
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEasy and fast in situ functionalization of exfoliated 2D black phosphorus with gold nanoparticles†
Salvatore
Moschetto
 *a,
Andrea
Ienco
b,
Gabriele
Manca
*b,
Manuel
Serrano-Ruiz
b,
Maurizio
Peruzzini
b,
Alessio
Mezzi
c,
Marco
Brucale
a,
Margherita
Bolognesi
*a and
Stefano
Toffanin
a
*a,
Andrea
Ienco
b,
Gabriele
Manca
*b,
Manuel
Serrano-Ruiz
b,
Maurizio
Peruzzini
b,
Alessio
Mezzi
c,
Marco
Brucale
a,
Margherita
Bolognesi
*a and
Stefano
Toffanin
a
aNational Research Council – Institute for the Study of Nanostructured Materials (CNR-ISMN), Via P. Gobetti, 101, 40129 Bologna, Italy. E-mail: salvatore.moschetto@ismn.cnr.it; margherita.bolognesi@cnr.it
bNational Research Council – Institute for the Chemistry of OrganoMetallic Compounds (CNR-ICCOM), Via Madonna del Piano 10, 50019 Sesto Fiorentino, Italy. E-mail: gabriele.manca@iccom.cnr.it
cNational Research Council - Institute for the Study of Nanostructured Materials (CNR-ISMN), Via salaria km 29.3, 00015 Monterotondo Stazione (Rome), Italy
First published on 2nd August 2021
Abstract
Heterostructures of single- and few-layer black phosphorus (2D bP) functionalized with gold nanoparticles (Au NPs) have been recently reported in the literature, exploiting their intriguing properties and biocompatibility for catalytic, therapeutical and diagnostic applications. However, a deeper insight on the structural and electronic properties at the interface of the 2D bP/Au NP heterostructure is still lacking. In this work, 2D bP is functionalized with Au nanoparticles (NPs) through in situ deposition–precipitation heterogeneous reaction. The smallest realized Au NPs have a diameter around 10 nm as revealed by atomic-force and scanning electron microscopy, and are partially positively charged as revealed by X-ray Photoelectron Spectroscopy (XPS). XPS, UV-vis and Raman spectroscopy, supported by density functional theory (DFT) calculations, confirmed that while the structural and electronic properties of 2D bP are overall preserved, a soft-pairing between P atoms at the surface of 2D bP and Au atoms at the surface of Au NPs occurs, leading to a partial charge transfer at the 2D bP/Au interface, with a positive charge being localized on the Au atoms directly bonded to 2D bP. DFT calculations also predicted a band gap lowering, by 0.8 eV, for phosphorene functionalized with a tetranuclear Au cluster. Larger effects are expected as the Au cluster nuclearity (and coverage) increases.
Introduction
Single- and few-layer black phosphorus (2D bP) is a promising novel 2D material that can be easily obtained by either micromechanical cleavage (Scotch tape method1,2) or liquid exfoliation3–10 of bulk bP, the most stable allotrope of elemental phosphorus at room temperature.11 Since its discovery, this new P-based 2D material has attracted enormous interest. In fact, it is endowed with a direct and tunable band gap ranging from 0.3 eV (bulk) up to 2.2 eV (monolayer) and a high carrier mobility (up to 6000 cm2 V−1 s−1),12 which prompted its application towards the production of nanoelectronic devices, such as field effect transistors (FETs).13–15 In any case, the material has to be protected in order to prevent its degradation toward oxygen and atmospheric moisture,16–18 although in recent years also some oxidized forms of 2D bP have attracted attention on them.19Additional intriguing properties can be obtained through the deposition of metal atoms, molecular fragments or nanoparticles on the surface, which may cause interesting structural and electronic variations.20 A systematic study through first-principles calculations on the binding energy, geometry, magnetic moment, and electronic structure of several metal adatoms adsorbed on 2D bP has been carried out, predicting that the immobilization of transition metal atoms on the surface of 2D bP is feasible, preserving its structural integrity.21–28 More recently, a detailed guideline has been provided for an efficient covalent functionalization of the exfoliated bP with transition metal fragments in different coordination modes taking into account all the possible electronic/steric features.29 An alternative way for the achievement of covalent functionalization could follow from the selective transfer of chalcogenide atoms (O, S) from suitable sources to the 2D bP material, in order to anchor the metal complexes and/or nanoparticles.30
On the other hand, many experimental studies report on the functionalization of 2D bP with metal nanoparticles (NPs) for a variety of applications including energy generation31 and conversion,32 sensing33 or catalysis.34 Much of the experimental work on heterostructures made of 2D bP and NPs deals with gold Au NPs generally addressing the biocompatibility of such systems,35 together with the properties of singlet oxygen generation ability and localized surface plasmon resonance (LSPR), for diagnostic or therapeutic36 or sensing applications.37,38 Other works deal with the synthesis of 2D bP/Au NP heterostructures and their catalytic applications.39 The efficiency of the system has been attributed to the coupling of the intrinsic catalytic activity of both 2D bP40 and Au NPs and to the prevented aggregation of NPs (typically resulting in inhibited catalytic performance), thus a synergic effect can be modulated through an accurate engineering of the heterostructure.39,41
However, in all the experimental works dealing with the decoration of 2D bP with Au NPs very little attention has been devoted to the detailed analysis of the structural and electronic properties at the interface of such heterostructure. In particular, the size control and the growth process of the metal clusters on top of the 2D bP surface is a key step for their application in different technological fields.42–44
This work aims at studying 2D bP functionalized in situ with Au NPs through a deposition–precipitation solid–liquid heterogeneous reaction starting from an Au(I) precursor, which upon reduction reaction may form small nanoparticles. In fact, it is known that a smaller size of Au NPs enhances their catalytic properties due to the larger surface area.45,46 In this view, we looked throughout in situ functionalization to obtain nm-sized NPs (diameter of ∼10 nm) and to eliminate any stabilizing agent that could interfere with the interaction with 2D bP. Moreover, the in situ functionalization provides a strong intimacy of contact between 2D bP and the metal-precursor, leading well-distributed and homogenous morphology at the nanoscale, which is optimal for investigating the 2D bP/Au interface. Spectroscopic (UV-vis absorption spectroscopy, X-ray photoelectron Spectroscopy, or XPS) and morphological characterization (atomic force microscopy, or AFM), supported by density functional theory (DFT) calculations, are herein reported to provide a deeper understanding on the cross-correlated electronic, structural and morphological characteristics of the 2D bP/Au NPs heterostructure.
Experimental
Materials
Bulk bP was synthesized following Lange's procedure.47 Chloro(dimethylsulfide)gold(I), [(Me2S)AuCl], and solvents (isopropanol, acetone, anhydrous chloroform) were purchased from Sigma-Aldrich. Si/SiO2 (with thermally grown SiO2, thickness 300 nm) and quartz substrates were washed with two cycles of acetone and two of isopropanol in ultrasonic bath before use.Samples preparation
We employed the micromechanical cleavage method with blue tape to exfoliate bulk bP to obtain few layers of 2D bP onto Si/SiO2 substrates or quartz. The substrates resulted covered on ≈4% of their total area by 2D bP flakes with mean lateral size of 1 μm and thickness ranging from 50 nm down to few nm.48 Quartz/2D bP and Si/SiO2/2D bP samples were functionalized with gold nanoclusters by dipping them in a 10−3 M solution of [(Me2S)AuCl] in anhydrous CHCl3. The functionalization was carried out under an inert atmosphere of N2, in the glove box, in the dark and at ambient temperature. Immersion times of 3, 18 and 72 hours were tested. Samples were finally gently rinsed with degassed and anhydrous chloroform.Samples characterization
Results and discussion
For the 2D bP functionalization with Au NPs, we choose the direct addition of an Au(I) complex solution on the surface of bP. The precursor complex, [(Me2S)AuCl], with one Cl− ligand and a labile dimethylsulfide ligand, easily reduces to gold NPs when added to a suspension of few layers 2D bP in CHCl3. We expressly avoided to use Au NPs stabilized with organic surfactants to avoid covering of the 2D bP surface by the surfactant through non-covalent interactions,48 as it occurs also for graphene,54,55 which could hinder the P-Au interaction. The solid–liquid heterogeneous reaction was carried out for a range of reaction times (see Experimental section), in order to find the best compromise between a good substrate coverage and minimum size of the NPs.Spectroscopic characterization
Raman spectroscopy was carried out on pristine 2D bP and on Au-functionalized samples (Fig. 1). Both spectra show the three typical Raman peaks of bP at 360, 435 and 460 cm−1, attributed to the A1g, B2g, and A2g vibrational modes, respectively.56–58 This confirms that the orthorhombic crystalline structure of 2D bP is maintained after Au-functionalization, at least in the atomic-thin layers of 2D bP under the functionalized surface. In fact, as reported in the literature, covalent surface functionalization of 2D bP causes only a slight weakening of all bP Raman features (in particular of the A1g mode) due to the partial disruption of intra-layer phosphorus bonding.59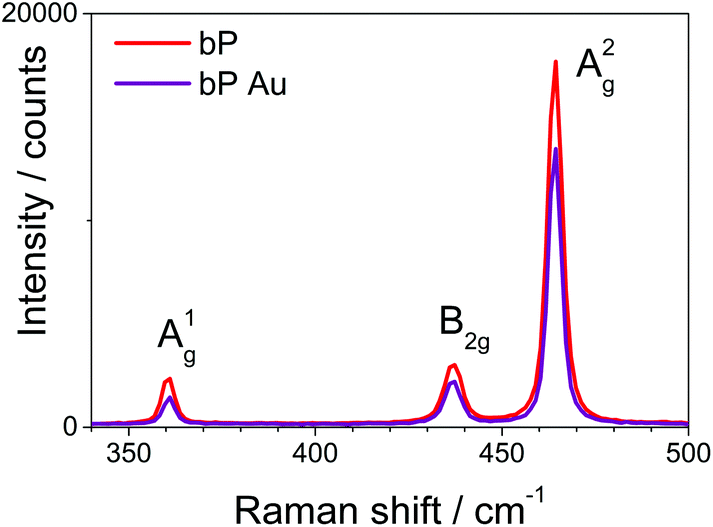 | ||
| Fig. 1 Raman spectra of pristine 2D bP (red) and Au-functionalized 2D bP (purple) on Si/SiO2 substrates. | ||
UV-vis spectroscopy on pristine 2D bP and Au-functionalized samples on quartz (incubation time 3 h, Fig. 2) resulted in broad and featureless absorption spectra for both samples, typical of bP. The expected peak showing plasmonic effects, (at about 520 nm, typical of Au NPs with size <10 nm as described in the following morphological characterization),32 is not present. This may be either ascribed to: (i) a low coverage of Au nanoclusters (ii) quenching by 2D bP, or, (iii) a combination of both factors. These assumptions are supported by literature, where it is supposed that the realization of 2D bP-anchored Au nanoclusters follows a kinetically controlled process,60 and quenching effects occur also for graphene and other 2D materials.61,62 Forming Au nanoclusters on the surface of 2D bP under experimental conditions featuring a low ratio between the Au precursor and the 2D bP (down to 2.5%) might enhance the collision probability between Au(I) atoms from the precursor and P centers from 2D bP, enabling an easier study of the heterostructure due to the large fraction of Au–P binding sites. However, we cannot exclude a-priori that the increase of the absorption spectrum of the 2D bP/Au-functionalized sample in the 400–475 nm spectral range is due to scattering effects.
 | ||
| Fig. 2 Absorption spectra of mechanically exfoliated pristine bP (black) and bP functionalized with gold NPs (red) on quartz substrates. | ||
As a surface-sensitive mass spectroscopic technique, XPS was used to shed light on the elemental composition, the stoichiometry, the chemical and electronic state of the elements present in our heterostructure systems. The XPS analysis of bulk bP, from which the 2D bP was exfoliated, evidenced the presence of elemental phosphorus characterized by the typical spin–orbit doublet P2p3/2–P2p1/2, separated by 0.9 eV, and positioned at BE = 130.0 eV (see Fig. S1 and Table S1 in the ESI†).
In the case of Au-functionalized sample (incubation time 3 h), the XPS measurements were performed on different areas of the sample. Since the lateral resolution of the XPS equipment is approximately of 20 μm, it was not possible to focus the X-ray beam on a single 2D bP flake, which, on average, presents typically shorter lateral dimension (see the optical microscopy images in Fig. S2 in the ESI†). However, it was possible to distinguish between two categories of regions, depending on their elemental composition, namely regions “A” and “B” respectively (see Table 1). In regions A, signals from Au and P atoms were found, while in regions B, signals from Au and Cl atoms were found. Both kind of regions were characterized also by signals from Si and O atoms, deriving from the silicon/silicon oxide substrate, and C atoms, ascribed to surface contamination with organic impurities or scotch tape residuals from the mechanical exfoliation process.
| Signal | Peak BE (eV) | Atomic % | Bond |
|---|---|---|---|
| Region A | |||
| Au4f7/2 | 84.3 | 0.3 | Au+ |
| C1s − 1 | 285.0 | 9.9 | C–C |
| C1s − 2 | 287.3 | 1.6 | C–O |
| O1s | 533.0 | 54.4 | SiO2 |
| P2p | 129.9 | 2.3 | P |
| Si2p | 103.6 | 31.6 | SiO2 |
| Region B | |||
| Au4f7/2 – 1 | 85.2 | 0.23 | Au+ |
| Au4f7/2 – 2 | 87.7 | 0.21 | Au3+ |
| C1s – 1 | 285.0 | 11.5 | C–C |
| C1s – 2 | 286.7 | 2.7 | C–O |
| C1s – 3 | 289.1 | 1.0 | –COOH |
| Cl2p3/2 | 199.2 | 1.2 | Cl− |
| N1s | 400.9 | 0.9 | Ammonium |
| O1s | 533.2 | 48.4 | SiO2 |
| Si2p – 1 | 103.9 | 32.7 | SiO2 |
| Si2p – 2 | 101.5 | 1.2 | Si–O |
The P 2p peak detected in the A regions was positioned at BE = 130.0 eV, characteristic for elemental P, as it was found in the pristine bP reference sample. Therefore, it can be attributed to 2D bP63 (Fig. 3). In addition, the Au 4f7/2 peak was positioned at BE = 84.4 ÷ 84.8 eV and it was assigned to Au arranged in NPs. As it is well known, the typical Au4f7/2 peak of bulk gold (Au0) is positioned at BE = 84.0 eV. Thus, the observed chemical shift strongly suggests an electronic interaction occurring between Au atoms and the 2D bP surface.
 | ||
| Fig. 3 XPS spectra, acquired in regions A and B of samples of 2D bP flakes functionalized with Au NPs on a Si/SiO2 substrate. | ||
In the B regions, where 2D bP was absent (no P 2p signal was detected), the Au 4f signal showed two doublets Au4f7/2–Au4f5/2, assigned to two different electronic configurations of Au centers (i.e. Au+ and Au3+).64 In the same point, also the Cl 2p signal was detected at peak position with BE = 199.2 eV. This value is characteristic for Cl−, most probably due to the presence of Cl− counteranions necessary to electronically stabilize Au+ and Au3+ atoms on the NPs surface in the absence of 2D bP.
Morphological characterization
Topographical AFM investigation of bare 2D bP before functionalization confirmed the expected planar and relatively featureless morphology (Fig. 4a) with occasional sharp steps of several Å (the smallest measuring 6.6 ± 1.0 Å, see Fig. 4e), suggesting the good quality of the 2D bP flakes prepared through the micromechanical exfoliation method. Controlled oxidation of 2D bP in air led initially to the formation on the surface of raised features similar to bubbles, which progressively grew in size upon further oxidation (see Fig. S3†). The oxidation bubbles were clearly discernible from the undamaged substrate via the Peak Force phase contrast, which attests a substantial difference between the nanomechanical characteristics of pristine bP and oxidized zones. | ||
| Fig. 4 (a–d): representative AFM micrographs of bP flakes immediately after mechanical cleavage (a) and after incubation with [(Me2S)AuCl] for 3 h (b), 18 h (c) and 72 h (d). All scale bars are 500 nm. Insets of panel (c) and (d) show 4× magnifications of the larger Au clusters found on the corresponding sample. (e): height profile of a typical step found on pristine bP measured along the dashed line shown in panel (a). (f–h): maximum vertical size distributions of Au NPs and clusters with respect to the BP plane in samples incubated for 3 h (f), 18 h (g) and 72 h (h). | ||
Fig. 4b, c and d show the surface morphology of 2D bP flakes incubated with [(CH3)2S]AuCl for respectively 3, 18 and 72 h. In all cases, the incubation process did not induce appreciable oxidation damage on bP (as it is observed instead for 2D bP in air, see Fig. S3†) thus the Au NPs coverage is preserved on the 2D bP (see for comparison bare 2D bP in Fig. S2b in the ESI†). The sample incubated for 3 h is largely decorated by isolated spherical Au NPs with an average size of = 10.2 ± 3.5 nm (Fig. 4f). Longer incubation times led to the formation of larger Au NPs clusters in addition to those observed on the sample incubated for 3 h. At 18 h, the average diameter of these larger aggregates is 27.0 ± 10.4 nm (Fig. 4g), while at 72 h the distribution further broadens to include aggregates up to ∼100 nm in diameter (Fig. 4h). All the larger Au clusters appear to be constituted by smaller grains having a diameter of ∼10 nm (see insets of Fig. 4c and d).
Considering all these observations, we can assess that the chosen functionalization procedure is effective in producing nm-sized NPs with a high affinity towards the 2D bP surface, in perfect accordance with the optical, Raman and XPS spectroscopic characterizations.
The morphology and size distribution of Au nanoclusters on the 2D bP surface were further confirmed by SEM results (Fig. S4†).
Computational studies
In order to shed light on the absorption sites, energy and bonding nature of the Au nanoclusters on 2D bP evidenced by spectroscopic and microscopic investigations, a detailed Density Functional Theory (DFT) computational study was carried out. The optimization of the single layer of the black phosphorus (monolayer 2D bP, or phosphorene) reproduced the experimental structure with two in-plane short P–P distances of 2.24 Å and one longer out of plane of 2.29 Å with a calculated channel width of 3.63 Å.The absorption of a single Au(0) atom on the phosphorene surface revealed a non-symmetric interaction between Au and three P centres, with one shorter (2.36 Å) and two longer (2.55 Å) bond lengths, caused by different reciprocal orientations of the lone pairs of P atoms.29 The absorption energy (Eabs) was calculated as large as −2.5 eV. This Eabs value is somewhat overestimated comparing to the one reported in the literature (−1.61 eV) due to the employment of different hybrid functionals (B3LYP rather than PBE0), whereas from a structural point of view the two results are in perfect agreement.21 In any case as shown in Fig. 5, the absorption of the metal atom causes a distortion of the structure, never put in evidence by the precedent investigation where the attention was focused only on the out of-plane P–P distance rather than on the width of the channels. In this regards, our calculations revealed channels wider by around 0.25 Å compared to the naked phosphorene (3.63 Å), a trend similar to what already obtained for the predicted functionalization of the phosphorene with AuCl fragment although the asymmetry in the Au–P bond is more pronounced.29 Otherwise in our calculations, the variation of the out-of-plane P–P distance (between P1–P4 atoms, Fig. 5) is only 0.03 Å larger compared to the naked phosphorene layer.
 | ||
| Fig. 5 Optimized structure of the black phosphorus monolayer after the absorption of a single gold atom. | ||
The Mulliken analysis revealed a +0.35 charge for the Au atom without any significant variations between the three involved P atoms. Recent publications revealed the oxidation of an adatom upon the interaction with the phosphorene surface for the case of a single copper atom, which donates its unpaired electron into the conduction band of the material.65 Our calculations highlighted an electronic population for the Au center of 0.49 e− less than the expected value for an Au atom at 0 oxidation state, suggesting a somewhat degree of electron donation of the metal towards the phosphorene layer.
On this basis, the adsorption on the 2D bP surface of the simplest seed of an Au nanocluster, namely two bonded Au atoms (Au1–Au2), was computed. For the Au1–Au2 assembly, two different arrangements on the 2D bP surface were investigated, as reported in Fig. 6.
 | ||
| Fig. 6 Optimized different configurations for the Au1-Au2 moiety on phosphorene: (a) parallel to the channel and (b) over the channel with a leaning structure (one gold on the channel and bonded to another Au, interacting with a fourth phosphorus of the surface), respectively. | ||
The optimization revealed that the most stable conformation is the one shown in Fig. 6b, with Eabs energy of −2.4 eV, more stable by 0.7 eV compared to the conformation parallel to the channel (Fig. 6a). From a structural viewpoint, the Au center in the channel (Au1) is now bonded to a P1 with a bond length of 2.28 Å and to the other Au center (Au2), with a metal-to-metal distance of 2.53 Å and P1–Au1–Au2 angle of 162°. The other two Au1–P bond distances elongate by 0.4 Å, as compared to the single Au atom on phosphorene, reaching a limit value of 2.91 Å. Differently from Au1, the Au2 center only weakly interacts with the surface through a single P atom being the Au2–P5 distance 2.76 Å. Such an interaction in any case causes a structural deformation since the P5–P6 bond results elongated up to 2.41 vs. 2.31 Å, similarly to the recent chalcogen atom transfer from a molecule of stibine chalcogenide to phosphorene.30
The absorption of the Au2 cluster on the phosphorene causes a less pronounced deformation being the ditch larger by only 0.18 Å. Concerning the charge distribution, also in this case a charge transfer is observed, especially from the Au1 center, which seems to be partially oxidized being its population 0.36 e− less than the atomic Au(0), while the Au2 remains very close to the Au(0) electronic configuration. The effect of the elongation of the P5–P6 bond is due to a somewhat donation of electron population from Au to the P–P σ* level being the P5–P6 overlap population halved than the pristine naked phosphorene.
In a scale-up approach, the interaction between a cluster made of four Au atoms (Au1,2,3,4) with the 2D bP surface was computed. Five alternative arrangements of the Au1, Au2, Au3, Au4 moiety were investigated, i.e. along, orthogonal or outside the channels. Some configuration were discharged since causing a partial distortion/disruption of the phosphorene crystallinity, which sharply contrasts with XPS results. The most stable configuration is the one shown in Fig. 7, featuring an adsorption energy as large as −3.7 eV. In this configuration, two Au atoms (Au1 and Au4) interact with two P ones (P1 and P5, respectively) with a short distance of 2.31 and 2.37 Å. The possible interaction of Au1 with other P atoms has to be discarded in view of their long distances (around 3.00 Å). While Au2 is only bounded to its neighboring Au1 and Au3 centers, an additional weak interaction is revealed between the gold center (Au3 or Au4) and the underlying phosphorene (bond lengths of 2.76 Å and 2.68 Å respectively). Interestingly, Au2, Au3 and Au4 centers attain an almost linear coordination with an angle of ca. 160°. As occurs for the binuclear Au1–Au2 also in this case an elongation of P5–P6 bond by ca. 0.14 Å is observed due to a somewhat degree of electron donation from the Au assembly into the σ* level.
 | ||
| Fig. 7 Optimized structure of phosphorene surface with Au4 cluster. | ||
From calculations, also a slight positive charge (+0.2) localized on the Au1 and Au4 atoms directly bonded to the P centers of phosphorene was revealed. This, together with the observation of the positive overlap population of the Au–P interactions, is in agreement with XPS measurements, indicating that Au NPs and nanoclusters present a fraction of Au atoms (most probably the superficial ones) in an oxidation state higher than 0.
Although the simulated cluster is far from being comparable, in dimensions, with the Au NPs experimentally obtained, the computed system was proven to be a good model for extrapolating energetic and structural information on the experimental one.
From an energetic point of view, simulations confirmed the propensity of 2D bP to bind the Au NPs by fixing a fraction of Au atoms at their periphery through a covalent bond. From a structural viewpoint, simulations also confirmed that the 2D bP crystalline structure is perfectly preserved after the adsorption of Au NPs. Finally, a partial charge transfer between P and Au atoms is revealed, with a positive charge being localized on the Au atoms directly bonded to the 2D bP. This may also explain the quenching of the plasmonic band of the Au NPs in the 2D bP/Au NPs heterostructure, as shown by absorption spectroscopy.
The adsorption of gold nanoclusters causes some significant electronic variation in particular concerning the band gap. In this respect, the naked phosphorene has an estimated direct band gap of 2.20 eV29 while the more is the metal content in the nanocluster and the smaller becomes the band gap. Such a behavior could be reasonably traced back to the presence of filled d orbitals of the metal at the top of the valence band. In a different way, the conduction band, mainly formed by the σ* P–P bond, is only slightly influenced in view of limited metal back-donation into empty P–P σ* bonding. Thus, the band gap becomes progressively smaller as the nuclearity of the particle increases. In this regards, the band gap starting from the 2.20 eV for the naked phosphorene reaches the lowest limit of 1.41 eV for phosphorene functionalized with the tetranuclear gold cluster. The greater is the size of the gold cluster and the smaller becomes the bandgap. The great contribution from the gold atoms at the top of the valence band is mainly attributed to gold–gold bond mediated by a significant contribution from the phosphorus, as shown by the Density of States (DOS) in ESI, Fig. S5 and S6† for the material functionalized with Au2 and Au4 moieties, respectively.
Conclusions
This work aimed at deeply investigating the electronic, structural and morphological properties of a heterostructure formed by 2D bP and Au NPs. The Au NPs, formed in situ through a solid–liquid heterogeneous reaction, have nanometric size (diameter ∼10 nm, ∼27 nm and ∼100 nm) as evidenced by AFM, and do not present plasmonic features due to quenching by 2D bP. In the heterostructure, 2D bP fully preserves its crystalline structure beneath the interface with Au NPs, as evidenced by Raman spectroscopy. The formation of the heterostructure involves the soft-pairing/coordination between P atoms from the surface of 2D bP to the Au atoms on the surface of the Au NPs. A partial charge transfer at the 2D bP/Au interface occurs, with a positive charge being localized on the Au atoms directly bonded to 2D bP, as predicted by calculations and confirmed by XPS experiments. Calculations finally predict, for Au-functionalized 2D bP, a band gap lowering which is more pronounced as the nuclearity of the Au clusters increases: i.e. a band gap decrease by 0.8 eV is calculated for phosphorene functionalized with a tetranuclear Au cluster. We expect that for a further increase of the cluster size, up to nanoparticles with >100 atoms, the more efficient is the charge transfer, as well as the smaller is the band gap of phosphorene. The results here presented give a theoretical insight and an overall deeper understanding of the interface in the heterostructures based on 2D bP/Au NPs, already widely used for optical and electrochemical sensing66 applications, or in plasmonic- or photonic- based biological applications.67 On the other hand, the specific heterostructure here designed and studied, obtained through an easy and in situ method leading to a “stabilizer-free” surface and optimal nanostructuring, holds promise for improved performance in specific applications, such as electro-, photo- or photoelectro-chemical catalysis, within others.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Mr. Franco Corticelli (CNR-IMM, Bologna) for the SEM-EDX measurements, and Mr. Vincenzo Ragona (CNR-ISMN, Bologna) and Dr. Federico Prescimone (CNR-ISMN) for the technical support. Thanks are expressed to the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (Grant Agreement No. 670173) for funding the project PHOSFUN by an ERC Advanced Grant to M.P.Notes and references
- L. Li, Y. Yu, G. J. Ye, Q. Ge, X. Ou, H. Wu, D. Feng, X. H. Chen and Y. Zhang, Nat. Nanotechnol., 2014, 9, 372–377 CrossRef CAS PubMed.
- H. Liu, A. T. Neal, Z. Zhu, Z. Luo, X. Xu, D. Tománek and P. D. Ye, ACS Nano, 2014, 8, 4033–4041 CrossRef CAS PubMed.
- M. Serrano-Ruiz, M. Caporali, A. Ienco, V. Piazza, S. Heun and M. Peruzzini, Adv. Mater. Interfaces, 2016, 3, 1500441 CrossRef PubMed.
- J. Kang, J. D. Wood, S. A. Wells, J.-H. Lee, X. Liu, K.-S. Chen and M. C. Hersam, ACS Nano, 2015, 9, 3596–3604 CrossRef CAS PubMed.
- P. Yasaei, B. Kumar, T. Foroozan, C. Wang, M. Asadi, D. Tuschel, J. E. Indacochea, R. F. Klie and A. Salehi-Khojin, Adv. Mater., 2015, 27, 1887–1892 CrossRef CAS PubMed.
- A. H. Woomer, T. W. Farnsworth, J. Hu, R. A. Wells, C. L. Donley and S. C. Warren, ACS Nano, 2015, 9, 8869–8884 CrossRef CAS PubMed.
- H. Wang, X. Yang, W. Shao, S. Chen, J. Xie, X. Zhang, J. Wang and Y. Xie, J. Am. Chem. Soc., 2015, 137, 11376–11382 CrossRef CAS PubMed.
- Z. Guo, H. Zhang, S. Lu, Z. Wang, S. Tang, J. Shao, Z. Sun, H. Xie, H. Wang, X.-F. Yu and P. K. Chu, Adv. Funct. Mater., 2015, 25, 6996–7002 CrossRef CAS.
- Z. Sun, H. Xie, S. Tang, X.-F. Yu, Z. Guo, J. Shao, H. Zhang, H. Huang, H. Wang and P. K. Chu, Angew. Chem., Int. Ed., 2015, 54, 11526–11530 CrossRef CAS PubMed.
- E. A. Lewis, J. R. Brent, B. Derby, S. J. Haigh and D. J. Lewis, Chem. Commun., 2017, 53, 1445–1458 RSC.
- C. E. Housecroft and A. G. Sharpe, Inorganic Chemistry, Pearson, Prentice Hall, 2nd edn, 2004, p. 392 Search PubMed.
- L. Li, F. Yang, G. J. Ye, Z. Zhang, Z. Zhu, W. Lou, X. Zhou, L. Li, K. Watanabe, T. Taniguchi, K. Chang, Y. Wang, X. H. Chen and Y. Zhang, Nat. Nanotechnol., 2016, 11, 593–597 CrossRef CAS PubMed.
- S. Das, W. Zhang, M. Demarteau, A. Hoffmann, M. Dubey and A. Roelofs, Nano Lett., 2014, 14, 5733–5739 CrossRef CAS PubMed.
- V. Tran, R. Soklaski, Y. Liang and L. Yang, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 235319 CrossRef.
- A. Castellanos-Gomez, J. Phys. Chem. Lett., 2015, 6, 4280–4291 CrossRef CAS.
- M. Bolognesi, S. Moschetto, M. Trapani, F. Prescimone, C. Ferroni, G. Manca, A. Ienco, S. Borsacchi, M. Caporali, M. Muccini, M. Peruzzini, M. Serrano-Ruiz, L. Calucci, M. A. Castriciano and S. Toffanin, ACS Appl. Mater. Interfaces, 2019, 11, 22637–22647 CrossRef CAS.
- E. Passaglia, F. Cicogna, G. Lorenzetti, S. Legnaioli, M. Caporali, M. Serrano-Ruiz, A. Ienco and M. Peruzzini, RSC Adv., 2016, 6, 53777–53783 RSC.
- G. Abellán, V. Lloret, U. Mundloch, M. Marcia, C. Neiss, A. Görling, M. Varela, F. Hauke and A. Hirsch, Angew. Chem., Int. Ed., 2016, 55, 14557–14562 CrossRef PubMed.
- S. Moschetto, M. Bolognesi, F. Prescimone, M. Brucale, A. Mezzi, L. Ortolani, M. Caporali, P. Pingue, M. Serrano-Ruiz, D. Pisignano, M. Peruzzini, L. Persano and S. Toffanin, ACS Appl. Nano Mater., 2021, 4, 3476–3485 CrossRef CAS.
- J. Plutnar, Z. Sofer and M. Pumera, ACS Nano, 2018, 12, 8390–8396 CrossRef CAS PubMed.
- V. V. Kulish, O. I. Malyi, C. Persson and P. Wu, Phys. Chem. Chem. Phys., 2014, 17, 992–1000 RSC.
- T. Hu and J. Hong, J. Phys. Chem. C, 2015, 119, 8199–8207 CrossRef CAS.
- Y. Jing, X. Zhang and Z. Zhou, WIREs Comput. Mol. Sci., 2016, 6, 5–19 CrossRef CAS.
- P. Rastogi, S. Kumar, S. Bhowmick, A. Agarwal and Y. S. Chauhan, arXiv:1503.04296 [cond-mat].
- L. Seixas, A. Carvalho and A. H. Castro Neto, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 155138 CrossRef.
- X. Sui, C. Si, B. Shao, X. Zou, J. Wu, B.-L. Gu and W. Duan, J. Phys. Chem. C, 2015, 119, 10059–10063 CrossRef CAS.
- H. Wang, S. Zhu, F. Fan, Z. Li and H. Wu, J. Magn. Magn. Mater., 2016, 401, 706–710 CrossRef CAS.
- M. Vanni, M. Serrano-Ruiz, F. Telesio, S. Heun, M. Banchelli, P. Matteini, A. M. Mio, G. Nicotra, C. Spinella, S. Caporali, A. Giaccherini, F. D'Acapito, M. Caporali and M. Peruzzini, Chem. Mater., 2019, 31, 5075–5080 CrossRef CAS.
- A. Ienco, G. Manca, M. Peruzzini and C. Mealli, Dalton Trans., 2018, 47, 17243–17256 RSC.
- A. Ienco, M. Peruzzini and G. Manca, Dalton Trans., 2020, 49, 15072–15080 RSC.
- C. J. An, Y. H. Kang, C. Lee and S. Y. Cho, Adv. Funct. Mater., 2018, 28, 1800532 CrossRef.
- J. Hu, Z. Guo, P. E. Mcwilliams, J. E. Darges, D. L. Druffel, A. M. Moran and S. C. Warren, Nano Lett., 2016, 16, 74–79 CrossRef CAS PubMed.
- A. Yang, D. Wang, X. Wang, D. Zhang, N. Koratkar and M. Rong, Nano Today, 2018, 20, 13–32 CrossRef CAS.
- M. Caporali, M. Serrano-Ruiz, F. Telesio, S. Heun, G. Nicotra, C. Spinella and M. Peruzzini, Chem. Commun., 2017, 53, 10946–10949 RSC.
- G. Qu, T. Xia, W. Zhou, X. Zhang, H. Zhang, L. Hu, J. Shi, X.-F. Yu and G. Jiang, Chem. Rev., 2020, 120, 2288–2346 CrossRef CAS PubMed.
- J. Wang, H. Zhang, X. Xiao, D. Liang, X. Liang, L. Mi, J. Wang and J. Liu, Acta Biomater., 2020, 107, 260–271 CrossRef CAS PubMed.
- D. Zhang, X. Lin, S. Lan, H. Sun, X. Wang, X. Liu, Y. Zhang and Y. Zeng, Part. Part. Syst. Charact., 2018, 35, 1800010 CrossRef.
- D. Huang, Z. Zhuang, Z. Wang, S. Li, H. Zhong, Z. Liu, Z. Guo and W. Zhang, Appl. Surf. Sci., 2019, 497, 143825 CrossRef CAS.
- Q. Wu, M. Liang, S. Zhang, X. Liu and F. Wang, Nanoscale, 2018, 10, 10428–10435 RSC.
- T. H. Lee, S. Y. Kim and H. W. Jang, Nanomaterials, 2016, 6, 194 CrossRef PubMed.
- R. Xu, J. Yang, Y. Zhu, H. Yan, J. Pei, Y. W. Myint, S. Zhang and Y. Lu, Nanoscale, 2015, 8, 129–135 RSC.
- G. J. Hutchings and M. Haruta, Appl. Catal., A, 2005, 291, 2–5 CrossRef CAS.
- D. C. Lim, I. Lopez-Salido and Y. D. Kim, Surf. Sci., 2005, 598, 96–103 CrossRef CAS.
- G. Barcaro and A. Fortunelli, New J. Phys., 2007, 9, 22–22 CrossRef.
- D. Latha, P. Prabu, G. Gnanamoorthy, S. Munusamy, S. Sampurnam, C. Arulvasu and V. Narayanan, SN Appl. Sci., 2018, 1, 134 CrossRef.
- P. Suchomel, L. Kvitek, R. Prucek, A. Panacek, A. Halder, S. Vajda and R. Zboril, Sci. Rep., 2018, 8, 4589 CrossRef.
- S. Lange, P. Schmidt and T. Nilges, Inorg. Chem., 2007, 46, 4028–4035 CrossRef CAS PubMed.
- M. Bolognesi, M. Brucale, A. Lorenzoni, F. Prescimone, S. Moschetto, V. V. Korolkov, M. Baldoni, M. Serrano-Ruiz, M. Caporali, F. Mercuri, E. Besley, M. Muccini, M. Peruzzini, P. H. Beton and S. Toffanin, Nanoscale, 2019, 11, 17252–17261 RSC.
- D. Nečas and P. Klapetek, Cent. Eur. J. Phys., 2012, 10, 181–188 Search PubMed.
- R. Dovesi, V. R. Saunders, C. Roetti, R. Orlando, C. M. Zicovich-Wilson, F. Pascale, B. Civalleri, K. Doll, N. M. Harrison, I. J. Bush, P. D'Arco, M. Llunell, M. Causà, Y. Noël, L. Maschio, A. Erba, M. Rerat and S. Casassa, CRYSTAL17, CRYSTAL17 User's Manual, University of Torino, Torino, 2017 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- R. Weihrich and I. Anusca, Z. Anorg. Allg. Chem., 2006, 632, 335–342 CrossRef CAS.
- T. Zhang, Z. Cheng, Y. Wang, Z. Li, C. Wang, Y. Li and Y. Fang, Nano Lett., 2010, 10, 4738–4741 CrossRef CAS PubMed.
- A. Ciesielski, S. Haar, M. El Gemayel, H. Yang, J. Clough, G. Melinte, M. Gobbi, E. Orgiu, M. V. Nardi, G. Ligorio, V. Palermo, N. Koch, O. Ersen, C. Casiraghi and P. Samorì, Angew. Chem., Int. Ed., 2014, 126, 10523–10529 CrossRef.
- S. Liu, N. Huo, S. Gan, Y. Li, Z. Wei, B. Huang, J. Liu, J. Li and H. Chen, J. Mater. Chem. C, 2015, 3, 10974–10980 RSC.
- J. Lin, L. Liang, X. Ling, S. Zhang, N. Mao, N. Zhang, B. G. Sumpter, V. Meunier, L. Tong and J. Zhang, J. Am. Chem. Soc., 2015, 137, 15511–15517 CrossRef CAS PubMed.
- Y. Feng, J. Zhou, Y. Du, F. Miao, C.-G. Duan, B. Wang and X. Wan, J. Phys.: Condens. Matter, 2015, 27, 185302 CrossRef PubMed.
- C. R. Ryder, J. D. Wood, S. A. Wells, Y. Yang, D. Jariwala, T. J. Marks, G. C. Schatz and M. C. Hersam, Nat. Chem., 2016, 8, 597–602 CrossRef CAS PubMed.
- H. Wu, X. Huang, M. Gao, X. Liao and B. Shi, Green Chem., 2011, 13, 651–658 RSC.
- V. Amendola, Phys. Chem. Chem. Phys., 2016, 18, 2230–2241 RSC.
- G. T. Forcherio and D. K. Roper, Adv. Opt. Mater., 2016, 4, 1288–1294 CrossRef CAS.
- W. Luo, D. Y. Zemlyanov, C. A. Milligan, Y. Du, L. Yang, Y. Wu and P. D. Ye, Nanotechnology, 2016, 27, 434002 CrossRef.
- S. Peters, S. Peredkov, M. Neeb, W. Eberhardt and M. Al-Hada, Surf. Sci., 2013, 608, 129–134 CrossRef CAS.
- S. P. Koenig, R. A. Doganov, L. Seixas, A. Carvalho, J. Y. Tan, K. Watanabe, T. Taniguchi, N. Yakovlev, A. H. Castro Neto and B. Özyilmaz, Nano Lett., 2016, 16, 2145–2151 CrossRef CAS PubMed.
- J. Xu, X. Qiao, Y. Wang, Q. Sheng, T. Yue, J. Zheng and M. Zhou, Microchim. Acta, 2019, 186, 238 CrossRef PubMed.
- H. Zhao, W. Zhang, Z. Liu, D. Huang, W. Zhang, B. Ye, G. Hu, H. Zhong, Z. Zhuang and Z. Guo, Nanophotonics, 2018, 7, 1651–1662 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1dt02123k |
| This journal is © The Royal Society of Chemistry 2021 |