
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Helical donor–acceptor platinum complexes displaying dual luminescence and near-infrared circularly polarized luminescence†
Pablo
Vázquez-Domínguez
a,
Océane
Journaud
b,
Nicolas
Vanthuyne
c,
Denis
Jacquemin
 *d,
Ludovic
Favereau
*b,
Jeanne
Crassous
*b and
Abel
Ros
*a
*d,
Ludovic
Favereau
*b,
Jeanne
Crassous
*b and
Abel
Ros
*a
aInstitute for Chemical Research (CSIC-US), C/Américo Vespucio 49, E-41092 Seville, Spain. E-mail: abel.ros@iiq.csic.es
bUniv Rennes, CNRS, ISCR-UMR 6226, F-35000 Rennes, France. E-mail: ludovic.favereau@univ-rennes1.fr; jeanne.crassous@univ-rennes1.fr
cAix Marseille University, CNRS, Centrale Marseille, iSm2, Marseille, France
dLaboratoire CEISAM, UMR 6230, CNRS, Université de Nantes, Nantes, France. E-mail: Denis.Jacquemin@univ-nantes.fr
First published on 13th September 2021
Abstract
A series of chiral platina[5]helicenes displaying dual luminescence, i.e., fluorescence between 450 and 600 nm and red/NIR phosphorescence between 700 and 900 nm, has been synthesised, characterised and studied by first-principle calculations. This unusual behavior has been attributed to limited electronic interactions between the d orbitals of the metal and the π-orbitals of the organic ligand on the excited-state. Accordingly, the electron richness of the donor group on the helical ligand does not affect the energy of the phosphorescence process but does play a role on its efficiency. Interestingly, near-infrared circularly polarized luminescence can be obtained for the three complexes with dissymmetry factors up to 3 × 10−3 at 750 nm.
The design of chiral π-conjugated materials that are able to interact specifically with a circularly polarized light (CP-Light) has recently attracted considerable attention due to the potential of the latter in the fields of (chir)optoelectronics including stereoscopic displays and organic light-emitting diodes (OLEDs), optical information processing, as well as in bio-imaging and chiral sensing.1 In this context, chiral lanthanide and chromium complexes have been intensively investigated for their high CP luminescence (CPL) intensity owing to their magnetically allowed transitions, resulting in luminescence dissymmetry factors, i.e., glum = 2(IL − IR)/(IL + IR), above unity,2 and enable access to efficient near-infrared CPL emitters.3 Recently, chiral organic and organometallic materials have received more attention as potential CP-Light absorbers and emitters due to their readily tuneable photo-physical and chiro-optical properties and their relatively simple integration in optoelectronic devices such as CP-OLEDs, chiral photovoltaics and transistors.4 Although this class of compounds often displays higher luminescence quantum yields than lanthanide and chromium complexes thanks to their electronically allowed transitions, their CPL intensity dramatically decreases with typical glum falling in the 10−4 to 10−2 range.5
Accordingly, one of the major challenges for such CPL emitters is to identify the key electronic and structural factors controlling the CPL intensity, which would allow establishing molecular design rules leading to higher glum values.6 Indeed, supramolecular assembly,7 and other intermolecular approaches such as energy transfer, charge transfer and excimer involving chiral molecules have resulted in promising to impressive CPL values,8 with glum up to 0.15,9 opening new opportunities for chiral organic and organometallic materials.5d Despite this achievement, the development of far-red and near-infrared (NIR) molecular chiral emitters for bio-imaging or optical data transmission remains a considerable challenge compared with the more classical blue, green and yellow CPL emitters.10 Indeed, non-radiative deactivation pathways become more efficient as the gap becomes smaller, an effect commonly dubbed ‘energy gap law’ that is critical when reaching the NIR region. Several chiral compounds displaying chiro-optical properties beyond 600 nm have been reported, based for instance on extended helical π-systems;11 however, only a few have shown a CPL over 700 nm (compounds A & B, Fig. 1).12
 | ||
| Fig. 1 Top: Chemical structures of the molecular CPL emitters displaying CPL maxima beyond 700 nm in solution (see ref. 8 and 9). Bottom: Chemical structures of the investigated helical donor–acceptor platinum(II) complexes in this report. | ||
Moreover, the measured glum values in such emitters hardly exceed 1.0 × 10−3, owing to the difficulty to efficiently combine a highly delocalized π-conjugated system with a chiral environment. In fact, the molecular CPL emitters displaying the most red-shifted response rely on a carbohelicene- or Schiff-platinum(II) complex, which afford glum of ±0.3 × 10−3 at 720 nm and ±2.8 × 10−3 at 731 nm, respectively (C & D, Fig. 1).13 Indeed, using Pt complexes to generate NIR phosphorescence in chiral systems appears as a good strategy to generate NIR CPL activity.13b,14 Furthermore, cyclometalated phosphorescent d8 metal complexes containing π-conjugated ligands with N and/or C donor atoms have been extensively studied and used in the development of high-performance OLEDs.13b,14c,15
Following a previously developed method by some of us using cycloplatination to generate helical platinacycles displaying chirality from the helix and room temperature phosphorescence from the platinum,16 herein, we explore a new approach towards the development of far-red and NIR chiral luminescent material based on a helical donor–acceptor π-conjugated structure incorporating a platinum metallic ion, namely, a push–pull platina[5]helicene. This new family of chiral organometallic complexes shows a dual emission process with a modulation of the chiro-optical and photo-physical properties resulting from the charge-transfer character of the helical organic ligand. Interestingly, increasing the electric dipole moment of the latter by tuning the donor substituent strength increases dramatically the efficiency of the phosphorescence process, which shows an emission maximum at 770 nm associated with a glum of 3.0 × 10−3. Such intensity of NIR CPL is among the highest achieved so far at the molecular level and provides a platform for designing future chiral dyes showing responses in the low-energy region of the spectrum.
Synthesis and structural analysis (experimental and theoretical)
For the synthesis of the Pt-helicene complexes, a general cyclo-platination/ligand substitution two-step process was followed (Scheme 1). First, the cyclo-platination reaction of the axially chiral arylisoquinolines 1–317 with [Pt(DMSO)2Cl2] under Na2CO3 (2 eq.)/toluene (reflux) conditions,16c gives the corresponding [Pt(C,N)(DMSO)Cl] complexes 4–5, which can then be subjected to DMSO → PPh3 ligand substitution to afford the desired platinahelicenes [Pt(C,N)PPh3Cl]18 in 71%–92% yields after column chromatography on silica gel. The racemic mixtures were resolved using semi-preparative chiral HPLC separation to give the enantiopure complexes with ee's up to 99% (further details can be found in the ESI†).
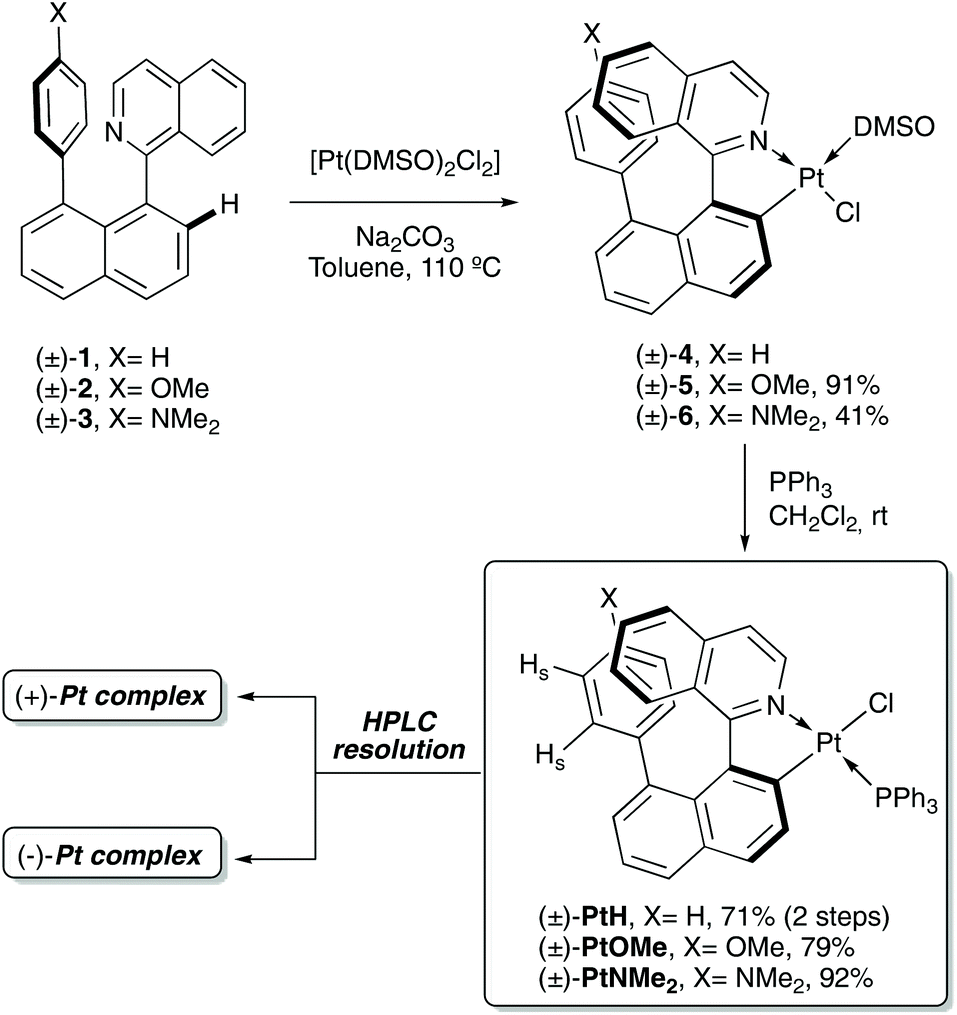 | ||
| Scheme 1 Synthesis of [Pt(C,N)(PPh3)Cl] complexes PtH, PtOMe and PtNMe2. | ||
Unfortunately, none of these complexes could be obtained as single crystal to be analysed by X-ray diffraction; however, the nuclear magnetic resonance (NMR) spectroscopy analysis in solution confirmed the formation of the expected complexes with a trans-N,P configuration. In more details, the 1H NMR spectra show the deshielding of the isoquinoline ortho-C–H protons, which appear as a doublet of doublet at 9.50 ppm with a 3JH,P ∼ 3.4 Hz (see ESI†), and the 31P NMR spectra exhibit a singlet peak around 22.7 ppm flanked by 195Pt satellites signals with 1JPtP ∼ 4300 Hz, in accordance with similar [Pt(C,N)(PPh3)Cl] complexes presenting a trans configuration between the phosphine moiety and the nitrogen atom.19 Moreover, the spectra of the three complexes display a shielding of the aryl protons assigned to the substituent at the 8-position of the naphthalene fragment of the ligand (HS, Scheme 1), owing to the π–π stacking with the isoquinoline moiety (see the ESI†). In the case of complex PtH, the formation of some amount of the cis-N,P isomer was observed (ca. 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 trans/cis mixture in CDCl3). Curiously, when this NMR sample in CDCl3 was heated at 40 °C, a slow evolution to the cis-N,P isomer was observed. The same isomerization was instantly achieved by dissolving the trans-complex in acetone, as evidenced by the corresponding 1H NMR spectra (see the ESI†).20 The reverse cis-to-trans N,P isomerization could also be observed by dissolving the cis-N,P isomer in CD2Cl2 and heating at 40 °C for two days.21
:1 trans/cis mixture in CDCl3). Curiously, when this NMR sample in CDCl3 was heated at 40 °C, a slow evolution to the cis-N,P isomer was observed. The same isomerization was instantly achieved by dissolving the trans-complex in acetone, as evidenced by the corresponding 1H NMR spectra (see the ESI†).20 The reverse cis-to-trans N,P isomerization could also be observed by dissolving the cis-N,P isomer in CD2Cl2 and heating at 40 °C for two days.21
Photo-physical and chiro-optical properties (experimental and theoretical)
Fig. 2 shows the ultraviolet–visible (UV–vis) absorption spectra of the helical platinum complexes, which display a similar pattern with two dominant bands between 300 and 400 nm (ε ∼ 5–10 × 103 M−1 cm−1) and below 300 nm (ε ∼ 15–30 × 103 M−1 cm−1) that are assigned to π → π* transitions and to an additional mixture of metal-to-ligand (ML) and intra-ligand (IL) charge-transfer excitations for the low-energy one (for comparison, the UV-vis spectra of the corresponding organic ligands are depicted in Fig. S4†). The weaker band between 400 and 550 nm (ε ∼ 1.5–2.5 × 103 M−1 cm−1), corresponding to the lowest energy excitation, involves charge-transfer (CT) transitions arising mainly from the metal to the isoquinoline ligand, with also a contribution of the naphthyl-substituted unit (ILCT transitions, Fig. 2). In the case of the latter, replacing a phenyl ring with a stronger N,N-dimethylaniline donor group greatly increases the intensity of the absorption throughout the whole spectrum and induces a small redshift of ca. 20 nm of the lowest energy band, which confirms the occurrence of ILCT for this excitation in addition to the classical MLCT. According to Time-Dependent Density Functional Theory (TD-DFT) calculations, the lowest vertical excitation of PtH, PtOMe, and PtNMe2 appear at 403, 408, and 421 nm, respectively. These values are blue-shifted as compared with the experimental results, which is the logical consequence of neglecting the vibronic couplings in the calculation. More importantly, the trend in the series with the ca. 20 nm redshift between PtH and PtOMe is reproduced. The electron density difference (EDD) plots for these lowest transitions are displayed in Fig. 2. As can be seen, the transitions involve both the ligand and the metal, the amino group playing a non-negligible donating role in PtNMe2.
 | ||
| Fig. 2 Top: UV-Vis and ECD spectra for the (+) and (−) enantiomers of PtH, PtOMe and PtNMe2 in dichloromethane solutions. Bottom: EDD plots (contour 0.002 au) for the lowest dipole-allowed transition of PtH, PtOMe and PtNMe2 with the crimson (blueberry) lobes representing increase (decrease) of electron density upon photo-excitation (the difference between the total excited state and ground state densities, as computed with TD-DFT and DFT, respectively). | ||
Electronic circular dichroism (ECD) of the investigated complexes further highlights the differences related to the nature of the substituent at the 8-position of the naphthalene fragment.
Although the three complexes show similar ECD signals in the high energy region, with a positive → negative couplet at 260 nm for the (+)-enantiomers (Fig. 2) attributed to the binaphthyl ECD-type signature,22 the low energy part of the spectra (between 300 and 550 nm), appears more redshifted for (+)-PtNMe2 than for (+)-PtOMe and (+)-PtH. In fact, the set of three signals at 320, 375 and 450 nm for the two latter compounds, respectively positive, negative and positive, are found at 350, 415, and 484 nm in the case of (+)-PtNMe2, which evidences the impact of the donor strength in these optical transitions.
Overall, this combined experimental and theoretical study shows that the metal significantly interacts with the helical π-conjugated system in the ground-state, explaining their optical properties. Expectedly, the role of the platinum atom is not only to stabilise the helical configuration but also to modify the electronic and chiro-optical properties of the organic donor–acceptor system.16 Such aspect is also clearly observed in the emissive properties of these new chiral derivatives. Indeed, the three complexes display a dual emission process with a fluorescence emission between 450 and 600 nm and red/NIR phosphorescence between 700 and 900 nm in diluted degassed solutions. Further experimental evidences of both fluorescence and phosphorescence emissions have been obtained by recording the luminescence response of the complexes under air atmosphere, which shows only a decrease of the NIR signal due to the quenching of the emitting triplet state by molecular oxygen. In addition, lifetime measurements indicate a decay of few nanoseconds for the high energy emission and of several hundred nanoseconds for the lower energy emission (Table 1 and ESI†). All these aspects confirm the assignments of the fluorescence and phosphorescence emissions.
| Complex (+)-PtH | Complex (+)-PtOMe | Complex (+)-PtNMe2 | |
|---|---|---|---|
| a In dichloromethane at 298 K. b In toluene at 298 K. c The experimental S1–T1 gaps of the complexes have been estimated from the luminescence spectra with the values of S1 and T1 obtained from the onsets of the fluorescence and phosphorescence bands, respectively (Fig. 4). | |||
|
λ
absa/nm (ε/M−1 cm−1) |
458 (1530), 373 (2265), 310 (5273), 266 (15615) |
465 (2294), 373 (3800), 310 (8055), 266 (25501) |
475 (2430), 373 (6968), 336 (10963), 310 (12430), 264 (32682) |
|
λ
ECDa/nm (Δε/M−1 cm−1) |
454 (+2), 377 (−4), 312 (+23), 271 (+31), 243 (−48) | 448 (+1), 381 (−0.5), 305 (+11), 274 (+21), 241 (−41) | 484 (+4), 415 (−2), 350 (+14), 296 (−1), 274 (+44), 238 (−46) |
|
λ
emb (nm)/ΔESTc |
520Fl, 765Ph/0.75 | 565Fl, 770Ph/0.73 | 590Fl, 780Ph/0.65 |
|
ϕ
Lumb (%) |
<1 | <1 | <1 |
|
τ
Lumb [ns] |
4.0 ns (28%) & 18.5 ns (72%) (560 nm, fluo.); 22.0 ns (26%) & 248.9 ns (74%) (770 nm, phos.) | 3.2 ns (45%) & 23.4 ns (55%) (560 nm, fluo.); 19.4 ns (4%) & 382.7 ns (96%) (770 nm, phos.) | 4.2 ns (580 nm, fluo.); 411 ns (770 nm, phos.) |
| |glum|b (×103) | 2.5 × 10−3 | 3.0 × 10−3 | 3.0 × 10−3 |
Such behaviour has been previously observed for platinum complexes exhibiting a weak electronic interaction between the d orbitals of the metal and the π-orbitals of the organic ligand.23 The observation of this dual luminescence suggests that this feature is present in the excited state of the present complexes. The theoretical calculations are consistent with these observations since vertical emission wavelengths at 523, 523, and 543 nm were obtained for PtH, PtOMe, and PtNMe2, respectively, by TD-DFT while U-DFT calculations predict vertical phosphorescence at 872, 862, and 871 nm for the same three derivatives. According to the calculations, these triplets are mainly ligand-centred with trifling role of the substituent (H, OMe, NMe2, see Fig. 3), which explains why the phosphorescence energies of the three dyes are more similar than their fluorescence counterparts. Interestingly, the intensity of the NIR phosphorescence increases significantly when going from the phenyl to the N,N-dimethylaniline donor substituent, suggesting an important role of the CT character of the ligand in the efficiency of this emission. Low photoluminescence quantum yields have been measured for the three complexes (>1%, Table 1), owing to probably high non-radiative rates arising from the presence of the monodentate ligands, along with the classically ‘energy gap law’ effect.
 | ||
| Fig. 3 Spin density difference plots (contour 0.005 au) of the lowest triplet state of PtH, PtOMe and PtNMe2 at its optimal geometry. | ||
To gain further insights on this luminescence behaviour, theoretical calculations were performed. It is generally admitted that inter-system crossing (ISC) takes place after relaxation to the lowest S1 state. At the optimal S1 geometry, there is only one triplet lower in energy than S1 according to theory. The experimental S1–T1 gaps have been experimentally estimated to 0.75, 0.73 and 0.65 eV for PtH, PtOMe and PtNMe2, respectively, following the trend obtained for the computed ones, i.e., 0.92, 0.92, and 0.59 eV for PtH, PtOMe and PtNMe2, respectively, indicating that ISC should be facilitated for PtNMe2 as the S–T gap is clearly the smallest. The computed spin–orbit coupling matrix elements (SOCMEs) for the S1–T1 transition are 17, 14 and 7 cm−1 in PtH, PtOMe and PtNMe2, respectively. While remaining moderate for a Pt-complex, which is consistent with the limited involvement of the metal in the excitation, these values are clearly large enough to induce ISC. This can be viewed as an intermediate situation, in which the rather large gaps make the ISC sufficiently slow to allow for radiative decay from S1 (fluorescence), while the non-trifling but not large SOCMEs allow for ISC. This is consistent with the experimental findings of dual emission. At the optimal T1 geometry, from which phosphorescence occurs, the SOCMEs for the T1–S0 process are very similar for the three dyes: 27, 24 and 24 cm−1 for PtH, PtOMe and PtNMe2, respectively, consistent with the density plots displayed in Fig. 3; therefore, the different behaviours noted experimentally should originate from various ISC kinetics rather than from different efficiencies in the actual phosphorescence radiative process.
The circularly polarized luminescence for each enantiomer of PtH, PtOMe and PtNMe2 was recorded in degassed toluene solutions, affording mirror-image signals corresponding only to the NIR phosphorescence emission, which can be probably explained by the weak efficiency of the fluorescence process (Fig. 4).
 | ||
| Fig. 4 (a) Luminescence and CPL spectra of the PtH (black line), PtOMe (blue line) and PtNMe2 (red line) complexes measured in toluene solution. Note that the raise of the signal at 820 nm is due to the limited sensitivity of the fluorimeter photodetector. | ||
On the basis of the obtained reliable signals, similar glum factors of ca. 2.5–3 × 10−3 for the three complexes were obtained. The three complexes exhibit a negative CPL response for the (+)-enantiomer, and their lowest ECD signals are positive, which indicates that different transitions are involved in the absorption and emission phenomena. Although the recorded glum values are similar to those of classical molecular CPL emitters,1c,5a,c the obtained NIR spectra are among the most red-shifted reported to date for chiral luminophores based on organic and platinum complexes. Especially, the combination of a strong push–pull organic ligand with metallic complexes appears an interesting approach for reaching far red and NIR emission.
Conclusions
In conclusion, we described here the synthesis of a new family of helical platinum(II) complexes bearing donating or accepting substituents within the helical organic ligand, and the joint experimental and theoretical investigation of their chiro-optical and photo-physical properties. We showed that the combination of platinum ion with an organic ligand possessing intramolecular charge-transfer abilities results in a dual luminescence behaviour with phosphorescence emission in the NIR region. Interestingly, the strength of the donor group on the helical ligand does not affect the energy of this emission process but rather plays a role on its efficiency through the tuning of the S–T gap and therefore of the ISC process. The three complexes exhibit corresponding CP phosphorescence with glum of 3 × 10−3, which is a significant value for this low energy region. We hope that this investigation may offer new opportunities to design innovative and efficient NIR CPL emitters.Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. F. and J. C. acknowledge the Ministère de l'Education Nationale, de la Recherche et de la Technologie and the Centre National de la Recherche Scientifique (CNRS). A. R. acknowledges the Spanish Ministerio de Ciencia e Innovación (Grants PID2019-106358GB-C21 and PID2019-106358GB-C22), European funding (ERDF), Junta de Andalucía (FQM-263 group). The PRISM core facility (Biogenouest©, UMS Biosit, Université de Rennes 1 – Campus de Villejean – 35043 RENNES Cedex, FRANCE) is acknowledged for the NMR characterizations and ECD measurements. The Caphter facility (ScanMAT, UMS 2001, Université de Rennes 1 – Campus de Beaulieu) is acknowledged for the photoluminescence characterizations of the compounds. This work used the computational resources of the CCIPL center installed in Nantes.Notes and references
- (a) M. Lindemann, G. Xu, T. Pusch, R. Michalzik, M. R. Hofmann, I. Žutić and N. C. Gerhardt, Nature, 2019, 568, 212–215 CrossRef CAS PubMed; (b) H. Wang, L. Liu and C. Lu, Procedia Comput. Sci., 2018, 131, 511–519 CrossRef; (c) J. M. Han, S. Guo, H. Lu, S. J. Liu, Q. Zhao and W. Huang, Adv. Opt. Mater., 2018, 6, 1800538 CrossRef; (d) T. Novikova, A. Pierangelo, S. Manhas, A. Benali, P. Validire, B. Gayet and A. D. Martino, Appl. Phys. Lett., 2013, 102, 241103 CrossRef; (e) B. Kunnen, C. Macdonald, A. Doronin, S. Jacques, M. Eccles and I. Meglinski, J. Biophotonics, 2015, 8, 317–323 CrossRef PubMed; (f) R. Carr, N. H. Evans and D. Parker, Chem. Soc. Rev., 2012, 41, 7673–7686 RSC; (g) L. E. MacKenzie and R. Pal, Nat. Rev. Chem., 2020, 5, 109–124 CrossRef.
- (a) F. Zinna and L. Di Bari, Chirality, 2015, 27, 1–13 CrossRef CAS PubMed; (b) E. R. Neil and D. Parker, RSC Adv., 2017, 7, 4531–4540 RSC; (c) S. Shuvaev, M. A. Fox and D. Parker, Angew. Chem., Int. Ed., 2018, 57, 7488–7492 CrossRef CAS PubMed; (d) F. Zinna, M. Pasini, F. Galeotti, C. Botta, L. Di Bari and U. Giovanella, Adv. Funct. Mater., 2017, 27, 1603719 CrossRef; (e) J. R. Jiménez, B. Doistau, C. M. Cruz, C. Besnard, J. M. Cuerva, A. G. Campaña and C. Piguet, J. Am. Chem. Soc., 2019, 141, 13244–13252 CrossRef PubMed; (f) J. R. Jiménez, M. Poncet, S. Miguez-Lago, S. Grass, J. Lacour, C. Besnard, J. M. Cuerva, A. G. Campaña and C. Piguet, Angew. Chem., 2021, 60, 10095–10102 CrossRef PubMed.
- (a) C. Dee, F. Zinna, W. R. Kitzmann, G. Pescitelli, K. Heinze, L. Di Bari and M. Seitz, Chem. Commun., 2019, 55, 13078–13081 RSC; (b) R. S. Dickins, J. A. K. Howard, C. L. Maupin, J. M. Moloney, D. Parker, J. P. Riehl, G. Siligardi and J. A. G. Williams, Chem. – Eur. J., 1999, 5, 1095–1105 CrossRef CAS; (c) C. L. Maupin, R. S. Dickins, L. G. Govenlock, C. E. Mathieu, D. Parker, J. A. G. Williams and J. P. Riehl, J. Phys. Chem. A, 2000, 104, 6709–6717 CrossRef CAS; (d) F. Zinna, L. Arrico and L. Di Bari, Chem. Commun., 2019, 55, 6607–6609 RSC; (e) B. Lefeuvre, C. A. Mattei, J. F. Gonzalez, F. Gendron, V. Dorcet, F. Riobé, C. Lalli, B. Le Guennic, O. Cador, O. Maury, S. Guy, A. Bensalah-Ledoux, B. Baguenard and F. Pointillart, Chem. – Eur. J., 2021, 27, 7362–7366 CrossRef CAS PubMed.
- (a) J. Gilot, R. Abbel, G. Lakhwani, E. W. Meijer, A. P. H. J. Schenning and S. C. J. Meskers, Adv. Mater., 2010, 22, E131–E134 CrossRef CAS PubMed; (b) Y. Yang, R. C. da Costa, M. J. Fuchter and A. J. Campbell, Nat. Photonics, 2013, 7, 634–638 CrossRef CAS; (c) Y. Yang, R. C. da Costa, D.-M. Smilgies, A. J. Campbell and M. J. Fuchter, Adv. Mater., 2013, 25, 2624–2628 CrossRef CAS PubMed; (d) J. R. Brandt, X. Wang, Y. Yang, A. J. Campbell and M. J. Fuchter, J. Am. Chem. Soc., 2016, 138, 9743–9746 CrossRef CAS PubMed; (e) S. Feuillastre, M. Pauton, L. Gao, A. Desmarchelier, A. J. Riives, D. Prim, D. Tondelier, B. Geffroy, G. Muller, G. Clavier and G. Pieters, J. Am. Chem. Soc., 2016, 138, 3990–3993 CrossRef CAS PubMed; (f) M. Schulz, M. Mack, O. Kolloge, A. Lutzen and M. Schiek, Phys. Chem. Chem. Phys., 2017, 19, 6996–7008 RSC; (g) P. Josse, L. Favereau, C. Shen, S. Dabos-Seignon, P. Blanchard, C. Cabanetos and J. Crassous, Chem. – Eur. J., 2017, 23, 6277–6281 CrossRef CAS PubMed; (h) J. R. Brandt, F. Salerno and M. J. Fuchter, Nat. Rev. Chem., 2017, 1, 0045 CrossRef CAS; (i) L. Frederic, A. Desmarchelier, L. Favereau and G. Pieters, Adv. Funct. Mater., 2021, 31, 2010281 CrossRef CAS; (j) K. Dhbaibi, L. Favereau, M. Srebro-Hooper, C. Quinton, N. Vanthuyne, L. Arrico, T. Roisnel, B. Jamoussi, C. Poriel, C. Cabanetos, J. Autschbach and J. Crassous, Chem. Sci., 2020, 11, 567–576 RSC; (k) K. Dhbaibi, L. Abella, S. Meunier-Della-Gatta, T. Roisnel, N. Vanthuyne, B. Jamoussi, G. Pieters, B. Racine, E. Quesnel, J. Autschbach, J. Crassous and L. Favereau, Chem. Sci., 2021, 12, 5522–5533 RSC; (l) D. W. Zhang, M. Li and C. F. Chen, Chem. Soc. Rev., 2020, 49, 1331–1343 RSC; (m) L. Zhou, G. Xie, F. Ni and C. Yang, Appl. Phys. Lett., 2020, 117, 130502 CrossRef CAS; (n) B. Doistau, J. R. Jimenez and C. Piguet, Front. Chem., 2020, 8, 555 CrossRef CAS PubMed.
- (a) H. Tanaka, Y. Inoue and T. Mori, ChemPhotoChem, 2018, 2, 386–402 CrossRef CAS; (b) W.-L. Zhao, M. Li, H.-Y. Lu and C.-F. Chen, Chem. Commun., 2019, 55, 13793–13803 RSC; (c) L. Arrico, L. Di Bari and F. Zinna, Chem. – Eur. J., 2021, 27, 2920–2934 CrossRef CAS PubMed; (d) J. Greenfield, J. Wade, J. Brandt, X. Shi, T. Penfold and M. Fuchter, Chem. Sci., 2021, 12, 8589–8602 RSC.
- (a) G. Longhi, E. Castiglioni, J. Koshoubu, G. Mazzeo and S. Abbate, Chirality, 2016, 28, 696–707 CrossRef CAS PubMed; (b) H. Tanaka, M. Ikenosako, Y. Kato, M. Fujiki, Y. Inoue and T. Mori, Commun. Chem., 2018, 1, 38 CrossRef; (c) C. Schaack, L. Arrico, E. Sidler, M. Gorecki, L. Di Bari and F. Diederich, Chem. – Eur. J., 2019, 25, 8003–8007 CrossRef CAS PubMed; (d) Y. Liu, Q. Xu, J. Sun, L. Wang, D. He, M. Wang and C. Yang, Spectrochim. Acta, Part A, 2020, 239, 118475 CrossRef CAS PubMed; (e) K. Tani, R. Imafuku, K. Miyanaga, M. E. Masaki, H. Kato, K. Hori, K. Kubono, M. Taneda, T. Harada, K. Goto, F. Tani and T. Mori, J. Phys. Chem. A, 2020, 124, 2057–2063 CrossRef CAS PubMed.
- (a) J. N. Wilson, W. Steffen, T. G. McKenzie, G. Lieser, M. Oda, D. Neher and U. H. F. Bunz, J. Am. Chem. Soc., 2002, 124, 6830–6831 CrossRef CAS PubMed; (b) Y. Geng, A. Trajkovska, S. W. Culligan, J. J. Ou, H. M. P. Chen, D. Katsis and S. H. Chen, J. Am. Chem. Soc., 2003, 125, 14032–14038 CrossRef CAS PubMed; (c) H. Tsumatori, T. Nakashima and T. Kawai, Org. Lett., 2010, 12, 2362–2365 CrossRef CAS PubMed; (d) D. Di Nuzzo, C. Kulkarni, B. Zhao, E. Smolinsky, F. Tassinari, S. C. J. Meskers, R. Naaman, E. W. Meijer and R. H. Friend, ACS Nano, 2017, 11, 12713–12722 CrossRef CAS PubMed; (e) M. Gon, R. Sawada, Y. Morisaki and Y. Chujo, Macromolecules, 2017, 50, 1790–1802 CrossRef CAS; (f) C. Kulkarni, D. Di Nuzzo, E. W. Meijer and S. C. J. Meskers, J. Phys. Chem. B, 2017, 121, 11520–11527 CrossRef CAS PubMed.
- (a) T. Zhao, J. Han, P. Duan and M. Liu, Acc. Chem. Res., 2020, 53, 1279–1292 CrossRef CAS PubMed; (b) L. Ji, Y. Sang, G. Ouyang, D. Yang, P. Duan, Y. Jiang and M. Liu, Angew. Chem., Int. Ed., 2019, 58, 844–848 CrossRef CAS PubMed; (c) J. Han, D. Yang, X. Jin, Y. Jiang, M. Liu and P. Duan, Angew. Chem., Int. Ed., 2019, 58, 7013–7019 CrossRef CAS PubMed; (d) D. Yang, P. Duan and M. Liu, Angew. Chem., Int. Ed., 2018, 57, 9357–9361 CrossRef CAS PubMed; (e) J. Han, P. Duan, X. Li and M. Liu, J. Am. Chem. Soc., 2017, 139, 9783–9786 CrossRef CAS PubMed; (f) A. Homberg, E. Brun, F. Zinna, S. Pascal, M. Górecki, L. Monnier, C. Besnard, G. Pescitelli, L. Di Bari and J. Lacour, Chem. Sci., 2018, 9, 7043–7052 RSC; (g) F. Zinna, E. Brun, A. Homberg and J. Lacour, in Circularly Polarized Luminescence of Isolated Small Organic Molecules, ed. T. Mori, Springer Singapore, Singapore, 2020, pp. 273–292, DOI:10.1007/978-981-15-2309-0_12.
- J. Wade, J. R. Brandt, D. Reger, F. Zinna, K. Y. Amsharov, N. Jux, D. L. Andrews and M. J. Fuchter, Angew. Chem., 2021, 60, 222–227 CrossRef CAS PubMed.
- X. Li, Y. Xie and Z. Li, Adv. Photonics Res., 2021, 2, 2000136 CrossRef.
- (a) K. Dhbaibi, L. Favereau, M. Srebro-Hooper, M. Jean, N. Vanthuyne, F. Zinna, B. Jamoussi, L. Di Bari, J. Autschbach and J. Crassous, Chem. Sci., 2018, 9, 735–742 RSC; (b) K. Dhbaibi, C. Shen, M. Jean, N. Vanthuyne, T. Roisnel, M. Górecki, B. Jamoussi, L. Favereau and J. Crassous, Front. Chem., 2020, 8, 237–237 CrossRef CAS PubMed; (c) R. Duwald, J. Bosson, S. Pascal, S. Grass, F. Zinna, C. Besnard, L. Di Bari, D. Jacquemin and J. Lacour, Chem. Sci., 2020, 11, 1165–1169 RSC; (d) J. Bosson, G. M. Labrador, C. Besnard, D. Jacquemin and J. Lacour, Angew. Chem., 2021, 60, 8733–8738 CrossRef CAS PubMed; (e) R. Tarrieu, I. Hernandez Delgado, F. Zinna, V. Dorcet, S. Colombel-Rouen, C. Crévisy, O. Basle, J. Bosson and J. Lacour, Chem. Commun., 2021, 57, 3793–3796 RSC; (f) J. Bosson, G. M. Labrador, S. Pascal, F. A. Miannay, O. Yushchenko, H. Li, L. Bouffier, N. Sojic, R. C. Tovar, G. Muller, D. Jacquemin, A. D. Laurent, B. Le Guennic, E. Vauthey and J. Lacour, Chem. – Eur. J., 2016, 22, 18394–18403 CrossRef CAS PubMed; (g) I. H. Delgado, S. Pascal, A. Wallabregue, R. Duwald, C. Besnard, L. Guenee, C. Nancoz, E. Vauthey, R. C. Tovar, J. L. Lunkley, G. Muller and J. Lacour, Chem. Sci., 2016, 7, 4685–4693 RSC; (h) R. Duwald, S. Pascal, J. Bosson, S. Grass, C. Besnard, T. Bürgi and J. Lacour, Chem. – Eur. J., 2017, 23, 13596–13601 CrossRef CAS PubMed.
- (a) J. Feng, L. Fu, H. Geng, W. Jiang and Z. Wang, Chem. Commun., 2020, 56, 912–915 RSC; (b) J. Jiménez, C. Díaz-Norambuena, S. Serrano, S. C. Ma, F. Moreno, B. L. Maroto, J. Bañuelos, G. Muller and S. de la Moya, Chem. Commun., 2021, 57, 5750–5753 RSC.
- (a) T. Biet, T. Cauchy, Q. Sun, J. Ding, A. Hauser, P. Oulevey, T. Bürgi, D. Jacquemin, N. Vanthuyne, J. Crassous and N. Avarvari, Chem. Commun., 2017, 53, 9210–9213 RSC; (b) G. Fu, Y. He, W. Li, B. Wang, X. Lü, H. He and W.-Y. Wong, J. Mater. Chem. C, 2019, 7, 13743–13747 RSC.
- (a) Y. Zhang, Y. Wang, J. Song, J. Qu, B. Li, W. Zhu and W.-Y. Wong, Adv. Opt. Mater., 2018, 6, 1800466 CrossRef; (b) A. Zampetti, A. Minotto and F. Cacialli, Adv. Funct. Mater., 2019, 29, 1807623 CrossRef; (c) K. Zhang, T. Y. Wang, T. W. Wu, Z. M. Ding, Q. Zhang, W. G. Zhu and Y. Liu, J. Mater. Chem. C, 2021, 9, 2282–2290 RSC.
- K. Tuong Ly, R.-W. Chen-Cheng, H.-W. Lin, Y.-J. Shiau, S.-H. Liu, P.-T. Chou, C.-S. Tsao, Y.-C. Huang and Y. Chi, Nat. Photonics, 2017, 11, 63–69 CrossRef CAS.
- (a) L. Norel, M. Rudolph, N. Vanthuyne, J. A. G. Williams, C. Lescop, C. Roussel, J. Autschbach, J. Crassous and R. Réau, Angew. Chem., Int. Ed., 2010, 49, 99–102 CrossRef CAS PubMed; (b) N. Saleh, C. Shen and J. Crassous, Chem. Sci., 2014, 5, 3680–3694 RSC; (c) C. Shen, E. Anger, M. Srebro, N. Vanthuyne, K. K. Deol, T. D. Jefferson, G. Muller, J. A. G. Williams, L. Toupet, C. Roussel, J. Autschbach, R. Reau and J. Crassous, Chem. Sci., 2014, 5, 1915–1927 RSC; (d) E. Anger, M. Rudolph, L. Norel, S. Zrig, C. Shen, N. Vanthuyne, L. Toupet, J. A. G. Williams, C. Roussel, J. Autschbach, J. Crassous and R. Réau, Chem. – Eur. J., 2011, 17, 14178–14198 CrossRef CAS PubMed.
- Z. Domínguez, R. López-Rodríguez, E. Álvarez, S. Abbate, G. Longhi, U. Pischel and A. Ros, Chem. – Eur. J., 2018, 24, 12660–12668 CrossRef PubMed.
- (a) V. V. Sivchik, A. I. Solomatina, Y.-T. Chen, A. J. Karttunen, S. P. Tunik, P.-T. Chou and I. O. Koshevoy, Angew. Chem., Int. Ed., 2015, 54, 14057–14060 CrossRef CAS PubMed.
- 1 J PtP ∼ 4300 Hz values for phosphine ligand being trans to the N ligating atom, and 1JPtP ∼ 2050 Hz for the phosphine ligand trans to C. Some examples: (a) H. Samouei, M. Rashidi and F. W. Heinemann, J. Iran. Chem. Soc., 2014, 11, 1207–1216 CrossRef CAS; (b) M. S. Sangari, M. G. Haghighi, S. M. Nabavizadeh, A. Pfitzner and M. Rashidi, New J. Chem., 2018, 42, 8661–8671 RSC.
- (a) Y.-J. Kim, J.-I. Park, S.-C. Lee, K. Osakada, M. Tanabe, J.-C. Choi, T.-a. Koizumi and T. Yamamoto, Organometallics, 1999, 18, 1349–1352 CrossRef CAS; (b) A. Polo, J. Duran, R. Juanola, J. Real, J. Benet-Buchholz, M. Solà and A. Poater, New J. Chem., 2017, 41, 3015–3028 RSC.
- (a) D. Mendola, N. Saleh, N. Vanthuyne, C. Roussel, L. Toupet, F. Castiglione, T. Caronna, A. Mele and J. Crassous, Angew. Chem., Int. Ed., 2014, 53, 5786–5790 CrossRef CAS PubMed; (b) D. Mendola, N. Saleh, N. Hellou, N. Vanthuyne, C. Roussel, L. Toupet, F. Castiglione, F. Melone, T. Caronna, F. Fontana, J. Martí-Rujas, E. Parisini, L. Malpezzi, A. Mele and J. Crassous, Inorg. Chem., 2016, 55, 2009–2017 CrossRef CAS PubMed.
- (a) L. Di Bari, G. Pescitelli and P. Salvadori, J. Am. Chem. Soc., 1999, 121, 7998–8004 CrossRef CAS; (b) N. Berova, L. D. Bari and G. Pescitelli, Chem. Soc. Rev., 2007, 36, 914–931 RSC.
- (a) Y. Liu, H. Guo and J. Zhao, Chem. Commun., 2011, 47, 11471–11473 RSC; (b) Y. Y. Chia and M. G. Tay, Dalton Trans., 2014, 43, 13159–13168 RSC; (c) F. Geist, A. Jackel and R. F. Winter, Dalton Trans., 2015, 44, 3974–3987 RSC; (d) F. Geist, A. Jackel and R. F. Winter, Inorg. Chem., 2015, 54, 10946–10957 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1dt02184b |
| This journal is © The Royal Society of Chemistry 2021 |