see above). In contrast to 1, 3 and other compounds, the silver compound 2 also demonstrates a fluorescence band at ca. 460 nm (τ < 5 ns), i.e. with a small Stokes shift, which dominates the PL at ambient temperature (Fig. 5). For all binuclear complexes, the emission intensity strongly decreases upon warming the samples up to room temperature. Accordingly, their PL efficiency at 295 K was estimated to be less than 1%. The trinuclear copper complex 4 also shows PLE spectra with an onset at ca. 450 nm and phosphorescence at ca. 500 nm (Fig. 6).
Its companion 4a only differs by the absence of a solvent (acetonitrile) molecule coordinated to the Cu3 chain in 4. As expected, the PL spectra of both compounds are very similar in terms of the shape (PLE) and spectral position (PLE and PL). Interestingly, the vibronic pattern in the low-temperature PL spectra of 4 (contributed by a ∼1300 cm−1 vibration) is absent in 4a. Accordingly, the vibronic “modulation” of the emission of 4 can be tentatively attributed to the coordinated acetonitrile molecule. In comparison to 4, the PLE/PL spectra of 5 are blue shifted to ca. 360/460 nm (at 20 K). Similar to 1–3, the emission (phosphorescence) intensity of trinuclear complexes, in particular of 5, strongly decreases by increasing the temperature above 100 K. This decrease roughly correlates with shortening of the PL lifetimes.
Compounds 4, 4a and 5 thus represent examples of complexes with metallophilic interactions, but very weak PL at ambient temperature, apparently due to efficient nonradiative excited state relaxation.
Theoretical investigation
The character of the underlying excited states is assessed using time-dependent density-functional theory, employing the PBE0 functional.82,83 The dhf-TZVPP basis set was used for Cu, Ag, Au, including suitable effective core potentials (ECPs) for the latter two. The dhf-SVP basis set was used for all other elements.84–86 All calculations were done with the TURBOMOLE program suite.87–89 Further computational details are given in the ESI.† Calculated triplet excitation energies in ground state geometry already follow the experimentally observed trends (see ESI†). However, due to the high computational cost involved with triplet state relaxation, we limit ourselves to a discussion of the absorption spectra. These are presented in Fig. 7. A rough comparison between calculated absorption spectra and experimental PLE shows both to be in reasonable agreement, with the calculated spectra appearing blue-shifted by approximately 0.5 eV, compared to the onset of the PLE. Some of this discrepancy may be due to a neglect of intermolecular interactions present in the solid state, which are not easily taken into account from a computational point of view. For the binuclear complexes 1–3 the absorption onset is observed to increase slightly in the order Cu, Au, Ag. According to a Mulliken population analysis90 the character of the first absorption band of 1 features significant contributions from the Cu 3d (13%) and nitrogen 3p (5%) or phosphorus 4p (2%) states. Upon excitation, the electron density is partly transferred to the π*-orbitals of the N–C–N subunit, as well as to the π*-orbitals of the phenyl groups. The bands therefore include a metal-to-ligand charge transfer (MLCT) character. The binuclear silver compound (2) shows a slightly lower d-character while the N, P p-character increases accordingly (4% and 9%). While overall of similar character, the bands appear blue shifted compared to 1. The binuclear gold complex (3) differs from the former two systems by a substantial structural change (see above). The P–M–P angles increase to approximately 160° in the optimized geometry. Excited states involving Au 5d admixtures are further shifted towards higher energies. | ||
| Fig. 7 Simulated absorption spectra for 1–5 obtained by superimposing Gaussian functions (corresponding oscillator strengths are depicted in green) with a full width at half maximum of 0.3 eV at the 100 lowest-lying singlet excitations. The non-relaxed transition densities are presented for selected regions as indicated by grey dotted lines, where a density gain is depicted by blue contours, while a density loss is shown in red. | ||
This is also evident from the non-relaxed difference densities depicted in Fig. 7.91 Experimentally observed trends for 1–3 could hence be well reproduced by these calculations. The simulated absorption spectra of the cationic trinuclear compounds 4, 4a and 5 are blue shifted compared to the spectra of 1–3. Compounds 4 and 4a formally only differ in an additional MeCN unit at the (outer) Cu1 atom, resulting in an increase in the respective Cu 3d-character of the transitions involved, when compared to 4a (e.g. Cu1 and Cu2 10%, Cu3 4% for the first band). In accordance with the crystal structure, additional two THF equivalents, coordinated to the outer silver atoms, were included, for cationic complex 5. The excited states are found at significantly higher energies when compared to 4 or 4a mirroring the experimental results. From the non-relaxed difference densities, the density gain is seen to be primarily located at the π-system of the phenyl groups and remains otherwise similar in nature.
Conclusions
Herein we have presented the synthesis and optical properties of homometallic bi- and trinuclear complexes of the coinage metals Cu, Ag and Au featuring the tetradentate PNNP ligand dpfam. The concept of hard and soft acids and bases applies remarkably well for complexes 1–3.92,93 While the copper and silver atoms in 1 and 2 occupy two of each (phosphorus and nitrogen) donor sites in a distorted tetrahedral coordination environment, the gold cations in 3 exclusively coordinate to the diphenyl phosphine moieties (in solid state), leading to a charge separation. A higher metal loading can be achieved by simple variation in equivalents used, resulting in the trinuclear copper and silver complexes 4, 4a and 5. All compounds show photoluminescence (mostly phosphorescence) at low temperatures which is still observed (except 5) at ambient temperatures. The varying MLCT character of the systems was investigated by TDDFT calculations, involving a transfer of electron density from the metal d orbitals into the π* ligand orbitals. The tetradentate pincer ligand system offers a multitude of possibilities for different metal loadings, resulting in distinct optical properties. Especially the binuclear gold complex, with its unusual charge separation, is being used to explore different kinds of mixed complexes in ongoing complementary work.Author contributions
The manuscript was written through contributions of all authors. M. D. synthesized and analysed all presented compounds under the supervision of P. W. R. Solid state PL was conducted by M. D. and S. L. and the data interpreted by S. L. and M. M. K. Theoretical investigations were done by M. K. under the supervision of W. K. The original idea was from P. W. R. who supervised the work and interpreted the data. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr C. Schoo and Dr M. T. Gamer are thankfully acknowledged for helping with the X-ray data refinement. Thanks go also to Ms H. Berberich for the NMR measurements and Dr T. Simler for his support on the interpretation of the NMR spectra and to Dr T. P. Seifert for fruitful discussion. Financial support by the DFG funded transregional collaborative research centre SFB/TRR 88 “Cooperative Effects in Homo and Heterometallic Complexes (3MET)” is gratefully acknowledged (projects C1, C3 and C7). M. D. thanks the “Fonds der Chemischen Industrie” for their generous fellowship.Notes and references
- M. Bardajía and A. Laguna, Eur. J. Inorg. Chem., 2003, 2003, 3069–3079 CrossRef.
- M. J. Calhorda, C. Ceamanos, O. Crespo, M. Concepción Gimeno, A. Laguna, C. Larraz, P. D. Vaz and M. D. Villacampa, Inorg. Chem., 2010, 49, 8255–8269 CrossRef CAS PubMed.
- P. Pyykkö, J. Li and N. Runeberg, Chem. Phys. Lett., 1994, 218, 133–138 CrossRef.
- P. Ai, A. A. Danopoulos, P. Braunstein and K. Y. Monakhov, Chem. Commun., 2014, 50, 103–105 RSC.
- S. Raju, H. B. Singh and R. J. Butcher, Dalton Trans., 2020, 49, 9099–9117 RSC.
- A. Burini, J. P. Fackler, R. Galassi, T. A. Grant, M. A. Omary, M. A. Rawashdeh-Omary, B. R. Pietroni and R. J. Staples, J. Am. Chem. Soc., 2000, 122, 11264–11265 CrossRef CAS.
- B. Singh, A. Thakur, M. Kumar and D. Jasrotia, Mater. Chem. Phys., 2017, 196, 52–61 CrossRef CAS.
- R. J. Oeschger and P. Chen, J. Am. Chem. Soc., 2017, 139, 1069–1072 CrossRef CAS PubMed.
- C. Janiak and R. Hoffmann, J. Am. Chem. Soc., 1990, 112, 5924–5946 CrossRef CAS.
- J. Echeverría, Chem. Commun., 2018, 54, 6312–6315 RSC.
- C. Yin, F. Huo, W. Wang, R.-H. Ismayiloy, G. Lee, C. Yeh, S. Peng and P. Yang, Chin. J. Chem., 2009, 27, 1295–1299 CrossRef CAS.
- J. Zhang and L.-G. Zhu, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2012, 42, 1071–1077 CrossRef CAS.
- Z. Zhu, R. C. Fischer, J. C. Fettinger, E. Rivard, M. Brynda and P. P. Power, J. Am. Chem. Soc., 2006, 128, 15068–15069 CrossRef CAS PubMed.
- E. Laurila, L. Oresmaa, J. Hassinen, P. Hirva and M. Haukka, Dalton Trans., 2013, 42, 395–398 RSC.
- L. Oresmaa, M. A. Moreno, M. Jakonen, S. Suvanto and M. Haukka, Appl. Catal., A, 2009, 353, 113–116 CrossRef CAS.
- E. Laurila, R. Tatikonda, L. Oresmaa, P. Hirva and M. Haukka, CrystEngComm, 2012, 14, 8401–8408 RSC.
- M. A. Ciriano, S. Sebastián, L. A. Oro, A. Tiripicchio, M. T. Camellini and F. J. Lahoz, Angew. Chem., Int. Ed. Engl., 1988, 27, 402–403 CrossRef.
- C. Tejel, M. A. Ciriano, J. A. López, F. J. Lahoz and L. A. Oro, Angew. Chem., Int. Ed., 1998, 37, 1542–1545 CrossRef CAS PubMed.
- E. J. Fernández, P. G. Jones, A. Laguna, J. M. López-de-Luzuriaga, M. Monge, J. Pérez and M. E. Olmos, Inorg. Chem., 2002, 41, 1056–1063 CrossRef PubMed.
- E. M. Gussenhoven, M. M. Olmstead, J. C. Fettinger and A. L. Balch, Inorg. Chem., 2008, 47, 4570–4578 CrossRef CAS PubMed.
- M. Kim, T. J. Taylor and F. P. Gabbaï, J. Am. Chem. Soc., 2008, 130, 6332–6333 CrossRef CAS PubMed.
- A. Aliprandi, D. Genovese, M. Mauro and L. D. Cola, Chem. Lett., 2015, 44, 1152–1169 CrossRef CAS.
- X.-Y. Chang, G.-T. Xu, B. Cao, J.-Y. Wang, J.-S. Huang and C.-M. Che, Chem. Sci., 2017, 8, 7815–7820 RSC.
- M. Alcarazo, K. Radkowski, G. Mehler, R. Goddard and A. Fürstner, Chem. Commun., 2013, 49, 3140–3142 RSC.
- M. Jansen, Angew. Chem., Int. Ed. Engl., 1987, 26, 1098–1110 CrossRef.
- P. K. Mehrotra and R. Hoffmann, Inorg. Chem., 2002, 17, 2187–2189 CrossRef.
- V. W.-W. Yam and K. Kam-Wing Lo, Chem. Soc. Rev., 1999, 28, 323–334 RSC.
- V. W.-W. Yam and E. C.-C. Cheng, Chem. Soc. Rev., 2008, 37, 1806–1813 RSC.
- V. W.-W. Yam and K. M.-C. Wong, Chem. Commun., 2011, 47, 11579–11592 RSC.
- V. W.-W. Yam, Pure Appl. Chem., 2013, 85, 1321–1329 CAS.
- V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS PubMed.
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370–412 RSC.
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2008, 37, 1931–1951 RSC.
- T. A. C. A. Bayrakdar, T. Scattolin, X. Ma and S. P. Nolan, Chem. Soc. Rev., 2020, 49, 7044–7100 RSC.
- T. P. Seifert, V. R. Naina, T. J. Feuerstein, N. D. Knöfel and P. W. Roesky, Nanoscale, 2020, 12, 20065–20088 RSC.
- P. Pyykkö, Angew. Chem., Int. Ed., 2004, 43, 4412–4456 CrossRef PubMed.
- V. W.-W. Yam and E. Chung-Chin Cheng, in Photochemistry and photophysics of coordination compounds II, ed. V. Balzani, S. Campagna and A. Barbieri, Springer, Berlin, Heidelberg, 2007, vol. 281, pp. 269–309 Search PubMed.
- H. Schmidbaur and A. Schier, Angew. Chem., Int. Ed., 2015, 54, 746–784 CrossRef CAS PubMed.
- H. L. Hermann, G. Boche and P. Schwerdtfeger, Chem. – Eur. J., 2001, 7, 5333–5342 CrossRef CAS.
- M. B. Brands, J. Nitsch and C. F. Guerra, Inorg. Chem., 2018, 57, 2603–2608 CrossRef CAS PubMed.
- J. Li and P. Pyykkö, Chem. Phys. Lett., 1992, 197, 586–590 CrossRef CAS.
- R. L. White-Morris, M. M. Olmstead and A. L. Balch, J. Am. Chem. Soc., 2003, 125, 1033–1040 CrossRef CAS PubMed.
- N. L. Coker, J. A. Krause Bauer and R. C. Elder, J. Am. Chem. Soc., 2004, 126, 12–13 CrossRef CAS PubMed.
- J. R. Shakirova, E. V. Grachova, V. V. Gurzhiy, I. O. Koshevoy, A. S. Melnikov, O. V. Sizova, S. P. Tunik and A. Laguna, Dalton Trans., 2012, 41, 2941–2949 RSC.
- Q.-M. Wang, Y.-A. Lee, O. Crespo, J. Deaton, C. Tang, H. J. Gysling, M. Concepción Gimeno, C. Larraz, M. D. Villacampa, A. Laguna and R. Eisenberg, J. Am. Chem. Soc., 2004, 126, 9488–9489 CrossRef CAS PubMed.
- S. Schäfer, M. T. Gamer, S. Lebedkin, F. Weigend, M. M. Kappes and P. W. Roesky, Chem. – Eur. J., 2017, 23, 12198–12209 CrossRef PubMed.
- M. T. Dau, J. R. Shakirova, A. J. Karttunen, E. V. Grachova, S. P. Tunik, A. S. Melnikov, T. A. Pakkanen and I. O. Koshevoy, Inorg. Chem., 2014, 53, 4705–4715 CrossRef CAS PubMed.
- N. Tsukada, O. Tamura and Y. Inoue, Organometallics, 2002, 21, 2521–2528 CrossRef CAS.
- L. Wesemann, H. Schubert, H. Mayer and S. Wernitz, DE102011079857A12013.01.31, 2011.
- K.-S. Son, D. M. Pearson, S.-J. Jeon and R. M. Waymouth, Eur. J. Inorg. Chem., 2011, 4256–4261 CrossRef CAS.
- S. Tanaka, A. Yagyu, M. Kikugawa, M. Ohashi, T. Yamagata and K. Mashima, Chemistry, 2011, 17, 3693–3709 CrossRef CAS PubMed.
- Y. Yamaguchi, K. Yamanishi, M. Kondo and N. Tsukada, Organometallics, 2013, 32, 4837–4842 CrossRef CAS.
- C. Zovko, S. Bestgen, C. Schoo, A. Görner, J. M. Goicoechea and P. W. Roesky, Chem. – Eur. J., 2020, 26, 13191–13202 CrossRef CAS PubMed.
- G. S. Day, B. Pan, D. L. Kellenberger, B. M. Foxman and C. M. Thomas, Chem. Commun., 2011, 47, 3634–3636 RSC.
- L.-C. Liang, P.-S. Chien, J.-M. Lin, M.-H. Huang, Y.-L. Huang and J.-H. Liao, Organometallics, 2006, 25, 1399–1411 CrossRef CAS.
- F. A. Cotton, X. Feng, M. Matusz and R. Poli, J. Am. Chem. Soc., 1988, 110, 7077–7083 CrossRef CAS.
- A. C. Lane, M. V. Vollmer, C. H. Laber, D. Y. Melgarejo, G. M. Chiarella, J. P. Fackler, X. Yang, G. A. Baker and J. R. Walensky, Inorg. Chem., 2014, 53, 11357–11366 CrossRef CAS PubMed.
- Y. Hattori, M. Nishikawa, T. Kusamoto, S. Kume and H. Nishihara, Inorg. Chem., 2014, 53, 2831–2840 CrossRef CAS PubMed.
- M. S. Balakrishna, R. Venkateswaran and S. M. Mobin, Inorg. Chim. Acta, 2009, 362, 271–276 CrossRef CAS.
- A. Cingolani, Effendy, F. Marchetti, C. Pettinari, R. Pettinari, B. W. Skelton and A. H. White, Inorg. Chem., 2004, 43, 4387–4399 CrossRef CAS PubMed.
- A. Pidcock, Chem. Commun., 1968, 92–92 RSC.
- R. H. Crabtree, The organometallic chemistry of the transition metals, Wiley, Hoboken, New Jersey, 6th edn, 2014 Search PubMed.
- B. K. Tate, C. M. Wyss, J. Bacsa, K. Kluge, L. Gelbaum and J. P. Sadighi, Chem. Sci., 2013, 4, 3068–3074 RSC.
- R. Usón, A. Laguna, M. Laguna, E. Fernandez, M. D. Villacampa, P. G. Jones and G. M. Sheldrick, J. Chem. Soc., Dalton Trans., 1983, 1679–1685 RSC.
- S. Bestgen, M. T. Gamer, S. Lebedkin, M. M. Kappes and P. W. Roesky, Chem. – Eur. J., 2015, 21, 601–614 CrossRef CAS PubMed.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC.
- M. Stollenz, Chem. – Eur. J., 2019, 25, 4274–4298 CrossRef CAS PubMed.
- W.-H. Chan, S.-M. Peng and C.-M. Che, J. Chem. Soc., Dalton Trans., 1998, 2867–2872 RSC.
- T. Zhang, C. Chen, Y. Qin and X. Meng, Inorg. Chem. Commun., 2006, 9, 72–74 CrossRef CAS.
- Y. Nakajima, Y. Shiraishi, T. Tsuchimoto and F. Ozawa, Chem. Commun., 2011, 47, 6332–6334 RSC.
- T.-H. Huang and M.-H. Zhang, Aust. J. Chem., 2014, 67, 887–894 CrossRef CAS.
- M. Abdul Jalil, T. Yamada, S. Fujinami, T. Honjo and H. Nishikawa, Polyhedron, 2001, 20, 627–633 CrossRef CAS.
- A. Ghisolfi, C. Fliedel, P. de Frémont and P. Braunstein, Dalton Trans., 2017, 46, 5571–5586 RSC.
- E. Tomás-Mendivil, R. García-Álvarez, S. E. García-Garrido, J. Díez, P. Crochet and V. Cadierno, J. Organomet. Chem., 2013, 727, 1–9 CrossRef.
- S. Martínez de Salinas, Á. L. Mudarra, J. Benet-Buchholz, T. Parella, F. Maseras and M. H. Pérez-Temprano, Chem. – Eur. J., 2018, 24, 11895–11898 CrossRef PubMed.
- C. S. Browning, D. H. Farrar and D. C. Frankel, Z. Kristallogr. - New Cryst. Struct., 1997, 212, 201–202 CAS.
- S. Porcel and A. M. Echavarren, Angew. Chem., Int. Ed., 2007, 46, 2672–2676 CrossRef CAS PubMed.
- Y. Yuan, H.-L. Han, S. Lin, Y.-Z. Cui, M. Liu, Z.-F. Li, Q.-H. Jin, Y.-P. Yang and Z.-W. Zhang, Polyhedron, 2016, 119, 184–193 CrossRef CAS.
- F. Hung-Low, A. Renz and K. K. Klausmeyer, Eur. J. Inorg. Chem., 2009, 2994–3002 CrossRef CAS.
- T. Simler, P. Braunstein and A. A. Danopoulos, Dalton Trans., 2016, 45, 5122–5139 RSC.
- J. M. Jenkins and B. L. Shaw, J. Chem. Soc. A, 1966, 770–775 RSC.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- F. Weigend and A. Baldes, J. Chem. Phys., 2010, 133, 174102 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- D. Figgen, G. Rauhut, M. Dolg and H. Stoll, Chem. Phys., 2005, 311, 227–244 CrossRef CAS.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- F. Furche, R. Ahlrichs, C. Hättig, W. Klopper, M. Sierka and F. Weigend, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 91–100 CAS.
- TURBOMOLE V7.4.1 2019, TURBOMOLE GmbH, 1989–2007, University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, http://www.turbomole.com, access date.
- R. S. Mulliken, J. Chem. Phys., 1955, 23, 1833–1840 CrossRef CAS.
- M. Kühn and F. Weigend, J. Chem. Phys., 2014, 141, 224302 CrossRef PubMed.
- R. G. Pearson, Coord. Chem. Rev., 1990, 100, 403–425 CrossRef CAS.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2041033–2041038 and 2094052. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt02226a |
| This journal is © The Royal Society of Chemistry 2021 |